

SUMMARY
The dynamic evolution of chromatin state patterns during metastasis, their relationship with bona fide genetic drivers, and their therapeutic vulnerabilities are not completely understood. Combinatorial chromatin state profiling of 46 melanoma samples reveals an association of NRAS mutants with bivalent histone H3 lysine 27 trimethylation (H3K27me3) and Polycomb repressive complex 2. Reprogramming of bivalent domains during metastasis occurs on master transcription factors of a mesenchymal phenotype, including ZEB1, TWIST1, and CDH1. Resolution of bivalency using pharmacological inhibition of EZH2 decreases invasive capacity of melanoma cells and markedly reduces tumor burden in vivo, specifically in NRAS mutants. Coincident with bivalent reprogramming, the increased expression of pro-metastatic and melanocyte-specific cell-identity genes is associated with exceptionally wide H3K4me3 domains, suggesting a role for this epigenetic element. Overall, we demonstrate that reprogramming of bivalent and broad domains represents key epigenetic alterations in metastatic melanoma and that EZH2 plus MEK inhibition may provide a promising therapeutic strategy for NRAS mutant melanoma patients.
Graphical abstract
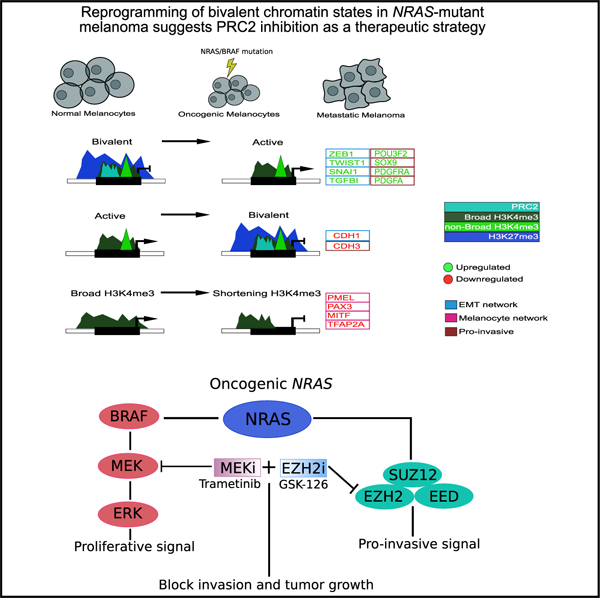
In brief
Terranova et al. provide a comprehensive epigenome resource for melanoma encompassing 284 chromatin maps. They find key regulatory roles for bivalent and broad domains in expression of pro-metastatic genes and identify EZH2 plus MEK inhibition as a therapeutic strategy for NRAS mutant melanomas.
INTRODUCTION
Melanoma is a deadly disease with an estimated 100,000 new cases each year in the United States (Lens and Dawes, 2004; Miller and Mihm, 2006). While targeted therapy and immunotherapy have become the standard of care with significant improvement in clinical response, thousands of patients still succumb to this disease per year due to primary or acquired resistance (Rosenberg et al., 2011; Tawbi et al., 2018).
Large-scale efforts from consortiums such as The Cancer Genome Atlas (TCGA) have provided deeper understanding of molecular aberrations in metastatic melanoma (Cancer Genome Atlas Network, 2015; Hodis et al., 2012; Tsao et al., 2012). These studies identified critical somatic mutations in this disease that likely occur due to UV exposure. Among these, somatic mutations in important bona fide oncogenes and tumor suppressors, such as BRAF, NRAS, NF1, INK/ARF, PTEN, and TP53, are well-chronicled drivers of this malignancy (Cancer Genome Atlas Network, 2015; Hodis et al., 2012; Tsao et al., 2012). One of the important findings from these studies was genetic aberrations in several key epigenetic regulators such as EZH2, IDH1/2, ARID2, KMT2C, and KMT2D (Cancer Genome Atlas Network, 2015; Hodis et al., 2012; Tsao et al., 2012), many of which are enzymes that control the covalent modifications of histones (Ceccarelli et al., 2016; Hodis et al., 2012; Ortega-Molina et al., 2015; Simon and Lange, 2008; Vallianatos and Iwase, 2015; Wu and Roberts, 2013). Although recent studies provide insight into the correlation of isolated chromatin modifiers and histone marks, there are a myriad of possible combinatorial patterns of histone modifications, and it is these combinatorial states—not individual modifications—that dictate epigenetic status of associated genomic loci (Lin et al., 2016; Lomberk et al., 2018; Roe et al., 2017). These observations suggest that epigenetic alterations, including those in histone modifications, may play important roles in melanoma progression. Indeed, specific functional roles have been assigned to histone variants such as macroH2A and H2A.z in melanoma (Kapoor et al., 2010; Vardabasso et al., 2015).
Taken together, these studies provide a strong rationale for systematic mapping of the epigenome to obtain a comprehensive understanding of regulatory elements that may act as driver events in particular melanoma tumors. This concept has been epitomized by DNA methylation studies in a large number of tumors profiled by TCGA. For example, a hypermethylation phenotype, termed CpG island methylation phenotype (CIMP), is a common epigenetic feature in various cancer types and tightly associated with critical driver gene mutations (i.e., IDH1) (Ceccarelli et al., 2016; Noushmehr et al., 2010; Weisenberger, 2014). Furthermore, other projects, such as ENCODE and Roadmap Epigenomics, have cataloged extensive histone modification in normal human tissues and cell lines. These studies provide critical information regarding tissue-specific regulatory elements, the link between relevant cell types and distinct human traits, and insight for evaluating the epigenetic basis of human diseases (ENCODE Project Consortium, 2012; Kundaje et al., 2015). Importantly, chromatin state mapping in a large number of tumors and cancer cell lines has the potential to enhance these concepts by identifying these regulatory features directly in a disease model (Corces et al., 2018). Since epigenetic aberrations are reversible by targeting their enzyme regulators, chromatin profiling can further identify therapeutic strategies in specific genetic or phenotypic contexts. For example, our previous study suggested histone deacetylase (HDAC) inhibitors could be a good strategy to block the initiation of pre-malignant to malignant melanoma in cell line systems (Fiziev et al., 2017).
During normal development histone modifications work in concert with other chromatin modifiers and transcription factors (TFs) to control the spatial and temporal regulation of gene expression patterns (Dambacher et al., 2010; Sarmento et al., 2004). In this context, the identity of each cell type, and its associated gene expression pattern, is maintained and subsequently inherited by daughter cells through mechanisms that do not alter the DNA sequence (Dambacher et al., 2010; Sarmento et al., 2004). Bivalent chromatin domains are characterized by the concurrent enrichment of active (i.e., Histone H3 lysine 4 trimethylation [H3K4me3]) and repressive (i.e., H3K27me3) histone modifications (Bernstein et al., 2006). During organogenesis, switches in bivalent domains can rapidly control the expression of critical lineage-specific genes that gain or lose these modifications as cells differentiate toward a particular phenotype (Bernstein et al., 2006; Voigt et al., 2013). In normal cells, bivalent marks are found at cancer-related genes (i.e., CDKN2A) with roles specific to the development of those tissues (Jadhav et al., 2016). Hence, it is plausible these genes were active during organogenesis, and an aberrant TF or epigenetic modification can leave them vulnerable to transcription in adult cells. Indeed, these studies have suggested tissue-specific mechanisms recruit histone methyltransferase Polycomb repressive complex 2 (PRC2) to place H3K27me3 at gene promoters in order to avoid unwanted mRNA expression (Jadhav et al., 2016). In melanoma tumors, H3K27me3 and Enhancer of zeste homolog 2 (EZH2) are predominantly expressed at the invasion front and have been linked to an invasive, epithelial-mesenchymal transition (EMT)-associated phenotype (Hoffmann et al., 2020). However, the role of bivalent H3K27me3 chromatin states and the involvement of PRC2 in metastatic melanoma have yet to be described.
In this study, we present a comprehensive chromatin state analysis of metastatic melanoma by performing chromatin immunoprecipitation sequencing (ChIP-seq) of 6 core histone modification marks using 46 melanoma tumors and cell lines. By integrative analysis of various omic datasets, we identify bivalent H3K27me3 and exceptionally wide H3K4me3 domains as potential drivers of a mesenchymal phenotype in metastatic melanoma, primarily in the NRAS mutant population. Mechanistically, the misappropriation of bivalent chromatin domains and increased levels of polycomb protein SUZ12 in NRAS mutant melanomas provide a plausible mechanism for the deregulation of pro-mesenchymal gene expression. Therapeutically, we demonstrate that the combination of EZH2 plus Mitogen-activated protein kinase kinase (MEK) inhibitors markedly reduces tumor burden in NRAS mutant cells, but not BRAF mutant cells. Thus, we provide evidence for genotype-dependent epigenome reprogramming in melanomas and show that disruption of PRC2 in combination with MEK inhibition may provide a promising therapeutic strategy for NRAS melanoma patients.
RESULTS
Bivalent H3K27me3 chromatin domains are enriched in metastatic melanoma
Chromatin state profiling remains a powerful tool for determining the regulatory status of annotated genes and identifying de novo elements in non-coding genomic regions (Kundaje et al., 2015). We identified enhancers (H3K27ac and H3K4me1), promoters (H3K4me3), actively transcribed loci (H3K79me2), polycomb silenced loci (H3K27me3), and heterochromatin (H3K9me3) elements in 46 melanoma samples by ChIP-seq using a high-throughput ChIP-seq protocol adapted for tumor tissues (Terranova et al., 2018). These constituted 20 metastatic melanoma tumors (TCGA [Cancer Genome Atlas Network, 2015]), 10 patient-derived melanoma short-term cultures (MSTCs; passage n < 10; profiled by internal effort at MD Anderson; unpublished data), and 16 established melanoma lines profiled by the Cancer Cell Line Encyclopedia/Sanger (CCLE) (Barretina et al., 2012) (Table S1). Using our cohort of 20 metastatic melanoma tumor samples, we computed multiple chromatin state models (2-state through 30-state models) with the ChromHMM algorithm (Figures 1A and S1A). We annotated an 18-state model since it is large enough to identify non-redundant functional elements encompassing canonical chromatin states that were not captured using lower state models (Figure S1A). These included such states as active promoters (E1); genic (E5, E6) and active enhancers (E7, E8) harboring high levels of H3K27ac and H3K4me1; and heterochromatic (E10) or polycomb (E14)-based repression harboring high levels of either H3K9me3 or H3K27me3, respectively (Figures 1A and S1B). We also observed two prominent bivalent/poised states: first, harboring both H3K4me3 and high levels of H3K9me3 (E12, annotated as “poised H3K9me3”); and second, H3K4me3 and high levels of H3K27me3 (E13, annotated as “poised H3K27me3”). Overall, the chromatin state profiles in metastatic melanoma tumors are associated with gene expression patterns (Figure 1B). As expected, active chromatin states (E1–E8) are associated with high gene expression levels, whereas repressed states (E10 and E15) are associated with low gene expression levels (Figure 1B).
Figure 1. Bivalent H3K27me3 chromatin states are enriched in metastatic melanoma.
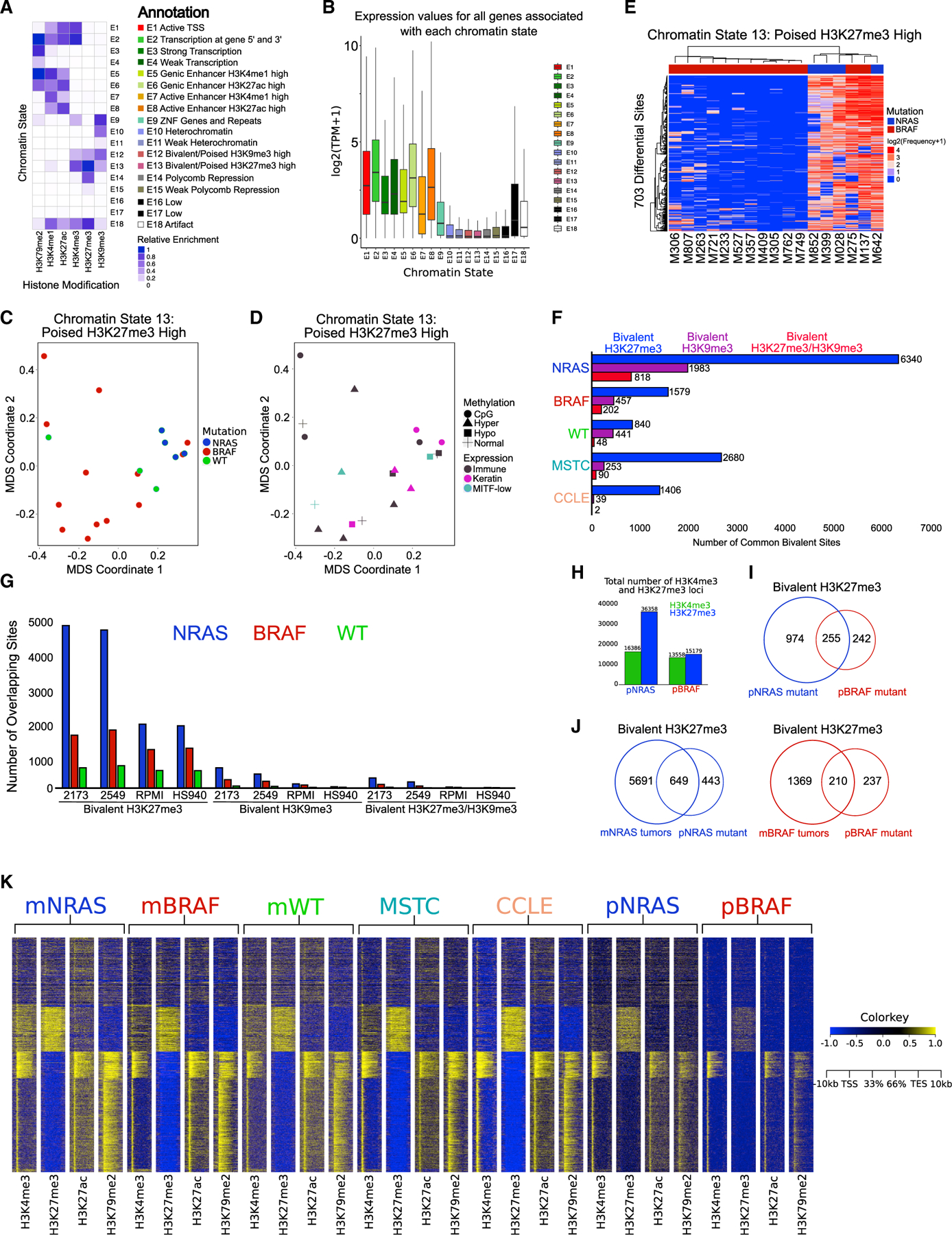
(A) Combinatorial chromatin state definitions and histone mark probabilities identified in 20 metastatic melanoma tumor samples using the ChromHMM algorithm.
(B) Boxplot illustrating mean gene expression levels from RNA-seq based on genomic regions overlapping with each chromatin state.
(C and D) MDS analysis of chromatin state E13 (poised H3K27me3 high) annotated by mutation (NRAS, BRAF, WT). (D) RNA expression (immune, keratin, MITF-low) and DNA methylation (normal, CpG, hyper, hypo) classifications from The Cancer Genome Atlas.
(E) Heatmap displaying differentially regulated regions (false discovery rate [FDR] < 0.05) of chromatin state E13 (poised H3K27me3 high) between NRAS and BRAF tumor subtypes. p values were calculated using a Wilcoxon test.
(F) Common bivalent H3K27me3 (H3K4me3 + H3K27me3), bivalent H3K9me3 (H3K4me3 + H3K9me3) and bivalent H3K27/H3K9me3 (H3K4me3 + H3K27me3 + H3K9me3) loci in melanoma tumor subtypes and melanoma short-term cultures (MSTCs) and CCLE lines.
(G) Co-occupancy analysis of common bivalent associated loci in melanoma tumor subtypes directly overlapping bivalent loci in representative MSTCs or CCLE lines.
(H) Barplot of H3K4me3 and H3K27me3 associated loci in isogenic mutant melanocytes harboring an NRAS (pNRAS) or BRAF (pBRAF) mutation.
(I) Venn diagram analysis of bivalent loci in pNRAS and pBRAF isogenic mutant melanocytes.
(J) Venn diagram analysis of bivalent loci in NRAS and BRAF mutant melanoma tumors with isogenic mutant melanocytes harboring an NRAS or BRAF mutation.
(K) Heatmap of H3K4me3 and H3K27me3 signal at −10 to +10 kb around transcription start sites (TSSs) of all ensemble genes in NRAS mutant melanoma tumors (mNRAS), BRAF mutant melanoma tumors (mBRAF), WT melanoma tumors (mWT), MSTCs, CCLE cell lines, and isogenic mutant melanocytes harboring an NRAS or BRAF mutation.
See also Figures S1–S3 and Table S1.
This TCGA tumor cohort is inclusive of well-described melanoma subgroups (Cancer Genome Atlas Network, 2015), including mutation (BRAF, NRAS, WT), transcriptomic (immune, keratin, Melanocyte inducing transcription factor [MITF]-low), and DNA methylation (CpG, hypermethlyated, hypomethylated, normal) subtypes (Figures S2A and S2B; Table S1). Projection of chromatin state data using multidimensional scaling (MDS) analysis revealed chromatin state E13 (poised H3K27me3) separated NRAS mutant tumors from BRAF mutant and WT samples in the first dimension (Figures 1C, 1D, and S2C). Moreover, differential analysis of bivalent chromatin states demonstrated NRAS enrichment for poised H3K27me3 domains (Figures 1E, S2D, and S2E). The identification of all potential bivalent combinations revealed a marked increase of bivalent H3K27me3, but not H3K9me3 or H3K27/H3K9me3, in tumor samples, MSTCs, CCLE lines, and isogenic mutant melanocytes harboring an NRASQ61K or BRAFV600E mutation (Fiziev et al., 2017; Garraway et al., 2005; Rai et al., 2015) (Figures 1F–1K and S3A–S3E). In accordance with our chromatin state analysis, NRAS mutants displayed the greatest number of bivalent H3K27me3 loci out of all the subgroups, whereas WT samples displayed the least number of bivalent loci on a global and meta-gene level (Figures 1F, 1G, and S3A–S3E). Similarly, NRASQ61K isogenic mutants (pNRAS) contained increased bivalent H3K27me3-associated loci compared with BRAFV600E mutants (pBRAF) (Figures 1H and 1I). Overlaps of bivalent domains in metastatic (m) mNRAS and mBRAF mutant tumors to those of isogenic mutants, as well as k-means clustering of H3K4me3 and H3K27me3 across all samples, showed enrichment of bivalent chromatin on both a genome-wide and meta-gene level (Figures 1J and 1K). Finally, we noted substantial overlaps between MSTC bivalent loci with the metastatic mNRAS and mBRAF mutant groups (Figures S3F and S3G), further demonstrating bivalent chromatin domains are enriched in NRAS mutant metastatic melanoma.
Bivalent domains are lost and gained on key mesenchymal genes
Previous studies have demonstrated various cancer-related genes (i.e., CDKN2A) maintain or acquire bivalent promoters in normal tissues (Jadhav et al., 2016); however, their role in melanoma progression has yet to be described. To determine how subtype-specific bivalent H3K27me3 losses and gains influence in melanoma progression, we calculated the overlaps of bivalent sites from mNRAS and mBRAF mutants to those in isogenic mutant melanocytes (pNRAS and pBRAF) and primary melanocytes (unmodified) from Roadmap data (Figures 2A and S3H–S3K). We observed a potential shift in bivalent domains as melanocytes progress toward melanoma, with a global decrease of bivalent loci in BRAF mutants and a global increase in NRAS mutants (Figures 2B and 2C). Average density analysis of these bivalent domains displayed low levels of active transcription marks (H3K27ac and H3K79me2) within these regions, further suggesting these domains are truly bivalent (Figures 2D–2F). We posited that removal of H3K27me3 mark from bivalent loci in melanocytes (“bivalent losses”) would lead to transcriptionally “active” loci in melanoma tumors, whereas gains of H3K27me3 mark on loci (“bivalent gains”) that harbor H3K4me3 in melanocytes (bivalent in tumors) will lead to transcriptional repression (Figure S3L). Determination of the gene targets (within −10 kilobases [kb] of TSSs to transcription end sites [TESs]) and subsequent pathway enrichment of bivalent gains and losses identified critical melanoma-associated “hallmark pathways” within each genetic subgroup (Figure 2G). For example, in NRAS mutants, losses of melanocyte-specific bivalency included genes associated with the “EMT” and “KRAS-signaling up,” while gains of tumor-specific bivalency included genes associated with “KRAS-signaling down” and “apical junction” (Figure 2G; Table S2). Apart from known activation of RAS-RAF pathway genes, various studies have demonstrated importance of mesenchymal driver genes in the invasive behavior of metastatic melanoma and other malignancies (Brabletz et al., 2018; Caramel et al., 2013; Kalluri and Weinberg, 2009).
Figure 2. Bivalent domains are lost and gained on mesenchymal genes.
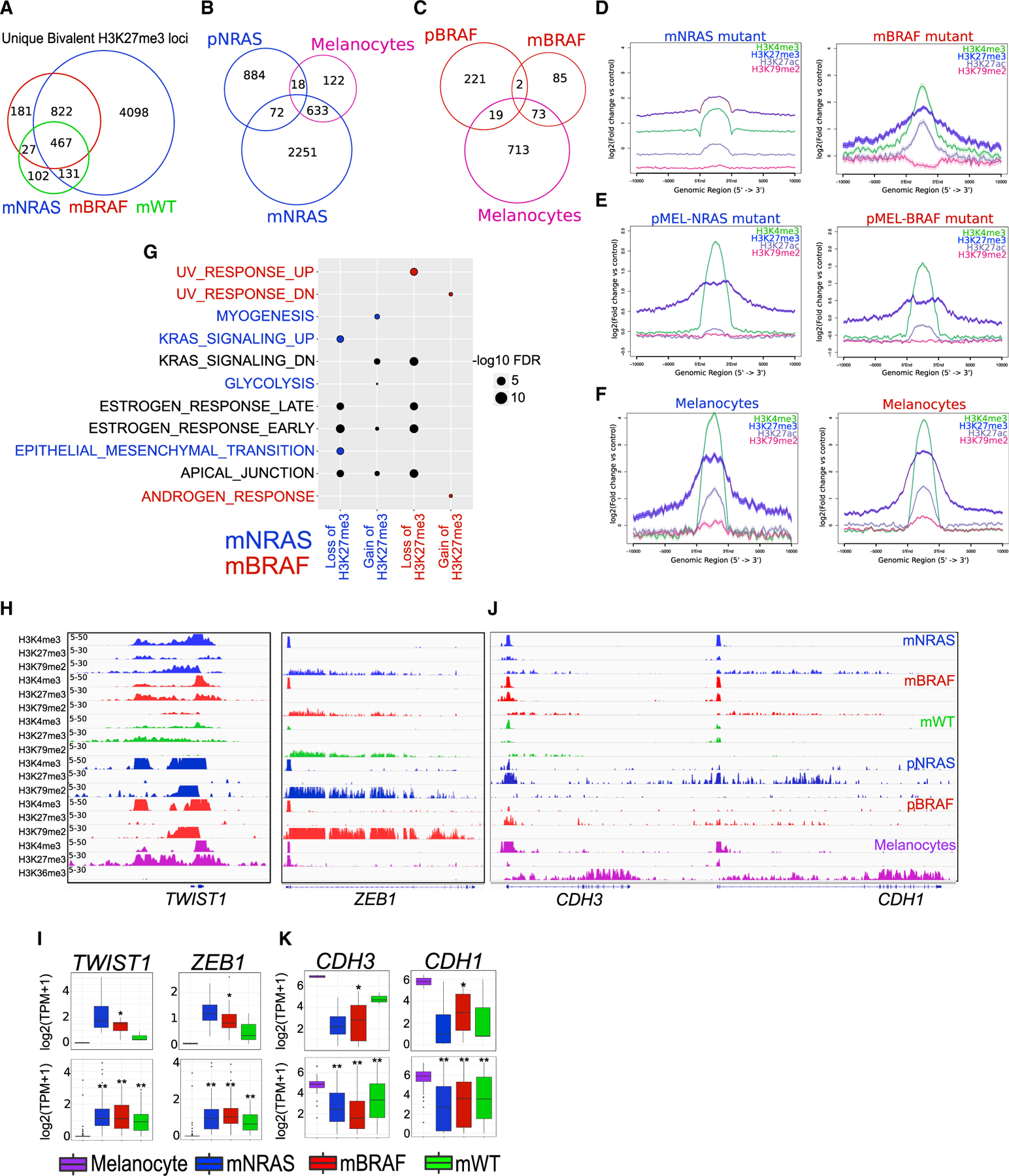
(A) Venn diagram analysis of bivalent H3K27me3 loci in mNRAS, mBRAF, and mWT tumor subtypes.
(B and C) Venn diagram analysis of unique bivalent H3K27me3 loci in mNRAS. (C) mBRAF tumor subtypes overlapping bivalent polycomb sites in isogenic mutant melanocytes and primary melanocytes from Roadmap.
(D–F) Average density profiles of H3K4me3, H3K27me3, H3K27ac, and H3K79me3 in mNRAS and mBRAF melanoma tumor subtypes. (E) Isogenic mutant melanocytes and (F) primary melanocytes from Roadmap.
(G) Top 5 significant MSigDB/GSEA HALLMARK pathways based on bivalent H3K27me3 loci that are lost and gained within ±10 kb TSS-TES of a gene in mNRAS and mBRAF tumor subtypes.
(H) Genome browser view of ChIP-seq tracks for H3K4me3, H3K27me3, and active transcription (H3K79me2/H3K36me3) on the TWIST1 and ZEB1 genes in mNRAS and mBRAF melanoma tumor subtypes, isogenic mutant melanocytes, and primary melanocytes from Roadmap.
(I) Boxplot displaying quantile normalized mean RNA expression profiles (log2 transcript per million [TPM]) of the TWIST1 and ZEB1 genes in melanocytes (n = 2) and melanoma tumor subtypes (NRAS = 4, BRAF = 13, WT = 3) with associated chromatin profiles (top) and in a large cohort of melanocytes (n = 86) and melanoma tumor subtypes (NRAS = 81, BRAF = 118, WT = 38 [bottom]). p values were calculated using a Wilcoxon test. *p < 0.05; **p < 0.0001.
(J) Genome browser view of ChIP-seq tracks for H3K4me3, H3K27me3, and active transcription (H3K79me2/H3K36me3) on the CDH3 and CDH1 genes in mNRAS and mBRAF melanoma tumor subtypes, isogenic mutant melanocytes, and primary melanocytes from Roadmap.
(K) Boxplot displaying quantile normalized mean RNA expression profiles (log2 TPM) of the CDH3 and CDH1 genes in melanocytes (n = 2) and melanoma tumor subtypes (NRAS = 4, BRAF = 13, WT = 3) with associated chromatin profiles (top) and in a large cohort of melanocytes (n = 86) and melanoma tumor subtypes (NRAS = 81, BRAF = 118, WT = 38 [bottom]). p values were calculated using a Wilcoxon test. *p < 0.05; **p < 0.0001.
See also Figures S3 and S4 and Table S2.
Identification of metastatic drivers that are subjected to bivalent regulation included key EMT-TFs ZEB1, TWIST1, SNAI1, and CDH1, which do not harbor genetic changes in melanoma (Figure S3M). On the TWIST1 and ZEB1 promoters, we found melanocytes harbor a bivalent configuration similar to that of embryonic stem cells (ESCs), which further switches to a transcriptionally active state upon isogenic mutation and remains active during metastasis (Figures 2H, 2I, S3N, S3O, S4A, and S4B). Genes such as SNAI1, however, continue to retain a bivalent configuration upon isogenic mutation and are transcriptionally active only in metastatic samples (Figures S3N and S3O). In contrast to bivalent losses, on genes such as CDH1 and CDH3 we found ESCs and melanocytes harbor transcriptionally active H3K4me3 and gain repressive bivalency upon isogenic mutation (Figures 2J, 2K, S4A, and S4B), suggesting EMT-TFs are dynamically regulated during melanoma progression. Importantly, these bivalentswitches are significantly correlated with mRNA expression levels in our primary cohort or in a large cohort of metastatic tumors from TCGA. Since bivalent domains in ESCs are known to be functional, allowing either rapid transcriptional activation or progression to stable silencing of gene expression, it is plausible these melanocyte-specific bivalent domains are functionally poised for activation.
A subset of melanocyte-specific bivalent genes transition to transcriptionally active broad H3K4me3
In contrast to typical H3K4me3 domains that are usually 200–1,000 bp long, broad H3K4me3 domains can span thousands of kilobases and have been implicated in various cellular processes, including increased gene expression, enhancer activity, and tumor-suppressor gene regulation (Benayoun et al., 2014; Chen et al., 2015; Dahl et al., 2016). Hence, we posited that genes losing bivalency in melanocytes may retain or acquire different signatures (broad or non-broad) of H3K4me3 in melanoma tumors. To this end, we systematically identified broad H3K4me3 domains by computing the overall width and density from MACS2 broad peaks in melanoma tumors and primary melanocytes (Figures 3A–3D). Globally, mNRAS and mBRAF mutant subtypes, as well as primary melanocytes, harbored the largest number of broad H3K4me3 peaks, in some cases spanning >30 kb (Figure 3A). We observed the broadest domains extended well beyond that of typical H3K4me3 (200–1,000 bp), with peaks reaching >4 kb in 15 of 17 of the individual samples (mNRAS and mBRAF) (Figures S4C and S4D). Separation of domains >4 kb to domains <4 kb revealed two distinct types of H3K4me3 in melanoma tumors, including broad domains spanning outside of the transcription start site (TSS) and non-broad domains localized within the promoter region (Figures 3E and S4E). Consistent with previous studies (Chen et al., 2015), we observed other epigenetic marks coincide with H3K4me3 domain width, in which active H3K27ac, H3K4me1, and H3K79me2 had similarly wide peaks at promoters harboring broad H3K4me3 (Figure 3E).
Figure 3. A subset of melanocyte-specific bivalent genes transition to transcriptionally active H3K4me3.
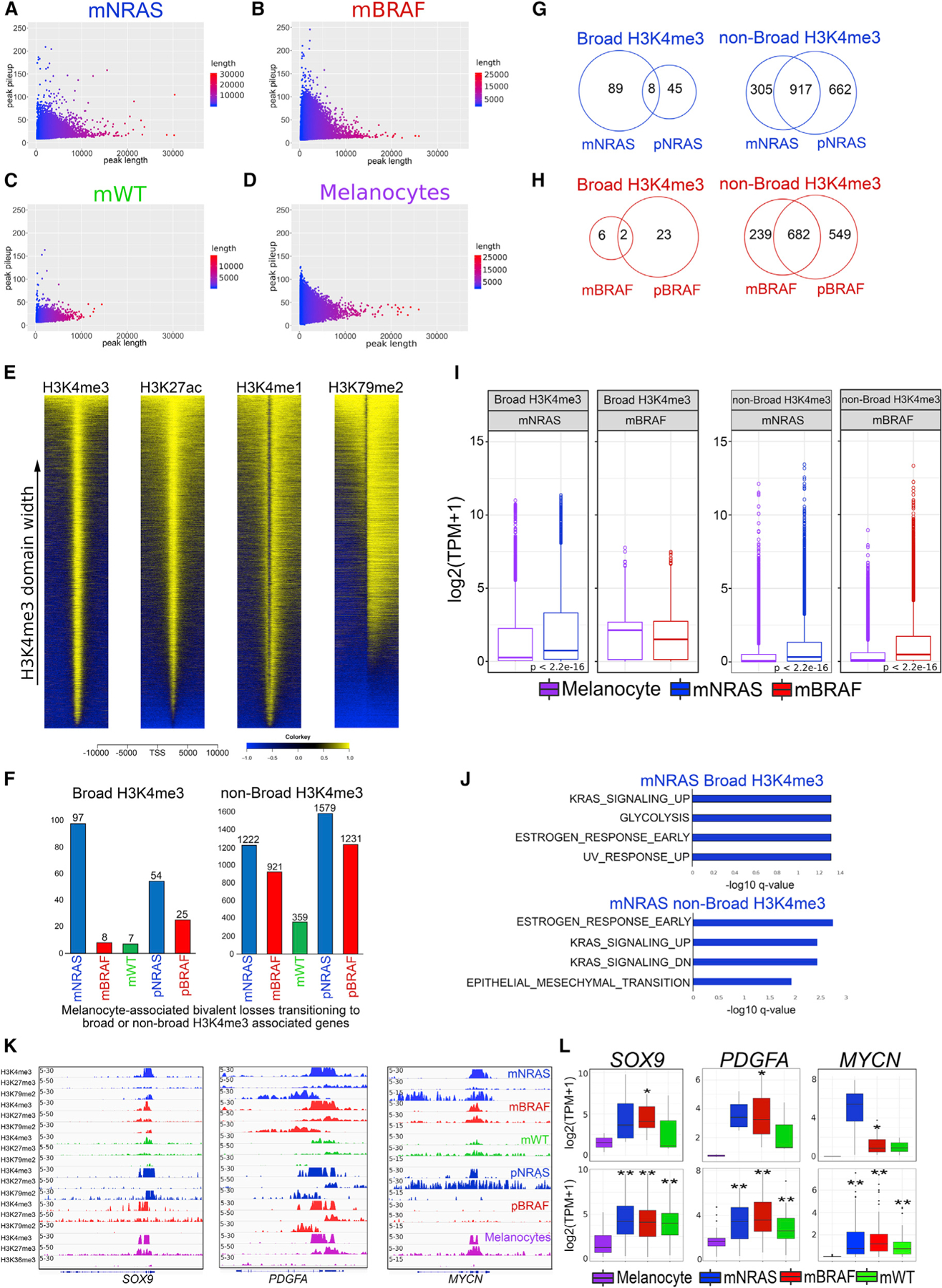
(A–D) Scatterplots of peak width (x axis) and height (y axis) from MACS2 broad peak calls (p value 1e–5) for H3K4me3 in mNRAS mutant tumors. (B) mBRAF mutant tumors, (C) mWT tumors, and (D) melanocytes from Roadmap.
(E) Heatmap of H3K4me3, H3K27ac, H3K4me1, and H3K79me2 signal at −10 to +10 kb around TSSs of ensemble genes based on H3K4me3 domain width in melanoma tumors.
(F) Barplot of bivalent H3K27me3 genes (±10 kb TSS-TES) that are lost in melanocytes and associated with broad or non-broad H3K4me3 in mNRAS, mBRAF, mWT melanoma tumor subtypes, and isogenic mutant melanocytes.
(G and H) Venn diagram of broad or non-broad H3K4me3 domains overlapping in mNRAS tumors and pNRAS melanocytes and (H) mBRAF tumors and pBRAF melanocytes.
(I) Boxplot displaying quantile normalized mean RNA expression profiles of mNRAS (n = 81) and mBRAF (n = 118) tumor subtypes and melanocytes (n = 86) based on broad or non-broad H3K4me3-associated genes identified in (G) and (H). p values were calculated using a Wilcoxon test.
(J) Top significant GSEA HALLMARK pathways based on bivalent H3K27me3 genes (±10 kb TSS-TES) that are lost in melanocytes and associated with broad or non-broad H3K4me3 domains in mNRAS tumor subtypes identified in (G).
(K) Genome browser views of ChIP-seq tracks for H3K4me3, H3K27me3, and active transcription (H3K79me2/H3K36me3) on the SOX9, PDGFA, and MYCN genes in mNRAS, mBRAF, mWT melanoma tumor subtypes, isogenic mutant melanocytes, and primary melanocytes from Roadmap.
(L) Boxplot displaying quantile normalized mean RNA expression profiles (log2 TPM) of the SOX9, PDGFA, and MYCN genes in melanocytes (n = 2) and melanoma tumor subtypes (NRAS = 4, BRAF = 13, WT = 3) with associated chromatin profiles (top) and in a large cohort of melanocytes (n = 86) and melanoma tumor subtypes (NRAS = 81, BRAF = 118, WT = 38 [bottom]). p values were calculated using a Wilcoxon test. *p < 0.05; **p < 0.0001.
To identify the subsets of melanocyte-specific bivalent genes that transition to transcriptionally active broad domains (and lose H3K27me3 in tumors), we further overlapped genes that harbor (1) bivalent domains uniquely in melanocytes (but not in tumors), (2) tumor-specific broad or non-broad H3K4me3 domains, (3) active transcription mark H3K79me2, and (4) gene expression (using TCGA RNA-seq data [Cancer Genome Atlas Network, 2015; Colaprico et al., 2016] from mNRAS [n = 81], mBRAF [n = 118], and mWT [n = 38] metastatic samples). Integrative analysis revealed mNRAS-specific broad domains were associated with increased gene expression (Figures 3F–3I; Table S3) and enriched for melanoma pathways such as “KRAS signaling up,” “UV response up,” and “Glycolysis” (Figure 3J). Genes displaying the transition to transcriptionally active broad H3K4me3 included critical metastatic drivers known to function in the switch to a mesenchymal/invasive state, including SOX9, PDGFA, PDGFRA, and MYCN (3.7 kb width) (Figures 3K, 3L, S4F, and S4G), suggesting a role for broad H3K4me3 domains in the regulation of mesenchymal genes in melanoma. In contrast to broad domains, we identified a relatively large and constant number of genes transitioning from a bivalent state to non-broad H3K4me3, many of which were shared in NRAS and BRAF mutants and associated with increased expression levels (Figures 3F–3I).
Broad H3K4me3 domain shortening correlates with transcriptional repression
Our results suggest that during the transition from a bivalent to an active state, a subset of genes that retain broad H3K4me3 domains are associated with increased transcriptional activation; however, this was only observed on a small number of genes (mNRAS = 89 and pNRAS = 45) (Figure 3G). We considered that another mechanism of gene activation could be spreading of the H3K4me3 signal, while gene repression may be associated with shortening of the broad H3K4me3 domains. On a genome-wide level and within gene-associated domains (±10 kb TSS), we observed preferential shortening (<2 kb) of H3K4me3 peaks in melanoma tumors relative to melanocytes (Figures 4A and 4B). The majority of genes that displayed shortening in metastatic tumors were shared with isogenic mutants and associated with a decrease in gene expression levels (Figures 4C–4E; Tables S4 and S5), suggesting broad H3K4me3 shortening is an early event in response to NRAS or BRAF oncogenic activation and a proxy for transcriptional activity. Promoters harboring some of the broadest H3K4me3 domains that displayed marked shortening and decreased gene expression included critical melanocyte-specific cell-identity genes, such as PMEL (also known as GP100), PAX3, MITF, and TFAP2A (Seberg et al., 2017) (Figures 4F, 4G, and S4H). During developmental progression, this broad H3K4me3 signature was not present in ESCs or germ layer cells (Figures 4H and 4I), indicating these domains are unique to fully differentiated somatic cells. Together, these results identify broad H3K4me3 domains as an epigenetic feature of melanoma progression.
Figure 4. Broad H3K4me3 domain shortening correlates with transcriptional repression.
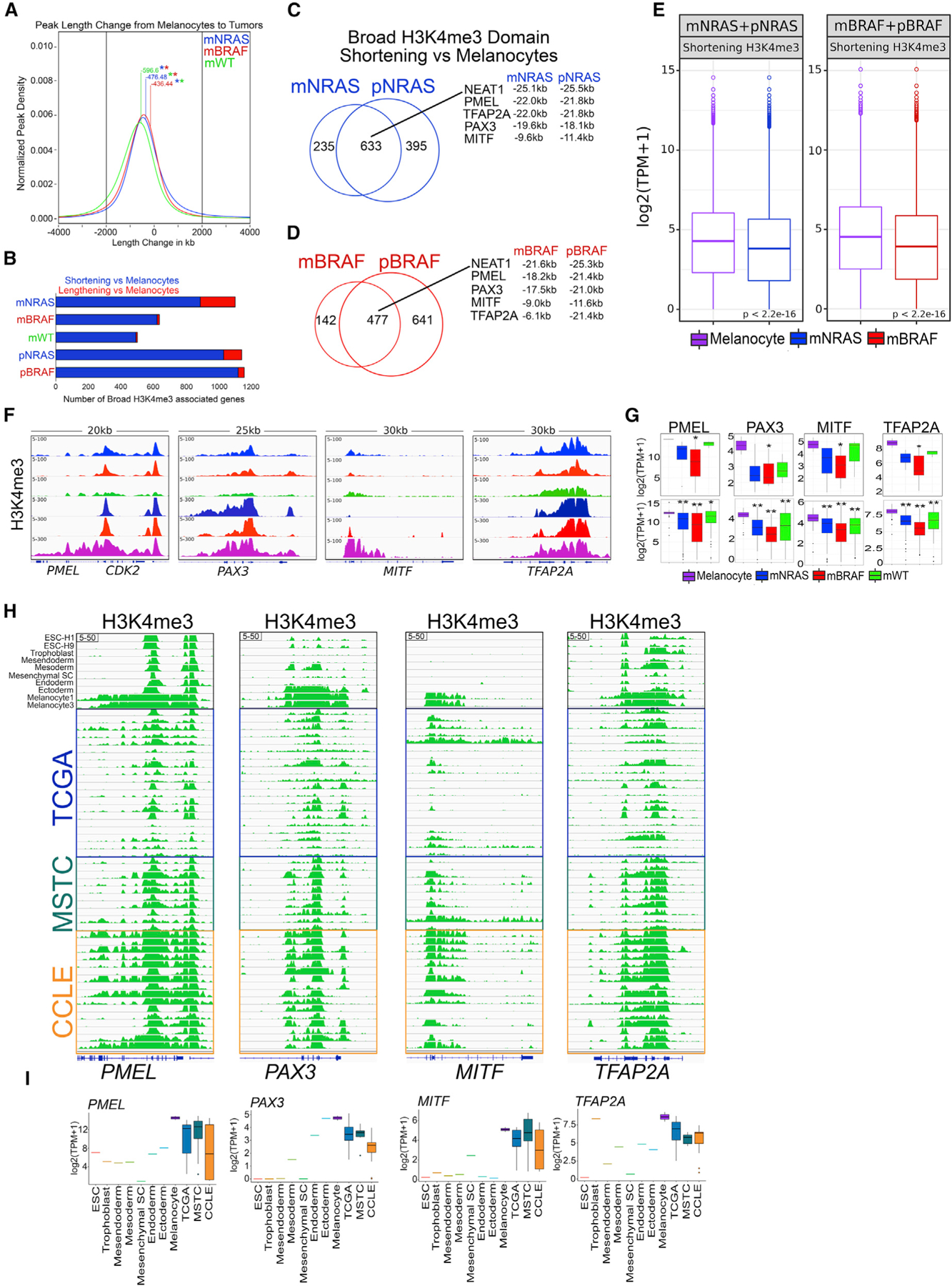
(A) Kernel density estimation plot displaying H3K4me3 peak length change (±2 kb) from melanocytes to melanoma tumor subtypes. Number denotes mean length change in kilobases between melanocytes and melanoma subtype. Asterisk denotes p value < 1e–50 between melanoma subtype length changes.
(B) Barplot of broad H3K4me3 promoter associated sites (−10 to +10 kb) displaying shortening or lengthening (±2 kb) in mNRAS, mBRAF, and mWT melanoma tumor subtypes and isogenic mutant melanocytes relative to primary melanocytes from Roadmap.
(C and D) Venn diagram analysis of broad H3K4me3 domains shortening (±2 kb) relative to melanocytes in mNRAS and pNRAS and (D) mBRAF and pBRAF samples.
(E) Boxplot displaying quantile normalized mean RNA-seq expression profiles from mNRAS (n = 81) and mBRAF (n = 118) melanoma tumor subtypes and melanocytes (n = 86) based on genes displaying shortening (±2 kb) of broad H3K4me3 domains identified in (C) and (D). p values were calculated using a Wilcoxon test.
(F) Genome browser view of ChIP-seq tracks displaying H3K4me3 on the PMEL, PAX3, MITF, and TFAP2A genes in mNRAS, mBRAF, and mWT melanoma tumor subtypes, isogenic mutant melanocytes, and primary melanocytes from Roadmap.
(G) Boxplot displaying quantile normalized mean RNA expression profiles (log2 TPM) of the PMEL, PAX3, MITF, and TFAP2A genes in melanocytes (n = 2) and melanoma tumor subtypes (NRAS = 4, BRAF = 13, WT = 3) with associated chromatin profiles (top) and in a large cohort of melanocytes (n = 86) and melanoma tumor subtypes (NRAS = 81, BRAF = 118, WT = 38 [bottom]). p values were calculated using a Wilcoxon test. *p < 0.05; **p < 0.0001.
(H) Genome browser view of ChIP-seq tracks for H3K4me3, on the PMEL, PAX3, MITF, and TFAP2A genes in ESC, trophoblast, mesendoderm, mesoderm, mesenchymal stem cells, endoderm, ectoderm, melanocytes from Roadmap and TCGA tumors, MSTCs, and CCLE cell lines.
(I) Boxplots displaying quantile normalized mean RNA expression profiles (log2 TPM) of the PMEL, PAX3, MITF, and TFAP2A genes in ESC, trophoblast, mesendoderm, mesoderm, mesenchymal stem cells, endoderm, ectoderm, melanocytes from Roadmap and TCGA tumors, MSTCs, and CCLE cell lines.
PRC2 complex regulates aberrant bivalent H3K27me3 domains
Trimethylation of H3K27 through an active PRC2 complex (EZH2, SUZ12, and EED) represses the expression of genes with acquired, tissue-restricted promoter bivalency (Jadhav et al., 2016). To determine whether PRC2 members are aberrantly expressed in metastatic melanoma, we first investigated their gene expression in tumor samples using TCGA RNA-seq data (Cancer Genome Atlas Network, 2015; Colaprico et al., 2016) from mNRAS (n = 81), mBRAF (n = 118), and mWT (n = 38). Here, NRAS mutant melanomas displayed increased expression of SUZ12 (p value = 0.037) compared with BRAF mutant samples (Figures 5A and S5A), and we further observed an increase of SUZ12 expression (p value = 0.053) in NRAS mutant melanoma in tumor dataset GSE15605 (Figures 5B and S5B) (Raskin et al., 2013). Furthermore, immunohistochemistry (IHC) of tissue microarrays (TMAs) harboring stages III and IV melanoma samples with annotated NRAS (patients = 7; samples = 14) and BRAF mutations (patients = 18; samples = 35) and western blotting analysis of MSTCs revealed that NRAS mutant, but not BRAF mutant, melanomas highly express SUZ12 (Figures 5C–5E and S5C–S5E). Consistently, we found that reducing NRAS levels in NRAS mutant MSTC-2125 using two different single guide RNAs (sgRNAs) decreased the expression of SUZ12, but not EZH2 (Figure 5F), further demonstrating an association between NRAS mutants and SUZ12 expression.
Figure 5. The PRC2 complex regulates aberrant bivalent H3K27me3 domains.
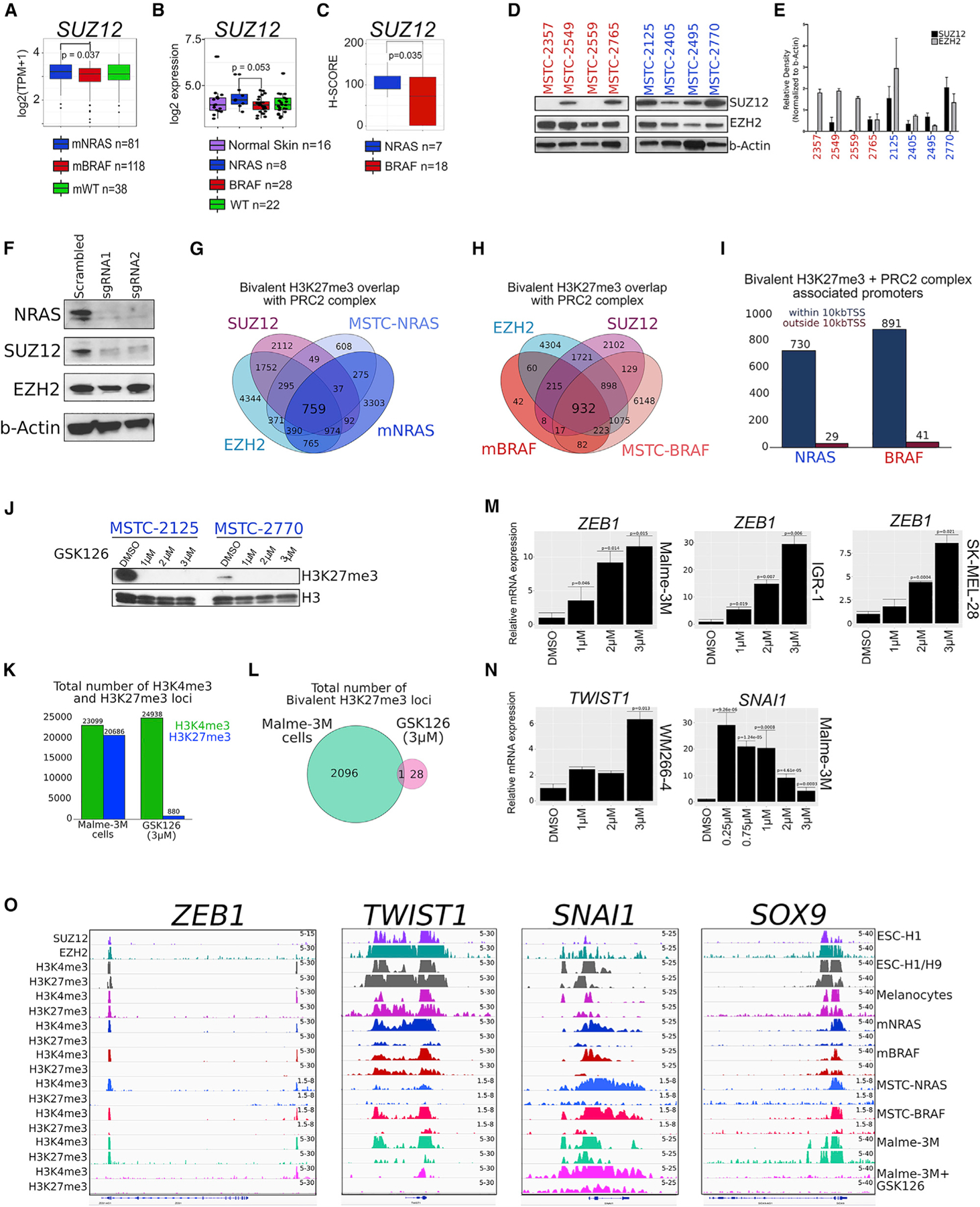
(A) Boxplot displaying quantile normalized mean RNA expression profiles (log2 TPM) for the SUZ12 gene in melanoma tumor subtypes (NRAS = 81, BRAF = 118, WT = 38) from the TCGA dataset.
(B) Boxplot displaying microarray profiles (log2) for the SUZ12 gene in melanoma tumor subtypes (normal skin = 16, NRAS = 8, BRAF = 28, WT = 22) from the GSE15605 dataset.
(C) Boxplot displaying H-SCORES from SUZ12 immunohistochemistry staining in patient TMA harboring an NRAS mutation (patients = 7; samples = 14) or BRAF mutation (patients = 18; samples = 35). p values were calculated using a t test based on H-SCORES in each mutational subtype.
(D and E) Western blotting analysis and (E) quantification for SUZ12 and EZH2 in MSTC harboring an NRAS or BRAF mutation. Relative density was quantified using ImageJ software, and the values of the target proteins were normalized to b-Actin. The results are representative of three independent biological replicates. Error bars represent mean ± SEM. NRAS and BRAF mutant MSTCs were run on the same gel and cropped for publication as two additional non-MSTCs were ran together.
(F) Western blotting analysis for NRAS, SUZ12, EZH2, and b-Actin in NRAS mutant MSTC-2125 expressing scrambled or two different sgRNAs targeting the NRAS gene.
(G and H) Venn diagram analysis of bivalent H3K27me3-associated loci in NRAS mutant and (H) BRAF mutant melanoma samples overlapping with PRC2 complex members SUZ12 and EZH2 in ESCs.
(I) Genomic localization of bivalent H3K27me3 + PRC2 complex associated loci.
(J) Western blotting analysis for H3K27me3 in MSTC harboring an NRAS mutation treated for 14 days with GSK-126 or DMSO.
(K) Barplot displaying total number of H3K4me3 and H3K27me3 associated loci in Malme-3M cells treated for 14 days with GSK-126 or DMSO.
(L) Venn diagram analysis of bivalent H3K27me3-associated loci in Malme-3M cells treated for 14 days with GSK-126 or DMSO.
(M) qRT-PCR for ZEB1 in Malme-3M, IGR-1, and SK-MEL-28 cell lines treated for 14 days with GSK-126 or DMSO. p values were calculated using a t test.
(N) qRT-PCR for TWIST1 WM2664 cell lines and SNAI1 in Malme-3M cell lines treated for 14 days with GSK-126 or DMSO. p values were calculated using a t test.
(O) Genome browser view of ChIP-seq tracks for SUZ12, EZH2, H3K4me3, and H3K27me3 in ESCs; H3K4me3 and H3K27me3 in melanocytes; mNRAS and mBRAF tumors; representative NRAS mutant MSTCs (MSTC-2125, MSTC-2770, MSTC-2495); and BRAF mutant MSTCs (MSTC-2549, MSTC-2765, MSTC-2357); and in Malme-3M cells treated for 14 days with GSK-126 or DMSO on the ZEB1, TWIST1, SNAI1, and SOX9 genes.
See also Figure S5.
We found that bivalent domains in melanoma associate with PRC2 members SUZ12 and EZH2, with 759 domains identified in NRAS mutants and 932 domains in BRAF mutants (Figures 5G and 5H). The majority of PRC2-associated bivalent domains localized within ±10 kb of a TSS (96.3% in NRAS mutants and 95.6% in BRAF mutants) (Figure 5I), suggesting these bivalent domains may be functional in regulating target gene expression. To examine the functional role of PRC2 in modulating bivalent H3K27me3-associated loci, we tested the impact of a potent EZH2 inhibitor, GSK-126 (McCabe et al., 2012; Verma et al., 2012), on these domains. As expected, EZH2 inhibition markedly decreased the global and site-specific levels of H3K27me3, as well as bivalent H3K27me3 associated loci, in both NRAS and BRAF mutant melanomas (Figures 5J–5L). The association of bivalent H3K27me3 domains with PRC2 and the disruption of H3K27me3-associated loci through EZH2 inhibition was observed on multiple EMT-TFs and critical developmental genes (Figures S5F–S5H). With these observations, we further postulated that bivalent domains may be functional in regulating the expression of EMT-TF genes in metastatic melanoma. In breast cancer, transforming growth factor β (TGF-β) enables the transition from a bivalent to active state at the ZEB1 promoter, a process associated with the removal of H3K27me3 (Chaffer et al., 2013). In melanoma, inhibiting H3K27me3 through GSK-126 treatment induced a dose-dependent increase of ZEB1 mRNA expression levels in multiple cell lines, including Malme-3M, IGR-1, and SK-MEL-28 (Figure 5M). An increase of the TWIST1 gene was further observed in WM266–4 cells in response to GSK-126 treatment, and SNAI1 was activated in Malme-3M cells, albeit this did not occur in a dose-dependent manner even at lower concentrations (Figure 5N). A dose-dependent increase of ZEB1 protein levels and an increase of SNAI1 protein levels were further observed in Malme-3M cells in response to GSK-126 treatment (Figure S5I). Consistently, we observed a loss or marked decrease of H3K27me3 on the ZEB1, TWIST1, SNAI1, SOX9, TGFBI, and PDFGRA promoters upon EZH2 inhibition (Figures 5O and S5F–S5H). With the ability of EZH2 inhibition to disrupt H3K27me3 on multiple EMT-TF genes harboring a bivalent domain, these data suggest genes involved in the switch to an invasive state may be regulated by shifts of bivalent chromatin in melanoma.
EZH2 and MEK combination therapy specifically abrogates tumor growth in NRAS mutant melanoma
Next, we sought to test the functional and therapeutic implications of our observations on PRC2-mediated increase in bivalent H3K27me3 levels in NRAS mutant melanomas. We first tested whether disruption of the PRC2 complex through EZH2 inhibition influenced the proliferative or invasive properties in NRAS mutant MSTC cells. While GSK-126 treatment (at 1, 2, and 3 µM) did not have a significant effect on cellular proliferation (Figures 6A and 6B), we observed a decrease in the invasive capacity of NRAS mutant cell lines (MSTC-2125 and MSTC-2770), but not in BRAF mutant cell lines (MSTC-2549 and MSTC-2765) (Figures 6C–6F). As expected, we observed loss in the global levels of H3K27me3 in both subtypes (Figures 5J, S6A, and S6B). Consistent with GSK-126 results, treatment of NRAS mutant cell lines with an additional EZH2 inhibitor, 3-deazaneplanocin A (DZNep) (Miranda et al., 2009), induced a global loss of H3K27me3 levels and a decrease in their invasive capacity (Figures S6C–S6E). This is consistent with the effects of bivalent domain reprogramming on mesenchymal driver genes. Since EZH2 inhibition abrogated invasion, but not proliferation, we tested whether EZH2 inhibitors could be combined with proliferation blocking US Food and Drug Administration (FDA)-approved MEK inhibitors to block tumor growth in melanoma. We treated tumors derived from two NRAS mutant cultures (MSTC-2125 and MSTC-2770) or BRAF mutant cultures (MSTC-2765 and MSTC-2549) in NUDE mice (n = 5 per condition) with EZH2 inhibitor GSK-126 (150 mg/kg), MEK inhibitor (1 mg/kg), or a combination of EZH2 plus MEK inhibitors. While monotherapy EZH2i or MEKi only showed modest effects, the combination treatment of EZH2i and MEKi drastically reduced the tumor burden of NRAS mutant melanoma cells (Figures 6G and 6H). By contrast, we did not observe a combinatorial effect of EZH2i plus MEKi on tumor volumes in BRAF mutant melanomas (Figures 6I and 6J). All of these treatments had minimal effects on the body weights of the treated mice (Figure S6F), demonstrating low toxicity from these combinatorial treatments. Importantly, we observed a significant decrease on tumor volume in BRAF mutants using BRAFV600E inhibitor vemurafenib (Figure S6G), demonstrating these samples are not inherently resistant to traditional therapies.
Figure 6. EZH2i and MEKi combination therapy decreases tumor burden in NRAS mutant melanoma.
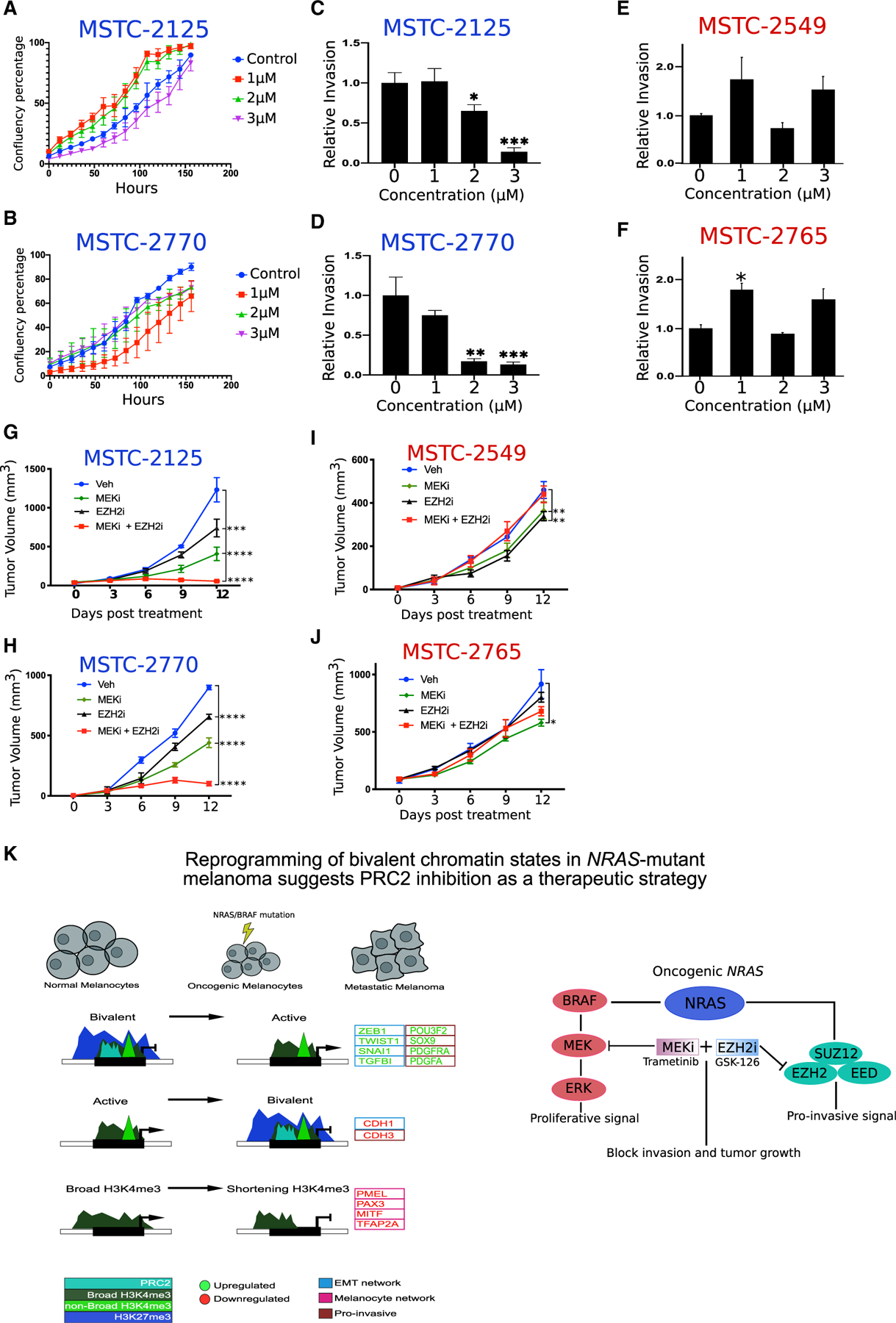
(A and B) Proliferation assay representing confluence percentage in NRAS mutant MSTC cell lines 2125 and (B) 2770 treated for 14 days with GSK-126 or DMSO. Error bars represent mean ± SEM.
(C and D) Boyden chamber invasion assay in NRAS mutant MSTC cell lines 2125 and (D) 2770 treated for 14 days with GSK-126 or DMSO. p values were calculated using a t test. *p < 0.05; **p < 0.01; ***p < 0.001. Error bars represent mean ± SEM.
(E and F) Boyden chamber invasion assay in BRAF mutant MSTC cell lines 2549 and (F) 2765 treated for 14 days with GSK-126 or DMSO. p values were calculated using a t test. *p < 0.05. Error bars represent mean ± SEM.
(G–J) Tumor volume curves for NRAS mutant melanoma cultures (G) MSTC-2125 and (H) MTSC-2770 or BRAF mutant melanoma cultures (I) MSTC-2549 and (J) MSTC-2765, upon treatment with vehicle, MEK inhibitors (trametinib), EZH2 inhibitor (GSK-126), and a combination of MEK and EZH2 inhibitors (trametinib + GSK-126) (n = 5 for each arm). p values represent pairwise t test comparison between the experimental arm to vehicle treatment. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Error bars represent mean ± SEM.
(K) Model explaining potential mechanism in which switches of bivalent H3K27me3 domains on EMT-TFs, and shortening of H3K4me3 domains on melanocyte-specific cell-identity genes, control their expression during the switch from a proliferative to an invasive phenotype in metastatic melanoma. EMT-TFs and melanocyte-specific genes identified here are differentially expressed in alternate phenotypic states, with ZEB1, TWIST1, SNAI1, SOX9, PDGFRA, and PDGFA displaying high expression in a mesenchymal/invasive phenotype and CDH1, MITF, PAX3, PMEL, and TFAP2A displaying high expression in melanocytes/proliferative phenotype.
See also Figure S6.
To determine whether EZH2 inhibition impacts invasion, we first checked the lung, liver, and lymph nodes for any nodules from all the mice that were injected with the EZH2 inhibitor or vehicle control in MSTC-2125 tumors. Here, we did not find any evidence for discernible micro or macro metastatic growth in distant organs potentially due to the duration of the experiment. To determine whether EZH2 inhibition impacts invasion on a molecular level, we further evaluated the protein levels of invasive marker vimentin using IHC in vehicle, EZH2, MEK, or EZH2i + MEKi-treated tumors. Interestingly, we observed a significant decrease in the number of vimentin-positive cells upon treatment with EZH2i or EZH2 + MEKi compared with vehicle control (Figures S6H and S6I), together suggesting that while we didn’t observe any metastatic events at this time frame, the molecular events promoting invasion and metastasis are impacted by EZH2 inhibition. Together, these results suggest inhibition of PRC2 in combination with MEKi may be a promising therapeutic strategy for NRAS mutant melanoma patients that harbor increased PRC2 activity and bivalent H3K27me3 domains.
DISCUSSION
Using combinatorial chromatin state profiling coupled with mutation, gene expression, and DNA methylation data, we find that a network of genes encompassing EMT-TF and melanocyte-specific cell-identity genes may be regulated by bivalent H3K27me3 and broad H3K4me3 domains in metastatic melanoma. Together, our work provides insight toward four important conceptual advances regarding the role of the epigenome in melanoma and its therapeutic implications (Figure 6K). (1) Genetic events such as NRAS or BRAF mutations may utilize specific chromatin states to bring about transcriptional changes unique to that genotype. (2) Chromatin state switches involving bivalent H3K27me3 domains occur on critical mesenchymal genes associated with a metastatic phenotype, including the EMT. (3) Exceptionally wide H3K4me3 domains spanning tens of kilobases associate with pro-metastatic drivers and melanocyte-specific cell-identity genes, suggesting roles for this epigenetic element in melanoma. (4) Blocking invasive ability of melanoma via EZH2 inhibitors along with proliferative features using MEK inhibitors may be a key therapeutic principle in NRAS mutant melanomas. Overall, the current study encompasses the most comprehensive dataset from a large number of well-annotated samples that is useful for identifying principles of epigenome regulation in melanoma metastasis and suggestions of epigenetic therapies.
Bivalent chromatin domains have primarily been described in ESCs, in which they control the expression of critical lineage-specific genes that gain or lose these modifications as cells differentiate toward a particular phenotype (Bernstein et al., 2006; Voigt et al., 2013). In normal cells, bivalent marks are also found at genes with roles specific to the development of those tissues. Hence, is it plausible these genes were active during organogenesis and an aberrant TF or epigenetic modification can leave them vulnerable to transcription in adult cells. In the context of melanocyte development, migratory neural crest cells undergo an initial EMT to break away from the neural fold and move to distant regions of the embryo (Baker et al., 1997; Sinnberg et al., 2018). This cellular plasticity, or phenotype switching, allows melanocytes to emerge amidst a myriad of other signaling pathways by quickly activating and repressing key regulatory genes, including EMT-TFs. In melanoma, the concept of phenotype switching (proliferative to invasive) has been reported, and various EMT-TFs are critical for this process (Caramel et al., 2013; Goding, 2000; Li et al., 2015). Although melanocytes are not part of the epithelial lineage, upon activation of BRAF or NRAS oncogenes, the EMT-TF network undergoes a reorganization that is associated with the upregulation of ZEB1 and TWIST1, the loss of CDH1, increased invasion, and poor prognosis in melanoma patients (Caramel et al., 2013).
In this study, we observed bivalent H3K27me3 domains display dynamic changes and associated gene expression patterns on core members of the EMT-TF network (ZEB1, TWIST1, SNAI1, TGFBI, CDH3, and CDH1) during the transition from ESCs > melanocytes > isogenic mutant melanocytes > metastatic melanoma. All of the core EMT-TF genes identified (with the exception of SNAI1) display losses and gains of bivalent H3K27me3 upon isogenic activation of NRAS and BRAF, indicating this switch occurs early in response to mitogen-activated protein kinase (MAPK) misregulation, and these genes remain active during metastasis. In addition to EMT-TFs, integration of melanocyte-specific bivalent genes that transition to transcriptionally active H3K4me3 domains (and lose H3K27me3 in tumors) revealed NRAS mutant-associated broad domains (>4 kb) were enriched on genes critical for neural crest migration and the switch to an invasive state (SOX9, POU3F2, MYCN, PDGFRA, and PDGFA) (Caramel et al., 2013; Cheng et al., 2015; Li et al., 2015; Simmons et al., 2017; Theveneau and Mayor, 2012), further indicating that melanoma tumor progression may resemble a reactivated developmental program mediated by bivalent switches. Since metastasis involves several cell-fate transitions during its progression from the primary site to colonization, which likely use a transcriptional circuitry consisting of different proliferative or invasive genes, it is plausible shifts involving bivalent chromatin states could be favored as the preferred mode of gene regulation for EMT-TFs during the switch to a metastatic phenotype.
In contrast to genes that lose bivalency and retain H3K4me3, the majority of broad H3K4me3 domains (>4 kb) displayed marked shortening in metastatic melanoma tumors relative to melanocytes. A similar observation in NRAS and BRAF isogenic mutant melanocytes suggests this is an early event in response to MAPK activation. However, unlike bivalent H3K27me3 domains, broad H3K4me3 shortening was not associated with a specific mutational subtype (NRAS or BRAF). Previous studies have demonstrated broad H3K4me3 domains are associated with increased gene expression and tumor-suppressor gene regulation (Benayoun et al., 2014; Chen et al., 2015; Dahl et al., 2016). Broad domains that are conserved across normal cells may mark a set of pan-cancer tumor suppressors, whereas cell-type-specific broad H3K4me3 peaks are associated with cell-identity genes (Chen et al., 2015). In melanoma, we found that promoters harboring some of the broadest H3K4me3 domains were associated with melanocyte cell-identity genes known to be downregulated during the switch to an invasive state, including PMEL (aka GP100), PAX3, MITF, and TFAP2A (Denecker et al., 2014; Goding, 2000; Kemper et al., 2014; Levy et al., 2006; Li et al., 2015; Seberg et al., 2017). The broad H3K4me3 signature was not present in ESCs or germ cells, but instead specifically gained in melanocytes (with the exception of PAX3 in ectodermal cells), suggesting broad H3K4me3 domains mark cell-identity genes necessary for normal melanocyte function that are lost during oncogenic activation. Importantly, further investigation is needed to determine whether histone methyltransferases responsible for regulating the broad H3K4me3 signal, such as KMT2D in medulloblastoma (Dhar et al., 2018), are functional in melanoma and can be targeted therapeutically.
Mechanisms governing bivalent promoters include histone methyltransferases such as PRC2 (EZH2, SUZ12, and EED) (Ku et al., 2008; Voigt et al., 2013). Studies in normal cells suggest tissue-specific mechanisms recruit PRC2 to place H3K27me3 at genes in order to avoid unwanted mRNA expression (Jadhav et al., 2016). In melanoma tumors, EZH2 and H3K27me3 are predominantly expressed at the invasion front and have been linked to an invasive, EMT-associated phenotype (Hoffmann et al., 2020; Shields et al., 2017; Zingg et al., 2017). In this study, we observed NRAS mutant melanomas (a highly metastatic and aggressive phenotype [Devitt et al., 2011; Jakob et al., 2012; Thumar et al., 2014]) harbor increased bivalent H3K27me3 domains and highly express core PRC2 members SUZ12 and EZH2. The high co-localization of bivalent H3K27me3 with SUZ12 and EZH2 on multiple EMT-TF genes suggests these domains may represent functional chromatin elements. Indeed, the ability to active the expression of EMT-TFs by modulating bivalent chromatin structure using EZH2 inhibitors demonstrates some of these domains are functional in cancer. Importantly however, further investigation is needed to determine what other active modifications or TFs regulate these genes, and whether their activation through bivalent modulation can drive an invasive phenotype.
Recently, epigenetic drugs targeting EZH2 have been successfully applied in various preclinical models and are rapidly progressing into clinical trials. For example, a recent clinical trial in patients with urothelial cancer demonstrated synergistic effect of EZH2 treatment and anti-PD1 therapy (NCT03854474). In melanoma cells, inhibition of EZH2 activity affects their invasive capacity and counteracts tumor growth and metastatic spread in vivo (Zingg et al., 2015). Here, we found that EZH2 inhibition (GSK-126) decreased H3K27me3 expression, reduced the invasive capacity, and markedly inhibited tumor burden specifically in NRAS mutant melanoma cultures, but not BRAF mutant cultures, in combination with MEK inhibitor. MEK inhibitor is an FDA-approved therapy in metastatic melanoma and remains a prominent therapy in NRAS mutant patients (Boespflug et al., 2017). However, the responses are limited in majority of the treated patients (Devitt et al., 2011; Hoffmann et al., 2020; Jakob et al., 2012; Thumar et al., 2014). Our data suggest that inhibition of proliferation by MEKi (Johnson and Sosman, 2013; Zipser et al., 2011) along with inhibition of the invasive/mesenchymal phenotype by EZH2i could be the biological explanation for the observed effect of combinatorial therapy in NRAS mutant melanoma. Thus, these observations provide a conceptual advance for design of future therapeutics to focus on targeting these two processes in NRAS mutant melanomas.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kunal Rai (krai@mdanderon.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The ChIP sequencing data has been deposited in the NCBI GEO BioProject database with the following accession number GSE134043. ChIP-seq data for histone modifications in ESCs, germ cells and melanocytes were downloaded from ftp://ftp.ncbi.nlm.nih.gov/pub/geo/DATA/roadmapepigenomics/. ChIP-seq data for SUZ12 and EZH2 was downloaded from the NCBI GEO Bio-Project database with the following accession number GSE29611. Code used in this study is available on https://rpubs.com/cjt5 and https://github.com/sccallahan/ChromXploreR.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
Melanoma tumors were obtained from the Melcore tumor bank at MD Anderson Cancer Center as per an approved Institutional Review Board protocol (IRB #PA12–0305). The details of the patient samples are available in Table S1.
Animals
Female mice (strains: Crl:NU(Ico)-Foxn1nu Charles river, strain code 620) were purchased, bred in house, and maintained at 72°F ± 2°F on a 12h light/dark cycle. Four- to six-week-old aged females were randomly assigned with five mice housed together per cage. Animals were provided ad libitium access to food and water. All the animal studies were performed as per an approved Institutional Animal Care and Use Committee protocol (IACUC #00001411-RN01).
Cell lines
Melanoma short term cultures were generated from metastatic tumor specimens as part of the Adoptive T cell Therapy Clinical Program at the University of Texas, MD Anderson Cancer Center (LAB06–0755 and 2004–0069), as previously described (Oba et al., 2018; Besser et al., 2009; Park et al., 2017). Briefly a tumor specimen from metastatic tumor was collected and incubated with an enzymatic digestion cocktail (0.375% collagenase type I, 75 µg/ml hyaluronidase and 250 U/mL DNase I) in tumor digestion medium (RPMI, 10 mM of HEPES, 1x Penicillin-Streptomycin and 20 ug/mL of Gentamicin; GIBCO/Invitrogen) in a humidified incubator at 37°C with 5% CO2 and with a gentle rotation for 2–3h to obtain a single cell suspension. The tumor digest was filtered through a 70-µm filter, washed, and re-suspended in a serum free media, which was then plated in one well of a 6-well culture plate and incubated at 37°C. After 24h, the media was replaced with fresh tumor media, comprised of RPMI with GlutaMAX, 10% FBS, Penicillin/Streptomycin, Gentamicin, β-mercaptoethanol (50 uM, GIBCO/Invitrogen), HEPES (10 mM), and insulin-selenium-transferin (5 ug/ml, GIBCO/Invitrogen). Cells were grown in enriched DMEM/F12 culture media (GIBCO/Invitrogen) supplemented with all growth factors including 10% FBS, sodium pyruvate (1mM), insulin-selenium-transferin, MycoZap-PR (Lonza), HEPES (10mM) and β-Mercaptoethanol. Once enough cells were grown, the purity of the tumor was tested using a melanoma tumor surface marker (MCSP-1) by flow cytometry. Cultures were deemed established when the cells stained positive for a melanoma tumor marker (MCSP-1) and negative for a fibroblast marker (CD90). Appropriate serum starving was performed to eliminate fibroblasts. All cell lines were tested for mycoplasma using MycoAlert detection kit (Lonza), and fingerprinted by STR fingerprinting, and validated by comparing with matched blood samples. A few passages after a pure tumor line was established, the cells were cryopreserved and kept in stocks in liquid nitrogen until use.
Isogenic mutant melanocytes
BRAFV600E or NRASQ61K melanocytes were generated as described in Fiziev et al. (2017), Garraway et al. (2005), and Rai et al. (2015). Briefly, normal melanocytes were immortalized by TERT expression and partially-transformed by overexpression of dominant negative TP53 and CDK4R24C. Lentiviral exogenous BRAFV600E or NRASQ61K derivatives were then expressed to generate isogenic melanocyte pairs.
METHOD DETAILS
Chromatin immunoprecipitation assays
ChIP assays were performed as described previously (Terranova et al., 2018). Briefly, for tumor samples 50mg of tissue (~7mg/IP) were manually dissociated in 2mL of Hanks Balanced Salt Solution (HBSS) using a sterile razor blade. An additional 8mL of HBSS was added and tissue was further disassociated using a MACS homogenizer with the follow cycles; h_tumor_01.01, h_tumor_02.01, h_tumor_03.01 and m_heart_02.01. For cell lines, 2 × 107 cells were harvested by scraping. Tumors and cell lines were cross-linked with 1% (wt/vol) formaldehyde for 10 min at 37°C with shaking. After quenching with 150 mM glycine for 5 min at 37°C with shaking, cells were washed twice with ice-cold PBS and frozen at −80°C for further processing. Cross-linked pellets were thawed and lysed on ice for 30 min in ChIP harvest buffer (12 mM Tris-Cl, 1 × PBS, 6 mM EDTA, 0.5% SDS) with protease inhibitors (Sigma). Lysed cells were sonicated with a Bioruptor (Diagenode) to obtain chromatin fragments (~200–500 bp) and centrifuged at 15,000 × g for 15 min to obtain a soluble chromatin fraction. In parallel with cellular lysis and sonication, antibodies (5 µg/3 × 106 cells) were coupled with 30 µL of magnetic protein G beads in binding/blocking buffer (PBS + 0.1% Tween + 0.2% BSA) for 2h at 4°C with rotation. Antibodies used for ChIP included anti-H3K4me3 (Abcam; ab8580), anti-H3K4me1 (Abcam; ab8895), anti-H3K27ac (Abcam; ab4729), anti-H3K79me2 (Abcam; ab3594), anti-H3K27me3 (Abcam; ab6002) and anti-H3K9me3 (Abcam; ab8898). Soluble chromatin was diluted five times using ChIP dilution buffer (10 mM Tris-Cl, 140 mM NaCl, 0.1% DOC, 1% Triton X, 1 mM EDTA) with protease inhibitors and added to the antibody-coupled beads with rotation at 4°C overnight. After washing, samples were treated with elution buffer (10 mM Tris-Cl, pH 8.0, 5 mM EDTA, 300 mM NaCl, 0.5% SDS), RNase A, and Proteinase K, and cross-links were reversed overnight at 37. Immune complexes were then washed five times with cold RIPA buffer (10mM Tris–HCl, pH 8.0, 1mM EDTA, pH 8.0, 140mM NaCl, 1% Triton X-100, 0.1% SDS, 0.1% DOC), twice with cold high-salt RIPA buffer (10mM Tris–HCl, pH 8.0, 1mM EDTA, pH 8.0, 500mM NaCl, 1% Triton X-100, 0.1% SDS, 0.1% DOC), and twice with cold LiCl buffer (10mM Tris–HCl, pH 8.0, 1mM EDTA, pH 8.0, 250mM LiCl, 0.5% NP-40, 0.5% DOC). ChIP DNA was purified using AMPure XP beads (Agencourt) and quantified using the Qubit 2000 (Invitrogen) and Bioanalyzer 1000 (Agilent). Libraries for Illumina sequencing were generated following the New England BioLabs (NEB) Next Ultra DNA Library Prep Kit protocol. A total of 10 cycles were used during PCR amplification for the generation of all ChIP-seq libraries. Amplified ChIP DNA was purified using double-sided AMPure XP to retain fragments (~200–500 bp) and quantified using the Qubit 2000 and Bioanalyzer 1000 before multiplexing.
ChIP-seq data processing
Raw fastq reads for all ChIP-seq experiments were processed using a snakemake based pipeline https://github.com/crazyhottommy/pyflow-ChIPseq. Briefly, raw reads were first processed using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and uniquely mapped reads were aligned to the hg19 reference genome using Bowtie version 1.1.2 (Langmead et al., 2009). Duplicate reads were removed using SAMBLASTER (Faust and Hall, 2014) before compression to bam files. To directly compare ChIP-seq samples uniquely mapped reads for each mark were downsampled per condition to 18 million, sorted and indexed using samtools version 1.5 (Li et al., 2009). To visualize ChIP-seq libraries on the IGV genome browser, we used deepTools version 2.4.0 (Ramírez et al., 2016) to generate bigWig files by scaling the bam files to reads per kilobase per million (RPKM). Super ChIP-seq tracks were generated by merging bam files from each cancer type, sorting and indexing using samtools and scaling to RPKM using deepTools.
Chromatin state analysis
ChromHMM (Ernst and Kellis, 2012) was used to identify combinatorial chromatin state patterns based on the histone modifications studied. Normalized bam files were converted to bed files and binarized at a 1000bp resolution using the BinarizeBed command. We specified that ChromHMM should learn a model based on 18 chromatin states. As we considered models between 2 and 30 chromatin states, we annotated an 18-state model since it is large enough to identify non-redundant functional elements encompassing canonical chromatin states that were not captured using lower state models. To determine chromatin state differences between different groups we used a two-step process. First, using the segmentation calls from the ChromHMM output the entire genome is divided into non-overlapping windows of 10 Kb. We next count the number of times a chromatin state is observed in each of the 10 Kb windows and obtain a frequency matrix for each state in the ChromHMM model (E1-E18). In the second step, low variable genomic loci are removed from the frequency matrix and significant differences between two groups of samples types are calculated by using a nonparametric Mann Whitney Wilcoxon test with a P value < 0.05 for each state separately.
Correlation of copy number from ChIP-seq and SNP array
SKCM TCGA copynumber data were downloaded by TCGAbiolinks (Colaprico et al., 2016). Copy number analysis for ChIPseq was carried out using copywriter (Kuilman et al., 2015), which uses off-target reads for accurate copy number detection. The ChIP-seq input files were used, which represent low-pass whole genome sequencing datasets. Bin size of 200kb was used for analysis. The copy number of each gene was determined by overlapping the genes with the segmentation files from ChIPseq and SNP array, respectively. The Pearson correlation of the copy number of all genes among samples was calculated and plotted in heatmap by ComplexHeatmap.
Correlation of RNA expression and chromatin state data
SKCM TCGA RNaseq data were downloaded by TCGAbiolinks (Colaprico et al., 2016). TPM (transcript per million) value was calculated from raw counts by scaling to the gene length first and then the library size. For each gene in a sample, the transcription start site is overlapped with the chromatin state segmentation file to determine the state of that gene. The expression values (TPM) for all genes and all samples are then combined and spilt by categories (18) of chromatin states. A boxplot is plotted for each chromatin state.
Correlation of DNA methylation and chromatin state data
SKCM TCGA 450k DNA methylation data were downloaded by TCGAbiolinks (Colaprico et al., 2016). For each sample and each chromatin state segmentation bin, the average of beta values from the DNA methylation data were calculated for each bin. The average values of all bins from all samples are combined and then split by categories (18) of chromatin states. A boxplot is plotted for each chromatin state.
Identification and visualization of ChIP-seq associated loci
We used Model-based analysis of ChIP-seq (MACS) version 1.4.2 (Zhang et al., 2008) peak calling algorithm to identify H3K4me3 (p value of 1e-5) and MACS version 2.1.0 to identify H3K27me3 (p value of 1e-5) enrichment over “input” background. Bivalent sites were identified by overlapping H3K4me3 with H3K27me3 or H3K9me3 by a minimum of 1bp using intersectBed (Quinlan and Hall, 2010). Identification of H3K27me3 peaks in isogenic mutant melanocytes were identified using MACS version 1.4.2. Bivalent polycomb-heterochromatin regions were identified by overlapping the H3K4me3+H3K27me3 output with H3K9me3 by a minimum of 1bp. Common sites were identified using bedops (Neph et al., 2012) with the following command; cat *bed | sort-bed - | bedmap–count–echo–delim ‘\t’ - | uniq | awk ‘$1 > = x’ | cut -f2- > samplename_common.bed. This was based on peaks present in BRAF = 7/13, NRAS = 2/4, WT = 2/3, MSTC = 5/10, CCLE = 8/16 and melanocytes = 2/2 samples. Final peaksets used for downstream analysis were generated using mergeBed. Unique BRAF, NRAS, WT and melanocyte peaks for bivalent domains were identified using the concatenate, cluster and subtract tools from the Galaxy/Cistrome web-based platform (Liu et al., 2011). To identify sites that were bivalent in melanocytes but active in melanoma tumors, and visa-versa, common H3K27me3 sites were subtracted from the opposing bivalent site in each comparison.
Identification of broad H3K4me3 domains
Broad H3K4me3 domains for all samples were identified using MACS2.1.0 with the broad setting (p value of 1e-5) followed by merging adjacent peaks within 1kb using mergeBed (Quinlan and Hall, 2010; Subramanian et al., 2005). We determined the optimal distance to merge adjacent peaks based on the number of broad H3K4me3 domains at distance thresholds between 1kb through 10kb in each mutational subtype. Broad H3K4me3 domains were further classified as peaks that extended at least 4x (> 4kb) beyond that of a typical H3K4me3 domain (0.2kb-1kb). Final peaksets for common sites broad and non-broad H3K4me3 and H3K27me3 domains were defined as in identification and visualization of ChIP-seq associated loci (BRAF = 7/13, NRAS = 2/4, WT = 2/3 and melanocytes = 2/2).
Assigning associated loci to genes
A list of Ensembl genes was obtained from the UCSC Table browser (http://genome.ucsc.edu/). Promoters were defined as ± 10kb from the transcription start site (TSS) and genic regions were identified as +10kbTSS to the transcription end site (TES). Peaks were assigned to genes if they overlapped the promoter or genic region by a minimum of 1bp using intersectBed. Gene body heatmaps and average density plots were generated using ngs.plot (Shen et al., 2014).
Gene set enrichment analysis
Gene Set Enrichment Analysis (GSEA) was performed using GSEA/MsigDB (Subramanian et al., 2005) HALLMARK and KEGG pathways based on ensemble gene lists from peaks within −10kb-TES for bivalent domains and ± 10kbTSS for broad H3K4me3 domains. All pathways are significant based on FDR q-value.
RNA-sequencing data processing
For TCGA data, raw counts were obtained from TCGAbiolinks (Colaprico et al., 2016) in R (https://www.rstudio.com/) and transformed to TPM. For the melanoma tumor data in addition to TCGA, raw microarray expression levels were obtained from GSE15605. For Roadmap data, raw counts were obtained from http://www.roadmapepigenomics.org/ and transformed to TPM. For melanocyte data in addition to Roadmap, raw counts were obtained from PRJNA421623 and SRP002621. For all RNA-seq boxplots based on count data, gene expression values were normalized using the quantile normalization function in R.
Proliferation assays
Cell proliferation was measured and quantified using IncuCyte ZOOM system (Essen Biosciences). Briefly, after 14 days of GSK-126 (1 µM, 2 µM, 3 µM) or DMSO treatment melanoma short-term cultures MSTC-2125 and MSTC-2770 cells were seeded at a density of 5,000 cells per well in 96-well plates. Cell proliferation was measured for 155h and media for GSK-126 and DMSO treatment was replaced every third day. Cell proliferation is plotted as confluence percentage.
Boyden Chamber invasion assays
Boyden Chamber assays were performed according to manufacturer’s protocol after 14 days of GSK-126 (1 µM, 2 µM, 3 µM) or DMSO treatment of melanoma short-term cultures. For 3-Deazaneplanocin A (DZNep), MSTC-2125 were treated for 14 days (1 µM, 2 µM, 3 µM) and MSTC-2770 were treated for 10 days (1 µM, 2 µM). Briefly, cells were washed 3-times with “empty” media (serum and antibiotic free) and 100,000 cells were subjected to Matrigel-coated well inserts (08774122, Corning). Growth media was used as a chemoattractant for 24h in Matrigel wells. For reference wells 100,000 cells were plated in growth media. Cells in Matrigel and reference wells were washed with 1% PBS and incubated with 10% formalin for 10 minutes. Cells were washed again with 1% PBS and incubated in crystal violet staining for 10 minutes. For quantitation, the invaded cells and the control plated cells were destained in 10% acetic acid for 15 minutes and calorimetry was performed at 590nM. The ratio of invaded cells to control cells was plotted to graph relative invasion.
Immunoblot analysis
Melanoma short-term cultures (1 × 107 cells) were washed with ice-cold PBS then lysed for 30 minutes at 4°C with agitation in RIPA buffer (Boston BioProducts; BP-115) supplemented with a Roche cOmplete, Mini, EDTA-free protease inhibitor cocktail (Millipore Sigma; 11836170001). Malme-3M cells were treated for 14 days with GSK-126 (1 µM, 2 µM, 3 µM) or DMSO before lysis. Lysate was centrifuged at 4°C for 15 minutes at 15,000rpm. The pellet was discarded and protein was quantified using BCA assay. Samples were supplemented with 2x Laemmli buffer (Bio-Rad; 1610737) and 2-mercaptoethanol, heated for 5 min at 95°C and loaded on 4%–20% Criterion TGX Stain-Free gels (Bio-Rad; 5678094). Protein expression was examined by western blot using anti-EZH2 (Cell Signaling; 5246S), anti-SUZ12 (Active Motif; 39057), anti-ZEB1 (Millipore Sigma; ABD53), anti-SNAI1 (ab216347), anti-NRAS (ab77392), anti-H3 (CST; 4499S) Beta-actin (CST; 4967S), goat anti-rabbit IgG-HRP (CST; 7074S) and horse anti-mouse IgG-HRP (CST; 7076S). Reactive bands were detected by Amersham ECL Western Blotting Detection Reagent (RPN2106, GE Healthcare), SuperSignal West Pico Plus Chemiluminescent substrate (Thermo; 34577) or SuperSignal West Dura Extended Duration substrate (Thermo; 34075). The membranes were reprobed after incubation in Restore Western Blot stripping buffer (Thermo; 21063). ImageJ software was utilized to measure image densitometry of bands as previously described (Janes, 2015). Western blot results were representative of at least three independent biological replicates.
Acid extraction of histones
Melanoma short-term cultures (1 × 107 cells) were treated for 14 days with GSK-126 (1 µM, 2 µM, 3 µM) or DMSO and then washed with ice-cold PBS then lysed for 10 minutes at 4°C with agitation in Triton X-100 extraction buffer supplemented with a Roche cOmplete, Mini, EDTA-free protease inhibitor cocktail (Millipore Sigma; 11836170001). For 3-Deazaneplanocin A (DZNep), MSTC-2770 were treated for 10 days (1 µM, 2 µM). Lysate was centrifuged at 4°C for 10 minutes at 6,500rpm. Supernatant was discarded and the pellet was washed with TEB and centrifuged as above. Supernatant was discarded and the pellet was resuspended in 0.2N HCl and histones were extracted overnight at 4°C. Samples were again centrifuged and supernatant was neutralized with 2M NaOH at a 1:10 volume. Protein was quantified using Bradford assay and protein expression was examined by western blot using anti-H3K27me3 (Abcam; ab6002) and anti-H3 (CST; 4499S).
Stable cell line generation
For lenti packaging, 5 µg of NRAS sgRNA CRISPR/Cas9 All-in-one vector (ABM: 321511110595) or 5 µg of Scrambled sgRNA CRISPR/Cas9 All-in-one vector (ABM: K010) was transfected together with packaging plasmids (5 µg) in HEK293T cells using Lip-ofectamine 3000 according to manufacturer’s instructions. For transduction, lentivirus with DMEM media + 10 µg of polybrene was reversed transduced with MSTC-2125 cells and transgene selection was performed using 3 µg/mL of puromycin.
Quantitative PCR
qRT-PCR assays were performed after 14 days of GSK-126 (1 µM, 2 µM, 3 µM) or DMSO treatment of melanoma cell lines. Total RNA was isolated using RNeasy Mini Kit (QIAGEN) and 500ng of RNA from each sample was reverse transcribed using SuperScript VILO cDNA Synthesis Kit (Thermo Fisher). qRT-PCR was performed with 2 µL of undiluted cDNA in triplicate for each primer set. GAPDH was used as a housekeeping gene and data was plotted and quantified using https://peerj.com/articles/4473.
Immunohistochemistry for patient tissue microarrays
Immunohistochemical stains were performed on 6 stage III and IV melanoma tissue microarrays (TMA) (Ekmekcioglu et al., 2016; Iida et al., 2017). After heat mediated epitope retrieval at pH 8.0 for 20 min, the sections were incubated with mouse monoclonal SUZ12, Clone 3D10 (1:500, ThermoFisher) and mouse monoclonal EZH2, Clone AC22 (1:200, Cell signaling Technology). IHC staining was performed using a Leica Bond Max automated stainer (Leica Biosystems, Buffalo Grove, IL). The IHC reaction was performed using Leica Bond Polymer Refine detection kit (Leica Biosystems). Immunoreactive cells were visualized using diaminobenzidine (DAB) as chromogen followed by counterstaining with hematoxylin. All IHC slides were scanned using an Aperio AT Turbo (Leica Biosystems). The scoring was performed by a pathologist (RL) using direct microscope evaluation. EZH2 and SUZ12 expression was considered positive when nuclear staining was present on tumor cells and it was evaluated by H-score, which assesses the percentage of positive cells (0 to 100) multiplied by the intensity of staining (0 to 3+), with a total score ranging from 0 to 300. H-scores for EZH2 or SUZ12 in patients harboring an NRAS or BRAF mutation were compared using a t test.
Immunohistochemistry for murine tumor samples
Tumors were fixed in formalin for 24 hours, paraffin embedded, sectioned and stained according to standard procedures. Briefly, slides were baked at 60°C for 1 hour then deparaffinized and rehydrated. After antigen retrieval in citrate buffer, endogenous peroxidases were inactivated by 3% hydrogen peroxide. Non-specific signals were blocked using Rodent Block M (Biocare RBM961). Then slides were stained using VIM antibody (Cell Signaling; 5741) overnight at 4°C. After overnight incubation, the slides were washed and incubated with secondary antibody (HRP-polymers, Biocare RMR622) for 30 min at room temperature. The slides were washed and stained with DAB substrate (ThermoFisher Scientific). The slides were then counterstained with hematoxylin and mounted with mounting medium (Richard-Allan Scientific). Cells were counted using imageJ based on a minimum of 9 20x images from each sample. P values were calculated using a wilcoxon test.
In vivo assays
NRAS- and BRAF-mutant melanoma short term cultures (MSTC) were transplanted in the flanks of NUDE mice and observed for tumor induction. Once visible tumors were formed, the mice were randomly distributed in 4 groups and injected with Vehicle, GSK-126 (150mg/kg), MEK inhibitor (1mg/kg), or a combination of the two drugs intraperitoneally every other day in 5 mice per arm. For BRAF inhibitors, mice were injected with Vehicle or BRAF-inhibitor (50mg/kg) every other day in 4 mice per arm. Tumors were measured every third day and the volume was calculated using 0.5 × L × W × H formula. P values were calculated by a paired t test for each time point using graph pad prism.
QUANTIFICATION AND STATISTICAL ANALYSIS
For chromatin state analysis and all RNA-seq comparisons significant differences between groups was calculated using a Wilcoxon test. For western blotting analysis (Figures 5D and 5E), relative density was quantified using ImageJ software, and the values of the target proteins were normalized to b-Actin. Error bars represent mean ± SEM. For all in-vivo data, tumors were measured every third day and the volume was calculated using 0.5 × L × W × H formula. p values were calculated by a paired t test for each time point using graph pad prism. For immunohistochemistry of patient tissue microarrays, scoring was performed by a pathologist using direct microscope evaluation. EZH2 and SUZ12 expression was considered positive when nuclear staining was present on tumor cells and it was evaluated by H-score. H-scores for EZH2 or SUZ12 in patients harboring an NRAS or BRAF mutation were compared using a t test. For immunohistochemistry of murine tumor samples, cells were counted using imageJ based on a minimum of 9 20x images from each treatment condition. p values were calculated using a wilcoxon test.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| anti-EZH2 (Cell Signaling; 5246S) | Cell Signaling | CAT#5246S; RRID:AB_10694683 |
| anti-SUZ12 | Active Motif | CAT#39057; RRID:AB_2614929 |
| anti-ZEB1 | Millipore Sigma | CAT#ABD53; RRID:N/A |
| H3K4me3 | Abcam | CAT#ab8580; RRID:AB_306649 |
| H3K4me1 | Abcam | CAT#ab8895; RRID:AB_306847 |
| H3K9me3 | Abcam | CAT#ab8898; RRID:AB_306848 |
| H3K27ac | Abcam | CAT#ab4729; RRID:AB_2118291 |
| H3K27me3 | Abcam | CAT#ab6002; RRID:AB_305237 |
| H3K79me2 | Abcam | CAT#ab3594; RRID:AB_303937 |
| anti-H3 | Cell Signaling | CAT#4499S; RRID:AB_10544537 |
| anti-Beta-actin | Cell Signaling | CAT#4967S; RRID:AB_330288 |
| anti-SNAI1 | Abcam | CAT# ab216347 RRID:N/A |
| anti-NRAS | Abcam | CAT#ab77392 RRID:AB_1524048 |
| anti-Vimentin | Cell Signaling | CAT#5741 RRID:AB_10695459 |
| Anti-rabbit IgG, horseradish peroxidase (HRP)-linked | Cell Signaling | CAT#7074S; RRID:AB_2099233 |
| Horse anti-mouse IgG, HRP-linked | Cell Signaling | CAT#7076S RRID:AB_330924 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| GSK-126 1346574–57-9, 5 mg | Caymen Chemical | CAT#NC0761027 |
| GSK-126, 500mg | MedChemExpress | CAT# HY-13470 |
| Vemurafenib | MedChemExpress | CAT# HY-12057 |
| Trametinib | MedChemExpress | CAT# HY-10999A |
| 3-Deazaneplanocin A | Caymen Chemical | CAT#102052–95-9 |
| NRAS sgRNA CRISPR/Cas9 All-in-one vector | ABM | CAT# 321511110595 |
| Scrambled sgRNA CRISPR/Cas9 All-in-one vector | ABM | CAT#K010 |
| miniProtease inhibitor | Roche | CAT#11836153001 |
| Dynabeads Protein G for Immunoprecipitation | Thermo Fisher | CAT#10009D |
| Solid-Phase Reversible Immobilization (SPRI) beads; Reagent, SPRIselect, 450mL | Beckman-Coulter | CAT#B23319 |
| Dulbecco’s Phosphate Buffered Saline (DPBS) (Ca2+- and Mg2+-free) | Millipore Sigma | CAT#D8537–500ML |
| HBSS (Ca2+- and Mg2+-free) | Thermo Fisher | CAT#88284 |
| Invitrogen UltraPure 0.5M EDTA, pH 8.0 | Thermo Fisher | CAT#15575020 |
| L-Glutamine (200 mM) | Millipore Sigma | CAT#25030081 |
| HEPES solution | Millipore Sigma | CAT#H0887–100ML |
| Sodium dodecyl sulfate (SDS) | Sigma-Aldrich | CAT#74255 |
| Lithium Chloride | Sigma-Aldrich | CAT#746460 |
| Sodium Chloride | Sigma-Aldrich | CAT#S7653 |
| Tris-HCL 1M pH 8.0 | Teknova | CAT#T1080 |
| Glycine | Sigma-Aldrich | CAT#G8898 |
| Pierce 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher | CAT#28908 |
| Bovine Serum Albumin – IgG free | Millipore Sigma | CAT#A2058 5G |
| RNase A | Invitrogen | CAT#12091021 |
| Proteinase K | Invitrogen | CAT#100005393 |
| RIPA buffer | Boston BioProducts | CAT# BP-115 |
| 2x Laemmli buffer | Bio-Rad | CAT#1610737 |
| 4–20% Criterion TGX Stain-Free gels | Bio-Rad | CAT#5678094 |
| Amersham ECL Western Blotting Detection Reagent | GE Healthcare | CAT#RPN2106 |
| SuperSignal West Pico Plus Chemiluminescent substrate | Thermo Fisher | CAT#34577 |
| SuperSignal West Dura Extended Duration substrate | Thermo Fisher | CAT#34075 |
| Restore Western Blot stripping buffer | Thermo Fisher | CAT#21063 |
|
| ||
| Critical commercial assays | ||
|
| ||
| NEBNext® Multiplex Oligos (index primer - Set1) | New England Biolabs | CAT#E7335L |
| NEBNext® Multiplex Oligos (index primer - Set2) | New England Biolabs | CAT#E7500L |
| NEBNext® Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | CAT#E7645L |
| High Sensitivity D1000 Reagents | Agilent Technologies | CAT#5067–5585 |
| Invitrogen Qubit dsDNA HS Assay Kit | Thermo Fisher | CAT#Q32851 |
| TruSeq DNA LT Sample Prep Kit | Illumina | CAT# FC-121–2001 and FC-121–2002 |
| DynaMag – 96 Side Skirted | Invitrogen | CAT#120.27 |
| Matrigel-coated well inserts | Corning | CAT#08774122 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed data | This paper | GSE134043 |
| Human reference genome (UCSC hg19) | Illumina iGenome | http://bowtie-bio.sourceforge.net/index.shtml |
| Ensemble annotated human genome features | Ensembl | https://genome.ucsc.edu/cgi-bin/hgTables |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Melanoma Short Term Cultures | This paper | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Crl:NU(Ico)-Foxn1nu | Charles River | Strain code: 620 |
|
| ||
| Software and algorithms | ||
|
| ||
| Raw ChIP read processing: FastQC | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Genome alignment: Bowtie (v1.1.2) | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| Duplicate read removal: SAMBLASTER | Faust and Hall, 2014 | https://github.com/GregoryFaust/samblaster |
| Downsampling: samtools (v1.5) | Li et al., 2009 | http://samtools.sourceforge.net/ |
| ChIP-seq library visualization: deepTools (v2.4.0) | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| ChIP-seq peak calling: Model-based analysis of ChIP-seq (MACS) (v1.4.2) and (v2.1.0) | Zhang et al., 2008 | http://liulab.dfci.harvard.edu/MACS/00README.html |
| Galaxy/Cistrome | Liu et al., 2011 | http://cistrome.org/ap/root |
| Gene body heatmaps and average density plots: ngs.plot | Shen et al., 2014 | https://github.com/shenlab-sinai/ngsplot |
| Chromatin state patterns: ChromHMM | Ernst and Kellis, 2012 | http://compbio.mit.edu/ChromHMM/ |
| Gene Set Enrichment Analysis (MSigDB) | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp |
| RStudio | https://www.rstudio.com/ | |
| Bedtools | Quinlan and Hall., 2010 | https://bedtools.readthedocs.io/en/latest/ |
| Bedops | Neph et al., 2012 | https://bedops.readthedocs.io/en/latest/ |
Highlights.
NRAS and BRAF genotypes display differential patterns of bivalent and broad domains
Key EMT regulators are likely activated by shifts in bivalent domains
Broad H3K4me3 domains associate with pro-metastatic drivers and cell-identity genes
NRAS mutants show enhanced sensitivity to combination of EZH2 plus MEK inhibition
ACKNOWLEDGMENTS
We are grateful to Kadir C. Akdemir, Yonathan Lissanu Deribe, Anand Singh, Scott Callahan, Veena Kochat, Sharon Landers, Angela Bhalla, and Keila Torres for helpful discussions and proofreading the manuscript. The work described in this article was supported by grants from the National Institutes of Health (CA160578 to K.R.; CA016672 to SMF Core), Center for Cancer Epigenetics at MDACC (to C.J.T. and K.R.), and MD Anderson Cancer Center (start-up funds to K.R.). Short tandem repeat (STR) DNA fingerprinting was done by the UTMDACC CCSG-funded Characterized Cell Line Core, NCI CA016672. The molecular characterization of the short-term culture tumor lines was supported by generous philanthropic contributions to The University of Texas MD Anderson Moon Shots Program.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109410.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Baker CV, Bronner-Fraser M, Le Douarin NM, and Teillet MA (1997). Early- and late-migrating cranial neural crest cell populations have equivalent developmental potential in vivo. Development 124, 3077–3087. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D., et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, Devarajan K, Daugherty AC, Kundaje AB, Mancini E., et al. (2014). H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 158, 673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K., et al. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326. [DOI] [PubMed] [Google Scholar]
- Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Schallmach E, Kubi A, Shalmon B, Hardan I, Catane R., et al. (2009). Minimally cultured or selected autologous tumor-infiltrating lymphocytes after a lympho-depleting chemotherapy regimen in metastatic melanoma patients. J. Immunother 32, 415–423. [DOI] [PubMed] [Google Scholar]
- Boespflug A, Caramel J, Dalle S, and Thomas L (2017). Treatment of NRAS-mutated advanced or metastatic melanoma: rationale, current trials and evidence to date. Ther. Adv. Med. Oncol 9, 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T, Kalluri R, Nieto MA, and Weinberg RA (2018). EMT in cancer. Nat. Rev. Cancer 18, 128–134. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2015). Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramel J, Papadogeorgakis E, Hill L, Browne GJ, Richard G, Wierinckx A, Saldanha G, Osborne J, Hutchinson P, Tse G., et al. (2013). A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 24, 466–480. [DOI] [PubMed] [Google Scholar]
- Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, et al. (2016). Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 164, 550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, D’Alessio AC, Young RA, and Weinberg RA (2013). Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 154, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Chen Z, Wu D, Zhang L, Lin X, Su J, Rodriguez B, Xi Y, Xia Z, Chen X., et al. (2015). Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat. Genet 47, 1149–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng PF, Shakhova O, Widmer DS, Eichhoff OM, Zingg D, Frommel SC, Belloni B, Raaijmakers MI, Goldinger SM, Santoro R., et al. (2015). Methylation-dependent SOX9 expression mediates invasion in human melanoma cells and is a negative prognostic factor in advanced melanoma. Genome Biol 16, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I., et al. (2016). TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 44, e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, et al. (2018). The chromatin accessibility landscape of primary human cancers. Science 362, eaav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl JA, Jung I, Aanes H, Greggains GD, Manaf A, Lerdrup M, Li G, Kuan S, Li B, Lee AY, et al. (2016). Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature 537, 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dambacher S, Hahn M, and Schotta G (2010). Epigenetic regulation of development by histone lysine methylation. Heredity 105, 24–37. [DOI] [PubMed] [Google Scholar]
- Denecker G, Vandamme N, Akay O, Koludrovic D, Taminau J, Lemeire K, Gheldof A, De Craene B, Van Gele M, Brochez L., et al. (2014). Identification of a ZEB2-MITF-ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ 21, 1250–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devitt B, Liu W, Salemi R, Wolfe R, Kelly J, Tzen CY, Dobrovic A, and McArthur G (2011). Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res 24, 666–672. [DOI] [PubMed] [Google Scholar]
- Dhar SS, Zhao D, Lin T, Gu B, Pal K, Wu SJ, Alam H, Lv J, Yun K, Gopalakrishnan V., et al. (2018). MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes. Mol. Cell 70, 825–841.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekmekcioglu S, Davies MA, Tanese K, Roszik J, Shin-Sim M, Bassett RL Jr., Milton DR, Woodman SE, Prieto VG, Gershenwald JE, et al. (2016). Inflammatory Marker Testing Identifies CD74 Expression in Melanoma Tumor Cells, and Its Expression Associates with Favorable Survival for Stage III Melanoma. Clin. Cancer Res 22, 3016–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2012). ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust GG, and Hall IM (2014). SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiziev P, Akdemir KC, Miller JP, Keung EZ, Samant NS, Sharma S, Natale CA, Terranova CJ, Maitituoheti M, Amin SB, et al. (2017). Systematic Epigenomic Analysis Reveals Chromatin States Associated with Melanoma Progression. Cell Rep 19, 875–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J., et al. (2005). Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436, 117–122. [DOI] [PubMed] [Google Scholar]
- Goding CR (2000). Mitf from neural crest to melanoma: signal transduction and transcription in the melanocyte lineage. Genes Dev 14, 1712–1728. [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C., et al. (2012). A landscape of driver mutations in melanoma. Cell 150, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann F, Niebel D, Aymans P, Ferring-Schmitt S, Dietrich D, and Landsberg J (2020). H3K27me3 and EZH2 expression in melanoma: relevance for melanoma progression and response to immune checkpoint blockade. Clin. Epigenetics 12, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida Y, Ciechanover A, Marzese DM, Hata K, Bustos M, Ono S, Wang J, Salomon MP, Tran K, Lam S., et al. (2017). Epigenetic Regulation of KPC1 Ubiquitin Ligase Affects the NF-kB Pathway in Melanoma. Clin. Cancer Res 23, 4831–4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav U, Nalapareddy K, Saxena M, O’Neill NK, Pinello L, Yuan GC, Orkin SH, and Shivdasani RA (2016). Acquired Tissue-Specific Promoter Bivalency Is a Basis for PRC2 Necessity in Adult Cells. Cell 165, 1389–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob JA, Bassett RL Jr., Ng CS, Curry JL, Joseph RW, Alvarado GC, Rohlfs ML, Richard J, Gershenwald JE, Kim KB, et al. (2012). NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 118, 4014–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes KA (2015). An analysis of critical factors for quantitative immunoblotting. Sci. Signal 8, rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DB, and Sosman JA (2013). Update on the targeted therapy of melanoma. Curr. Treat. Options Oncol 14, 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, and Weinberg RA (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest 119, 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, Menendez S, Vardabasso C, Leroy G, Vidal CI, et al. (2010). The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 468, 1105–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper K, de Goeje PL, Peeper DS, and van Amerongen R (2014). Phenotype switching: tumor cell plasticity as a resistance mechanism and target for therapy. Cancer Res 74, 5937–5941. [DOI] [PubMed] [Google Scholar]
- Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, et al. (2008). Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4, e1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Velds A, Kemper K, Ranzani M, Bombardelli L, Hoogstraat M, Nevedomskaya E, Xu G, de Ruiter J, Lolkema MP, et al. (2015). CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol 16, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lens MB, and Dawes M (2004). Global perspectives of contemporary epidemiological trends of cutaneous malignant melanoma. Br. J. Dermatol 150, 179–185. [DOI] [PubMed] [Google Scholar]
- Levy C, Khaled M, and Fisher DE (2006). MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med 12, 406–414. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FZ, Dhillon AS, Anderson RL, McArthur G, and Ferrao PT (2015). Phenotype switching in melanoma: implications for progression and therapy. Front. Oncol 5, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, Haldipur P, Kawauchi D, Risch T, Warnatz HJ, et al. (2016). Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 530, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y., et al. (2011). Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol 12, R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomberk G, Blum Y, Nicolle R, Nair A, Gaonkar KS, Marisa L, Mathison A, Sun Z, Yan H, Elarouci N., et al. (2018). Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun 9, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A 3rd, Diaz E., et al. (2012). EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112. [DOI] [PubMed] [Google Scholar]
- Miller AJ, and Mihm MC Jr. (2006). Melanoma. N. Engl. J. Med 355, 51–65. [DOI] [PubMed] [Google Scholar]
- Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Marquez VE, and Jones PA (2009). DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther 8, 1579–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK, Rynes E, Maurano MT, Vierstra J, Thomas S., et al. (2012). BEDOPS: high-performance genomic feature operations. Bioinformatics 28, 1919–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. (2010). Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oba J, Kim SH, Wang WL, Macedo MP, Carapeto F, McKean MA, Van Arnam J, Eterovic AK, Sen S, Kale CR, et al. (2018). Targeting the HGF/MET Axis Counters Primary Resistance to KIT Inhibition in KIT-Mutant Melanoma. JCO Precis. Oncol 2018, PO.18.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I., et al. (2015). The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat. Med 21, 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Talukder AH, Lim SA, Kim K, Pan K, Melendez B, Bradley SD, Jackson KR, Khalili JS, Wang J., et al. (2017). SLC45A2: A Melanoma Antigen with High Tumor Selectivity and Reduced Potential for Autoimmune Toxicity. Cancer Immunol. Res 5, 618–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai K, Akdemir KC, Kwong LN, Fiziev P, Wu CJ, Keung EZ, Sharma S, Samant NS, Williams M, Axelrad JB, et al. (2015). Dual Roles of RNF2 in Melanoma Progression. Cancer Discov 5, 1314–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan D, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskin L, Fullen DR, Giordano TJ, Thomas DG, Frohm ML, Cha KB, Ahn J, Mukherjee B, Johnson TM, and Gruber SB (2013). Transcriptome profiling identifies HMGA2 as a biomarker of melanoma progression and prognosis. J. Invest. Dermatol 133, 2585–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe JS, Hwang CI, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, Maiorino L, Tiriac H, Young CM, Miyabayashi K., et al. (2017). Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 170, 875–888.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, et al. (2011). Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res 17, 4550–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmento OF, Digilio LC, Wang Y, Perlin J, Herr JC, Allis CD, and Coonrod SA (2004). Dynamic alterations of specific histone modifications during early murine development. J. Cell Sci 117, 4449–4459. [DOI] [PubMed] [Google Scholar]
- Seberg HE, Van Otterloo E, Loftus SK, Liu H, Bonde G, Sompallae R, Gildea DE, Santana JF, Manak JR, Pavan WJ, et al. (2017). TFAP2 paralogs regulate melanocyte differentiation in parallel with MITF. PLoS Genet 13, e1006636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, and Nestler E (2014). ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields BD, Mahmoud F, Taylor EM, Byrum SD, Sengupta D, Koss B, Baldini G, Ransom S, Cline K, Mackintosh SG, et al. (2017). Indicators of responsiveness to immune checkpoint inhibitors. Sci. Rep 7, 807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons JL, Pierce CJ, Al-Ejeh F, and Boyle GM (2017). MITF and BRN2 contribute to metastatic growth after dissemination of melanoma. Sci. Rep 7, 10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, and Lange CA (2008). Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res 647, 21–29. [DOI] [PubMed] [Google Scholar]
- Sinnberg T, Levesque MP, Krochmann J, Cheng PF, Ikenberg K, Meraz-Torres F, Niessner H, Garbe C, and Busch C (2018). Wnt-signaling enhances neural crest migration of melanoma cells and induces an invasive phenotype. Mol. Cancer 17, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, Khushalani NI, Lewis K, Lao CD, Postow MA, et al. (2018). Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. N. Engl. J. Med 379, 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terranova C, Tang M, Orouji E, Maitituoheti M, Raman A, Amin S, Liu Z, and Rai K (2018). An Integrated Platform for Genome-wide Mapping of Chromatin States Using High-throughput ChIP-sequencing in Tumor Tissues. J. Vis. Exp 134, 56972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E, and Mayor R (2012). Neural crest delamination and migration: from epithelium-to-mesenchyme transition to collective cell migration. Dev. Biol 366, 34–54. [DOI] [PubMed] [Google Scholar]
- Thumar J, Shahbazian D, Aziz SA, Jilaveanu LB, and Kluger HM (2014). MEK targeting in N-RAS mutated metastatic melanoma. Mol. Cancer 13, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao H, Chin L, Garraway LA, and Fisher DE (2012). Melanoma: from mutations to medicine. Genes Dev 26, 1131–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallianatos CN, and Iwase S (2015). Disrupted intricacy of histone H3K4 methylation in neurodevelopmental disorders. Epigenomics 7, 503–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardabasso C, Hake SB, and Bernstein E (2015). Histone variant H2A.Z.2: A novel driver of melanoma progression. Mol. Cell. Oncol 3, e1073417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, Romeril SP, Burgess JL, Grant SW, Brackley JA, et al. (2012). Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Med. Chem. Lett 3, 1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt P, Tee WW, and Reinberg D (2013). A double take on bivalent promoters. Genes Dev 27, 1318–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenberger DJ (2014). Characterizing DNA methylation alterations from The Cancer Genome Atlas. J. Clin. Invest 124, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JN, and Roberts CW (2013). ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov 3, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingg D, Debbache J, Schaefer SM, Tuncer E, Frommel SC, Cheng P, Arenas-Ramirez N, Haeusel J, Zhang Y, Bonalli M., et al. (2015). The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat. Commun 6, 6051. [DOI] [PubMed] [Google Scholar]
- Zingg D, Arenas-Ramirez N, Sahin D, Rosalia RA, Antunes AT, Haeusel J, Sommer L, and Boyman O (2017). The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep 20, 854–867. [DOI] [PubMed] [Google Scholar]
- Zipser MC, Eichhoff OM, Widmer DS, Schlegel NC, Schoenewolf NL, Stuart D, Liu W, Gardner H, Smith PD, Nuciforo P., et al. (2011). A proliferative melanoma cell phenotype is responsive to RAF/MEK inhibition independent of BRAF mutation status. Pigment Cell Melanoma Res 24, 326–333. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ChIP sequencing data has been deposited in the NCBI GEO BioProject database with the following accession number GSE134043. ChIP-seq data for histone modifications in ESCs, germ cells and melanocytes were downloaded from ftp://ftp.ncbi.nlm.nih.gov/pub/geo/DATA/roadmapepigenomics/. ChIP-seq data for SUZ12 and EZH2 was downloaded from the NCBI GEO Bio-Project database with the following accession number GSE29611. Code used in this study is available on https://rpubs.com/cjt5 and https://github.com/sccallahan/ChromXploreR.