

Abstract
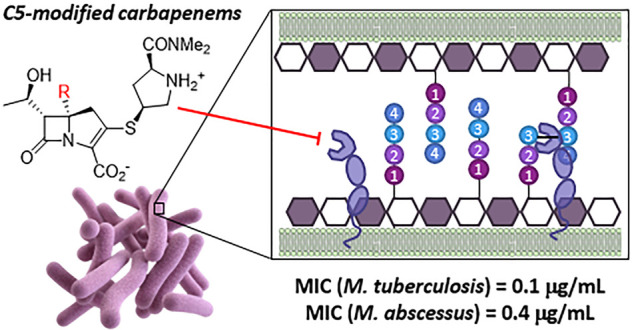
Commercial carbapenem antibiotics are being used to treat multidrug resistant (MDR) and extensively drug resistant (XDR) tuberculosis. Like other β-lactams, carbapenems are irreversible inhibitors of serine d,d-transpeptidases involved in peptidoglycan biosynthesis. In addition to d,d-transpeptidases, mycobacteria also utilize nonhomologous cysteine l,d-transpeptidases (Ldts) to cross-link the stem peptides of peptidoglycan, and carbapenems form long-lived acyl-enzymes with Ldts. Commercial carbapenems are C2 modifications of a common scaffold. This study describes the synthesis of a series of atypical, C5α modifications of the carbapenem scaffold, microbiological evaluation against Mycobacterium tuberculosis (Mtb) and the nontuberculous mycobacterial species, Mycobacterium abscessus (Mab), as well as acylation of an important mycobacterial target Ldt, LdtMt2. In vitro evaluation of these C5α-modified carbapenems revealed compounds with standalone (i.e., in the absence of a β-lactamase inhibitor) minimum inhibitory concentrations (MICs) superior to meropenem-clavulanate for Mtb, and meropenem-avibactam for Mab. Time-kill kinetics assays showed better killing (2–4 log decrease) of Mtb and Mab with lower concentrations of compound 10a as compared to meropenem. Although susceptibility of clinical isolates to meropenem varied by nearly 100-fold, 10a maintained excellent activity against all Mtb and Mab strains. High resolution mass spectrometry revealed that 10a acylates LdtMt2 at a rate comparable to meropenem, but subsequently undergoes an unprecedented carbapenem fragmentation, leading to an acyl-enzyme with mass of Δm = +86 Da. Rationale for the divergence of the nonhydrolytic fragmentation of the LdtMt2 acyl-enzymes is proposed. The observed activity illustrates the potential of novel atypical carbapenems as prospective candidates for treatment of Mtb and Mab infections.
Keywords: Mycobacterium; antimicrobial activity; carbapenems; l,d-transpeptidase
Tuberculosis (TB) is caused by the slow-growing intracellular pathogen Mycobacterium tuberculosis (Mtb), and currently infects an estimated 2 billion people, representing one-fourth of the world’s population. TB causes 1.5 million deaths annually and requires a challenging treatment regimen involving the administration of four antitubercular agents for a period of at least six months. The emergence of multidrug resistant (MDR), extensively resistant (XDR), and totally resistant (TDR) TB necessitates development of new antitubercular agents.1Mycobacterium abscessus (Mab), by contrast, is a rapidly growing nontuberculous mycobacterial (NTM) species that typically causes pulmonary infections, but can also cause soft-tissue, burn, and wound infections.2 Like Mtb, Mab requires long-term treatment with multiple antibiotics, frequently resulting in significant adverse side effects.3 Due to the intrinsic antimicrobial resistance of Mab, cure rates are <50%, and recurrence rates are 50%, underscoring the urgent need for new therapeutic agents.4,5
β-Lactam antibiotics are not currently included among the first line antitubercular agents isoniazid, rifampin, ethambutol, and pyrazinamide. The earliest studies of β-lactam antibiotics indicated little antitubercular activity.6−8 It was soon recognized that their apparent inactivity was due to the presence of mycobacterial β-lactamases.9 Penicillinase-resistant penicillins, such as oxacillin, and third-generation cephalosporins, which are poor substrates for the mycobacterial β-lactamase, were observed to possess activity.10,11 Later it was demonstrated that carbapenems and combinations of β-lactam antibiotics and β-lactamase inhibitors were also active antimycobacterial agents.12−14 The major Mtb β-lactamase, BlaC, is an endogenous Ambler class A serine penicillinase that has broad substrate specificity and is inhibited by clavulanic acid.15−17 A combination of clavulanic acid and the carbapenem meropenem was shown to be active against XDR-Mtb and recent studies compared in vitro efficacy of several β-lactam/β-lactamase inhibitor combinations against MDR-TB.18−20Mab also expresses an endogenous serine class A β-lactamase, BlaMab, with broad substrate specificity that hydrolyzes the conventional β-lactamase inhibitors, clavulanate, sulbactam, and tazobactam, but is inhibited by the newer diazabicyclooctanes (DBO, e.g., avibactam, relebactam) and boronic acid (e.g., vaborbactam) inhibitors.21−23 Carbapenems were found to be more effective against Mab in combination with other antibiotics, both β-lactam and non-β-lactam, rather than when used alone.24−33 Newly designed penems are in development to treat Mab.34
β-Lactam antibiotics inhibit the transpeptidases involved in cell wall biosynthesis. The cell wall component peptidoglycan (PG) is a cross-linked polymeric structure comprised of glycan polymers cross-linked by short peptide strands which protrude from these carbohydrates. Glycan strands of most bacteria are comprised of alternating β(1 → 4)linked N-acetylglucosamine (GlcNAc) and modified N-acetylmuramic acid (MurNAc) residues, and in both Mtb and Mab, the MurNAc is extensively (∼70%) replaced by N-glycolylmuramic acid (MurNGlyc).35−37 The muramic acid residues are covalently attached to short (3 to 5 residue) peptide side chains, which are cross-linked to provide structural stability, protecting the microorganism from the effects of turgor pressure. The stem pentapeptides consist of glycan-linked-l-Ala-D-γGlu-meso-DAP-d-Ala-d-Ala, where DAP = diaminopimelate. Two types of mycobacterial cross-links are observed: (1) 4 → 3 cross-links, formed by d,d-transpeptidase-catalyzed cleavage between the d-Ala-d-Ala linkage and joining the resultant acyl-enzyme carbonyl to the γ-amino group of DAP, and (2) 3 → 3 cross-links, formed by l,d-transpeptidase (Ldt)-catalyzed cleavage of the DAP-d-Ala linkage and joining the carbonyl group of the resultant acyl-enzyme with the same γ-amino group of DAP. The 4 → 3 cross-links were historically commonly observed, and d,d-transpeptidases have become known as penicillin-binding proteins (PBPs). By contrast, the Ldt-catalyzed 3 → 3 cross-links are predominant (70–80%) in the PG of both Mtb and Mab.38 These 3 → 3 cross-links have since been observed in numerous species, including Enterococcus faecium, Corynebacterium jeikeium, Escherichia coli, Clostridium difficile, Acinetobacter baylyi, and Acinetobacter baumannii.39−44 The acceptor substrate for an Ldt is a tetrapeptide stem, which has the terminal d-Ala residue cleaved from the d-Ala-d-Ala terminus of the stem pentapeptide, a transformation performed by a d,d-carboxypeptidase.39
The carbapenem class of β-lactam antibiotics exhibits high potency, broad spectrum activity, and stability to 20th century β-lactamases (e.g., TEM-1). Carbapenems have also demonstrated in vitro efficacy against Mtb, the ability to inhibit Mtb PBPs, activity in combination with β-lactamase inhibitors against XDR Mtb, as well as ability to inhibit LdtMt1 and LdtMt2.14,20,45−47 Additionally, it was recently discovered that meropenem inhibits the d,d-carboxypeptidase responsible for cleaving the terminal d-Ala-d-Ala linkage of the stem pentapeptides to provide the tetrapeptide substrates for Ldts.48 Thus, carbapenems interfere with Mtb cell wall biosynthesis at multiple steps, including PBP-catalyzed 4 → 3 cross-linking, d,d-carboxypeptidase-catalyzed cleavage of the terminal d-Ala residue, and Ldt-catalyzed 3 → 3 cross-linking.49−52
The carbapenem meropenem and the carbapenem/dehydropeptidase-I inhibitor combination, imipenem/cilastatin, have recently been included as add-ons in treatment of MDR-TB, when used in combination with the β-lactamase inhibitor clavulanic acid.53 Studies of clinical use of carbapenems, including meropenem, imipenem, and ertapenem, in the treatment of MDR-TB documented their safety and tolerability and improvement of success rates.54−57 Imipenem is included in British Thoracic Society guidelines as part of the regimen to treat NTM infections including Mab, and the US Cystic Fibrosis Foundation and European Cystic Fibrosis Society includes imipenem in the regimen for treating pulmonary NTM infections.58,59 Analysis of outcomes indicate improved success rates with NTM patients receiving imipenem, along with other agents.60
Structural development of the carbapenem class in the past 30 years has been limited to modification of the C2 position on the scaffold, despite the fact that multiple pathogens have evolved carbapenemases capable of hydrolyzing all commercial carbapenems.61−68 We recently began a program to reinvestigate atypical (i.e., substituted at positions other than C2) carbapenems with respect to their susceptibility to carbapenemase-mediated hydrolysis and antimicrobial efficacy against resistant pathogens. We reasoned that such atypical substitutions had potential to differentially affect interaction of the carbapenems to β-lactamases relative to PBPs, including noncovalent recognition, the rate of protein acylation, and, in the case of β-lactamases, the rate of acyl-enzyme hydrolysis. We realized that these differences had potential to render the new carbapenem antibiotics less susceptible to carbapenemase-mediated hydrolysis, while maintaining the ability to bind PBP and Ldt enzymes. Due to the synthetic challenges, the C5 position of the carbapenem scaffold is notably underexplored, although one early study, not including mycobacterial strains, of C5α substituted carbapenems indicated very modest activity against a few representative strains.69 Our study describes the synthesis and evaluation of C5α-substituted (C5α-methyl, 10a and 13a, and C5α-ethyl, 10b and 13b, see Schemes 1 and 2) carbapenems as antibacterial agents targeting Mtb and Mab. The C5α position substituent is hydrogen in all bicyclic commercial β-lactam antibiotics. We also noted that the substituted pyrrolidine C2 side chains of commercial carbapenems incorporated into 10a and 10b may not be optimal for Mtb, and thus, in addition to incorporating the meropenem pyrrolidine C2 side chain, we decided to synthesize and evaluate simplified C2 thioethyl analogues (13a and 13b) of the new atypical C5α-substituted carbapenems.70 In reported studies, the carbapenem with the minimal C2 thioethyl group was able to acylate the LdtMt1 protein eight times more efficiently than meropenem.70
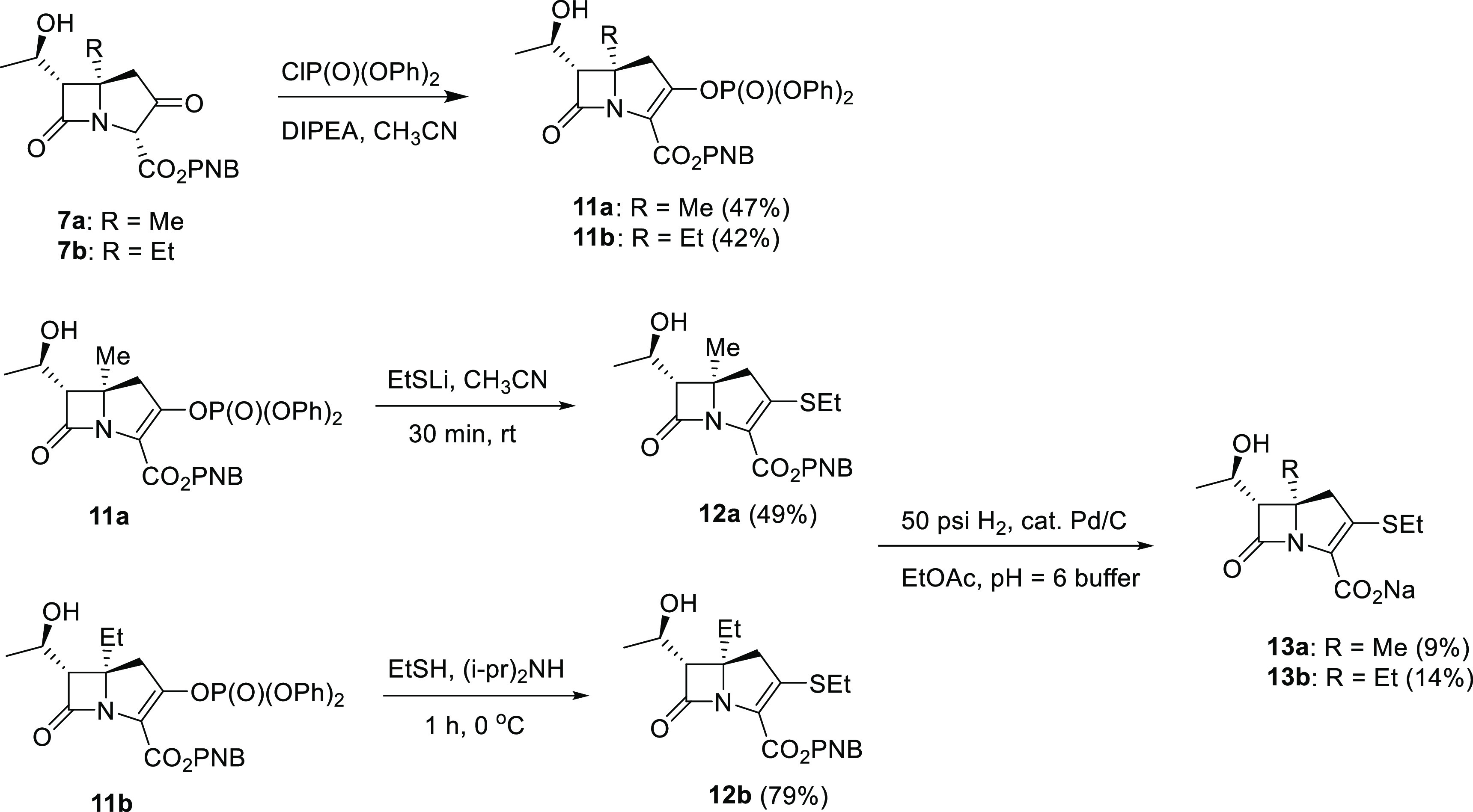
Scheme 1. Syntheses of C5α-Substituted Carbapenems 10a and 10b.
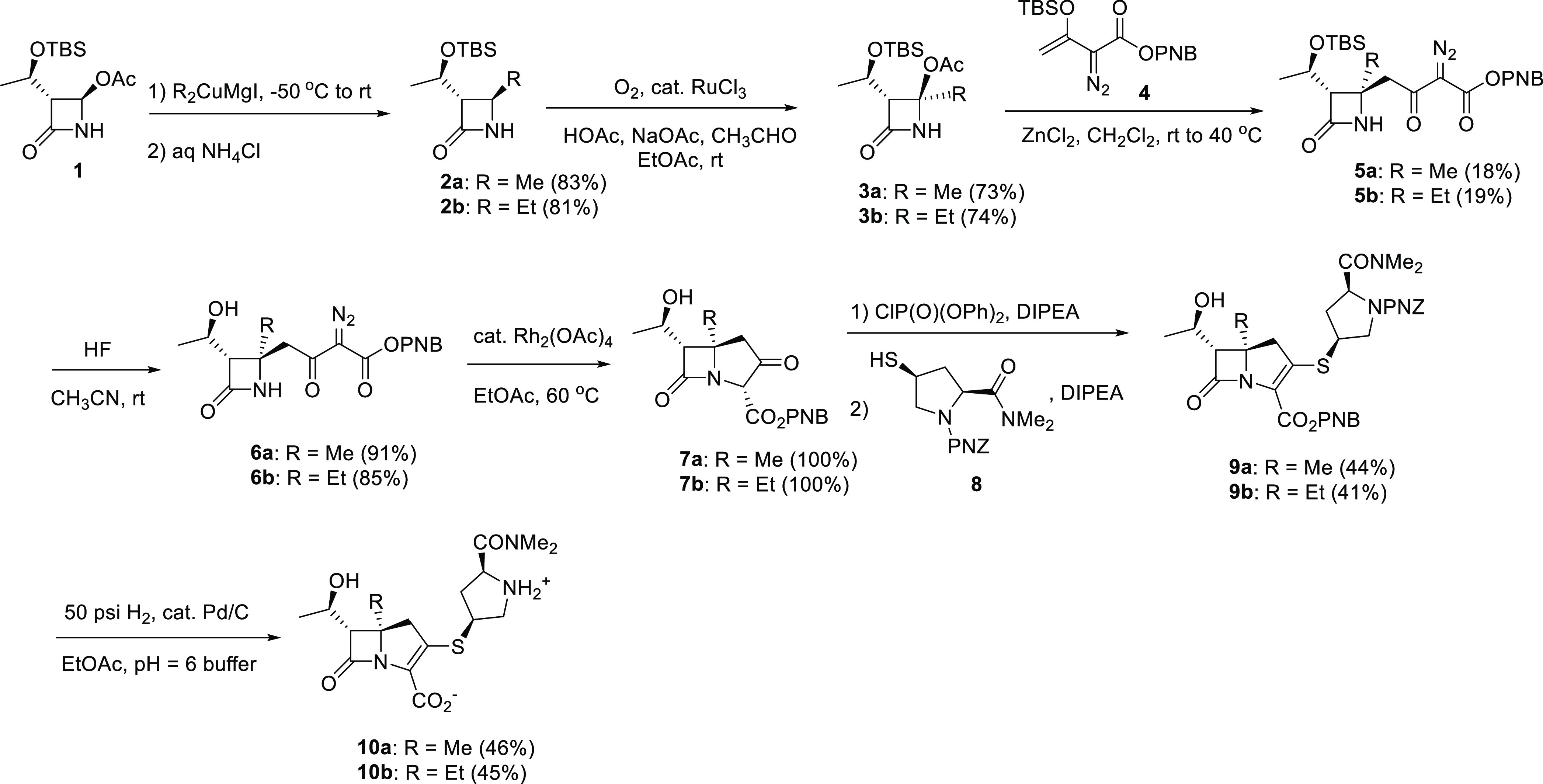
Scheme 2. Syntheses of C5α-Substituted Carbapenems 13a and 13b.
Results
Chemistry
The syntheses of C5α-methyl carbapenems 10a and 13a as well as the C5α-ethyl carbapenems 10b and 13b are illustrated in Schemes 1 and 2. In summary, the C4-alkylazetidinone 2a or 2b was prepared by reaction of commercially available 4-acetoxyazetidinone 1 with iodomagnesium dimethylcuprate or iodomagnesium diethylcuprate, respectively. The unstable tertiary acetate, 3a or 3b was prepared utilizing a ruthenium(III) chloride catalyzed oxidation process as shown.71 This compound was reacted with the highly functional TBS enol ether 4 in the presence of ZnCl2 to generate 5a or 5b with appropriate stereochemistry.72 The TBS protecting group was then removed from the side chain using aqueous HF in acetonitrile at room temperature and the cyclization of the (hydroxyethyl)diazodicarbonyl compound 6a or 6b performed using catalytic Rh2(OAc)4 in EtOAc to generate ketoester 7a or 7b. The meropenem pyrrolidine sulfide side chain was introduced by formation of the enol phosphate, and subsequent reaction with thiol 8 without isolation of the intermediate as shown in Scheme 1. Deprotection in a two-phase system using catalytic palladium under hydrogen pressure removed the two PNZ protecting groups and subsequent purification using a DiaionHP20 chromatography column and increasing ethanol/water concentration, followed by lyophilization produced antibiotic 10a or 10b.
As shown in Scheme 2, to introduce the C2 thioethyl side chain, we chose to first isolate enol phosphate 11a or 11b, and subsequently react with ethanethiol. In the case of 11a, we utilized a THF solution of preformed lithium thioethoxide, and in the case of 11b we utilized ethanethiol in the presence of DIPA to generate 12a or 12b, respectively, and then deprotected utilizing catalytic palladium under hydrogen pressure in a two-phase system, and subsequent purification as described above.
In Vitro Antimycobacterial Activity
Mtb and Mab produce extended spectrum β-lactamases BlaC and BlaMab, which are inhibited by clavulanate and avibactam, respectively.73 Since these enzymes are capable of hydrolyzing commercial carbapenems, the four atypical C5α-substituted carbapenems (10a, 10b, 13a, and 13b) were evaluated against these two mycobacterial species both alone and in the presence of the appropriate β-lactamase inhibitors as shown in Table 1, using commercial meropenem as reference. All C5α-substituted carbapenems retained activity against Mtb, with 10a exhibiting ∼10-fold enhanced potency compared to meropenem (Table 1). Replacement of the C2 substituted pyrrolidine group with a simplified thioethyl moiety in 13a and 13b resulted in decreased activity toward Mtb. Only meropenem showed a statistically significant decrease in MIC when combined with clavulanate.
Table 1. MIC Values (μg/mL) of C5α-Substituted Carbapenem Antibiotics against Mtb and Maba.
|
Mtb |
Mab |
|||
|---|---|---|---|---|
| carbapenem | alone | with clavulanate (5 μg/mL) | alone | with avibactam# (5 μg/mL) |
| meropenem* | 1.1 | 0.5 | 2.6 | 3.1 |
| 10a | 0.1 | 0.1 | 0.4 | 0.5 |
| 10b | 2.1 | 1.6 | 22.3 | 11.6 |
| 13a | 0.8 | 0.6 | 7.5 | 5.8 |
| 13b | 7.9 | 5.6 | NA | NA |
NA: No activity at the highest conc. (30.3 μg/mL). *P < 0.05, comparison of +/- clavulanate. #No statistical difference with +/- avibactam.
Similarly, when evaluated against Mab, 10a was the most potent C5-substituted analogue, while others showed little (10b, 13a) to no (13b) inhibition of Mab growth (Table 1). Inhibition of BlaMab with avibactam did not significantly alter the MIC of any tested compounds.
The activity of carbapenem 10a was further characterized in time-kill assays as shown in Figure 1. Concentration dependent killing of both Mtb and Mab by 10a, superior to that of meropenem, was observed at 2, 4, and 8 times the MIC.
Figure 1.

Concentration- and time-dependent bactericidal activity of 10a and meropenem against Mtb and Mab. The bacterial cultures were treated with different drug concentrations for 6 days with Mtb and for 72 h in the case of Mab and plated for CFU at indicated time points. Data is an average of four independent experiments with a total of 12 technical replicates with standard deviation error bars.
To confirm the selectivity of C5α-substituted carbapenems, 10a was tested for cytotoxicity against murine macrophage (J774) and hepatic (HepG2) cell lines using 2-fold dilution series of the compound. No toxicity was noted even at the highest concentration, indicating a selectivity index (SI = IC50/MIC) of >90.
We determined MIC values for 10a against five clinical strains of Mtb and Mab (Table 2) in comparison to reference strains CDC1551 (Mtb) and 390S (Mab). The slightly different MIC values for reference strains noted here likely stems from the use of different Mtb assay readouts (Table 2: resazurin assay vs Table 1: luciferase assay) and slight loss of compound activity during storage.
Table 2. MIC Values (μg/mL) against Mtb and Mab Clinical Strains.
| carbapenem | CI-1 | CI-2 | CI-3 | CI-4 | CI-5 | reference | |
|---|---|---|---|---|---|---|---|
| Mtb | meropenem | 4.6 | 12.5 | 5.1 | 0.7 | 5.9 | 2.6 |
| 10a | 0.7 | 0.7 | 0.8 | 0.2 | 1.0 | 0.7 | |
| Mab | meropenem | 5.2 | 9.7 | 5.4 | 16.0 | 96.1 | 4.4 |
| 10a | 1.1 | 0.8 | 1.1 | 0.9 | 1.3 | 0.8 |
Clinical isolates exhibited variable susceptibility to meropenem, most notably the enhanced sensitivity of Mtb CI-4 belonging to the Indo-Oceanic lineage 6 and dramatically decreased sensitivity of Mab CI-5 (MIC 21× higher than reference). However, 10a was largely unaffected by strain-specific differences in susceptibility.
Acylation of LdtMt2
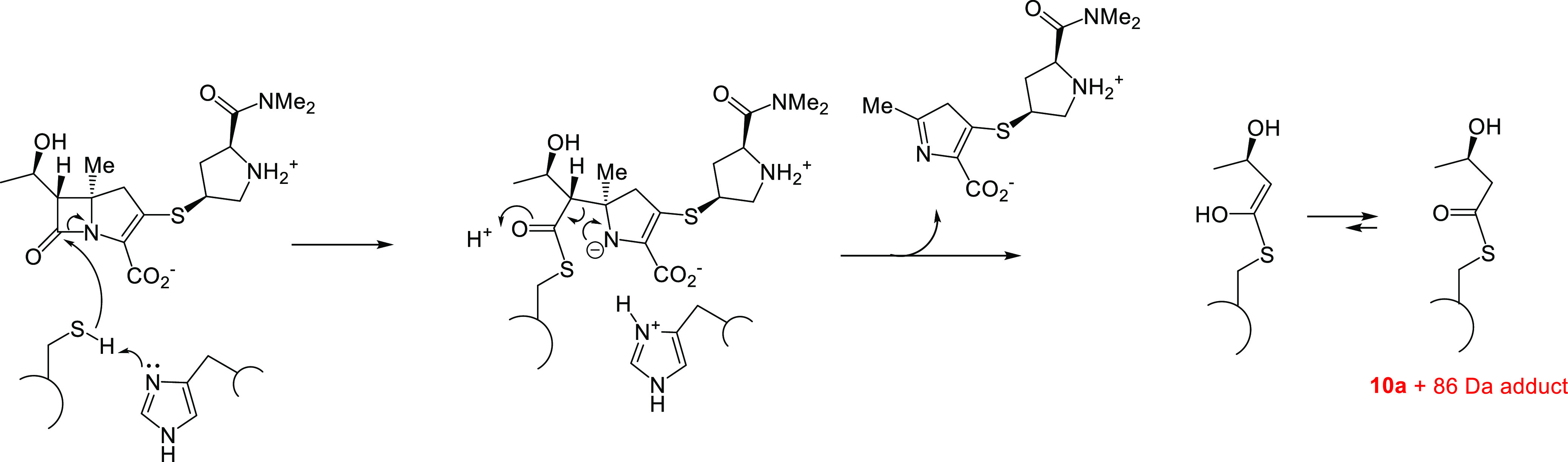
Mtb cross-links PG using four PBPs and the five Ldts, LdtMt1–LdtMt5. LdtMt2 is essential for virulence and has been proposed as a potential antituberculosis target.51 Carbapenems, penems, and cephalosporins are known to acylate class 2 Ldts, and thus we decided to evaluate the time-dependent interaction of our new C5α-alkylated carbapenems with LdtMt2 using high resolution mass spectrometry (HRMS).45 As shown in Figure 2, all four of the new carbapenems form stable adducts with LdtMt2, and all degrade to a Δm = +86 Da adduct over the course of the 5-h reaction time. Like other C1-unsubstituted carbapenems, it was possible to observe the intact carbapenem antibiotic bound to the enzyme after 1 h of incubation in the case of 10a and 10b, but fragmentation of the acyl-enzyme to the Δm = +86 Da adduct was more rapid for 13a and 13b than for 10a and 10b (Figure 2). Commercial carbapenems are observed to lose the C6-hydroxyethyl group, fragmenting to a Δm = +Antibiotic −44 Da adduct.74 This typical carbapenem-Ldt adduct was only transiently observed in the case of 10a as shown in Figure 2 and Scheme 3. With the exception of 13b, the antibiotics rapidly formed covalent adducts with LdtMt2, as witnessed by the absence of apo-enzyme at t = 1 h in the case of 10a, 10b, and 13a. In the case of carbapenem 13b, however, substantial apo-enzyme was observed at t = 1 h, and this species was completely replaced by the Δm = +86 Da adduct after 5 h of incubation. The mechanism for formation of the +86 Da adduct following acylation is proposed in Scheme 4.
Figure 2.

LdtMt2 time course showing that C5α-substituted carbapenems 10a (red traces), 10b (blue traces), and 13a (orange traces) fully acylate LdtMt2 after 1 h, and 13b (green traces) fully acylates LdtMt2 after 5 h as indicated by change in mass (Δm). All C5α-substituted carbapenems degrade to the Δm = +86 Da adduct on the enzyme.
Scheme 3. Mechanism of LdtMt2 Inactivation by 10a and Subsequent Loss of the C6 Hydroxyethyl Substituent.
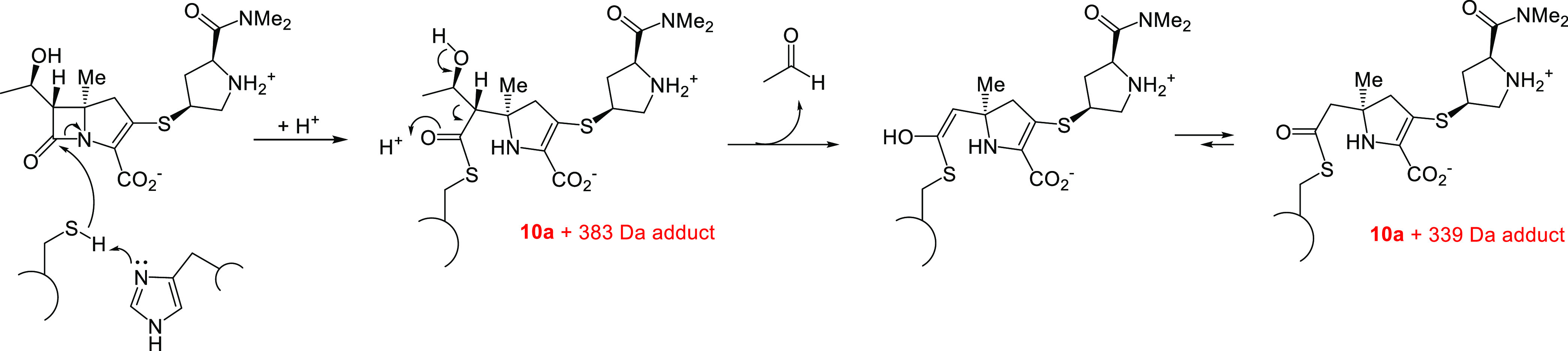
Scheme 4. Mechanism of C5α-Substituted Carbapenem Degradation to the Observed Δm = +86 Da Adduct.
Discussion
The in vitro activity data of the atypical carbapenems in Table 1 indicates that the C5α-methyl analogues 10a and 13a are superior to the C5α-ethyl analogues 10b and 13b, and that the meropenem-like C2 pyrrolidine side chain analogues 10a and 10b are superior to the less structurally complex C2 thioethyl analogues 13a and 13b, with respect to inhibition of growth of both Mtb and Mab. The effect of structure on activity was more pronounced in the case of Mab than with Mtb. As shown by the data in Table 1, the C5α-methylcarbapenem 10a has standalone activity superior to that of either meropenem alone or meropenem combined with β-lactamase inhibitor against either Mtb or Mab. Of interest is the lack of synergy with clavulanate of all the novel atypical carbapenems, as indicated in Table 1, indicating improved stability of these C5α-substituted carbapenem analogues toward BlaC-mediated hydrolysis. Meropenem, by contrast, is reported to be an extremely slow BlaC substrate as also confirmed by our data, Table 1, demonstrating meropenem/clavulanate synergy.75Table 1 data suggests that incorporation of the C5α-alkyl group further slows the BlaC-mediated hydrolysis of these new atypical carbapenems. Structure can dramatically effect the rate β-lactamase mediated hydrolysis, as indicated by the generation of BlaC-specific fluorogenic cephalosporins for detection of Mtb.76
HRMS analysis of the acylation of LdtMt2 by the atypical carbapenems indicates that all four of the novel atypical carbapenems acylate the transpeptidase and subsequently degrade by nonhydrolytic fragmentation of the acyl-enzyme to the smaller hydroxybutyryl cysteine derivative (Δm = +86 Da), rather than the larger Δm = +carbapenem −44 Da analogue uniformly observed for commercial carbapenem antibiotics, including meropenem.77 This hydroxybutyryl acyl-enzyme was also observed in the case of the interaction of LdtMt2 with the penem, faropenem, which has an identical C6α-hydroxyethyl side chain to meropenem (Figure 3), but has not previously been observed in the carbapenem series.46,78 A previous crystallographic study indicated that, in the case of the faropenem-LdtMt2 complex, the Δm = +86 Da intermediate exists as a stable thioester with the active site Cys354 covalently linked to the carbonyl of the β-lactam.78 In that study, the β-hairpin lid of the protein (residues 300–323) adopted a closed conformation and the carbon of the acylating carbonyl group was buried and inaccessible by solvent. Additionally, it has been reported that this hydroxybutyryl-LdtMt2 adduct is unreactive toward externally added amine receptors, including meso-DAP, further characterizing it as a highly stable acyl-enzyme, unable to liberate itself from the covalently attached moiety.78
Figure 3.

Structural classes of bicyclic β-lactam antibiotics
Why does the C5α structural modification of these atypical carbapenems alter the fate of the Ldt acyl-enzyme, relative to that of commercial carbapenems? Why do the Ldt acyl-enzymes undergo these retroaldol-type fragmentations? A recently published study sought to understand the mechanistic reasons for the varying nonhydrolytic fragmentation pathways of the varying classes of β-lactam antibiotics (Figure 3) on the LdtMt2 cysteine transpeptidase through comparisons of analogous interactions of these antibiotics with the serine carbapenemase mutants OXA-48 (S70C) and KPC-2 (S70C).74 That study observed that the retroaldol-type cleavages of the acyl-enzymes across C5–C6 (penicillin and penem) and C6–C7 (cephalosporin) occurred in the case of LdtMt2, and in the case of the two S70C carbapenemase mutants, but not in the corresponding wild type serine carbapenemases. These data led to the hypothesis that the differing outcome of the Ldt or carbapenemase interactions with the antibiotics was due to the differing pKa values of the corresponding, hypothetical product oxoester enolates (ester pKa ≈ 25) and observed product thioester enolates (thioester pKa ≈ 20). The acyl-enzyme fragments to form the less basic thioester enolate (Scheme 5A), whereas the oxoester does not (in the case of the wild type carbapenemases) fragment to the more basic oxoester enolate. The fact that the carbapenem thioester acyl-enzymes degrade via an alternate C8–C6 retroaldol fragmentation route (Scheme 5B) leading to a similar (less basic) thioester enolate suggests that the S1 position sulfur atom (and sulfur electron pairs) in the penicillins, cephalosporins, and penems plays a crucial role in directing the C5–C6 fragmentation of the thioester Ldt acyl-enzyme, which is absent in the case of the carbapenems. The current observation that the fragmentation of the atypical carbapenem Ldt acyl-enzyme can now be redirected to follow the C5–C6 bond cleavage suggests that the transition state has carbocation character as shown in Scheme 5C. This carbocation character is stabilized by the tertiary nature of the C5 carbon in the atypical analogues and is also stabilized by the sulfur electrons in the case of penems, penicillins, and cephalosporins (Scheme 5A). This carbocation stabilization is missing in the case of the commercial carbapenem antibiotics, which have a hydrogen atom at C5 and also lack a second adjacent heteroatom, thus directing the fragmentation to occur at the C8–C6 bond of the heteroatom-containing side chain (Scheme 5B).
Scheme 5. Observed LdtMt2 Acyl-Enzyme Nonhydrolytic Fragmentation Pattern for (A) Penems, (B) Commercial Carbapenems, and (C) Atypical C5α-Substituted Carbapenems.

What factors are responsible for the improved antimycobacterial activity of these structurally modified antibiotics? Efficacy of a β-lactam antibiotic is a complex function of transpeptidase target recognition and inhibition, together with ability to avoid β-lactamase-catalyzed hydrolysis, penetrate the cell envelope, and avoid efflux. Our data indicate that these modified carbapenems target LdtMt2 and avoid the BlaC Mtb carbapenemase. It is likely that these novel carbapenems are also interacting with mycobacterial PBPs. Mtb and Mab produce ten transpeptidases, including representatives of five of the six mycobacterial Ldt classes (LdtMt1–5), and at least five PBPs (PonA1, PonA2, PBPA, PBPB, and PBP-lipo), as well as a number of d,d-carboxypeptidases. The potentially nonredundant functions of these transpeptidases are under current investigation. A recently published study compared the efficacy of carbapenems with that of representative penicillins, cephalosporins, and β-lactamase inhibitors at binding to five Mab PBPs.79 The authors concluded that the carbapenems imipenem and meropenem inactivated the widest range of PBPs at low concentration, indicating it is likely that the most efficacious antimycobacterial β-lactam antibiotic or combination of antibiotics will inactivate a series of mycobacterial transpeptidases. For example, it has been determined that loss of the PBP PonA2, either by mutation or treatment with cephalosporins, increases sensitivity to meropenem, potentially involving increasing reliance on LdtMt2, a known meropenem target.80 One theory is that β-lactam mediated cell death occurs through an uncoupling of the transpeptidase and transglycosylase activity of the high molecular weight PBPs.
With the previously reported improvement in Ldt acylation efficacy of the minimal C2 thioethyl side chain, it is challenging to rationalize the higher MIC values of compounds 13a and 13b, relative to the meropenem-like C2 pyrollidine analogues 10a and 10b.70 In the case of Gram-negative pathogens, the basic C2 side chains of the commercial carbapenems are well-documented to enable porin-mediated transfer across the outer membrane, due to their overall charge similarity to the basic amino acid substrates of the porins, lysine and arginine.81,82 The mycobacterial outer membrane can serve as a permeability barrier, containing substrate specific water-filled porin channels to facilitate antibiotic uptake. MspA, an outer membrane porin of the rapidly growing Mycobacterium smegmatis has been observed to be selective for positively charged compounds and was found to facilitate the transport of zwitterionic β-lactams, like cephaloridine, as opposed to negatively charged cephalosporins, like cephalothin.83−85 While it is not yet clear that similar porins exist in slow-growing mycobacteria, such as Mtb, it has been observed that expression of the MspA porin in Mtb leads to decreased MIC values for zwitterionic β-lactam antibiotics as well as other antibiotics.86 This has relevance to the present series of compounds which contain both zwitterionic (10a and 10b) as well as negatively charged (13a and 13b) carbapenems. The decrease in in vitro potency observed in Table 1 of the 13 series, relative to the 10 series, is potentially due to a lessening of efficiency in penetrating the mycobacterial cell envelope, particularly in the case of the rapidly growing mycobacterium Mab, which may possess zwitterionic importing porins, to reach the transpeptidase targets on the surface of the cytoplasmic membrane.
Taken together, the data presented here highlight the potential of atypical carbapenems as drugs to treat mycobacterial infections. Atypical carbapenem 10a exhibited a surprising ability to maintain excellent in vitro antimycobacterial activity, even against strains which show high levels of resistance to meropenem. Further studies in determining the Ldt binding and acylation kinetics, stability to carbapenemases, and optimization of the atypical scaffold of these compounds are warranted and being explored.
Methods
Chemical Compounds
Carbapenems were stereospecifically synthesized as described in the Supporting Information.
Amikacin (AMK), rifampicin (RIF), and isoniazid (INH), meropenem, and clavulanic acid were purchased from Sigma-Aldrich. Avibactam was purchased from Med Chem Express.
Solution Preparations
Stock solutions of amikacin (AMK), rifampicin (RIF), and isoniazid (INH) were prepared according to the manufacturer’s instructions. Carbapenem compounds stocks were prepared in water at 10 mM concentration and then diluted to appropriate assay concentrations. Stocks of clavulanic acid and avibactam were made fresh every time and used at final concentrations of 5 μg/mL. All the stocks were stored at −80 °C.
Bacterial Strains and Culture Conditions
Bacterial strains Mtb CDC1551, 5 Mtb clinical isolates, Mab 390S, Mab subsp. abscessus clinical isolates (obtained from National Jewish Health, Colorado), were used in this study (Table S1). Mtb and Mab strains were cultured in Middlebrook 7H9 supplemented with 0.05% Tween80 and 10% oleic acid/albumin/dextrose/catalase (OADC) and incubated at 37 °C and 5% CO2.87−89 Kanamycin 50 μg/mL (KAN), cycloheximide 100 μg/mL, and amikacin 32 μg/mL (AMK) were added when appropriate. Kanamycin was included during routine culturing of luciferase reporter strains to ensure maintenance of this construct. Cycloheximide was included in all solid agar media to prevent fungal contamination.
Minimum Inhibitory Concentration (MIC) Assay
MICs of synthesized carbapenem compounds were determined using bioluminescent strains of Mab and Mtb(90,91) in solid white 384-well microtiter plates (Corning). A 16-point 2-fold serial dilution series of the compounds (starting at 40 μg/mL) was used to conduct MIC determination as described previously.90,92 MIC is defined as the lowest drug concentration at which more than 99% decrease in lux signal was observed as compared to the untreated control. MIC values reported are the average of 3 independent replicates with duplicate dose response curves on each plate (total of 6 replicates per sample).
Cytotoxicity Assay
Cytotoxicity was assessed using J774A.1 (murine macrophage-like) and HepG2 (human liver carcinoma) cell lines using 2-fold dilution of the compounds (starting at 40 μg/mL) as described previously.90,92
Time-Kill Kinetic Assay
We conducted a time-kill kinetic assay with the top C5α-substituted carbapenem (10a) and commercially available compound, meropenem. The assay is described in detail previously.90,92 Briefly, Mtb, CDC1551 cultures of OD600 of 0.01 were added to a solid-white 384-well plate containing compounds at final concentrations of 0, 1×, 4×, and 8× MIC in a total volume of 30 μL. The plate was incubated for 6 days for Mtb and an aliquot was taken at 0, 24 (Day 1), 72 (Day 3), and 144 h (Day 6) postinoculation, serially diluted and plated onto 7H10 quad-plates supplemented with OADC. Colonies were counted after 3–4 weeks of incubation at 37 °C and CFU/mL was calculated. Similarly, a time-kill assay was performed with Mab 390S culture where an aliquot was taken out at 0, 24, 48, and 72 h for plating. Colonies were counted after 5 days of incubation following plating. Our limit of detection by CFU/mL is 102 assuming at least 1 colony with the lowest dilution plated (101). The data in Figure 1 is an average from 4 independent experiments each with 3 technical replicates.
In Vitro Activity against Mtb and Mab Clinical Strains
Activity of compounds were evaluated against 5 clinical strains of Mtb from 5 different phylogenetic lineages.88 MIC determination was carried out in a 16 step 2-fold dilution series (starting at 40 μg/mL) as described previously.90,92 The screening plate was incubated for 5 days at 37 °C, thereafter resazurin dye was added at 1/10th of the total volume and incubated for 24 h. Fluorescence was measured at Ex/Em 530/590 with auto gain settings.
MIC of the compounds was also determined against Mab clinical strains using five bioluminescent clinical strains of Mab subsp. abscessus (Table S1) similarly to the MIC assay mentioned earlier and as described.90,92
LdtMt2-Adduct HRMS Analyses
A truncated version of LdtMt2 lacking amino acids 1–26 was purified as reported previously.77 Carbapenems were prepared as 20 mM or 40 mM stock solutions in ddH2O. LdtMt2 (10 μM) was incubated in the presence or absence of 200 μM 10a, 10b, 13a, or 13b in 50 mM HEPES, pH 7.5, for 1 or 5 h at 37 °C. Reactions were quenched with formic acid (0.1% final v/v). Samples were desalted following passage through a polyacrylamide spin column (Pierce). Desalted samples were analyzed by high-resolution mass spectrometry (HRMS) as previously reported.77 Briefly, UPLC-high resolution MS samples were analyzed on a Waters Acquity H-Class ultraperformance liquid chromatography (LC) system equipped with a multiwavelength UV-violet diode array detector (200–500 nm) in conjunction with a Waters Acquity BEH-300 ultraperformance LC column packed with a C4 stationary phase (2.1 × 50 mm; 1.7 μm) in tandem with high resolution MS analysis by a Waters Xevo-G2 quadrupole-TOF electrospray ionization mass spectrometer. Enzyme samples were resolved at 60 °C in order to improve peak resolution. The samples were resolved with a flow rate of 0.3 mL/min and the following mobile phase: 0–1 min 90% water, 10% ACN, 0.1% formic acid (FA); 1–7.5 min gradient up to 20% water, 80% ACN, 0.1% FA; 7.5–8.4 min 20% water, 80% ACN, 0.1% FA; 8.4–8.5 min gradient up to 90% water, 10% ACN, 0.1% FA; 8.5–10 min 90% water + 10% ACN 0.1% FA. Mass Spectra were deconvoluted using the maxEnt1 algorithm running as part of the Waters BioPharmaLynx data processing software package. Mass/intensity data were extracted from the processed data for further processing as necessary. Data were normalized and MS images were created using GraphPad Prism 7.
Acknowledgments
This work was supported by a grant from the National Institutes of Health to JDB (1R15AI142699).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.1c00185.
Description of synthetic procedures; Table of the strains (PDF)
Author Contributions
JDB, KHR, and LABB wrote the manuscript. JDB conceived the project and designed and synthesized the new carbapenems. LABB prepared samples for HRMS and interpreted the data. PM ran HRMS samples. RG and BSS conducted all antimicrobial assays.
The authors declare no competing financial interest.
Supplementary Material
References
- Migliori G. B.; Tiberi S.; Zumla A.; Petersen E.; Chakaya J. M.; Wejse C.; Munoz Torrico M.; Duarte R.; Alffenaar J. W.; Schaaf H. S.; Marais B. J.; Cirillo D. M.; Alagna R.; Rendon A.; Pontali E.; Piubello A.; Figueroa J.; Ferlazzo G.; Garcia-Basteiro A.; Centis R.; Visca D.; D’Ambrosio L.; Sotgiu G.; et al. (2020) MDR/XDR-TB management of patients and contacts: Challenges facing the new decade. The 2020 clinical update by the Global Tuberculosis Network. Int. J. Infect. Dis. 92, S15–S25. 10.1016/j.ijid.2020.01.042. [DOI] [PubMed] [Google Scholar]
- Lee M.-R.; Sheng W.-H.; Hung C.-C.; Yu C.-J.; Lee L.-N.; Hsueh P.-R. (2015) Mycobacterium abscessus complex infections in humans. Emerging Infect. Dis. 21, 1638–1646. 10.3201/2109.141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Zhao L.; Mao Y.; Ye M.; Guo Q.; Zhang Y.; Xu L.; Zhang Z.; Li B.; Chu H. (2019) Clinical Efficacy and Adverse Effects of Antibiotics Used to Treat Mycobacterium abscessus Pulmonary Disease. Front. Microbiol. 10, 1977. 10.3389/fmicb.2019.01977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skripconoka V.; Danilovits M.; Pehme L.; Tomson T.; Skenders G.; Kummik T.; Cirule A.; Leimane V.; Kurve A.; Levina K.; Geiter L. J.; Manissero D.; Wells C. D. (2013) Delamanid improves outcomes and reduces mortality in multidrug-resistant tuberculosis. Eur. Respir. J. 41, 1393–1400. 10.1183/09031936.00125812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakare R.; Soni I.; Dasgupta A.; Chopra S. (2015) Delamanid for the treatment of pulmonary multidrug-resistant tuberculosis. Drugs Today 51, 117–123. 10.1358/dot.2015.51.2.2245645. [DOI] [PubMed] [Google Scholar]
- Robinson H. J. (1943) Toxicity and efficacy of penicillin. J. Pharmacol. 77, 70–79. [Google Scholar]
- Smith M. I.; Emmart E. W. (1944) Action of penicillin extracts in experimental tuberculosis. Public Health Rep. 59, 417–423. 10.2307/4584828.19315967 [DOI] [Google Scholar]
- Bil’ko I. P.; Mil’chenko K. P.; Ginzburg T. S.; Sokalo S. V. (1982) Experimental study of the antibiotic activity of cefuroxime. Antibiotiki 27, 595–598. [PubMed] [Google Scholar]
- Iland C. N.; Baines S. (1949) The effect of penicillin on the tubercle bacillus; tubercle penicillinase. J. Pathol. Bacteriol. 61, 329–335. 10.1002/path.1700610303. [DOI] [Google Scholar]
- Kasik J. E. (1964) Activity of some semisynthetic penicillins on Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 10, 315–320. [PubMed] [Google Scholar]
- Vinogradova T. I.; Aleksandrova A. E.; Tschegoleva R. A. (1993) Cephalosporins as possible methods of etiotropic therapy of tuberculosis. Probl Tuberk 45–48. [PubMed] [Google Scholar]
- Chambers H. F.; Turner J.; Schecter G. F.; Kawamura M.; Hopewell P. C. (2005) Imipenem for treatment of tuberculosis in mice and humans. Antimicrob. Agents Chemother. 49, 2816–2821. 10.1128/AAC.49.7.2816-2821.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg T. B.; Cynamon M. H. (1987) Comparison of four β-lactamase inhibitors in combination with ampicillin against Mycobacterium tuberculosis. J. Antimicrob. Chemother. 19, 59–64. 10.1093/jac/19.1.59. [DOI] [PubMed] [Google Scholar]
- Chambers H. F.; Moreau D.; Yajko D.; Miick C.; Wagner c.; Hackbarth C.; Kocagoz S.; Rosenberg E.; Hadley W. K.; Nikaido H. (1995) Can penicillins and other β-lactam antibiotics be used to treat tuberculosis?. Antimicrob. Agents Chemother. 39, 2620–2624. 10.1128/AAC.39.12.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackbarth C. J.; Unsal I.; Chambers H. F. (1997) Cloning and sequence analysis of a class A β-lactamase from Mycobacterium tuberculosis H37Ra. Antimicrob. Agents Chemother. 41, 1182–1185. 10.1128/AAC.41.5.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voladri R. K.; Lakey D. L.; Hennigan S. H.; Menzies B. E.; Edwards K. M.; Kernodle D. S. (1998) Recombinant expression and characterization of the major beta-lactamase of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 42, 1375–1381. 10.1128/AAC.42.6.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugonnet J. E.; Blanchard J. S. (2007) Irreversible inhibition of the mycobacterium tuberculosis beta-lactarnase by clavulanate. Biochemistry 46, 11998–12004. 10.1021/bi701506h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; Wang Y. F.; Lu J.; Pang Y. (2016) In Vitro Activity of beta-Lactams in Combination with beta-Lactamase Inhibitors against Multidrug-Resistant Mycobacterium tuberculosis Isolates. Antimicrob. Agents Chemother. 60, 393–399. 10.1128/AAC.01035-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solapure S.; Dinesh N.; Shandil R.; Ramachandran V.; Sharma S.; Bhattacharjee D.; Ganguly S.; Reddy J.; Ahuja V.; Panduga V.; Parab M.; Vishwas K. G.; Kumar N.; Balganesh M.; Balasubramanian V. (2013) In vitro and in vivo efficacy of beta-lactams against replicating and slowly growing/nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 57, 2506–2510. 10.1128/AAC.00023-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugonnet J.-E.; Tremblay L. W.; Boshoff H. I.; Barry C. E. 3rd; Blanchard J. S. (2009) Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science 323 (5918), 1215–8. 10.1126/science.1167498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soroka D.; Dubee V.; Soulier-Escrihuela O.; Cuinet G.; Hugonnet J.-E.; Gutmann L.; Mainardi J.-L.; Arthur M. (2014) Characterization of broad-spectrum Mycobacterium abscessus class A β-lactamase. J. Antimicrob. Chemother. 69, 691–696. 10.1093/jac/dkt410. [DOI] [PubMed] [Google Scholar]
- Dubee V.; Bernut A.; Cortes M.; Lesne T.; Dorchene D.; Lefebvre A.-L.; Hugonnet J.-E.; Gutmann L.; Mainardi J.-L.; Herrmann J.-L.; Gaillard J.-L.; Kremer L.; Arthur M. (2014) β-lactamase inhibition by avibactam in Mycobacterium abscessus. J. Antimicrob. Chemother. 70, 1051–1058. 10.1093/jac/dku510. [DOI] [PubMed] [Google Scholar]
- Le Run E.; Atze H.; Arthur M.; Mainardi J.-L. (2019) Impact of relebactam-mediated inhibition of Mycobacterium abscessus BlaMab β-lactamase on the in vitro and intracellular efficacy of imipenem. J. Antimicrob. Chemother. 75, 379–383. 10.1093/jac/dkz433. [DOI] [PubMed] [Google Scholar]
- Story-Roller E.; Maggioncalda E. C.; Lamichhane G. (2019) Synergistic efficacy of β-lactam combinations against Mycobacterium abscessus pulmonary infection in mice. Antimicrob. Agents Chemother. 63, e00614–e00619. 10.1128/AAC.00614-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik A.; Ammerman N. C.; Lee J.; Martins O.; Kreiswirth B. N.; Lamichhane G.; Parrish N. M.; Nuermberger E. L. (2019) In Vitro Activity of the New beta-Lactamase Inhibitors Relebactam and Vaborbactam in Combination with beta-Lactams against Mycobacterium abscessus Complex Clinical Isolates. Antimicrob. Agents Chemother. 63, 1–25. 10.1128/AAC.02623-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey R.; Chen L.; Manca C.; Jenkins S.; Glaser L.; Vinnard C.; Stone G.; Lee J.; Mathema B.; Nuermberger E. L.; Bonomo R. A.; Kreiswirth B. N. (2019) Dual beta-Lactam Combinations Highly Active against Mycobacterium abscessus Complex In Vitro. mBio 10, e02895-18. 10.1128/mBio.02895-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik A.; Gupta C.; Fisher S.; Story-Roller E.; Galanis C.; Parrish N.; Lamichhane G. (2017) Combinations of avibactam and carbapenems exhibit enhanced potencies against drug-resistant Mycobacterium abscessus. Future Microbiol. 12, 473–480. 10.2217/fmb-2016-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik A.; Makkar N.; Pandey P.; Parrish N.; Singh U.; Lamichhane G. (2015) Carbapenems and Rifampin Exhibit Synergy against Mycobacterium tuberculosis and Mycobacterium abscessus. Antimicrob. Agents Chemother. 59, 6561–6567. 10.1128/AAC.01158-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Chauhan V.; Silva J. R. A.; Lameira J.; d’Andrea F. B.; Li S. G.; Ginell S. L.; Freundlich J. S.; Alves C. N.; Bailey S.; Cohen K. A.; Lamichhane G. (2017) Mycobacterium abscessus l,d-Transpeptidases Are Susceptible to Inactivation by Carbapenems and Cephalosporins but Not Penicillins. Antimicrob. Agents Chemother. 61, e00866-17. 10.1128/AAC.00866-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Run E.; Arthur M.; Mainardi J. L. (2018) In Vitro and Intracellular Activity of Imipenem Combined with Rifabutin and Avibactam against Mycobacterium abscessus. Antimicrob. Agents Chemother. 62, e00623-18. 10.1128/AAC.00623-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopeman R. C.; Harrison J.; Rathbone D. L.; Desai M.; Lambert P. A.; Cox J. A. G. (2020) Effect of Amoxicillin in combination with Imipenem-Relebactam against Mycobacterium abscessus. Sci. Rep. 10, 928–939. 10.1038/s41598-020-57844-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.; Cao X.; Yu J.; Zhan Q.; Yang J.; Wu X.; Wan B.; Liu Y.; Yu F. (2020) Antimicrobial susceptibility of Mycobacterium abscessus complex clinical isolates from a Chinese Tertiary Hospital. Infect. Drug Resist. 13, 2001–2010. 10.2147/IDR.S252485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei S.; Ihara H.; Togo S.; Nakamura A.; Fujimoto Y.; Watanabe J.; Kurokawa K.; Shibayama K.; Sumiyoshi I.; Ochi Y.; Iwai M.; Okabe T.; Chonan M.; Misawa S.; Ohsaka A.; Takahashi K. (2020) The synergetic effect of Imipenem-clarithromycin combination in the Mycobacteroides abscessus complex. BMC Microbiol. 20, 316. 10.1186/s12866-020-02000-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelder H. R.; Story-Roller E.; Lloyd E. P.; Kaushik A.; Bigelow K. M.; Maggioncalda E. C.; Nuermberger E. L.; Lamichhane G.; Townsend C. A. (2020) Development of a penem antibiotic against Mycobacteroides abscessus. Commun. Biol. 3, 741. 10.1038/s42003-020-01475-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahapatra S.; Scherman H.; Brennan P. J.; Crick D. C. (2005) N glycolylation of the nucleotide precursors of peptidoglycan biosynthesis of Mycobacterium spp. is altered by drug treatment. J. Bacteriol. 187, 2341–2347. 10.1128/JB.187.7.2341-2347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra A.; Munshi T.; Healy J.; Martin L. T.; Vollmer W.; Keep N. H.; Bhakta S. (2019) Cell wall peptidoglycan in Mycobacterium tuberculosis: an Achilles’ heel for the TB-causing pathogen. FEMS Microbiol. Rev. 43, 548–575. 10.1093/femsre/fuz016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavollay M.; Fourgeaud M.; Herrmann J. L.; Dubost L.; Marie A.; Gutmann L.; Arthur M.; Mainardi J. L. (2011) The Peptidoglycan of Mycobacterium abscessus Is Predominantly Cross-Linked by L,D-Transpeptidases. J. Bacteriol. 193, 778–782. 10.1128/JB.00606-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wietzerbin J.; Das B. C.; Petit J. F.; Lederer E.; Leyh-Bouille M.; Ghuysen J. M. (1974) Occurrence of D-alanyl-(D)-meso-diaminopimelic acid and meso-diaminopimelyl-meso-diaminopimelic acid interpeptide linkages in the peptidoglycan of Mycobacteria. Biochemistry 13, 3471–3476. 10.1021/bi00714a008. [DOI] [PubMed] [Google Scholar]
- Mainardi J.-L.; Fourgeaud M.; Hugonnet J.-E.; Dubost L.; Brouard J.-P.; Ouazzani J.; Rice L. B.; Gutmann L.; Arthur M. (2005) A Novel Peptidoglycan Cross-linking Enzyme for a β-Lactam-resistant Transpeptidation Pathway. J. Biol. Chem. 280, 38146–38152. 10.1074/jbc.M507384200. [DOI] [PubMed] [Google Scholar]
- Lavollay M.; Arthur M.; Fourgeaud M.; Dubost L.; Marie A.; Riegel P.; Gutmann L.; Mainardi J.-L. (2009) The β-lactam-sensitive D,D-carboxypeptidase activity of Pbp4 controls the L,D and D,D transpeptidation pathways in Corynebacterium jeikeium. Mol. Microbiol. 74, 650–661. 10.1111/j.1365-2958.2009.06887.x. [DOI] [PubMed] [Google Scholar]
- Sanders A. N.; Pavelka M. S. (2013) Phenotypic analysis of Escherichia coli mutants lacking L,D-transpeptidases. Microbiology (London, U. K.) 159, 1842–1852. 10.1099/mic.0.069211-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltier J.; Courtin P.; El Meouche I.; Lemee L.; Chapot-Chartier M.-P.; Pons J.-L. (2011) Clostridium difficile Has an Original Peptidoglycan Structure with a High Level of N-Acetylglucosamine Deacetylation and Mainly 3–3 Cross-links. J. Biol. Chem. 286, 29053–29062. 10.1074/jbc.M111.259150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey J.; Cass J.; Gasper J.; Ngo N.-D.; Wiggins P.; Manoil C. (2019) Essential gene deletions producing gigantic bacteria. PLoS Genet. 15, e1008195 10.1371/journal.pgen.1008195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang K. N.; Kazi M. I.; Bovermann H.; Ausman J.; Boutte C. C.; Boll J. M.; Biboy J.; Vollmer W.; Gray J. (2021) Septal Class A Penicillin-Binding Protein Activity and ld-Transpeptidases Mediate Selection of Colistin-Resistant Lipooligosaccharide-Deficient Acinetobacter baumannii. mBio 12, e02185-20 10.1128/mBio.02185-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. S.; Kim J.; Im H. N.; Yoon J. Y.; An D. R.; Yoon H. J.; Kim J. Y.; Min H. K.; Kim S.-J.; Lee J. Y.; Han B. W.; Suh S. W. (2013) Structural basis for the inhibition ofMycobacterium tuberculosisL,D-transpeptidase by Meropenem, a drug effective against extensively drug-resistant strains. Acta Crystallogr., Sect. D: Biol. Crystallogr. 69, 420–431. 10.1107/S0907444912048998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Kaushik A.; Lloyd E. P.; Li S.-G.; Mattoo R.; Ammerman N. C.; Bell D. T.; Perryman A. L.; Zandi T. A.; Ekins S.; Ginell S. L.; Townsend C. A.; Freundlich J. S.; Lamichhane G. (2017) Non-classical transpeptidases yield insight into new antibacterials. Nat. Chem. Biol. 13, 54–61. 10.1038/nchembio.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubée V.; Triboulet S.; Mainardi J.-L.; Ethève-Quelquejeu M.; Gutmann L.; Marie A.; Dubost L.; Hugonnet J.-E.; Arthur M. (2012) Inactivation of Mycobacterium tuberculosis l,d-transpeptidase LdtMt1 by carbapenems and cephalosporins. Antimicrob. Agents Chemother. 56 (8), 4189–4195. 10.1128/AAC.00665-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Arora K.; Lloyd J. R.; Lee Y.; Nair V.; Fischer E.; Boshoff H. I. M.; Barry C. E. (2012) Meropenem inhibits D,D-carboxypeptidase activity in Mycobacterium tuberculosis. Mol. Microbiol. 86, 367–381. 10.1111/j.1365-2958.2012.08199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordillot M.; Dubée V.; Triboulet S.; Dubost L.; Marie A.; Hugonnet J.-E.; Arthur M.; Mainardi J.-L. (2013) In vitro cross-linking of Mycobacterium tuberculosis peptidoglycan by L,D-transpeptidases and inactivation of these enzymes by carbapenems. Antimicrob. Agents Chemother. 57, 5940–5945. 10.1128/AAC.01663-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdemli S. B.; Gupta R.; Bishai W. R.; Lamichhane G.; Amzel L. M.; Bianchet M. A. (2012) Targeting the cell wall of Mycobacterium tuberculosis: structure and mechanism of L,D-transpeptidase 2. Structure (Oxford, U. K.) 20, 2103–2115. 10.1016/j.str.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R.; Lavollay M.; Mainardi J.-L.; Arthur M.; Bishai W. R.; Lamichhane G. (2010) The Mycobacterium tuberculosis protein LdtMt2 is a nonclassical transpeptidase required for virulence and resistance to amoxicillin. Nat. Med. 16, 466–469. 10.1038/nm.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dousa K. M.; Kurz S. G.; Taracila M. A.; Bethel C. R.; Barnes M. D.; Bonomo R. A.; Taracila M. A.; Barnes M. D.; Bonomo R. A.; Bonfield T.; Selvaraju S.; Abdelhamed A. M.; Kreiswirth B. N.; Boom W. H.; Kasperbauer S. H.; Daley C. L.; Bonomo R. A.; Bonomo R. A.; Bonomo R. A.; Bonomo R. A.; Bonomo R. A.; Bonomo R. A.; Bonomo R. A. (2020) Insights into the L,D-transpeptidases and D,D-carboxypeptidase of Mycobacterium abscessus: ceftaroline, imipenem and novel diazabicyclooctanes inhibitors. Antimicrob. Agents Chemother. 64, e00098-20. 10.1128/AAC.00098-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . (2019) WHO Guidelines Approved by the Guidelines Review Committee. In WHO Consolidated Guidelines on Drug-Resistant Tuberculosis Treatment, World Health Organization, Geneva. [Google Scholar]
- Sotgiu G.; D’Ambrosio L.; Centis R.; Tiberi S.; Esposito S.; Dore S.; Spanevello A.; Migliori G. B. (2016) Carbapenems to treat multidrug and extensively drug-resistant tuberculosis: a systematic review. Int. J. Mol. Sci. 17, 373. 10.3390/ijms17030373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiberi S.; Sotgiu G.; D’Ambrosio L.; Centis R.; Abdo Arbex M.; Arrascue E. A.; Alffenaar J. W.; Caminero J. A.; Gaga M.; Gualano G.; Skrahina A.; Solovic I.; Sulis G.; Tadolini M.; Guizado V. A.; De Lorenzo S.; Arias A. J. R.; Scardigli A.; Akkerman O. W.; Aleksa A.; Artsukevich J.; Auchynka V.; Bonini E. H.; Marin F. A. C.; Lopez L. C.; de Vries G.; Dore S.; Kunst H.; Matteelli A.; Moschos C.; Palmieri F.; Papavasileiou A.; Payen M.-C.; Piana A.; Spanevello A.; Vasquez D. V.; Viggiani P.; White V.; Zumla A.; Migliori G. B. (2016) Comparison of effectiveness and safety of imipenem/clavulanate- versus Meropenem/clavulanate-containing regimens in the treatment of MDR- and XDR-TB. Eur. Respir. J. 47, 1758–1766. 10.1183/13993003.00214-2016. [DOI] [PubMed] [Google Scholar]
- Tiberi S.; Payen M.-C.; Sotgiu G.; D’Ambrosio L.; Guizado V. A.; Alffenaar J. W.; Arbex M. A.; Caminero J. A.; Centis R.; De Lorenzo S.; Gaga M.; Gualano G.; Arias A. J. R.; Scardigli A.; Skrahina A.; Solovic I.; Sulis G.; Tadolini M.; Akkerman O. W.; Arrascue E. A.; Aleska A.; Avchinko V.; Bonini E. H.; Marin F. A. C.; Lopez L. C.; de Vries G.; Dore S.; Kunst H.; Matteelli A.; Moschos C.; Palmieri F.; Papavasileiou A.; Spanevello A.; Vasquez D. V.; Viggiani P.; White V.; Zumla A.; Migliori G. B. (2016) Effectiveness and safety of Meropenem/clavulanate-containing regimens in the treatment of MDR- and XDR-TB. Eur. Respir. J. 47, 1235–1243. 10.1183/13993003.02146-2015. [DOI] [PubMed] [Google Scholar]
- Tiberi S.; D’Ambrosio L.; De Lorenzo S.; Viggiani P.; Centis R.; Sotgiu G.; Alffenaar J. W. C.; Migliori G. B. (2016) Ertapenem in the treatment of multidrug-resistant tuberculosis: first clinical experience. Eur. Respir. J. 47, 333–336. 10.1183/13993003.01278-2015. [DOI] [PubMed] [Google Scholar]
- Haworth C. S.; Floto R. A.; Banks J.; Capstick T.; Fisher A. J.; Gorsuch T.; Laurenson I. F.; Leitch A.; Loebinger M. R.; Wilson R.; Milburn H. J.; Nightingale M.; Ormerod P.; Shingadia D.; Smith D.; Whitehead N.; Floto R. A. (2017) British Thoracic Society guidelines for the management of non-tuberculous mycobacterial pulmonary disease (NTM-PD). Thorax 72, ii1–ii64. 10.1136/thoraxjnl-2017-210927. [DOI] [PubMed] [Google Scholar]
- Flume P. A. (2016) US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis. J. Cystic Fibrosis 15, 139–140. 10.1016/S1569-1993(16)00018-7. [DOI] [PubMed] [Google Scholar]
- Kwak N.; Park J.; Yim J.-J.; Dalcolmo M. P.; Gayoso R.; Daley C. L.; Eather G.; Hasegawa N.; Jhun B. W.; Koh W.-J.; Namkoong H.; Thomson R.; van Ingen J.; Zweijpfenning S. M. H. (2019) Mycobacterium abscessus pulmonary disease: individual patient data meta-analysis. Eur. Respir. J. 54, 1801991. 10.1183/13993003.01991-2018. [DOI] [PubMed] [Google Scholar]
- Elshamy A. A.; Aboshanab K. M. (2020) A review on bacterial resistance to carbapenems: epidemiology, detection and treatment options. Future Sci. OA 6, FSO438. 10.2144/fsoa-2019-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomo R. A.; Burd E. M.; Conly J.; Limbago B. M.; Poirel L.; Segre J. A.; Westblade L. F. (2018) Carbapenemase-producing organisms: a global scourge. Clin. Infect. Dis. 66, 1290–1297. 10.1093/cid/cix893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K. (2018) Past and Present Perspectives on β-Lactamases. Antimicrob. Agents Chemother. 62, e01076-18. 10.1128/AAC.01076-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codjoe F. S.; Codjoe F. S.; Donkor E. S. (2018) Carbapenem Resistance: A Review. Med. Sci. 6, 1. 10.3390/medsci6010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naas T.; Dortet L.; Iorga B. I. (2016) Structural and functional aspects of class A carbapenemases. Curr. Drug Targets 17, 1006–1028. 10.2174/1389450117666160310144501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docquier J.-D.; Mangani S. (2016) Structure-Function Relationships of Class D Carbapenemases. Curr. Drug Targets 17, 1061–1071. 10.2174/1389450116666150825115824. [DOI] [PubMed] [Google Scholar]
- Jeon J. H.; Lee J. H.; Lee J. J.; Park K. S.; Karim A. M.; Lee C.-R.; Jeong B. C.; Lee S. H. (2015) Structural basis for carbapenem-hydrolyzing mechanisms of carbapenemases conferring antibiotic resistance. Int. J. Mol. Sci. 16, 9654–9692. 10.3390/ijms16059654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp-Wallace K. M.; Endimiani A.; Taracila M. A.; Bonomo R. A. (2011) Carbapenems: past, present, and future. Antimicrob. Agents Chemother. 55, 4943–4960. 10.1128/AAC.00296-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoue H.; Narukawa Y. (1989) Synthesis and antibacterial activity of 5-methylcarbapenems. J. Antibiot. 42, 1100–13. 10.7164/antibiotics.42.1100. [DOI] [PubMed] [Google Scholar]
- Iannazzo L.; Soroka D.; Triboulet S.; Fonvielle M.; Compain F.; Dubée V.; Mainardi J. L.; Hugonnet J. E.; Braud E.; Arthur M.; Etheve-Quelquejeu M. (2016) Routes of Synthesis of Carbapenems for Optimizing Both the Inactivation of L,D-Transpeptidase LdtMt1 of Mycobacterium tuberculosis and the Stability toward Hydrolysis by β-Lactamase BlaC. J. Med. Chem. 59, 3427–3438. 10.1021/acs.jmedchem.6b00096. [DOI] [PubMed] [Google Scholar]
- Murahash S.; Saito T.; Naota T.; Kumobayashi H.; Akutagawa S. (1991) Ruthenium-catalyzed oxidation of β-lactams with molecular oxygen and aldehydes. Tetrahedron Lett. 32, 5991–5994. 10.1016/S0040-4039(00)79446-9. [DOI] [Google Scholar]
- Ueda Y., and Roberge G. (1986) Carbapenem intermediates. GB2173801A.
- Soroka D.; Ourghanlian C.; Compain F.; Fichini M.; Dubee V.; Mainardi J.-L.; Hugonnet J.-E.; Arthur M. (2016) Inhibition of β-lactamases of mycobacteria by avibactam and clavulanate. J. Antimicrob. Chemother. 72, 1081–1088. 10.1093/jac/dkw546. [DOI] [PubMed] [Google Scholar]
- Lohans C. T.; Chan H. T. H.; Malla T. R.; Kumar K.; Kamps J. J. A. G.; McArdle D. J. B.; van Groesen E.; de Munnik M.; Tooke C. L.; Spencer J.; Paton R. S.; Brem J.; Schofield C. J. (2019) Non-Hydrolytic β-Lactam Antibiotic Fragmentation by l,d-Transpeptidases and Serine β-Lactamase Cysteine Variants. Angew. Chem., Int. Ed. 58, 1990–1994. 10.1002/anie.201809424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow C.; Xu H.; Blanchard J. S. (2013) Kinetic characterization of hydrolysis of nitrocefin, cefoxitin, and Meropenem by β-lactamase from Mycobacterium tuberculosis. Biochemistry 52, 4097–4104. 10.1021/bi400177y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.; Xie H.; Sule P.; Hassounah H.; Graviss E. A.; Kong Y.; Cirillo J. D.; Rao J. (2014) Fluorogenic Probes with Substitutions at the 2 and 7 Positions of Cephalosporin are Highly BlaC-Specific for Rapid Mycobacterium tuberculosis Detection. Angew. Chem., Int. Ed. 53, 9360–9364. 10.1002/anie.201405243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi T. A.; Marshburn R. L.; Stateler P. K.; Brammer Basta L. A. (2019) Phylogenetic and Biochemical Analyses of Mycobacterial l,d-Transpeptidases Reveal a Distinct Enzyme Class That Is Preferentially Acylated by Meropenem. ACS Infect. Dis. 5, 2047–2054. 10.1021/acsinfecdis.9b00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner E. M.; Schneider G.; Schnell R. (2017) Binding and processing of β-lactam antibiotics by the transpeptidase LdtMt2 from Mycobacterium tuberculosis. FEBS J. 284, 725–741. 10.1111/febs.14010. [DOI] [PubMed] [Google Scholar]
- Sayed A. R. M.; Shah N. R.; Basso K. B.; Kamat M.; Jiao Y.; Moya B.; Sutaria D. S.; Lang Y.; Tao X.; Liu W.; Shin E.; Zhou J.; Werkman C.; Louie A.; Drusano G. L.; Bulitta J. B. (2020) First penicillin-binding protein occupancy patterns for 15 β-lactams and β-lactamase inhibitors in Mycobacterium abscessus. Antimicrob. Agents Chemother. 65, e01956-20 10.1128/AAC.01956-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wivagg C. N.; Wellington S.; Gomez J. E.; Hung D. T. (2016) Loss of a class A penicillin-binding protein alters β-lactam susceptibilities in Mycobacterium tuberculosis. ACS Infect. Dis. 2, 104–110. 10.1021/acsinfecdis.5b00119. [DOI] [PubMed] [Google Scholar]
- Trias J.; Nikaido H. (1990) Outer membrane protein D2 catalyzes facilitated diffusion of carbapenems and penems through the outer membrane of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 34 (1), 52–7. 10.1128/AAC.34.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trias J.; Nikaido H. (1990) Protein D2 channel of the Pseudomonas aeruginosa outer membrane has a binding site for basic amino acids and peptides. J. Biol. Chem. 265 (26), 15680–4. 10.1016/S0021-9258(18)55452-1. [DOI] [PubMed] [Google Scholar]
- Trias J.; Benz R. (1994) Permeability of the cell wall of Mycobacterium smegmatis. Mol. Microbiol. 14, 283–90. 10.1111/j.1365-2958.1994.tb01289.x. [DOI] [PubMed] [Google Scholar]
- Stephan J.; Mailaender C.; Etienne G.; Daffe M.; Niederweis M. (2004) Multidrug resistance of a porin deletion mutant of Mycobacterium smegmatis. Antimicrob. Agents Chemother. 48, 4163–4170. 10.1128/AAC.48.11.4163-4170.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilchanka O.; Pavlenok M.; Niederweis M. (2008) Role of porins for uptake of antibiotics by Mycobacterium smegmatis. Antimicrob. Agents Chemother. 52, 3127–3134. 10.1128/AAC.00239-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailaender C.; Reiling N.; Engelhardt H.; Bossmann S.; Ehlers S.; Niederweis M. (2004) The MspA porin promotes growth and increases antibiotic susceptibility of both Mycobacterium bovis BCG and Mycobacterium tuberculosis. Microbiology (London, U. K.) 150, 853–864. 10.1099/mic.0.26902-0. [DOI] [PubMed] [Google Scholar]
- Fleischmann R. D.; Alland D.; Eisen J. A.; Carpenter L.; White O.; Peterson J.; DeBoy R.; Dodson R.; Gwinn M.; Haft D.; Hickey E.; Kolonay J. F.; Nelson W. C.; Umayam L. A.; Ermolaeva M.; Salzberg S. L.; Delcher A.; Utterback T.; Weidman J.; Khouri H.; Gill J.; Mikula A.; Bishai W.; Jacobs W. R. Jr; Jr; Venter J. C.; Fraser C. M. (2002) Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J. Bacteriol. 184, 5479–5490. 10.1128/JB.184.19.5479-5490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homolka S.; Niemann S.; Russell D. G.; Rohde K. H. (2010) Functional genetic diversity among Mycobacterium tuberculosis complex clinical isolates: delineation of conserved core and lineage-specific transcriptomes during intracellular survival. PLoS Pathog. 6, e1000988. 10.1371/journal.ppat.1000988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard S. T.; Rhoades E.; Recht J.; Pang X.; Alsup A.; Kolter R.; Lyons C. R.; Byrd T. F. (2006) Spontaneous reversion of Mycobacterium abscessus from a smooth to a rough morphotype is associated with reduced expression of glycopeptidolipid and reacquisition of an invasive phenotype. Microbiology 152, 1581–1590. 10.1099/mic.0.28625-0. [DOI] [PubMed] [Google Scholar]
- Rodrigues Felix C.; Gupta R.; Geden S.; Roberts J.; Winder P.; Pomponi S. A.; Diaz M. C.; Reed J. K.; Wright A. E.; Rohde K. H. (2017) Selective Killing Of Dormant Mycobacterium tuberculosis By Marine Natural Products. Antimicrob. Agents Chemother. 61, e00743-17. 10.1128/AAC.00743-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R.; Netherton M.; Byrd T. F.; Rohde K. H. (2017) Reporter-Based Assays for High-Throughput Drug Screening against Mycobacterium abscessus. Front. Microbiol. 8, 2204. 10.3389/fmicb.2017.02204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R.; Rodrigues Felix C.; Akerman M. P.; Akerman K. J.; Slabber C. A.; Wang W.; Adams J.; Shaw L. N.; Tse-Dinh Y. C.; Munro O. Q.; Rohde K. H. (2018) Evidence for Inhibition of Topoisomerase 1A by Gold(III) Macrocycles and Chelates Targeting Mycobacterium tuberculosis and Mycobacterium abscessus. Antimicrob. Agents Chemother. 62, e01696-17. 10.1128/AAC.01696-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.