

Abstract
Gleason score, a measure of prostate tumor differentiation, is the strongest predictor of lethal prostate cancer at the time of diagnosis. Metabolomic profiling of tumor and of patient serum could identify biomarkers of aggressive disease and lead to the development of a less-invasive assay to perform active surveillance monitoring. Metabolomic profiling of prostate tissue and serum samples was performed. Metabolite levels and metabolite sets were compared across Gleason scores. Machine learning algorithms were trained and tuned to predict transformation or differentiation status from metabolite data. A total of 135 metabolites were significantly different (Padjusted < 0.05) in tumor versus normal tissue, and pathway analysis identified one sugar metabolism pathway (Padjusted = 0.03). Machine learning identified profiles that predicted tumor versus normal tissue (AUC of 0.82 ± 0.08). In tumor tissue, 25 metabolites were associated with Gleason score (unadjusted P < 0.05), 4 increased in high grade while the remainder were enriched in low grade. While pyroglutamine and 1,5-anhydroglucitol were correlated (0.73 and 0.72, respectively) between tissue and serum from the same patient, no metabolites were consistently associated with Gleason score in serum. Previously reported as well as novel metabolites with differing abundance were identified across tumor tissue. However, a “metabolite signature” for Gleason score was not obtained. This may be due to study design and analytic challenges that future studies should consider.
Introduction
Gleason score, a pathologic measure of the degree of differentiation of prostate tumor tissue, is one of the strongest predictors of lethal prostate cancer (1). Many men diagnosed with low-grade Gleason score 6 tumors are treated with radical prostatectomy (RP) or radio-therapy, resulting in substantial overtreatment of the disease. Recently, more patients with low-grade prostate cancer have been recommended for active surveillance (2), a continued monitoring of potential disease progression with repeated PSA measurements and prostate biopsies. However, due to the multifocality and heterogeneity of prostate cancer, biopsies can miss higher grade tumors. Approximately 43%–65% of patients who are Gleason score 6 at diagnosis are upgraded at RP (3–5). PSA is currently the main biomarker used to detect the presence of high-grade disease, with a recent study showing that among men with Gleason score 6 at diagnosis, those with PSA 10–20 ng/μL had a 30% chance of being upgraded at RP (6). Commercially available genomic testing predicts outcome based on the lesion biopsied (7, 8) rather than detecting the presence of occult higher grade tumors. A nontissue-based, less-invasive assay would be an ideal method to address the heterogeneity and multifocality of prostate cancer to monitor patients and inform treatment decisions.
Metabolic rewiring appears to be specific to the genomic alterations that occur in tumor cells (9). Thus, metabolic profiling may help identify new biomarkers to differentiate high from low-grade prostate cancer as these differentiation states appear to be molecularly distinct (10–12). Metabolic markers have previously been shown to differ not only in patients with and without prostate cancer, but also in aggressive and nonaggressive disease (13). In tumor tissue, McDunn and colleagues (14) identified several metabolites associated with Gleason score, including palmitoyl sphingomyelin, citrate, choline, and ADP. Giskeodegard and colleagues (15) observed that high-grade tumors had lower levels of citrate and spermine when compared with low-grade tumors. Using in situ mass spectrometry imaging, lipid abundance has been shown to tightly correlate with increasing Gleason score (16), and citrate represents the predominant source for de novo lipogenesis that is responsible for lipid abundance. These data suggest that metabolomics could contribute molecular insights into the metabolic rewiring that underpins the biological behavior and differentiation of prostate tumors.
As tumors with higher Gleason score appear to produce and may excrete different metabolites or differing amounts from tumors with only lower grade present, we hypothesize that these metabolites could potentially be detected using serum metabolomics. This would be a necessary first step in the development of a less-invasive metabolomics-based assay to monitor active surveillance patients. Previous work in this field has had limited success. Fan and colleagues identified metabolites in serum that could distinguish prostate cancer from benign prostatic hyperplasia. However, this “signature” was not associated with Gleason score (17). The authors did observe that several metabolites were significantly different in the serum of patients with low versus those with intermediate/high Gleason score prostate cancer. Osl and colleagues attempted to develop a serum metabolite signature to distinguish men with Gleason score 6 from men with Gleason score 8–10, but did not identify any reliable candidate metabolites (18).
To identify metabolites associated with Gleason score in prostate tissue, we performed untargeted metabolomic profiling in prostate tumor and matched benign prostate tissue. We then examined metabolite levels in a large population of patients with prostate cancer to identify metabolite biomarkers of Gleason score, which could provide an additional less-invasive method to monitor men during active surveillance.
Materials and Methods
Cohort description
We utilized samples from the Dana-Farber Cancer Institute (DFCI)/Harvard Cancer Center SPORE Prostate Cancer Cohort. The cohort has been described previously (19). Briefly, the SPORE Prostate Cancer Cohort includes clinical information, blood and tissue samples from more than 6,000 patients with prostate cancer followed prospectively. Fresh-frozen RP specimens were selected from the DFCI Gelb Center biobank and database, as part of DFCI Protocols 01–045, 11–104, 17–000, and 09–171, approved by the DFCI/Harvard Cancer Center Institutional Review Board. Research was conducted in accordance with the U.S. Common Rule and written informed consent was obtained from each subject for use of clinical data and specimens for research purposes. For this study, we utilized fresh-frozen RP specimens and serum samples for metabolomics profiling. All serum samples were obtained from a blood sample taken after diagnosis but prior to RP. Patients’ fasting status prior to serum collection is unknown. We included tumor specimens from 124 patients, with matched normal prostate tissue from 105. We included serum from 94 patients (“Original” cohort), some of whom also had their tissue profiled (see numbers below). We additionally profiled serum from men (“Upgrading” cohort) who had Gleason score 6 at biopsy and remained Gleason score 6 at RP (N = 50), and from men with Gleason score 6 at biopsy who were upgraded to Gleason score 7 or higher at RP (N = 50). All Gleason scores described here for the “Upgrading” cohort are based on medical records. Comprehensive Gleason score rereview data (described below) was available for 141 of the total 194 patients included in the final serum analysis.
RP specimens and comprehensive pathology rereview
RP specimen were received fresh from the operating room, weighed without the seminal vesicles and all three dimensions [apical to basal (vertical), left to right (transverse), and anterior to posterior (sagittal)] recorded. Subsequently, the entire specimen was inked; apex to base slices (5–10 mm thick) were sliced transversely. Each slice was cut in quarters, and embedded in separate cassettes. Alternate transverse slices were formalin-fixed paraffin embedded (FFPE) and embedded in optimal cutting temperature (OCT) compound and stored in liquid nitrogen. Tissues were fixed for 24 hours in 4% neutral buffered formalin. A total of 5-μm-thick sections cut from FFPE and OCT blocks were stained with hematoxylin and eosin (H&E) and examined histologically. For all tissue analyses, Gleason score was assigned as the Gleason score of the tissue block utilized. One tumor focus per patient was chosen to best represent the Gleason score of the patient in the final report. To create a cleaner comparison of grade 3 versus grade 4/5, we compared Gleason score 6 with Gleason score ≥8.
All slides and tissue blocks (FFPE and frozen) for 141 of the 194 patients whose serum was included in this study were recovered. Study pathologists (H. El Fandy, F. Giunchi, R. Lis, H. Coulson, M. Loda) performed a comprehensive rereview of all recovered prostate tissue. The dimensions of the area of tumor for each tissue slice/quadrant were assessed, and the percentage of each Gleason grade in each area was noted (Supplementary Fig. S1). We determined the total tumor volume as described previously (20), with slice thickness estimated from the prostate measurements and total number of slices. We determined the percent of Gleason grade 3, 4, and 5 by multiplying the percent by the area for each region of tumor, and then summing across the slices and dividing by the total area. If these data suggested a different Gleason score than that from the pathology records, this new Gleason score was used for the serum analysis. For serum analyses, we compared patients with Gleason score 6 to those with Gleason score ≥7 as our goal was to detect the presence of any higher grade (≥4) tumor.
Metabolomic profiling
For each individual in the study, tissue sections of 5 μm thickness were prepared for subsequent staining with H&E. Areas of tumor and normal reflecting the Gleason score reported at signout were identified and cored. Approximately 1 mg of tissue, and 100 μL of serum were sent to Metabolon, Inc. Metabolon prepared the frozen tissue cores and serum samples for analysis using their proprietary solvent extraction method. The extracted supernatant was split into equal parts for analysis on the GC-MS and LC/MS-MS platforms. In addition, internal standards were added to each sample for normalization and quality control purposes. Identification of known chemical entities is based on comparison with metabolomic library entries of purified standards. Approximately 1,500 commercially available purified standard biochemicals had been acquired for both the LC and GC platforms for determination of their analytic characteristics. The combination of chromatographic properties and mass spectra gives an indication of a match to a specific compound or an isobaric entity. We previously published a detailed methodology (21).
Data preprocessing and normalization
Tissue and serum datasets were processed and normalized separately. Metabolites with more than 50% missing values were excluded from the analysis. After filtering, measurements were normalized to the run day median. Missing values were imputed using the k-nearest neighbors imputation algorithm (k = 5; ref. 22). Metabolomic measurements for samples that were profiled multiple times were averaged.
In tissue specimens, the number of metabolites detected by batch ranged from 307 to 390; 222 common metabolites were measured in all three batches. After filtering out sparse metabolites, 214 were retained for tissue analyses. In serum specimens, the number of metabolites detected by batch ranged from 376 to 455; 246 common metabolites were detected in all batches. After filtering out sparse metabolites, 238 were retained for serum analyses.
Statistical analysis
For tumor versus normal comparisons, we used a subset of 105 paired samples. Comparisons were performed using nonparametric Wilcoxon signed-rank test. For the analysis of Gleason score within tissue and serum, levels of each metabolite were compared across Gleason score categories with nonparametric Mann–Whitney rank-sum test. Unsupervised clustering for heatmaps was performed using Canberra distance applied to the metabolite abundance ranks across specimens.
Metabolomic pathway and superpathway annotations were provided by Metabolon. Annotations were subset to the list of observed metabolites. Pathways with less than five metabolites available in our data were removed from the analyses. The enriched pathways were identified using Mann–Whitney test, as implemented in limma (23), including metabolites according to their P values in the corresponding comparisons, or using Fisher exact test by cross-tabulating metabolites by significance status and pathway membership.
P values for individual metabolite and pathway results were corrected for multiple testing using the Benjamini–Hochberg FDR method (24) and reported as adjusted P values.
Machine learning
We considered three binary classification machine learning (ML) tasks: (i) distinguishing tumor tissue from normal tissue; (ii) distinguishing Gleason 6 tissue from Gleason ≥8 tissue; and (iii) distinguishing Gleason 6 serum from Gleason ≥7 serum. For each prediction task, data were randomly partitioned into training and testing sets according to a 75%/25% split. Models were fitted and tuned on the training set (using 5-fold cross-validation) and evaluated on the held-out testing set to assess model performance on unseen data.
For each task, logistic regression was used as a baseline model. We also evaluated L1-regularized least absolute shrinkage and selection operator (LASSO) regression (25) and L2-regularized ridge regression (26). Because of the high dimensionality of the data, we also considered support vector classifiers (SVC; ref. 27) and random forest (RF) classifiers(28). Tree-based ML models such as RF have the additional benefit of implicitly handling interactions between variables, making them well suited for metabolomics analyses where there are complex relationships between predictors (in general, a tree of depth n will capture interactions of order n). For both SVC and RF, grid search with 5-fold cross-validation was used to tune hyperparameters.
Finally, we employed automated ML (AutoML) methods to leverage recent advances in hyperparameter search and selection (29), implemented in Auto-Sklearn (30). More specifically, these AutoML methods consist of two phases: Bayesian hyperparameter optimization followed by greedy ensemble construction. Bayesian hyperparameter optimization (31) iteratively samples the search space according to an acquisition function designed to balance exploration of unsampled search space and exploitation of the search space near the top-performing hyperparameters, allowing efficient search of high-dimensional parameter space in an automated and empirical manner. We use balanced accuracy as the model performance metric during AutoML optimization to mitigate any potential problems due to class imbalance. Under the Auto-Sklearn paradigm, hyperparameters that are searched include not only numerical model parameters, but also the choice of preprocessing procedures and the choice of models themselves.
Throughout the course of hyperparameter selection, individual models are automatically saved and then ensemble selection is performed in a greedy manner [i.e., starting with an empty ensemble and iteratively adding the model that provides the greatest performance boost on held-out validation data (32)]. The AutoML fitting process was constrained to 24 hours of runtime, with a limit of 6 minutes per model.
Performance of ML models was recorded across several metrics: F1 score, AUC, accuracy, and balanced accuracy. Sensitivity and specificity were also computed to facilitate a possible clinical interpretation. To evaluate model robustness, we assessed model performance using 10-fold cross-validation. Data were randomly partitioned into 10 folds, stratified so as to maintain an even class balance among folds. Through 10 iterations, each fold was used once as the held-out validation set to assess the performance of the model when fit using the other 9 folds.
Results
The numbers of participants with Gleason score for each sample type are provided in Table 1. While all samples were included in data processing, only the number of samples described in the Materials and Methods section and below were included in each analysis.
Table 1.
Sample sizes and Gleason scores for each subcohort. Gleason grades are based on rereviewed values, where available, and abstracted from medical records for the remaining cases.
| 6 | 3+4 | 4+3 | 8+ | NA | |
|---|---|---|---|---|---|
| Tumor tissue | 53 | 15 | 16 | 33 | 7 |
| Normal tissuea | 43 | 12 | 15 | 31 | 5 |
| Serum samples (original) | 21 | 38 | 24 | 11 | 0 |
| Serum samples (upgrading) | 53 | 38 | 8 | 1 | 0 |
Gleason score is given for the block from which the adjacent normal tissue was sampled.
Comparisons of tumor and normal tissue metabolomics
Comparing tumor with normal tissue, of the 214 metabolites detected and tested, 135 had significantly (Padjusted < 0.05) different abundances (Fig. 1). The two largest unsupervised clusters were differentially enriched for tumor specimens (Fisher exact test, P < 10−4). The pathway analysis identified the fructose, mannose, galactose, starch, and sucrose metabolism pathway as differentially expressed (Padjusted = 0.03) while the amino acid superpathway was marginally significant (Padjusted = 0.06).
Figure 1.

Heatmap of top differential (unadjusted P < 0.05) metabolites between paired tumor and normal prostate tissue specimens.
Comparisons of high and low Gleason score tumor and normal tissue metabolomics
We compared the metabolome of 33 high (≥8) and 53 low (6) Gleason score tumor tissue. There were no differentially abundant metabolites at 0.05 FDR. Six metabolites were significant at Padjusted <0.2, including previously reported metabolites, such as citrate, spermine, and alpha-ketoglutarate. Figure 2 shows a heatmap for 25 metabolites with unadjusted P < 0.05. In a clustering analysis, there was no statistically significant difference between the distributions of Gleason grade of tumors belonging to two clusters (Fisher exact test P = 0.35). At the pathway level, while no pathways reached statistical significance, the top ranked pathway was the polyamine metabolism subpathway (Padjusted = 0.09). In normal prostate tissue, six metabolites were nominally significant in the comparison between normal specimens obtained from the same block as tumors with high and low Gleason score (unadjusted P < 0.05; Supplementary Fig. S2). At the pathway level, the peptide superpathway reached adjusted P value of 0.01.
Figure 2.
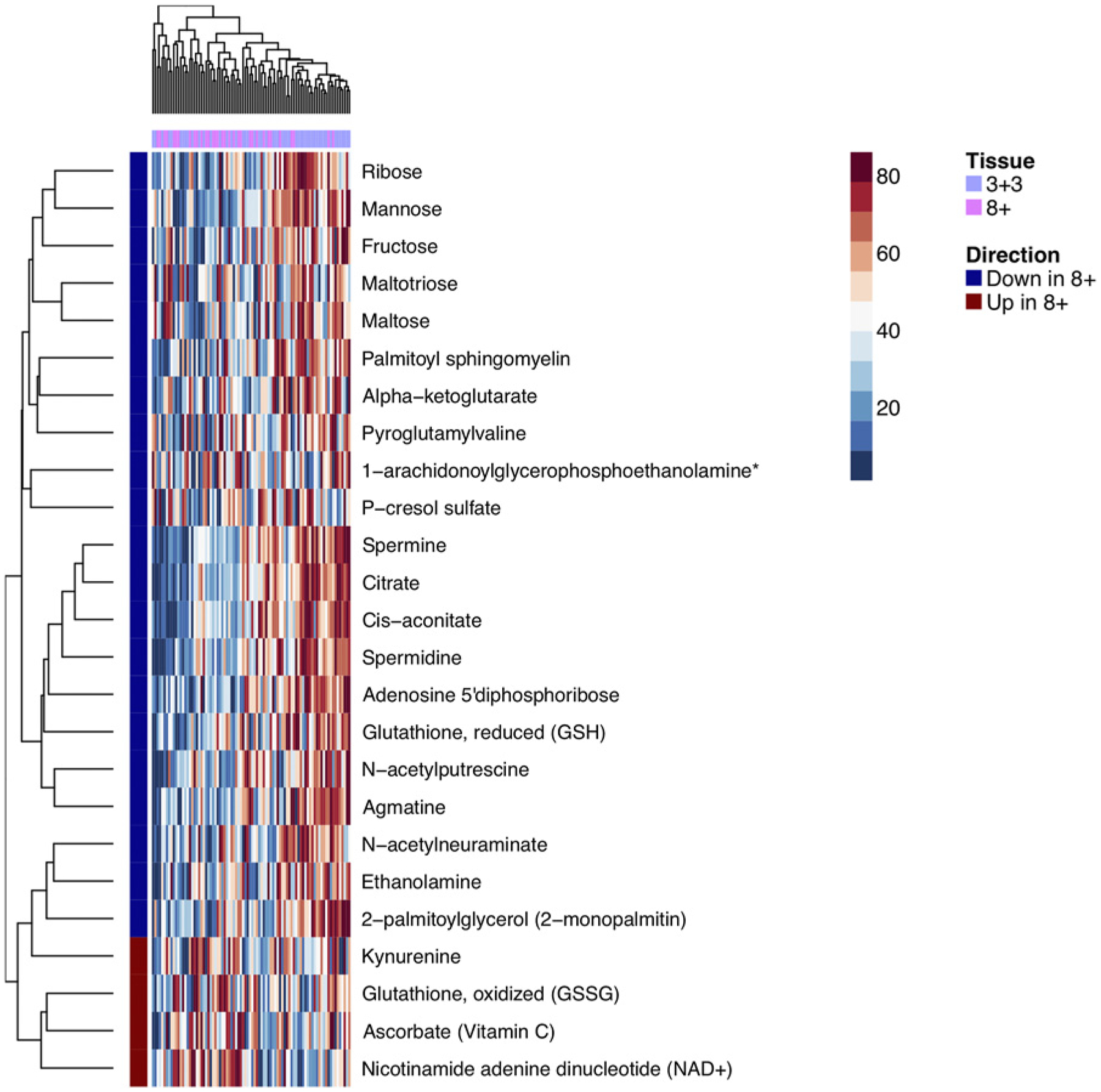
Heatmap of top differential (unadjusted P < 0.05) metabolites in tumor tissue between Gleason score 6 and Gleason score ≥8 tumors.
Metabolite comparisons in serum
To determine whether metabolites in serum were associated with the presence of Gleason grade 4 tumors, we assessed the association of metabolites with Gleason score 6 versus ≥7 in the “Original” and “Upgrading” serum cohorts separately and in the two populations combined. Across both cohorts, 238 metabolites were measured. In the “Original” cohort (21 Gleason 6 and 58 Gleason ≥7 cases), 12 metabolites had unadjusted P < 0.05 (Fig. 3A); at a pathway level, the amino acid superpathway had Padjusted = 0.001. In the “Upgrading” cohort (53 Gleason 6 and 47 Gleason ≥7 cases), 11 metabolites had unadjusted P < 0.05 (Fig. 3B); no pathways were enriched in this cohort. There was no intersection between the top metabolites identified in two cohorts.
Figure 3.

Heatmap of top differential (unadjusted P < 0.05) metabolites in serum between Gleason score 6 and Gleason score ≥7 tumors. A, Original cohort. B, Upgrading cohort.
Spearman correlations between total tumor volumes and abundances of metabolites detected in serum ranged between −0.23 and 0.14. Correlations between the percent of Gleason pattern 4 and the abundance of metabolites ranged between −0.26 and 0.14.
Correlation of metabolites across tissue and serum
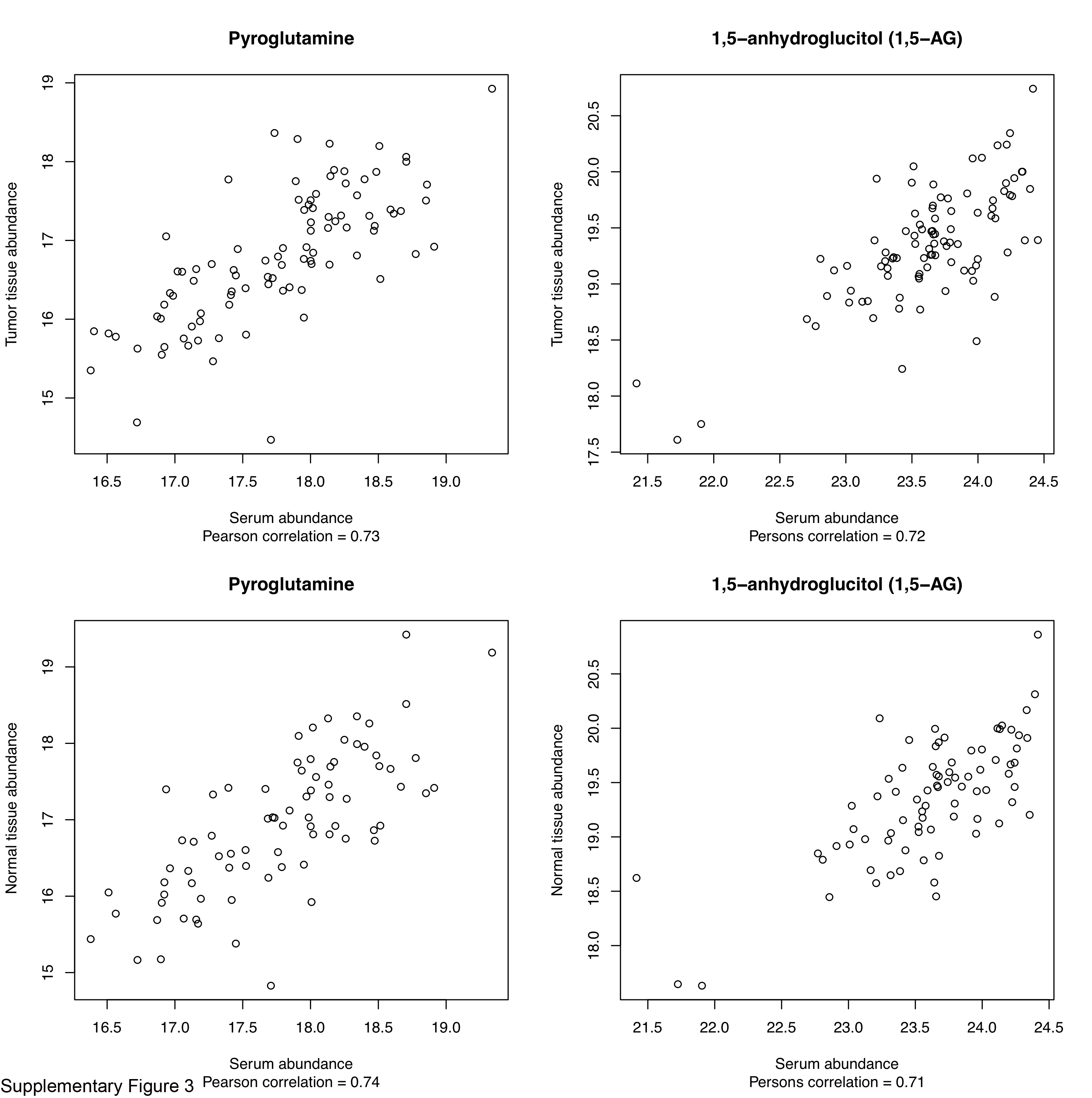
A subset of 94 patients had both tumor tissue and serum specimens with 119 common metabolites measured. While most of the metabolites had poor Pearson correlation between tumor tissue and serum (median correlation 0.08), two metabolites, pyroglutamine and 1,5-anhydroglucitol, had correlations of 0.73 and 0.72, respectively. Similar distribution of correlation coefficients was observed in 79 pairs of normal tissue and serum from the same patients (Supplementary Fig. S3). Pyroglutamine, a metabolite belonging to glutamate metabolism pathway, was associated with Gleason score (unadjusted P = 0.04) in one serum cohort. However, this metabolite was not associated with Gleason score in tissue. A potential explanation to this discrepancy between tissue and serum could be that the tissue block used for metabolomic profiling might not necessarily have contained tumor of the highest grade and might not solely contribute to the serum metabolome levels.
Development of classifiers using ML approach
To build classification models, we took a ML approach that did not rely on our findings from the previous sections. Performance metrics for each ML model are reported in Supplementary Table S1. Top performance across all tasks and models was achieved by AutoML for the tumor/normal classification on tissue data, with a cross-validation AUC of 0.82 ± 0.08 and a test-set AUC of 0.79 (Fig. 4). In contrast, models classifying high versus low Gleason score did not yield well-performing models, with a maximum cross-validation mean AUC of 0.67 for tissue and 0.56 for serum (Supplementary Table S1). These findings are consistent with those from univariate analyses, in that they indicate a lack of strong signal differentiating low-grade from high-grade cancer.
Figure 4.
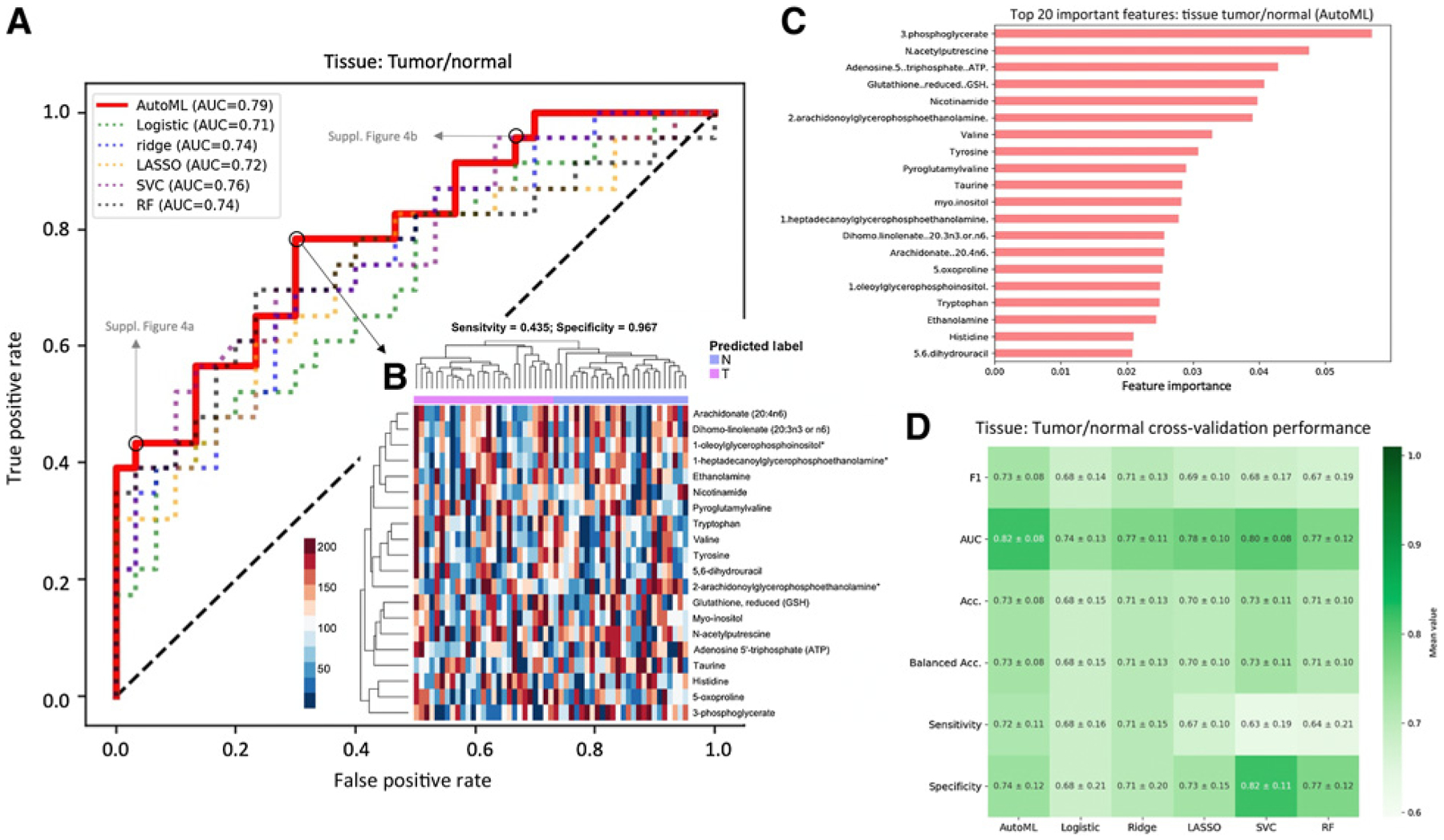
Model performances for tumor/normal tissue classification. A, ROC curves for all models, evaluated on a held-out test set. B, Heatmap of the top 20 metabolites as identified by permutation feature importance analysis of AutoML model. Predicted classes at the optimal cutoff (as determined by maximizing the sum of sensitivity and specificity; black circle in A) are shown above. Corresponding heatmaps at two other cut points (gray circles in A) are shown in Supplementary Materials. C, Feature importance of the top 20 important metabolites in the top-performing AutoML model. Feature importance is calculated as the difference in binary cross-entropy loss of model predictions when the feature is randomly permuted. D, Results of 10-fold cross-validation to assess stability of models across several performance metrics.
To further explore the ML results in search of a signature, we performed a permutation-based feature importance analysis to identify which metabolites were learned by the model to discriminate between tumor and normal tissue (Supplementary Fig. S4). The top five most important metabolites were 3-phosphoglycerate, N-acetylputrescine, ATP, glutathione (GSH), and nicotinamide (Fig. 4).
Discussion
Patients with low Gleason score prostate tumors are often treated with surgery or radiation, though current recommendations suggest many low-grade patients should be followed using active surveillance, monitoring for disease progression with repeated PSA measurements and prostate biopsies. These, however, can fail to identify higher grade tumors in a substantial percentage of cases. Here, we investigated tissue metabolomics to identify metabolites associated with Gleason score, as well as a serum-based metabolite analysis of Gleason score to guide development of a less-invasive assay to address the heterogeneity and multifocality of prostate cancer.
In this large comprehensive metabolomics study, we identified several previously reported and novel metabolites associated with Gleason score in tissue. This suggests that the tissue metabolomics data are of high quality and with larger sample sizes individual metabolites or pathways may have reached statistical significance. However, the identification of a metabolite with differential median abundance between groups does not guarantee that they are useful for discrimination between these groups, for example, due to high variances and large overlap in groupwise distributions of these metabolites. Many samples with similar ranges of metabolite levels could belong to different groups, making the classification success rate poor. We therefore performed a ML analysis, and demonstrate that this method may complement traditional pathway and metabolite set enrichment analysis. We trained several models to accurately and robustly discriminate tumor and normal tissue from metabolite profiles alone, with AutoML achieving the top performance. Notably, this method required no user interaction or manual tuning/composition of models. This demonstrates the utility of AutoML to efficiently search the high-dimensional space of models and hyperparameters and discover well-performing model configurations. Because ML models are fundamentally directed toward prediction, these approaches are naturally suited for the translation into the clinical setting where any implemented models must be able to reliably assess previously untested patients. We observe (i) an ample degree of biological plausibility of the top metabolites comparing tumor and normal tissue identified by feature importance analysis of the final AutoML ensemble, and (ii) notable overlap between these metabolites identified by AutoML and those identified in univariate comparisons.
We had previously reported that metabolic profiling could be accomplished in FFPE tissue (21). In this study, several metabolites were preserved but this depended largely on the metabolite class. We therefore compared the conservation of metabolites that differentiated tumor from normal and high from low Gleason score. Of the 135 metabolites significant for tumor versus normal, 96 were detectable in FFPE. For the 25 nominally significant metabolites from the tumor tissue Gleason score analysis, 16 were detectable in FFPE (Supplementary Table S2). This will render this type of analysis potentially amenable to FFPE tissue.
The top important metabolite, 3-phosphoglycerate, is involved in the glycolysis pathway and is the substrate of the PGAM1 enzyme which is upregulated in many cancers and thought to be involved in promotion of tumor growth (33–36). Serine, also observed as significantly different in tumor and normal tissue, is required for the synthesis of nucleotides, proteins, lipids, and one-carbon metabolism, all of which are important for cancer cell proliferation (37) and can be de novo synthesized from 3-phosphoglycerate in the serine biosynthetic pathway. An additional important metabolite is N-acetylputrescine, which was previously found to differentiate prostate cancer status in a study investigating metabolomic profiles in urine samples from 104 subjects (33). Furthermore, the nonacetylated compound putrescine is a precursor of spermidine and spermine. GSH and nicotinamide were also among the top five important metabolites and are both involved in the cellular response to oxidative stress: GSH as the primary cellular antioxidant, found to play a key role in carcino-genesis and modulation of cellular response to antineoplastic agents (34–39). This is more abundant in low than in high-grade tumors. Nicotinamide as a critical component of NAD, also found to be more abundant in high-grade tumors, which is used to transfer electrons in redox reactions. These findings are consistent with changes in oxidative stress pathways being linked with cancer (40). Triptophan, increased in tumor compared with normal prostate tissue, is metabolized to kynurenine, which is an immune suppressor, as well as to NAD. Both were found to be increased in high-grade Gleason. Interestingly, indoleamine-2, 3-dioxygenase, the enzyme that catalyzes triptophan to kynurenine, was found to be increased in xenografts of AT-2 rat prostate cancer cells (41). Dysregulation of the tryptophan/kynurinine is common in both bladder and renal and likely plays a role in tumor immune evasion (42, 43). Finally, we observe multiple fatty acids among the top 20 important metabolites, consistent with previous findings of altered fatty acid metabolism in cancer cells (44–46). These findings suggest that the ML methodology is a valid complement of the pathway analysis and is discriminating well on the basis of sensible features.
The novelty of our study lies in the overall design that included tumor and normal prostate tissue as well as serum metabolomics data. These data provide a metabolite profile of localized prostate cancer encompassing all Gleason grades. Our comprehensive pathology rereview ensured that we had the most accurate Gleason scoring, and we were able to assess and perform the analysis with the volume and percentage of high-grade disease. This is likely the correct measure to use when attempting to identify serum metabolites correlated with the presence of Gleason grade 4 and higher disease. We identified several metabolites and a prediction model for tumor versus normal prostate tissue, demonstrating that our data are of high quality. We attempted to develop prediction models using cutting-edge AI/ML methods. Our sample size was also large when compared with other metabolite studies, and the use of two sample sets for the serum analysis allowed us to attempt to confirm our findings.
Despite these many strengths, we observed few metabolites associated with Gleason score in serum and none were significant across our two serum study populations. This could be due to the low enrichment of higher grade cases (4+3 and above) in the “Upgrading” cohort. All other cell types from the prostate could also be contributing to the secretion and might have different profiles. Therefore, this might provide an additional explanation as to why we found only a mild correlation between tumor and serum profiles. Furthermore, association with grade and therefore disease aggressiveness could be masked.
In addition, we were unable to develop a strong prediction model for Gleason score in serum despite the strengths of the AutoML method. This may be due to several limitations of metabolomics studies performed in an existing patient population. We did not have fasting status or other potentially important information about the participants at the time of blood collection. These factors likely have a large impact on metabolite levels in serum—likely much larger than the impact of Gleason score 4 disease—and therefore could make it difficult to detect signal when we are unable to adjust for these factors. We also noted large differences across batches, with several metabolites detected in one batch but then not found in the next. This suggests that there is batch-to-batch variation, and while we did normalize metabolites across batches this variation could have created some of the differences observed between the “Original” and “Upgrading” cohort as these were assayed at different time points. Metabolomics data also have large dynamic ranges with high variability between patients leading to lower power and making differential analyses and construction of reproducible predictive models challenging. It is also possible that associations of some metabolic profiles with phenotypes could be nonlinear calling for more sophisticated and flexible ML approaches as implemented in AutoML framework. These challenges likely contribute to the difficulties we experienced in developing a predictive model of Gleason score in the serum samples.
It is additionally possible that metabolomics in serum may be associated more with tumor genomic alterations or with other tumor characteristics, such as proliferation, than with differentiation. While we have seen associations of genomic alterations with metabolites in tumor tissue (9), we have not examined these associations in serum. Future studies of metabolomics of Gleason score in serum should consider the limitations we describe above to design a study best able to determine whether there are serum metabolites associated with the presence of high-grade prostate tumors.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Implications:
Metabolic profiling can distinguish benign and neoplastic tissues. A novel unsupervised machine learning method can be utilized to achieve this distinction.
Acknowledgments
M. Loda is supported by NIH grants R01CA131945, DoD PC160357, DoD PC180582, P50CA211024, P50CA090381 and the Prostate Cancer Foundation. K. L. Penney was supported by a Prostate Cancer Foundation Young Investigator Award. G. Zadra was supported by DoD PC150263.
Authors’ Disclosures
S. Borgstein reports grants from NCI during the conduct of the study. R. Lis reports personal fees and nonfinancial support from Janssen outside the submitted work. R. Umeton reports consulting from Health Catalyst, reports patents/IP (devices, algorithms, and solutions for medical diagnosis) from eye scans and drug targets, genomic characterizations, and drug formulations for diagnosis and treatment of various vascular, neurologic, immuno-mediated diseases. No disclosures were reported by the other authors.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
References
- 1.Stark JR, Perner S, Stampfer MJ, Sinnott JA, Finn S, Eisenstein AS, et al. Gleason score and lethal prostate cancer: does 3 + 4 = 4 + 3? J Clin Oncol 2009;27:3459 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahal BA, Butler S, Franco I, Spratt DE, Rebbeck TR, D’Amico AV, et al. Use of active surveillance or watchful waiting for low-risk prostate cancer and management trends across risk groups in the United States, 2010–2015. JAMA 2019; 321:704–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porcaro AB, Siracusano S, de Luyk N, Corsi P, Sebben M, Tafuri A, et al. Low-risk prostate cancer and tumor upgrading in the surgical specimen: analysis of clinical factors predicting tumor upgrading in a contemporary series of patients who were evaluated according to the modified gleason score grading system. Curr Urol 2017;10:118–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caster JM, Falchook AD, Hendrix LH, Chen RC. Risk of pathologic upgrading or locally advanced disease in early prostate cancer patients based on biopsy gleason score and PSA: a population-based study of modern patients. Int J Radiat Oncol Biol Phys 2015;92:244–51. [DOI] [PubMed] [Google Scholar]
- 5.Schiffmann J, Wenzel P, Salomon G, Budaus L, Schlomm T, Minner S, et al. Heterogeneity in D’Amico classification-based low-risk prostate cancer: Differences in upgrading and upstaging according to active surveillance eligibility. Urol Oncol 2015;33:329.e13–9. [DOI] [PubMed] [Google Scholar]
- 6.Yang DD, Mahal BA, Muralidhar V, Vastola ME, Boldbaatar N, Labe SA, et al. Pathologic outcomes of gleason 6 favorable intermediate-risk prostate cancer treated with radical prostatectomy: implications for active surveillance. Clin Genitourin Cancer 2018;16:226–34. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen HG, Welty CJ, Cooperberg MR. Diagnostic associations of gene expression signatures in prostate cancer tissue. Curr Opin Urol 2015;25:65–70. [DOI] [PubMed] [Google Scholar]
- 8.Martin NE. New developments in prostate cancer biomarkers. Curr Opin Oncol 2016;28:248–52. [DOI] [PubMed] [Google Scholar]
- 9.Priolo C, Pyne S, Rose J, Regan ER, Zadra G, Photopoulos C, et al. AKT1 and MYC induce distinctive metabolic fingerprints in human prostate cancer. Cancer Res 2014;74:7198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Penney KL, Sinnott JA, Fall K, Pawitan Y, Hoshida Y, Kraft P, et al. mRNA expression signature of Gleason grade predicts lethal prostate cancer. J Clin Oncol 2011;29:2391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sinnott JA, Peisch SF, Tyekucheva S, Gerke T, Lis R, Rider JR, et al. Prognostic utility of a new mRNA expression signature of gleason score. Clin Cancer Res 2017;23:81–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015;163:1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly RS, Vander Heiden MG, Giovannucci E, Mucci LA. Metabolomic biomarkers of prostate cancer: prediction, diagnosis, progression, prognosis, and recurrence. Cancer Epidemiol Biomarkers Prev 2016;25:887–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McDunn JE, Li Z, Adam KP, Neri BP, Wolfert RL, Milburn MV, et al. Metabolomic signatures of aggressive prostate cancer. Prostate 2013;73:1547–60. [DOI] [PubMed] [Google Scholar]
- 15.Giskeodegard GF, Bertilsson H, Selnaes KM, Wright AJ, Bathen TF, Viset T, et al. Spermine and citrate as metabolic biomarkers for assessing prostate cancer aggressiveness. PLoS One 2013;8:e62375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Randall EC, Zadra G, Chetta P, Lopez BGC, Syamala S, Basu SS, et al. Molecular characterization of prostate cancer with associated gleason score using mass spectrometry imaging. Mol Cancer Res 2019;17:1155–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan Y, Murphy TB, Byrne JC, Brennan L, Fitzpatrick JM, Watson RW. Applying random forests to identify biomarker panels in serum 2D-DIGE data for the detection and staging of prostate cancer. J Proteome Res 2011;10:1361–73. [DOI] [PubMed] [Google Scholar]
- 18.Osl M, Dreiseitl S, Pfeifer B, Weinberger K, Klocker H, Bartsch G, et al. A new rule-based algorithm for identifying metabolic markers in prostate cancer using tandem mass spectrometry. Bioinformatics 2008;24:2908–14. [DOI] [PubMed] [Google Scholar]
- 19.Oh WK, Hayes J, Evan C, Manola J, George DJ, Waldron H, et al. Development of an integrated prostate cancer research information system. Clin Genitourin Cancer 2006;5:61–6. [DOI] [PubMed] [Google Scholar]
- 20.Chen ME, Johnston D, Reyes AO, Soto CP, Babaian RJ, Troncoso P. A streamlined three-dimensional volume estimation method accurately classifies prostate tumors by volume. Am J Surg Pathol 2003;27:1291–301. [DOI] [PubMed] [Google Scholar]
- 21.Cacciatore S, Zadra G, Bango C, Penney KL, Tyekucheva S, Yanes O, et al. Metabolic profiling in formalin-fixed and paraffin-embedded prostate cancer tissues. Mol Cancer Res 2017;15:439–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Do KT, Wahl S, Raffler J, Molnos S, Laimighofer M, Adamski J, et al. Characterization of missing values in untargeted MS-based metabolomics data and evaluation of missing data handling strategies. Metabolomics 2018; 14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benjamini Y, Hochberg Y. On the adaptive control of the false discovery rate in multiple testing with independent statistics. J Educ Behav Stat 2000;25:60–83. [Google Scholar]
- 25.Tibshirani R Regression shrinkage and selection via the lasso. J R Stat Soc Ser B 1996;58:267–88. [Google Scholar]
- 26.Hoerl AE, Kennard RW. Ridge regression: biased estimation for nonorthogonal problems. Technometrics 1970;12:55–67. [Google Scholar]
- 27.Cortes C, Vapnik V. Support-vector networks. Machine Learning. 1995;20: 273–97. [Google Scholar]
- 28.Liaw A, Wiener M. Classification and regression by randomForest. R news 2002; 2:18–22. [Google Scholar]
- 29.Waring J, Lindvall C, Umeton R. Automated machine learning: review of the state-of-the-art and opportunities for healthcare. Artif Intell Med 2020;104: 101822. [DOI] [PubMed] [Google Scholar]
- 30.Feurer M, Klein A, Eggensperger K, Springenberg J, Blum M, Hutter F. Efficient and robust automated machine learning. Adv Neural Inf Proc Sys 28 (NIPS 2015) 2015;28:2962–70. [Google Scholar]
- 31.Hutter F, Hoos HH, Leyton-Brown K. Sequential model-based optimization for general algorithm configuration. Springer; New York, NY: 2011. p. 507–23. [Google Scholar]
- 32.Caruana R, Niculescu-Mizil A, Crew G, Ksikes A. Ensemble selection from libraries of models. In Proceedings of the 21st International Conference on Machine Learning; 21st ICML Proceedings, Banff, Canada, 2004. p. 18. [Google Scholar]
- 33.Fernandez-Peralbo MA, Gomez-Gomez E, Calderon-Santiago M, Carrasco-Valiente J, Ruiz-Garcia J, Requena-Tapia MJ, et al. Prostate cancer patients-negative biopsy controls discrimination by untargeted metabolomics analysis of urine by LC-QTOF: upstream information on other omics. Sci Rep 2016;6: 38243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balendiran GK, Dabur R, Fraser D. The role of glutathione in cancer. Cell Biochem Funct 2004;22:343–52. [DOI] [PubMed] [Google Scholar]
- 35.Arrick BA, Nathan CF. Glutathione metabolism as a determinant of therapeutic efficacy: a review. Cancer Res 1984;44:4224–32. [PubMed] [Google Scholar]
- 36.Bansal A, Simon MC. Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol 2018;217:2291–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Estrela JM, Ortega A, Obrador E. Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci 2006;43:143–81. [DOI] [PubMed] [Google Scholar]
- 38.Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev 2013;2013:972913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolf CR, Lewis AD, Carmichael J, Adams DJ, Allan SG, Ansell DJ. The role of glutathione in determining the response of normal and tumor cells to anticancer drugs. Biochem Soc Trans 1987;15:728–30. [DOI] [PubMed] [Google Scholar]
- 40.Grek CL, Tew KD. Redox metabolism and malignancy. Curr Opin Pharmacol 2010;10:362–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hagiwara A, Nakamura Y, Nishimoto R, Ueno S, Miyagi Y. Induction of tryptophan hydroxylase in the liver of s.c. tumor model of prostate cancer. Cancer Sci 2020;111:1218–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hornigold N, Dunn KR, Craven RA, Zougman A, Trainor S, Shreeve R, et al. Dysregulation at multiple points of the kynurenine pathway is a ubiquitous feature of renal cancer: implications for tumour immune evasion. Br J Cancer 2020;123:137–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pasikanti KK, Esuvaranathan K, Hong Y, Ho PC, Mahendran R, Raman Nee Mani L, et al. Urinary metabotyping of bladder cancer using two-dimensional gas chromatography time-of-flight mass spectrometry. J Proteome Res 2013;12: 3865–73. [DOI] [PubMed] [Google Scholar]
- 44.Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab 2013;18:153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis 2006;9:230–4. [DOI] [PubMed] [Google Scholar]
- 46.Zadra G, Loda M. Metabolic vulnerabilities of prostate cancer: diagnostic and therapeutic opportunities. Cold Spring Harb Perspect Med 2018;8: a030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.