

Abstract
Mucopolysaccharidosis (MPS) IIIB is a neuropathic lysosomal storage disease characterized by the deficient activity of a lysosomal enzyme obligate for the degradation of the glycosaminoglycan (GAG) heparan sulfate (HS). The pathogenesis of neurodegeneration in MPS IIIB is incompletely understood. Large animal models are attractive for pathogenesis and therapeutic studies due to their larger size, outbred genetics, longer lifespan, and naturally occurring MPS IIIB disease. However, the temporospatial development of neuropathologic changes has not been reported for canine MPS IIIB. Here we describe lesions in 8 brain regions, cervical spinal cord, and dorsal root ganglion (DRG) in a canine model of MPS IIIB that includes dogs aged from 2 to 26 months of age. Pathological changes in the brain included early microscopic vacuolation of glial cells initially observed at 2 months, and vacuolation of neurons initially observed at 10 months. Inclusions within affected cells variably stained positively with PAS and LFB stains. Quantitative immunohistochemistry demonstrated increased glial expression of GFAP and Iba1 in dogs with MPS IIIB compared to age-matched controls at all time points, suggesting neuroinflammation occurs early in disease. Loss of Purkinje cells was initially observed at 10 months and was pronounced in 18 and 26-month- old dogs with MPS IIIB. Our results support the dog as a replicative model of MPS IIIB neurologic lesions and detail the pathologic and neuroinflammatory changes in the spinal cord and DRG of MPS IIIB-affected dogs.
Keywords: Animal models of human disease, Dogs, Cerebrospinal fluid, Gliosis, Histology, Lysosomal storage disease, Mucopolysaccharidosis IIIB, Neuroinflammation, Physiopathology, Sanfilippo syndrome
The mucopolysaccharidoses (MPSs) are a heterogeneous group of lysosomal storage diseases characterized by the deficient activity of individual lysosomal enzymes obligate for the complete degradation of glycosaminoglycans (GAGs). All the MPSs are characterized clinically by either severe somatic/bone (coarse facial features, hypertelorism, dystostosis multiplex, hypertrichosis) and/or neurological disease, and at the cell level by lysosomal accumulation of GAGs resulting in cellular death and/or dysfunction.46 One of the MPS types observed in humans, MPS III (Sanfilippo syndrome) is characterized clinically by severe neurodevelopmental and neurodegenerative impairment, with mild somatic manifestations, and biochemically by the storage of only one GAG subtype, heparan sulfate (HS).46 There are four clinically similar but genetically and biochemically distinct subtypes of MPS III, designated as IIIA-IIID in humans. In mice there is a fifth MPS III form (MPS IIIE),38 which is suspected in dogs47 and is as yet undiscovered in humans. In the aggregate, Sanfilippo syndrome has been documented to be the MPS type with the greatest incidence, at 1:72.9 thousand births,43 and among the subtypes MPS IIIB is one of the most common subtypes with an incidence rate geographically ranging from 1 in every 139,000 to 500,000 live births.37,43
Each MPS III subtype in humans is clinically characterized by childhood-onset (typically 1-3 years of age), progressive neurocognitive (speech impairment, decreased intelligence quotient) and behavioral (aggression, hyperactivity, sleeping disorder, and/or autistic-like behavior) abnormalities. Neurocognitive symptoms are progressive, leading to seizures, dementia, and eventually death (typically in the second decade of life).28 Each MPS III subtype is further characterized on a molecular level by deficient activity of one of four lysosomal enzymes obligate for the complete degradation of HS.46 As with most of the MPSs, MPS IIIB (OMIM:252920), is an autosomal recessive disorder. It is caused by a loss-of-function mutation in the α-N-acetylglucosamindase (NAGLU) gene, which codes for the NAGLU enzyme (EC: 3.2.1.50). Over 100 novel mutations in the NAGLU gene have been documented as the cause of MPS IIIB in humans as homozygous or compound heterozygous genotypes.16,4 Heterozygosity for a rare missense NAGLU mutation also confers a form of autosomal dominant axonal Charcot-Marie-Tooth disease type 2V.58 Currently, there is no therapy for the devastating neurological course of MPS IIIB.
Potential therapeutic options for MPS include hematopoietic stem cell transplant, enzyme replacement therapy, substrate reduction therapy, and gene therapy. Thus far, these therapies have exhibited variable success in translation from animal models to humans and have been limited in their efficacy.9 Hematopoietic stem cell transplantation fails to correct neurologic disease.55,62 Intravenous enzyme replacement therapy to correct neurologic signs of MPS III are met with substantial physiologic challenges that involve crossing the blood-brain barrier and activation of the adaptive immune system.54 Intrathecal and intracerebroventricular enzyme replacement therapy protocols have been developed; however, there have been issues concerning maintenance of drug delivery devices (i.e. intrathecal and intracerebroventricular implants) and the potential for infection.54,14 Even in the event of stable therapeutic efficacy of centrally administered enzyme replacement therapy, such therapy would be lifelong, increasing the chance of untoward sequela and increasing economic burdens. Gene therapy using viral vectored enzyme replacement has improved significantly over the years, but is still met with several challenges regarding the development of immunity to the vector and/or enzyme.53,8,19,29,45 Despite advances in the experimental treatment of MPS IIIB, current therapeutic options are limited, and as such, reliable modeling of MPS IIIB disease remains a necessary step in the search for new therapies. To gain a clearer understanding of the underlying pathologic processes of MPS IIIB, a well-characterized animal model that is genetically, physiologically, and anatomically similar to humans is required.
To date, animal models for MPS IIIB include murine,40canine,12 avian,50,20 bovine,33 and porcine66 models. Of these, only the mouse and canine models have extensively been utilized. The murine model of MPS IIIB was developed by disrupting exon 6 of the NAGLU gene.40 Affected mice demonstrated altered behavior (increased anxiety, reduced daily activity, and altered circadian rhythm), altered coordination, hearing deficits, vision loss, and significantly shortened lifespan. In some reports, mice with MPS IIIB also demonstrated skeletal dysmorphism, ventriculomegaly, organomegaly, and ocular abnormalities.22 In addition, histopathologic changes in the brains of affected mice are similar to those observed in humans, with vacuolation of neurons and glial cells and accumulation of both HS and gangliosides.28
The canine model of MPS IIIB evaluated in the current study is a naturally occurring disease that was originally identified in a family of Schipperke dogs.12 The identified mutation is an insertion in exon 6 of the NAGLU gene.51 Two 4-5 year old Schipperke dogs were evaluated for tremors and ataxia. Neurologic examination identified findings consistent with severe cerebellar dysfunction (dysmetria, ataxia, truncal sway). Radiologic findings did not indicate somatic abnormalities. Cytologic examination of blood smears and CSF identified intracytoplasmic inclusions in white blood cells, similar to those described in humans.56 Urine GAG analysis demonstrated significantly increased excretion of HS, and fluorometric analysis of Naglu enzymatic activity demonstrated a significant reduction in activity (4.3% to 9.2% of normal values).12 These biochemical results are in agreement with those found in human IIIB patients.56,23 Postmortem examination of these two animals demonstrated a pattern of vacuolation of neurons and glial cells and accumulation of HS and gangliosides in the brain similar to that observed in humans and the murine model.40,23,12,13
Thus far, most pathogenesis and therapeutic studies of MPS IIIB have been carried out in mouse models. The mouse model of MPS IIIB has proved invaluable, however, it does have limitations.6,25 The genetic and immunologic background of the mouse is different than humans, MPS IIIB does not naturally occur in the mouse, and the affected mice have limited overt neurological phenotypes.40,6,44 Large animal models of MPS IIIB are naturally occurring and have a heterogeneous genetic background similar to humans, thus better mirroring pathologic processes occurring in human disease. Of the naturally occurring large animal models, the canine model is optimal because of body size, litter size, ease of handling, depth of clinical veterinary knowledge and expertise, and physiologic and pathologic similarities to humans. An MPS IIIA canine model has been characterized and is currently being used to successfully test emerging therapies.36,7,35,34,12 The MPS IIIB canine model has been used in gene therapeutic studies, but longitudinal studies of lesions and disease kinetics of the canine model would improve its utility.12 In the current study, we have characterized the clinicopathological findings and temporospatial distribution of neurologic lesions in a canine model of MPS IIIB.
Materials and Methods
Animals and Tissue Processing
This work was performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Iowa State University IACUC. The original NAGLU founder mutation was described in a family of Schipperke dogs.13 A colony was established at the University of Pennsylvania by outcrossing MPS IIIB-affected Schipperkes with unaffected beagles and cross-bred hound dogs. This colony is currently maintained and bred at Iowa State University and all animals (control and MPS IIIB-affected) included in this study were derived from this colony. Animals in the current study were diagnosed at birth by PCR for the mutant NAGLU allele. Control animals were either heterozygous for the NAGLU mutation or homozygous normal at NAGLU. Neurologic examination was not conducted on any animals included in this study. The natural disease course observed in these animals is such that they are apparently normal until 18-30 months of age. After 18-30 months, these dogs typically develop nystagmus, ataxia, and other symptoms of cerebellar dysfunction. Animals succumb to severe neurologic issues at 4 to 5 years of age. 13,12
Dogs were euthanized by intravenous sodium pentobarbital overdose at selected time points of 2 (n=1 control, 1 MPS IIIB), 4 (n=2 control, 2 MPS IIIB), 10 (n=2 control, 2 MPS IIIB), 18 (n=1 control, 4 MPS IIIB), and 26 (n=1control, 5 MPS IIIB) months of age (Table 1). Cerebrospinal fluid (CSF) (n=15) and blood for analysis (n=4) were collected ante-mortem or immediately post-mortem from a subset of dogs. CSF (n=15) was collected from the cisterna magna using a 1-1/2 inch 18- gauge needle on a 12 mL Luer-lock syringe and placed into an EDTA tube. Examined CSF and blood samples were processed as described in the clinical pathology methods.
Table 1.
Signalment and genotype of 14 dogs with mucopolysaccharidosis IIIB and 7 controls.a
| Unaffected | ||||
|---|---|---|---|---|
| Animal ID# |
Genotype |
Sex |
Age (months) |
|
| I614" | IIIB normal | M | 1.8 | |
| B592* | IIIB carrier | M | 3.8 | |
| B617** | IIIB carrier | F | 4.1 | |
| B574^ | IIIB normal | M | 10.3 | |
| B575^ | IIIB normal | M | 10.3 | |
| B717 | IIIB carrier | M | 18.0 | |
| N20 | IIIB carrier | M | 26.0 | |
| MPS IIIB | ||||
| B765 | IIIB Affected | M | 2.1 | |
| B590* | IIIB Affected | M | 3.8 | |
| B618** | IIIB Affected | F | 4.1 | |
| B571^ | IIIB Affected | M | 10.3 | |
| B573^ | IIIB Affected | M | 10.3 | |
| B727 | IIIB Affected | M | 17.9 | |
| B734^^ | IIIB Affected | F | 18.0 | |
| B737^^ | IIIB Affected | F | 18.0 | |
| B738^^ | IIIB Affected | M | 18.0 | |
| B693*** | IIIB Affected | M | 25.8 | |
| B694*** | IIIB Affected | M | 25.8 | |
| B699*** | IIIB Affected | M | 25.8 | |
| B702*** | IIIB Affected | F | 26.1 | |
| B703*** | IIIB Affected | F | 26.1 |
Superscripts represent sibling relationships.
All animals (n=21) were necropsied immediately following euthanasia, and a full set of tissues was collected into 4% paraformaldehyde. Fresh brains (n=21) were placed in chilled physiologic saline for 30 minutes prior to sectioning. After chilling, brains were placed into a specialized canine brain matrix (ASI Instruments, DBM-1000C) for sectioning into consistent 4-mm-thick transverse sections. The initial section was made at the level of the optic chiasm, and sections were made every 4 mm both rostrally and caudally from the optic chiasm until the entire brain was completely sectioned. Seventeen sections of brain were generated from each animal (Supplemental Figs. S1 and S2). For every animal, the brain sections were laid out in order from #1- #17 on a tray, assessed for consistency of neuroanatomical regions present, and photographed (Supplemental Fig. S2). Fresh frozen samples were collected using a 4 mm biopsy punch from the following anatomical regions: cerebral cortex at level of caudate nucleus, centrum semiovale, caudate nucleus, thalamus, cerebellar vermis, and cerebellar white matter. Brain sections were bisected mid-sagittally and the left halves were placed in 4% paraformaldehyde and the right halves frozen over a bed of dry ice. Formalin fixed tissue was used for all microscopic evaluation in the current study. Frozen tissue was archived for future studies. Spinal cords (n=21) were removed using a dorsal approach that consisted of transecting the vertebral laminae using a Stryker autopsy saw and removing the vertebral arch. The spinal cords were removed by transecting the spinal rootlets with a 3-inch dissecting scissors. After removal, the dura was transected sagittally with scissors and the entire cervical spinal cord was placed into 4% paraformaldehyde. After 24 hours of fixation, cervical spinal cords were transversely sectioned into 4mm sections between C1-C2, C2-C3, C3-C4, and C4-C5. Dorsal root ganglia (DRG) (n=13) from rootlets C2, C3, and C4 were harvested bilaterally and fixed in 4% paraformaldehyde for 24 hours. After 24 hours fixation, all formalin-fixed tissues were processed by routine histologic methods and embedded in paraffin. Serial sections (5-μm-thick) were cut from formalin-fixed paraffin-embedded (FFPE) tissues for histochemical staining and immunohistochemistry.
Regions of interest (ROIs) in the brain were selected based on CNS lesions reported in IIIB-affected humans.25,60,10,24 Additional regions (hippocampus, cerebellar peduncle, vestibular nucleus, lateral funiculus) were selected for completeness of the survey study. All ROIs were identified with the aid of a canine histologic brain atlas.49 Three transverse sections from each brain (brain sections #6,10,14) (n=21) (Supplemental Figs. S3-S6), one transverse section from each cervical spinal cord (between C2-C3 segement) (n=21), and one section from each available DRG (from C2 rootlets) (n=12) were selected for examination. Each section of brain was examined to ensure it contained the following ROIs: cerebral cortical layers II/III of the postcruciate gyrus (at the level of the optic chiasm-brain section #6), centrum semiovale (at the level of the optic chiasm- brain section #6) (Supplemental Fig. S7), caudate nucleus (at the level of the optic chiasm- brain section #6) (Supplemental Fig. S8), CA2 region of the hippocampus (brain section #10), dorsal pulvinar thalamic nucleus (brain section #10) (Supplemental Fig. S9), superior cerebellar peduncle (brain section #14), lateral vestibular nucleus (brain section #14), and 3rd cerebellar folium (brain section#14) (Supplemental Fig. S10). ROIs in the cervical spinal cord included the ventral horn and left lateral funiculus between C2-C3 segment. Whole dorsal root ganglia (n=13) originating from C2-C3 spinal cord segments were selected.
Clinical Pathology
CSF total protein was determined using a refractometer (TS Meter; American Optical, Southbridge, MA, USA). Total nucleated cell counts (TNCC) and red blood cell (RBC) counts were determined manually on a hemocytometer. Cytocentrifugation of CSF was performed using a Shandon Cytospin3 (Thermo Scientific, Waltham, MA, USA) at 72g for 10 min (low acceleration). Air-dried slides were stained with modified Wright's on an automated stainer (Siemens Hematek; Siemens Corporation). Determination of differential cell counts was done under 1000× magnification by counting 300 nucleated cells by a board-certified veterinary clinical pathologist (SJH) without knowledge of animal genotype or age at time of collection. Complete blood counts (CBC) and serum chemistries were run on a subset of dogs (n=4). Whole blood for CBCs was collected in EDTA and analyzed with the Advia 2120 Hematology System (Siemens Healthineers) and serum chemistries were analyzed using an automated clinical chemistry system (VITROS®, Ortho Clinical Diagnostics).
Histochemistry and Histopathology
Histologic sections were deparaffinized in xylene and rehydrated through graded concentrations of ethanol. Sections were incubated in either hematoxylin and eosin (HE), periodic acid—Schiff (PAS), or Luxol fast blue stains (LFB). For HE staining, sections were incubated in freshly filtered Mayer’s hematoxylin solution (BBC Biochemical, 3580) for 10 minutes and alcoholic eosin solution (American Mastertech, STE0457) for 2 minutes. For PAS staining, sections were incubated in 1% periodic acid solution (Fisher Scientific, AAB2043318) for 10 minutes and Schiff reagent (Fisher Scientific, SS32-500) for 20 minutes with Mayer’s hematoxylin counterstain. For LFB staining, sections were incubated in 0.1% luxol fast blue stain (Sigma, S3382) for 16 hours at 56° Celsius. Sections were differentiated individually in 0.05% lithium carbonate (Fisher, L119-500) and 70% ethanol, counterstained with 0.1% cresyl violet acetate solution (Sigma, C5042) for 6 minutes, and dehydrated through a graded ethanol series, cleared in xylene, coverslipped and examined using a BX-41 Olympus Trinocular brightfield microscope with an Olympus DP73 digital colored camera and cellSens imaging software (v1.15, Olympus Corporation).
Linear Cell Density
One HE-stained section from each animal (n=21) was evaluated for cell density. Each evaluated section was required to contain cerebellar linguae, lateral vestibular nucleus, trapezoid bodies, and pyramidal fibers. Cerebellar Purkinje cells were manually counted in five contiguous, nonoverlapping 200x fields (1.185 mm2) from each of the following neuroanatomical regions: the dorsal-most paravermis folium, ventral hemisphere folium (just dorsal to anisform lobe), dorsal vermis, and ventral vermis (Supplemental Fig. S10). Counts were completed on one HE-stained section per animal utilizing a similar approach as previously described.41,27 In the Purkinje cell layer, any large cell with a nucleus and prominent eosinophilic cytoplasm was interpreted and counted as a Purkinje cell. The sum of Purkinje cells from each ROI was calculated and the Purkinje cell layers in each region were measured in μm using the line tool in HALO image analysis software (v2.0.1145.19, Indica Labs). Linear cell densities in each of the cerebellar ROIs were determined by using the following equation:
Immunohistochemistry
Immunohistochemical analyses were performed to assess astroglial and microglial density and morphology changes observed in MPS IIIB-affected animals. Glial fibrillary acidic protein (GFAP) was used as a marker of astrocytes, and ionizing calcium binding molecule 1 (Iba1) was used as a marker of microglia. Tissue sections for immunohistochemistry underwent antigen retrieval in citrate buffer (10 mM, pH 6.0) for 20 minutes, endogenous peroxidase blocking in 3% H2O2 /tris buffered saline solution for 20 minutes, and endogenous Fc receptor blocking in normal horse serum for 30 minutes. Sections were incubated at 4°C overnight with the following primary antibodies: rabbit anti-GFAP (Z0334, Dako; 1:1000) and rabbit anti-Iba-1 (ab178847, Abcam; 1:500) Slides were washed and incubated with a pre-diluted HRP-conjugated horse anti-mouse/-rabbit secondary antibody (MP7500, Vector Laboratories) for 14-16 h at 4°C. After washing, sections were labelled with NovaRED HRP substrate (SK4800, Vector Laboratories) according to kit instructions, and counterstained with Mayer’s hematoxylin. Sections were dehydrated through a graded ethanol series and cleared in xylene. Both GFAP (Z0334, Dako; 1:1000) and Iba1 (ab178847, Abcam; 1:500) antibodies were commercially available and validated for mouse tissues. In addition, the GFAP antibody was validated for canine brain tissues. FFPE 5-μm-thick mouse brain sections served as positive controls for both GFAP and Iba1 IHC. Positive controls were visually inspected for appropriate histomorphologic labeling of both astrocytes and microglia based on GFAP and Iba1 immunolabeling, respectively. Negative controls consisted of primary antibody omission and were visually inspected to confirm negative labeling.
Image analysis
Sections from all animals (n=21) immunolabelled for GFAP and Iba1 IHC were imaged as follows. Three contiguous, nonoverlapping 400x images encompassing a total area of 0.711 mm2 from each neuroanatomical ROI (Supplemental Figs S7-S10) per animal were captured as above and analyzed using Area Quantification v1.0 module in HALO image analysis software (v2.0.1145.19, Indica Labs). Using the 18-month-old control animal, GFAP and Iba1 algorithms were developed by using the Area Quantification v1.0 module. Algorithms were created by designating NovaRED chromogen and hematoxylin counterstains as stain 1 and 2, respectively. Deconvolution of these stains were confirmed visually and manually adjusted to optimally recognize deposition of the chromogen in the tissues. GFAP algorithm was standardly applied to all GFAP image sets and the Iba1 algorithm was standardly applied to all Iba1 image sets. Manual thresholding was carried out on images where the Quantification module recognized background artifact. Total deposition of chromogen in the labeled tissues was quantified by the quantification module as % total positive tissue.
Statistical Analysis
Data are shown as mean ± standard error of the mean (SEM). Means and SEMs were calculated, and graphs were constructed using Graphpad Prism version 7.0 (Graphpad Software) for Macintosh.
Results
Clinical and CSF Cytologic Findings
A summary of tissues used from each dog and analyses performed is provided in supplemental table S1. The CSF was evaluated in 15 of the 21 dogs included in the study at 10 (n=2 control, n=2 MPS IIIB), 18 (n=1 control, n=4 MPS IIIB), and 26 (n=1 control, n=5 MPS IIIB) months of age. The CSF from all IIIB-affected dogs (n=11) contained inclusions in mononuclear cells consistent with lysosomal storage disease (Fig. 1-4). The inclusions appeared as aggregates of amphophilic to basophilic material within cytoplasmic vacuoles. The aggregates were round to linear in shape and varied in staining intensity. Subjectively, the inclusions appeared to increase in size and number with increasing age. The CSF cytology from three of the four control dogs had no evidence of storage disease. One control dog had low numbers of mononuclear cells containing indistinct material within cytoplasmic vacuoles (interpreted as equivocal by the pathologist). Total nucleated cell count (TNCC) ranged from 2 to 12/ μl (reference interval 0-5/ μl) for all CSF samples (cases and controls). RBC counts were low (0-8/ μl) in the majority of CSF samples (13/15). Two of the sixteen samples had elevated RBC counts: one control (208/ μl) and one MPS IIIB-affected (222/ μl). The corresponding TNCC for these two samples were both within reference interval (1/ μl in each case). A 300-cell differential count was performed on all samples with adequate cytospin cellularity. In cases with less than 300 cells on the cytospin, all nucleated cells available were counted in the differential. No significant differences between control and MPS IIIB dogs were identified for TNCC, RBC, and cell differentials (data not shown).
Figures 1-4.

Cerebrospinal fluid (CSF) cytocentrifuge preparations, dog. Modified Wright’s stain. Figure 1. Normal CSF, control, 26 months old. Mononuclear cells appear normal with no appreciable cytoplasmic vacuoles or inclusions. Figure 2. Mucopolysaccharidosis (MPS) IIIB, 10 months old. Distinct vacuoles and basophilic inclusions are observed in the cytoplasm of mononuclear cells. Figure 3. MPS IIIB, 18 months old. Subjectively, there are increased numbers of cytoplasmic vacuoles and basophilic inclusions within mononuclear cells. Figure 4. MPS IIIB, 26 months old. Cytoplasmic vacuoles and basophilic inclusions in mononuclear cells subjectively appear larger in the 26 months old dogs with MPS IIIB when compared to the 10 and 18-month-old dogs with MPS IIIB (Figs. 2 and 3 respectively).
CBC and serum chemistry analysis were run on a subset of dogs (two 10 mo. control, two 10 mo. MPS IIIB). There were similar mild electrolyte abnormalities noted in both controls and dogs with MPS IIIB. Sodium values ranged from 139-140 mEq/L (reference range: 141-151mEq/L) and chloride ranged from 107-108 mEq/L (reference range: 112-121 mEq/L).
Histopathologic Findings
Histologic sections of brain and cervical spinal cord from MPS IIIB cases and age-matched controls were examined at 2, 4, 10, 18, and 26 months of age. Examined regions of the brain and spinal cord are listed in Table 2.
Table 2.
Temporospatial patterns of vacuolation and staining observed in dogs with mucopolysaccharidosis IIIB.
| 2-mos-old (n=1) |
4- mos-old (n=2) |
10- mos-old (n=2) |
18- mos-old (n=4) |
26- mos-old (n=5) |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vac | Vac | Vac | Vac | Vac | ||||||||||||||||
| ROIs |
G |
N |
PAS |
LFB |
G |
N |
PAS |
LFB |
G |
N |
PAS |
LFB |
G |
N |
PAS |
LFB |
G |
N |
PAS |
LFB |
| PCG | + | − | − | − | + | − | − | + | +++ | + | + | + | +++ | ++ | ++ | ++ | +++ | ++ | ++ | ++ |
| Cd | − | − | − | − | + | − | − | + | ++ | − | − | + | + | + | ++ | + | + | + | + | + |
| Hp | − | − | − | + | + | − | + | + | + | − | + | + | ++ | ++ | ++ | ++ | ++ | + | ++ | ++ |
| DTN | − | − | − | − | + | − | − | − | ++ | + | + | − | ++ | ++ | ++ | + | +++ | ++ | +++ | ++ |
| PKC | − | − | − | − | − | − | + | + | − | + | + | + | + | ++ | ++ | + | ++ | ++ | ++ | ++ |
| VN | − | − | − | − | + | − | − | − | + | + | − | − | ++ | ++ | ++ | + | +++ | +++ | +++ | +++ |
| VH | − | − | − | + | − | − | + | + | − | − | + | + | + | ++ | + | + | ++ | ++ | ++ | ++ |
| DRG | − | − | − | − | NC | NC | NC | NC | NC | NC | NC | NC | + | + | − | + | ++ | + | ++ | ++ |
Abbreviations: ROIs, regions of interest; Vac, vacuolation; G, glial cells; N, neurons; PAS, periodic acid Schiff; LFB, luxol fast blue; PCG, posterior cruciate gyrus of cerebral cortex; Cd, caudate nucleus; Hp, hippocampus; DTN, dorsal thalamic nucleus; PKC, purkinje cell; VN, vestibular nucleus; VH, ventral horn, DRG, dorsal root ganglion; NC, not collected.
One 0.237 mm2 area was assessed in each region of interest. (−) = No positive staining or vacuolated cells/0.237mm2, (+) = < 25% of cell/0.237mm2, (++) = 25-50% of cells/0.237mm2, (+++) = > 50% of cells/0.237mm2.
Different cell populations accrued storage material at different rates (Table 2). Initial evidence of lysosomal storage was observed on an HE-stained section from the 2-month-old dogs with MPS IIIB. Perivascular and perikaryal glial cells (identified as cells surrounding neuronal bodies) of the cerebral cortex were mildly to moderately swollen and vacuolated in 2 and 4-month- old dogs (Fig.6 and 7) when compared to control dogs (Fig. 5). Cytoplasmic vacuolation of glial cells was more severe and more widely distributed in the brains and spinal cords of the older dogs with MPS IIIB (Figs. 8-10; Table 2). In addition, the perivascular spaces of the 18-month-old (n=4) and 26-month-old (n=5) dogs with MPS IIIB were expanded by severely vacuolated glial cells (Fig. 11a), which were determined to be microglia based on Iba1 immunolabeling (Fig. 11c).
Figures 5-10.

Cerebral cortex (postcruciate gyrus) at the level of the caudal nucleus, dog. Hematoxylin and eosin. Figure 5. Control, 26 months old. Vacuolated cells are not present. Figure 6. Mucopolysaccharidosis (MPS) IIIB, 2 months old. There is cytoplasmic vacuolation of scattered glial cells (arrows). Cytoplasmic vacuoles peripheralize nuclei, and are large, clear, and variably contain lacy to granular material (inset). Figure 7. MPS IIIB, 4 months old. Scattered vacuolated glial cells (arrows) are observed. Figures 8-10. MPS IIIB, cerebral cortex, dogs, 10 months old (Fig. 8), 18 months old (Fig. 9), and 26 months old (Fig. 10). Compared to the younger dogs, there is a marked increase in the number of vacuolated glial cells (arrows), as well as cytoplasmic vacuolation of neurons (arrowheads).
Figures 11-14.

Mucopolysaccharidosis IIIB, dog, 26 months old. Figure 11. Cerebral cortex (postcruciate gyrus). (a) Moderate to severe cytoplasmic vacuolation of perivascular glia. Cytoplasmic vacuoles are large and clear. Hematoxylin and eosin (HE). (b) The vacuoles contain small periodic acid-Schiff (PAS)-positive inclusions (arrows). (c) Perivascular glial cells are compatible with microglia based on diffuse cytoplasmic immunolabelling for Iba1. Figure 12. Cerebellar cortex. (a) Severe cytoplasmic vacuolation of Purkinje cells. Cytoplasmic vacuoles are up to 1-μm-diameter, clear, and variably contain eosinophilic inclusions. HE. (b,c) The vacuoles contain PAS-positive (b) and Luxol fast blue-positive (LFB) (c) inclusions. Figure 13. Lateral vestibular nucleus. (a) There are cytoplasmic vacuoles that are small, punctate to granular, and located eccentrically. HE. (b-c) Neurons contain inclusions that stain positively with PAS (b) and LFB (c). Figure 14. Dorsal root ganglion. (a) Cytoplasmic vacuolation (arrowheads) of a neuron. Satellite glial cells (*) contain large, clear cytoplasmic vacuoles. HE. (b,c) The vacuoles contain PAS-positive (b) and LFB-positive (c) inclusions.
Initial evidence of lysosomal storage within neurons was observed at 10 months of age (n=2) on HE-stained sections (Table 2). Neurons in several of the ROIs were swollen and mildly expanded by small, round, granular to fine cytoplasmic vacuoles. Vacuoles were most commonly observed at the periphery of the cell body. Neuronal vacuolation was most severe in the 18 (n=4) and 26 (n=5) month old dogs with MPS IIIB, with severe distention of the cytoplasm (ballooning) and peripheralization of Nissl substance and nuclei. Neuronal vacuolation was also observed in the spinal cord of dog with MPS IIIB (Table 2). The degree of individual neuronal vacuolation was most pronounced in the cerebellum (Fig. 12a), brainstem nuclei (Fig. 13a), and spinal cord. Neurons in these regions contained larger cytoplasmic vacuoles that variably contained eosinophilic inclusions. Neuronal vacuolation was not observed in any of the control dogs (n=7).
The DRG were not available for examination from 4 and 10-month-old dogs and were only examined from the 2, 18, and 26-month-old dogs. No differences in neuronal or satellite glial cell morphology were identified in the DRG from the 2-month-old dog with MPS IIIB (n=1) compared to the 2-month-old control dog (n=1). Ganglion neurons in 18-month-old (n=4) and 26-month-old (n=5) dogs with MPS IIIB were mildly to moderately swollen and contained cytoplasmic vacuoles (Fig. 14a). In addition, cytoplasmic vacuoles were observed in satellite glial cells and fibrocytes. GFAP and Iba1 immunohistochemistry demonstrated localization of the vacuoles to satellite glial cells and Iba1-positive cells in the supporting connective tissue (Figs. 31, 33).
Figures 30-33.

Dorsal root ganglion (DRG), dog. Immunohistochemistry. Figures 30. DRG, control, 26 months old. Glial fibrillary acidic protein (GFAP) immunolabeling (brown) is limited to satellite glial cells. Figure 31. Mucopolysaccharidosis (MPS) IIIB, DRG, 26 months old. There is intense GFAP labeling of satellite glial cells. GFAP immunolabeling highlights large clear cytoplasmic vacuoles (arrow, inset). Figure 32. DRG, control dog, 26 months old. Ionized calcium binding adaptor molecule 1 (Iba1) immunolabeling (brown) in the DRG was minimal in control dogs. Iba1-positive cells were limited to the supporting connective tissue and were commonly admixed with satellite glia. Figure 33. MPS IIIB, DRG, 26- month-old. Iba1 expression is markedly increased. Numerous Iba1-positive cells are expanded by large clear cytoplasmic vacuoles that variably impinge upon neurons (arrow, inset).
Loss of Purkinje cells was the most striking difference in dogs with MPS IIIB when compared to control dogs. Histologically, there was a detectable reduction in Purkinje cell density in 18-month-old (n=4) and 26-month-old (n=5) dogs with MPS IIIB compared to control dogs (n=1, 18-month-old and n=1, 26-month-old) (Figs. 15-18). Purkinje cell density was quantified in all dogs to determine the degree and distribution of Purkinje cell loss (Fig. 19). Decreased Purkinje cell linear density was initially observed in all evaluated regions of the cerebellum in the 10-month-old dogs with MPS IIIB (n=2) when compared to age-matched control dogs (n=2). Purkinje cells loss was greatest in the 26-month-old dogs with MPS IIB when compared to the age-matched control dog and all other age groups.
Figures 15-18.

Cerebellum at the level of cerebellar linguae, dog. Hematoxylin and eosin. Figure 15. Dorsal cerebellar hemisphere folium, control, 26 months old. Adequate Purkinje cell density. Figure 16. Mucopolysaccharidosis (MPS) IIIB, dorsal cerebellar hemisphere folium, 26 months old. Severe reduction in Purkinje cell density. Figure 17. Dorsal vermis, control dog, 26 months old. Adequate Purkinje cell density. Figure 18. MPS IIIB, dorsal vermis, 26 months old. Severe reduction in Purkinje cell density.
Figure 19.
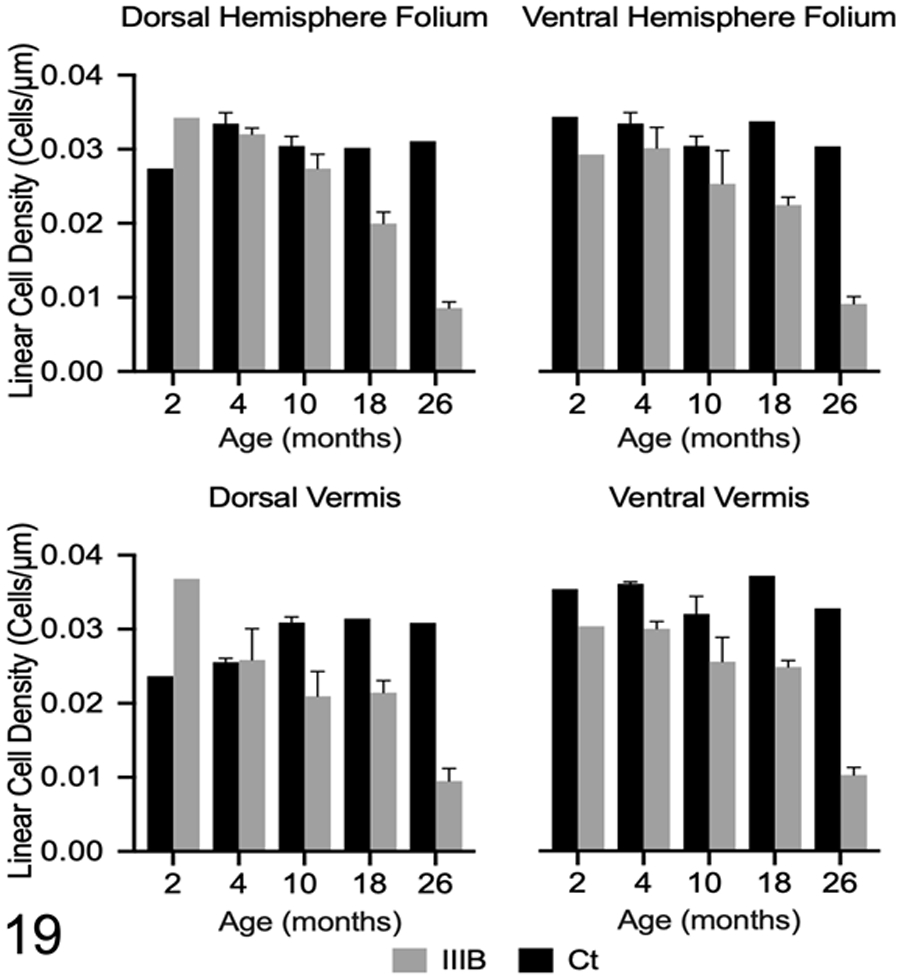
Linear Purkinje cell densities in control dogs (Ct) and dogs with Mucopolysaccharidosis IIIB (IIIB) were calculated at each time point in four regions of the cerebellum. Mean linear cell density is decreased in three of the four examined regions of the cerebellum at 4 months of age and decreased in all examined regions at 10 months of age. Loss of Purkinje cells were not observed in any control dogs. Data are shown as mean ± SEM.
Histochemical stains were used to further characterize the storage material within vacuoles of neurons and glial cells. PAS was used to characterize neutral glycolipid/glycoprotein/GAG content, and LFB was used to characterize lipid/lipoprotein content in storage vacuoles.3,39 Inclusions within perivascular and perikaryal glial cells were PAS-positive (Fig. 11b). The earliest PAS-positive inclusions were observed in the 4-month-old dogs with MPS IIIB (n=2). Glial cells containing PAS-positive inclusions increased in number and distribution over time (Table 2). PAS-positive inclusions were not observed in glial cells from any of the control dogs (n=7). LFB staining was not appreciated in glial cells of either dogs with IIIB or control dogs at any time point. Neuronal inclusions stained positive with both PAS and LFB (Figs. 12 b-c, 13 b-c, 14 b-c). The earliest PAS and LFB positive neuronal inclusions were observed in the 4-month-old dogs with MPS IIIB (n=2) and increased in number and distribution over time (Table 2). PAS and LFB-positive inclusions were not observed in neurons from any of the control dogs (n=7).
Immunohistochemical Findings
Changes in astrocyte morphology were qualitatively assessed based on GFAP-labeled sections of the postcruciate gyrus. Parameters for reactive astrogliosis in this study were subjectively based on degree of hypertrophy (mild, moderate, marked). Mild reactive astrogliosis (increased GFAP labeling of astrocytic process with little intermingling of processes) was observed in the 2-month-old (n=1) and 4-month-old (n=2) dogs with MPS IIIB (Supplemental Fig. S12). Moderate astrogliosis (increased GFAP labeling of astrocytic bodies and processes with intermingling of the processes) was observed in the 10-month-old (n=2) and 18-month-old (n=4) dogs with MPS IIIB (Supplemental Fig. S13), and severe astrogliosis (increased GFAP labeling of astrocytic bodies and processes with substantial intermingling of processes) was observed in the 26-month-old dogs with MPS IIIB (n=5) (Supplemental Fig. S14).
Quantitative IHC was used to evaluate GFAP immunoreactivity in the brain and spinal cord of dogs with MPS IIIB and control dogs at all time points. Mean GFAP immunoreactivity in dogs with MPS IIIB was increased over age-matched controls in all brain ROIs at all time points, with the exception of the centrum semiovale and hippocampus at 2- months (Figs. 20-23, 28). Increased GFAP immunoreactivity in MPS IIIB brains compared to control brains was greatest at 26 months. In the spinal cord, mean GFAP immunoreactivity was also increased over age-matched controls at all time points, and increased over time (Figs. 24-27, 29).
Figure 20-27.

Mucopolysaccharidosis IIIB, cerebral cortex (Figs 20-23) and cervical spinal cord (Figs 24-27), dog. Immunohistochemistry for glial fibrillary acidic protein (GFAP). Samples are from dogs aged 4 (Fig. 20, 24), 10 (Fig. 21, 25), 18 (Fig. 22, 26), and 26 (Fig. 23, 27) months. Subjectively, there is increased GFAP immunolabeling (brown) in both the cerebral cortex and cervical spinal cord observed at each of the increasing time points with the greatest immunolabeling being observed at 26 months of age.
Figure 28.
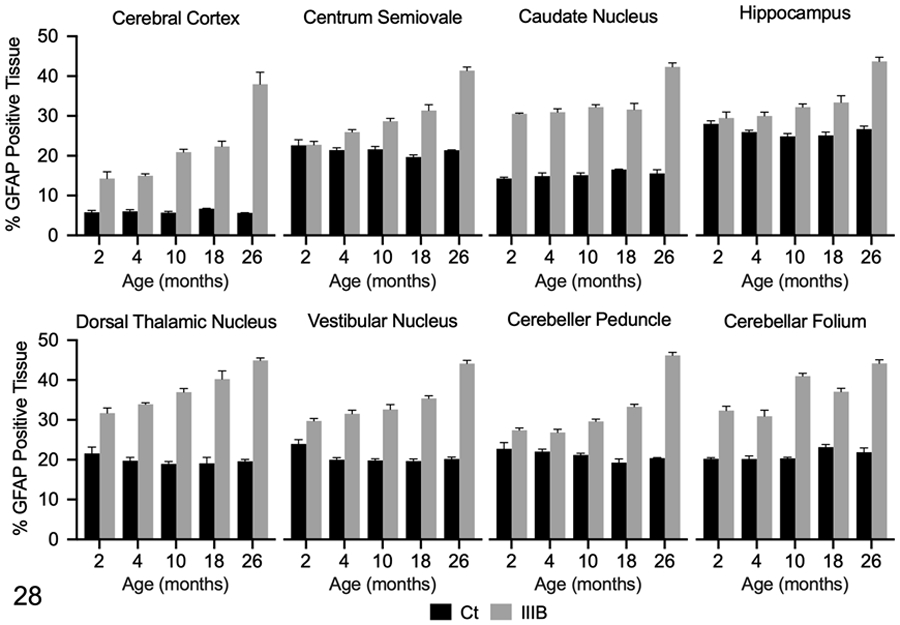
Glial fibrillary acidic protein (GFAP) immunolabeling in the brain of control (Ct) and mucopolysaccharidosis (MPS) IIIB (IIIB) dogs. The data shown represents GFAP immunolabeling in each region of interest as determined by pixel-based analysis (% positive tissue labeled). Mean GFAP immunolabeling was increased in all brain regions at all time points in dogs with MPS IIIB, with the exception of the centrum semiovale and hippocampus at 2 months of age. Immunolabeling was increased in these regions by 4 months of age in dogs with MPS IIIB.
Figure 29.
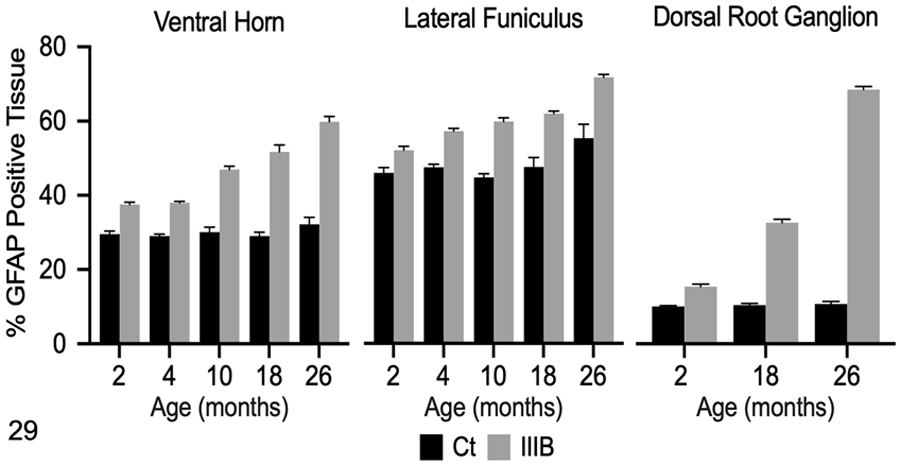
Glial fibrillary acidic protein (GFAP) immunolabeling in the cervical spinal cord and dorsal root ganglion (DRG) of control (Ct) and mucopolysaccharidosis (MPS) IIIB (IIIB) dogs. The data shown represents GFAP immunolabeling in each region of interest as determined by pixel-based analysis (% positive tissue labeled). Mean immunolabeling for GFAP was increased in the ventral horn and lateral funiculus of the spinal cord in dogs with MPS IIIB at all time points compared to control dogs. Mean GFAP immunoreactivity was increased in the DRG of dogs with MPS IIIB compared to control dogs at all evaluated time points (2, 18, 26 months of age).
Dorsal root ganglia from dogs with MPS IIIB and age-matched controls were only collected at the 2 (n=1 control, 1 MPS IIIB), 18 (n=1 control, 4 MPS IIIB), and 26 (n=1 control, 5 MPS IIIB) month time points. GFAP immunoreactivity in the DRG of controls was limited to satellite cells (Fig. 30). A similar pattern of GFAP immunoreactivity was observed in dogs with MPS IIIB; however, there was visibly increased labeling of satellite cells in the 18 (n=4) and 26 (n=5) month old dogs with MPS IIIB (Fig. 31). In addition, GFAP-labeled satellite cells variably contained 1-2 μm in diameter, clear vacuoles (Fig. 31, inset). Quantitative IHC demonstrated increased GFAP immunoreactivity in dogs with MPS IIIB compared to age-matched control dog at all evaluated time points (Fig. 29).
Changes in microglia morphology were qualitatively assessed based on Iba1-labeled sections of the postcruciate gyrus. The first and most striking microglial change observed in dogs with MPS IIIB was cytoplasmic vacuolation. No vacuolated microglia were observed in any of the control dogs (n=7). Microglia were characterized by mild, moderate, or severe vacuolation with either retention of a quiescent ramified state or reactive ameboid state (Supplemental Figs. S15-S18). Scattered and perikaryal, mildly vacuolated microglia were observed in the cerebral cortex, predominately layers II-V, and expanding perivascular spaces in 2-month-old (n=1) and 4-month-old (n=2) dogs with MPS IIIB (Supplemental Fig. S16). Microglia in 10-month-old dogs with MPS IIIB demonstrated moderate vacuolation and reduced ramified projections (Supplemental Fig. S17). Microglial vacuolation was severe in 18-month-old (n=4) and 26-month-old (n=5) dogs with MPS IIIB, and a majority demonstrated a reactive ameboid morphology (Supplemental Fig. S18).
Quantitative IHC was used to assess Iba1 immunoreactivity in the brain and spinal cord of dogs with MPS IIIB and control dogs at all time points. Mean Iba1 immunoreactivity was increased over age-matched controls in all brain ROIs at all time points, with the exception of the centrum semiovale at 2 months (Figs. 34-37, 42). The most prominent labeling was observed at 18 and 26 months. In the spinal cord, mean Iba1 immunoreactivity was initially increased over the age-matched control in the lateral funiculus at 2 months (n=1), and in the ventral horn at 4 months (n=2) (Fig. 38-41, 43). Immunoreactivity progressively increased from 10 (n=2) to 26 (n=5) months in the dogs with IIIB (Fig. 43).
Figures 34-41.

Mucopolysaccharidosis IIIB, cerebral cortex (Figs 34-37) and cervical spinal cord (Figs. 38-41), dog. Immunohistochemistry for ionized calcium binding adaptor molecule 1 (Iba1). Samples are from dogs aged 4 (Fig. 34, 38), 10 (Fig. 35, 39), 18 (Fig. 36, 40), and 26 (Fig. 37, 41) months. Subjectively, there is increased Iba1 immunolabeling (brown) in both the cerebral cortex and cervical spinal cord observed at each of the increasing time points with the greatest immunolabeling being observed at 26-months of age.
Figures 42.
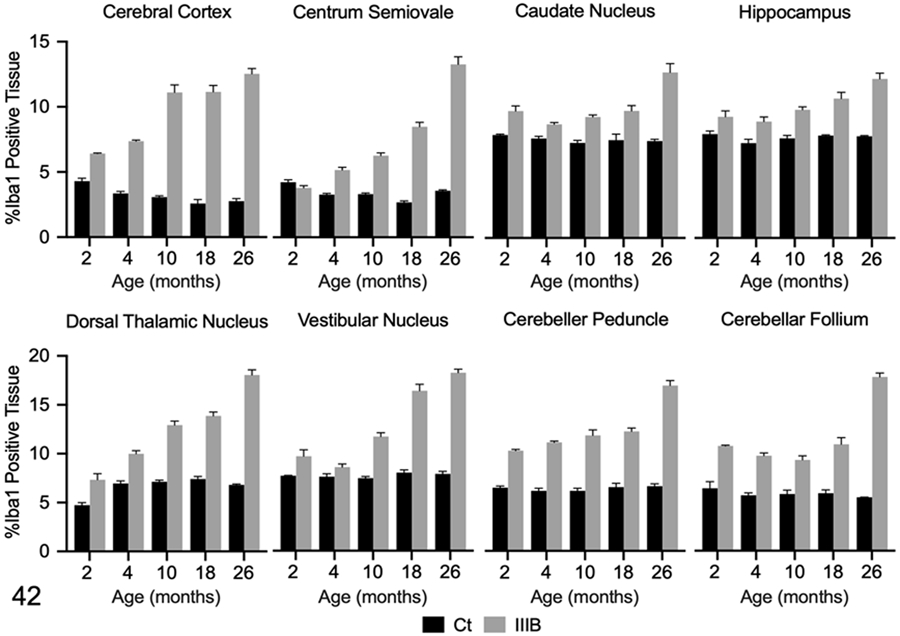
Ionized calcium binding adaptor molecule (Iba)1 immunolabeling in the brain of control (Ct) and mucopolysaccharidosis (MPS) IIIB (IIIB) dogs. The data shown represents Iba1 immunolabeling in each region of interest as determined by pixel-based analysis (% positive tissue labeled). Mean Iba1 immunolabeling was increased in all ROIs, except the centrum semiovale, in the 2-month- old dog with MPS IIIB. Iba-1 immunolabeling was increased in all ROIs of the 4- month- old dogs with MPS IIIB. Increased Iba-1 immunolabeling was observed at all the additional time points (10, 18, and 26 months of age).
Figure 43.
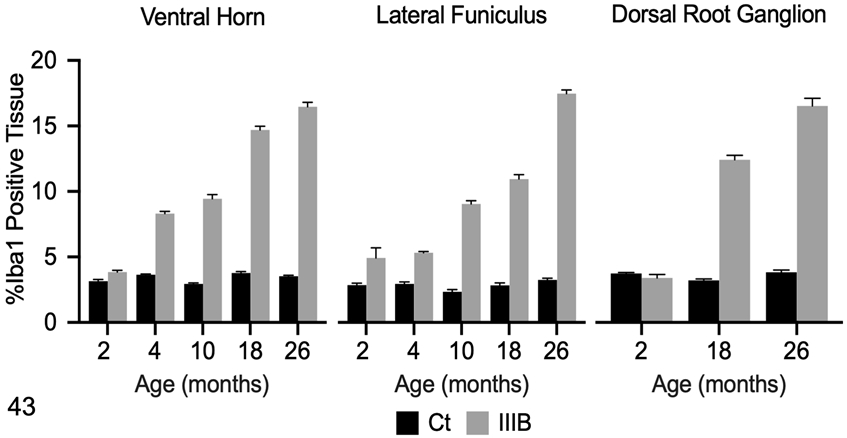
Ionized calcium binding adaptor molecule 1 (Iba1) immunolabeling in the cervical spinal cord and dorsal root ganglion (DRG) of control (Ct) and mucopolysaccharidosis (MPS) IIIB (IIIB) dogs. The data shown represents Iba1 immunolabeling in each ROI as determined by pixel-based analysis (% positive tissue labeled). Mean Iba1 immunolabeling was increased in both the ventral horn and lateral funiculus of the cervical spinal cord in 4-month-old dogs with MPS IIIB and was increased in all subsequent time points. No difference in Iba1 immunolabeling was observed in the DRG between the control dog and the 2 months old dog with MPS IIIB. Iba1 immunoreactivity was markedly increased in 18- and 26-month-old dogs with MPS IIIB.
Iba1 immunoreactivity in the DRG from control dogs was minimal (Fig. 32). In control dogs, Iba1-positive cells were present directly around ganglion neurons, admixed with satellite cells, and within the supportive connective tissue. In 18-month-old (n=4) and 26-month-old (n=5) dogs with MPS IIIB, Iba1-positive cells were markedly expanded by cytoplasmic vacuoles (Fig. 33). There was no difference in mean Iba1 immunoreactivity between the 2-month-old dog with MPS IIIB (n=1) and its age-matched control (n=1) (Fig. 43). There was a marked increase in Iba1 immunoreactivity in dogs with MPS IIIB over controls at 18 (n=4) and 26 (n=5) months (Fig. 43).
Discussion
In the present work, we have described the character and distribution of neuropathologic lesions associated with MPS IIIB in the brain, spinal cord and dorsal root ganglion in a canine model, as well as cytologic changes observed in the CSF from 10 to 26 months. Our results are comparable to those reported in human MPS IIIB patients and the murine model of MPS IIIB, indicating the dog could serve a model for the nervous system disease associated with MPS IIIB.
Evaluation of the CSF revealed numerous distended cytoplasmic vacuoles containing discrete inclusions within mononuclear cells of MPS IIIB-affected dogs. These inclusions are a common finding in CSF mononuclear cells of MPS IIIB-affected animals and humans.1 The inclusions are thought to consist of HS as well as secondary gangliosides. Cytochemical staining was not performed in the current study due to limited sample. Phagocytosed myelin and cellular debris within the CSF can appear similar to the inclusions described here, but usually to a much lesser extent.
Mild changes were identified on CBC and serum chemistry in a subset of both affected and control dogs. The electrolyte disturbances can be attributed to mild transient diarrhea that was noted in a small number of control and dogs with MPS IIIB during the study.
Histologic examination of postmortem brain specimens from MPS III human patients are limited, but demonstrated severe gross and histologic lesions.5 Gross pathologic findings in the CNS consist of bilateral ventriculomegaly, cerebral atrophy, and variable degrees of meningeal thickening.30,56,21,23 Grossly, dogs with MPS IIIB demonstrated a similar ventriculomegaly and atrophy of the cerebrum and cerebellum at an advanced stage of the disease (4-5 years of age).13
Histologic features of brains from human MPS IIIB patients are characterized by cytoplasmic vacuolation and distension of neurons and glia due to excessive storage of both HS and gangliosides (GM2 and GM3), and variable degrees of gliosis.24,23,63 In the present study, histologic examination of this canine model demonstrated vacuolation of neurons in cerebral cortex, diencephalon, cerebellum, brainstem, and peripheral ganglia (DRG). Vacuolated neurons in this model were initially observed in the 10-month-old dogs with MPS IIIB but were more severe and widely distributed in the 18 and 26-month-old dogs with MPS IIIB, indicating a progressive process. In addition, vacuolation of neurons in the brainstem nuclei, cerebellum, and spinal cord was more severe than other examined regions. This pattern of neuronal vacuolation (basal ganglia, thalamus, brainstem) can be observed in human MPS IIIB patients; however, storage in humans appears to be more variable in severity and distribution.23,56,21,57
Cytoplasmic vacuolation of glial cells was the first noticeable histologic lesion identified in dogs with MPS IIIB in this study. Glial cell vacuolation was initially observed at 2 months and became more prominent and widely distributed in the older MPS IIIB-affected dogs. Immunolabeling for astrocytes (GFAP) and microglia (Iba1) demonstrated that cytoplasmic vacuolation was limited to microglia. Vacuolation of microglia/macrophages in the brain/meninges have been documented in humans MPS III patients.24,10,32,23 Vacuolation of astrocytes and oligodendrocytes have variably been reported in human MPS III patients56,21; however, we did not observe vacuolation in either cell type. In addition, vacuolated microglia expanded perivascular spaces within the cerebral cortex and cortical white matter, especially in the 18 and 26-month-old dogs with MPS IIIB. Expanded perivascular spaces are commonly observed in human MPS IIIB patients and can be appreciated both radiographically (magnetic resonance imaging) and histologically,10,24 and are likely the result of GAG accumulation, infiltration of vacuolated microglia, and potentially failure of the blood-brain barrier. 24,5,23
To further characterize storage material, numerous MPS III studies have employed the use of special stains. Storage inclusions in human MPS III patients typically stain positive with periodic acid-Schiff (neutral GAGs), Alcian blue (acidic GAGs), colloidal iron (sulfated GAGs), toluidine blue (acidic GAG), Sudan black (lipid), Oil red O (lipid), and Luxol fast blue (lipid/lipoprotein/myelin) stains.56,23,61 In this study, PAS and LFB stains were employed, and inclusions within vacuolated and swollen neurons were positive for both. LFB and PAS-positive inclusions became more prominent and numerous in the 18 and 26-month-old dogs with MPS IIIB. The histochemical staining patterns of the inclusions are suggestive of primary GAG (HS)/polysaccharide accumulation and secondary ganglioside/glycolipid accumulation within neurons.13 A previous study utilizing dogs from the same MPS IIIB colony, demonstrated significantly increased brain content of total sulfated GAGs, and GM2 and GM3 gangliosides in dogs with MPS IIIB.13 PAS-positive inclusions were appreciated in vacuolated glial cells, but LFB staining was not. In the MPS IIIC murine model, electron microscopy demonstrated electron lucent storage material in cytoplasm of glial cells, which was suggestive of GAG accumulation only. It is possible that glial cells in this canine model of MPS IIIB accumulate only GAG and not gangliosides, resulting in the LFB-negative staining.
In addition to vacuolar degeneration of neurons and glial cells, reduced neuronal density and gliosis can also be observed in human MPS IIIB patients at advanced stages of the disease. Neuronal loss is most severe the cerebral cortex, olivary nucleus, substantia nigra, thalamus, cerebellar cortex, striatum, and raphe nucleus of the pons.5,24,61,10,23 Decreased Purkinje cell density was observed in the current study; however, neuronal densities in other brain regions were not assessed. Thorough morphometric analysis of neuronal cell density from these MPS IIIIB-affected dogs would be required for a more complete comparison of the model. Gliosis is commonly observed in the cerebral white matter (centrum semiovale), globus pallidus, thalamus, olivary nucleus, substantia nigra, and brain stem nuclei of human MPS IIIB patients.24 In this canine model, we observed moderate to severe gliosis in the cerebral cortex, centrum semiovale, caudate nucleus, thalamus, hippocampus, and brainstem nuclei, which appears consistent with that reported in humans.
Purkinje cell vacuolation and loss is a prominent and well-documented pathologic feature of MPS III in both humans and animals.28,23,15,13,31,17,27 We calculated linear cell density based on previous methods27, 41 to better quantify Purkinje cell density in the cerebellum. Purkinje cell loss in the dorsal and ventral aspects of the cerebellar hemisphere and vermis were compared. Linear cell density was decreased in dogs with MPS IIIB compared to age-matched control dogs. Purkinje cell loss has been documented in the canine model of MPS IIIA with loss occurring around 40 months of age, which is much later than that observed in the MPS IIIB model.27 Purkinje cell loss was also a prominent feature observed in both Schipperke dogs from which the colony used in this study originated, but these animals were 4-5 years of age.13 Purkinje cell lesions and loss in humans with MPS has also been described; however, the severity of Purkinje cell lesions in humans is more variable than that observed in the canine model.63,23,32 Despite this inconsistency, the canine model may be useful for investigating mechanisms of Purkinje cell loss and development of cerebellar dysfunction.
This study is the first to describe pathological changes in the dorsal root ganglia of dogs with MPS IIIB. Vacuolated ganglion neurons (sympathetic, myenteric, submucosal) have been documented in post-mortem histologic examination of human MPS IIIB patients.56 However, a detailed description of DRG lesions in human MPS IIIB patients has not been reported. Histologic examination of the DRG in this model showed significant pathologic changes associated with storage disease. Ganglion neurons were swollen and expanded by numerous cytoplasmic vacuoles. Intraneuronal cytoplasmic inclusions stained positively with PAS and LFB, suggestive of HS and ganglioside accumulation. Satellite cells were immunolabeled for GFAP in both controls and MPS IIIB-affected dogs, which is consistent with a previous study examining immunohistochemical labeling of the DRG in the dog.60 GFAP immunolabeling aided in the identification of cytoplasmic vacuoles within satellite cells, and GFAP and Iba1 immunoreactivity were both increased in MPS IIIB-affected dogs compared to controls indicating glial reactivity. DRG lesions in this model are consistent with those reported in the murine MPS IIIB model, where the authors demonstrated evidence of lysosomal storage in ganglion neurons, satellite cells, and Schwann cells.18 A clear understanding of the contribution of pathologic changes in the peripheral nervous system to MPS IIIB is lacking, however, its elucidation may prove valuable. It is possible that peripheral neuropathies in MPS IIIB patients result in sensory system impairment (decreased tactile and thermal perception), as highlighted by research carried out in the murine MPS IIIB model.18 A recent study examining a dominant NAGLU mutation demonstrated that affected individuals suffer from an adult-onset painful polyneuropathy.59 Tetreault and others59 speculate that lesions in the DRG may contribute to the explosive behavior seen in some MPS IIIB patients, as manifestation of a pain syndrome, but no such link has been elucidated. Further investigation into the contribution of DRG lesions to the clinical presentation of MPS IIIB is needed.
Neuroinflammation is a widely reported finding in the MPS IIIB murine model, with well-documented astrogliosis, microgliosis, and increased levels of pro-inflammatory cytokines/chemokines and oxidative agents.64,42 Varying degrees of astrogliosis and microgliosis are observed in human MPS IIIB patients.10,23,24 In this model, neuroinflammation, evidenced by increased astrocytic GFAP and microglial Iba1 expression, was increased early in the disease course, which is in agreement with other animal studies of MPS.2,48,42,11,65 Elevations in GFAP have similarly been demonstrated in the MPS IIIB murine model and the MPS IIIA canine model.64,65 Increased GFAP expression in this model occurred in several brain regions where gliosis in humans has been reported (i.e., centrum semiovale, diencephalon, brainstem).23,56,21 Morphological changes of reactive astrogliosis, characterized by hypertrophy of cell bodies and projections, were first observed in the 2 and 4-month-old dogs with MPS IIIB. In the 26-month-old dogs with MPS IIIB, GFAP immunoreactivity was markedly elevated compared to the age-matched control, and astrocytes were moderate to severely hypertrophied with numerous overlapping cellular projections, indicative of a reactive state. Astrogliosis has been observed in many lysosomal storage diseases52; however, the exact role astrocytes play in the pathogenesis of MPS IIIB remains unclear.
Increased Iba1 expression and vacuolated microglia were observed in several brain regions (excluding the centrum semiovale and dorsal thalamic nucleus) and the cervical spinal cord of the 2-month-old dog with MPS IIIB. Vacuolation of microglia and Iba1 expression became more prominent in the older dogs when compared to age matched controls. In addition, vacuolated microglia observed in the 26-month-old dogs with MPS IIIB demonstrated a predominately ameboid morphology, indicating a reactive or inflammatory phenotype. Our findings indicate that neuroinflammation precedes the development of neuronal lesions. Studies using other models have come to similar conclusions on the kinetics of neuroinflammation in MPS.64,2,11,48,65 One study in particular utilizing a canine model of MPS IIIA demonstrated early microgliosis in all brain regions examined (excluding the dentate gyrus) in affected dogs starting at 2 weeks of age.65 In the MPS IIIA canine model, microglia demonstrated ameboid morphology and microgliosis early in the disease process, but vacuolation of microglia was not reported.65 In this model, vacuolation of microglia was observed in the 2-month-old dog with MPS IIIB, but ameboid morphology was not a prominent feature until 10 months of age.
Studies utilizing the canine MPS IIIA model65, murine MPS IIIB model42, and the current study, demonstrate that neuroinflammation develops early in the disease process prior to the development of neuronal lesions. Considering the similarities of the canine and murine MPS III models to humans, it is possible that similar neuroinflammatory processes occur early in human MPS IIIB disease, but it is unclear if neuroinflammation contributes to the onset of clinical signs. Clinical signs were not observed in association with early onset neuroinflammation in the canine MPS IIIA model,65 which may suggest that neuroinflammation does not contribute to neurologic impairment early in the disease course. In addition, a Toll-like receptor 4 and MyD88 double-knockout MPS IIIB murine model demonstrated decreased early onset neuroinflammation but persistence of neurodegeneration2, which may indicate neuroinflammation and neurodegeneration develop independently early in disease. To conclude, the exact relationship between neuroinflammation and its contribution to neuronal lesions and clinical signs remains unclear in any of the animal models. Further research is imperative to characterize the extent and kinetics of neuroinflammation as they apply to astrocyte and microglial activation and clinical disease.
The development of a well-characterized large animal model is vital to the understanding of MPS IIIB pathogenesis and would aid in the screening and development of new therapeutics. The importance of the canine model has been highlighted by the success of canine models of other MPSs.31,36,35,34,12 The current study provides a detailed characterization of canine MPS IIIB disease kinetics with regard to neurologic lesions and neuroinflammation that will serve as foundational data for future pathogenesis and therapeutic studies in dog models of MPS IIIB.
Supplementary Material
Acknowledgements
The authors thank E. M. Snella, J. K. Jens, and V. Montgomery for excellent technical assistance. We acknowledge the excellent care provided by the veterinary and animal care staff of the ISU Laboratory Animal Resources. Additionally, the care and production of these animals is made possible by a capable and dedicated cohort of ISU undergraduate students. The foundation and maintenance of the colony was possible by a grant from the National MPS Society, and via an unrestricted gift from Shire Human Genetic Therapies Inc. (currently a division of Takeda). Affected animals were supported in part by a grant from Alexion Pharmaceuticals, Inc. Normal control tissues were derived from animals supported in part by Grants from the NIH (NS088766 and NS085381 to Patricia I. Dickson).
References
- 1.Federico GC A, Cecio A,D'Auria N,Di Iorio G,Ronsisvalle L,and Di Natale P. Sanfilippo B Syndrome (MPS III B): Case Report with Analysis of CSF Mucopolysaccharides and Conjunctival Biopsy. Journal of Neurology. 1981. [DOI] [PubMed] [Google Scholar]
- 2.Ausseil J, Desmaris N, Bigou S, et al. Early neurodegeneration progresses independently of microglial activation by heparan sulfate in the brain of mucopolysaccharidosis IIIB mice. PLoS One. 2008;3: e2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bancroft JD, Layton C. 12 - Connective and other mesenchymal tissues with their stains. In: Suvarna SK, Layton C, Bancroft JD, eds. Bancroft's Theory and Practice of Histological Techniques (Eighth Edition). Content Repository Only!; 2019:153–175. [Google Scholar]
- 4.Beesley CE, Jackson M, Young EP, Vellodi A, Winchester BG. Molecular defects in Sanfilippo syndrome type B (mucopolysaccharidosis IIIB). J Inherit Metab Dis. 2005;28: 759–767. [DOI] [PubMed] [Google Scholar]
- 5.Bigger BW, Begley DJ, Virgintino D, Pshezhetsky AV. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol Genet Metab. 2018;125: 322–331. [DOI] [PubMed] [Google Scholar]
- 6.Casal M, Haskins M. Large animal models and gene therapy. Eur J Hum Genet. 2006;14: 266–272. [DOI] [PubMed] [Google Scholar]
- 7.Crawley AC, Marshall N, Beard H, et al. Enzyme replacement reduces neuropathology in MPS IIIA dogs. Neurobiol Dis. 2011;43: 422–434. [DOI] [PubMed] [Google Scholar]
- 8.Cressant A, Desmaris N, Verot L, et al. Improved behavior and neuropathology in the mouse model of Sanfilippo type IIIB disease after adeno-associated virus-mediated gene transfer in the striatum. J Neurosci. 2004;24: 10229–10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Pasquale V, Sarogni P, Pistorio V, Cerulo G, Paladino S, Pavone LM. Targeting Heparan Sulfate Proteoglycans as a Novel Therapeutic Strategy for Mucopolysaccharidoses. Mol Ther Methods Clin Dev. 2018;10: 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dekaban AS, Constantopoulos G. Mucopolysaccharidosis type I, II, IIIA and V. Pathological and biochemical abnormalities in the neural and mesenchymal elements of the brain. Acta Neuropathol. 1977;39: 1–7. [DOI] [PubMed] [Google Scholar]
- 11.DiRosario J, Divers E, Wang C, et al. Innate and adaptive immune activation in the brain of MPS IIIB mouse model. J Neurosci Res. 2009;87: 978–990. [DOI] [PubMed] [Google Scholar]
- 12.Ellinwood NM, Ausseil J, Desmaris N, et al. Safe, efficient, and reproducible gene therapy of the brain in the dog models of Sanfilippo and Hurler syndromes. Mol Ther. 2011;19: 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellinwood NM, Wang P, Skeen T, et al. A model of mucopolysaccharidosis IIIB (Sanfilippo syndrome type IIIB): N-acetyl-alpha-D-glucosaminidase deficiency in Schipperke dogs. J Inherit Metab Dis. 2003;26: 489–504. [DOI] [PubMed] [Google Scholar]
- 14.Fecarotta S, Gasperini S, Parenti G. New treatments for the mucopolysaccharidoses: from pathophysiology to therapy. In: Ital J Pediatr. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrer I, Cusi V, Pineda M, Galofre E, Vila J. Focal dendritic swellings in Purkinje cells in mucopolysaccharidoses types I, II and III. A Golgi and ultrastructural study. Neuropathol Appl Neurobiol. 1988;14: 315–323. [DOI] [PubMed] [Google Scholar]
- 16.Ficko-Blean E, Stubbs KA, Nemirovsky O, Vocadlo DJ, Boraston AB. Structural and mechanistic insight into the basis of mucopolysaccharidosis IIIB. In: Proc Natl Acad Sci U S A. 2008:6560–6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer A, Carmichael KP, Munnell JF, et al. Sulfamidase deficiency in a family of Dachshunds: a canine model of mucopolysaccharidosis IIIA (Sanfilippo A). Pediatr Res. 1998;44: 74–82. [DOI] [PubMed] [Google Scholar]
- 18.Fu H, Bartz JD, Stephens RL Jr., McCarty DM. Peripheral nervous system neuropathology and progressive sensory impairments in a mouse model of Mucopolysaccharidosis IIIB. PLoS One. 2012;7: e45992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu H, DiRosario J, Kang L, Muenzer J, McCarty DM. Restoration of central nervous system alpha-N-acetylglucosaminidase activity and therapeutic benefits in mucopolysaccharidosis IIIB mice by a single intracisternal recombinant adeno-associated viral type 2 vector delivery. J Gene Med. 2010;12: 624–633. [DOI] [PubMed] [Google Scholar]
- 20.Genger SC, Mizukami K, Martin MP, Applegate JR Jr., Barnes HJ, Giger U. Mucopolysaccharidosis IIIB (Sanfilippo syndrome B) in a commercial emu (Dromaius novaehollandiae) flock. Avian Pathol. 2018;47: 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghatak NR, Fleming DF, Hinman A. Neuropathology of sanfilippo syndrome. Annals of Neurology. 1977;2: 161–166. [Google Scholar]
- 22.Gografe SI, Garbuzova-Davis S, Willing AE, Haas K, Chamizo W, Sanberg PR. Mouse model of Sanfilippo syndrome type B: relation of phenotypic features to background strain. Comp Med. 2003;53: 622–632. [PubMed] [Google Scholar]
- 23.Hadfield MG, Ghatak NR, Nakoneczna I, et al. Pathologic findings in mucopolysaccharidosis type IIIB (Sanfilippo's sydnrome B). Arch Neurol. 1980;37: 645–650. [DOI] [PubMed] [Google Scholar]
- 24.Hamano K, Hayashi M, Shioda K, Fukatsu R, Mizutani S. Mechanisms of neurodegeneration in mucopolysaccharidoses II and IIIB: analysis of human brain tissue. Acta Neuropathol. 2008;115: 547–559. [DOI] [PubMed] [Google Scholar]
- 25.Haskins M Gene therapy for lysosomal storage diseases (LSDs) in large animal models. Ilar j. 2009;50: 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haskins ME, Giger U, Patterson DF. Animal models of lysosomal storage diseases: their development and clinical relevance. In: Mehta A, Beck M, Sunder-Plassmann G, eds. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006. [PubMed] [Google Scholar]
- 27.Hassiotis S, Jolly RD, Hemsley KM. Development of cerebellar pathology in the canine model of mucopolysaccharidosis type IIIA (MPS IIIA). Mol Genet Metab. 2014;113: 283–293. [DOI] [PubMed] [Google Scholar]
- 28.Heldermon CD, Hennig AK, Ohlemiller KK, et al. Development of Sensory, Motor and Behavioral Deficits in the Murine Model of Sanfilippo Syndrome Type B. In: PLoS One. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heldermon CD, Ohlemiller KK, Herzog ED, et al. Therapeutic efficacy of bone marrow transplant, intracranial AAV-mediated gene therapy, or both in the mouse model of MPS IIIB. Mol Ther. 2010;18: 873–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jensen OA. Mucopolysaccharidosis type 3 (Sanfilippo's syndrome). Acta Pathol Microbiol Scand A. 1971;79: 257–273. [DOI] [PubMed] [Google Scholar]
- 31.Jolly RD, Allan FJ, Collett MG, Rozaklis T, Muller VJ, Hopwood JJ. Mucopolysaccharidosis IIIA (Sanfilippo syndrome) in a New Zealand Huntaway dog with ataxia. N Z Vet J. 2000;48: 144–148. [DOI] [PubMed] [Google Scholar]
- 32.Jones MZ, Alroy J, Rutledge JC, et al. Human mucopolysaccharidosis IIID: clinical, biochemical, morphological and immunohistochemical characteristics. J Neuropathol Exp Neurol. 1997;56: 1158–1167. [PubMed] [Google Scholar]
- 33.Karageorgos L, Hill B, Bawden MJ, Hopwood JJ. Bovine mucopolysaccharidosis type IIIB. J Inherit Metab Dis. 2007;30: 358–364. [DOI] [PubMed] [Google Scholar]
- 34.King B, Hassiotis S, Rozaklis T, et al. Low-dose, continuous enzyme replacement therapy ameliorates brain pathology in the neurodegenerative lysosomal disorder mucopolysaccharidosis type IIIA. J Neurochem. 2016;137: 409–422. [DOI] [PubMed] [Google Scholar]
- 35.King B, Marshall N, Beard H, et al. Evaluation of enzyme dose and dose-frequency in ameliorating substrate accumulation in MPS IIIA Huntaway dog brain. J Inherit Metab Dis. 2015;38: 341–350. [DOI] [PubMed] [Google Scholar]
- 36.King B, Marshall NR, Hassiotis S, et al. Slow, continuous enzyme replacement via spinal CSF in dogs with the paediatric-onset neurodegenerative disease, MPS IIIA. J Inherit Metab Dis. 2017;40: 443–453. [DOI] [PubMed] [Google Scholar]
- 37.Kingma SD, Bodamer OA, Wijburg FA. Epidemiology and diagnosis of lysosomal storage disorders; challenges of screening. Best Pract Res Clin Endocrinol Metab. 2015;29: 145–157. [DOI] [PubMed] [Google Scholar]
- 38.Kowalewski B, Lamanna WC, Lawrence R, et al. Arylsulfatase G inactivation causes loss of heparan sulfate 3-O-sulfatase activity and mucopolysaccharidosis in mice. Proc Natl Acad Sci U S A. 2012;109: 10310–10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Layton C, Bancroft JD. 13 - Carbohydrates. In: Suvarna SK, Layton C, Bancroft JD, eds. Bancroft's Theory and Practice of Histological Techniques (Eighth Edition). Content Repository Only!; 2019:176–197. [Google Scholar]
- 40.Li HH, Yu WH, Rozengurt N, et al. Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding alpha-N-acetylglucosaminidase. Proc Natl Acad Sci U S A. 1999;96: 14505–14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Louis ED, Babij R, Lee M, Cortés E, Vonsattel JPG. Quantification of Cerebellar Hemispheric Purkinje Cell Linear Density: 32 ET Cases vs. 16 Controls. Mov Disord. 2013;28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martins C, Hulkova H, Dridi L, et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain. 2015;138: 336–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. Jama. 1999;281: 249–254. [DOI] [PubMed] [Google Scholar]
- 44.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172: 2731–2738. [DOI] [PubMed] [Google Scholar]
- 45.Murrey DA, Naughton BJ, Duncan FJ, et al. Feasibility and safety of systemic rAAV9-hNAGLU delivery for treating mucopolysaccharidosis IIIB: toxicology, biodistribution, and immunological assessments in primates. Hum Gene Ther Clin Dev. 2014;25: 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neufeld EM J The mucopolysaccharidoses. In: Scriver CB, AL. Sly WS. Valle D, ed. The metabolic and molecular basis of inherited disease. 7 ed. New York: McGraw-Hill; 2001:3421–3452. [Google Scholar]
- 47.Nolte A, Bello A, Drogemuller M, et al. Neuronal ceroid lipofuscinosis in an adult American Staffordshire Terrier. Tierarztl Prax Ausg K Kleintiere Heimtiere. 2016;44: 431–437. [DOI] [PubMed] [Google Scholar]
- 48.Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci U S A. 2003;100: 1902–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palazzi X The Beagle Brain in Stereotaxic Coordinates. 2011. [Google Scholar]
- 50.Palmieri C, Giger U, Wang P, Pizarro M, Shivaprasad HL. Pathological and biochemical studies of mucopolysaccharidosis type IIIB (Sanfilippo syndrome type B) in juvenile emus (Dromaius novaehollandiae). Vet Pathol. 2015;52: 160–169. [DOI] [PubMed] [Google Scholar]
- 51.Raj K, Ellinwood NM, Giger U. An exonic insertion in the NAGLU gene causing Mucopolysaccharidosis IIIB in Schipperke dogs. Sci Rep. 2020;10: 3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rama Rao KV, Kielian T. Astrocytes and Lysosomal Storage Diseases. Neuroscience. 2016;323: 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ribera A, Haurigot V, Garcia M, et al. Biochemical, histological and functional correction of mucopolysaccharidosis type IIIB by intra-cerebrospinal fluid gene therapy. Hum Mol Genet. 2015;24: 2078–2095. [DOI] [PubMed] [Google Scholar]
- 54.Scarpa M, Orchard PJ, Schulz A, et al. Treatment of brain disease in the mucopolysaccharidoses. Mol Genet Metab. 2017;122s: 25–34. [DOI] [PubMed] [Google Scholar]
- 55.Shapiro EG, Lockman LA, Balthazor M, Krivit W. Neuropsychological outcomes of several storage diseases with and without bone marrow transplantation. J Inherit Metab Dis. 1995;18: 413–429. [DOI] [PubMed] [Google Scholar]
- 56.Shimamura K, Hakozaki H, Takahashi K, Kimura A, Fujino J. Sanfilippo B syndrome. A case report. Acta Pathol Jpn. 1976;26: 739–764. [DOI] [PubMed] [Google Scholar]
- 57.Tamagawa K, Nakajima S, Terauchi A, et al. [A clinical study of the siblings of Sanfilippo B syndrome (MPS III B)]. No To Hattatsu. 1983;15: 57–66. [PubMed] [Google Scholar]
- 58.Tessitore A, Villani GR, Di Domenico C, Filocamo M, Gatti R, Di Natale P. Molecular defects in the alpha-N-acetylglucosaminidase gene in Italian Sanfilippo type B patients. Hum Genet. 2000;107: 568–576. [DOI] [PubMed] [Google Scholar]
- 59.Tetreault M, Gonzalez M, Dicaire MJ, et al. Adult-onset painful axonal polyneuropathy caused by a dominant NAGLU mutation. Brain. 2015;138: 1477–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tongtako W, Lehmbecker A, Wang Y, Hahn K, Baumgärtner W, Gerhauser I. Canine dorsal root ganglia satellite glial cells represent an exceptional cell population with astrocytic and oligodendrocytic properties. In: Sci Rep. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van Dessel G, Lagrou A, Martin JJ, Ceuterick C, Dierick W. Two cases of mucopolysaccharidosis type III (Sanfilippo). A biochemical study. J Neurol Sci. 1979;40: 77–86. [DOI] [PubMed] [Google Scholar]
- 62.Vellodi A, Young E, New M, Pot-Mees C, Hugh-Jones K. Bone marrow transplantation for Sanfilippo disease type B. J Inherit Metab Dis. 1992;15: 911–918. [DOI] [PubMed] [Google Scholar]
- 63.Wallace BJ, Kaplan D, Adachi M, Schneck L, Volk BW. Mucopolysaccharidosis type 3. Morphologic and biochemical studies of two siblings with Sanfilippo syndrome. Arch Pathol. 1966;82: 462–473. [PubMed] [Google Scholar]
- 64.Wilkinson FL, Holley RJ, Langford-Smith KJ, et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS One. 2012;7: e35787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Winner LK, Marshall NR, Jolly RD, et al. Evaluation of Disease Lesions in the Developing Canine MPS IIIA Brain. In: JIMD Rep. 2019:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang Q, Zhao X, Xing Y, et al. A model of mucopolysaccharidosis type IIIB in pigs. Biol Open. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.