

Abstract
Overproduction of reactive oxygen species (ROS) plays an important role in the pathogenesis of hypertension. The dopamine D5 receptor (D5R) is known to decrease ROS production, but the mechanism is not completely understood. In HEK293 cells overexpressing D5R, fenoldopam, an agonist of the two D1-like receptors, D1R and D5R, decreased the production of mitochondria-derived ROS (mito-ROS). The fenoldopam-mediated decrease in mito-ROS production was mimicked by Sp-cAMPS but blocked by Rp-cAMPS. In human renal proximal tubule cells with DRD1 gene silencing to eliminate the confounding effect of D1R, fenoldopam still decreased mito-ROS production. By contrast, Sch23390, a D1R and D5R antagonist, increased mito-ROS production in the absence of D1R, D5R is constitutively active. The fenoldopam-mediated inhibition of mito-ROS production may have been related to autophagy because fenoldopam increased the expression of the autophagy hallmark proteins, autophagy protein 5 (ATG5), and the microtubule-associated protein 1 light chain (LC)3-II. In the presence of chloroquine or spautin-1, inhibitors of autophagy, fenoldopam further increased ATG5 and LC3-II expression, indicating an important role of D5R in the positive regulation of autophagy. However, when autophagy was inhibited, fenoldopam was unable to inhibit ROS production. Indeed, the levels of these autophagy hallmark proteins were decreased in the kidney cortices of Drd5−/− mice. Moreover, ROS production was increased in mitochondria isolated from the kidney cortices of Drd5−/− mice, relative to Drd5+/+ littermates. In conclusion, D5R-mediated activation of autophagy plays a role in the D5R-mediated inhibition of mito-ROS production in the kidneys.
Keywords: Autophagy, Dopamine D5 receptor, Hypertension, Mitochondria, Reactive oxygen species
Introduction
Reactive oxygen species (ROS) are a family of chemically derived reactive molecules from oxygen [1–3]. In addition to their established role in killing invading microorganisms, ROS, via direct oxidative damage or activation of cellular signaling pathways, are implicated in the pathogenesis of a wide variety of disorders, including hypertension [1–3]. There are many sources of ROS, e.g., NADPH oxidase [1–3], and mitochondria-derived ROS (mito-ROS) play an important role in the pathophysiology of hypertension [1–4].
Dopamine, produced in the kidney and via activation of dopamine receptors, regulates sodium balance and blood pressure [5–7]. Dopamine receptors are classified into D1-like (D1R and D5R) receptors that stimulate adenylyl cyclases and D2-like (D2R, D3R, and D4R) receptors that inhibit adenylyl cyclases [5–7]. Abnormalities in renal dopamine production and D1-like and D2-like receptors are associated with hypertension and/or salt sensitivity in several ethnic groups [5–7]. The loci of human D5R (4p15.1–16.1) and its pseudogenes (1q21.1 and 2p11.1–p11.2) are linked to human hypertension [8, 9]. Germline deletion of the Drd5 gene (Drd5−/−) in mice causes hypertension [10]. However, the mechanisms responsible for the increased blood pressure in Drd5−/− mice are not completely understood.
In addition to genetic factors, many cellular factors, including ROS, are involved in the pathogenesis and maintenance of hypertension [1–4]. We have reported that ROS production in the kidneys and brain is greater in Drd5−/− mice than in their Drd5+/+ littermates and that D5R exerts its anti-oxidant properties in part by inhibiting NADPH oxidase activity [10]. NADPH oxidase is an important source of ROS production in renal tubule cells [1–3, 10–12], and the results of experiments inhibiting this enzyme complex with specific or nonspecific inhibitors support its importance in the regulation of blood pressure [1–3, 12, 13]. Recently, mitochondria-generated ROS have been reported to contribute to oxidative stress-mediated hypertension [1–4], but the mechanism is not known.
There is accumulating evidence linking autophagy and hypertension in response to oxidative stress [14]. Autophagy, an intracellular self-degradative process that delivers cytoplasmic constituents to lysosomes, has long been considered a cellular physiological process involved in the turnover of subcellular organelles and proteins [15, 16]. However, recent studies have indicated that autophagy plays important roles in the pathogeneses of human diseases, including those caused by oxidative stress [17]. Furthermore, autophagy can mediate inflammatory responses that induce oxidative stress [18, 19]. Oxidative stress increases the activity of both autophagosomes and auto-lysosomes; inhibition of autophagy attenuates autophagic flux [14], but autophagy protects against oxidative injury [20]. Moreover, autophagy is important in the removal of damaged mitochondria and polyubiquitinated proteins and in protection from damage caused by oxidative stress [14, 17]. D5R can regulate the expression and activity of proteins that increase oxidative stress, for example, by triggering the ubiquitination and subsequent degradation of angiotensin II (Ang II) type 1 receptor (AT1R) [10, 21–23]. However, investigations linking D5R to autophagy and mitochondrial oxidative stress are sparse.
We investigated the potential role(s) of D5R in mito-ROS production by determining whether the D5R-mediated inhibition of renal cellular ROS production is caused in part by a decrease in mito-ROS production through an increase in autophagy.
Methods
Reagents
MitoTracker Green, MitoSOX Red (Mito-HE), and Amplex Red were purchased from Molecular Probes (Eugene, OR), and anti-autophagy protein 5 (ATG5) (Cat. No. 12994) and anti-LC3-II (Cat. No. 3868) antibodies were purchased from Cell Signaling Technology (Danvers, MA). The origins of the anti-D1R and anti-D5R antibodies in renal proximal tubule (RPT) cells and rat kidneys have been reported previously, and the antibodies have been verified in D1R−/− and D5R−/− mice [24–28]. Anti-Bax and anti-Bcl-xL antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); anti-Mn-SOD and anti-Cu/Zn-SOD antibodies were purchased from Abcam (Cambridge, MA); an anti-prohibitin antibody was purchased from GeneTex (Irvine, CA); and anti-calnexin, anti-histone B4, and anti-GM130 antibodies were purchased from BD Transduction Laboratories (Lexington, KY). Culture media, fetal bovine serum (FBS), glutamine, and Lipofectamine transfection reagent were purchased from Invitrogen (Gaithersburg, MD). Percoll was purchased from GE Healthcare (Piscataway, NJ). NADPH was purchased from MP Biomedicals (Solon, OH). An adenosine triphosphate (ATP) bioluminescence assay kit was purchased from Roche Applied Science (Indianapolis, IN). Spautin-1 was purchased from Griffin Biotech (Hong Kong, China). Fenoldopam, Sch23390, Rp-cAMPS, Sp-cAMPS, chloroquine, antimycin A, and other reagents were purchased from Sigma (St. Louis, MO).
Cell culture, siRNA, and transfection
Human embryonic kidney 293 (HEK293) cells expressing D5R (D5R-HEK293) and human RPT cells were cultured as previously described [23, 25]. Empty vector (EV)-transfected HEK293 and D5R expression vector-transfected HEK293 cells (D5R-HEK293 cells) were maintained in culture with 10 μg/mL blasticidin. The stable protein expression of D5R in D5R-HEK293 cells was confirmed before the actual experiments were performed (Supplementary Fig. 1). The RPT origins of human RPT cells were verified by staining with antibodies against γ-glutamyl transpeptidase, as previously described [25]. The cells were cultured in a 1:1 mixture of DMEM and Ham’s F-12 medium supplemented with 5% FBS, selenium (5 ng/mL), insulin (5 μg/mL), transferrin (5 μg/mL), hydrocortisone (36 ng/mL), triiodothyronine (4 pg/mL), and epidermal growth factor (10 ng/mL).
Specific DRD1, DRD5 (Santa Cruz Biotechnology), ATG5, and LC3-II (Cell Signaling Technology) siRNAs and their control siRNA constructs were transfected into human RPT cells or D5R-HEK293 cells using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer’s instructions and our published procedure [29].
NADPH oxidase activity
NADPH oxidase activity was measured as previously described [10]. Equal amounts of cell membranes were incubated with lucigenin (5 μmol/L) for 10 min at 37 °C in a final volume of 1 mL of assay buffer. Recording of the dynamic chemiluminescence traces was started upon injection of NADPH (final concentration: 100 μmol/L) and continued for 180 s (Autolumet Plus LB953, EG&G Berthold, Bad Wildbad, Germany). The activity levels are expressed as arbitrary light units (ALUs) and were corrected for the protein concentration and duration of the experiment (ALU/s/mg protein).
Superoxide anion production
Superoxide anion production was measured using a cytochrome c assay, as previously described [10]. Cell homogenates in Hank’s balanced salt solution (final concentration 1 mg/mL) were distributed in 96-well plates (final volume 200 μL/well). Cytochrome c (500 μmol/L) was added, and the homogenates were incubated in the presence or absence of superoxide dismutase (SOD, 200 U/mL) at room temperature for 30 min. Cytochrome c reduction was measured by reading the absorbance at 550 nm on a microplate reader.
Confocal fluorescence microscopy
Confocal fluorescence microscopy was performed as previously described [29, 30]. The mito-ROS signals in live cells stained with MitoSOX Red were monitored by laser scanning confocal microscopy (LSM 510, Carl Zeiss). The cells were treated with vehicle or the D1-like receptor agonist fenoldopam (1 μM, 15 min) in the presence or absence of the D1-like receptor antagonist Sch23390 (1 μM, 45 min). After washing the cells with Hank’s balanced salt solution, the cells were incubated with 5 μM MitoSOX Red for 10 min. Images were captured with excitation at 405 nm and emission at 590 nm under a Plan-Apochromat 63×/1.4 Oil NA objective. The MitoSOX Red fluorescence density was semiquantified in 3–5 random fields from 25–40 cells [30]. MitoTracker Green was used to identify mitochondria. Endogenous light chain (LC)3-II was immunostained with an anti-LC3-II antibody (Cell Signaling Technology). The fluorescence intensity was estimated from the range within the initial linear phase to minimize potential problems arising from mitochondrial dye saturation and leakage.
Western blotting
Western blotting was performed as previously described [10, 25]. Samples (D5R-HEK293, human RPT cells, kidney cortices, and isolated mitochondria) were adjusted to the same protein concentration. The proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred onto a nitrocellulose membrane, and then probed with primary antibodies and appropriate horseradish peroxidase-conjugated secondary antibodies. The images were visualized by chemiluminescence.
ATP assay
ATP concentrations were quantified as previously described [30, 31]. In brief, human RPT cells were grown in 25-cm tissue culture dishes. After harvesting the cells by centrifugation, the cell pellets were sonicated in 800 μL of lysis buffer. The ATP released in 300 μL of buffer (100 mM Tris-Cl, pH 7.75; 4 mM EDTA) was boiled for 2 min. ATP bioluminescence was monitored by a microplate lumen-ometer (Centro LB 960, Berthold Technologies, Bad Wildbad, Germany).
Generation of Drd5−/− mice
The method used for generation of Drd5−/− mice has been reported [10, 23]. F6 generation Drd5−/− mice on a C57Bl/6 (>98% congenic) background and their sex-matched wild-type littermates were used in this study. Fenoldopam (1 mg/kg body weight, 0.5 mL) or vehicle (saline, for control) was injected intraperitoneally daily for 7 days. Subsequently, the kidneys were collected, and their cortices were excised and homogenized for immunoblotting of autophagy marker proteins. Mitochondria were isolated and purified for H2O2 production assays.
The animal protocols were reviewed and approved by the Georgetown University, Children’s National Medical Center, and University of Maryland School of Medicine Animal Care and Use Committees.
Mitochondria isolation and H2O2 measurement
Mitochondria were isolated from D5R-HEK293 cells, human RPT cells, and mouse kidney cortices by Percoll density gradient centrifugation as previously described [32] with modifications. In brief, cell pellets or minced kidney cortices were homogenized. The homogenates were suspended in a buffer (pH 7.4) containing 225 mM mannitol, 75 mM sucrose, 5 mM HEPES, 1 mM ethylene glycol-bis (2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), and 1 mg/mL fatty acid-free bovine serum albumin and centrifuged at 1300 × g for 3 min The pelleted material was resuspended in the above buffer and centrifuged at 1300 × g for 3 min. The combined supernatants were then centrifuged at 21,000 × g for 10 min. The pellets were suspended in 15% Percoll and gently transferred onto the surface of 23% Percoll without touching the 40% Percoll. The gradients were centrifuged at 29,000 × g for 8 min The mitochondria were isolated from the interface of the 23 and 40% Percoll. The isolated mitochondria, which had no measurable NADPH oxidase activity and were characterized with pyruvate/malate (5 mM) and succinate (4 mM) in a medium containing 125 mM KCl, 2 mM K2HPO4, 3 mM HEPES, 4 mM MgCl2, 3 mM ATP, and 0.2% fatty acid-free bovine serum albumin (pH 7.2), showed coupling of oxidative phosphorylation and normal respiration. The purity of the mitochondrial fraction was confirmed by immunoblotting markers of organelles.
Mito-ROS production by isolated mitochondria was measured with Amplex Red in the presence of exogenous superoxide dismutase (40 U/mL) and horseradish peroxidase (10 U/mL), as described previously [33] with modifications. The fluorescence intensity was measured with a microplate reader in 96-well plates at an excitation wavelength of 530 nm and an emission wavelength of 590 nm. The rate of H2O2 production was linear for a fluorescence density up to 3 μmol/L in the standard curve. ROS production is expressed in picomoles of H2O2 per mg of mitochondrial protein/min.
Statistical analysis
The results are expressed as the mean ± standard deviation, as indicated. Significant differences among groups (n > 2) were determined by one-way factorial ANOVA and the Newman–Keuls test, and significant differences between two groups were determined by Student’s t test. P < 0.05 was considered to indicate statistical significance (SigmaStat 3.0, SPSS Inc, Chicago, IL).
Results
Activation of D5R decreased ROS production in mitochondria from D5R-HEK293 cells
Consistent with our previous report [10], stimulation of D5R with fenoldopam decreased both cellular superoxide (O2−•) production (0.296 ± 0.033 ΔOD550/min/mg vs vehicle 0.493 ± 0.024 ΔOD550/min/mg, P < 0.05), as quantified by cytochrome c reduction (Supplementary Fig. 2A), and NADPH oxidase activity (2183 ± 123 ALU/min/mg vs vehicle 2449 ± 52 ALU/min/mg, P < 0.01), as quantified by lucigenin assay (Supplementary Fig. 2B), in D5R-HEK293 cells. However, O2− production was lower in the presence of both fenoldopam and apocynin (10 μM), an NADPH oxidase inhibitor, than in the presence of apocynin alone (0.187 ± 0.012 ΔOD550/min/mg vs apocynin alone 0.358 ± 0.045 ΔOD550/min/mg, P < 0.01) (Supplementary Fig. 2A). By contrast, under the same experimental conditions, fenoldopam did not enhance the apocynin-mediated inhibition of NADPH oxidase activity (1890 ± 147 ALU/min/mg vs apocynin alone 1906 ± 77 ALU/min/mg, P > 0.05) (Supplementary Fig. 2B). These results indicate that fenoldopam decreased ROS production by an additional mechanism or mechanisms other than inhibition of NADPH oxidase activity.
Because mitochondria are important sources of cellular ROS production in the kidneys [1, 3, 4], we next determined whether mitochondria are the additional sources of ROS targeted by D5R, as observed in Supplementary Fig. 2A. Mitochondria isolated from D5R-HEK293 cells (Fig. 1A) produced less hydrogen peroxide (H2O2) than those from EV-transfected HEK293 cells (Fig. 1B). Fenoldopam treatment further decreased H2O2 production in D5R-HEK293 cells but had no effect on EV-transfected HEK293 cells (Fig. 1B). In intact D5R-HEK293 cells, fenoldopam decreased mito-ROS production, as detected with MitoSOX Red, a hydroethidine (HE)-derived fluorescent dye (Fig. 1C). The colocalization of MitoSOX Red staining with MitoTracker Green staining indicated the mitochondrial origin of the generated ROS (Fig. 1C). The inhibitory effect of fenoldopam on ROS production was prevented by Sch23390, a D1-like receptor antagonist, in both isolated mitochondria (Fig. 1B) and intact cells (Fig. 1C, D). These findings indicate that fenoldopam, a D1R/D5R agonist, decreased ROS production by stimulating D5R (D1R is not expressed in D5R-HEK293 cells). Sch23390, a D1R/D5R antagonist but a D5R antagonist in the absence of D1R, itself increased ROS production (Fig. 1B–D), which could be due to its blockade of D5R’s constitutive activity [10] because, as stated earlier, HEK293 cells do not express D1R. H2O2 production was increased by antimycin A, a Qi site inhibitor of mitochondrial electron transport chain (ETC) Complex III and prevented the inhibitory effect of fenoldopam (Fig. 1B). By contrast, rotenone, a Complex I inhibitor (Supplementary Fig. 3), had no effect on the inhibitory effect of fenoldopam, indicating that Complex III, not Complex I, is the target of the D5R-mediated decrease in mito-ROS production. H2O2 production, as expected, was decreased by catalase despite the presence of antimycin A (Fig. 1B).
Fig. 1.
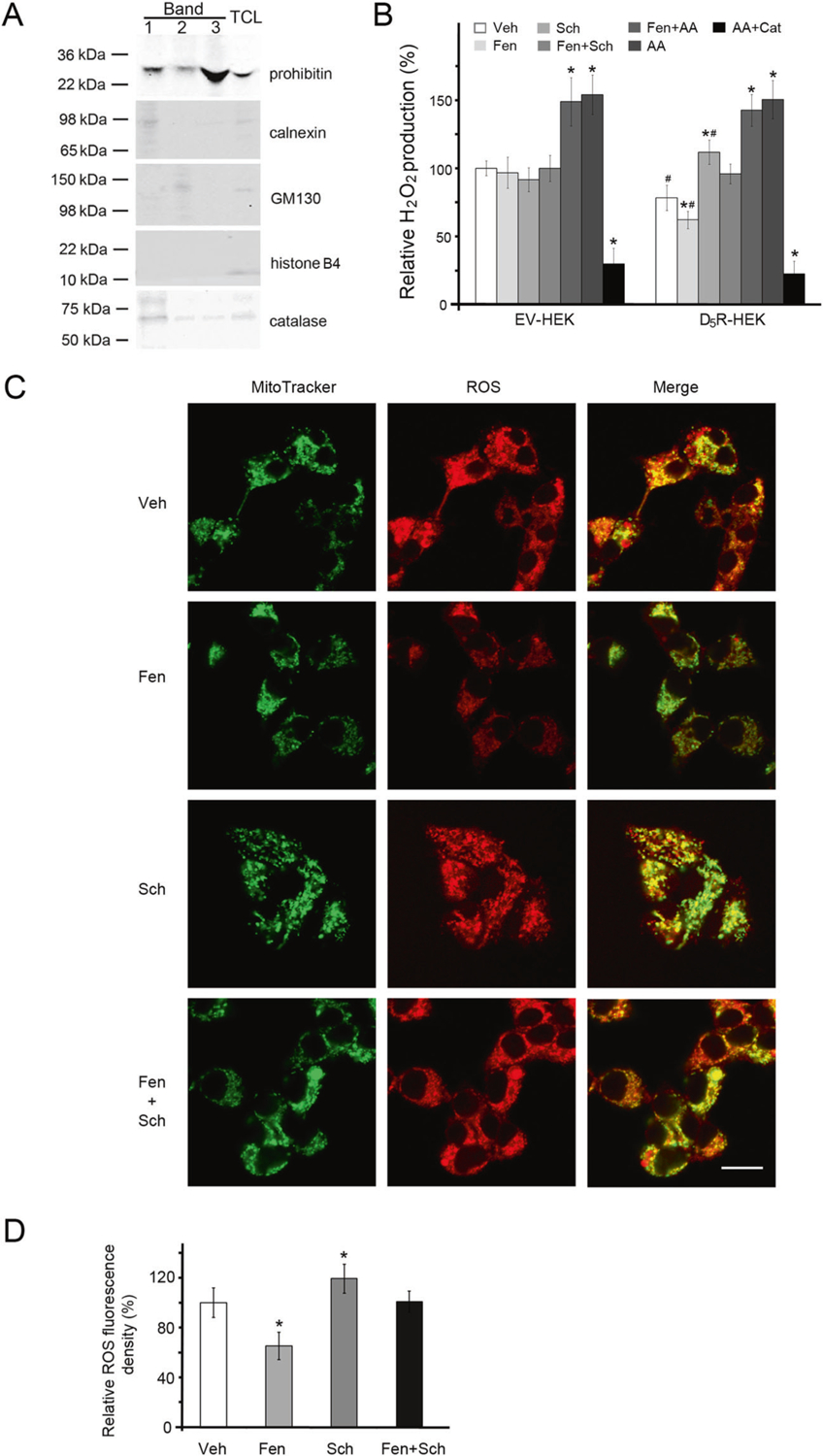
Mito-ROS production in D5R-HEK293 cells. A Characterization of purified mitochondria (Band 3, as described in Supplementary Fig. 7) by immunoblotting with antibodies against markers for mitochondria (prohibitin), endoplasmic reticula (calnexin), Golgi bodies (GM130), nuclei (histone B4), and peroxisomes (catalase). TCL total cell lysate. B H2O2 production in isolated mitochondria, with pyruvate (5 mM) and malate (5 mM) as the substrates. Veh vehicle, Fen fenoldopam (D1R/D5R agonist, 1.0 μM, 12 h), Sch Sch23390 (D1R/D5R antagonist 1.0 μM, 12 h), AA antimycin A (Qi site inhibitor of mitochondrial ETC Complex III, 0.75 μM, 2 h), Cat catalase (10 μM, 30 min prior to AA treatment). Open rectangles, empty vector-transfected HEK293 (EV-HEK) cells; filled rectangles, D5R-overexpressing HEK293 (D5R-HEK) cells. n = 4/group, *P < 0.05 vs Veh, #P < 0.05 vs EV-HEK. C D5-RHEK293 cells were treated with Veh or Fen (1.0 μM, 12 h) in the absence or presence of Sch (1.0 μM, 12 h). Mitochondria were stained with MitoTracker Green; mito-ROS were monitored with MitoSOX Red. Bar, 20 μm. The images are from one of three independent experiments. D The MitoSOX Red fluorescence density was obtained in 3–5 random fields from 25–40 D5R-HEK293 cells in three independent experiments, as described in C; *P < 0.05 vs Veh
Activation of D5R, similar to activation of D1R, stimulates cAMP and protein kinase A (PKA) [5–7]. To determine whether the D5R-mediated inhibition of mito-ROS is cAMP-dependent, D5R-HEK293 cells were pretreated with Rp-cAMPS (Fig. 2), a selective inhibitor of PKA [34]. Rp-cAMPS alone did not affect H2O2 production but prevented the inhibitory effect of fenoldopam. Sch23390 itself increased H2O2 production; as indicated above, this effect was probably mediated by inhibition of the constitutive activity of D5R [10]. This stimulatory effect of Sch23390 was also inhibited by Rp-cAMPS. Sp-cAMPS, a PKA activator, decreased mito-ROS production (Fig. 2), an effect that was prevented by Rp-cAMPS, indicating the specificity of the effect of Sp-cAMPS on the cAMP pathway. All these results indicate that the D5R-mediated inhibition of mito-ROS production is cAMP-dependent.
Fig. 2.
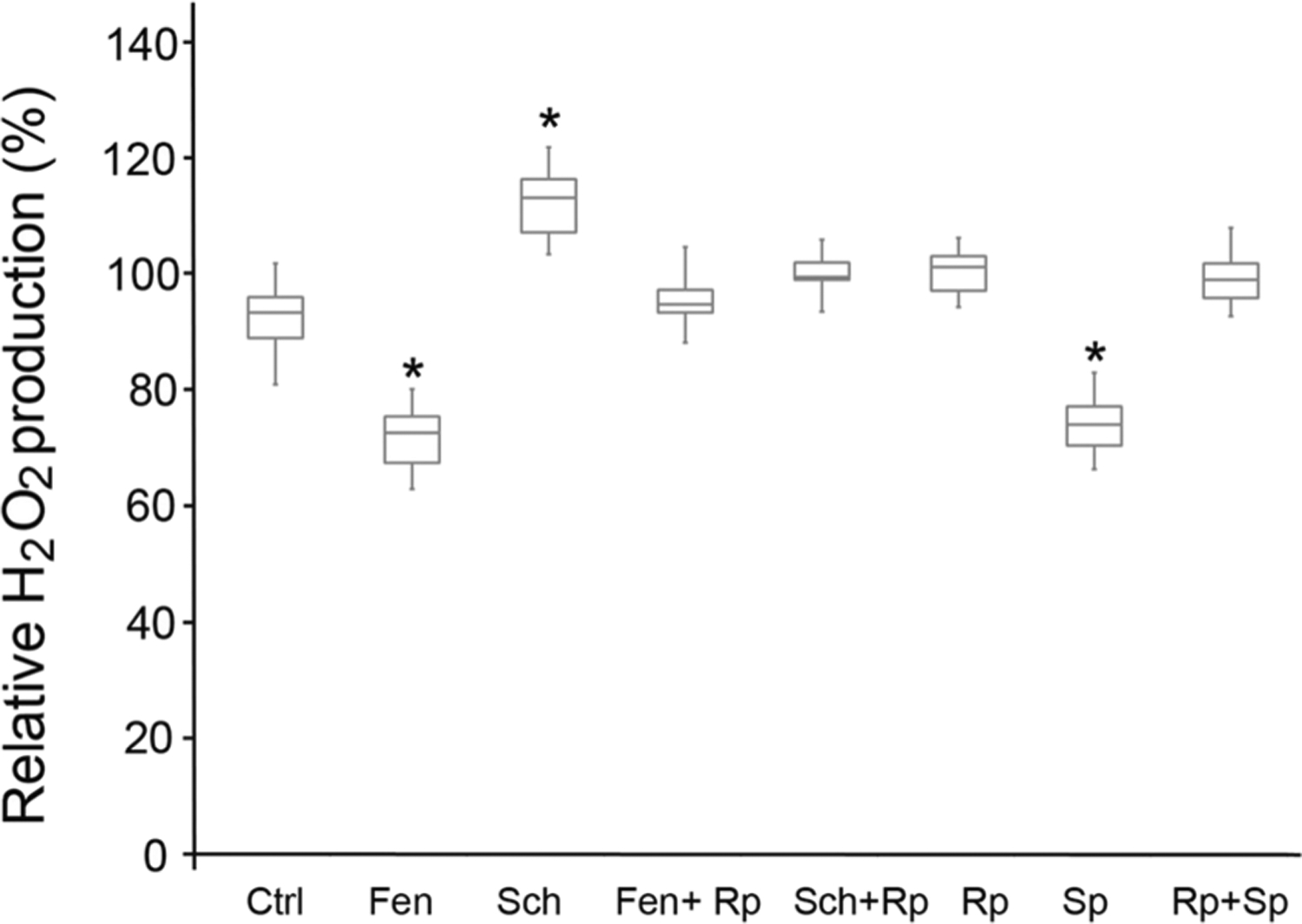
cAMP dependence of D5R-mediated inhibition of mito-ROS production. D5R-HEK293 cells were preincubated for 30 min with vehicle (Ctrl), Fen (fenoldopam, D1R/D5R agonist, 1.0 μM), Sch (Sch23390, D1R/D5R antagonist, 1.0 μM), Fen + Rp (Rp-cAMPS, PKA inhibitor, 50 μM), Sch + Rp, Rp alone, Sp (Sp-cAMPS, PKA activator, 10 μM) alone, or Rp + Sp. n = 6/group, *P < 0.05 vs Ctrl
Activation of D5R decreased ROS production in mitochondria from human RPT cells
To study further the effect of D5R on mito-ROS production in connection with its potential role in regulating renal sodium transport and ultimately blood pressure [10, 23], we studied mito-ROS production in human RPT cells [21]. Similar to the observations in D5R-HEK293 cells (Fig. 1), fenoldopam decreased mito-ROS production in human RPT cells. This effect was inhibited by Sch23390, a D1R/D5R antagonist, which increased mito-ROS production when used alone (Fig. 3A).
Fig. 3.
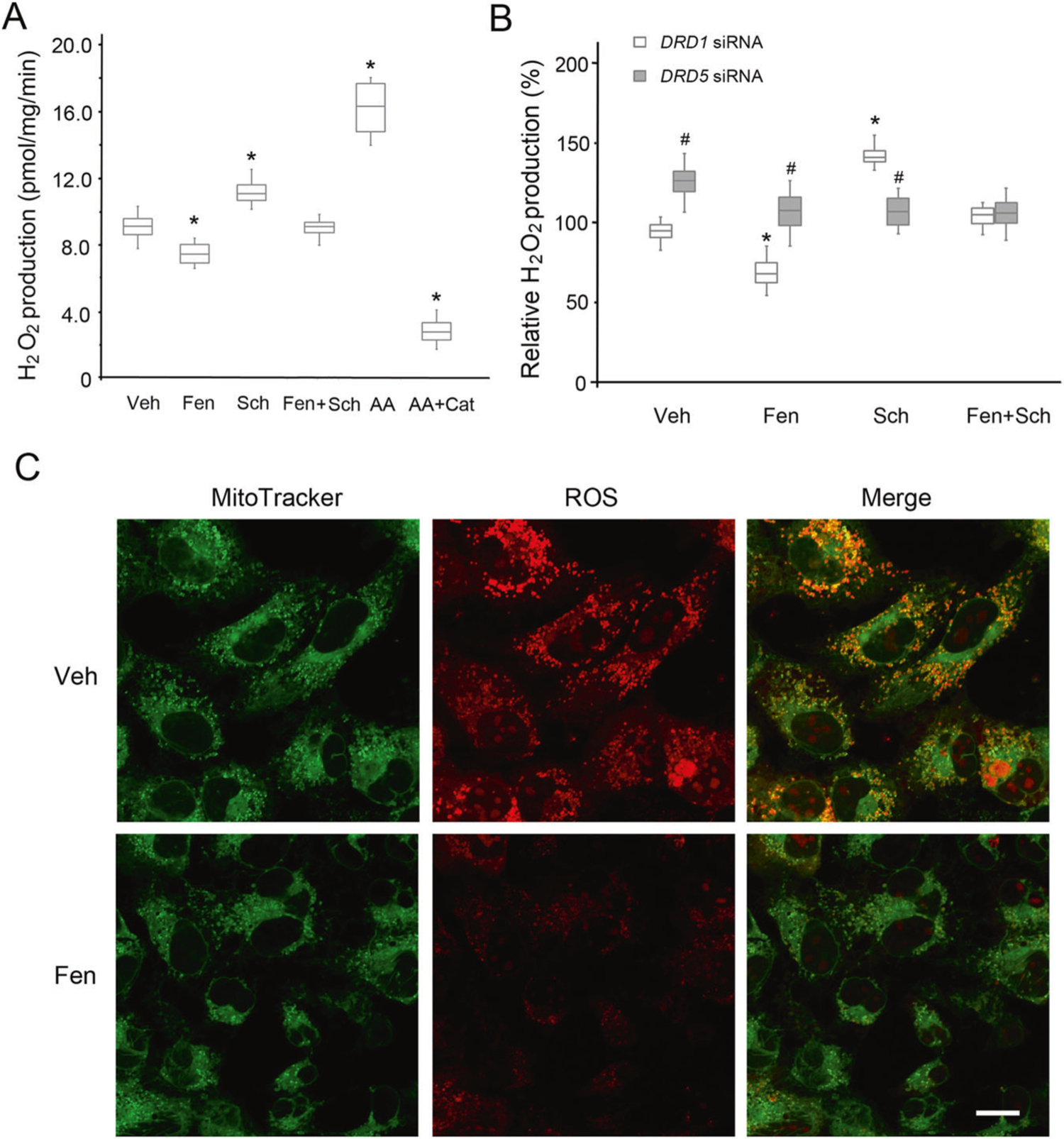
Mito-ROS production in human RPT cells. A H2O2 production from mitochondria isolated from human RPT cells, with pyruvate and malate as the substrates. Veh vehicle, Fen fenoldopam (D1R/D5R agonist, 1.0 μM, 12 h), Sch Sch23390 (D1R/D5R antagonist, 1.0 μM, 12 h), AA antimycin A (Qi site inhibitor of mitochondrial ETC Complex III, 0.75 μM, 2 h), Cat catalase (10 μM, 30 min prior to AA treatment). n = 4/group, *P < 0.05 vs Veh. B H2O2 production from mitochondria isolated from DRD1- or DRD5-silenced human RPT cells. The cells were treated with vehicle (Veh), Fen (1.0 μM, 12 h), or Sch (1.0 μM, 12 h). n = 4/group, *P < 0.05 vs Veh, #P < 0.05 vs DRD1-silenced RPT cells. C Mito-ROS staining in DRD1-silenced intact human RPT cells treated with Veh or Fen (1.0 μM, 12 h), as indicated. Bar, 10 μm. The images are from one of three independent experiments
Since no commercially available antagonist or agonist can distinguish the effect of D5R from that of D1R, both of which are expressed endogenously in human RPT cells, we silenced the expression of D1R or D5R by transfection with either DRD1- or DRD5-specific siRNA (Supplementary Fig. 4). With DRD1 silencing, fenoldopam still decreased mito-ROS production in both isolated mitochondria (Fig. 3B) and intact human RPT cells (Fig. 3C). There was no effect of fenoldopam when DRD5 was silenced, indicating that D5R, not D1R, inhibits ROS production in the mitochondria. Similar to the D5R-HEK293 results, the inhibitory effect of fenoldopam on ROS production was reversed by the D1-like receptor antagonist Sch23390 in human RPT cells with intact D1R (Fig. 3A) or silenced D1R (Fig. 3B), affirming that the effect of fenoldopam is exerted via D5R.
Similar to the findings in D5R-HEK293 cells (Fig. 1), Sch23390 alone increased mito-ROS production in D1R-intact (Fig. 3A) and D1R-silenced (Fig. 3B) human RPT cells. DRD5 silencing alone also increased H2O2 production (Fig. 3B), similar to the situation observed with Sch23390 in D5R-HEK293 cells (Fig. 2). These results support the notion that D5R is constitutively active [10]. By contrast, fenoldopam failed to decrease and Sch23390 failed to increase mito-ROS production in human RPT cells with silenced DRD5 (Fig. 3B), indicating a minimal role of D1R in the regulation of mito-ROS in human RPT cells.
Activation of D5R inhibited mito-ROS through an increase in autophagy
Recent evidence suggests that oxidative stress regulates autophagy [14, 16, 17] and that products of dopamine oxidation stimulate autophagy in a neuroblastoma cell line [35]. Therefore, we investigated whether D5R stimulation regulates autophagy. The D1-like receptor agonist fenoldopam inhibited ROS production (Figs. 1–3) [10]. In D5R-HEK293 cells, fenoldopam increased cellular autophagy, as shown by the increase in LC3-II immunostaining (Supplementary Fig. 5). Consistently, fenoldopam also increased the protein expression of ATG5 and LC3-II (Fig. 4A), both of which are involved in autophagy. In the presence of chloroquine, an inhibitor of autophagosome-lysosome fusion and subsequently lysosome-mediated proteolysis [36], the ATG5 and LC3-II protein expression levels were increased further by fenoldopam. These results indicate that the fenoldopam-induced increases in the expression of ATG5 and LC3-II protein were due to an increase in autophagic flux rather than to blockade of the degradation of these proteins. Consistent with this interpretation is the demonstration that Sch23390, an antagonist of D5R (in the absence of D1R), decreased the protein expression of ATG5 and LC3-II by blocking D5R activation. This effect was blocked by chloroquine (Fig. 4B), probably because chloroquine prevented the degradation of ATG5 and LC3-II protein [36].
Fig. 4.
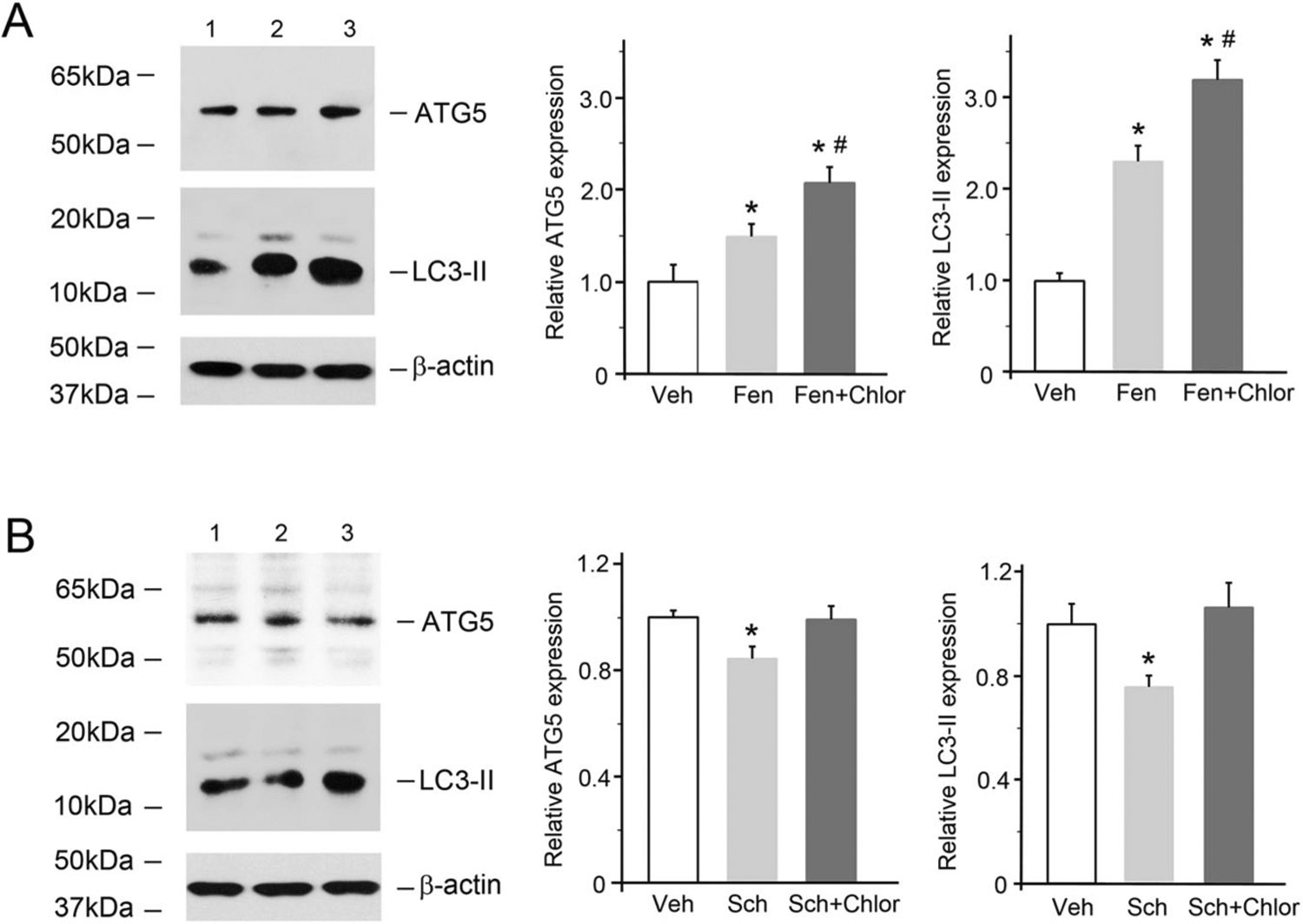
D5R-mediated increases in the levels of autophagy-related proteins in D5R-HEK293 cells. A The cells were treated with fenoldopam (Fen, 1.0 μM, 12 h) in the absence or presence of chloroquine (an inhibitor of autophagosome-lysosome fusion/lysosome-mediated proteolysis, 10 μM, 12 h). Immunoblots from one of five independent experiments are shown. Lane 1, vehicle (Veh); lane 2, fenoldopam (Fen); lane 3, fenoldopam plus chloroquine (Fen + Chlor). The β-actin protein level was used to measure the amount of sample loaded in each lane. n = 5/group, *P < 0.05 vs Veh, #P < 0.05 vs Fen. B D5R-HEK293 cells were treated with Sch (Sch23390, 1.0 μM, 12 h) in the absence or presence of chloroquine (Chlor). Immunoblots from one of five independent experiments are shown. Lane 1, Veh; lane 2, Sch; lane 3, Fen + Chlor. The β-actin protein level was used to measure the amount of sample loaded in each lane. n = 5/group, *P < 0.05 vs others
In DRD1-silenced human RPT cells, fenoldopam also increased the protein expression of ATG5 and LC3-II (Fig. 5A) but not the expression of the proapoptotic protein Bax or the antiapoptotic protein Bcl-xL (Fig. 5B). These results were associated with decreases in cellular ATP concentrations (Fig. 5C), indicating that D5R activation increased autophagy but not apoptosis, which is related to a decrease in mitochondrial energy production in human RPT cells. We then studied whether autophagy involves D5R-mediated inhibition of mito-ROS production using chloroquine. Chloroquine alone slightly but significantly increased mito-ROS production in D1R-silenced human RPT cells but did not affect mito-ROS production in D5R-silenced human RPT cells (Fig. 5D). Similar results were observed with spautin-1, another autophagy inhibitor (Supplementary Fig. 6). Indeed, when autophagy was inhibited by either chloroquine (Fig. 5E) or spautin-1 (Supplementary Fig. 6), fenoldopam was unable to decrease mito-ROS in human RPT cells with intact D5R when DRD1 was silenced. These results indicate that stimulation of autophagy is involved in the D5R-mediated decrease in mito-ROS production in human RPT cells. Moreover, in the presence of apocynin, an NADPH oxidase inhibitor, fenoldopam was still able to decrease mito-ROS production in DRD5-intact and DRD1-silenced (Fig. 5E) human RPT cells, confirming the involvement of a non-NADPH oxidase-mediated mechanism, presumably related to mito-ROS, similar to that observed in D5R-HEK293 cells (Supplementary Fig. 2A).
Fig. 5.
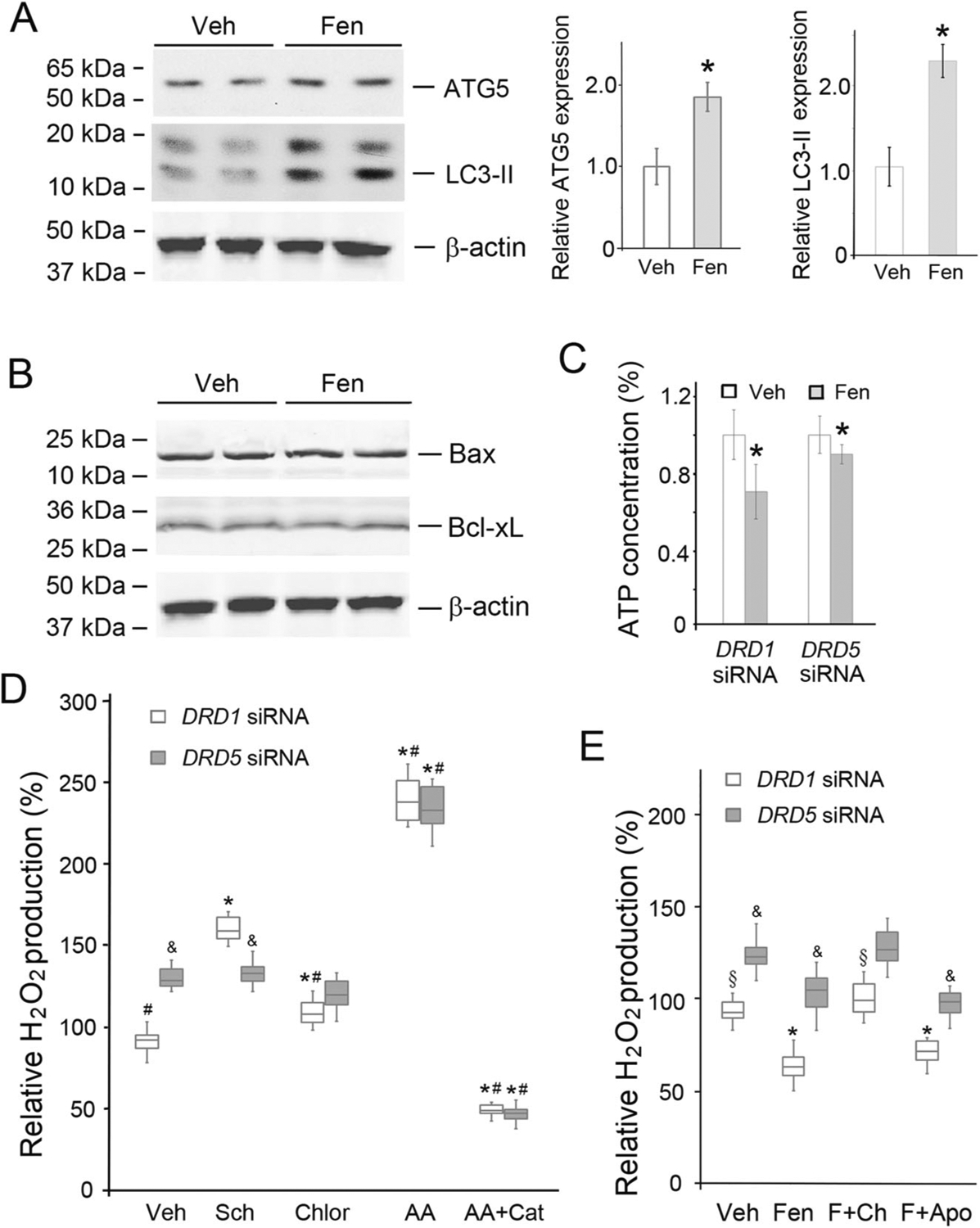
D5R-mediated alteration of autophagy-related protein expression and function. A Autophagy marker protein expression in DRD1-silenced human RPT cells treated with Veh (vehicle) or Fen (fenoldopam, 1.0 μM, 12 h). The immunoblots show that the levels of the autophagy marker proteins ATG5 and LC3-II increased with Fen treatment. n = 4/group, *P < 0.05 vs Veh. B Proapoptotic and antiapoptotic protein (Bax and Bcl-xL) expression in DRD1-silenced human RPT cells. The human RPT cells were treated, as described in A. C ATP concentrations were measured using a bioluminescence assay kit (Roche). The ATP concentration in human RPT cells treated with vehicle was set to 1. Veh vehicle, Fen fenoldopam (1.0 μM, 12 h). n = 5/group, *P < 0.05 vs Veh. D H2O2 production in isolated mitochondria from DRD1- or DRD5-silenced human RPT cells treated as indicated. Veh vehicle, Sch Sch23390 (1.0 μM, 12 h), Chlor chloroquine (inhibitor of autophagosome-lysosome fusion/lysosome-mediated proteolysis, 10 μM, 12 h), AA antimycin A (Qi site inhibitor of mitochondrial ETC Complex III, 0.75 μM, 2 h), Cat catalase (10 μM, 30 min prior to AA treatment). n = 5–6/group, *P < 0.05 vs Veh, #P < 0.05 vs Sch, &P < 0.05 vs DRD1 siRNA. E H2O2 production in isolated mitochondria from human RPT cells, as described in D. Veh vehicle, Fen or F fenoldopam (1.0 μM, 12 h), Ch chloroquine (10 μM, 12 h), Apo apocynin A (10 μM, 12 h). n = 8/group, *P < 0.05 vs Veh, §P < 0.05 vs Fen, &P < 0.05 vs DRD1 siRNA. There was no significant difference between the Fen and Fen + Apo groups in terms of human RPT cells lacking D1R (DRD1 siRNA) or D5R (DRD5 siRNA)
Autophagy protein expression and mito-ROS production in Drd5−/− mouse kidneys
The protein expression levels of ATG5 and LC3-II in Drd5−/− mouse kidney cortices were lower than those in kidney cortices obtained from wild-type Drd5+/+ littermates (Fig. 6A), indicating that the kidney cortices of Drd5−/− mice have decreased autophagy. Mitochondria from the kidney cortices of these mice were isolated (Supplementary Fig. 7) for determination of H2O2 production. As shown in Fig. 6B, H2O2 production in mitochondria isolated from the kidney cortices of Drd5−/− mice was significantly higher than in mitochondria isolated from the kidney cortices of Drd5+/+ mice.
Fig. 6.
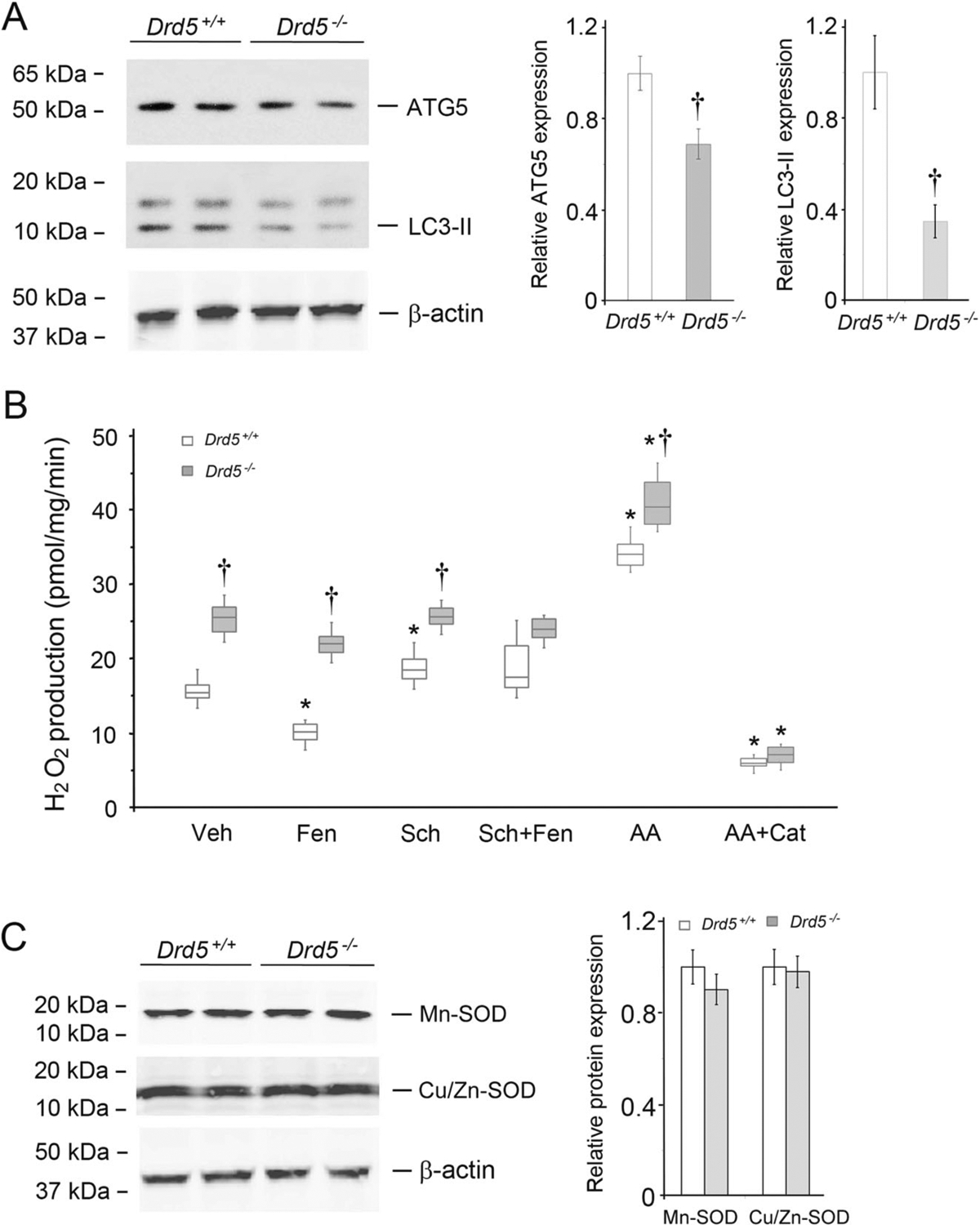
Autophagy protein expression and mito-ROS production in mouse kidney cortices. A Autophagy marker protein expression in kidney cortices of Drd5−/− mice and their wild-type (Drd5+/+) littermates. The immunoblots show decreased protein expression of the autophagy marker proteins ATG5 and LC3-II in the kidney cortices of Drd5−/− mice relative to Drd5+/+ littermates. n = 4/group, †P < 0.05 vs Drd5+/+. B H2O2 production in isolated mitochondria from kidney cortices of Drd5−/− mice and their Drd5+/+ littermates. Veh vehicle, Fen fenoldopam (1.0 μM, 12 h), Sch Sch23390 (1.0 μM, 12 h), AA antimycin A (0.75 μM, 2 h), Cat catalase (10 μM, 30 min prior to AA treatment). n = 6/group, *P < 0.05 vs Veh, †P < 0.05 vs Drd5+/+. C Mitochondrial superoxide dismutase (SOD) protein (Mn-SOD and Cu/Zn-SOD) expression in isolated mitochondria from kidney cortices of Drd5−/− mice and their Drd5+/+ littermates. n = 4/group. There were no significant differences in SOD protein expression between Drd5+/+ and Drd5−/− mice
Treatment of Drd5+/+ mice with fenoldopam significantly decreased mitochondrial H2O2 production (Fig. 6B) and increased ATG5 and LC3-II protein expression in kidney cortices (Supplementary Fig. 8A). By contrast, fenoldopam treatment of Drd5−/− mice did not change H2O2 production by isolated renal mitochondria (Fig. 6B) or the protein expression of ATG5 and LC3-II (Supplementary Fig. 8B). These findings indicated that the fenoldopam-mediated increase in autophagy and decrease in mito-ROS were D5R- but not D1R-dependent in vivo, which corroborated the in vitro observations.
As expected, antimycin A increased H2O2 production in mitochondria isolated from kidney cortices from both Drd5−/− and Drd5+/+ mice, and catalase prevented the stimulatory effect of antimycin A in both Drd5−/− and Drd5+/+ mice (Fig. 6B), consistent with the results of DRD5 silencing studies in human RPT cells (Fig. 5D). Neither Mn-SOD nor Cu/Zn-SOD protein expression in kidney cortices differed between Drd5−/− and Drd5+/+ mice (Fig. 6C).
Discussion
Oxidative stress is implicated in many pathological conditions, such as hypertension, atherosclerosis, and chronic kidney diseases [1–5, 10, 12–14, 19, 20, 37]. Mitochondria generate more than half of cellular ROS in the basal state [4]. Dopamine, through D1-like (D1R and D5R) and D2-like (D2R, D3R and D4R) receptors, decreases ROS production in RPT cells and kidneys from humans and animals [1, 5–7, 10, 21, 23]. D5R negatively regulates ROS production, in part through inhibition of NADPH oxidase expression and activity [10]. The current study demonstrates that fenoldopam, a D5R agonist (in the absence of D1R), decreases mito-ROS production in D5R-HEK293 and human RPT cells and the kidneys of Drd5+/+ mice but not in EV-HEK293 cells lacking D5R, DRD5-silenced human RPT cells, or the kidneys of Drd5−/− mice.
Mitochondria have long been recognized as a major source of ROS in most eukaryotic cells [4]. However, the roles of mitochondrial dysfunction in the pathogeneses of cardiovascular disorders [38] and the importance of mito-ROS in the pathogenesis of hypertension are not as well appreciated as the roles of ROS produced by NADPH oxidase, partly due to the difficulty in accurately measuring mito-ROS production [37, 39–41]. Cytochrome c reduction assays, high-performance liquid chromatography, electron paramagnetic resonance spectroscopy, nitroblue tetrazolium assays, chemiluminescence-based assays, and immunospin trapping have all been used to detect cellular or extracellular superoxide [39–41]. However, due to cumbersome sample preparation methods, low sensitivity, or the need for expensive instruments, these available techniques are not widely used for measurement of mito-ROS in living cells [39–41]. Fluorescence microscopy with a fluorescence-based probe targeting mitochondria enables sensitive, real-time monitoring of mito-ROS signals in individual live cells. Mito-HE, a derivative of HE with a hexyl linker to triphenylphosphonium that accumulates preferentially in mitochondria by several hundred fold [42, 43], was used to monitor mito-ROS production in intact cells. Fluorescent 2-hydroxyethidium, which is generated by the reaction of Mito-HE with ROS, is a relatively reliable indicator of mito-ROS signaling in a biological context [43]. Using Mito-HE (which is also known as MitoSOX Red) as the probe for ROS, we found that activation of D5R attenuated mitochondrial oxidative stress in live D5R-HEK293 and human RPT cells. To avoid the potential technical limitations of Mito-HE [42] and cross-talk with NADPH oxidase-derived ROS in intact cells, additional experiments were performed in mitochondria isolated from D5R-HEK293 and human RPT cells in which the other D1-like receptor, D1R, was silenced and in kidney cortices of Drd5−/− and Drd5+/+ mice. All results of these studies consistently show that D5R, in the absence of D1R, negatively regulates mito-ROS production.
The production of ROS by mitochondria is caused by leakage of electrons from the ETC, with Complex I and Complex III conventionally recognized as the major sites [44, 45]. In vitro studies have demonstrated that Complex I is the major site for ROS production when cells do not produce ATP with high NADH/NAD+ and CoQH2/CoQ ratios and high proton motive force [44]. However, under most conditions, mitochondria in cells synthesize ATP. In this situation, Complex III and other sites are important sites for ROS production in mitochondria [44, 45]. Our in vitro data demonstrated that ROS production was significantly increased upon inhibition of the Complex III Qi site with antimycin A but not upon inhibition of Complex I with rotenone. Moreover, antimycin A, but not rotenone, prevented the inhibitory effect of fenoldopam on ROS production in intact D5R-HEK cells, indicating that D5R may directly or indirectly interact with Complex III rather than Complex I. The site(s) of ROS production within the mitochondria depend(s) on the cell bioenergetic status and the substrate being oxidized [44–46]. Whether D5R targets Complex III in vivo under different bioenergetic conditions or mechanisms requires further investigation.
Mitochondria and NADPH oxidases are the two main sources of cellular ROS, and it is believed there is a vicious cycle between these two sources of ROS; NADPH oxidase-derived ROS increase mito-ROS formation, and mito-ROS activate NADPH oxidase [47]. We have reported that D1-like receptor activation decreases renal ROS production in part through inhibition of NADPH oxidase in the kidneys [10, 48]. Yang et al. have also reported that D1-like receptors decrease NADPH oxidase-mediated ROS production in human RPT cells [49]. However, D5R, not D1R, decreases ROS production, in part by increasing paraoxonase 2 protein levels [49]. Our present study demonstrates that D5R, not D1R, decreases Mito-ROS production, which is consistent with a recent report that paraoxonase 2 decreases mitochondrial ROS production in cardiomyocytes [50]. The decrease in mito-ROS production mediated by the D1R/D5R agonist fenoldopam was not abrogated by the presence of the NADPH oxidase inhibitor apocynin in human RPT cells deficient in D1R but with intact D5R. This result indicates that the D5R-mediated decrease in mito-ROS production is not downstream of its inhibition of NADPH oxidase activity.
Dopamine increases LC3-II expression and autophagy in neuroblastoma cells [35]. D3R induces autophagy under hyperammonia [51]. By contrast, inhibition of D2R activity induces autophagy in neuroblastoma cells [52] and cardiomyocytes [53]. However, inhibition of D4R activity also disrupts autophagosome-lysosomal fusion [54]. In our study, we demonstrated that activation of D5R using fenoldopam at concentrations that inhibit ROS production [10] increased autophagic protein expression and activation in human RPT cells, which was confirmed in the kidneys of Drd5+/+ mice, indicating that D5R, like D3R and D4R, is involved in the regulation of autophagy. Of note, all dopamine receptors are expressed in the kidneys, and their expression is different at different stages of development [25–27, 55–57]. Thus, it is possible that the role of D5R in autophagy could be different from that of other dopamine receptors in different nephron segments at different stages of development.
Autophagy can be both a cause and consequence of oxidative stress [14, 17]. Mitochondria are both the major sources and targets of ROS; therefore, mitochondria could play critical roles in the cellular responses to oxidative stress. On the one hand, mito-ROS, via multiple signaling pathways, can lead to downregulation or upregulation of autophagy [14, 17, 58]. On the other hand, autophagy regulates ROS signaling or cellular consequences in response to oxidative stress [14]. Autophagic deficiency in mouse embryonic fibroblasts protects against H2O2-induced cell death [59]. However, hepatocyte-specific deletion of Atg5 in mice induces oxidative stress in these cells [60]; pancreas-specific disruption of Atg5 in mice also increases ROS production in this organ [61]. Intestinal epithelial cell-specific deletion of Atg5 in mice decreases the basal intestinal epithelial cell mitochondrial membrane potential and increases ROS production [62], suggesting that deficiency of autophagy could lead to an increase in ROS production that may be related to mitochondrial dysfunction.
Mild oxidative stress induces autophagy, while severe oxidative stress inhibits autophagy [17, 58], and high concentrations of dopamine increase autophagy via oxidative products of dopamine [35]. High, cytotoxic concentrations of dopamine (>100 μM) increase oxidative stress, but low, noncytotoxic concentrations of dopamine decrease oxidative stress [63, 64]. Stimulation of D1-like receptors by the D1-like receptor agonist fenoldopam decreases ROS production [10], but the effect of fenoldopam on autophagy has not been reported. Ang II promotes autophagy, which is mediated by mito-ROS in podocytes [65, 66]. D5R activation promotes the ubiquitination and proteasomal degradation of AT1R [21–23], and germline deletion of Drd5 in mice increases renal AT1R expression [23]. By contrast, AT1R blockade with losartan or germline deletion of Agtr1 in mice increases D5R protein expression [7, 67]. Therefore, treatment with an AT1R blocker should increase the inhibitory effect of fenoldopam on mito-ROS production but negate the stimulatory effect of fenoldopam on autophagy. However, fenoldopam decreases ROS production and stimulates autophagy, indicating that the effect of fenoldopam on autophagy is direct (vide infra) and not mediated through ROS. It is also known that enhancement of ubiquitination or inhibition of the proteasome pathway plays an important role in the induction of autophagy associated with mitochondrial oxidative stress [68–70]. However, D5R activates both the ubiquitination and proteasome pathways in addition to inhibiting ROS production, as described previously [23]. Studies by others have shown that the increase in autophagy caused by ROS is mediated by AMPK [71], which can be stimulated by cAMP [71, 72], the levels of which are increased by D5R [5–7]. The presence of soluble adenylyl cyclase both outside and inside the mitochondria suggests that phosphorylation of mitochondrial proteins may be a major regulatory mechanism for mitochondrial function [73]. cAMP-PKA signaling can regulate all steps of autophagy [74]. Therefore, cAMP signaling could act as a bridge between D5R in the cell membrane and mitochondria to regulate mitochondrial function. However, depending on the local microenvironments of tissues and cells, activation of cAMP signaling could induce [75, 76], reduce [77], or have no effect [78] on autophagy. Thus, while both D1R and D5R increase cAMP production and decrease ROS production [5–7, 10, 48], these two D1-like receptors have different effects on autophagy and ROS production: D5R, but not D1R, stimulates autophagy and inhibits the production of ROS in mitochondria. D1R increases adenylyl cyclase type 5/6 proteins in lipid rafts, while D5R increases adenylyl cyclase type 5/6 proteins in nonlipid rafts [79]. We speculate that these differential effects of D5R and D1R could be due to their differential expression in lipid and nonlipid raft microenvironments, their coupling to different isoforms of adenylyl cyclases [79], or other unknown factors; these possibilities warrant further study. Nevertheless, our current study shows that D5R-mediated decreases in ROS production are probably due to increases in cAMP production and subsequent induction of autophagy.
In summary, activation of D5R, independent of D1R decreased mito-ROS production in D5R-HEK293 and RPT cells. This effect was mimicked by the PKA activator Sp-cAMPS and was attenuated by the PKA inhibitor Rp-cAMPS and the autophagy inhibitors chloroquine and spautin-1. ROS production was increased in mitochondria isolated from the kidney cortices of Drd5−/− mice relative to their wild-type littermates. Furthermore, the renal protein expression of autophagy hallmark proteins was increased by fenoldopam, a D5R agonist (in the absence of D1R), in vitro and in vivo. These results suggest that D5R negatively regulates ROS production in a cAMP- and autophagy-dependent manner.
Supplementary Material
Acknowledgements
This study was supported in part by grants from the National Institutes of Health (HL074940, HL023081, DK039308, HL092196, DK119652, and DK090918), the Children’s National Medical Center Intramural Avery Award, the National Natural Science Foundation of China (81670698, 91739119, 81670406, 30971186), the Second Hospital of Dalian Medical University Start-up Funds, and the Department of Education of Liaoning Province Grant (L2016020).
Footnotes
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41440-021-00646-w.
Conflict of interest The authors declare no competing interests.
References
- 1.Araujo M, Wilcox CS. Oxidative stress in hypertension: role of the kidney. Antioxid Redox Signal. 2014;20:74–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loperena R, Harrison DG. Oxidative stress and hypertensive diseases. Med Clin North Am. 2017;101:169–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montezano AC, Dulak-Lis M, Tsiropoulou S, Harvey A, Briones AM, Touyz RM. Oxidative stress and human hypertension: vascular mechanisms, biomarkers, and novel therapies. Can J Cardiol. 2015;31:631–41. [DOI] [PubMed] [Google Scholar]
- 4.Addabbo F, Montagnani M, Goligorsky MS. Mitochondria and reactive oxygen species. Hypertension. 2009;53:885–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang MZ, Harris RC. Antihypertensive mechanisms of intra-renal dopamine. Curr Opin Nephrol Hypertens. 2015;24:117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asghar M, Tayebati SK, Lokhandwala MF, Hussain T. Potential dopamine-1 receptor stimulation in hypertension management. Curr Hypertens Rep. 2011;13:294–302. [DOI] [PubMed] [Google Scholar]
- 7.Zeng C, Jose PA. Dopamine receptors: important antihypertensive counterbalance against hypertensive factors. Hypertension. 2011; 57:11–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allayee H, de Bruin TW, Michelle Dominguez K, Cheng LS, Ipp E, Cantor RM, et al. Genome scan for blood pressure in Dutch dyslipidemic families reveals linkage to a locus on chromosome 4p. Hypertension. 2001;38:773–8. [DOI] [PubMed] [Google Scholar]
- 9.Cohn DH, Shohat T, Yahav M, Ilan T, Rechavi G, King L, et al. A locus for an autosomal dominant form of progressive renal failure and hypertension at chromosome 1q21. Am J Hum Genet. 2000; 67:647–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Z, Asico LD, Yu P, Wang Z, Jones JE, Escano CS, et al. D5 dopamine receptor regulation of reactive oxygen species production, NADPH oxidase, and blood pressure. Am J Physiol Regul Integr Comp Physiol. 2006;290:R96–104. [DOI] [PubMed] [Google Scholar]
- 11.Saez F, Hong NJ, Garvin JL. Luminal flow induces NADPH oxidase 4 translocation to the nuclei of thick ascending limbs. Physiol Rep. 2016;4:e12724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Q, Wu FR, Wang JN, Gao L, Jiang L, Li HD, et al. Nox4 in renal diseases: an update. Free Radic Biol Med. 2018;124:466–72. [DOI] [PubMed] [Google Scholar]
- 13.Haque MZ, Majid DS. Reduced renal responses to nitric oxide synthase inhibition in mice lacking the gene for gp91phox subunit of NAD(P)H oxidase. Am J Physiol Ren Physiol. 2008;295: F758–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryter SW, Bhatia D, Choi ME. Autophagy: a lysosome-dependent process with implications in cellular redox homeostasis and human disease. Antioxid Redox Signal. 2019;30:138–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–64. [DOI] [PubMed] [Google Scholar]
- 17.Woodall BP, Gustafsson AB. Autophagy—a key pathway for cardiac health and longevity. Acta Physiol. 2018;20:e13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanderson RD, Elkin M, Rapraeger AC, Ilan N, Vlodavsky I. Heparanase regulation of cancer, autophagy and inflammation: new mechanisms and targets for therapy. FEBS J. 2017;284:42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peña-Oyarzun D, Bravo-Sagua R, Diaz-Vega A, Aleman L, Chiong M, Garcia L, et al. Autophagy and oxidative stress in noncommunicable diseases: a matter of the inflammatory state. Free Radic Biol Med. 2018;124:61–78. [DOI] [PubMed] [Google Scholar]
- 20.Wible DJ, Bratton SB. Reciprocity in ROS and autophagic signaling. Curr Opin Toxicol. 2018;7:28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gildea JJ, Wang X, Jose PA, Felder RA. Differential D1 and D5 receptor regulation and degradation of the angiotensin type 1 receptor. Hypertension. 2008;51:360–6. [DOI] [PubMed] [Google Scholar]
- 22.Jean-Charles PY, Snyder JC, Shenoy SK. Ubiquitination and deubiquitination of G protein-coupled receptors. Prog Mol Biol Transl Sci. 2016;141:1–55. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Armando I, Yu P, Escano C, Mueller SC, Asico L, et al. Dopamine 5 receptor mediates Ang II type 1 receptor degradation via a ubiquitin-proteasome pathway in mice and human cells. J Clin Investig. 2008;118:2180–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hollon TR, Bek MJ, Lachowicz JE, Ariano MA, Mezey E, Ramachandran R, et al. Mice lacking D5 dopamine receptors have increased sympathetic tone and are hypertensive. J Neurosci. 2002;22:10801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanada H, Jose PA, Hazen-Martin D, Yu PY, Xu J, Bruns DE, et al. Dopamine-1 receptor coupling defect in renal proximal tubule cells in hypertension. Hypertension. 1999;33:1036–42. [DOI] [PubMed] [Google Scholar]
- 26.O’Connell DP, Botkin SJ, Ramos SI, Sibley DR, Ariano MA, Felder RA, et al. Localization of dopamine D1A receptor protein in rat kidneys. Am J Physiol. 1995;268:F1185–97. [DOI] [PubMed] [Google Scholar]
- 27.Ennis RC, Asico LD, Armando I, Yang J, Feranil JB, Jurgens JA, et al. Dopamine D1-like receptors regulate the α1A-adrenergic receptor in human renal proximal tubule cells and D1-like dopamine receptor knockout mice. Am J Physiol Ren Physiol. 2014;307:F1238–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Li F, Jose PA, Ecelbarger CM. Reduction of renal dopamine receptor expression in obese Zucker rats: role of sex and angiotensin II. Am J Physiol Ren Physiol. 2010;299:F1164–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Li HF, Felder RA, Periasamy A, Jose PA. Actin cytoskeleton-dependent Rab GTPase-regulated angiotensin type I receptor lysosomal degradation studied by fluorescence lifetime imaging microscopy. J Biomed Opt. 2008;13:031206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee H, Abe Y, Lee I, Shrivastav S, Crusan AP, Huttemann M, et al. Increased mitochondrial activity in renal proximal tubule cells from young spontaneously hypertensive rats. Kidney Int. 2014;85:561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee I, Salomon AR, Ficarro S, Mathes I, Lottspeich F, Grossman LI, et al. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J Biol Chem. 2005;280: 6094–100. [DOI] [PubMed] [Google Scholar]
- 32.Polster BM, Nicholls DG, Ge SX, Roelofs BA. Use of potentiometric fluorophores in the measurement of mitochondrial reactive oxygen species. Methods Enzymol. 2014;547:225–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Votyakova TV, Reynolds IJ. Detection of hydrogen peroxide with Amplex Red: interference by NADH and reduced glutathione auto-oxidation. Arch Biochem Biophys. 2004;431:138–44. [DOI] [PubMed] [Google Scholar]
- 34.Lochner A, Moolman JA. The many faces of H89: a review. Cardiovasc Drug Rev. 2006;24:261–74. [DOI] [PubMed] [Google Scholar]
- 35.Gimenez-Xavier P, Francisco R, Santidrian AF, Gil J, Ambrosio S. Effects of dopamine on LC3-II activation as a marker of autophagy in a neuroblastoma cell model. Neurotoxicology. 2009; 30:658–65. [DOI] [PubMed] [Google Scholar]
- 36.Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirkman DL, Muth BJ, Ramick MG, Townsend RR, Edwards DG. Role of mitochondria-derived reactive oxygen species in microvascular dysfunction in chronic kidney disease. Am J Physiol Ren Physiol. 2018;314:F423–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonora M, Wieckowski MR, Sinclair DA, Kroemer G, Pinton P, Galluzzi L. Targeting mitochondria for cardiovascular disorders: therapeutic potential and obstacles. Nat Rev Cardiol. 2019;16: 33–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, et al. Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: a scientific statement from the American Heart Association. Circ Res. 2016;119:e39–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee R, Margaritis M, Channon KM, Antoniades C. Evaluating oxidative stress in human cardiovascular disease: methodological aspects and considerations. Curr Med Chem. 2012;19:2504–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mason RP. Imaging free radicals in organelles, cells, tissue, and in vivo with immuno-spin trapping. Redox Biol. 2016;8:422–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalyanaraman B, Dranka BP, Hardy M, Michalski R, Zielonka J. HPLC-based monitoring of products formed from hydroethidine-based fluorogenic probes–the ultimate approach for intra- and extracellular superoxide detection. Biochim Biophys Acta. 2014;1840:739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, et al. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci USA. 2006;103:15038–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bleier L, Drose S. Superoxide generation by complex III: from mechanistic rationales to functional consequences. Biochim Biophys Acta. 2013;1827:1320–31. [DOI] [PubMed] [Google Scholar]
- 46.Wong HS, Dighe PA, Mezera V, Monternier PA, Brand MD. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J Biol Chem. 2017;292:16804–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daiber A, Di Lisa F, Oelze M, Kröller-Schön S, Steven S, Schulz E, et al. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br J Pharm. 2017;174:1670–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H, Han W, Villar VA, Keever LB, Lu Q, Hopfer U, et al. D1-like receptors regulate NADPH oxidase activity and subunit expression in lipid raft microdomains of renal proximal tubule cells. Hypertension. 2009;53:1054–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang S, Yang Y, Yu P, Yang J, Jiang X, Villar VA, et al. Dopamine D1 and D5 receptors differentially regulate oxidative stress through paraoxonase 2 in kidney cells. Free Radic Res. 2015;49:397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sulaiman D, Li J, Devarajan A, Cunningham CM, Li M, Fishbein GA, et al. Paraoxonase 2 protects against acute myocardial ischemia-reperfusion injury by modulating mitochondrial function and oxidative stress via the PI3K/Akt/GSK-3β RISK pathway. J Mol Cell Cardiol. 2019;129:154–64. [DOI] [PubMed] [Google Scholar]
- 51.Li Z, Ji X, Wang W, Liu J, Liang X, Wu H, et al. Ammonia induces autophagy through dopamine receptor D3 and mTOR. PLoS ONE. 2016;11:e0153526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shin JH, Park SJ, Kim ES, Jo YK, Hong J, Cho DH. Sertindole, a potent antagonist at dopamine D2 receptors, induces autophagy by increasing reactive oxygen species in SH-SY5Y neuroblastoma cells. Biol Pharm Bull. 2012;35:1069–75. [DOI] [PubMed] [Google Scholar]
- 53.Yan H, Li WL, Xu JJ, Zhu SQ, Long X, Che JP. D2 dopamine receptor antagonist raclopride induces non-canonical autophagy in cardiac myocytes. J Cell Biochem. 2013;114:103–10. [DOI] [PubMed] [Google Scholar]
- 54.Dolma S, Selvadurai HJ, Lan X, Lee L, Kushida M, Voisin V, et al. Inhibition of dopamine receptor D4 impedes autophagic flux, proliferation, and survival of glioblastoma stem cells. Cancer Cell. 2016;29:859–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jose PA, Eisner GM, Drago J, Carey RM, Felder RA. Dopamine receptor signaling defects in spontaneous hypertension. Am J Hypertens. 1996;9:400–5. [DOI] [PubMed] [Google Scholar]
- 56.Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. [DOI] [PubMed] [Google Scholar]
- 57.Gildea JJ, Shah I, Weiss R, Casscells ND, McGrath HE, Zhang J, et al. HK-2 human renal proximal tubule cells as a model for G protein-coupled receptor kinase type 4-mediated dopamine 1 receptor uncoupling. Hypertension. 2010;56:505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012; 441:523–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pyo JO, Nah J, Kim HJ, Lee HJ, Heo J, Lee H, et al. Compensatory activation of ERK1/2 in Atg5-deficient mouse embryo fibroblasts suppresses oxidative stress-induced cell death. Autophagy. 2008;4:315–21. [DOI] [PubMed] [Google Scholar]
- 60.Tian Y, Kuo CF, Sir D, Wang L, Govindarajan S, Petrovic LM, et al. Autophagy inhibits oxidative stress and tumor suppressors to exert its dual effect on hepatocarcinogenesis. Cell Death Differ. 2015;22:1025–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Diakopoulos KN, Lesina M, Wormann S, Song L, Aichler M, Schild L, et al. Impaired autophagy induces chronic atrophic pancreatitis in mice via sex- and nutrition-dependent processes. Gastroenterology. 2015;148:626–38. [DOI] [PubMed] [Google Scholar]
- 62.Harada S, Nakagawa T, Yokoe S, Edogawa S, Takeuchi T, Inoue T, et al. Autophagy deficiency diminishes indomethacin-induced intestinal epithelial cell damage through activation of the ERK/Nrf2/HO-1 pathway. J Pharm Exp Ther. 2015;355:353–61. [DOI] [PubMed] [Google Scholar]
- 63.Jones DC, Gunasekar PG, Borowitz JL, Isom GE. Dopamine-induced apoptosis is mediated by oxidative stress and is enhanced by cyanide in differentiated PC12 cells. J Neurochem. 2000;74: 2296–304. [DOI] [PubMed] [Google Scholar]
- 64.Leng ZG, Lin SJ, Wu ZR, Guo YH, Cai L, Shang HB, et al. Activation of DRD5 (dopamine receptor D5) inhibits tumor growth by autophagic cell death. Autophagy. 2017;13:1404–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yadav A, Vallabu S, Arora S, Tandon P, Slahan D, Teichberg S, et al. Ang II promotes autophagy in podocytes. Am J Physiol Cell Physiol. 2010;299:C488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sachse A, Wolf G. Angiotensin II-induced reactive oxygen species and the kidney. J Am Soc Nephrol. 2007;18:2439–46. [DOI] [PubMed] [Google Scholar]
- 67.Zeng C, Yang Z, Wang Z, Jones J, Wang X, Altea J, et al. Interaction of angiotensin II type 1 and D5 dopamine receptors in renal proximal tubule cells. Hypertension. 2005;45:804–10. [DOI] [PubMed] [Google Scholar]
- 68.Haller M, Hock AK, Giampazolias E, Oberst A, Green DR, Debnath J, et al. Ubiquitination and proteasomal degradation of ATG12 regulates its proapoptotic activity. Autophagy. 2014;10: 2269–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang S, Park DW, Gao Y, Ravi S, Darley-Usmar V, Abraham E, et al. Participation of proteasome-ubiquitin protein degradation in autophagy and the activation of amp-activated protein kinase. Cell Signal. 2015;27:1186–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Livnat-Levanon N, Glickman MH. Ubiquitin-proteasome system and mitochondria - reciprocity. Biochim Biophys Acta. 2011;1809:80–7. [DOI] [PubMed] [Google Scholar]
- 71.Omar B, Zmuda-Trzebiatowska E, Manganiello V, Göransson O, Degerman E. Regulation of AMP-activated protein kinase by cAMP in adipocytes: roles for phosphodiesterases, protein kinase B, protein kinase A, Epac and lipolysis. Cell Signal. 2009;21:760–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Decara J, Rivera P, Arrabal S, Vargas A, Serrano A, Pavón FJ, et al. Cooperative role of the glucagon-like peptide-1 receptor and β3-adrenergic-mediated signalling on fat mass reduction through the downregulation of PKA/AKT/AMPK signalling in the adipose tissue and muscle of rats. Acta Physiol. 2018;222:e13008. [DOI] [PubMed] [Google Scholar]
- 73.Valsecchi F, Ramos-Espiritu LS, Buck J, Levin LR, Manfredi G. cAMP and mitochondria. Physiology. 2013;28:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Torres-Quiroz F, Filteau M, Landry CR. Feedback regulation between autophagy and PKA. Autophagy. 2015;11:1181–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee YJ, Shu MS, Kim JY, Kim YH, Sim KH, Sung WJ, et al. Cilostazol protects hepatocytes against alcohol-induced apoptosis via activation of AMPK pathway. PLoS ONE. 2019;14:e0211415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen ML, Yi L, Jin X, Liang XY, Zhou Y, Zhang T, et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy. 2013; 9:2033–45. [DOI] [PubMed] [Google Scholar]
- 77.Akabane S, Uno M, Tani N, Shimazaki S, Ebara N, Kato H, et al. PKA regulates PINK1 stability and parkin recruitment to damaged mitochondria through phosphorylation of MIC60. Mol Cell. 2016;62:371–84. [DOI] [PubMed] [Google Scholar]
- 78.Wolter S, Kloth C, Golombek M, Dittmar F, Försterling L, Seifert R. cCMP causes caspase-dependent apoptosis in mouse lymphoma cell lines. Biochem Pharm. 2015;98:119–31. [DOI] [PubMed] [Google Scholar]
- 79.Yu P, Sun M, Villar VA, Zhang Y, Weinman EJ, Felder RA, et al. Differential dopamine receptor subtype regulation of adenylyl cyclases in lipid rafts in human embryonic kidney and renal proximal tubule cells. Cell Signal. 2014;26:2521–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.