

Abstract
Protein aggregation is associated with a large number of human protein-misfolding diseases, yet FDA-approved drugs are currently not available. Amylin amyloid and plaque depositions in the pancreas are hallmark features of type 2 diabetes. Moreover, these amyloid deposits are implicated in the pathogenesis of diabetic complications such as neurodegeneration. We recently discovered that catechols and redox-related quinones/anthraquinones represent a broad class of protein aggregation inhibitors. Further screening of a targeted library of natural compounds in complementary medicine that were enriched with catechol-containing compounds identified rosmarinic acid (RA) as a potent inhibitor of amylin aggregation (estimated inhibitory concentration IC50 = 200–300 nM). Structure–function relationship analysis of RA showed the additive effects of the two catechol-containing components of the RA molecule. We further showed that RA does not reverse fibrillation back to monomeric amylin but rather lead to nontoxic, remodeled protein aggregates. RA has significant ex vivo efficacy in reducing human amylin oligomer levels in HIP rat sera as well as in sera from diabetic patients. In vivo efficacy studies of RA treatment with the diabetic HIP rat model demonstrated significant reduction in amyloid islet deposition and strong mitigation of diabetic pathology. Our work provides new in vitro molecular mechanisms and in vivo efficacy insights for a model nutraceutical agent against type 2 diabetes and other aging-related protein-misfolding diseases.
Keywords: amylin amyloid, type 2 diabetes, protein aggregation inhibitor, rosmarinic acid, catechol-containing natural compounds, inhibition mechanisms
Protein-misfolding diseases include a broad spectrum of neurodegenerative and related aging diseases such as Alzheimer’s disease (AD), Parkinson’s disease, prion disease, and type 2 diabetes (T2D).1−5 The pathological hallmarks of this class of diseases are amyloid fibrils: structurally conserved intracellular and extracellular insoluble proteinaceous deposits.6 Amyloid formation proceeds through a nucleation-dependent aggregation process. Monomeric and oligomeric aggregates form amyloid “seeds” that initiate a cascade that results in equilibrium between mature amyloid fibrils and their small precursor aggregates.3,7 Over the last two decades, increasing evidence has indicated that the primary pathological amyloid species are these small nonfibrillar precursor aggregates, ranging from unstructured oligomers to β-sheet rich aggregates or protofibrils.3,5,8,9
Amylin, also referred to as islet amyloid polypeptide or IAPP, is a 37 amino acid (AA) peptide hormone co-secreted with insulin by pancreatic β-cells.1,10 Physiologically, amylin plays important roles in glycemic regulation.1,11 Amylin is one of the most cytotoxic amyloidogenic proteins known, even more toxic than the well-studied amyloid β-peptide, Aβ42 (Figure S1).12 A state of hyperamylinemia is associated with the compensatory increased insulin secretion that occurs during the development of insulin resistance and T2D due to the cosecretion of amylin and insulin hormones. Toxic amylin oligomer/amyloid formation and plaque deposition in the pancreas are hallmark features of T2D.1,13−15 Experimental evidence shows that hyperamylinemia also induces toxicity in other organs, including the heart, kidneys, and the brain.16−19 The accumulation of amylin is associated with vascular and tissue damage to the heart and brain.17,20 While the rodent amylin sequence is not amyloidogenic,1,21 a “humanized” transgenic diabetic rat model overexpressing human amylin (HIP rat) has provided strong causal evidence that amylin oligomers/amyloid contribute to heart dysfunction and AD-mimicking neurological deficits in addition to diabetes.18,20,22−24 Amylin oligomers were detected in the serum of HIP rats, and the accumulation of amylin deposits in the hearts and the brains of these animals was also observed.18,24
Currently, there are no cures for any protein amyloid diseases. Progress toward managing protein-misfolding diseases in general has been hampered by the failure to develop effective disease-modifying drugs. This is partly due to our limited understanding of amyloidogenic proteins as well as their interactions with small molecules/inhibitors. Identification of effective amyloid inhibitors is challenging because of the intrinsic structural disorder of the protein targets of amyloid assembly; however, there are recent significant advances in the structural biology research of amyloid proteins, such as cryogenic electron microscopy (cryo-EM) characterization of their key fragments, which may enable structure-based inhibitor design in the future.6,25−29
There are multiple therapeutic strategies for identifying disease-modifying agents for neutralizing toxic protein amyloids.30−32 One of the current strategies aimed at identifying lead compounds focuses on inhibiting amyloid aggregation by (i) inhibiting toxic amyloid formation or stabilizing its native form from further aggregation and (ii) remodeling or degrading toxic amyloid oligomers and/or insoluble fibrils. For natural-product-based amyloid inhibitor identification, one source of information from epidemiological studies suggests that preventative effects against neurodegenerative dementia or diabetes may be associated with diets containing a high intake of flavonoids and polyphenolic compounds.33,34 The importance of diet has been well-recognized as dietary risks are ranked as one of the highest risk factors in aging diseases. Past epidemiological work as well as alternative medicine studies led to testable hypotheses and experimental efforts that have successfully identified numerous natural compound amyloid inhibitors, including the well-studied polyphenol EGCG.21,35−43
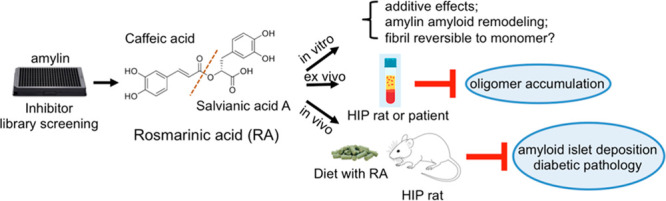
In order to gain insight into general classes of chemical structures of amyloid inhibitors, we recently screened an NIH Clinical Collection (NIHCC) drug library of 700 small-molecule compounds with diverse chemotypes against three prototype amyloidogenic proteins, amylin, Aβ, and tau. We identified catechols and redox-related quinones/anthraquinones as a broad class of protein aggregation inhibitors.21 To identify new, potent, small-molecule inhibitors of amylin amyloids, we further screened a targeted library of natural products used in alternative medicine enriched with catechol-containing compounds. This effort identified rosmarinic acid (RA), a catechol-containing natural product, as a potent inhibitor. This dual-catechol-containing molecule demonstrated an additive effect, attributing the activities of the molecule to two active components, caffeic acid (CA) and salvianic acid A (SAA). Our inhibitor-induced amyloid remodeling data provided further evidence suggesting that amyloid remodeling is rapid and uses a pathway different from insoluble amyloid dissolution or fibril to monomer disaggregation. RA showed ex vivo efficacy in reducing human amylin oligomers in the sera from transgenic diabetic HIP rats and from that of diabetic patients. In vivo animal studies with the HIP rats demonstrated that RA potently reduced amyloid islet deposition and mitigated HIP rat diabetic pathology. Our work provides new in vitro molecular mechanisms and in vivo efficacy insights for a model nutraceutical agent against T2D and other aging-related protein-misfolding diseases.
Methods
Peptides, Chemicals, and Sera
Synthetic amidated human amylin was purchased from AnaSpec Inc. (Fremont, CA), and the peptide quality was further validated by the Virginia Tech Mass Spectrometry Incubator. Hexafluoroisopropanol (HFIP) and thioflavin T (ThT) were purchased from Sigma-Aldrich (St. Louis, MO). RA, CA, and SAA were purchased from Toronto Research Chemicals (North York, ON, Canada), Fisher Scientific Inc. (Hampton, NH), and Sigma-Aldrich Corp. (St. Louis, MO) respectively. Dulbecco’s phosphate-buffered saline (DBPS, pH 7.4) was purchased from Lonza (Walkersville, MD). Black 96-well nonstick clear-bottomed plates and optically clear sealing film were purchased from Greiner Bio-one (Frickenhausen, Germany) and Hampton Research (Aliso Viejo, CA), respectively. We purchased 300 mesh Formvar carbon-coated copper grids and uranyl acetate replacement solution from Electron Microscopy Sciences (Hatfeild, PA).
Lyophilized amylin powder (0.5 mg) was initially dissolved in 100% HFIP at a final concentration of 1–2 mM. An additional lyophilizing step was employed to eliminate traces of organic solvents, which have been shown to affect amylin aggregation. Aliquots were either lyophilized again prior to use in cell-based assays or dissolved directly into DPBS (10 mM phosphate buffer, pH 7.4) or 20 mM Tris-HCl (pH 7.4) for all amylin-amyloid-related in vitro assays. All remaining 1–2 mM stocks in 100% DMSO were stored at −80 °C until later use. The lyophilized powder from all compounds and ThT were dissolved in DMSO (10 mM) and distilled water or relevant buffer (1–4 mM). These stocks were stored at −20 °C until later use. Residual DMSO in the final samples used for all in vitro assays ranged from 0 to 9.5%. We determined that these DMSO concentrations had negligible effects on amylin amyloid aggregation as reflected by ThT fluorescence, transmission electron microscopy (TEM) assays, and inhibitor-induced amyloid remodeling assays.
HIP rat sera and diabetic patient sera samples are generous gifts from Prof. Florian Despa (University of Kentucky). Detailed sample collection and institutional approval of usage were described in the literature.18,23
Cell Culture
Rat pancreatic INS-1 cells and mouse neuroblastoma Neuro2A cells were generously provided by Profs. Pierre Maechler (University of Geneva) and Deborah Good (Virginia Tech), respectively. The SH-SY5Y cells were purchased from ATCC (no. CRL-2266). Vascular smooth muscle cells (VSMCs) were primary cells isolated from mouse aorta using the treatment of types I and II collagenases and type III elastase. INS-1, Neuro2A, and SHSY-5Y cells were cultured per vendor’s instruction. Briefly, INS-1 cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum and supplemented with 11.1 mM glucose, 1 mM sodium pyruvate, 10 mM HEPES, 50 μM β-mercaptoethanol, and 23.8 mM NaHCO3. The medium was changed every other day until the cells became confluent. Neuro2A cells were cultured in DMEM containing 1% nonessential amino acids, 1% l-glutamate, and 10% FBS. SH-SY5Y cells were cultured in EMEM with 1% nonessential amino acids and 10% FBS. VSMCs were grown in DMEM containing 20% FBS.
Cytotoxicity Assay and Inhibitor Activity Quantification
An MTT-based cell viability assay was used. The INS-1, Neuro-2A, and SH-SY5Y cells were seeded in 96-well plate at a density of 4 × 104 cells/well. After 24 h of incubation, cells were treated with human amylin (3.75 μM) with or without specified natural compounds. Following another 24 h of incubation, 0.9 mM MTT was added to each well. The reduced insoluble MTT formazan product was then dissolved in SDS-HCl lysis buffer (5 mM HCl, 5% SDS) at 37 °C. Cell viability was determined by measurement of absorbance change at 570 nm using a spectrometric plate reader. Cells treated with peptides dissolved in buffer were used as the positive control and taken as 100%, and cells treated with 0.5% Triton X-100 at the start of the incubation period with test peptides were used as the negative control and taken as 0%.
ThT Fluorescence Assay
Fluorescence experiments were carried out using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA). All kinetic reads were taken at 25 °C in nonbinding all black clear-bottomed Greiner 96-well plates covered with optically clear films and stirred for 10 s prior to each reading. ThT fluorescence was measured at 444 and 491 nm (excitation and emission wavelengths, respectively). Each kinetic assay consisted of final concentrations of 30 μM amylin and 10 μM ThT. The amount of time required reaching the half-maximum ThT intensity (t1/2) and inhibitory concentration (IC50) values for dose–response curves were estimated by multiparameter logistic nonlinear regression analysis. The transition from the lag-phase to the growth phase was estimated as when the first measurable ThT fluorescence/time (slope) value exceeded ≥5-fold of the previous measured slope. All experiments were repeated three times using peptide stock solutions from the same lot. The natural product library used was as described.39 The NIH Clinical Collection (NIHCC) library contains a total of 700 FDA-approved drugs and investigational compounds with diverse chemical structures. The library was supplied in 96-well plates in DMSO. For screening, a 384-well plate was used. A Bravo liquid handler with a 96-channel disposable tip head (Agilent Technologies, Wilmington, DE) was used to aid sample transfer. The total volume in each well was 15 μL. Residual DMSO (<5.4%) in each well had a negligible effect on the fluorescent signals.
TEM Analysis
TEM images were obtained with a JEOL 1400 microscope operating at 120 kV. Samples consisting of 30 μM amylin (20 mM Tris-HCl, 2% DMSO, pH 7.4) in the presence of drug or vehicle control were incubated for ≥48 h at 37 °C with agitation. Prior to imaging, a small aliquot of each sample was blotted on a 200-mesh Formvar carbon-coated grid for 5 min and then stained with uranyl acetate (1%). Both sample and stain solutions were wicked dry (sample dried before addition of stain) with filter paper. Qualitative assessments of the amount of fibrils or oligomers observed were made by taking representative images following a careful survey of each grid.
Gel-Based Remodeling Assays
Vehicle control or specified compounds were spiked into freshly dissolved amylin samples (containing amylin and buffer). Thereafter, amylin aggregation was allowed to proceed for 3 days. The final amylin concentration was 15 μM, which included 45 μM compound (drug/amylin molar ratio was 3:1). After 3 days, these samples were vacuum-dried and redissolved in 6.5 M urea containing 15 mM Tris and 1× SDS Laemmeli sample buffer, boiled at 95 °C for 5–10 min, and subjected to SDS-PAGE followed by Western blot analysis with anti-amylin primary antibody (T-4157, 1:5000, Peninsula Laboratories, San Carlos, CA). All gel-based amyloid remodeling assays were repeated at least twice.
Western Blot and Dot Blot Analyses
Standard procedures were followed. For Western blots, either nitrocellulose or PVDF membranes were used. For dot blots, grids were drawn by pencil on nitrocellulose membrane to indicate the blotting regions. Samples (2 μL) were spotted onto the nitrocellulose membrane at the center of each grid, and the membrane was allowed to dry. For both blotting analyses, the membranes were blocked with 5% nonfat dry milk in PBS-T buffer (0.1% Tween-20 in PBS) for 1 h followed by overnight incubation at 4 °C with primary antibody. After three washes with PBS-T buffer, the membrane was incubated with secondary antibody conjugated with HRP for 1 h at room temperature. The membranes were incubated with Amersham ECL reagents (GE Life Sciences) for signal development after PBS-T and PBS washes. Densitometry was used for quantifying Western bands where applicable.
Animal Studies
HIP rats (RIP-HAT) and wild-type Sprague–Dawley (SD) control rats were purchased from Charles River Laboratories (Wilmington, MA). Dietary supplementation approach was used to study the in vivo effects of RA on HIP rats amylin amyloid islet deposition, serum oligomer formation, and animal diabetic pathology. Dietary supplemental treatments were initiated when HIP rats were at prediabetic stage (5–6 months old), and the preventative treatment lasted for 4 months. Diet supplementation with RA (at a dosage of 0.5% w/w) was prepared by Envigo Inc. (Madison, WI). Bulk RA was purchased from Toronto Research Chemicals, and its identity and purity were further validated at Virginia Tech Mass Spectrometry Incubator Facilities. Standard rodent chow diet (without RA incorporation) was used for the untreated HIP rats and wild-type SD control rats groups. All animals were housed in temperature- (23 ± 2 °C) and light-controlled, pathogen-free animal quarters and were provided ad libitum access to diet. Animal protocol was approved by institutional IACUC committee. Sera samples for glucose and insulin analysis and amylin oligomer Western blotting analysis were collected from the tail veins of the rats. Glucose levels were measured with an Agamatrix presto meter (AgaMatrix Inc., Salem, NH). Insulin levels were detected and quantified by a rat insulin ELISA kit (Mercodia AB, Uppsala, Sweden).
Islet Immunohistochemistry and Amyloid Staining
At the end of animal studies, rats were euthanized, and the pancreas tissues were dissected and fixed in 4% (v/v) formaldehyde buffer (pH 7.2). Pancreas samples were embedded in paraffin and sectioned by AML Laboratories Inc. (St. Augustine, FL). A series of tissue sections (5 μm thickness at 200 μm interval) were prepared, mounted on glass slides, and stained with Congo Red dye (Sigma-Aldrich, St. Louis, MO) or with rabbit anti-amylin primary antibody (T-4157, Peninsula Laboratories, San Carlos, CA) followed by a rabbit ImmPRESS HRP anti-rabbit IgG (peroxidase) polymer detection kit and Vector NovaRED substrate kit (Vector Laboratories). Images were visualized by microscopy (Zeiss Axio Observer, Thornwood, NY) or Nikon Eclipase Ti–U and were analyzed with Zeiss 2 (blue edition) or NIS-Elements AR 411.00.
Statistical Analysis
All data are presented as the mean ± SEM, and the differences were analyzed with a one-way analysis of variance followed by Holm–Sidak’s multiple comparisons (amylin kinetics) or unpaired Student’s t-test. These tests were implemented within GraphPad Prism software (version 6.0). The p-values of <0.05 were considered significant.
Molecular Dynamics (MD) Simulations
Starting Structure
In order to simulate trimer formation of amylin in a reasonable time scale using MD simulations, the principal amyloidogenic region (residues 20–29) was extracted from an equilibrated MD structure of full length (residues 1–37) human amylin (Protein Data Bank (PDB) ID 2L86).44 This PDB structure was chosen to best represent biologically active, monomeric amylin given the presence of the disulfide bound between Cys2–Cys7 and C-terminal amidation. However, 2L86 was solved in an SDS micelle environment, and to better mimic experimental procedures, we sought to use a structure of amylin that mimicked solution conditions (water and 150 mM NaCl). Simulations of full-length amylin were constructed by placing the structure of 2L86 in a cubic box with a minimum solute–box distance of 1.0 nm. The SPC water model was used to solvate the box,45 and Na+ and Cl– ions were added to reach a final concentration of 150 mM NaCl and maintain a net-neutral system. All simulations were done using the GROMACS software package, version 4.6.0,46,47 and the GROMOS53a6 force field.48 Energy minimization was carried out using the steepest descent method, with all protein heavy atoms being restrained during equilibration and later released during MD simulation. Equilibration was carried out in two sequential steps, NVT and NPT. Three replicates starting with different random starting velocities in NVT were carried out. NVT was carried out for 100 ps and maintained at 310 K using the Berendsen weak coupling method.49 Following NVT, NPT dynamics analysis using a Nosé–Hoover thermostat50,51 and a Parrinello–Rahman barostat52,53 to maintain temperature (310 K) and pressure (1 bar) was carried out for 100 ps. Following equilibration, MD simulations were run using three-dimensional periodic boundary conditions, with short-range cutoffs of 1.4 nm being applied to all nonbonded interactions. Long-range interactions were calculated using the smooth particle mesh Ewald (PME method)54,55 using cubic interpolation and Fourier grid spacing of 0.16 nm. Bond lengths were constrained by P-LINCS56 using an integration time step of 0.2 fs. Simulations were run until the criteria of convergence was met, which consisted of stabilization of 100 ns for both backbone root-mean-square deviation (RMSD) and protein secondary structure, as determined by the DSSP algorithm.57 RMSD clustering was used to produce a representative structure of the last 100 ns of each replicate.58 The center structure of the replicate cluster structure whose average secondary structure was closest to the set average was chosen as the template for simulations of trimer formation of residues 20–29 of amylin (amylin(20–29)).
Trimer Formation
Amylin(20–29) was extracted from the representative structure generated by MD simulations of full-length amylin in water and 150 mM NaCl. This was done in order to have native, monomeric fragments of this region rather than the preformed fibrils. Acetyl and amide groups were added to cap the ends of the 10-residue fragment to negate spurious effects from having charges at the termini. The sequence simulated of the amylin(20–29) fragment is as follows: Ace-SNNFGAILSS-NH2. Three amylin(20–29) fragments were randomly placed at least 2.0 nm away from each other in a cubic box with dimensions of 11 × 11 × 11 nm3, with a solute–box distance of 1.0 nm. For control simulations, no small molecules were added to the system. For simulations with RA, CA, and SAA, five molecules were randomly placed in the system at least 2.0 nm away from the amylin(20–29) fragments and other small molecules. Topologies for RA, CA, and SAA were generated using the PRODRG2 server59 and refined by using charges and atom types found in functional groups within the GROMOS53a6 force field. For the ester functional group, parameters were taken from literature60 and modified to account for the double bond in the “R” position. Parameters and charges for all small molecules used in this study are available upon request. RA, CA, and SAA were deprotonated to mimic their protonation state at pH 7.4 (net charge −1). The following systems were generated and are identified in the following way: Control: 3 amylin(20–29) fragments and no small molecules; RA: 3 amylin(20–29) fragments and five molecules of RA; CA: 3 amylin(20–29) fragments and five molecules of CA; and SAA: 3 amylin(20–29) fragments and five molecules of SAA. These systems were then equilibrated and were run in MD simulations in the same way as those for full-length amylin. Simulations were run to 600 ns, which was found to be an adequate amount of time for trimer formation and β-strand structure to remain stabilized in the control simulations. Backbone RMSD and secondary structure analyses were also used to determine convergence. In total, simulations were carried out for 600 ns, for three replicates for each system, totaling 7.2 μs. Analysis was carried out over the last 100 ns of converged simulation, totaling 300 ns of aggregate sample time per system and 1.2 μs of simulation data that was analyzed. Analysis was carried out using programs in the GROMACS package and over the last 100 ns of simulation time (e.g., 500–600 ns) in order to report on stabilized trimer structures or what their corresponding trimer structure looks like in the presence of small molecules. RMSD clustering with a cutoff of 0.2 nm was used to generate the structures. PyMOL was used for molecular visualization and statistical analysis was done using a two-tailed t-test, with significance determined if p < 0.05.
Results
Identification of Rosmarinic Acid
We previously screened a NIHCC drug-repurposing library of drugs and investigational compounds of diverse chemical structures and discovered that catechol-containing compounds are a broad class of amyloid inhibitors.21 On the basis of this finding, we further rapidly screened a small library of natural compounds (∼100 compounds) that are enriched in catechol-containing compounds used in complementary medicine for their antidiabetic, anti-inflammatory, or neuroprotective effects. Epigallocatechin gallate (EGCG) was used as a positive control as it is extensively reported in literature for its strong antiamyloid effects (Figure 1A,B).61−65 Several other compounds including morin, baicalein, salvianiolic acid B, resveratrol, CA, and oleuropein were confirmed in our screen for their inhibitory effects against amylin amyloid formation (Figure 1A).39,40,66−69 RA was discovered to be one of the highest-ranking inhibitors in blocking amylin amyloid formation (Figure 1A). To minimize potential false positives from ThT fluorescence-based primary screening,67,70 we further validated RA in multiple secondary assays. Using TEM analysis, we found that RA significantly reduced amyloid fibril formation (Figure 1B; estimated to be 50%), while at the same time enhancing the formation of nontoxic aggregates or amorphous aggregates at a 3:1 drug/amylin molar ratio (Figure 1B). This potency is similar to the control compound EGCG. On the basis of ThT fluorescence assay, we further determined that the inhibitory concentration IC50 for RA is 200–300 nM, several folds stronger than that of EGCG (estimated to be 1 μM) (Figure 2A). Consistent with these data, RA also neutralized amylin-amyloid-induced cytotoxicity in pancreatic INS-1 cells and neuronal Neuro2A cells (Figure 2B). As a control experiment, RA itself has no effects on cell viability for both INS-1 cells and Neuro2A cells (Figures 2B and S2). RA further significantly delayed amylin amyloid formation. This was evidenced by RA causing a dose-dependent kinetic delay in the observed t1/2 values (Figure 2C).
Figure 1.

Identification of RA as a potent amylin amyloid inhibitor. (A) Identification of RA as strong amylin amyloid inhibitor from a natural product library enriched with catechol-containing compounds, a class of broad amyloid inhibitors we recently discovered, using a ThT fluorescence-based screening. EGCG was used as a positive control. Amylin concentration was 5 μM, and the molar ratio of compound to amylin was 2:1. Chemical structure of RA is shown. (B) TEM images of human amylin amyloid with and without inhibitor treatment (1:20 amylin/drug molar ratio). Mature fibrils and amorphous aggregates are indicated by the red and blue arrows, respectively. Buffer treatment sample served as control (100% mature fibril). RA treatment resulted in an estimated 50% mature fibrils and 50% amorphous aggregates, whereas treatment with EGCG, a known strong inhibitor, resulted in similar partial fibrils and partial amorphous aggregates.
Figure 2.

Characterizations of RA as a strong amylin amyloid inhibitor. (A) Amylin–ThT fluorescence-based inhibition assay. RA is estimated to have an IC50 between 200 and 300 nM. IC50 curve for control EGCG is also shown (1 μM) as a comparison. (B) Neutralization of amylin-amyloid-induced cytotoxicity by RA in pancreatic INS-1 cells and neuronal Neuro2A cells. Amylin concentration was 3.75 μM, and the ratio of inhibitor/amylin was 5:1. RA significantly detoxified amylin-amyloid-induced cytotoxicity in both cell lines, as indicated by asterisks (p < 0.05). (C) RA-induced dose-dependent kinetic delays in amyloid formation. Left panel shows the measurements of t1/2 of amylin amyloid formation by amylin–ThT fluorescence based assays, as indicated by the arrows. Concentration of amylin was 10 μM. Time points for t1/2 are shown as arrowed. At least three repeats have been carried out. The right panel shows corresponding quantification and display in bar graph format of t1/2 values with each treatment. With increased concentration of RA treatment, t1/2 becomes significantly delayed (**, p < 0.05; ***, p < 0.01; ****, p < 0.001).
Additive Effects of Rosmarinic Acid
Because RA consists of two catechol-containing components, CA and SAA, with a link of an ester bond (Figure 3A), we hypothesized that each component, CA and SAA, would also inhibit amyloid formation and that their added effects would be similar to that of RA. To test this hypothesis, we first carried out ThT fluorescence assays, in which CA and SAA both showed significant amyloid inhibition activities, but neither as potently as RA. A mixture of CA and SAA at a 1:1 molar ratio showed inhibitory activity similar to that of RA (Figure 3A). We further quantitatively determined the IC50 values for RA, CA, and SAA as 200–300 nM, 7.2 μM, and 90 μM, respectively (Figure 3B). We also investigated the remodeling capacities of RA, CA, SAA, and a mixture of CA and SAA after the ThT fluorescent signal of amylin amyloid reached a plateau. Rapid drops in ThT signals were observed when preformed amylin amyloids were spiked with RA, CA, SAA, and CA+SAA (a mixture of CA and SAA in an equimolar ratio; Figure 3C). The relative amyloid remodeling potencies depicted by these data are consistent with the antiamyloid additive effects demonstrated by RA, CA, and SAA (Figure 3A). This additive effect was also manifested in amylin-amyloid-induced cytotoxicity rescue assays. At the inhibitor/amylin ratio of 3:1 (amylin concentration of 15 μM), RA fully neutralized amylin-induced neurotoxicity, while CA and SAA only partially rescued the cell viability (Figure 3D).
Figure 3.

Additive effects of RA components in amylin amyloid inhibition. (A) ThT fluorescence-based amyloid inhibition assay by RA and its hydrolytic components CA and SAA. Inhibition from a 1:1 molar ratio mixture of CA and SAA is similar to that of RA, but significantly stronger than those from CA or SAA alone. (B) IC50 measurements by ThT fluorescence based inhibition assays. IC50 is estimated to be 200–300 nM for RA, 7.2 μM for CA, and 90 μM for SAA. (C) ThT fluorescence-based amyloid remodeling assay shows amyloid remodeling after spiking of buffer control, RA, CA, SAA, and CA+SAA (1:1 molar ratio) after aggregation reaches a plateau (t = 17 h). (D) Neutralization of amyloid-induced toxicity by RA, CA, and SAA in Neuro2A cells. Amylin concentration was 2.5 μM, and the ratio of the inhibitor/amylin is 5:1. Compared to the control, RA, and to lesser degrees, CA and SAA, significantly neutralize amyloid-induced toxicity. *, p < 0.05; **, p < 0.01.
In parallel with biochemical and cell biology evaluation, we carried out molecular dynamics (MD) simulation studies to test the additive effect hypothesis. Simulations were carried out using fragments of amylin consisting of residues 20–29; this fragment has been shown to be the core sequence that drives fibril formation.10,71 We first confirmed that these fragments form distinct β-sheet structures during MD simulation that mimic experimental nuclear magnetic resonance studies (PDB ID: 2KIB). We carried out simulations using three human amylin (20–29) fragments in the absence of any inhibitor and in the presence of RA, CA, or SAA (5 molecules inhibitor/system). Significantly less β-strand structure was observed in the simulations in the presence of RA (28 ± 11%) in comparison with that of the no-drug control (58 ± 11%). The amount of β-strand structure was also reduced in the presence of CA (37 ± 17%) and SAA (45 ± 20%), though not to the extent of RA (Table S1). Therefore, the MD studies showed collaborative results in terms of effectiveness in preventing aggregate formation as the experimental studies. Furthermore, analysis of interpeptide interactions revealed a reduction between peptide fragments in the presence of RA, CA, and SAA as evaluated from the average hydrogen bond presence (Table S2). Consistently, the reduction of interactions was more pronounced in the case of RA than with both CA and SAA, as represented by the nondiagonal peaks in the peptide fragment interaction “heat maps” (Figure 4), which additionally show a reduction of parallel and antiparallel β-strand structures. Reduction of amylin fragment β-strand stacking is also evidenced by increased solvent accessible surface areas of amylin fragments in the presence of RA, CA, or SAA (Table S3). It is hypothesized that RA is disrupting the peptide–peptide hydrogen bond network and contributing to the observed decrease in β-strand structure as compared to that of the control simulations. This is in agreement with previous literature,72 which highlights the ability of hydroxyl groups on polyphenols to disrupt the stabilization of amylin oligomers. Collectively, these molecular simulation studies provided atomic-level mechanistic insights into the interactions between RA, CA, SAA and the amyloidogenic fragment (residues 20–29) of amylin. These findings also were consistent with our experimental results that demonstrated the additive effect of RA described above. Additionally, these MD studies provided us with a model system to study amylin aggregation and the effect of small molecules on these pathways, as well as a correlate at the atomistic level compared to assays carried out in this study.
Figure 4.

Additive effects of RA components in amylin amyloid inhibition with MD simulation studies. Dominant morphologies of amylin20–29 trimer formation (A) with and without the presence of RA (B), CA (C), and SAA (D) shown in the upper portions of each panel. Individual amylin20–29 peptide fragments are shown in cartoon and colored red (peptide 1), orange (peptide 2), and yellow (peptide 3). RA, CA, and SAA are shown as gray sticks, colored by element. For the panels of CA (C) and SAA (D), only CA and SAA molecules within 1.0 nm of the amylin20–29 trimer are shown for clarity. Significantly less β-structure is observed in the presence of RA (B), and a reduction in β-structure is observed in the cases of CA (C) and SAA (D). Residue–residue interaction plots for corresponding drug treatment are shown as “heat” maps in the lower portions of each panel. x- and y-axis labels are in peptide fragment length (residues 20–29), with each repeating number (e.g., 20) being a separate peptide. Significantly less intermolecular interactions between amylin fragments is observed in the presence of RA in comparison with the control. Reduction in interactions is observed in the cases of CA and SAA treatment.
Inhibitor-Induced Amyloid Remodeling
RA demonstrated significant amyloid remodeling function similar to EGCG in orthogonal ThT fluorescence assays and gel-based remodeling assays (Figure 5).61,73,74 Such similarity was also seen in the morphology of the remodeled products under TEM (Figure 1B): Formation of amorphous aggregates described in the literature as “soft and disordered clusters” that are structurally distinct from untreated regular rigid, long fibrils.75 Importantly, RA neutralized amylin-amyloid-induced cytotoxicity (Figure 3D) and itself had no significant effects on cell viability for a variety of cell lines (Figure S2). RA remodeled amylin amyloid into nontoxic aggregates, presumably via off-pathway channels as exemplified by EGCG.61,62,73,74 We found that mixing RA with amylin amyloid led to sodium dodecyl sulfate (SDS)-resistant, remodeled aggregates spanning a broad range of molecular weight that are nontoxic, as has been observed with EGCG in the literature (Figure 5B,C).74 Remodeled molecular species were experimentally captured as quickly as 45 min (boxed in the red rectangle; Figure 5B). Amylin amyloid remodeling may also be shown in ThT fluorescence assays. Spiking RA or EGCG rapidly remodeled the preformed amylin amyloid (Figure 5A). The same general steep drop in ThT fluorescence was observed when RA and EGCG were spiked at the plateau phase of amyloid formation (t = 17 h) and after mature fibrils were formed (t = 3 days). No significant fluorescent signals for amyloid formation were observed when these compounds were spiked in at t = 0 or before the amyloid growth phase (boxed in blue rectangle; Figure 5A).
Figure 5.

Amylin amyloid remodeling induced by RA as probed by ThT fluorescence-based assays and orthogonal gel-based assays. (A) ThT fluorescence-based assay shows amylin amyloid remodeling after spiking of buffer control, RA, and EGCG before amylin amyloid is formed (t = 0 h), after amyloid has been formed (t = 17 h) or mature fibrils are formed (t = 3 days). (B) Single-fraction analysis of inhibitor-induced amyloid remodeling. Incubation of amylin amyloid with buffer control, EGCG, and RA leads to broad molecular weight range, SDS-resistant remodeled protein aggregates. Samples were separated by SDS-PAGE and probed by an antibody specific for human amylin (T-4157). The urea-dissolved, broadly distributed molecular weight of amylin-inhibitor aggregates were observed, while only monomer (indicated as “m”) and dimer bands (indicated as “d”) of amylin were seen in untreated samples. While with the quenching with denaturing conditions (6.5 M urea), both EGCG and RA induced broad molecular weight aggregates. SDS-resistant aggregates may be captured starting from 45 min of incubation (boxed in the red rectangle). (C) “Two-fractions” experiments demonstrated that EGCG and RA interact with monomeric amylin to form both soluble and insoluble aggregates of a broad molecular weight range, but such interactions with preformed fibrils lead to only insoluble aggregates. The experimental settings were the same as those described in panel B, except that the pellet and the supernatant fractions (two fractions) of each sample were separated by ultracentrifugation before the samples were separated by SDS-PAGE and probed by an antibody specific for human amylin (T-4157). Spiking of buffer control, EGCG, and RA was initiated either at 0 h (left panel) or after mature fibrils were formed (t = 3 days; right panel) as depicted in the schemes drawn on the left of each panel.
The mechanisms of remodeling by small-molecule inhibitors and the physical basis of the related species changes and molecular interactions are not fully understood. The rapid drop in ThT fluorescence has been proposed to reflect disaggregation of insoluble mature amyloid fibrils in some cases.62 In other cases, this signal drop was proposed to reflect ThT binding site competition, altered stability, and seeding capacity of the remodeled fibril masses but not their solubility.74
Next, we used a “two-fraction” gel-based remodeling assay to assess if potential remodeling activities against preformed mature amyloid fibrils would result in their dissolution or disaggregation of fibrils to monomers. Herein, we spiked RA or EGCG with either freshly dissolved, unaggregated amylin or with preformed mature amyloid fibrils, and after 3 days (sufficient time for mature fibril formation), we analyzed the insoluble and soluble fractions from both conditions by Western blotting analysis using amylin-specific antibody T-4157 (Figure 5C). As expected for amyloid inhibitors, all compounds maintained a significant amount of peptide mass within the soluble fraction (highlighted in the red rectangle; Figure 5C left panel) when spiked into solutions initially containing freshly dissolved unaggregated amylin (i.e., a significant amount of insoluble amyloid formation was prevented). Moreover, both RA and EGCG induced amyloid remodeling toward denaturant-resistant aggregates, whether compounds were spiked into solutions initially containing unaggregated amylin or preformed amylin fibrils (Figure 5C). However, no compound was able to dissolve preformed amyloid fibrils nor did any compound disaggregate fibrils to amylin monomers, as reflected by the absence of any detectable amylin mass or monomeric amylin band in the soluble fraction of amylin fibril samples treated with the compounds (highlighted in the red rectangle; Figure 5C, right panel). Our data demonstrated that amylin amyloid remodeling by RA/EGCG is not simply the reverse pathway of amyloid formation; remodeling uses a pathway different from disaggregation of fibrils to monomers. Furthermore, both RA- and EGCG-induced amyloid remodeling showed differential solubility effects of remodeled off-pathway aggregates that is amyloid formation stage-dependent: EGCG and RA interact with monomeric amylin to form both soluble and insoluble aggregates of a broad molecular weight range, but such that interactions with preformed fibrils lead to only insoluble aggregates (Figure 5C), suggesting irreversibility once mature fibrils are formed. Conceivably, fibril formation may conceal certain RA interaction sites. To our knowledge, such differential solubility effects of inhibitor-remodeled aggregates have not been reported. This finding may have significant implication practically in selecting effective inhibitors to reduce pre-existing fibrils/plaques for therapeutic development for amyloid disease treatment.
Ex Vivo Efficacies of RA
Because rodent amylin is not amyloidogenic,1,21 a “humanized” diabetic rat model (HIP rats), which overexpresses human amylin, has been established for mechanistic and translational applications.18,76 Past studies have confirmed that sera samples from HIP rats contain preformed amylin oligomers.18,20 Thus, as a step toward testing the in vivo efficacy studies of RA, we obtained sera from HIP rats and then treated them with either RA or vehicle buffer control. These samples were then analyzed via Western blots using amylin-specific antibody T-4157. In contrast to vehicle-loaded controls, RA effectively reduced amylin oligomers (estimated to be 72, 84, and 96 kDa) in a dose-dependent manner (Figure 6A). Amylin oligomers of similar sizes from HIP rats have been reported in the literature.18 Further dot blot analyses showed that RA efficacies were most apparent to the fibrillar oligomer species recognized by OC antibody (Figure S3).
Figure 6.

Ex vivo experiments demonstrate that RA significantly reduces human amylin oligomers in the sera samples from HIP rats and diabetic patients. (A) Western blot analyses of HIP rat sera treated with increased concentrations of RA as indicated. Samples were incubated with RA at indicated concentrations for 3 h at 37 °C. Equal amount of sera were loaded in each well as demonstrated by equal intensities of Western blot bands of control GAPDH. Lanes labeled as 0 μM are the no-treatment controls. Anti-human-amylin blots were probed with amylin-specific antibody (T-4157). Estimated sizes of ∼72, ∼84, and ∼96 kDa of human amylin oligomers are indicated. Quantification of the Western blot results by densitometry are shown in the lower panel. (B) Western blot analyses of diabetic patient sera samples treated with 50 μM RA as indicated over a time course of 0, 2, 8, and 24 h. Equal amounts of sera were loaded in each well as demonstrated by equal intensities of Western blot bands of control GAPDH. Anti-human-amylin blots were probed with amylin-specific antibody (T-4157). Estimated size of the human amylin oligomer (∼52 kDa) is indicated. Western blot results were quantified by densitometry, shown in the lower panel.
To test whether RA has therapeutic potential for future usage in humans, we investigated RA with sera from diabetic patients, as amylin oligomers had been detected in such clinical samples.77 We showed that RA effectively reduced amylin oligomers (estimated to be 52 kDa) in sera from diabetic patients at a treatment concentration of 50 μM (Figure 6B). As expected, reduction of amylin oligomer band intensity from diabetic patient sera was shown to be incubation-time-dependent. The difference in molecular weights of the amylin oligomers detected between HIP rats and patient sera samples may be due to variation of amylin oligomer size in different hosts and/or different degrees of post-translational modifications. Overall, RA was effective at the incubation time range of 3–24 h and at concentration ranges of 10–50 μM in sera from both HIP rats and diabetic patients.
In Vivo Efficacy Studies of RA
HIP rats had been established as an excellent model to study amylin-amyloid-induced islet pathology and to test novel approaches to the prevention and treatment of T2D and related complications.18,20,22 Hemizygous HIP rats spontaneously developed midlife diabetes (6–12 month of age) associated with islet amyloids.76 To test RA in reducing amylin amyloid oligomer formation in sera, pancreatic amyloid deposition, and in ameliorating islet diabetic pathology, we carried out in vivo efficacy tests in hemizygous HIP rats with RA treatment via diet supplementation at a dose of 0.5% (w/w). The dose was chosen based on literature where several phenolic compounds (RA included) were tested in against Aβ amyloids in Tg2576 mice and showed in vivo efficacies.78 The dietary treatment was initiated when HIP rats were at prediabetic stage (at the age of 6 month),76 and the treatment lasted for 4 months. Due to significantly delayed phenotype in female rats, only male HIP rats were used.24 While EGCG is an excellent control for in vitro biochemical studies, it has not shown in vivo efficacy and failed in a recent clinical trial against α-synuclein aggregation (multiple system atrophy).79 Therefore, EGCG was not chosen to be included in our animal studies.
Our results demonstrated that RA showed highly effective antiamyloid formation and antidiabetic activities in vivo that were consistent with its in vitro activities described in this work: (1) As expected, diabetic HIP rats (10–12 months old) showed severe amylin amyloid deposition in the pancreatic islets as visualized by Congo Red staining (Figure 7D, middle panel). The extensive amyloid islet deposition was reminiscent of a clinical thioflavin S staining of the islets from a human with noninsulin dependent diabetes mellitus or T2D.14 In contrast, wild-type age-matched Sprague–Dawley (SD) rats showed no amyloid deposition in the islets (Figure 7D, left panel). Amylin amyloid deposition in islets was significantly reduced in RA-treated, age-matched HIP rats (Figure 7D, right panel). However, residual amyloid deposition was evident, suggesting that RA treatment regimen did not completely block amylin aggregation and deposition on the pancreas. (2) Immunohistochemistry analysis of islet tissues provided collaborative evidence that amylin deposition levels were significantly higher in the islets of untreated HIP rats in comparison with those of wild-type SD rats and RA-treated HIP rats (Figure 7E). (3) We also evaluated sera amylin oligomer levels by Western blots using amylin-specific antibody T-4157. In contrast to wild-type SD rat controls that showed insignificant levels of amylin oligomers, untreated HIP rats showed strong intensities of amylin oligomers (estimated to be 32 and 52 kDa, respectively). With RA treatment, corresponding oligomer levels were either significantly reduced (52 kDa bands) or fully abrogated (32 kDa bands) (Figure 7C). Amylin oligomers of similar sizes from HIP rats have been reported in the literature.18 (4) We further evaluated diabetic conditions of each cohort of rats. Wild-type SD rats have an average nonfasting blood glucose level of 146.8 mg/dL. Untreated HIP rats have an average nonfasting blood level of 242.3 mg/dL that is evidently diabetic. Such spontaneous diabetes development in HIP rats and hyperglycemia are consistent with what have been reported.18,76 Noticeably, RA-treated HIP rats have a significantly reduced average nonfasting blood glucose level of 181.0 mg/dL (Figure 7A). Consistently, serum insulin levels were significantly lower in untreated HIP rats than those in wild-type SD rats and RA-treated HIP rats at the age of 10 months (Figure 7B). Hypoinsulinemia state of HIP rats at the age of 10 months had been reported.80 We do not find any significant differences between RA-treated versus nontreatment HIP groups on body weight and food intake (data not shown).
Figure 7.

In vivo experiments demonstrate that RA potently reduced amylin amyloid islet deposition and ameliorated diabetic pathology in HIP rats. Prediabetic HIP rats (6 month old) were treated (HIP-T) or untreated (HIP) with 0.5% w/w RA in regular chow diet for 4 months. Age-matched wild-type Sprague–Dawley rats with regular chow diet served as negative controls. At the end of the treatments, rats were euthanized, and pancreatic tissues and sera were collected for immunohistochemistry and biochemical analysis. (A) Serum glucose levels were quantified for each group of rats. Significant reduction of serum glucose concentration was observed, indicated by asterisks (p < 0.05) comparing the untreated HIP rats with those of RA-treated HIP rats and control SD rats. (B) Insulin levels were measured for each group of rats (n = 4). Statistically lower levels of insulin were observed and indicated by asterisks (p < 0.05) comparing the untreated HIP rats with those of RA-treated HIP rats and control SD rats. (C) Western blot analysis of serum amylin oligomer levels with amylin-specific antibody (T-4157). GAPDH served as loading control. Compared with the untreated HIP rat sera, a significant reduction of the 52 kDa amylin oligomer bands and fully abrogated 32 kDa oligomer bands were observed with RA-treated HIP sera. There were essentially no intensities of these oligomer bands in control SD rat sera. (D) Representative Congo red staining demonstrated strong amyloid staining in untreated HIP rat pancreatic tissue slices (orange-red color in the islets, middle panel). Congo red staining was significantly reduced in the islets of RA-treated HIP rats (right panel). No Congo red staining was observed in islets in control wild-type SD rats (left panel). All pancreatic islets are marked by red asterisks. These qualitative results represent multiple samples from RA-treated or untreated HIP rats or WT Sprague–Dawley controls (n = 4), and at least 50 islets from pancreatic head, body, and tail for each group were examined. Scale bar in each panel is 100 μm. (E) Representative amylin deposition in the pancreatic tissue slice using amylin antibody staining from different rat groups. Pancreatic tissue slices were immune-stained with amylin-specific antibody (T-4157) followed by secondary antibody and NovaRED substrate (Vector Lab) treatment. Stained islets are shown in dark red. Islets in the pancreatic slices from untreated HIP rats were intensely stained, whereas those from RA-treated and control SD rats were only weakly stained. At least 10 tissue slices were observed from each group. Scale bar in each panel as indicated is 1 mm.
In summary, we demonstrated in vivo efficacy of RA in mitigating diabetic pathology and reducing amylin amyloid deposition in the pancreas of HIP rats. To our knowledge, this is the first report describing in vivo effectiveness of RA against amylin aggregation. We also discovered additive effects of its two active components, CA and SAA, on amylin fibrillation by a combination of biochemical, cell-based and computational approaches. We showed that RA does not reverse fibrillation back to monomeric amylin, but rather leads to nontoxic, remodeled protein aggregates.
Discussion
Several lines of research including ours have shown that catechol groups in individual catechol-containing polyphenols and flavonoids play key roles in amyloid inhibition.39,40,81,82 Extending from individual compounds, we recently demonstrated that catechols and redox-related quinones/anthraquinones are a broad class of amyloid inhibitors.21 The catechol moiety is a common structural component of many natural products, including scores of flavonoids and polyphenols, many of which were shown to have antidiabetic, neuroprotective, and antiaging activities.33,34,37,83 RA, as a natural product containing dual catechol components, provides an outstanding example supporting our theory that catechol-containing compounds are amyloid inhibitors by way of demonstrating mechanistic additive effects. Catechol-containing flavonoids and polyphenols, with the advantages of low cost and few side effects as natural products, may have significant potential in translational applications as disease-preventating nutraceuticals.
RA is a natural compound identified from our library screening. With rational design using medicinal chemistry, more potent synthetic RA analog inhibitors may be engineered. The additive effect of RA provided an opportunity for us to further improve inhibitor potencies via a hybrid strategy. Our past work has demonstrated that such a strategy is effective in providing more potent analogs from initial natural product leads.84,85 RA and its analog inhibitors will be useful not only for preclinical translational applications, but also as unique chemical biology probes in mechanistic studies, such as in understanding amylin-amyloid-induced islet pathogenesis.
Orally administrated RA may present as glucuronide- or sulfate-conjugated or methylated forms in serum and may be metabolized to conjugated forms of CA and ferulic acid (FA);86 thus, active entities in serum after RA administration may include free and conjugated RA, methyl-RA, CA, and FA. RA may have off-target effects or polypharmacology. For example, RA can interact with GABA transaminase and inhibit cyclooxygenase and acetylcholinesterase. While we did not observe any significant adverse effects in the HIP rat model, relevant biological responses may need to be evaluated when we design next-generation amylin amyloid inhibitors and proceed with expanded in vivo studies.
The mechanisms of amyloid inhibition by small molecules are not fully understood. This and our past work led to several testable hypotheses: Does amylin amyloid inhibition by RA and other catechol-containing compounds occur via the mechanism of autoxidized o-quinone intermediates? Does RA conjugate with amylin through amine-groups of Lys1 or Arg11 or the amino-terminal amine? We propose, as one potential mechanism, that the catechol functional groups in RA can be autoxidized to o-quinone intermediates and then covalently reacted with amylin to block amyloid growth.21,39 Quinones may be readily conjugated with the amine groups in Lys1 and/or Arg11 or at the amino terminal in human amylin and form Schiff base or Michael addition conjugates.39,82 We demonstrated such Schiff base conjugates in the case of another specific example of catechol-containing compound, baicalein.39 However, detection of such conjugates may be largely dependent on the conjugation efficiency of individual compounds. Such general covalent conjugation mechanisms, if validated, will have broad implications for both amyloid inhibitor design and mechanisms of inhibition for multiple amyloid systems in protein-misfolding diseases. Our work, however, does not exclude other mechanisms, such as noncovalent inhibition mechanisms as proposed in the literature.34,69,74
Using EGCG as a benchmark, we provided strong evidence that both RA and control compound EGCG “remodeled” human amylin oligomers to form nontoxic amorphous aggregates, which had the characteristic broad range molecular weight “smears” in SDS-PAGE gel-based experiments (Figure 5B,C). To further understand the mechanisms of inhibitor-induced amyloid remodeling, high-resolution characterizations of bound species, using approaches such as ion mobility spectrometry-mass spectrometry,87 are necessary to define the interactions between RA and amylin. At cellular and physiological levels, additional factors, such as lipid membranes, pH, transition metal ions, and insulin, may play significantly roles in amylin aggregation.41,65 For example, zinc ion (Zn2+), ordinarily chelating with insulin, which co-secretes with amylin, may boost EGCG’s amylin amyloid inhibition.41 Effects on transition metal ions such as Zn2+ on RA remain to be determined.
Amylin amyloid deposition in the pancreas is a well-recognized hallmark feature of T2D.1,14,15 Recently, amylin amyloid deposition was also found in the brains of T2D patients with dementia or Alzheimer’s disease, as well as in the brains of diabetic HIP animal models, raising the possibility that amylin amyloid may be a new amyloid in the brain.17,18,24,88,89 The exact mechanism of such diabetes-induced neurodegeneration remains elusive. However, validating this new hypothesis will be highly significant, as it opens a new field by identifying a specific molecular link between diabetes and neurodegeneration. Such validation will support the clinical and epidemiological observations that obese/diabetic individuals are more prone to dementia/neurodegeneration. RA, its analogues, or other effective amyloid inhibitors could provide useful chemical biology tools to validate this new hypothesis. It will be interesting to extend future investigations of amylin-amyloid-induced pathology from the pancreas and diabetes to the brain and neurodegeneration.
Acknowledgments
This work was supported in part by the Hatch Program of the National Institute of Food and Agriculture, USDA (B.X. and D.R.B.), Commonwealth Health Research Board Grant 208-01-16 (B.X., D.R.B., and S.Z.), NIH grants R03AG061531 (B.X.) and R01AG058673 (S.Z.), Diabetes Action Research and Education Foundation Grant (B.X.), Awards No. 16-1, 18-2 and 18-4 from the Commonwealth of Virginia’s Alzheimer’s and Related Diseases Research Award Fund (B.X., L.W., and S.Z.), Alzheimer’s Association/Michael J. Fox Foundation grant BAND-19-614848 (B.X.), and Alzheimer’s Drug Discovery Foundation 20150601 (S.Z.). We thank Prof. Florian Despa (University of Kentucky) for generously providing HIP rat and diabetic patient sera samples. We thank Ms. Kathy Lowe at Virginia-Maryland Regional College of Veterinary Medicine for her excellent technical assistance in collecting TEM data.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00028.
Average secondary structure of trimer fragment formation, average hydrogen bond presences, average total solvent-accessible surface areas, cytotoxicity of amylin amyloid on various cell lines, control experiments demonstrating that RA has no effects on cell viability, ex vivo and in vivo efficacies of RA by dot blot analyses (PDF)
Author Contributions
◆ L.W. and P.V. contributed equally to this work. L.W. and P.V. carried out the experiments and acquired and analyzed the data. A.M.B. carried out the molecular simulation studies and analyzed the data. Y.W. assisted with the diabetic animal studies and tissue analyses. D.L. contributed to the design of animal studies and diabetic animal characterization. D.R.B. designed the molecular simulation experiments and analyzed the data. S.Z. contributed to drug discovery experimental design. B.X. conceived, organized, designed the experiments, and analyzed the data. B.X., L.W., P.V., A.B., and D.R.B. wrote the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Westermark P.; Andersson A.; Westermark G. T. (2011) Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 91, 795–826. 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- Eisenberg D.; Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203. 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles T. P.; Vendruscolo M.; Dobson C. M. (2014) The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396. 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Goedert M. (2015) NEURODEGENERATION. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 349, 1255555. 10.1126/science.1255555. [DOI] [PubMed] [Google Scholar]
- Chiti F.; Dobson C. M. (2017) Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 86, 27–68. 10.1146/annurev-biochem-061516-045115. [DOI] [PubMed] [Google Scholar]
- Eisenberg D. S.; Sawaya M. R. (2017) Strucvtural Studies of Amyloid Proteins at the Molecular Level. Annu. Rev. Biochem. 86, 69–95. 10.1146/annurev-biochem-061516-045104. [DOI] [PubMed] [Google Scholar]
- Holmes B. B.; Furman J. L.; Mahan T. E.; Yamasaki T. R.; Mirbaha H.; Eades W. C.; Belaygorod L.; Cairns N. J.; Holtzman D. M.; Diamond M. I. (2014) Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. U. S. A. 111, E4376–4385. 10.1073/pnas.1411649111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieschke J.; Herbst M.; Wiglenda T.; Friedrich R. P.; Boeddrich A.; Schiele F.; Kleckers D.; Lopez del Amo J. M.; Grüning B. A.; Wang Q.; Schmidt M. R.; Lurz R.; Anwyl R.; Schnoegl S.; Fändrich M.; Frank R. F.; Reif B.; Günther S.; Walsh D. M.; Wanker E. E. (2012) Small-molecule conversion of toxic oligomers to nontoxic β-sheet-rich amyloid fibrils. Nat. Chem. Biol. 8, 93–101. 10.1038/nchembio.719. [DOI] [PubMed] [Google Scholar]
- Kayed R.; Lasagna-Reeves C. A. (2012) Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimer's Dis. 33 (1), S67–78. 10.3233/JAD-2012-129001. [DOI] [PubMed] [Google Scholar]
- Cao P.; Abedini A.; Raleigh D. P. (2013) Aggregation of islet amyloid polypeptide: from physical chemistry to cell biology. Curr. Opin. Struct. Biol. 23, 82–89. 10.1016/j.sbi.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A. N.; Leighton B.; Todd J. A.; Cockburn D.; Schofield P. N.; Sutton R.; Holt S.; Boyd Y.; Day A. J.; Foot E. A. (1989) Molecular and functional characterization of amylin, a peptide associated with type 2 diabetes mellitus. Proc. Natl. Acad. Sci. U. S. A. 86, 9662–9666. 10.1073/pnas.86.24.9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedini A.; Schmidt A. M. (2013) Mechanisms of islet amyloidosis toxicity in type 2 diabetes. FEBS Lett. 587, 1119–1127. 10.1016/j.febslet.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark P. (1972) Quantitative studies on amyloid in the islets of Langerhans. Upsala J. Med. Sci. 77, 91–94. 10.1517/03009734000000014. [DOI] [PubMed] [Google Scholar]
- Verchere C. B.; D’Alessio D. A.; Palmiter R. D.; Weir G. C.; Bonner-Weir S.; Baskin D. G.; Kahn S. E. (1996) Islet amyloid formation associated with hyperglycemia in transgenic mice with pancreatic beta cell expression of human islet amyloid polypeptide. Proc. Natl. Acad. Sci. U. S. A. 93, 3492–3496. 10.1073/pnas.93.8.3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höppener J. W.; Ahrén B.; Lips C. J. (2000) Islet amyloid and type 2 diabetes mellitus. N. Engl. J. Med. 343, 411–419. 10.1056/NEJM200008103430607. [DOI] [PubMed] [Google Scholar]
- Gong W.; Liu Z. H.; Zeng C. H.; Peng A.; Chen H. P.; Zhou H.; Li L. S. (2007) Amylin deposition in the kidney of patients with diabetic nephropathy. Kidney Int. 72, 213–218. 10.1038/sj.ki.5002305. [DOI] [PubMed] [Google Scholar]
- Jackson K.; Barisone G. A.; Diaz E.; Jin L. W.; DeCarli C.; Despa F. (2013) Amylin deposition in the brain: A second amyloid in Alzheimer disease?. Ann. Neurol. 74, 517–526. 10.1002/ana.23956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srodulski S.; Sharma S.; Bachstetter A. B.; Brelsfoard J. M.; Pascual C.; Xie X. S.; Saatman K. E.; Van Eldik L. J.; Despa F. (2014) Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol. Neurodegener. 9, 30. 10.1186/1750-1326-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma N.; Ly H.; Liu M.; Chen J.; Zhu H.; Chow M.; Hersh L. B.; Despa F. (2016) Intraneuronal Amylin Deposition, Peroxidative Membrane Injury and Increased IL-1β Synthesis in Brains of Alzheimer’s Disease Patients with Type-2 Diabetes and in Diabetic HIP Rats. J. Alzheimer's Dis. 53, 259–272. 10.3233/JAD-160047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S.; Margulies K. B.; Chen L.; Knowlton A. A.; Havel P. J.; Taegtmeyer H.; Bers D. M.; Despa F. (2012) Hyperamylinemia contributes to cardiac dysfunction in obesity and diabetes: a study in humans and rats. Circ. Res. 110, 598–608. 10.1161/CIRCRESAHA.111.258285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velander P., Wu L., Hildreth S. B., Vogelaar N. J., Mukhopadhyay B., Zhang S., Helm R. F., and Xu B. (October 13, 2020) Catechol-containing compounds are a broad class of amyloid inhibitors: redox state is a key determinant of the inhibitory activities. bioRxiv (Pharmacology and Toxicology), 2020.10.08.873620, ver. 3, DOI: 10.1101/2020.10.08.873620. [DOI] [PMC free article] [PubMed]
- Matveyenko A. V.; Butler P. C. (2006) Islet amyloid polypeptide (IAPP) transgenic rodents as models for type 2 diabetes. ILAR J. 47, 225–233. 10.1093/ilar.47.3.225. [DOI] [PubMed] [Google Scholar]
- Despa S.; Sharma S.; Harris T. R.; Dong H.; Li N.; Chiamvimonvat N.; Taegtmeyer H.; Margulies K. B.; Hammock B. D.; Despa F. (2014) Cardioprotection by controlling hyperamylinemia in a “humanized” diabetic rat model. J. Am. Heart Assoc. 3, e001015 10.1161/JAHA.114.001015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly H.; Verma N.; Wu F.; Liu M.; Saatman K. E.; Nelson P. T.; Slevin J. T.; Goldstein L. B.; Biessels G. J.; Despa F. (2017) Brain microvascular injury and white matter disease provoked by diabetes-associated hyperamylinemia. Ann. Neurol. 82, 208–222. 10.1002/ana.24992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R. (2015) Amyloid polymorphism: structural basis and neurobiological relevance. Neuron 86, 632–645. 10.1016/j.neuron.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick A. W. P.; Falcon B.; He S.; Murzin A. G.; Murshudov G.; Garringer H. J.; Crowther R. A.; Ghetti B.; Goedert M.; Scheres S. H. W. (2017) Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–90. 10.1038/nature23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krotee P.; Rodriguez J. A.; Sawaya M. R.; Cascio D.; Reyes F. E.; Shi D.; Hattne J.; Nannenga B. L.; Oskarsson M. E.; Philipp S.; Griner S.; Jiang L.; Glabe C. G.; Westermark G. T.; Gonen T.; Eisenberg D. S. (2017) Atomic structures of fibrillar segments of hIAPP suggest tightly mated β-sheets are important for cytotoxicity. eLife 6, e19273 10.7554/eLife.19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidler P. M.; Boyer D. R.; Rodriguez J. A.; Sawaya M. R.; Cascio D.; Murray K.; Gonen T.; Eisenberg D. S. (2018) Structure-based inhibitors of tau aggregation. Nat. Chem. 10, 170–176. 10.1038/nchem.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon B.; Zhang W.; Murzin A. G.; Murshudov G.; Garringer H. J.; Vidal R.; Crowther R. A.; Ghetti B.; Scheres S.; Goedert M. (2018) Structure of filaments from Pick’s disease reveal a novel tau protein fold. Nature 561, 137–140. 10.1038/s41586-018-0454-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L.; Liu C.; Leibly D.; Landau M.; Zhao M.; Hughes M. P.; Eisenberg D. S. (2013) Structure-based discovery of fiber-binding compounds that reduce the cytotoxicity of amyloid beta. eLife 2, e00857 10.7554/eLife.00857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele Y. S.; Monteiro C.; Fearns C.; Encalada S. E.; Wiseman R. L.; Powers E. T.; Kelly J. W. (2015) Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discovery 14, 759–780. 10.1038/nrd4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habchi J.; Chia S.; Limbocker R.; Mannini B.; Ahn M.; Perni M.; Hansson O.; Arosio P.; Kumita J. R.; Challa P. K.; Cohen S. I.; Linse S.; Dobson C. M.; Knowles T. P.; Vendruscolo M. (2017) Systematic development of small molecules to inhibit specific microscopic steps of Aβ42 aggregation in Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 114, E200–E208. 10.1073/pnas.1615613114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M.; Ono K.; Hamaguchi T.; Noguchi-Shinohara M. (2015) Natural Phenolic Compounds as Therapeutic and Preventive Agents for Cerebral Amyloidosis. Adv. Exp. Med. Biol. 863, 79–94. 10.1007/978-3-319-18365-7_4. [DOI] [PubMed] [Google Scholar]
- Velander P.; Wu L.; Henderson F.; Zhang S.; Bevan D. R.; Xu B. (2017) Natural product-based amyloid inhibitors. Biochem. Pharmacol. 139, 40–55. 10.1016/j.bcp.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K.; Hasegawa K.; Naiki H.; Yamada M. (2004) Curcumin has potent anti-amyloidogenic effects for Alzheimer’s beta-amyloid fibrils in vitro. J. Neurosci. Res. 75, 742–750. 10.1002/jnr.20025. [DOI] [PubMed] [Google Scholar]
- Rigacci S.; Guidotti V.; Bucciantini M.; Parri M.; Nediani C.; Cerbai E.; Stefani M.; Berti A. (2010) aglycon prevents cytotoxic amyloid aggregation of human amylin. J. Nutr. Biochem. 21, 726–735. 10.1016/j.jnutbio.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Ono K.; Li L.; Takamura Y.; Yoshiike Y.; Zhu L.; Han F.; Mao X.; Ikeda T.; Takasaki J.; Nishijo H.; Takashima A.; Teplow D. B.; Zagorski M. G.; Yamada M. (2012) Phenolic compounds prevent amyloid β-protein oligomerization and synaptic dysfunction by site-specific binding. J. Biol. Chem. 287, 14631–14643. 10.1074/jbc.M111.325456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardah M. T.; Paleologou K. E.; Lv G.; Menon S. A.; Abul Khair S. B.; Lu J. H.; Safieh-Garabedian B.; Al-Hayani A. A.; Eliezer D.; Li M.; El-Agnaf O. M. (2015) Ginsenoside Rb1 inhibits fibrillation and toxicity of alpha-synuclein and disaggregates preformed fibrils. Neurobiol. Dis. 74, 89–101. 10.1016/j.nbd.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velander P.; Wu L.; Ray W. K.; Helm R. F.; Xu B. (2016) Amylin Amyloid Inhibition by Flavonoid Baicalein: Key Roles of Its Vicinal Dihydroxyl Groups of the Catechol Moiety. Biochemistry 55, 4255–4258. 10.1021/acs.biochem.6b00578. [DOI] [PubMed] [Google Scholar]
- Wu L.; Velander P.; Liu D.; Xu B. (2017) Olive Component Oleuropein Promotes β-Cell Insulin Secretion and Protects β-Cells from Amylin Amyloid-Induced Cytotoxicity. Biochemistry 56, 5035–5039. 10.1021/acs.biochem.7b00199. [DOI] [PubMed] [Google Scholar]
- Lee Y.; Lin Y.; Cox S. J.; Kinoshita M.; Sahoo B. R.; Ivanova M.; Ramamoorthy A. (2019) Zinc boosts EGCG’s hIAPP amyloid inhibition both in solution and membrane. Biochim. Biophys. Acta, Proteins Proteomics 1867, 529–536. 10.1016/j.bbapap.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox S. J.; Rodriguez Camargo D. C.; Lee Y.; Dubini R. C. A.; Rovo P.; Ivanova M. I.; Padmini V.; Reif B.; Ramamoorthy A. (2020) Small molecule induced toxic human-IAPP species characterized by NMR. Chem. Commun. (Cambridge, U. K.) 56, 13129–13132. 10.1039/D0CC04803H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyung S.; Detoma A. S.; Brender J. R.; Lee S.; Vivekanandan S.; Kochi A.; Choi J.; Ramamoorthy A.; Ruotolo B. T.; Lim M. H. (2013) Insights into antiamyloidogenic properties of the green tea extract (−)-epigallocatechin-3-gallate toward metal-associated amyloid-I species. Proc. Natl. Acad. Sci. U. S. A. 110, 3743–3748. 10.1073/pnas.1220326110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanga R. P.; Brender J. R.; Vivekanandan S.; Ramamoorthy A. (2011) Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim. Biophys. Acta, Biomembr. 1808, 2337–2342. 10.1016/j.bbamem.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., and Hermans J. (1981) Interaction Models for Water in Relation to Protein Hydration, in Intermolecular Forces. The Jerusalem Symposia on Quantum Chemistry and Biochemistry (Pullman B., Ed.), pp 331–42, Springer, Dordrecht. [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. (2008) GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 4, 435–447. 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Pronk S.; Páll S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; van der Spoel D.; Hess B.; Lindahl E. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostenbrink C.; Villa A.; Mark A. E.; van Gunsteren W. F. (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 25, 1656–1676. 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Nosé S. (2002) A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 100, 191–198. 10.1080/00268970110089108. [DOI] [Google Scholar]
- Hoover W. G. (1985) Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A: At., Mol., Opt. Phys. 31, 1695–1697. 10.1103/PhysRevA.31.1695. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. (1981) Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190. 10.1063/1.328693. [DOI] [Google Scholar]
- Nosé S.; Klein M. L. (1983) Constant pressure molecular dynamics for molecular systems. Mol. Phys. 50, 1055–1076. 10.1080/00268978300102851. [DOI] [Google Scholar]
- Darden T.; York D.; Pedersen L. (1993) Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Hess B. (2008) P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 4, 116–122. 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- Kabsch W.; Sander C. (1983) Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Daura X.; Gademann K.; Jaun B.; Seebach D.; van Gunsteren W. F.; Mark A. E. (1999) Peptide Folding: When Simulation Meets Experiment. Angew. Chem., Int. Ed. 38, 236–240. . [DOI] [Google Scholar]
- Schuttelkopf A. W.; van Aalten D. M. (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 1355–1363. 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Horta B. A. C.; Fuchs P. F. J.; van Gunsteren W. F.; Hünenberger P. H. (2011) New Interaction Parameters for Oxygen Compounds in the GROMOS Force Field: Improved Pure-Liquid and Solvation Properties for Alcohols, Ethers, Aldehydes, Ketones, Carboxylic Acids, and Esters. J. Chem. Theory Comput. 7, 1016–1031. 10.1021/ct1006407. [DOI] [PubMed] [Google Scholar]
- Ehrnhoefer D. E.; Bieschke J.; Boeddrich A.; Herbst M.; Masino L.; Lurz R.; Engemann S.; Pastore A.; Wanker E. E. (2008) EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 15, 558–566. 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- Meng F.; Abedini A.; Plesner A.; Verchere C. B.; Raleigh D. P. (2010) The flavanol (−)-epigallocatechin 3-gallate inhibits amyloid formation by islet amyloid polypeptide, disaggregates amyloid fibrils, and protects cultured cells against IAPP-induced toxicity. Biochemistry 49, 8127–8133. 10.1021/bi100939a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pithadia A.; Brender J. R.; Fierke C. A.; Ramamoorthy A. (2016) Inhibition of IAPP Aggregation and Toxicity by Natural Products and Derivatives. J. Diabetes Res. 2016, 2046327. 10.1155/2016/2046327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovych N.; Brender J. R.; Soong R.; Vivekanandan S.; Hartman K.; Basrur V.; MacDonald P. M.; Ramamoorthy A. (2012) Site specific interaction of the polyphenol EGCG with the SEVI amyloid peptide PAP(248–286). J. Phys. Chem. B 116, 3650–3658. 10.1021/jp2121577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R.; Vivekanandan S.; Brender J. R.; Abe Y.; Naito A.; Ramamoorthy A. (2012) NMR characterization of monomeric and oligomeric conformations of human calcitonin and its interaction with EGCG. J. Mol. Biol. 416, 108–120. 10.1016/j.jmb.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B.; Liu X.; Gong H.; Huang L.; Chen H.; Zhang X.; Li C.; Yang M.; Ma B.; Jiao L.; Zheng L.; Huang K. (2011) Coffee components inhibit amyloid formation of human islet amyloid polypeptide in vitro: possible link between coffee consumption and diabetes mellitus. (2011). J. Agric. Food Chem. 59, 13147–13155. 10.1021/jf201702h. [DOI] [PubMed] [Google Scholar]
- Noor H.; Cao P.; Raleigh D. P. (2012) Morin hydrate inhibits amyloid formation by islet amyloid polypeptide and disaggregates amyloid fibers. Protein Sci. 21, 373–382. 10.1002/pro.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B.; Gong H.; Li X.; Sun Y.; Chen H.; Zhang X.; Wu Q.; Zheng L.; Huang K. (2013) Salvianolic acid B inhibits the amyloid formation of human islet amyloid polypeptide and protects pancreatic beta-cells against cytotoxicity. Proteins: Struct., Funct., Genet. 81, 613–621. 10.1002/prot.24216. [DOI] [PubMed] [Google Scholar]
- Tu L. H.; Young L. M.; Wong A. G.; Ashcroft A. E.; Radford S. E.; Raleigh D. P. (2015) Mutational analysis of the ability of resveratrol to inhibit amyloid formation by islet amyloid polypeptide: critical evaluation of the importance of aromatic-inhibitor and histidine-inhibitor interactions. Biochemistry 54, 666–676. 10.1021/bi501016r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y.; Brender J. R.; Hartman K.; Ramamoorthy A.; Marsh E. N. (2012) Alternative pathways of human islet amyloid polypeptide aggregation distinguished by 19F NMR-detected kinetics of monomer consumption. Biochemistry 51, 8154–8162. 10.1021/bi3012548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark P.; Engström U.; Johnson K. H.; Westermark G. T.; Betsholtz C. (1990) Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. U. S. A. 87, 5036–5040. 10.1073/pnas.87.13.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedumpully-Govindan P.; Kakinen A.; Pilkington E. H.; Davis T. P.; Chun Ke P.; Ding F. (2016) Stabilizing Off-pathway Oligomers by Polyphenol Nanoassemblies for IAPP Aggregation Inhibition. Sci. Rep. 6, 19463. 10.1038/srep19463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieschke J.; Russ J.; Friedrich R. P.; Ehrnhoefer D. E.; Wobst H.; Neugebauer K.; Wanker E. E. (2010) EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. U. S. A. 107, 7710–7715. 10.1073/pnas.0910723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palhano F. L.; Lee J.; Grimster N. P.; Kelly J. W. (2013) Toward the molecular mechanism(s) by which EGCG treatment remodels mature amyloid fibrils. J. Am. Chem. Soc. 135, 7503–7510. 10.1021/ja3115696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakinen A.; Adamcik J.; Wang B.; Ge X.; Mezzenga R.; Davis T. P.; Ding F.; Ke P. C. (2018) Nanoscale inhibition of polymorphic and ambidextrous IAPP amyloid aggregation with small molecules. Nano Res. 11, 3636–3647. 10.1007/s12274-017-1930-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A. E.; Jang J.; Gurlo T.; Carty M. D.; Soeller W. C.; Butler P. C. (2004) Diabetes due to a progressive defect in beta-cell mass in rats transgenic for human islet amyloid polypeptide (HIP Rat): a new model for type 2 diabetes. Diabetes 53, 1509–1516. 10.2337/diabetes.53.6.1509. [DOI] [PubMed] [Google Scholar]
- Liu M.; Verma N.; Peng X.; Srodulski S.; Morris A.; Chow M.; Hersh L. B.; Chen J.; Zhu H.; Netea M. G.; Margulies K. B.; Despa S.; Despa F. (2016) Hyperamylinemia Increases IL-1β Synthesis in the Heart via Peroxidative Sarcolemmal Injury. Diabetes 65, 2772–2783. 10.2337/db16-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi T.; Ono K.; Murase A.; Yamada M. (2009) Phenolic compounds prevent Alzheimer’s pathology through different effects on the amyloid-beta aggregation pathway. Am. J. Pathol. 175, 2557–2565. 10.2353/ajpath.2009.090417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin J.; Maaß S.; Schuberth M.; Giese A.; Oertel W. H.; Poewe W.; Trenkwalder C.; Wenning G. K.; Mansmann U.; Südmeyer M.; et al. (2019) Safety and efficacy of epigallocatechin gallate in multiple system atrophy (PROMESA): a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 18, 724–735. 10.1016/S1474-4422(19)30141-3. [DOI] [PubMed] [Google Scholar]
- Matveyenko A. V.; Butler P. C. (2006) Beta-cell deficit due to increased apoptosis in the human islet amyloid polypeptide transgenic (HIP) rat recapitulates the metabolic defects present in type 2 diabetes. Diabetes 55, 2106–2114. 10.2337/db05-1672. [DOI] [PubMed] [Google Scholar]
- Caruana M.; Högen T.; Levin J.; Hillmer A.; Giese A.; Vassallo N. (2011) Inhibition and disaggregation of α-synuclein oligomers by natural polyphenolic compounds. FEBS Lett. 585, 1113–1120. 10.1016/j.febslet.2011.03.046. [DOI] [PubMed] [Google Scholar]
- Sato M.; Murakami K.; Uno M.; Nakagawa Y.; Katayama S.; Akagi K.; Masuda Y.; Takegoshi K.; Irie K. (2013) Site-specific inhibitory mechanism for amyloid β42 aggregation by catechol-type flavonoids targeting the Lys residues. J. Biol. Chem. 288, 23212–23224. 10.1074/jbc.M113.464222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P.; Raleigh D. P. (2012) Analysis of the inhibition and remodeling of islet amyloid polypeptide amyloid fibers by flavanols. Biochemistry 51, 2670–2683. 10.1021/bi2015162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chojnacki J. E.; Liu K.; Yan X.; Toldo S.; Selden T.; Estrada M.; Rodríguez-Franco M. I.; Halquist M. S.; Ye D.; Zhang S. (2014) Discovery of 5-(4-hydroxyphenyl)-3-oxo-pentanoic acid [2-(5-methoxy-1H-indol-3-yl)-ethyl]-amide as a neuroprotectant for Alzheimer’s disease by hybridization of curcumin and melatonin. ACS Chem. Neurosci. 5, 690–699. 10.1021/cn500081s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L.; Jiang Y.; Liu K.; Gomez-Murcia V.; Ma X.; Torrecillas A.; Chen Q.; Zhu X.; Lesnefsky E.; Gomez-Fernandez J. C.; Xu B.; Zhang S. (2018) Insights into the Impact of a Membrane-Anchoring Moiety on the Biological Activities of Bivalent Compounds As Potential Neuroprotectants for Alzheimer’s Disease. J. Med. Chem. 61, 777–790. 10.1021/acs.jmedchem.7b01284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba S.; Osakabe N.; Natsume M.; Terao J. (2004) Orally administered rosmarinic acid is present as the conjugated and/or methylated forms in plasma, and is degraded and metabolized to conjugated forms of caffeic acid, ferulic acid and m-coumaric acid. Life Sci. 75, 165–178. 10.1016/j.lfs.2003.11.028. [DOI] [PubMed] [Google Scholar]
- Young L. M.; Saunders J. C.; Mahood R. A.; Revill C. H.; Foster R. J.; Tu L. H.; Raleigh D. P.; Radford S. E.; Ashcroft A. E. (2015) Screening and classifying small-molecule inhibitors of amyloid formation using ion mobility spectrometry-mass spectrometry. Nat. Chem. 7, 73–81. 10.1038/nchem.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H.; Tao Q.; Ang T. F. A.; Massaro J.; Gan Q.; Salim S.; Zhu R. Y.; Kolachalama V. B.; Zhang X.; Devine S.; Auerbach S. H.; DeCarli C.; Au R.; Qiu W. Q. (2019) Association of Plasma Amylin Concentration With Alzheimer Disease and Brain Structure in Older Adults. JAMA Netw Open. 2, e199826 10.1001/jamanetworkopen.2019.9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa F.; Goldstein L. B.; Biessels G. J. (2020) Amylin as a Potential Link between Type 2 Diabetes and Alzheimer Disease. Ann. Neurol. 87, 486. 10.1002/ana.25668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.