

Abstract
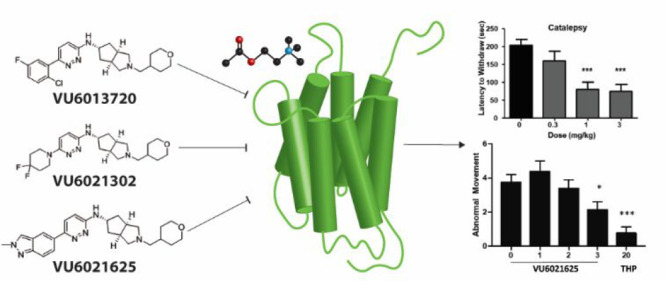
Nonselective antagonists of muscarinic acetylcholine receptors (mAChRs) that broadly inhibit all five mAChR subtypes provide an efficacious treatment for some movement disorders, including Parkinson’s disease and dystonia. Despite their efficacy in these and other central nervous system disorders, antimuscarinic therapy has limited utility due to severe adverse effects that often limit their tolerability by patients. Recent advances in understanding the roles that each mAChR subtype plays in disease pathology suggest that highly selective ligands for individual subtypes may underlie the antiparkinsonian and antidystonic efficacy observed with the use of nonselective antimuscarinic therapeutics. Our recent work has indicated that the M4 muscarinic acetylcholine receptor has several important roles in opposing aberrant neurotransmitter release, intracellular signaling pathways, and brain circuits associated with movement disorders. This raises the possibility that selective antagonists of M4 may recapitulate the efficacy of nonselective antimuscarinic therapeutics and may decrease or eliminate the adverse effects associated with these drugs. However, this has not been directly tested due to lack of selective antagonists of M4. Here, we utilize genetic mAChR knockout animals in combination with nonselective mAChR antagonists to confirm that the M4 receptor activation is required for the locomotor-stimulating and antiparkinsonian efficacy in rodent models. We also report the synthesis, discovery, and characterization of the first-in-class selective M4 antagonists VU6013720, VU6021302, and VU6021625 and confirm that these optimized compounds have antiparkinsonian and antidystonic efficacy in pharmacological and genetic models of movement disorders.
Keywords: acetylcholine, muscarinic, Parkinson’s disease, dopamine, cholinergic, dystonia
Dopamine (DA) release and signaling in the basal ganglia (BG) are critical for fine-tuned motor control and locomotor ability.1−4 When DA release or signaling is diminished, such as in Parkinson’s disease (PD) following the death of DA-releasing cells or in genetic forms of dystonia, aberrant motor behaviors are present.1−6 In many of these disease states, especially PD, treatment often centers around boosting DA levels in the brain through administration of the dopamine prodrug levodopa (L-DOPA), preventing the breakdown of DA, or directly activating DA receptors.7 However, these treatments can often lead to severe side effects such as dyskinesia and are not effective at treating all of the symptoms in the disease state, and their efficacy is unreliable with disease progression or chronic use.8 The development of non-DA-based therapies with different mechanisms of action that do not directly target the DA system could meet a large unmet clinical need in several movement disorders.9,10
One possible non-DA-based treatment mechanism is through the targeting of muscarinic acetylcholine receptors (mAChRs).11,12 Acetylcholine (ACh) acting through mAChRs has powerful neuromodulatory actions on the BG motor circuit.10,13,14 Activation of mAChRs induces several actions to oppose DA release, DA signaling, as well as related motor behaviors. Consistent with these multiple actions on DA release and signaling, anti-mAChR therapy that targets each of the five mAChR subtypes (M1–M5) equally has efficacy in reducing the primary motor symptoms of PD and dystonia.7,9,11−13,15−19 However, like DA-targeted therapies, despite their efficacy, nonselective anti-mAChR therapy can lead to serious on-target adverse effects that limit their tolerability by patients.11,20
Recent pharmacological and genetic studies have made it possible to define unique roles of individual mAChR subtypes in motor disorders.10,17 These data raise the possibility that the targeting of individual mAChR subtypes with truly selective and specific pharmacological ligands may maintain the efficacy but eliminate or reduce the adverse effects associated with nonselective mAChR pharmacological agents.10 Several of the peripheral adverse effects associated with nonselective antimuscarinic agents are likely mediated by M2 and M3, and central side effects centering around memory and cognition may be due to M1.9,10,17,21,22 However, recent studies using first-in-class M4 positive allosteric modulators (PAMs) along with genetic approaches suggest that potentiation of M4 signaling opposes DA signaling in the BG motor circuit via multiple mechanisms. For example, potentiation of M4 activation in the dorsal striatum can cause a sustained inhibition of DA release.19 Furthermore, M4 potentiation on BG direct pathway terminals in the substantia nigra pars reticulata (SNr), the primary BG output nucleus in rodents, directly opposes dopamine receptor subtype 1 (D1) signaling in these cells, leading to tonic inhibition of BG direct pathway activity and reduced locomotion.15 Genetic deletion of M4 either globally or in D1DA-expressing spiny projection neurons (D1-SPNs), which form the BG direct pathway, recapitulates many of our pharmacological findings as well.16,18 Mice with selective M4 deletion in D1-SPNs or globally are also more sensitive to the prolocomotor effects of psychomotor stimulants, have elevated basal DA levels, and are hyperlocomotive.16,18 These genetic and pharmacological studies of mAChR subtypes suggest that M4 may be the dominant mAChR in the regulation of DA and locomotor activity and that M4-selective inhibitors may maintain the efficacy seen with nonselective anti-mAChR therapeutics in movement disorders while reducing or eliminating their side effects.9
Directly testing the hypothesis that M4 antagonism will have efficacy in movement disorders has been greatly limited due to the lack of truly selective M4 inhibitors.9 To evaluate whether M4 underlies the efficacy of nonselective mAChR antagonists, we report the discovery of a first-in-class series of M4-selective antagonists. Utilizing knockout (KO) animals in conjunction with these novel, selective M4 antagonists, our data support the role of M4 in movement disorders and demonstrate the antiparkinsonian and antidystonic efficacy of M4 antagonists in preclinical models.
Results
M4 Underlies the Antiparkinsonian and Prolocomotor Efficacy of Anti-mAChR Compounds
To determine which mAChR subtype or subtypes underlie the locomotor-stimulating effects of the anti-mAChR agent scopolamine, we first assessed the effects of multiple doses of scopolamine on locomotor activity in wild-type (WT) mice.23 Mice were placed into an open field chamber and allowed to habituate for 90 min while activity (total distance traveled in cm/5 min bins) was recorded. After 90 min, scopolamine (0.1–3 mg/kg, 10 mL/kg 10% Tween 80, intraperitoneal (i.p.)) was injected, and total distance traveled was recorded (Figure 1A). Scopolamine increased total distance traveled from 90 to 150 min in a dose-dependent manner, with the 3 mg/kg dose showing maximal efficacy (Figure 1A,B; one-way ANOVA with Dunnett’s posthoc test; F4,25 = 9.064; p < 0.0001). The administration of 0.1 or 0.3 mg/kg (i.p.) of scopolamine did not significantly increase locomotor activity (1346.0 ± 385.9 cm for vehicle, 509.7 ± 193.5 cm for 0.1 mg/kg scopolamine, 1322.0 ± 324.9 cm for 0.3 mg/kg scopolamine; Figure 1A,B; p > 0.05). In contrast, 1 and 3 mg/kg of scopolamine induced a significant increase in locomotor activity from 90 to 150 min (1346.0 ± 385.9 cm for vehicle, 5272.0 ± 726.4 cm for 1 mg/kg scopolamine, 6796.0 ± 1870.0 cm for 3 mg/kg scopolamine; Figure 1A,B; p < 0.05 for 1 mg/kg; p < 0.01 for 3 mg/kg). We then repeated this procedure with 3 mg/kg scopolamine (i.p.) in M1 or M4 global KO mice and compared responses to their littermate controls. Similar to the previous experiment, 3 mg/kg scopolamine significantly increased distance traveled in WT littermate mice compared to vehicle-injected WT littermate control mice (1786.0 ± 256.6 cm from 90 to 150 min for vehicle, 8952.0 ± 789.8 cm for 3 mg/kg scopolamine; one-way ANOVA with Tukey’s multiple comparison posthoc test; F5,72 = 26.7; Figure 1C,E; p < 0.001). In M1 knockout animals, 3 mg/kg scopolamine (i.p.) elicited a hyperlocomotor response as compared to vehicle-treated M1 KO mice (2093.0 ± 388.1 cm for vehicle-treated M1 KO, 7357.0 ± 1427.0 cm for M1 KO after 3 mg/kg scopolamine; Figure 1C,E; p < 0.001). In contrast, the scopolamine-induced increase in locomotor activity observed in WT mice was largely and significantly absent in M4 global KO animals (1492.0 ± 248.7 cm for vehicle-treated M4 KO mice, 3574.0 ± 407.3 cm for M4KO administered 3 mg/kg scopolamine; Figure 1D,E; p > 0.05). When compared to WT animals dosed i.p. with 3 mg/kg scopolamine, M4 KO mice administered scopolamine had a significantly reduced distance traveled from 90 to 150 min (Figure 1E, p < 0.001). This suggests that M4 plays a dominant role in mediating the scopolamine-induced increase in locomotor activity.
Figure 1.

M4 underlies the antiparkinsonian and locomotor-stimulating effects of the muscarinic acetylcholine antagonist scopolamine. Scopolamine induces a dose-dependent increase in locomotor activity (A, B), which persists in M1 knockout mice (C, E) but is largely absent in M4 knockout mice (D, E). Scopolamine significantly reverses haloperidol-induced catalepsy in wild-type mice (F), an effect that is absent in M4 knockout mice (G) but not in M1 knockout mice (H). N = 6–12 per group. One-way ANOVA with Dunnett’s or Tukey’s multiple comparison posthoc test; * p < 0.05, ** p < 0.01, *** p < 0.001.
We also examined if M4 was responsible for the antiparkinsonian efficacy of scopolamine in a model of parkinsonism with predictive validity for antiparkinsonian efficacy, haloperidol-induced catalepsy (HIC).24 Similar to the locomotor assay, we first performed a dose–response study to find the dose of scopolamine that was maximally efficacious in reducing catalepsy. 2 h after injection with haloperidol (1 mg/kg, 10 mL/kg, 0.25% lactic acid in water, i.p.) and a dose of scopolamine (15 min before testing, 1–10 mg/kg, 10 mL/kg, 10% Tween 80, i.p.), the latency of WT mice to remove their forepaws from an elevated bar was assessed. Scopolamine induced a dose-dependent reversal of HIC (Figure 1F, one-way ANOVA with Dunnett’s posthoc test; F3,27 = 34.8, p < 0.0001, Figure 1F). In vehicle-treated mice, the mean latency to withdraw was 210.9 ± 35.9 s. The administration of scopolamine at all doses tested significantly reduced the mean latency to withdraw in these mice, and the maximally efficacious dose was again 3 mg/kg (23.4 ± 6.7 s for 1 mg/kg, 4.4 ± 2.3 s for 3 mg/kg, 2.9 ± 1.8 s for 10 mg/kg; Figure 1F, *** p < 0.0001). This dose of scopolamine was then used in the same assay in global M4 KO mice and WT littermate controls. In WT littermate controls, 3 mg/kg scopolamine (i.p.) significantly reversed catalepsy (155.9 ± 25.3 s for vehicle, 6.1 ± 1.4 s for 3 mg/kg scopolamine; one-way ANOVA with Dunnett’s; F3,34 = 8.7, p = 0.0002; Figure 1G, ***p < 0.001). Vehicle-treated M4 global KO mice demonstrated a similar cataleptic behavior to vehicle-treated WT littermates (151.9 ± 28.9 s for M4KO vs 155.9 ± 25.3 s for WT vehicle; Figure 1G, p > 0.05). The administration of 3 mg/kg of scopolamine to M4 KO animals did not significantly reduce the mean latency to withdraw compared to WT vehicle-treated mice (113 ± 29.5 s for M4KO; p < 0.05, Figure 1G). We additionally repeated these experiments in M1 KO animals and littermate controls. 3 mg/kg scopolamine significantly reversed the cataleptic phenotype in both littermate controls and M1 KO animals (WT, 281.3 ± 37.5 s for vehicle, 9.8 ± 5.9 s for scopolamine, M1 266.7 ± 51.72 s for vehicle, 76.33 ± 111.4 s for scopolamine; one-way ANOVA with Dunnett’s; F3,19 = 17.34, p < 0.0001; Figure 1H, ***p < 0.001). Taken together, these data indicate that M4 is the primary mAChR responsible for the antiparkinsonian efficacy of the nonselective anti-mAChR antagonist, scopolamine, in HIC.
Discovery and Synthesis of First-in-Class M4-Selective Antagonists
Our data implicating M4 as the primary mAChR subtype responsible for the locomotor-stimulating and antiparkinsonian effects of scopolamine gave clear rationale for the development of the first truly selective M4 antagonist tool compounds. Utilizing a recently reported partially selective antimuscarinic compound PCS1055,25 a human M4 preferring antagonist with potent acetylcholinesterase activity, as a starting point, we devoted significant medicinal chemistry efforts to discover a novel, first-in-class series of selective M4 antagonists. Here, we report novel, M4 antagonists with balanced activity at both human and rat M4 receptors that were devoid of acetylcholinesterase activity and engendered in vitro and in vivo drug metabolism and pharmacokinetic (DMPK) profiles suitable for in vivo proof of concept studies. Final compounds were prepared as described in Scheme 1, and structures are reported in Figure 2. Briefly, exo-amine (1) (prepared as previously reported26) underwent nucleophilic aromatic substitution (SNAr) with 3,6-dichloropyridazine (2) to give chloropyridazine 3. Boc-deprotection, followed by reductive amination with tetrahydro-2H-pyran-4-carbaldehyde (4), gave 5, which was then substituted via either Suzuki–Miyaura coupling to give 6 (VU6013720) and 7 (VU6021625) or SNAr to give 8 (VU6021302). Analogues can also be synthesized by first substituting chloropyridazine 5 (under identical conditions for the generation of 6–8), followed by Boc-deprotection and reductive amination. Detailed methods, 1H NMR, and 13C NMR for each final compound (6–8) and intermediates are described in the Supporting Information.
Scheme 1. Final Compounds.

Figure 2.

Structure of highly optimized M4 antagonists. Chemical structures of novel M4 antagonists.
Pharmacological Characterization of VU6013720, VU6021302, and VU6021625
To understand the potency and selectivity of our novel series of compounds, we tested VU6013720, VU6021302, and VU6021625 in a calcium mobilization assay in cell lines that express individual mAChR receptors.27,28 First, we examined the potency of each compound in blocking the effects of an EC80 concentration of ACh, using calcium mobilization in CHO cell lines that express either rat or human M4 as a readout. The receptor density of each mAChR subtype in our stable cell lines is shown in Table S1. Each of the novel M4 antagonists inhibited the response to ACh with the IC50 values listed for VU6013720 (rM4IC50 = 20 nM, hM4IC50 = 0.59 nM), VU6021302 (rM4IC50 = 70 nM, hM4IC50 = 1.8 nM), and VU6021625 (rM4IC50 = 57 nM, hM4IC50 = 0.44 nM) (Figure 3A–C and Table 1). Interestingly, this series of compounds is more potent at human M4 than rat M4.
Figure 3.

Potency, selectivity, and binding affinity of VU6013720, VU6021302, and VU6021625. Potencies were determined by adding a concentration–response curve of M4 antagonist followed by an EC80 of acetylcholine in human, rat, or mouse M4-expressing CHO cells. (A–C) VU6013720, VU6021302, and VU6021625 induced a concentration-dependent inhibition of the release of calcium. The selectivity of these M4 antagonists was evaluated by adding a concentration–response curve of compound followed by an EC80 of acetylcholine in M1, M2, M3, M4, or M5 expressing CHO cells. Competition binding assay of this series of compounds with [3H]-NMS in rat (G) or mouse (H) M4-expressing CHO cell membranes. Data represent the mean ± SEM of 3 independent experiments performed in duplicate.
Table 1. Fold Selectivity of VU6013720, VU6021302, and VU6021625; Fold Selectivity of M4 Antagonists over Other Muscarinic Acetylcholine Receptors and a Summary of IC50 Values (Data Derived from Graphs in Figure 3).
| compound | human M4 | rat M4 | rat M1 | rat M2 | rat M3 | rat M5 | |
|---|---|---|---|---|---|---|---|
| VU6013720 | IC50 (nM) [pIC50 ± SEM] | 0.59 [9.23 ± 0.04] | 20 [7.71 ± 0.19] | 1700 [5.76 ± 0.04] | 670 [6.17 ± 0.04] | >10 000 | >10 000 |
| fold selectivity | 85 | 34 | >100 | >100 | |||
| VU6021302 | IC50 (nM) [pIC50 ± SEMI] | 1.8 [8.75 ± 0.05] | 70 [7.15 ± 0.10] | >10 000 | 2500 [5.61 ± 0.10] | >10 000 | >10 000 |
| fold selectivity | >100 | 36 | >100 | >100 | |||
| VU6021625 | IC50 (nM) [pIC50 ± SEM] | 0.44 [9.36 ± 0.13] | 57 [7.25 ± 0.06] | 5500 [5.26 ± 0.16] | 3200 [5.50 ± 0.02] | >10 000 | >10 000 |
| fold selectivity | 96 | 56 | >100 | >100 |
We then repeated these calcium mobilization assays in cell lines that express the rat M1, M2, M3, or M5 receptors to examine the selectivity of these compounds relative to other mAChR subtypes. VU6013720, VU6021302, and VU6021625 all show IC50 values >10 000 nM at rM3 and rM5 in calcium mobilization assays, and each compound shows functional selectivity >100 fold for rM4 relative to rM3 and rM5 (Figure 3D–F and Table 1). At rM1, VU6013720 has an IC50 = 1700 nM displaying ∼85 fold selectivity, VU6021302 an IC50 > 10 000 nM displaying >100 fold selectivity, and VU6021625 an IC50 = 5500 nM displaying ∼96 fold selectivity (Figure 3D–F and Table 1). The greatest challenge for this series of compounds was selectivity with regard to M2. At rat M2, VU6013720 has an IC50 = 670 nM displaying ∼34 fold selectivity, VU6021302 an IC50 = 2500 nM displaying ∼36 fold selectivity, and VU6021625 an IC50 = 3200 nM displaying ∼56 fold selectivity over M2 (Figure 3D–F and Table 1).
The binding properties of these M4 antagonists at the rat M4 receptor were evaluated by equilibrium [3H]-NMS competition assays using membranes harvested from CHO cells stably expressing the rat M4 receptor (Figure 3G). Similar to atropine, VU6021625, VU6021302, and VU6013720 all fully displaced [3H]-NMS binding from the M4 receptors in a concentration-dependent manner with Ki values of 11.4 ± 2.24, 11.7 ± 2.01, and 1.5 ± 0.15 nM, respectively. VU6021625 was also evaluated at the M4 mouse receptor (Figure 3H) demonstrating an affinity similar to rat of 9.6 ± 1.17 nM. These data also suggest that this series of compounds may interact competitively with the orthosteric binding site, although additional studies will be required to confirm this, and will be included in a subsequent publication.
Due to the overall selectivity, especially with regard to M2, and potency profile of VU6021625, we chose this as our lead tool compound and submitted this compound for ancillary pharmacology profiling. We examined VU6021625 in the Eurofins radioligand binding panel that utilizes 78 separate G protein-coupled receptors (GPCRs), ion channels, and transporters. With this assay, which screened VU6021625 binding to this panel of targets at 10 μM, we observed little off target binding (see Table S2). Outside of mAChRs, only the histamine H3 receptor showed appreciable binding (88% radioligand displacement), and modest binding to nicotinic α3β4 and serotonin 5-HT2B receptor was also observed (55% and 53% displacement, respectively, Table S2). With this overall potency, selectivity, and specificity profile, VU6021625 represents an excellent first-in-class tool compound to examine the effects of M4 antagonism.
VU6021625 ex Vivo Blocks mAChR-Induced Decreases in D1-SPNs Activity and DA Signaling
Previously, using M4 positive allosteric modulators and global or conditional M4 KO animals in ex vivo electrophysiology studies, we have shown that M4 activation can tonically inhibit the BG direct pathway/D1-SPNs as and induce a sustained inhibition of DA release.15,19 Using our new tool M4 antagonist compound VU6021625, we examined its ability to reverse electrophysiological measures of M4 inhibition of direct pathway activity and DA release. First, as previously described, we used whole-cell electrophysiology to patch into GABAergic cells of the SNr and recorded miniature inhibitory postsynaptic currents (mIPSCs) to examine direct pathway neurotransmitter release probability.15 After recording a baseline period of 5 min, we bath applied 10 μM VU6021625 and recorded mIPSC frequency. Inhibition of M4 activity in the direct pathway increased mIPSC frequency by ∼40%, indicating that M4 tonically inhibits the BG direct pathway and that removal of this inhibition increases direct pathway output, as would be predicted by our previous work (Figure 4A,B, paired t test, *p < 0.05). To further evaluate effects of VU6021625 on mAChR mediated inhibition of transmission from D1-SPN terminals, we again recorded mIPSCs but bath applied 10 μM of the nonselective mAChR agonist oxotremorine-M (Oxo-M), which decreased D1-SPN mIPSC frequency by ∼20% (Figure 4C,D). We have previously shown that the effects of Oxo-M at this synapse are mediated exclusively by M4.15 However, it is possible, although unlikely, that there are non-M4 mediated mechanisms at this synapse that are attributable to the modest off-target binding of VU6021625 to other receptors such as the histone H3 receptor (Table S2). Bath application of VU6021625 completely blocked this effect, indicating that M4 antagonism can block tonic activation of M4 by endogenous ACh as well as M4-mediated effects on inhibitory transmission in the direct pathway that are induced by exogenously added agonists (Figure 4C,D, paired t test, **p < 0.01).
Figure 4.

VU6021625 reversed muscarinic-induced deficits in dopamine release and signaling. (A) Sample traces of miniature inhibitory postsynaptic currents (mIPSCs) during baseline (top) and following the bath application of 10 μM VU6021625 (bottom). (B) Inhibition of M4 activity by VU6021625 significantly increased mIPSC frequency (paired t test, *p < 0.05, n = 5 cells). (C) Sample traces of mIPSCs during baseline (top), bath application of the nonselective mAChR agonist oxotremorine-M (Oxo-M) (middle), or bath application of 10 μM Oxo-M + VU6021625 (bottom). Bath application of VU6021625 completely blocked the change in frequency induced by Oxo-M, paired t test, **p < 0.01 (C–D, n = 5–7 cells/group). Sample traces (E) and time-courses (F) of Oxo-M-induced inhibition of DA release in the absence or presence of the M4-selective antagonist VU6021625. (G) Bar graph summary depicting the % inhibition of DA release observed under different conditions from 30 to 40 min (n = 6–7, * p < 0.005, two tailed Mann–Whitney test, n = 5–7 slices per group). (H) Time-course of M4-mediated effects obtained from subtracting the mean values for the time-course in absence of antagonist from the mean values of the time-course in the presence of VU6021625.
Application of the M4-Selective Antagonist VU6021625 Unmasks a Robust Oxo-M-Dependent Increase in DA Release
Beyond the effects of M4 inhibition of direct pathway activity, we also previously found that M4 potentiation causes a sustained inhibition of DA release in the dorsal striatum.19 To determine the ability of the M4 receptor to modulate striatal DA release, we monitored Oxo-M mediated changes in electrically evoked DA via fast-scan cyclic voltammetry in the dorsolateral striatum in the absence or presence of VU6021625 (Figure 4E–G). All experiments reported here were performed in the presence of nicotinic acetylcholine receptor (nAChR) antagonist (DhβE; 1 μM) to remove nAChR-mediated DA release. In the absence of M4 antagonism, we saw a transient increase in DA when Oxo-M was applied followed by a sustained inhibition after drug washout (sustained inhibition of −12.64 ± 3.18% of baseline DA release). The inclusion of VU6021625 (3 μM) caused an increase in the amount of DA release (Figure 4F) and significantly reversed the direction of Oxo-M effects on DA release observed after Oxo-M washout resulting in a net increase in DA release (sustained increase of 9.45 ± 4.28% of baseline DA release; Figure 4G). To determine the specific contribution of M4 receptor activation on DA release, we subtracted the mean values of the Oxo-M time-course obtained in the absence of any antagonist from the mean values obtained in the presence of VU6021625 (Figure 4H), which revealed a rapid onset of M4-mediated inhibition of DA release that was sustained at time-points well after Oxo-M had washed out of the bath.
The results obtained with VU6021625 in fast-scan cyclic voltammetry support our previous finding that M4 receptor activation can induce a sustained inhibition of DA release in the dorsolateral striatum in the presence of nAChR antagonists.17 Unlike our previous report where Oxo-M induced a suppression at all time-points examined, we found that 30 μM Oxo-M alone caused a transient increase in DA release followed by a sustained inhibition. However, in the presence of VU6021625, an Oxo-M-induced increase in DA release was observed at all time-points (Figure 4). While this is consistent with M4 receptor activation leading to a sustained inhibition of DA release, the identity of the mAChR subtype mediating this increase in DA release is still not known. Studies in the nucleus accumbens have shown that M5 receptors can mediate an enhancement of DA release in this brain area raising the possibility that M5 receptors may also be able to increase DA release in the dorsolateral striatum. These results suggest that there could be a competition between different mAChR-subtypes in the striatum with activation of some subtypes leading to inhibition of DA release while activation of other mAChR subtypes could lead to a potentiation. Future studies will be needed to gain critical insights into what mAChR subtypes mediate these increases in DA release in the dorsolateral striatum and elucidate if there are physiological conditions that favor the activation of DA inhibiting mAChRs and DA potentiating mAChRs.
VU6021625 Possesses DMPK Properties Suitable for in Vivo Use
Before use in animal models of parkinsonism and dystonia, we assessed the DMPK properties of VU6021625 to determine if VU6021625 was suitable for in vivo use in rodents and to inform the dosing paradigms for behavioral studies. Mouse plasma and brain pharmacokinetics (PK) were obtained over a 7 h time-course following a single 1 mg/kg i.p. dose (vehicle: 20% β-cyclodextrin 80% water [w/v]; 10 mL/kg body weight) to male, C57Bl/6J mice (n = 3 per time point) (Figure 5A). The maximum total concentration of the compound in plasma (Cmax,total) was 170 ng/mL (393 nM) with a time-to-reach Cmax (Tmax) of 0.25 h. In brain, a Cmax,total of 31.6 ng/mL (73.0 nM) was observed with a Tmax of 1 h. The distribution to brain from plasma on an area-under-the-curve (AUC)-based total concentration basis (Kp) was moderate (0.25, Figure 5A).
Figure 5.

In vivo pharmacokinetics of VU6021625. Plasma and brain concentrations of VU6021625 were measured following systemic administration (1 mg/kg, 10 mL/kg, i.p.) in mice (A) and (10 mg/kg, 10 mL/kg, p.o.) in rats (B). SD = Sprague–Dawley. N = 3 animals per group.
Rat plasma and brain PK were obtained over a 5 h time-course following a single 10 mg/kg per oral (p.o.) dose (vehicle: 0.5% methylcellulose 99.5% water [w/v]; 10 mL/kg body weight) to male, Sprague–Dawley rats (n = 3 per time point) (Figure 5B). The total Cmax in plasma was 586 ng/mL (1350 nM) with a Tmax of 5 h. The total Cmax in the brain was 282 ng/mL (652 nM) with a Tmax of 5 h. The AUC-based total brain distribution was moderate (Kp = 0.35) while unbound distribution was low (Kp,uu = 0.13) based on in vitro rat plasma and brain homogenate binding data (fuplasma = 0.563, fubrain = 0.206) from equilibrium dialysis assays.
In light of the low brain:plasma Kp,uu observed in the rat study, potential P-glycoprotein (P-gp) substrate activity of VU6021625 was evaluated using an in vitro bidirectional efflux assay with MDCK-MDR1 cells (performed via contract by Absorption Systems LLC [Exton, PA]). This experiment revealed a high efflux ratio (ER) of 71 indicating that the compound is subject to P-gp-mediated active efflux (Table 2). However, an absolute unbound brain concentration (134 nM) >2-fold higher than the compound’s in vitro rat M4 potency was achieved in the rat PK study thereby demonstrating its in vivo utility despite P-gp efflux.
Table 2. Pharmacokinetic Properties and Summary of in Vivo and in Vitro Characteristics of VU6021625.
| property | value |
|---|---|
| MDCK-MDR1 (efflux ratio) | 71 |
| fu brain (r) | 0.206 |
| fu plasma (r) | 0.563 |
| kinetic solubility (μM) | 95.1 ± 10.3 (pH 2.2) |
| 90.8 ± 9.62 (pH 6.8) |
VU6021625 Does Not Induce Hyperlocomotion
To test if VU6021625 could induce a hyperlocomotor state similar to that of scopolamine, we utilized both mice and rats and performed experiments similar to those in Figure 1A–E. After a habituation period of 90 min in WT mice (Figure 6A,B) or 30 min in Sprague–Dawley Rats (Figure 6C,D), rodents were injected i.p. with 3 mg/kg scopolamine or 3 mg/kg VU6021625, and locomotion was observed. Similar to what we reported in Figure 1A–E, scopolamine in both mice and rats caused marked, significant increases in locomotion (Figure 6B, mice vehicle 2440 ± 712.9 cm versus 10 951 ± 3057 cm for scopolamine with Dunnett’s posthoc test, F2,35 = 67.73, p < 0.0001; rats vehicle 5121 ± 766.7 cm versus 8666 ± 2995 cm for scopolamine, one-way ANOVA with Dunnett’s posthoc test, F2,21 = 14.55, p = 0.0001, **p < 0.01, ***p < 0.001). However, VU6021625, unlike scopolamine, did not induce a hyperlocomotor-like state in either mice or rats (Figure 6B, mice vehicle 2440 ± 712.9 cm versus 2787 ± 1574 cm for VU6021625 with Dunnett’s posthoc test, F2,35 = 67.73, p > 0.05; rat vehicle 5121 ± 766.7 cm versus 3746 ± 1034 cm for VU6021625, one-way ANOVA with Dunnett’s posthoc test, F2,21 = 14.55, p = 0.0001; NS, not significant). Additionally, unlike scopolamine, VU6021625 did not alter spontaneous locomotion in mice or rats (Figure S1). This suggests that M4 may be necessary but not sufficient to stimulate locomotion, a more nuanced role for M4 in influencing locomotion, or that M4 may preferentially interact with basal ganglia motor loops that are disturbed in the disease state.
Figure 6.

VU6021625 does not increase locomotor activity in normal, drug naïve rodents. Unlike scopolamine (3 mg/kg, i.p.), VU6021625 (3 mg/kg, i.p.) does not increase locomotor activity in either mice (A, B) or rats (C, D) (A, C, time activity curve; B, D, total distance traveled after vehicle, VU6021625, or scopolamine administration). Mouse N = 12 per group. Rat N = 8 per group. One way ANOVA with Dunnett’s posthoc test **p < 0.01, ***p < 0.001; NS, not significant.
VU6021625 Has Antiparkinsonian Efficacy
To test if VU6021625 has antiparkinsonian efficacy as expected from our data presented in Figure 1, we utilized the HIC animal model of parkinsonian motor deficits. Vehicle-treated mice displayed a mean latency to withdraw their forepaws of 202.4 ± 16.6 s. Administration of 0.3 mg/kg VU6021625 did not significantly reduce catalepsy in these mice (159.0 ± 27.3 s, Figure 7A,B, 10 mL/kg, 20% (2-hydroxypropyl)-β-cyclodextrin (HPBCD), i.p; one-way ANOVA with Dunnett’s posthoc test, F3,44 = 8.6, p > 0.05). However, administration of 1 or 3 mg/kg VU6021625 i.p. significantly reversed cataleptic behavior (60.7% and 63.3%, respectively) when compared to vehicle-treated mice (79.6 ± 20.2 s for 1 mg/kg, 74.3 ± 19.5 for 3 mg/kg, Figure 7A,B; 10 mL/kg, 20% HPBCD, i.p; one-way ANOVA with Dunnett’s posthoc test, F3,44 = 8.6, p < 0.0001). To benchmark these values obtained with VU6021625 with the nonspecific mAChR antagonist, scopolamine, we repeated catalepsy testing in WT animals. The maximally efficacious dose of scopolamine of 3 mg/kg significantly reversed the cataleptic phenotype by 92.7% (Figure S2A, 197.2 ± 102.2 s for vehicle, 14.33 ± 8.897 s for 3 mg/kg scopolamine, i.p., two-way t test, t = 6, df = 22, p < 0.001). Additionally, to ensure that VU6021625 had on-target efficacy, we repeated catalepsy testing in a small number of M4 KO mice. Similar to what was observed in WT mice, VU6021625 significantly reversed HIC in the M4 littermate control mice. However, we did not observe a significant reversal of catalepsy between vehicle- and VU6021625-treated groups in M4 KO mice (Figure S2B, 142.2 ± 80.74 s for vehicle, 193.8 ± 131.1 s for 3 mg/kg VU6021625, i.p., one-way ANOVA with Dunnett’s posthoc test, F3,38 = 4.135, p > 0.05). This suggests that the majority of efficacy seen with scopolamine and VU6021625 can be attributed to on-target inhibition of M4.
Figure 7.

VU6021625 displays antiparkinsonian efficacy. Systemic administration of VU6021625 demonstrates dose-dependent efficacy in the haloperidol-induced catalepsy animal model of parkinsonian motor deficits in both mice (A, B) and rats (C, D) with a minimal effective dose of 1 mg/kg in both species. N = 8–10, per group. One-way ANOVA with Dunnett’s posthoc test; *** p < 0.001.
In order to demonstrate that this was not a species-specific effect, VU6021625 was also tested in a rat HIC model. In rats, VU6021625 reduced the cataleptic phenotype in this model at the same doses needed for efficacy in mice. Vehicle-treated rats exhibited a mean latency to withdraw of 43.6 ± 4.8 s. The administration of 0.3 mg/kg VU6021625 did not significantly reduce mean latency to withdraw in these rats (30.9 ± 5.7 s, Figure 7C,D, 1 mL/kg, 10% Tween 80/0.5% methylcellulose, i.p.; one-way ANOVA with Dunnett’s posthoc test, F3,36 = 10.9, p < 0.05). The administration of 1 or 3 mg/kg significantly reversed cataleptic behavior when compared to vehicle-treated rats (15.0 ± 2.7 s for 1 mg/kg, 13.7 ± 3.3 for 3 mg/kg; Figure 7C,D, 1 mL/kg, 10% Tween 80/0.5% methylcellulose, i.p.; one-way ANOVA with Dunnett’s posthoc test, F3,36 = 10.9, ***p < 0.001). This equated to a significant reversal of catalepsy with 1 mg/kg or 3 mg/kg VU6021625 (29.1 ± 13.0% reversal for 0.3 mg/kg, 65.6 ± 6.2% reversal for 1 mg/kg, 68.8 ± 7.7% reversal for 3 mg/kg; one-way ANOVA with Dunnett’s posthoc test, Figure 7D, ***p < 0.001). The minimum effective dose for VU6021625 in this assay was also 1 mg/kg. Additionally, we collected plasma and brain samples at the end of the rat behavioral study to determine the concentration of VU6021625 by LC-MS/MS analysis. At the 1 and 3 mg/kg dose levels, the compound reached unbound brain concentrations of 12.3 and 34.2 nM, respectively (Table 3). These concentrations align well with the potency of this compound at rat M4, are well below the potency of the other four mAChR subtypes (Table 1), and indicate that VU6021625 has antiparkinsonian efficacy in mouse and rat models at exposure levels selective for M4.
Table 3. Pharamacokinetic–Pharmacodynamic Relationship of VU6021625a.
| VU6021625 HIC dose response | |
|---|---|
| dose (mg/kg) | brain unbound (nM) |
| 0.3 | 4.8 |
| 1 | 12.3 |
| 3 | 34.2 |
VU6021625 Has Antidystonic Efficacy
Trihexyphenidyl, a nonselective mAChR antagonist, is the only oral medication for dystonia that has been proven effective in a double-blind, placebo-controlled clinical trial.11,12 To determine if VU6021625 is also effective, we tested a genetic model of DOPA-responsive dystonia (DRD) that carries a point mutation in the mouse tyrosine hydroxylase gene that diminishes DA synthesis and release and is synonymous with human forms of the disease.29 The abnormal dystonic movements in DRD mice are ameliorated by trihexyphenidyl, similar to human patients.29 Administration of VU6021625 (1–3 mg/kg, 10 mL/kg, 1% Tween80, i.p.) significantly reduced the dystonic movements observed in DRD mice at the 3 mg/kg dose (Figure 8, repeated measures ANOVA, F3,21 = 4.85, Dunnett’s multiple comparisons post hoc test, *p < 0.05), providing similar efficacy to trihexyphenidyl (paired t test, ***p < 0.001). These data demonstrate that VU6021625 has antidystonic efficacy in this mouse model, as well as antiparkinsonian efficacy.
Figure 8.
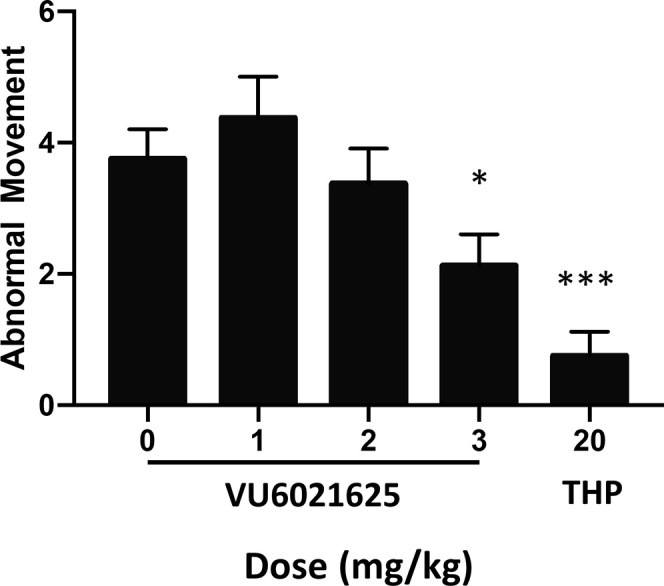
VU6021625 displayed antidystonic efficacy. Using a genetic model of DOPA-responsive dystonia (DRD), we demonstrated that VU6021625 significantly reduced abnormal movements observed in these mice with a minimal effective dose of 3 mg/kg (1–3 mg/kg, 10 mL/kg, 1% Tween 80, i.p.; repeated measures ANOVA with Dunnett’s multiple comparisons post hoc test; * p < 0.05), providing similar efficacy to trihexyphenidyl (THP, paired t test compared to vehicle, *** p < 0.001, N = 8).
Discussion
We report the discovery and characterization of a novel class of M4 antagonists that mark a major advance in the selectivity and specificity over previously reported compounds. For example, tropicamide, widely reported as M4-selective in the literature, has equal binding to M2 and approximately displays about a half log of selectivity over M1, M3, and M5.30 Other more recently reported compounds, such as PD102807, show >100 fold selectivity with respect to binding but have not demonstrated this level of selectivity in functional assays.31 Additionally, PCS1055, while showing modest to good selectivity in functional and binding assays, possesses off target activity as an antagonist of acetylcholinesterase.25 The most potent and selective of our compounds, VU6021625, has greatly improved pharmacological properties over these previously reported compounds with increased functional and binding selectivity with only micromolar-level off-target activity. Importantly, VU6021625 exhibits the ability to reverse effects of mAChR agonists on BG activity and DA release in mouse brain slices and to exert antiparkinsonian and antidystonic efficacy in pharmacological and genetic animal models of movement disorders. Furthermore, in these models, we have established a PK–PD relationship that demonstrates that, at doses required for efficacy, unbound brain concentrations of VU6021625 are well within the selectivity range that VU6021625 possesses over other mAChRs. These findings, as well as our data utilizing genetic KO mice of M4 with nonselective compounds, implicate M4 as the primary mAChR subtype that mediates the antiparkinsonian and antidystonic effects of nonselective anti-mAChR antagonists. However, it is possible that other mAChRs, namely M1, may also play a modulatory role to muscarinic antiparkinsonian efficacy as well.32
The ability of VU6021625 to reverse the cataleptic phenotype in HIC studies suggests that M4-selective antagonists may be beneficial in removing hypokinetic aspects of parkinsonian motor deficits, such as bradykinesia and rigidity. Testing the role of M4-selective antagonists in other rodent models as well as other aspects of parkinsonian motor impairments including tremor and gait disturbances will be key to understanding the extent of antiparkinsonian effects of M4 antagonists. Nonselective mAChR antagonists have also shown efficacy in reducing the prevalence of some of these other parkinsonian motor symptoms. A mechanistic determination of the specific role of M4 antagonism in the efficacy of these nonselective compounds in treating other parkinsonian symptoms will be critical to understanding the potential utility of M4 antagonists as a monotherapy in PD. For example, M4 potentiation using VU0467154 has been previously shown to relieve dyskinesia induced by chronic L-DOPA administration,33 suggesting that, in treatment-induced abnormal movements in PD, M4 activity may be low in the dyskinetic state.9 Whether M4 antagonists would further exacerbate dyskinesia, or if repeated administration of M4 antagonists could cause dyskinesia, remains to be tested. A greater understanding of the ability of M4 to cause or alter abnormal movements will be important to understanding the full potential utility of M4 antagonists in the treatment of movement disorders.
While the etiology of dystonia is complex,5 a number of genes have been shown to be causative for dystonia.34 Some of these genes, such as the genes which are causative for DRD, have clear links to the synthesis, degradation, or signaling of DA.6,29,35 However, several genetic forms of dystonia, such as Dyt1, have no clear link to DA, although some mouse models of these genetic forms of dystonia do show DA deficits.36,37 Similar to trihexyphenidyl, which is used clinically to treat movement disorders, VU6021625 reduces abnormal movements in one of these DA-focused models of dystonia. Whether VU6021625 can relieve behavioral and pathological correlates of dystonia in other forms of genetic dystonia not linked to DA, as well as in other DA linked models, has yet to be determined but will be critical to our understanding of the range of utility of M4 antagonism in dystonia as well as mechanistically understanding the etiology of different forms of dystonia.
Previously, we have utilized M4-selective positive allosteric modulators in conjunction with genetic animal models to elucidate the possible mechanisms through which M4 inhibition may be exerting antiparkinsonian and antidystonic efficacy.9,15,19,27,38 We, and others, have identified a number of pathways within the basal ganglia that are powerfully modulated by M4 activity and have been implicated in the pathological changes associated with PD and dystonia. These include M4 activation leading to a sustained inhibition of DA release,19 directly inhibiting D1DA receptor signaling,15 decreasing corticostriatal glutamate release,39 and modulating striatal plasticity.33 VU6021625 removing or reducing M4 activity at these synapses and circuits, as suggested through our ex vivo electrophysiology experiment, may mechanistically underlie the efficacy observed in our models of parkinsonian or dystonic motor deficits. However, this will need rigorous testing in multiple animal models of movement disorders.
Our data using nonselective mAChR antagonists in conjunction with M4 knockout animals, as well as the use of VU6021625 in wild-type rodents, raise interesting questions on the role of M4 on influencing locomotion. While it is clear from the knockout studies that genetic removal of M4 reduces the hyperlocomotor response to scopolamine, VU6021625 does not increase locomotor activity upon administration of doses that do have antiparkinsonian or antidystonic efficacy. This raises several possibilities. One possibility is that M4 is necessary but not sufficient to induce locomotion. Also, it may be possible that the role of M4 in regulating basal ganglia motor loops is more nuanced with a role in sustaining or permitting locomotion, but not necessarily initiating locomotion.
One important consideration is the possible on-target adverse effects that inhibition of M4 may elicit.9 Beyond the possibility of M4 antagonists exacerbating or inducing dyskinesia in PD patients, there are a number of theoretical on-target adverse effects that could be induced through M4-selective antagonists. While M1 appears to be the mAChR subtype that underlies the majority of procognitive efficacy observed with mAChR agonism,40 we have seen procognitive efficacy with M4PAMs that are likely related to a role for M4 in modulating cortical circuits.27,41 This raises the possibility that M4 antagonism may cause decreased cognitive ability or modulate other aspects of cognition, learning, or memory. Another possibility is that, through relieving M4 inhibition of dopamine release, this could cause an abuse liability with M4 antagonists. However, similar to mechanistically understanding how VU6021625 relieves parkinsonian and dystonic motor deficits, understanding the on-target adverse effects will require further rigorous testing.
VU6021625 represents a major advance in the discovery of selective M4 antagonists and provides the first highly selective M4 antagonist tool compound that can be used to explore the role that M4 plays in movement disorders in vivo. Our initial data support the hypothesis that M4 blockade may underlie a majority of the efficacy observed with nonselective anti-mAChR therapeutics in movement disorders. The development of this in vivo tool compound enables a number of studies that will help with understanding the role of M4 in the treatment and etiology of several movement disorders. These data will provide a critical preclinical rationale for the further development and optimization of M4 antagonists, and the selective blockade of M4 represents a potential novel treatment mechanism to meet the huge unmet clinical need across several movement disorders.
Methods
General Chemistry Methods
All reactions were carried out employing standard chemical techniques under an inert atmosphere. Solvents used for extraction, washing, and chromatography were HPLC grade. All reagents were purchased from commercial sources and were used without further purification. Low-resolution mass spectra were obtained on an Agilent 6120 or 6150 with UV detection at 215 and 254 nm along with ELSD detection and electrospray ionization, with all final compounds showing >95% purity and a parent mass ion consistent with the desired structure. All NMR spectra were recorded on a 400 MHz Brüker AV-400 instrument. 1H chemical shifts are reported as δ values in ppm relative to the residual solvent peak (MeOD = 3.31). Data are reported as follows: chemical shift, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, dd = doublet of doublets, ddd = doublet of doublet of doublets, td = triplet of doublets, dt = doublet of triplets, m = multiplet), coupling constant, and integration. 13C chemical shifts are reported as δ values in ppm relative to the residual solvent peak (MeOD = 49.0). Automated flash column chromatography was performed on a Biotage Isolera 1 and a Teledyne ISCO Combi-Flash system. Microwave synthesis was performed in a Biotage Initiator microwave synthesis reactor. RP-HPLC purification of final compounds was performed on a Gilson preparative LC system. For detailed instructions on the synthesis of each compound, see Scheme S1.
Calcium Mobilization Assays
Compound-evoked decreases to an EC80 concentration of ACh in intracellular calcium were measured using Chinese hamster ovary (CHO) cells stably expressing mouse, rat, or human muscarinic receptors (M1–M5; M2 and M4 cells were cotransfected with Gqi5). Cell culture reagents were purchased from Gibco-ThermoFisher Scientific (Waltham, MA) unless otherwise noted. Cells (15 000 cells/20 μL/well) were plated in Greiner black wall/clear bottom 384-well plates in F12 medium containing 10% FBS, 20 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), and 1× antibiotic/antimycotic. The cells were grown overnight at 37 °C in the presence of 5% CO2. The next day, the medium was removed and replaced with 20 μL of 1 μM Fluo-4, AM (Invitrogen, Carlsbad, CA) prepared as a 2.3 mM stock in dimethyl sulfoxide (DMSO) and mixed in a 1:1 ratio with 10% (w/v) pluronic acid F-127 (Invitrogen, Carlsbad, CA) and diluted in assay buffer [Hank’s balanced salt solution (HBSS), 20 mM HEPES, 4.16 mM sodium bicarbonate (Sigma-Aldrich, St. Louis, MO), and 2.5 mM probenecid (Sigma-Aldrich, St. Louis, MO)] for 50 min at room temperature. Dye was removed and replaced with 20 μL of assay buffer. Compounds were serially diluted 1:3 into 10 point concentration response curves in DMSO using the AGILENT Bravo liquid handler (Atlantic Lab Equipment, Santa Clara, CA), transferred to daughter plates using an Echo acoustic plate reformatter (Labcyte, Sunnyvale, CA), and diluted in assay buffer to a 2× final concentration. Ca2+ flux was measured using a Functional Drug Screening System 6000 or 7000 (FDSS6000/7000, Hamamatsu, Japan). After the establishment of a fluorescence baseline for 2–3 s (2–3 images at 1 Hz; excitation, 480 ± 20 nm; emission, 540 ± 30 nm), 20 μL of test compound was added to the cells, and the response was measured. 140 s later, 10 μL (5×) of an EC20 concentration of ACh (Sigma-Aldrich, St. Louis, MO) was added to the cells, and the response of the cells was measured. Approximately 125 s later, an EC80 concentration of ACh was added. Calcium fluorescence was recorded as fold over basal fluorescence, and raw data were normalized to the maximal response to agonist. Potency (IC50) and maximum response (% ACh Max) for compounds were determined using a four-parameter logistical equation using GraphPad Prism (La Jolla, CA) or the Dotmatics software platform:
| 1 |
where A is the molar concentration of the compound; bottom and top denote the lower and upper plateaus of the concentration–response curve; Hillslope is the Hill coefficient that describes the steepness of the curve; and EC50 is the molar concentration of compound required to generate a response halfway between the top and bottom.
Radioligand Binding Assays
Membranes were made from CHO cells stably expressing the rat M4 receptor (coexpressing Gqi5). Radioligand competition binding assays were performed as previously described (Anderson et al., 2002) with minor modifications. In brief, M4 antagonists were serially diluted into assay buffer and added to each well of a 96-well plate, along with 10 μg/well cell membrane and approximately 100 pM [3H]-NMS (PerKinElmer, Boston, MA). Following a 3 h incubation period on a shaker at room temperature, the membrane-bound ligand was separated from free ligand by filtration through glass fiber 96-well filter plates (Unifilter-96, GF/B; PerkinElmer, Boston, MA). 40 μL of scintillation fluid was added to each well, and the membrane-bound radioactivity was determined by scintillation counting (TopCount; PerkinElmer Life and Analytical Sciences, Boston, MA). Nonspecific binding was determined using 10 μM atropine.
Ancillary Pharmacology Screening
VU6021625 was tested at a concentration of 10 μM in the Eurofins Panlabs lead profiling screen, a radioligand binding assay panel consisting of 78 GPCRs, ion channels, and transporters. Displacement of ≥50% radioligand binding at a panel target was considered significant.
Brain Slice Preparation
C57Bl6/J mice were anesthetized by continuous isoflurane (5%) and then transcardially perfused with a cold solution of 92 mM N-methyl-d-glucamine (NMDG), 2.5 mM KCl, 1.2 mM NaH2PO4, 30 mM NaHCO3, 20 mM HEPES, 25 mM d-glucose, 5 mM sodium ascorbate, 2 mM thiourea, 3 mM sodium pyruvate, 10 mM MgSO4, and 0.5 mM CaCl2 (pH 7.3) at 295–300 mOsm. Mice were then decapitated, and the brains were removed. 300 μm coronal sections of the SNr or striatum were made on a Leica Vibratome 1200S instrument. After sectioning, coronal slices were submerged for 10–15 min at 32 °C in the same NMDG solution as above. Following this recovery period, slices were transferred to ACSF containing 126 mM NaCl, 2.5 mM KCl, 26.2 mM NaHCO3, 1.25 mM NaH2PO4, 2 mM CaCl2, 1.5 mM MgSO4, 10 mM d-glucose, and 5 mM sodium ascorbate.
Whole Cell Patch Clamp Electrophysiology
The whole cell voltage-clamp signal was amplified using Axon Multiclamp 700B amplifiers with appropriate electrode-capacitance compensation. Patch pipets were prepared from borosilicate glass using a Sutter instruments P-100 Flaming/Brown micropipet puller and had a resistance of 3–6 MΩ when filled with the following intracellular solution (mM): 130 CsCl, 10 NaCl, 0.25 CaCl2, 2 MgCl2, 5 EGTA, 10 HEPES, 10 glucose, 2 Mg-ATP. The pH of the pipet solution was adjusted to 7.3 with 1 M CsOH, and osmolarity was adjusted to 285–290. Whole cell recordings were made from visually identified cells in the SNr under an Olympus BX50WI upright microscope (Olympus, Lake Success, NY). A low-power objective (4×) was used to identify the SNr, and a 40× water immersion objective coupled with Hoffman optics was used to visualize the individual neurons. GABAergic cells of the SNr were identified by previously determined membrane characteristics and firing rates.15,42
To isolate mIPSCs, slices were perfused continuously at a rate of ∼2.0 mL/min with an oxygenated solution containing (in mM) 126 mM NaCl, 2.5 mM KCl, 26.2 mM NaHCO3, 1.25 mM NaH2PO4, 2 mM CaCl2, 1.5 mM MgSO4, and 10 mM d-glucose, pH 7.35, with 0.5 μM tetrodotoxin (TTX), 5 μM AMPA receptor 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), and 1 μM NMDA receptor antagonist DL-2-amino-4-methyl-5-phosphono-3-pentenoic acid (AP-5). Slices were perfused with this solution at 25 °C for at least 15 min following the establishment of electrical access. Access resistances were <15 MΩ. mIPSCs were recorded from GABAergic cells of the SNr held at −70 mV in GAP free mode. All drugs were bath-applied with the complete exchange of the external solution not exceeding 30 s. Data were acquired using Digidata 1440A and pClamp 9.2 and analyzed with Mini Analysis software (Synaptosoft).
Fast Scan Cyclic Voltammetry
Electrically evoked DA overflow was monitored with carbon fiber electrodes with a 5 μm diameter as previously described.19 A triangular voltage wave (−400 to +1000 mV at 300 V/s) was applied to fresh cut carbon fiber electrodes every 100 ms. When monitoring electrically evoked DA transients, stimulating electrodes were placed 75 μm deep into the dorsolateral striatum, and slices were electrically stimulated (30–600 μA) every 2.5 min via a bipolar stimulating electrode placed ∼100 μm from the carbon fiber. For typical experiments, the stimulation intensity used was 200–400 μA so as to induce both nAChR-dependent and nAChR-independent DA release. Current was acquired using a Clampex9.2/Digidata1440A system with a low pass Bessell filter at 10 kHz and digitized at 100 kHz. Background-subtracted cyclic voltammograms served both to calibrate the electrodes and to identify dopamine as the substance that was released following electrical stimulation. The best-fit simulation of electrically evoked dopamine overflow was found by nonlinear regression. All time-course data are presented as the mean ± SEM for individual time points. Sustained inhibition was defined as the inhibition that was observed 10–20 min after Oxo-M has washed out. These data were analyzed using a two-tailed Mann–Whitney test, and statistical significance was determined as p < 0.05.
Pharmacokinetics (PK)
The mouse PK study was performed in adult male C57Bl/6J mice (n = 3 per time point). In this study, VU6021625 was formulated (0.1 mg/mL) in 20% β-cyclodextrin 80% water (w/v) and administered at 10 mL/kg body weight. Nonserial sampling of plasma and brain was performed at multiple time points (0.25, 0.5, 1, 2, 4, and 7 h) postadministration. The rat PK study was performed in adult male Sprague–Dawley rats (n = 3 per time point). In this study, VU6021625 was formulated (1 mg/mL) in 0.5% methylcellulose 99.5% water (w/v) and administered at 10 mL/kg body weight. Nonserial sampling of plasma and brain was performed at multiple time points (0.25, 0.5, 1, 3, and 5 h) postadministration. In both studies, brain:plasma Kp was determined by division of the brain AUC0.25-last by the plasma AUC0.25-last. In the rat study, unbound brain:unbound plasma Kp,uu was determined by division of the Kp by the [fuplasma/fubrain] ratio, which was obtained via equilibrium dialysis binding assays with rat plasma (n = 1 performed in triplicate) and rat brain homogenate (n = 1 performed in triplicate). Plasma and brain homogenate binding assays, bioanalytical sample preparation, and quantitation by LC-MS/MS were performed essentially as described previously.43 Data are presented (Figure 5A,B) as means ± SD. The evaluation of VU6021625 for P-gp efflux potential was performed via contract with Absorption Systems LLC (Exton, PA) using a bidirectional transwell assay (n = 1 performed in duplicate) with a 5 μM substrate concentration and their standard in-house assay and quantitation procedures.
Animal Housing
Animals were group-housed under a 12/12 h light–dark cycle with food and water available ad libitum. All animal experiments were approved by the Vanderbilt University or Emory University Animal Care and Use Committee, and experimental procedures conformed to guidelines established by the National Research Council “Guide for the Care and Use of Laboratory Animals”.
Open Field Locomotor Assay
Locomotor activity was tested in wild-type and M4 or M1 KO mice (all congenic on C57Bl6/J background), 8–12 weeks old, using an open field system (OFA-510, MedAssociates, St. Albans, VT) with three 16 × 16 arrays of infrared photobeams. Scopolamine-induced locomotor activity was assessed with the following paradigm: animals were habituated for 90 min in the open field before being injected with vehicle (10% Tween 80, 10 mL/kg, i.p.) or scopolamine (0.1, 0.3, 1, or 3 mg/kg, 10 mL/kg, 10% Tween 80 i.p.), and locomotor activity was recorded for an additional 60 min (150 min total session length). VU6021625-induced locomotor activity was assessed with the following paradigm: animals were habituated for 90 min in the open field before being injected with vehicle (20% HPBCD, 10 mL/kg, i.p.), scopolamine (3 mg/kg, 10 mL/kg, 10% Tween 80, i.p.), or VU6021625 (3 mg/kg, 10 mL/kg, 20% HPBCD, i.p.), and locomotor activity was recorded for an additional 60 min (150 min total session length). Data were analyzed using the activity software package (MedAssociates, St. Albans VT) and expressed as total distance traveled in cm per 5 min bin.
Locomotor activity was tested in adult male Sprague–Dawley rats. The animals were placed in the activity chambers (KinderScientific, San Diego, CA) to habituate to the chambers for 30 min. The animals were then administered VU6021625 (3 mg/kg, 1 mL/kg, 20% HPBCD, i.p.), scopolamine (3 mg/kg, 1 mL/kg, 10% Tween 80, i.p.), or vehicle (20% HPBCD, 10 mL/kg, i.p.) and returned to the activity chambers. Locomotor activity was recorded for an additional 60 min (90 min total session length). Data were analyzed using the activity software package (KinderScientific, San Diego, CA) and expressed as total distance traveled in cm per 5 min bin.
Haloperidol-Induced Catalepsy
Wild-type C57Bl6/J, littermate controls, M4, or M1 global knockout animals (both congenic on C57Bl6/J background) were administered haloperidol (1 mg/kg, 0.25% lactic acid in water, i.p.). For the scopolamine experiments, the mice were administered vehicle (10% Tween80) or scopolamine (i.p.) 105 min after haloperidol. For the VU6021625 experiment, the animals were administered vehicle (20% HPBCD) or VU6021625 (0.3–3 mg/kg, i.p.) 105 min after haloperidol. They were tested 15 min later by placing their forelimbs on a raised bar and recording the latency for the animals to remove their forelimbs from the bar with a cutoff of 300 s. Adult male Sprague–Dawley rats were injected with 1.5 mg/kg haloperidol i.p. 1 h later, the animals were administered 0.3–3 mg/kg of VU6021625, or vehicle. Cataleptic behavior was determined 30 min later as previously described34 by placing the forelimbs on a bar raised 6 cm above the table and recording the amount of time it takes for the rat to withdraw the forelimbs with a cutoff of 60 s. Data are expressed as mean latency to withdraw + SEM or percent inhibition of catalepsy + SEM. At the end of the study (0.5 h postadministration of VU6021625), plasma and brain samples from each rat (all dose groups) were collected and stored (−80 °C) for bioanalysis by LC-MS/MS as described previously.35,39,40
Abnormal Involuntary Movement Scoring in DRD Mice
The dystonia exhibited by knockin mice bearing the c.1160C>A Th mutation (DRD mice), which causes DOPA-responsive dystonia in humans, was assessed after treatment with VU6021625. DRD mice were produced as F1 hybrids of C57BL/6J + /ThDRD × DBA/2J + /ThDRD to avoid the perinatal death associated with inbred C57BL/6J DRD mice.29 The c.1160C>A mutation in Th is coisogenic on C57BL/6 and congenic on DBA/2J. DRD mice were maintained and genotyped as previously described and tested at 16–21 weeks of age.29 A behavioral inventory was used to define the type of abnormal movement, including tonic flexion or tonic extension (limbs, trunk, head), clonus (limbs), and twisting (trunk, head), as described.44−46 Homozygous DRD mice (n = 8) were habituated to the test cages overnight, and experiments started at 8 am, when the abnormal movements are most severe in DRD mice.29 DRD mice were challenged with vehicle or VU6021625 (i.p.) in 1% Tween-80 in a volume of 10 mL/kg. Trihexyphenidyl was used as a positive control. Behavioral assessments began 15 min after compound administration. Abnormal movements were scored for 30 s at 10 min intervals for 60 min. Mice were tested in a repeated measures design with a pseudorandom order of drug doses and vehicle; each mouse received every dose only once within an experiment. Mice were given at least 4 days between challenges. Observers were blinded to treatment. An arithmetic sum of the disability scores was used to calculate a total score for the entire 1 h session. These data approximate a continuous variable when total scores from one animal are added together.
Acknowledgments
Experiments were performed in part through the use of the Neurobehavior Core lab at the Vanderbilt University Medical Center. The authors would like to thank the following funding sources for their generous support of this work: Ancora Innovation, LLC contract to P.J.C. and C.W.L., Michael J. Fox Foundation Target Advancement Program Award 13445 to M.S.M., and NIH grants R01NS08852 to E.J.H., R01MH073676 to P.J.C., K99NS110878 and R00NS110878 to M.S.M., and DoD grant W81XWH-19-1-0355 to P.J.C.
Glossary
Abbreviations
- μM
micromolar
- 5-HT2B
serotonin receptor subtype 2B ACh, acetylcholine
- ANOVA
analysis of variance
- AUC
area under the curve
- BG
basal ganglia
- cm
centimeter
- D1
dopamine receptor subtype 1
- D1-SPN
direct pathway spiny projection neuron
- DA
dopamine
- DMPK
drug metabolism and pharmacokinetics
- DS
dorsal striatum
- DRD
DOPA-responsive dystonia
- EC80
effective concentration of 80% maximal response
- ER
efflux ratio
- GABA
γ-aminobutyric acid
- GPCR
G-protein coupled receptor
- HIC
haloperidol-induced catalepsy
- IC50
inhibitory concentration of 50% maximal response
- i.p.
intraperitoneal, KO, knockout
- L-DOPA
l-3,4-dihydroxyphenylalanine
- M1
muscarinic acetylcholine receptor subtype 1
- M2
muscarinic acetylcholine receptor subtype 2
- M3
muscarinic acetylcholine receptor subtype 3
- M4
muscarinic acetylcholine receptor subtype 4
- M5
muscarinic acetylcholine receptor subtype 5
- mAChR
muscarinic acetylcholine receptors
- mIPSC
miniature inhibitory post synaptic currents
- nAChR
nicotinic acetylcholine receptor
- nM
nanomolar
- NMR
nuclear magnetic resonance
- Oxo-M
oxotremorine
- PK
pharmacokinetic
- PD
pharmacodynamic
- PD
Parkinson’s disease
- p.o.
per oral
- s.c.
subcutaneous
- SNc
substantia nigra pars compacta
- SNr
substantia nigra pars reticulata
- WT
wild-type
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00162.
Additional synthesis, NMR details, binding data, and experimental evaluation of VU6013720, VU6021302, and VU6021625 (PDF)
Author Contributions
M.S.M., A.M.B., D.J.F., J.W.D., A.Q., H.P.C., W.P., T.M.B., Z.B., S.C., K.J.W., J.C.O., Y.D., J.L.E., and L.P. performed experiments. M.S.M., A.M.B., D.J.F., J.W.D., W.P., A.Q., H.P.C., Z.B., T.M.B., A.L.R., C.M.N., and J.M.R. analyzed data. M.S.M., D.J.F., H.P.C., C.M.N., C.W.L., E.J.H., P.J.C., and J.M.R. designed experiments. M.S.M. and J.M.R. wrote the manuscript with the input of all authors.
The authors declare the following competing financial interest(s): C.M.N., J.W.D., C.W.L., P.J.C., T.M.B., A.M.B., J.L.E., and J.M.R. are inventors on applications for composition of matter patents that protect several series of M4 antagonists.
Supplementary Material
References
- Albin R. L.; Young A. B.; Penney J. B. (1989) The functional anatomy of basal ganglia disorders. Trends Neurosci. 12 (10), 366–75. 10.1016/0166-2236(89)90074-X. [DOI] [PubMed] [Google Scholar]
- DeLong M. R. (1990) Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 13 (7), 281–5. 10.1016/0166-2236(90)90110-V. [DOI] [PubMed] [Google Scholar]
- DeLong M.; Wichmann T. (2009) Update on models of basal ganglia function and dysfunction. Parkinsonism & related disorders 15 (3), S237–40. 10.1016/S1353-8020(09)70822-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson A. B.; Kreitzer A. C. (2014) Reassessing models of basal ganglia function and dysfunction. Annu. Rev. Neurosci. 37, 117–35. 10.1146/annurev-neuro-071013-013916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breakefield X. O.; Blood A. J.; Li Y.; Hallett M.; Hanson P. I.; Standaert D. G. (2008) The pathophysiological basis of dystonias. Nat. Rev. Neurosci. 9 (3), 222–234. 10.1038/nrn2337. [DOI] [PubMed] [Google Scholar]
- Karimi M.; Perlmutter J. S. (2020) The role of dopamine and dopaminergic pathways in dystonia: insights from neuroimaging. Tremor and other hyperkinetic movements (New York, N.Y.) 5, 280. 10.5334/tohm.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korczyn A. D. (2004) Drug treatment of Parkinson’s disease. Dialogues Clin Neurosci 6 (3), 315–322. 10.31887/DCNS.2004.6.3/akorczyn. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanvi B.; Lo N.; Robinson T. (2007) Levodopa-induced dyskinesia in Parkinson’s disease: clinical features, pathogenesis, prevention and treatment. Postgrad. Med. J. 83 (980), 384–8. 10.1136/pgmj.2006.054759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehle M. S.; Conn P. J. (2019) Mov. Disord. 34 (8), 1089–1099. 10.1002/mds.27740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. J.; Jones C. K.; Lindsley C. W. (2009) Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol. Sci. 30 (3), 148–55. 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S.; Burke R.; Stern Y. (1990) Antimuscarinic drugs in the treatment of movement disorders. Prog. Brain Res. 84, 389–97. 10.1016/S0079-6123(08)60922-X. [DOI] [PubMed] [Google Scholar]
- Burke R. E.; Fahn S.; Marsden C. D. (1986) Torsion dystonia: a double-blind, prospective trial of high-dosage trihexyphenidyl. Neurology 36 (2), 160–4. 10.1212/WNL.36.2.160. [DOI] [PubMed] [Google Scholar]
- Aosaki T.; Miura M.; Suzuki T.; Nishimura K.; Masuda M. (2010) Acetylcholine-dopamine balance hypothesis in the striatum: an update. Geriatrics & gerontology international 10 (1), S148–57. 10.1111/j.1447-0594.2010.00588.x. [DOI] [PubMed] [Google Scholar]
- Benarroch E. E. (2012) Effects of acetylcholine in the striatum. Recent insights and therapeutic implications. Neurology 79 (3), 274–81. 10.1212/WNL.0b013e31825fe154. [DOI] [PubMed] [Google Scholar]
- Moehle M. S.; Pancani T.; Byun N.; Yohn S. E.; Wilson G. H. 3rd; Dickerson J. W.; Remke D. H.; Xiang Z.; Niswender C. M.; Wess J.; Jones C. K.; Lindsley C. W.; Rook J. M.; Conn P. J. (2017) Cholinergic Projections to the Substantia Nigra Pars Reticulata Inhibit Dopamine Modulation of Basal Ganglia through the M(4) Muscarinic Receptor. Neuron 96 (6), 1358–1372. 10.1016/j.neuron.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomeza J.; Zhang L.; Kostenis E.; Felder C.; Bymaster F.; Brodkin J.; Shannon H.; Xia B.; Deng C.; Wess J. (1999) Enhancement of D1 dopamine receptor-mediated locomotor stimulation in M(4) muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. U. S. A. 96 (18), 10483–10488. 10.1073/pnas.96.18.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wess J.; Eglen R. M.; Gautam D. (2007) Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat. Rev. Drug Discovery 6 (9), 721–733. 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- Jeon J.; Dencker D.; Wörtwein G.; Woldbye D. P. D.; Cui Y.; Davis A. A.; Levey A. I.; Schütz G.; Sager T. N.; Mørk A.; Li C.; Deng C.-X.; Fink-Jensen A.; Wess J. (2010) A subpopulation of neuronal M4 muscarinic acetylcholine receptors plays a critical role in modulating dopamine-dependent behaviors. J. Neurosci. 30 (6), 2396–2405. 10.1523/JNEUROSCI.3843-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster D. J.; Wilson J. M.; Remke D. H.; Mahmood M. S.; Uddin M. J.; Wess J.; Patel S.; Marnett L. J.; Niswender C. M.; Jones C. K.; Xiang Z.; Lindsley C. W.; Rook J. M.; Conn P. J. (2016) Antipsychotic-like Effects of M4 Positive Allosteric Modulators Are Mediated by CB2 Receptor-Dependent Inhibition of Dopamine Release. Neuron 91 (6), 1244–1252. 10.1016/j.neuron.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloud L. J.; Jinnah H. A. (2010) Treatment strategies for dystonia. Expert Opin. Pharmacother. 11 (1), 5–15. 10.1517/14656560903426171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokland A.; Sambeth A.; Prickaerts J.; Riedel W. J. (2016) Why an M1 Antagonist Could Be a More Selective Model for Memory Impairment than Scopolamine. Frontiers in neurology 7, 167. 10.3389/fneur.2016.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson K.-E.; Campeau L.; Olshansky B. (2011) Cardiac effects of muscarinic receptor antagonists used for voiding dysfunction. Br. J. Clin. Pharmacol. 72 (2), 186–196. 10.1111/j.1365-2125.2010.03813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller K.; Peel J. L. (1990) Scopolamine produces locomotor stereotypy in an open field but apomorphine does not. Pharmacol., Biochem. Behav. 36 (3), 613–7. 10.1016/0091-3057(90)90264-I. [DOI] [PubMed] [Google Scholar]
- Duty S.; Jenner P. (2011) Animal models of Parkinson’s disease: a source of novel treatments and clues to the cause of the disease. British journal of pharmacology 164 (4), 1357–91. 10.1111/j.1476-5381.2011.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croy C. H.; Chan W. Y.; Castetter A. M.; Watt M. L.; Quets A. T.; Felder C. C. (2016) Characterization of PCS1055, a novel muscarinic M4 receptor antagonist. Eur. J. Pharmacol. 782, 70–6. 10.1016/j.ejphar.2016.04.022. [DOI] [PubMed] [Google Scholar]
- Cho T. P.; Long Y. F.; Gang L. Z.; Yang W.; Jun L. H.; Yuan S. G.; Hong F. J.; Lin W.; Liang G. D.; Lei Z.; Jing L. J.; Shen G. A.; Hong S. G.; Dan W.; Ying F.; Ke Y. P.; Ying L.; Jun F.; Tai M. X. (2010) Synthesis and biological evaluation of azobicyclo[3.3.0] octane derivatives as dipeptidyl peptidase 4 inhibitors for the treatment of type 2 diabetes. Bioorg. Med. Chem. Lett. 20 (12), 3565–3568. 10.1016/j.bmcl.2010.04.120. [DOI] [PubMed] [Google Scholar]
- Bubser M.; Bridges T. M.; Dencker D.; Gould R. W.; Grannan M.; Noetzel M. J.; Lamsal A.; Niswender C. M.; Daniels J. S.; Poslusney M. S.; Melancon B. J.; Tarr J. C.; Byers F. W.; Wess J.; Duggan M. E.; Dunlop J.; Wood M. W.; Brandon N. J.; Wood M. R.; Lindsley C. W.; Conn P. J.; Jones C. K. (2014) Selective activation of M4 muscarinic acetylcholine receptors reverses MK-801-induced behavioral impairments and enhances associative learning in rodents. ACS Chem. Neurosci. 5 (10), 920–42. 10.1021/cn500128b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady A. E.; Jones C. K.; Bridges T. M.; Kennedy J. P.; Thompson A. D.; Heiman J. U.; Breininger M. L.; Gentry P. R.; Yin H.; Jadhav S. B.; Shirey J. K.; Conn P. J.; Lindsley C. W. (2008) Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J. Pharmacol. Exp. Ther. 327 (3), 941–53. 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose S. J.; Yu X. Y.; Heinzer A. K.; Harrast P.; Fan X.; Raike R. S.; Thompson V. B.; Pare J. F.; Weinshenker D.; Smith Y.; Jinnah H. A.; Hess E. J. (2015) A new knock-in mouse model of l-DOPA-responsive dystonia. Brain 138 (10), 2987–3002. 10.1093/brain/awv212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong G. Z.; Kameyama K.; Rinken A.; Haga T. (1994) Ligand binding properties of muscarinic acetylcholine receptor subtypes (m1-m5) expressed in baculovirus-infected insect cells. Jpn. J. Pharmacol. 274 (1), 378–84. 10.1016/S0021-5198(19)50375-4. [DOI] [PubMed] [Google Scholar]
- Böhme T. M.; Augelli-Szafran C. E.; Hallak H.; Pugsley T.; Serpa K.; Schwarz R. D. (2002) Synthesis and pharmacology of benzoxazines as highly selective antagonists at M(4) muscarinic receptors. J. Med. Chem. 45 (14), 3094–102. 10.1021/jm011116o. [DOI] [PubMed] [Google Scholar]
- Kharkwal G.; Brami-Cherrier K.; Lizardi-Ortiz J. E.; Nelson A. B.; Ramos M.; Del Barrio D.; Sulzer D.; Kreitzer A. C.; Borrelli E. (2016) Parkinsonism Driven by Antipsychotics Originates from Dopaminergic Control of Striatal Cholinergic Interneurons. Neuron 91 (1), 67–78. 10.1016/j.neuron.2016.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W.; Plotkin J. L.; Francardo V.; Ko W. K.; Xie Z.; Li Q.; Fieblinger T.; Wess J.; Neubig R. R.; Lindsley C. W.; Conn P. J.; Greengard P.; Bezard E.; Cenci M. A.; Surmeier D. J. (2015) M4Muscarinic Receptor Signaling Ameliorates Striatal Plasticity Deficits in Models of L-DOPA-Induced Dyskinesia. Neuron 88 (4), 762–73. 10.1016/j.neuron.2015.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeek D. S.; Gasser T. (2017) Unmet Needs in Dystonia: Genetics and Molecular Biology-How Many Dystonias?. Frontiers in neurology 7, 241. 10.3389/fneur.2016.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs T.; Saunders-Pullman R.; Masuho I.; Luciano M. S.; Raymond D.; Factor S.; Lang A. E.; Liang T. W.; Trosch R. M.; White S.; Ainehsazan E.; Hervé D.; Sharma N.; Ehrlich M. E.; Martemyanov K. A.; Bressman S. B.; Ozelius L. J. (2013) Mutations in GNAL cause primary torsion dystonia. Nat. Genet. 45 (1), 88–92. 10.1038/ng.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; DeCuypere M.; LeDoux M. S. (2008) Abnormal motor function and dopamine neurotransmission in DYT1 DeltaGAG transgenic mice. Exp. Neurol. 210 (2), 719–30. 10.1016/j.expneurol.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs A. M.; Fan X.; Donsante C.; Jinnah H. A.; Hess E. J. (2019) Trihexyphenidyl rescues the deficit in dopamine neurotransmission in a mouse model of DYT1 dystonia. Neurobiol. Dis. 125, 115–122. 10.1016/j.nbd.2019.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancani T.; Foster D. J.; Moehle M. S.; Bichell T. J.; Bradley E.; Bridges T. M.; Klar R.; Poslusney M.; Rook J. M.; Daniels J. S.; Niswender C. M.; Jones C. K.; Wood M. R.; Bowman A. B.; Lindsley C. W.; Xiang Z.; Conn P. J. (2015) Allosteric activation of M4 muscarinic receptors improve behavioral and physiological alterations in early symptomatic YAC128 mice. Proc. Natl. Acad. Sci. U. S. A. 112 (45), 14078–14083. 10.1073/pnas.1512812112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancani T.; Bolarinwa C.; Smith Y.; Lindsley C. W.; Conn P. J.; Xiang Z. (2014) M4 mAChR-mediated modulation of glutamatergic transmission at corticostriatal synapses. ACS Chem. Neurosci. 5 (4), 318–24. 10.1021/cn500003z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C. K.; Brady A. E.; Davis A. A.; Xiang Z.; Bubser M.; Tantawy M. N.; Kane A. S.; Bridges T. M.; Kennedy J. P.; Bradley S. R.; Peterson T. E.; Ansari M. S.; Baldwin R. M.; Kessler R. M.; Deutch A. Y.; Lah J. J.; Levey A. I.; Lindsley C. W.; Conn P. J. (2008) Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J. Neurosci. 28 (41), 10422–33. 10.1523/JNEUROSCI.1850-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould R. W.; Grannan M. D.; Gunter B. W.; Ball J.; Bubser M.; Bridges T. M.; Wess J.; Wood M. W.; Brandon N. J.; Duggan M. E.; Niswender C. M.; Lindsley C. W.; Conn P. J.; Jones C. K. (2018) Cognitive enhancement and antipsychotic-like activity following repeated dosing with the selective M(4) PAM VU0467154. Neuropharmacology 128, 492–502. 10.1016/j.neuropharm.2017.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F. M.; Lee C. R. (2011) Intrinsic and integrative properties of substantia nigra pars reticulata neurons. Neuroscience 198, 69–94. 10.1016/j.neuroscience.2011.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthur C. J.; Morrison R.; Felts A. S.; Smith K. A.; Engers J. L.; Byers F. W.; Daniels J. S.; Emmitte K. A.; Conn P. J.; Lindsley C. W. (2013) Discovery of (R)-(2-fluoro-4-((−4-methoxyphenyl)ethynyl)phenyl) (3-hydroxypiperidin-1-yl)methanone (ML337), an mGlu3 selective and CNS penetrant negative allosteric modulator (NAM). J. Med. Chem. 56 (12), 5208–12. 10.1021/jm400439t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devanagondi R.; Egami K.; LeDoux M. S.; Hess E. J.; Jinnah H. A. (2007) Neuroanatomical substrates for paroxysmal dyskinesia in lethargic mice. Neurobiol. Dis. 27 (3), 249–57. 10.1016/j.nbd.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raike R. S.; Pizoli C. E.; Weisz C.; van den Maagdenberg A. M. J. M.; Jinnah H. A.; Hess E. J. (2013) Limited regional cerebellar dysfunction induces focal dystonia in mice. Neurobiol. Dis. 49, 200–210. 10.1016/j.nbd.2012.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirley T. L.; Rao L. M.; Hess E. J.; Jinnah H. A. (2008) Paroxysmal dyskinesias in mice. Mov. Disord. 23 (2), 259–64. 10.1002/mds.21829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.