

Abstract
Background/Objectives.
While outcomes for pediatric T-cell acute lymphoblastic leukemia (T-ALL) are favorable, there are few widely accepted prognostic factors, limiting the ability to risk-stratify therapy.
Design/Methods.
DFCI Protocols 05-001 and 11-001 enrolled pediatric patients with newly diagnosed B- or T-ALL from 2005-2011 and 2012-2015, respectively. Protocol therapy was nearly identical for patients with T-ALL (N=123), who were all initially assigned to the high risk arm. End-induction minimal residual disease (MRD) was assessed by RT-PCR or next generation sequencing (NGS) but was not used to modify post-induction therapy. Early T-cell precursor (ETP) status was determined by flow cytometry. Cases with sufficient diagnostic DNA were retrospectively evaluated by targeted NGS of known genetic drivers of T-ALL, including Notch, PI3K, and Ras pathway genes.
Results.
The 5-year event free survival and overall survival (OS) for patients with T-ALL was 81% [95% CI, 73-87%] and 90% [95% CI, 83-94%], respectively. ETP phenotype was associated with failure to achieve complete remission, but not with inferior OS. Low end-induction MRD (<10−4) was associated with superior disease-free survival (DFS). Pathogenic mutations of the PI3K pathway were mutually exclusive of ETP phenotype and were associated with inferior 5-year DFS and OS.
Conclusions.
Together, our findings demonstrate that ETP phenotype, end-induction MRD and PI3K pathway mutation status are prognostically relevant in pediatric T-ALL and should be considered for risk classification in future trials. DFCI Protocols 05-001 and 11-001 are registered at www.clinicaltrials.gov as NCT00400946 and NCT01574274, respectively.
Keywords: T-ALL; Clinical Trials; Pediatric Oncology; ALL, Molecular Diagnosis and Therapy; Minimal Residual Disease
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) comprises approximately 10 to 15% of newly diagnosed pediatric acute lymphoblastic leukemia (ALL). Historically, T-ALL has been associated with an inferior prognosis, but with more contemporary therapy, outcomes for T-ALL and B-cell ALL (B-ALL) are similar.1,2 However, while clinical presenting features and key genetic perturbations have been identified as independent risk factors in B-ALL 3-12, there are few widely accepted or validated prognostic factors in T-ALL, except for treatment response, including glucocorticoid prophase response13-15, induction remission status16,17, and minimal residual disease (MRD) levels at end-induction and at a second later timepoint.10,18,19
There is controversy regarding the prognostic significance of several factors in T-ALL, including early T-cell precursor (ETP) phenotype and various recurrent genomic aberrations. ETP subtype, occurring in approximately 15% of T-ALL cases, was first identified by gene expression profiling and subsequently linked to a distinctive immunophenotype.20 In initial reports, ETP status was associated with high rates of induction failure, early relapse, and poor overall survival.18,20,21 However, in more recent reports, despite an association with slow early response, ETP status has lacked prognostic significance.22-24
While multiple recurrent genomic aberrations have been identified in T-ALL25,26, few have been reproducibly associated with prognosis and none are prospectively used in risk-stratification. Activating mutations of the Notch pathway are found in over 50% of T-ALL cases25,27,28, and have been associated with favorable outcomes in pediatric and adult T-ALL studies, albeit inconsistently.28-30 Additionally, mutational dysregulation of PI3K and Ras pathways have been associated with poor outcomes in childhood T-ALL.31-35 In a study of adult patients with T-ALL published by the Group for Research in Adult Acute Lymphoblastic Leukemia (GRAALL), the presence of a NOTCH1 activating mutation combined with the absence of PTEN/RAS alterations predicted a favorable outcome.36 The French Acute Lymphoblastic Leukaemia (FRALLE) Study Group proposed a new pediatric T-ALL risk stratification paradigm that combined MRD assessment and genetic analysis of NOTCH1, FBXW7, PTEN, KRAS, NRAS.37 To date, this risk stratification scheme has not been validated in another pediatric population.
On Dana-Farber Cancer Institute (DFCI) ALL Consortium Protocols 05-001 and 11-001, patients with T-ALL were treated with a uniform high risk (HR) regimen.38 Here we present the results of patients with T-ALL treated on these protocols, assess the impact of end-induction MRD, ETP immunophenotype, and mutational status of Notch, PI3K, and Ras pathways on outcome, and further investigate validation of the novel FRALLE risk classification in our cohort.
Methods
Study Design
DFCI 05-001 (NCT00165087) and DFCI 11-001 (NCT01574274) were consecutive Phase III open-label, randomized trials that enrolled patients with newly diagnosed ALL aged 1-18 years between 2005-201138, and aged 1-21 years between 2012-201539, respectively. DFCI served as the study sponsor and coordinating center. Each protocol was approved by the Institutional Review Board at participating institutions. Written informed consent from each participant’s parent/legal guardian, as well as patient assent, when appropriate, was obtained prior to enrollment and initiation of therapy.
Protocol Treatment
Details of DFCI 05-001 therapy have been previously published.38 Protocol therapy, which was nearly identical in each study, is summarized in Table S1. All patients with T-ALL received multiagent induction chemotherapy, including anthracycline. Patients with KMT2A-rearrangements or hypodiploidy were changed to the very high risk (VHR) group; those with Philadelphia chromosome-positive (Ph+) ALL were removed from protocol treatment by end-induction but were followed for outcome analyses. All remaining patients continued treatment on the HR arm. Complete remission (CR) status was assessed at Day 32; patients meeting definition of induction failure were removed from study therapy. Cranial radiation was administered to all patients with T-ALL regardless of CNS status. Therapy was completed 24 months after CR date. Because there was no difference in toxicity or outcome between native E.coli L-asparaginase and pegaspargase on DFCI 05-001 and between calaspargase pegol and pegaspargase on DFCI 11-001 results have been combined.38,39
MRD Assessment
Marrow samples were collected to prospectively assess end-induction MRD by reverse transcription polymerase chain reaction (RT-PCR), as previously described40; results were not used to modify post-induction therapy. Retrospective MRD assessment was performed using an NGS assay41 for patients with available stored samples whose MRD was inevaluable by RT-PCR. As studies have shown high concordance between these two methodologies41,42, MRD results were combined for analyses.
Identification of Early T-cell precursor (ETP) ALL
On DFCI 05-001, ETP status was retrospectively determined by assessment of diagnostic flow cytometry and was defined as absence of CD1a and CD8, weak CD5 expression, and coexpression of myeloid and/or stem cell markers.20 On DFCI 11-001, ETP status was determined prospectively through central review at DFCI; those who achieved CR by Day 32 were changed to VHR group (N=2) and received two additional cycles of consolidation chemotherapy (Table S1).
Targeted Next Generation Sequencing and Analysis
Genomic DNA (gDNA) was extracted from viably frozen, banked diagnostic specimens and targeted exome sequencing and analysis was performed at the Center for Cancer Genome Discovery at DFCI using an Illumina sequencing platform for all protein-coding exons of the genes in Table S2, as previously described.43 Fourteen T-ALL diagnostic samples underwent targeted NGS and analysis using the clinical OncoPanel platform at DFCI, as previously described.44
Mutation calls were made for variants predicted to result in a non-synonymous amino acid alteration, frameshift mutation, stop codon, or splice site alteration, and for variants with at least ten reads of the mutant allele. Germline variant filters were applied and those represented in the Genome Aggregation Database (gnomADv.2.0) at > 0.01% frequency at time of analysis were excluded.
Analysis of mutations was restricted to the following previously implicated pathogenic genes in T-ALL: NOTCH1, FBXW7, PTEN, AKT, PIK3R1, KRAS, NRAS, NF1, PTPN11. Pathogenic mutations were defined based on the following criteria: missense mutations with experimental evidence demonstrating gain-of-function or loss-of-function effects; truncating or frameshift mutations of tumor suppressors; C-terminal truncating or frameshift mutations predicted to delete the C-terminal negative regulatory domains of NOTCH1 (PEST domain), or the PI3K regulatory subunit PIK3R1; missense mutations of pathogenic residues in closely related paralogs.45
Statistical Methods
Induction failure was defined as ≥ 5% marrow blasts and/or residual extramedullary disease at end-induction. Event-free survival (EFS) was defined as the time from diagnosis to first outcome event; overall survival (OS) was calculated from the time of diagnosis to death from any cause; these analyses included all 123 enrolled subjects. Disease-free survival (DFS) was calculated for those who achieved a complete remission (N=108) from time of remission to relapse or death. The Wilcoxon rank sum test compared continuous measures and the Fisher exact test compared categorical variables. Time-to-event outcomes were estimated using the Kaplan-Meier method and compared using a log-rank test. Univariate and multivariable logistic regression models of achievement of CR and a Cox proportional-hazards model of EFS were investigated with presenting clinical characteristics as covariates. P values are two-sided, and values less than 0.05 were considered significant.
Results
Overall T-ALL Outcomes
A total of 123 patients with T-ALL were enrolled and considered evaluable (Fig. 1); presenting characteristics are presented in Table 1. There were three induction deaths and 12 induction failures (Table S3); 108 subjects (88%) achieved CR (Table 2). In both univariate and multivariable analyses, the only presenting features associated with failure to achieve CR were CNS-3 and ETP status (Table S4). When the logistic regression model was applied to patients with non-ETP ALL, there were no significant predictors of failure to achieve CR. Of the 108 patients achieving CR, one withdrew from protocol therapy prior to final risk group assignment, three were classified as VHR (one for KMT2A-rearrangement and two on 11-001 for ETP immunophenotype), one had Ph+ T-ALL and was subsequently removed from protocol therapy for hematopoietic stem cell transplant (HSCT); the remaining 103 patients were classified as HR. Six patients relapsed, one died in remission, and one died of a secondary malignancy 11 months after achievement of CR (Table S5). With a median follow-up of 5.3 years (range 0.1 to 10 years), the 5-year EFS for all 123 patients was 81% [95% CI, 73-87%] and the 5-year OS was 90% [95% CI, 83-94%] (Fig. 2A). Age, presenting leukocyte count, sex and presence of a mediastinal mass were not significantly associated with EFS or OS (Table 3). When a Cox proportional-hazards model was applied, ETP status was the only characteristic associated with EFS (Table S6).
FIGURE 1.
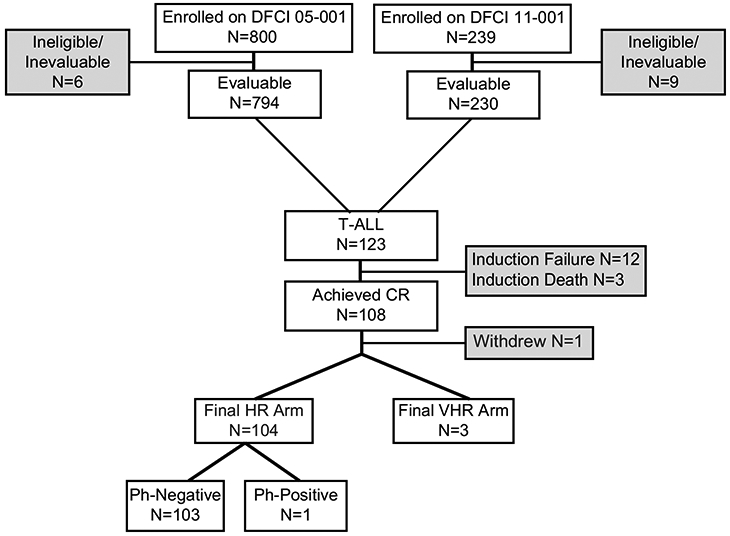
DFCI 05-001 and 11-001 patient enrollment and risk group stratification for patients with T-ALL All patients with T-ALL were enrolled to the initial HR arm and received a multiagent induction regimen. Final risk stratification was performed post-induction chemotherapy for patients who achieved CR. Three patients with T-ALL were upstaged to the final VHR arm (one participant on the basis of a KMT2A-rearrangement and two participants with ETP-ALL). Details of protocol therapy are provided in Supplementary Information Table S1. CR, complete remission; HR, high risk; VHR, very high risk; Ph, Philadelphia chromosome.
TABLE 1.
Presenting clinical characteristics of patients with T-ALL on DFCI 05-001 and 11-001
| Patient Characteristic | N (%) | non ETP T-ALL N (%) |
ETP-ALL* N (%) |
p-value† |
|---|---|---|---|---|
| No. Pts. | 123‡ | 100 | 21 | |
| Protocol | ||||
| 05-001 | 97 (79) | 78 (78) | 17 (81) | 0.99 |
| 11-001 | 26 (21) | 22 (22) | 4 (19) | |
| Age | ||||
| <10 yrs | 67 (54) | 58 (58) | 7 (33) | 0.054 |
| ≥10 yrs | 56 (46) | 42 (42) | 14 (67) | |
| median (range) yrs. | 8.5 (1.2, 17.9) | 8.2 (1.2, 18) | 11.1 (2.9, 17.9) | 0.086 |
| Sex | ||||
| Male | 91 (74) | 77 (77) | 13 (62) | 0.17 |
| Female | 32 (26) | 23 (23) | 8 (38) | |
| WBC | ||||
| <50 K | 53 (43) | 35 (35) | 17 (81) | 0.0002 |
| ≥50 K | 70 (57) | 65 (65) | 4 (19) | |
| median (range) | 62.4 (0.9, 905.0) | 81.1 (1.1, 905.0) | 13.4 (1.0, 259.2) | 0.0001 |
| Mediastinal Mass | ||||
| Yes | 55 (45) | 48 (48) | 6 (29) | 0.15 |
| No | 68 (55) | 52 (52) | 15 (71) | |
| CNS Status | ||||
| 1 | 71 (58) | 59 (59) | 11 (52) | 0.76 |
| 2 | 42 (34) | 33 (33) | 8 (38) | |
| 3 | 10 (8) | 8 (8) | 2 (10) | |
Includes 1 case of probable ETP, which was not assessed for CD1a status
Comparison of non ETP-ALL and ETP-ALL
Of the 123 patients with T-ALL enrolled, sufficient data to evaluate ETP status was available for 121 patients
TABLE 2.
Outcome of patients with T-ALL on DFCI 05-001 and 11-001
| N (%) | non ETP T-ALL N (%) |
ETP-ALL† N (%) |
p-value | |
|---|---|---|---|---|
| Number of Patients | 123 | 100 | 21 | |
| Induction Death | 3 (2) | 2 (2) | 1 (5) | 0.44 |
| Induction Failure* | 12 (10) | 5 (5) | 7 (33) | 0.0008 |
| M1 Marrow w/ persistent CNS blasts | 1 | 1 | 0 | |
| M2 Marrow | 5 | 3 | 2 | |
| M3 Marrow | 5 | 1 | 4 | |
| Unknown‡ | 1 | 0 | 1 | |
| Achieved CR* | 108 (88) | 93 (93) | 13 (62) | 0.0007 |
| MRD < 10−3 | 69 (75) | 66 (76) | 1 (8) | |
| MRD ≥ 10−3 | 23 (25) | 21 (24) | 2 (15) | |
| MRD < 10−4 | 45 (49) | 43 (49) | 1 (8) | |
| MRD ≥ 10−4 | 47 (51) | 44(51) | 2 (15) | |
| Indeterminate/Unknown MRD | 16 (15) | 6 (6) | 10 (77) | |
| Relapse§ | 6 | 4 | 1 | |
| Remission Death | 1 | 1 | 0 | |
| Second Malignant Neoplasm║ | 1 | 1 | 0 | |
| 5 yr EFS (%) [95% CI] | 81 [73-87] | 87 [79-92] | 54 [29-74] | 0.0006 |
| 5 yr OS (%) [95% CI] | 90 [83-94] | 92 [84-96] | 85 [61-95] | 0.31 |
Assessed at end of first month of treatment (Day 32)
Includes 1 case of probable ETP, which was not assessed for CD1a status
Includes 1 subject that was taken off study on D18 due to progressive disease, no D32 marrow was assessed
Includes 1 subject with unknown ETP status
This subject subsequently died in remission 11 months following achievement of CR
FIGURE 2.
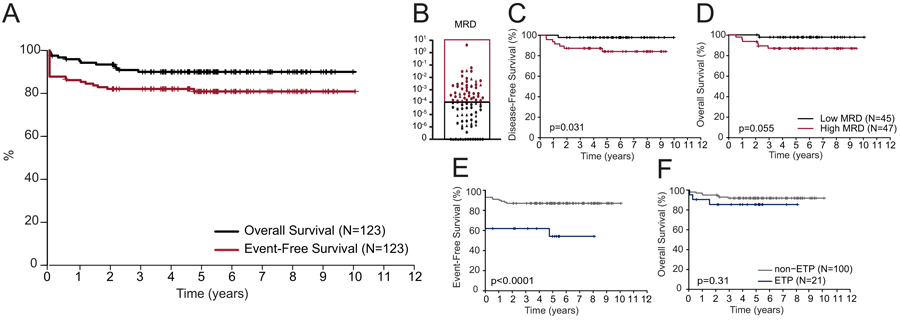
Outcomes for patients with T-ALL treated on DFCI 05-001 and 11-001. (A) Kaplan-Meier analysis of overall (OS) and event-free (EFS) survival for all 123 patients with T-ALL treated on DFCI 05-001 and 11-001. (B) End-induction minimal residual disease (MRD) results for T-ALL participants treated on DFCI Protocols 05-001 and 11-001. Kaplan-Meier analysis of (C) disease-free survival (DFS) and (D) OS for patients with low (<10−4) and high (≥10−4) MRD; and (E) EFS and (F) OS for ETP vs. non-ETP ALL. A two-sided log rank (Mantel-Cox) test was used to test for differences in survival between groups in panels B-F. ■ denotes MRD assessment by RT-PCR; ▲ denotes MRD assessment by NGS of Ig and TCR rearrangements. MRD, Minimal residual disease.
TABLE 3.
Presenting characteristics, molecular analysis and outcome for participants with T-ALL on DFCI 05-001 and 11-001
| N (%) | 5-yr DFS%* [95% CI] |
p-value | 5-yr EFS% [95% CI] |
p-value | 5-yr OS% [95% CI] |
p-value | |
|---|---|---|---|---|---|---|---|
| No. Pts. | 123 | 81 [73-87] | 90 [83-94] | ||||
| Age | |||||||
| <10 yrs. | 67 (54) | 83 [72-90] | 0.49 | 91 [80-96] | 0.75 | ||
| ≥10 yrs. | 56 (46) | 78 [64-87] | 89 [78-95] | ||||
| WBC | |||||||
| < 50K | 53 (43) | 79 [64-88] | 0.60 | 92 [81-97] | 0.48 | ||
| ≥ 50K | 70 (57) | 83 [72-90] | 88 [78-94] | ||||
| Sex | |||||||
| Male | 91 (74) | 81 [72-88] | 0.97 | 90 [81-95] | 0.99 | ||
| Female | 32 (26) | 80 [60-90] | 91 [74-97] | ||||
| CNS Status | |||||||
| CNS 1 | 71 (58) | 87 [76-93] | 0.052 | 94 [85-98] | 0.17 | ||
| CNS 2 | 42 (34) | 76 [60-86] | 86 [71-95] | ||||
| CNS 3 | 10 (8) | 60 [25-83] | 80 [41-93] | ||||
| Mediastinal Mass | |||||||
| Yes | 55 (45) | 85 [73-92] | 0.27 | 89 [77-95] | 0.77 | ||
| No | 68 (55) | 77 [65-86] | 91 [81-96] | ||||
| ETP Phenotype | |||||||
| Yes | 100 (81) | 54 [29-74] | 0.0006 | 85 [61-95] | 0.31 | ||
| No | 21 (17) | 87 [79-92] | 92 [84-96] | ||||
| Unknown | 2 (2) | --- | ---- | ||||
| End-induction MRD | |||||||
| <10−3 | 69 (75) | 94 [85-98] | 0.079 | --- | 94 [85-98] | 0.24 | |
| ≥10−3 | 23 (25) | 79 [51-92] | --- | 86 [65-96] | |||
| <10−4 | 45 (49) | 98 [85-99] | 0.031 | --- | 98 [85-99] | 0.055 | |
| ≥10−4 | 47 (51) | 84 [69-92] | --- | 87 [73-94] | |||
| Post-Induction Asparaginase† | |||||||
| E.coli | 59 (48) | 93 [83-97] | 0.68 | 93 [82-97] | 0.81 | ||
| Pegaspargase | 37 (30) | 88 [72-95] | 92 [76-97] | ||||
| Erwinia | 2 (2) | 100 [NA] | 100 [NA] | ||||
| Calaspargase pegol | 9 (7) | 100 [NA] | 100 [NA] | ||||
| Gene/Pathway Mutations‡ | |||||||
| NOTCH1 gene | |||||||
| Present | 59 (51) | 92 [80-97] | 0.84 | 86 [73-93] | 0.26 | 95 [85-98] | 0.080 |
| Absent | 56 (49) | 91 [79-97] | 78 [65-87] | 85 [72-92] | |||
| Notch Pathway§ | |||||||
| Present | 66 (57) | 90 [78-95] | 0.39 | 84 [72-91] | 0.43 | 92 [83-97] | 0.34 |
| Absent | 49 43) | 95 [82-99] | 79 [65-88] | 87 [74-94] | |||
| PI3K Pathway║ | |||||||
| Present | 18 (16) | 81 [52-93] | 0.054 | 72 [46-87] | 0.19 | 77 [49-91] | 0.034 |
| Absent | 97 (84) | 94 [86-97] | 84 [75-90] | 93 [85-96] | |||
| Ras Pathway | |||||||
| Present | 13 (11) | 90 [84-96] | 0.73 | 69 [37-87] | 0.15 | 85 [51-96] | 0.43 |
| Absent | 103 (89) | 92 [84-96] | 84 [75-90] | 91 [83-95] | |||
DFS calculated for those who achieved CR and had evaluable MRD for outcome assessment by end-induction MRD or who achieved CR and had evaluable NGS for outcome assessment by genetic alterations
15 subjects did not achieve a CR and were not randomized; one participant withdrew from study therapy after achievement of CR, but before randomization
N=115 participants for whom diagnostic samples was available for NGS
Includes NOTCH1 and FBXW7 mutations
Includes PTEN, AKT, PIK3R1
Includes KRAS, NRAS, NF1, and PTPN11
Prognostic Impact of End-Induction MRD
Of the 108 patients who achieved CR at end-induction, MRD was evaluable in 92 patients; 71 patients had MRD evaluated prospectively using RT-PCR and 21 patients had MRD analyzed retrospectively using NGS. Therapy was not modified based on MRD results. MRD thresholds of ≥10−3 and ≥10−4 to define high MRD were both analyzed. At ≥10−3, there was a trend toward inferior 5-year DFS, but no difference in OS (Table 3). However, using a ≥10−4 threshold, there was an inferior 5-year DFS and OS for patients with high MRD (Fig. 2B-D; Table 3).
Outcomes of ETP-ALL
ETP status was evaluable in 121 patients, 21 of whom (17%) were classified as ETP. Patients with ETP-ALL had lower presenting leukocyte counts (p=0.0001). There was a non-significant trend toward older age at presentation and absence of a mediastinal mass for patients with ETP-ALL. All other presenting characteristics were similar (Table 1).
ETP status was a significant predictor of failure to achieve CR (p=0.0007), primarily due to induction failure (p=0.0008), resulting in an inferior EFS (p=0.0006) (Table 2 and Fig. 2E). There was no significant difference in OS between ETP and non-ETP groups (Table 2 and Fig. 2F). Of the seven patients with ETP-ALL and induction failure, six patients achieved CR following salvage chemotherapy before proceeding to HSCT; one patient died of progressive disease without achieving remission (Table S3). With a median follow-up of 5.03 years, five of six patients with ETP-ALL who proceeded to HSCT in CR1 remained alive at last follow-up; one patient relapsed and died following HSCT. MRD was evaluable in only 3 of 13 patients with ETP-ALL who achieved CR after initial induction, limiting the ability to analyze the prognostic value of end-induction MRD within this subset.
Outcomes by Pathogenic Mutations
Targeted NGS analysis was performed in 115 patients; 150 mutations in 80 patients were identified within the genes of interest (Table S7). Pathogenic mutations within Notch, PI3K, and Ras pathways were identified in 57%, 16% and 11% of patients, respectively (Fig. 3 and Table S7).
FIGURE 3.
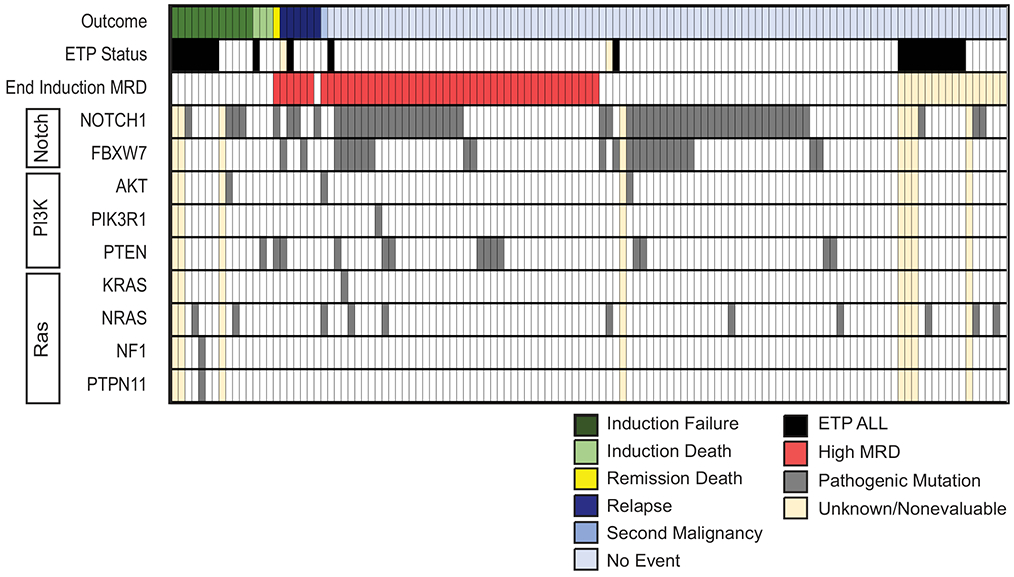
Mutational landscape and clinical outcomes of patients with T-ALL on DFCI 05-001 and 11-001. Summary of pathogenic mutations identified within Notch, PI3K, and Ras signaling pathways that are known to drive T-ALL and clinical outcome in patients treated on DFCI 05-001 and 11-001. ETP, Early T-cell Precursor; MRD, Minimal residual disease.
Ras pathway mutations were associated with younger age at presentation (17% <10 years vs. 4% ≥10 years, p=0.036); this was particularly notable for NRAS mutations (12% <10 years vs. 0% ≥10 years, p=0.037). FBXW7 mutations were associated with male sex (26% of males vs. 7% of females, p=0.035) and presence of a mediastinal mass (31% of patients with a mediastinal mass vs. 13% without a mediastinal mass, p=0.022). PTEN mutations were associated with a higher presenting leukocyte count (17% of patients with leukocyte count ≥50k cells/ul vs. 4% of patients with leukocyte count <50k cells/ul; p=0.043). Activating Notch pathway mutations, and more specifically NOTCH1 mutations were associated with non-ETP immunophenotype (62% of non-ETP vs. 25% of ETP had Notch pathway mutations, p=0.012; 56% of non-ETP vs. 19% of ETP had NOTCH1 mutations, p=0.006). PI3K pathway mutations and ETP immunophenotype were mutually exclusive. There were no other significant associations with presenting clinical characteristics.
Patients with T-ALL and low end-induction MRD were more likely to have activating NOTCH1 mutations (65% of children with MRD <10−3 (n=68), p=0.016; 68% of children with MRD <10−4 (n=44), p=0.056). However, there was no significant association between Notch pathway status and outcome, although there was a trend toward superior OS for children with activating NOTCH1 mutations (Table 3). Conversely, patients who harbored pathogenic PI3K pathway mutations had significantly inferior DFS and OS (Table 3). There was no significant association between outcome and Ras pathway mutation status.
The FRALLE Study Group recently proposed an oncogenetic risk stratification schema for pediatric T-ALL, which incorporated a 4-gene panel (NOTCH1, FBXW7, PTEN, RAS) to define low- and high-risk subtypes. In the FRALLE study, patients with low-risk subtype (defined as the presence of a NOTCH1 or FBXW7 mutation and the absence of a RAS or PTEN mutation) achieved a 5-year DFS of 88.6%, compared with a 5-year DFS of 60.4% for those classified as genetic high-risk (defined as the presence of a PTEN or RAS mutation regardless of NOTCH1/FBXW7 status, or absence of NOTCH1/FBXW7 and RAS/PTEN mutations).37 On our studies, patients meeting the FRALLE low-risk subtype criteria had a 5-year EFS of 86% [95% CI, 73-93%] and OS of 94% [95% CI, 83-98%]. Those meeting the FRALLE high-risk subtype criteria had 5-year EFS of 79% [95% CI, 66-87%] and OS of 87% [95% CI, 75-93%], not significantly different than patients meeting low-risk criteria (p=0.25 and 0.16, respectively) (Table S8 and Fig. S1). Similarly, when analyzing outcome for each genetic subgroup by end-induction MRD, using a threshold of 10−4 based on the FRALLE definition, there was no difference in EFS or OS for our T-ALL cohort (Table S8).
Discussion
Children and adolescents with T-ALL enrolled on DFCI 05-001 and 11-001 achieved a favorable outcome, with results similar to those reported for children and adolescents with B-ALL treated on DFCI 05-0013, confirming that T-cell immunophenotype, when treated on a HR ALL regimen, is no longer associated with inferior outcome in pediatric ALL. The EFS and OS reported here are similar to those recently reported for T-ALL patients treated on the COG AALL0434 study (5-year EFS 83.8% and OS 89.5%)46 and compare favorably to those reported by other groups.37,47 Significant prognostic factors associated with outcome in our T-ALL cohort included CNS status, ETP immunophenotype, end-induction MRD, and mutational status, most notably of PI3K pathway.
The prognostic significance of ETP-ALL remains a subject of debate. Our findings indicate that patients with ETP-ALL are more likely to have higher levels of end-induction residual disease compared with the non-ETP subgroup. While other studies have reported similar findings24,48, the rate of induction failure for ETP-ALL on our studies is higher than previous reports, which may in part be due to differences in definitions of this endpoint. In our analysis, induction failure was defined as ≥5% marrow blasts at Day 32, while other cooperative groups have used higher cut-offs (≥25% marrow blasts) and/or marrow status at later time points to define induction failure. Additional factors, such as the corticosteroid agent used during induction, may also influence rates of induction failure between protocols. Despite the high rate of induction failure, very few relapses were observed in patients with ETP-ALL who achieved CR and the OS for the ETP subgroup in our cohort was not significantly different than for non-ETP-ALL, indicating a high salvage rate. While the majority of our patients with ETP-ALL and induction failure were transplanted in CR1, others have shown that patients with ETP-ALL with suboptimal induction response can fare well with chemotherapy-only regimens.22,49 However, on COG AALL0434 patients with T-ALL and an M3 marrow after induction achieved a 4-year DFS of only 54.8% when treated with intensified chemotherapy.50 While this included patients with ETP and non-ETP ALL, there was a disproportionally high percentage of patients with ETP-ALL in this group. Given the high frequency of slow early response in ETP-ALL and the poor outcome associated with initial induction failure in T-ALL, investigation of novel strategies to improve initial induction response in ETP patients is warranted. For instance, preclinical data has demonstrated that ETP-ALL is dependent on BCL-2 signaling51, suggesting that these patients may benefit from the addition of BCL-2 inhibitors to sensitize leukemic cells to the effects of cytotoxic chemotherapy, potentially improving early response.
Unlike B-ALL, the prognostic implications of recurrent genetic mutations in T-ALL are not well defined. Activating NOTCH1 mutations are known to play a prominent role in the pathogenesis of T-ALL27, but their prognostic significance remains controversial. Results from the ALL-BFM 2000 study indicated that NOTCH1 mutations were associated with low MRD and superior relapse-free survival30; the GRAALL-2003 study similarly demonstrated that NOTCH1 mutations were a favorable prognostic factor.36 Conversely, a study from the Dutch Childhood Oncology Group and EORTC groups failed to demonstrate a clinically significant difference in outcome for patients harboring NOTCH1 mutations.52,53 Here we demonstrate that the presence of NOTCH1 activating mutations was associated with non-ETP status and more frequent in patients with low end-induction MRD. However, there was no statistically significant survival difference for patients based on NOTCH1 status. In contrast, we observed significantly lower DFS and OS in patients with PI3K pathway mutations, consistent with results from the FRALLE group and others.32,34,37 While we validated the excellent outcomes of T-ALL patients who met FRALLE definition of genetic low-risk, patients with genetic high-risk lesions had better outcomes on our trials than on the FRALLE study, which may be attributable to differences in therapy. Further work is needed to identify other genetic lesions that may more robustly predict outcomes in T-ALL to refine risk stratification and to develop novel targeted therapies.
We demonstrated the prognostic significance of end-induction MRD, confirming previous findings.10,19, 54 Our analysis indicates that an end-induction MRD threshold of 10−4 has greater prognostic value than a 10−3 cut-off, which has historically been used by several groups to risk-stratify patients. The 5-year DFS was 98% for patients with end-induction MRD <10−4 (approximately 50% of T-ALL patients in our cohort), defining a subset of patients with an extremely favorable prognosis. On COG ALL0434, which utilized a similar 4-drug induction, the EFS for those with end-induction MRD <0.01% (59% of patients) was 89% compared with 76% for those with MRD ≥0.01%48, while we observed a 5-year DFS of 84% for those with MRD ≥10−4. On COG AALL0434, patients were randomized to receive either Capizzi-methotrexate (escalating methotrexate dosing with pegaspargase) or high-dose methotrexate. Subset analyses indicated that that the use of Capizzi-methotrexate may benefit patients with high end-induction MRD. For patients with slow early response (defined as M2/M3 marrow at Day 15 or Day 29 MRD ≥0.1%) the 5-year DFS was 88.7% on the Capizzi-methotrexate arm compared with 78.1% on the high-dose methotrexate arm (p=0.06); there was no significant difference in outcome for patients with rapid early response.46 COG AALL0434 also demonstrated superiority of adding nelarabine to the augmented BFM backbone50; similar subgroup analyses based on end-induction MRD response for nelarabine randomization may add key information as to the potential benefit of nelarabine for patients with a slow early response, an intervention we are also testing on our ongoing trial, DFCI 16-001.
On the AIEOP-BFM ALL 2000 study, MRD negativity at end-induction was also the most favorable prognostic factor for patients with T-ALL; however, a favorable outcome was also achieved by patients with high TP1 MRD who achieved MRD negativity at a second later timepoint (TP2, Day 78). Patients with the worst prognosis had high TP2 MRD.18 Thus, assessment of TP2 MRD is important for risk stratification for T-ALL and can be used to differentiate those with high TP1 MRD who may fare well with standard therapy versus those who require alternative or novel treatment approaches to improve outcome. We have incorporated assessment of TP2 MRD in risk stratification for T-ALL in the currently open DFCI Consortium trial.
Historically, interpretation of the prognostic significance of MRD in T-ALL on our studies was limited by the high proportion of inevaluable results using the RT-PCR assay, primarily resulting from failure to identify specific immunoglobulin (Ig) or T-cell receptor (TCR) gene rearrangements. This is particularly problematic for ETP-ALL, which is derived from early thymic precursor cells that have not undergone TCR gene rearrangement.20,21 Flow cytometry is successfully used by many groups to quantify MRD in T-ALL, and has the advantage of faster turnaround time and a lower failure rate compared with PCR-based assay; studies have demonstrated a high concordance between the two assays.54 More recently, an NGS-based MRD assay has been developed to assess clonal Ig and TCR gene rearrangements, which has a lower reported failure rate and higher level of sensitivity than either PCR or flow-based MRD.41 Here, we retrospectively assessed end-induction MRD in patients with T-ALL for whom MRD was previously inevaluable by RT-PCR. NGS MRD assessment was successfully performed in 21 of 23 non-ETP samples (91%) for which RT-PCR MRD results were indeterminate. MRD assessment remains challenging in ETP-ALL, however. Of the three patients with ETP-ALL and indeterminate MRD by RT-PCR who had available samples, none were found to have trackable clones by NGS. For patients with ETP-ALL, flow cytometric techniques may provide a better assessment of MRD given the distinctive immunophenotype.20
While the outcome for children with T-ALL on DFCI 05-001 and 11-001 was relatively favorable, all were treated with higher intensity therapy, including high cumulative doxorubicin dosage (300 mg/m2) and cranial radiation, both of which are associated with adverse late effects. All patients with T-ALL on our studies received dexrazoxane prior to each dose of doxorubicin given our previous findings that the use of dexrazoxane minimized late echocardiographic changes in patients with HR ALL without impacting relapse rates or risk of second malignant neoplasms.55-58 The use of cranial radiation for all patients with T-ALL on DFCI 05-001 and 11-001 was based on previous data suggesting that these patients had a significantly higher risk of CNS relapse when treated without radiation59-61 and, in fact, we did not observe any CNS relapses. Recent studies indicate, however, that cranial radiation may not be necessary in T-ALL therapy.62,63 A multivariable and meta-regression analysis demonstrated no difference in EFS on studies that used cranial radiation for all patients, for only CNS-positive patients, or for no patients.63 Additionally, a retrospective review of over 16,000 patients with ALL treated on 10 cooperative study group trials demonstrated no significant survival advantage with the use of cranial radiation for any ALL subset (including T-ALL), except for patients with CNS-3 status for whom cranial radiation was associated with a superior EFS, but not OS.62 Together with results from our cohort indicating that CNS-3 status was an independent predictor of inferior EFS, the currently open DFCI ALL Consortium trial omits cranial radiation for patients with T-ALL, except for those who are CNS-3 at diagnosis.
In conclusion, our study confirms the prognostic significance of end-induction MRD, ETP status and PI3K pathway mutational status and supports further investigation of these factors. Patients with end-induction MRD <10−4 have an excellent prognosis; given the long-term risks associated with T-ALL therapy, further studies should explore whether de-intensification of therapy in this group may reduce risk of late effects while preserving overall outcome. ETP immunophenotype identifies a subgroup at higher risk of slow early response for whom novel interventions during induction should be investigated, but intensification of post-induction therapy based exclusively on ETP status may not be necessary. Finally, our results suggest that PI3K pathway mutations are associated with inferior outcome. Whether this or other genetic findings identify high-risk patients who may benefit from new post-induction treatment strategies awaits further validation and highlights the urgent need for further comprehensive genomic and transcriptomic studies of childhood T-ALL.
Supplementary Material
TABLE S1 Protocol treatment for participants with T-ALL on DFCI Protocols 05-001 and 11-001.
TABLE S2 Genes whose protein-coding exons were sequenced in each targeted exome sequencing cohort.
TABLE S3 Patients with T-ALL and induction failure on DFCI 05-001 and 11-001.
TABLE S4 Logistic regression modeling for the achievement of complete remission for patients with known ETP status.
TABLE S5 Post-induction events for patients with T-ALL on DFCI 05-001 and 11-001.
TABLE S6 Cox proportional-hazards model of EFS for patients with known ETP and mutational status.
TABLE S8 FRALLE risk stratification analysis of participants with T-ALL on DFCI 05-001 and 11-001.
FIGURE S1 Analysis of DFCI 05-001 and 11-001 patients with T-ALL using the FRALLE oncogenetic classification. Kaplan-Meier analysis of (A) EFS and (B) OS of patients with low- or high-risk subtype T-ALL. Low-risk is defined according to the FRALLE publication as the presence of a Notch pathway activating mutation and the absence of a PTEN/RAS mutation. High-risk is defined as the presence of PTEN/RAS mutation regardless of NOTCH1/FBXW7 mutation status, or the absence of NOTCH1, FBXW7, PTEN, or RAS mutations. A two-sided log rank (Mantel-Cox) test was used to test for differences in survival between groups.
TABLE S7 Pathogenic mutations identified in the analysis of childhood T-ALL on DFCI 05-001 and 11-001.
Acknowledgements
We thank the patients and families who participated in this study, and Annette Dalton, Jeffrey Kutok, Albert Moghrabi, Yvan Samson, and Cindy Schwartz for their contributions to this work. We also thank the Center for Cancer Genome Discovery at DFCI for their contributions to the Oncopanel sequencing and assistance with data analysis/interpretation. This work was supported by grants from the National Institutes of Health, National Cancer Institute (5P01CA068484 and 1R01CA193651) and Enzon Pharmaceuticals.
Abbreviations Key:
- ALL
Acute lymphoblastic leukemia
- B-ALL
B-cell acute lymphoblastic leukemia
- CNS
Central nervous system
- CR
Complete remission
- DFCI
Dana-Farber Cancer Institute
- DFS
Disease-free survival
- EFS
Event-free survival
- EORTC
Children Leukemia Group of the European Organisation for Research and Treatment of Cancer
- ETP
Early T-cell precursor
- FRALLE
The French Acute Lymphoblastic Leukaemia Study Group
- gDNA
Genomic DNA
- GRAAL
The Group for Research in Adult Acute Lymphoblastic Leukemia
- HR
High risk
- HSCT
Hematopoietic stem cell transplant
- Ig
Immunoglobulin
- MRD
Minimal residual disease
- NGS
Next generation sequencing
- OR
Odds ratio
- OS
Overall survival
- Ph+
Philadelphia chromosome positive
- RT-PCR
Reverse transcription polymerase chain reaction
- T-ALL
T-cell acute lymphoblastic leukemia
- TCR
T-cell receptor
- TP1
Time point 1
- TP2
Time point 2
- VHR
Very high risk
Footnotes
Data Sharing Statement
DFCI Protocols 05-001 and 11-001 are registered at clinicaltrials.gov as NCT00165087 and NCT01574274, respectively. Full study protocols may be requested from the corresponding author. Targeted next generation sequencing data of diagnostic T-ALL samples will be deposited in a publicly available repository.
Conflict of Interest Statement: All other authors declare no competing financial interests.
References
- 1.Goldberg JM, Silverman LB, Levy DE, et al. Childhood T-cell acute lymphoblastic leukemia: the Dana-Farber Cancer Institute acute lymphoblastic leukemia consortium experience. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2003;21(19):3616–3622. [DOI] [PubMed] [Google Scholar]
- 2.Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30(14):1663–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vrooman LM, Blonquist TM, Harris MH, et al. Refining risk classification in childhood B acute lymphoblastic leukemia: results of DFCI ALL Consortium Protocol 05-001. Blood Adv. 2018;2(12):1449–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith M, Arthur D, Camitta B, et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1996;14(1):18–24. [DOI] [PubMed] [Google Scholar]
- 5.Conter V, Bartram CR, Valsecchi MG, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood. 2010;115(16):3206–3214. [DOI] [PubMed] [Google Scholar]
- 6.Moorman AV, Robinson H, Schwab C, et al. Risk-directed treatment intensification significantly reduces the risk of relapse among children and adolescents with acute lymphoblastic leukemia and intrachromosomal amplification of chromosome 21: a comparison of the MRC ALL97/99 and UKALL2003 trials. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(27):3389–3396. [DOI] [PubMed] [Google Scholar]
- 7.Heerema NA, Carroll AJ, Devidas M, et al. Intrachromosomal amplification of chromosome 21 is associated with inferior outcomes in children with acute lymphoblastic leukemia treated in contemporary standard-risk children's oncology group studies: a report from the children's oncology group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(27):3397–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clappier E, Grardel N, Bakkus M, et al. IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: results of the EORTC Children's Leukemia Group study 58951. Leukemia. 2015;29(11):2154–2161. [DOI] [PubMed] [Google Scholar]
- 9.Borowitz MJ, Wood BL, Devidas M, et al. Prognostic significance of minimal residual disease in high risk B-ALL: a report from Children's Oncology Group study AALL0232. Blood. 2015;126(8):964–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pieters R, de Groot-Kruseman H, Van der Velden V, et al. Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol. 2016;34(22):2591–2601. [DOI] [PubMed] [Google Scholar]
- 11.Tran TH, Harris MH, Nguyen JV, et al. Prognostic impact of kinase-activating fusions and IKZF1 deletions in pediatric high-risk B-lineage acute lymphoblastic leukemia. Blood Adv. 2018;2(5):529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel B, Rai L, Buck G, et al. Minimal residual disease is a significant predictor of treatment failure in non T-lineage adult acute lymphoblastic leukaemia: final results of the international trial UKALL XII/ECOG2993. British journal of haematology. 2010;148(1):80–89. [DOI] [PubMed] [Google Scholar]
- 13.Lauten M, Moricke A, Beier R, et al. Prediction of outcome by early bone marrow response in childhood acute lymphoblastic leukemia treated in the ALL-BFM 95 trial: differential effects in precursor B-cell and T-cell leukemia. Haematologica. 2012;97(7):1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffin TC, Shuster JJ, Buchanan GR, Murphy SB, Camitta BM, Amylon MD. Slow disappearance of peripheral blood blasts is an adverse prognostic factor in childhood T cell acute lymphoblastic leukemia: a Pediatric Oncology Group study. Leukemia. 2000;14(5):792–795. [DOI] [PubMed] [Google Scholar]
- 15.Arico M, Basso G, Mandelli F, et al. Good steroid response in vivo predicts a favorable outcome in children with T-cell acute lymphoblastic leukemia. The Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP). Cancer. 1995;75(7):1684–1693. [DOI] [PubMed] [Google Scholar]
- 16.Goldberg JM, Silverman LB, Levy DE, et al. Childhood T-Cell Acute Lymphoblastic Leukemia: The Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia Consortium Experience. Journal of Clinical Oncology. 2003;21(19):3616–3622. [DOI] [PubMed] [Google Scholar]
- 17.Schrappe M, Hunger SP, Pui CH, et al. Outcomes after induction failure in childhood acute lymphoblastic leukemia. N Engl J Med. 2012;366(15):1371–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schrappe M, Valsecchi MG, Bartram CR, et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood. 2011;118(8):2077–2084. [DOI] [PubMed] [Google Scholar]
- 19.O'Connor D, Enshaei A, Bartram J, et al. Genotype-Specific Minimal Residual Disease Interpretation Improves Stratification in Pediatric Acute Lymphoblastic Leukemia. J Clin Oncol. 2018;36(1):34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coustan-Smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10(2):147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gutierrez A, Dahlberg SE, Neuberg DS, et al. Absence of biallelic TCRgamma deletion predicts early treatment failure in pediatric T-cell acute lymphoblastic leukemia. J Clin Oncol. 2010;28(24):3816–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patrick K, Wade R, Goulden N, et al. Outcome for children and young people with Early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br J Haematol. 2014;166(3):421–424. [DOI] [PubMed] [Google Scholar]
- 23.Fuhrmann S, Schabath R, Moricke A, et al. Expression of CD56 defines a distinct subgroup in childhood T-ALL with inferior outcome. Results of the ALL-BFM 2000 trial. Br J Haematol. 2018;183(1):96–103. [DOI] [PubMed] [Google Scholar]
- 24.Conter V, Valsecchi MG, Buldini B, et al. Early T-cell precursor acute lymphoblastic leukaemia in children treated in AIEOP centres with AIEOP-BFM protocols: a retrospective analysis. Lancet Haematol. 2016;3(2):e80–86. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Easton J, Shao Y, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269–271. [DOI] [PubMed] [Google Scholar]
- 28.Fogelstrand L, Staffas A, Wasslavik C, et al. Prognostic implications of mutations in NOTCH1 and FBXW7 in childhood T-ALL treated according to the NOPHO ALL-1992 and ALL-2000 protocols. Pediatr Blood Cancer. 2014;61(3):424–430. [DOI] [PubMed] [Google Scholar]
- 29.Asnafi V, Buzyn A, Le Noir S, et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood. 2009;113(17):3918–3924. [DOI] [PubMed] [Google Scholar]
- 30.Breit S, Stanulla M, Flohr T, et al. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood. 2006;108(4):1151–1157. [DOI] [PubMed] [Google Scholar]
- 31.Bandapalli OR, Zimmermann M, Kox C, et al. NOTCH1 activation clinically antagonizes the unfavorable effect of PTEN inactivation in BFM-treated children with precursor T-cell acute lymphoblastic leukemia. Haematologica. 2013;98(6):928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gutierrez A, Sanda T, Grebliunaite R, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009;114(3):647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonnet M, Loosveld M, Montpellier B, et al. Posttranscriptional deregulation of MYC via PTEN constitutes a major alternative pathway of MYC activation in T-cell acute lymphoblastic leukemia. Blood. 2011;117(24):6650–6659. [DOI] [PubMed] [Google Scholar]
- 34.Paganin M, Grillo MF, Silvestri D, et al. The presence of mutated and deleted PTEN is associated with an increased risk of relapse in childhood T cell acute lymphoblastic leukaemia treated with AIEOP-BFM ALL protocols. Br J Haematol. 2018;182(5):705–711. [DOI] [PubMed] [Google Scholar]
- 35.Szarzynska-Zawadzka B, Kunz JB, Sedek L, et al. PTEN abnormalities predict poor outcome in children with T-cell acute lymphoblastic leukemia treated according to ALL IC-BFM protocols. Am J Hematol. 2019;94(4):E93–E96. [DOI] [PubMed] [Google Scholar]
- 36.Trinquand A, Tanguy-Schmidt A, Ben Abdelali R, et al. Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: a Group for Research in Adult Acute Lymphoblastic Leukemia study. J Clin Oncol. 2013;31(34):4333–4342. [DOI] [PubMed] [Google Scholar]
- 37.Petit A, Trinquand A, Chevret S, et al. Oncogenetic mutations combined with MRD improve outcome prediction in pediatric T-cell acute lymphoblastic leukemia. Blood. 2018;131(3):289–300. [DOI] [PubMed] [Google Scholar]
- 38.Place AE, Stevenson KE, Vrooman LM, et al. Intravenous pegylated asparaginase versus intramuscular native Escherichia coli L-asparaginase in newly diagnosed childhood acute lymphoblastic leukaemia (DFCI 05-001): a randomised, open-label phase 3 trial. Lancet Oncol. 2015;16(16):1677–1690. [DOI] [PubMed] [Google Scholar]
- 39.Vrooman LM, Blonquist TM, Supko JG, et al. Efficacy and toxicity of pegaspargase and calaspargase pegol in childhood acute lymphoblastic leukemia/lymphoma: Results of DFCI 11-001. Journal of Clinical Oncology. 2019;37(15_suppl):10006–10006. [DOI] [PubMed] [Google Scholar]
- 40.Zhou J, Goldwasser MA, Li A, et al. Quantitative analysis of minimal residual disease predicts relapse in children with B-lineage acute lymphoblastic leukemia in DFCI ALL Consortium Protocol 95-01. Blood. 2007;110(5):1607–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ladetto M, Brüggemann M, Monitillo L, et al. Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia. 2014;28(6):1299–1307. [DOI] [PubMed] [Google Scholar]
- 42.Faham M, Zheng J, Moorhead M, et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012;120(26):5173–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burns MA, Liao ZW, Yamagata N, et al. Hedgehog pathway mutations drive oncogenic transformation in high-risk T-cell acute lymphoblastic leukemia. Leukemia. 2018;32(10):2126–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sholl LM, Do K, Shivdasani P, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI insight. 2016;1(19):e87062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aoki Y, Niihori T, Kawame H, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nature genetics. 2005;37(10):1038–1040. [DOI] [PubMed] [Google Scholar]
- 46.Winter SS, Dunsmore KP, Devidas M, et al. Improved Survival for Children and Young Adults With T-Lineage Acute Lymphoblastic Leukemia: Results From the Children's Oncology Group AALL0434 Methotrexate Randomization. J Clin Oncol. 2018;36(29):2926–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moricke A, Zimmermann M, Valsecchi MG, et al. Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood. 2016;127(17):2101–2112. [DOI] [PubMed] [Google Scholar]
- 48.Wood BL, Winter SS, Dunsmore KP, et al. T-Lymphoblastic Leukemia (T-ALL) Shows Excellent Outcome, Lack of Significance of the Early Thymic Precursor (ETP) Immunophenotype, and Validation of the Prognostic Value of End-Induction Minimal Residual Disease (MRD) in Children’s Oncology Group (COG) Study AALL0434. Blood. 2014;124(21):1–1.24993873 [Google Scholar]
- 49.Bond J, Graux C, Lhermitte L, et al. Early Response-Based Therapy Stratification Improves Survival in Adult Early Thymic Precursor Acute Lymphoblastic Leukemia: A Group for Research on Adult Acute Lymphoblastic Leukemia Study. J Clin Oncol. 2017;35(23):2683–2691. [DOI] [PubMed] [Google Scholar]
- 50.Dunsmore KP, Winter S, Devidas M, et al. COG AALL0434: A randomized trial testing nelarabine in newly diagnosed t-cell malignancy. Journal of Clinical Oncology. 2018;36(15_suppl):10500–10500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chonghaile TN, Roderick JE, Glenfield C, et al. Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov. 2014;4(9):1074–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zuurbier L, Homminga I, Calvert V, et al. NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on DCOG or COALL protocols. Leukemia. 2010;24(12):2014–2022. [DOI] [PubMed] [Google Scholar]
- 53.Clappier E, Collette S, Grardel N, et al. NOTCH1 and FBXW7 mutations have a favorable impact on early response to treatment, but not on outcome, in children with T-cell acute lymphoblastic leukemia (T-ALL) treated on EORTC trials 58881 and 58951. Leukemia. 2010;24(12):2023–2031. [DOI] [PubMed] [Google Scholar]
- 54.Modvig S, Madsen HO, Siitonen SM, et al. Minimal residual disease quantification by flow cytometry provides reliable risk stratification in T-cell acute lymphoblastic leukemia. Leukemia. 2019;33(6):1324–1336. [DOI] [PubMed] [Google Scholar]
- 55.Barry EV, Vrooman LM, Dahlberg SE, et al. Absence of secondary malignant neoplasms in children with high-risk acute lymphoblastic leukemia treated with dexrazoxane. J Clin Oncol. 2008;26(7):1106–1111. [DOI] [PubMed] [Google Scholar]
- 56.Silverman LB, Stevenson KE, O'Brien JE, et al. Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985-2000). Leukemia. 2010;24(2):320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lipshultz SE, Scully RE, Lipsitz SR, et al. Assessment of dexrazoxane as a cardioprotectant in doxorubicin-treated children with high-risk acute lymphoblastic leukaemia: long-term follow-up of a prospective, randomised, multicentre trial. Lancet Oncol. 2010;11(10):950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vrooman LM, Neuberg DS, Stevenson KE, et al. The low incidence of secondary acute myelogenous leukaemia in children and adolescents treated with dexrazoxane for acute lymphoblastic leukaemia: a report from the Dana-Farber Cancer Institute ALL Consortium. Eur J Cancer. 2011;47(9):1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cherlow JM, Steinherz PG, Sather HN, et al. The role of radiation therapy in the treatment of acute lymphoblastic leukemia with lymphomatous presentation: a report from the Childrens Cancer Group. Int J Radiat Oncol Biol Phys. 1993;27(5):1001–1009. [DOI] [PubMed] [Google Scholar]
- 60.Conter V, Schrappe M, Arico M, et al. Role of cranial radiotherapy for childhood T-cell acute lymphoblastic leukemia with high WBC count and good response to prednisone. Associazione Italiana Ematologia Oncologia Pediatrica and the Berlin-Frankfurt-Munster groups. J Clin Oncol. 1997;15(8):2786–2791. [DOI] [PubMed] [Google Scholar]
- 61.Laver JH, Barredo JC, Amylon M, et al. Effects of cranial radiation in children with high risk T cell acute lymphoblastic leukemia: a Pediatric Oncology Group report. Leukemia. 2000;14(3):369–373. [DOI] [PubMed] [Google Scholar]
- 62.Vora A, Andreano A, Pui CH, et al. Influence of Cranial Radiotherapy on Outcome in Children With Acute Lymphoblastic Leukemia Treated With Contemporary Therapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2016;34(9):919–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kelly MJ, Trikalinos TA, Dahabreh IJ, Gianferante M, Parsons SK. Cranial radiation for pediatric T-lineage acute lymphoblastic leukemia: a systematic review and meta-analysis. Am J Hematol. 2014;89(10):992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steinherz PG, Redner A, Steinherz L, Meyers P, Tan C, Heller G. Development of a new intensive therapy for acute lymphoblastic leukemia in children at increased risk of early relapse. The Memorial Sloan-Kettering-New York-II protocol. Cancer. 1993;72(10):3120–3130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Protocol treatment for participants with T-ALL on DFCI Protocols 05-001 and 11-001.
TABLE S2 Genes whose protein-coding exons were sequenced in each targeted exome sequencing cohort.
TABLE S3 Patients with T-ALL and induction failure on DFCI 05-001 and 11-001.
TABLE S4 Logistic regression modeling for the achievement of complete remission for patients with known ETP status.
TABLE S5 Post-induction events for patients with T-ALL on DFCI 05-001 and 11-001.
TABLE S6 Cox proportional-hazards model of EFS for patients with known ETP and mutational status.
TABLE S8 FRALLE risk stratification analysis of participants with T-ALL on DFCI 05-001 and 11-001.
FIGURE S1 Analysis of DFCI 05-001 and 11-001 patients with T-ALL using the FRALLE oncogenetic classification. Kaplan-Meier analysis of (A) EFS and (B) OS of patients with low- or high-risk subtype T-ALL. Low-risk is defined according to the FRALLE publication as the presence of a Notch pathway activating mutation and the absence of a PTEN/RAS mutation. High-risk is defined as the presence of PTEN/RAS mutation regardless of NOTCH1/FBXW7 mutation status, or the absence of NOTCH1, FBXW7, PTEN, or RAS mutations. A two-sided log rank (Mantel-Cox) test was used to test for differences in survival between groups.
TABLE S7 Pathogenic mutations identified in the analysis of childhood T-ALL on DFCI 05-001 and 11-001.