

Abstract
Duchenne muscular dystrophy (DMD) is an X-linked neuromuscular disease characterized by progressive wasting of skeletal muscles. The neuromuscular junction (NMJ) is a synapse between motor neurons and skeletal muscle fibers, critical for the control of muscle contraction. The NMJ decline is observed in DMD patients, but the mechanism is unclear. LRP4 serves as a receptor for agrin, a proteoglycan secreted by motor neurons to induce NMJ, and plays a critical role in NMJ formation and maintenance. Interestingly, we found that protein levels of LRP4 were reduced both in muscles of the DMD patients and DMD model mdx mice. We explored whether increasing LRP4 is beneficial for DMD and crossed muscle-specific LRP4 transgenic mice with mdx mice (mdx; HSA-LRP4). The LRP4 transgene increased muscle strength, together with improved neuromuscular transmission in mdx mice. Furthermore, we found the LRP4 expression mitigated NMJ fragments and denervation in mdx mice. Mechanically, we showed that overexpression of LRP4 increased the activity of MuSK and expression of dystrophin-associated glycoprotein complex proteins in the mdx mice. Overall, our findings suggest that increasing LRP4 improves both function and structure of NMJ in the mdx mice and Agrin signaling might serve as a new therapeutic strategy in DMD.
Introduction
Duchenne muscular dystrophy (DMD) is the most common and severe form of muscular dystrophy, affecting 1 in 3500 to 5000 boys (1). It is an X-linked neuromuscular disease caused by the absence of dystrophin protein (2), a component of the dystrophin-associated glycoprotein complex (DGC) that connects the cytoskeleton of muscle fibers to the extracellular matrix (3,4). In DMD patients, mutated dystrophin leads to a rapid progression of muscle degeneration, loss of ambulation and premature fatality. One therapeutic strategy for DMD is to restore dystrophin expression by exon skipping, read-through therapy, CRISPR/Cas9-mediated gene editing, vector- or virus-mediated gene therapy and cell therapy. In addition, DMD patients could benefit from anti-inflammatory, -fibrotic and -oxidant treatments and means to inhibit myostatin pathway, to enhance the nitric oxide synthase and to increase utrophin expression (5–10). However, the efficacies of these therapeutic strategies are still limited and ambiguous.
Muscle contraction is controlled by the neuromuscular junction (NMJ), the synapse between motor neurons and muscle fibers. NMJ formation requires the agrin-LRP4-MuSK signaling. Agrin is a factor released from motor neurons via binding to LRP4, a transmembrane protein of the LDL receptor family, to activate the receptor tyrosine kinase MuSK. Subsequent signaling events, including increased E3 ligase activity of Rapsyn, promote the clustering of AChRs and NMJ formation (11–13). The agrin-LRP4-MuSK pathway is also critical for NMJ maintenance. After NMJ formation, conditional deletion of agrin, MuSK or LRP4 leads to NMJ disintegration and dysfunction (14–18). Dystrophin accumulates at the NMJ and has been implicated in its maintenance (19,20). In DMD patients, NMJs are dysfunctional with reduced synaptic fold size, partial innervation, reduced compound muscle action potential (CMAP) amplitude and more sensitive to AChR blockers (21,22). The mdx mice are a widely used DMD model because of the lack of dystrophin that displays reduced grip strength, muscle fiber degeneration and regenerated fibers. Besides, NMJs in mdx mice are fragmented and junctional folds reduced; concomitantly, there is a reduction in the amplitudes of miniature endplate potentials (mEPP) and in AChR density (20,23–26). However, pathological mechanisms of NMJ decline in DMD are not well understood.
In the present study, we investigated the impact of dystrophin deletion on NMJ and agrin-LRP4-MuSK pathway. We found LRP4 was reduced in muscles of DMD and Becker muscular dystrophy (BMD) patients. BMD is a rare disease also caused by a genetic defect in the dystrophin gene leading to a less but not complete absence of functional dystrophin protein and accompanied by a less severe phenotype. Moreover, LRP4 reduction was showed to be prior to the appearance of NMJ deficits in mdx mice. Remarkably, muscle-specific expression of LRP4 in mdx mice diminished NMJ deficits and improved muscle strength. Mechanistically, LRP4 expression increased agrin signaling and expression of dystrophin-associated glycoprotein complex proteins in muscles of mdx mice. Our results suggest that increasing LRP4 and agrin signaling might serve as a new therapeutic strategy in DMD.
Results
Reduced LRP4 in muscles of DMD patients and mdx mice
NMJ is declined in DMD patients, and agrin signaling is critical for NMJ maintenances (14–18). However, it is unclear whether agrin signaling is involved in DMD. Here, we first detected mRNA levels of LRP4, MUSK, DOK7, RAPSYN and CHRNG in the bicep muscle of DMD and BMD patients by RT-qPCR. As shown in Figure 1A, compared with unaffected controls, CHRNG was increased and MUSK was reduced in DMD patients, consistent with results from DMD model mice (27,28). Interestingly, LRP4 is the only gene that was down-regulated in both BMD and DMD patients, compared with unaffected controls (Fig. 1A). Western blot also revealed that LRP4 protein level was reduced in BMD and DMD patients (Fig. 1B and C).
Figure 1 .
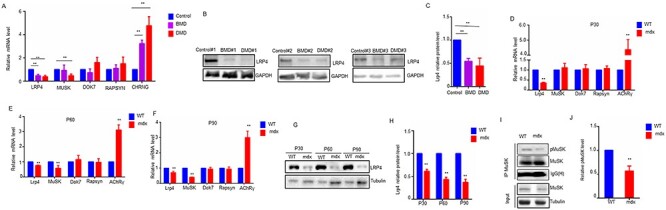
LRP4 reduction in muscles of DMD patients and mdx mice. (A) qRT-PCR of agrin signaling genes in muscles from DMD (n = 3), BMD patients (n = 6) and healthy control (n = 3). (B) Western analysis of LRP4 in muscle from DMD, BMD patients and healthy control. Three independent replicated experiments were performed. (C) Quantification of data in B (D–F) qRT-PCR of agrin signaling genes in muscles from mdx mice at P30, P60 and P90. N = 3 or 5 mice per group. (G) Western analysis of LRP4 in muscle from mdx mice P30, P60 and P90. (H) Quantification of data in E. N = 3 or 5 mice per group. (I) Phospho-MuSK was reduced in P 30-mdx muscles. (J) Quantification of data in F. N = 3 or 5 mice per group. For all graphs, *P < 0.05, **P < 0.01 (unpaired t-test).
Next, to verify the impact of dystrophin deletion on agrin-LRP4-MuSK pathway in mice, we examined mRNA levels of Lrp4, MuSK, Dok7, Rapsyn and AChRγ in TA muscle of mdx mice at postnatal day (P) 30, P60 and P90. As reported before (28), AChRγ mRNA was increased in mdx muscles (Fig. 1D–F), validating our model. Lrp4 and MuSK were down-regulated in mdx mice at P60 and P90. Noticeably, Lrp4, but not MuSK, decreased as early as at P30, suggesting that Lrp4 may be the first gene of agrin pathway to be down-regulated in mdx mice muscles. In accord, western blot results showed that LRP4 protein was also decreased in muscles of mdx mice at the three different ages (Fig. 1G and H). Because of the reduced LRP4, we wondered whether agrin signaling was compromised in mdx muscles. To this end, we compared phospho-MuSK between mdx and control muscles. As shown in Figure 1I and J, phospho-MuSK was reduced in P30-mdx muscles. Taken together, these results indicate that LRP4 is reduced in muscles of DMD patients and mdx mice.
NMJ deficits appeared later than LRP4 reduction in mdx mice
Previous studies reported NMJ deficits in adult mdx mice (23–26). To investigate LRP4 reduction was a cause or consequence of NMJ deficits in mdx mice, we determined when NMJ deficits begin to occur by analyzing NMJ morphology at P30, P60 and P90. Tibialis anterior (TA), soleus (SOL) and diaphragm (DIA) muscles were stained with antibodies against neurofilament (NF) and synaptophysin (SYN) to visualize axons and nerve terminals and with rhodamine-conjugated α-bungarotoxin (R-BTX) to visualize AChR (18,29). As shown in Figure 2A, NMJs appeared as characteristic pretzel-like morphology at P30 in both WT and mdx mice; most AChR clusters were covered with anti-NF/SYN staining, suggesting proper innervation. Quantitatively, there was no difference in NMJ fragments and innervation of muscles between the WT and mdx mice. In contrast, at P60, some branches of AChR clusters in mdx mice were no longer continuous but became fragmented. In addition, AChR clusters were not in complete registry with presynaptic staining at P60; many AChR cluster fragments were not positive for NF/SYN, indicating poor innervation. No further impairment was observed between P60 and P90 mdx mice, indicating that most NMJ decline in mdx mice occurred before P60 (Fig. 2B–D). Together, these results demonstrate that the development of NMJ deficits in mdx mice is age dependent.
Figure 2 .

NMJ structure in mdx mice at age of P30, P60 and P90. (A) NMJ is fragmented and partially innervated after P 60, but normal at P 30. TA, soleus and diaphragm muscles were stained with R-BTX to label AChR (red) and NF/SYN to label nerve terminals. Scale bar = 10 μm (B–D) Quantification of data in A. N = 10 mice per group and 25–30 NMJs of each mouse were counted. *P < 0.05, **P < 0.01 (unpaired t-test).
To determine how morphological deficits impair neuromuscular transmission, we measured compound muscle action potentials (CMAPs) triggered by10 consecutive nerve stimuli (29–32). As shown in Figure 3A, at the age of P30, both WT and mdx mice, CMAP amplitudes between the 1st and 10th stimuli were similar, regardless of the stimulation frequency. However, at age of P60 and P90, CMAP amplitudes of mdx mice were significantly decreased at the 40 HZ stimulation frequency and CMAP reduction was detectable from the third stimulation at 40 HZ stimulation (Fig. 3B and C).
Figure 3 .

NMJ transmission in mdx mice at age of P30, P60 and P90. (A) (B and C) CMAPs are normal in mdx mice at P30, but impaired at P60 and P90. In all panels, the left are representative traces and right are the quantification of data. Shown were frequency dependent in CMAP amplitude and ratio of 2nd–10th CMAP amplitudes over the 1st CMAP amplitude at 40 Hz stimulations. (D–I) mEPP and EPP are normal in mdx mice at P30 but impaired at P60 and P90. In all panels, the left are representative traces and right are the quantification of data. N = 5 mice per group, *P < 0.05, **P < 0.01 (two-way ANOVA).
To investigate underlying cellular mechanisms of neuromuscular transmission deficits, we also measured miniature endplate potentials (mEPPs) and endplate potentials (EPPs). Mdx mice at age of P30 displayed similar mEPPs and EPPs as WT, suggesting normal transmission at this age (Fig. 3D and E). However, mEPP amplitude was reduced in P60 and P90 mdx mice, compared with WT controls, suggesting reduced AChR concentration at the NMJ, in agreement with previous reports (23,26). In addition, mEPP frequency was decreased in mdx mice than that in control mice at age of P60 and P90, in accord with reduced NF/SYN staining and suggesting presynaptic deficits (Fig. 3F and H). EPP amplitudes were reduced in P60 and P90 mdx mice, compared with control mice (Fig. 3G and I). Because normal EPPs are necessary to elicit action potentials in muscles to initiate muscle contraction, these results suggest that the ability to generate muscle action potentials could be compromised in mdx mice. Together, results demonstrate that both morphological and functional deficits of NMJs are age dependent, not detectable at P30, but detectable at P60 and after. These results suggest that LRP4 was reduced prior to NMJ deficits in mdx mice, identifying LRP4 may be a potential pathological mechanism of NMJ decline in DMD.
Increased muscle strength by transgenic expression of LRP4
LRP4 is necessary for NMJ formation and maintenance (18,33). In light of the observations that LRP4 and phospho-MuSK were down-regulated in muscles of mdx mice, we tested the hypothesis that insufficient LRP4 and agrin signaling may be a mechanism of NMJ deficits in mdx mice. This hypothesis predicts that increasing LRP4 expression could diminish NMJ deficits in mdx mice. To test this, we crossed mdx mice with HSA-LRP4 mice that express LRP4 specifically in skeletal muscles (via the human skeletal muscle actin promoter) (Fig. 4A) (30,34,35). As shown in Figure 4B, LRP4 levels were increased in skeletal muscles of HSA-LRP4 and mdx; HSA-LRP4 mice, compared with WT or mdx mice. There were no differences in bodyweight among the four different genotypes: WT, HSA-LRP4, mdx, and mdx; HSA-LRP4 mice (Fig. 4C). Remarkably, the grip strength and hanging time of mdx; HSA-LRP4 mice were increased by 35.8 and 30.7%, respectively, compared with those of mdx mice (Fig. 4D and E). Moreover, treadmill test also showed that total running distance of mdx; HSA-LRP4 mice was increased by 31.7% (Fig. 4F). These results suggest that LRP4 expression could be beneficial for mdx mice. However, the strength of these mice remained significantly less than that of WT mice, suggesting that the muscle strength could not be completely improved by LRP4 expression. To further investigate whether LRP4 expression contributes to the muscle fiber, we quantified their cross-section area (CSA) and central nuclei, an indicator of muscle regeneration. Interestingly, muscle fiber CSA and central nuclei number were similar between mdx and mdx; HSA-LRP4 mice (Fig. 4G–I), suggesting that LRP4 expression had no effect on muscle fiber conditions.
Figure 4 .

LRP4 expression increased muscle strength in mdx mice. (A) Chematic diagrams of muscle-specific LRP4 transgenic mdx mice (mdx; HSA-LRP4). (B) LRP4 is overexpressed in the HSA-LRP4 and mdx; HSA-LRP4 mice, TA muscles were subjected to western blot with anti-LRP4, MuSK and tubulin antibodies. (C) Body weight is normal in mdx and LRP4 transgenic mice (N = 10 per genotype). (D–F) Grip strength, hanging time and running distance were increased after increasing LRP4 in the mdx mice. (N = 13–15 per genotype). (G) H&E staining showed degeneration of myofiber was not improved after increasing LRP4 in the mdx mice. Scale bar = 100 μm. (H and I) Quantification of central nuclei fibers (CNF) and cross-section area (CSA) showed no difference between mdx and mdx; HSA-LRP4 mice (N = 3 per genotype). (J) Scheme of in vivo muscle twitch and tetanic force measurement by nerve stimulation. (K and L) Comparable single twitch force and tetanic force between mdx and mdx; HSA-LRP4 mice by nerve stimulation (N = 5 per genotype). For all graphs, *P < 0.05, **P < 0.01 (unpaired t-test).
Nerve stimulation requires appropriate neuromuscular conduction to cause muscle contraction. Next, we asked whether LRP4 expression improved muscle strength in mdx mice by nerve stimulation. To this end, we measured muscle contractions in response to nerve stimulation (Fig. 4J). Intriguingly, muscle twitch and tetanic forces by nerve stimulation in mdx; HSA-LRP4 mice were significantly stronger than those in mdx mice (Fig. 4K and L), indicating that LRP4 expression increased nerve stimulation-induced muscle strength. Together, these results suggest that muscle strength improvement in mdx mice by LRP4 expression may be due to repaired neuromuscular transmission.
Improved NMJ transmission in mdx mice
To determine whether LRP4 expression ameliorated transmission deficits in mdx mice, we recorded CMAP, mEPPs and EPP at age of P90. As expected, CMAPs were improved in mdx muscles after LRP4 expression. The 10th to 1st ratios of CMAP at 40 Hz of stimulation was 86.2% group, which was increased to 93.2% in mdx; HSA-LRP4 group (Fig. 5A and C). At 40 Hz, CMAP reduction observed in mdx was also diminished (Fig. 5B). Reduction in mEPP frequency and amplitude was also attenuated in mdx; HSA-LRP4 mice by 12.7 and 12.9%, respectively, suggesting improved neuromuscular transmission by LRP4 expression (Fig. 5D–F). In accord, EPP amplitudes in mdx; HSA-LRP4 mice were higher than those of mdx mice (Fig. 5G and H). These results indicated that neuromuscular transmission deficits in mdx mice could be diminished by increasing LRP4 expression. A parsimonious explanation of these observations is that LPR4 expression improves muscle contraction by improving neuromuscular transmission.
Figure 5 .

LRP4 expression improved the NMJ transmission in mdx mice. (A) Representative CMAP traces. (B) Frequency dependent in CMAP amplitude. (C) Ratio of 2nd-10th CMAP amplitudes over the 1st CMAP amplitude at 40 Hz stimulations. N = 5 mice per group, *P < 0.05, **P < 0.01 (two-way ANOVA). (D) Representative mEPP traces. (E and F) Increased mEPP frequencies (B) and amplitudes (C) after increasing LRP4 in the mdx mice. (G) Representative EPP traces. (H) Increased EPP amplitude after increasing LRP4 in the mdx mice. N = 5 mice per group, *P < 0.05, **P < 0.01 (unpaired t-test).
Mitigated NMJ fragmentation and denervation in mdx mice
To investigate whether LRP4 expression diminished NMJ structural deficits in mdx mice, TA muscles were stained with R-BTX to label AChR clusters and with anti-NF/SYN antibodies to label nerve terminals. As described above and shown in Figure 2, NMJs in adult mdx mice displayed fragmentation and denervation. However, the numbers of fragmented NMJs were reduced in mdx; HSA-LRP4 mice (Fig. 6A and B). Meanwhile, partially innervated NMJs were also reduced in mdx; HSA-LRP4 mice (Fig. 6C). On the other hand, AChR intensity and presynaptic coverage (presynaptic area/postsynaptic area) were increased by 20.1 and 15.9%, respectively, but AChR area was not changed in mdx; HSA-LRP4 mice, compared with mdx mice (Fig. 6D–F). Together results were indicating that LRP4 expression may prevent NMJ from becoming fragmented and denervated.
Figure 6 .

LRP4 expression mitigated the NMJ fragmentation and denervation in the mdx mice. (A) R-BTX (red) and dystrophin (green) staining of TA muscles. In wild-type muscles, dystrophin protein enrichment was colocalized to AChR clusters at the NMJ. Overexpression of LRP4 showed more continuous AChR clusters in muscle of mdx mice. Scale bar = 10 μm. (B) R-BTX (red) and NF/SYN (green) staining of TA muscles. In mdx mice, AChR receptors that are partially covered by the nerve terminal suggesting partially innervated NMJs. Scale bar = 10 μm (C) Quantitative analysis revealed less fragmented AChR clusters after increasing muscle LRP4 in mdx mice. N = 10 mice per group *P < 0.05, **P < 0.01 (unpaired t-test). (D) Quantitative analysis revealed less partially innervated NMJ AChR clusters after increasing muscle LRP4 in mdx mice. (E–G) Quantitative analysis revealed AChR intensity (E) and presynaptic coverage (F) were increased but AChR area (G) was not altered after increasing muscle LRP4 in mdx mice. For all graphs, N = 10 mice per group and 25–30 NMJs of each mouse were counted, *P < 0.05, **P < 0.01 (unpaired t-test).
Increased MuSK phosphorylation and DGC proteins expression
Because of its critical role in agrin signaling and NMJ maintenance, we next studied the effect of LRP4 expression on MuSK activation by measuring its tyrosine phosphorylation. As described in Figure 1I, MuSK phosphorylation was reduced in mdx mice. However, phospho-MuSK was increased in mdx; HSA-LRP4 mice, compared with mdx mice (Fig. 7A and B). Agrin signaling is known to promote AChR clustering as well as its synthesis (36,37). To investigate how improved agrin signaling attenuates mdx NMJ deficits, we determined whether LRP4 expression increased the levels of MuSK, Dok7, Rapsyn and AChR subunits. Unexpectedly, their expressions were not changed in mdx; HSA-LRP4 mice, compared with mdx mice, suggesting that LRP4 expression does not affect agrin-induced AChR expression (Fig. 7C). Dystrophin complex has been implicated in NMJ integrity (24). Therefore, we tested whether LRP4 expression altered their expression. Utrophin is ubiquitously expressed autosomal paralogue of dystrophin gene. Its mRNA level was reported up-regulated in mdx mice, which also showed in our results. Interestingly, LRP4 expression increased utrophin mRNA level in mdx; HSA-LRP4 mice, compared with mdx mice (Fig. 7D). Moreover, dystroglycans and sarcoglycans (SGCA, SGCG and SGCD), which were parts of the dystrophin-associated glycoprotein complex (DGC) and reduced in mdx mice, were increased after LRP4 expressed (Fig. 7E). These may reveal that LRP4 expression does not increase AChR expression but induce AChR clustering by increasing expressing dystrophin-complex proteins in mdx mice.
Figure 7 .

LRP4 expression increased the activity of MuSK and expression of DAPs in the mdx mice. (A) MuSK immunoprecipitates (IP) from whole-tissue lysates (WTLs) of the TA muscle were immunoblotted for phosphotyrosine (pMuSK) and MuSK. (B) Quantification of data in A. N = 3 mice per group. *P < 0.05, **P < 0.01 (unpaired t-test). (C–E) mRNA expression of agrin signaling and DGC proteins in TA muscle of four genotyped mice. N = 3 or 5 mice per group. *P < 0.05, **P < 0.01 (unpaired t-test).
Discussion
Duchenne muscular dystrophy (DMD) is a severe type of muscular dystrophy neuromuscular disorder disease. Most current therapeutic studies have focused on muscle fragility and degeneration because dystrophin plays critical roles in stabilizing the sarcolemma of muscle (4). However, genetic evidence has also shown that dystrophin and dystrophin-associated glycoprotein complex (DGC) stabilize the NMJ (24,38,39). Alterations in pre- and postsynaptic NMJ structure and function can be observed in DMD model animals and patients. Moreover, experimental approaches that diminish NMJ decline in DMD models attenuate disease progression and improve functional outcomes (22,40–43), supporting that NMJ may be another therapeutic target of DMD. Recently, CRISPR/Cas9-mediated gene editing of dystrophin has been reported to treat DMD (44–46). It will be interesting to evaluate the NMJ function after gene editing treatment.
In the present study, we first revealed that MuSK and LRP4 were reduced in DMD patients (Fig. 1A–C), suggesting agrin signaling activity was attenuated in DMD. To our knowledge, it is for the first time to reveal the involvement of agrin signaling in DMD, indicating agrin signaling might serve as a new therapeutic target in DMD. Mdx mice lacking dystrophin showed mild symptoms of muscular dystrophy, which is different from DMD patients. However, the structural alterations and functional impairments of NMJ in mdx mice are similar to those in DMD patients, suggesting that mdx mice are a good model to study the dysfunctional NMJ of DMD. Our study provides evidence that agrin signaling is weakened in mdx mice, and increasing LRP4 signaling improves both structural and functional defects of NMJ. We find that the NMJ deficits in mdx mice are age dependent, only detectable after P60. The mRNA and protein levels of LRP4 were both decreased as early as P30 in the muscles of mdx mice, a time point when the NMJs still appeared with normal morphology and function. To answer whether increased expression of LRP4 could rescue NMJ deficits or not, we further generated a transgenic mouse overexpressing LRP4 in the muscles and crossed it with the mdx. Conditional overexpression of LRP4 in muscles increased muscle strength and improved NMJ transmission in mdx mice. Morphological analyses also revealed more continuous and innervated NMJ in the LRP4 transgenic mdx mice (summarized in Fig. 8). Mechanically, increased LRP4 activates MuSK and up-regulates DGC expression in the mdx mice. Together, these observations suggest that overexpressing LRP4 in muscles diminishes NMJ deficits in mdx mice.
Figure 8 .

Schematic diagram depicting main findings of the present study.
LRP4 is a type I single transmembrane protein of the LDLR family. As a receptor for agrin, it is required for inducing AChR clusters (33,47,48). However, treatment of muscle cells with agrin-induced AChR clusters had little effect on Lrp4 mRNA levels in vitro (49), suggesting additional mechanisms underlying regulation of Lrp4, independent of agrin signaling. Intriguingly, LRP4 mRNA is enriched at the central of muscle fibers (synaptic region) in embryonic and adult mice (33,50). However, mechanisms underlying this synapse-specific transcription are poorly understood. Our results showed loss of dystrophin leads to a reduction of LRP4, both in mRNA and protein levels (Fig. 1D–H). A future study will be needed to explore how dystrophin regulates the expression of LRP4, especially in mRNA levels. Meanwhile, defective agrin-LRP4-MuSK signaling is involved in many neuromuscular disorders and muscle weakness diseases. Our results revealed that agrin signaling was compromised in the NMJ of mdx mice (Fig. 1I and J), consistent with previous reports (27,51). However, it remains unclear whether defective agrin signaling causes or results from muscular dystrophy. In the present study, we found that decreased LRP4 levels appeared much earlier than the fragmented NMJ, suggesting that defective agrin signaling causes the impaired NMJ.
What causes NMJ fragmentation in DMD is not well understood. Some studies propose that these deficits are the consequence of muscle degeneration (24,26,52). However, delivery of truncated dystrophins, which rescue muscle degeneration, does not prevent NMJ fragmentation in mdx mice, suggesting that NMJ fragmentation and muscle fiber degeneration are not relevant (25). Our data showed that LRP4 is decreased prior to NMJ deficits and overexpression of LRP4 in muscles makes the NMJ more continuous in mdx mice. A recent paper has reported that overexpression of LRP4 mitigates NMJ deficits in the aged mice, which also display reduced LRP4 levels and fragmented NMJs (30). Deleting agrin in adult motor neurons or LRP4 in adult muscle leads to impaired transmission and NMJ fragmentation (17,18). Together, these results imply that NMJ fragmentation may be caused by weakened agrin-LRP4-MuSK signaling.
Here, we noticed that LRP4 level in HSA-LRP4 was increased many times than the endogenous LRP4 level in the WT mice, but LRP4 reduction in mdx mice is quite minor (Fig. 4B). That indicates LRP4 overexpression may be supraphysiological in HSA-LRP4 and mdx; HSA-LRP4 mice. Thus, we compared WT and HSA-LRP4 mice on all detected muscle strength and NMJ phenotypes and found HSA-LRP4 was similar to WT. This may suggest that the high expression of LRP4 does not affect normal physiological functions in WT mice.
In the present study, how increased LRP4 improves NMJ deficits in mdx mice is not clear. One possible mechanism is that increased LRP4 results in releasing more neuronal agrin into the synaptic basal lamina and activating more MuSK, compensating for the dystrophin signaling. In inducible muscle-specific LRP4 knockout mice, there is a time-dependent loss of synaptic agrin and the 90-kDa fragments before that of other pre- and postjunctional components, suggesting that LRP4 may regulate the stability of neuronal agrin on muscle fiber (18). Moreover, LRP4 is a critical muscle-derived retrograde signal for presynaptic differentiation (47,53). Our results showed that increased muscle LRP4 reduced NMJ denervation in the mdx mice (Fig. 6A, D and F), suggesting that increased muscle LRP4 may delay motor axon withdrawal. It will be interesting to investigate whether increased muscle LRP4 also delays denervation and improves neuromuscular function in ALS mice. Another possible mechanism is that overexpression of LRP4 induces AChR clustering at postsynaptic membrane of NMJ in mdx mice. Our electrophysiological results showed that the amplitude of mEPP was increased in mdx; HSA-LRP4 mice (Fig. 5D), indicating more functional AChRs, which was confirmed by increased AChR intensity (Fig. 6E). However, AChR expression was not changed upon LRP4 overexpression (Fig. 7C). These results suggest LRP4 only induces the AChR clustering without altering its expression. In vitro, LRP4 alone seemed unable to induce AChR cluster, but a decrease of LRP4 levels attenuates AChR clustering in muscle cells (49). The LRP4 reduction we observed in mdx mice may weaken the AChR clustering, which can be rescued by overexpression of LRP4.
Dystrophin-associated glycoprotein complex (DGC) is a transmembrane complex of proteins linking the actin cytoskeleton of the muscle fiber to the basal lamina. DGC proteins can be divided into three subcomplexes: (1) cytoskeleton linker proteins dystrophin and utrophin, (2) the transmembrane proteins dystroglycans, sarcoglycans and sarcospan and (3) cytoplasmic proteins dystrobrevin and syntrophin (12,13,30). Increasing genetic evidence showed that DGC is important for NMJ development and maintenance (24,54–57). Our present study showed that LRP4 increases the expression of dystrophin-complex proteins in mdx mice (Fig. 7D and E), suggesting that agrin signaling regulates the expression of DAPs. In agreement with our results, injection of AAV-MuSK-GFP into muscles of mdx mice increased expression of utrophin and β-DG throughout the sarcolemma (43). In aged mice, muscle LRP4 interacts with α-SG and α-SG prevents LRP4 degradation. α-SG expression mitigated AChR fragmentation and denervation and improved neuromuscular transmission in the aged mice (30).
Materials and methods
Muscle biopsies
Skeletal muscle biopsies were performed from the left bicep of control donors (n = 3) and patients. DMD (n = 3) and BMD (n = 6) patients were recruited at the neurological department of the first affiliated hospital of Nanchang University and diagnosed by genetic, biochemical and immunohistochemical analysis. All samples were obtained after a written consent signed by each individual in compliance with the bioethical laws of China as well as the Declaration of Helsinki. All research protocols were approved by the ethics committee of the first affiliated hospital of Nanchang University.
Mouse strains
HSA-LRP4 mice were described previously (30,34,35,47). The mdx mice were purchased from the Jackson Laboratory (stock # 001801). All mice were maintained on a C57BL/6J background. One–three months old male mice were characterized in the current study. Experimental procedures were approved by the Institutional Animal Care and Use Committee of the Nanchang University.
Measurement of muscle strength
Limb muscle strength was measured using an SR-1 hanging scale (American Weigh Scales) as described previously (18,29). Briefly, forelimbs were allowed to grip a square metal grid that was connected to the hanging scale. With hind limbs suspended, mice were gently pulled horizontally by the tail until grip was released. For the hanging time test, mice were placed on a 1.5-mm-diameter wire and measured the time remaining on the wire.
Treadmill test
The treadmill running distance test was performed using an Exer-3/6 treadmill (Columbus Instruments). Briefly, after 3 consecutive days of training, mice were placed on the track and started to run on the treadmill at 10 m/min for 5 min, with the gradual increased speed by 2 m/min every 2 min until they were exhausted. Exhaustion was determined when the mouse was unable to run on the treadmill for 10 s upon 5 times electric shock. The total running distance was recorded by Treadmill software (Columbus Instruments).
In vivo twitch and tetanic force measurement
Muscle twitch and tetanic force analysis were performed as previously reported (29). Briefly, mice were anesthetized with isoflurane continuously supplied by VetFlo anesthesia system (Kent Scientific) and placed on a 37°C heating pad. With gentle pressing the knee clamps, left feet were fixed onto the footplate that was connected to the servomotor (Aurora Scientific 1300A). For nerve stimulation, the sciatic nerve was exposed at thigh level and stimulated by two needle electrodes that were close to both sides of the nerve. The best position of muscle contraction was found by adjusting the distance between the footplate and the knee and measuring the muscle force by stimulating nerve with a single electrical stimulation (100 mA current, 0.2 ms pulse width). When the muscle force was no longer increasing, the position was the best position of muscle contraction. To identify best stimulation strength, a single muscle electrical stimulation was given starting at 100 mA current, 0.2 ms pulse width in the best position, the muscle force was measured every 30 mA increase, with an interval of 30s. When the muscle force was no longer increasing, the current was the best stimulation strength. In the best position and the best stimulation strength, twitch force was measured by stimulating muscle with a single electrical stimulation (0.2 ms pulse width), repeating 10 times with an interval of 30 s. Tetanic force was measured by stimulating nerve with 300 ms duration, 0.2 ms pulse width at a series of frequencies from 50 to 120 Hz (50, 80, 100 and 120 Hz) with an interval of 2 min. Twitch and tetanic forces were normalized by body weight.
Electrophysiological recording
For CMAPs, mice were anesthetized and fixed on the plastic plate. The right thigh fur was shaved and sterilized with 75% alcohol, and 1 cm incision in the skin was made over the lateral femur. The stimulation needle electrode was inserted near the exposed sciatic nerve. The reference needle electrode and the recording needle electrode were inserted into the Achilles tendon and the right gastrocnemius, respectively. The reference and recording electrodes were connected to MultiClamp 700B Amplifier. The sciatic nerve was stimulated with a series of 10 stimuli at 1, 5, 10, 20 and 40 Hz. CMAPs were recorded by Digidata 1550A and analyzed in Clampfit 10.5 software.
For mEPPs, left hemidiaphragms were isolated with ribs and phrenic nerves, pinned on Sylard gel and perfused at room temperature in oxygenated (95% O2, 5% CO2) Ringer’s solution (137 mm NaCl, 5 mm KCl, 12 mm NaHCO3, 1 mm NaH2PO4, 1 mm MgCl2, 2 mm CaCl2, 11 mm D-glucose, pH 7.3). mEPPs were recorded by piercing microelectrodes (20–40 mΩ, filled with 3 M KCl) into the center of muscle fibers. Three or more muscle fibers were recorded from each hemidiaphragm for 3 min or longer. To elicit EPPs, phrenic nerve stubs were held by electrodes via sucking and stimulated by platinum electrodes as described previously (18,29). Stimulation intensity was kept at ~130% or more of the action potential threshold. EPP recording of individual muscle fibers was repeated 2–3 times with 10-second intervals. Five muscle fibers were measured for each mouse. Conotoxin GIIIB (Bachem Americas) (1 M) was used to block nerve-induced muscle contraction. Data collected with a MultiClamp 700B Amplifier were digitized (10 kHz low-pass filtered) with Digidata 1550A and analyzed in Clampfit10.5 software.
Western blot and analysis
Muscles were lysed in modified RIPA buffer containing 50 mm Tris–HCl, pH 7.4, 150 mm NaCl, 1% NP-40, 2% sodium dodecyl sulfate (SDS), 2% deoxycholate, 1 mm PMSF (phenylmethanesulfonyl fluoride), 1 mm EDTA, 5 mm sodium fluoride, 2 mm sodium orthovanadate and protease inhibitors including 1 mm phenylmethylsulfonyl fluoride, 1 μg/μl pepstatin, 1 μg/μl leupeptin and 2 μg/ml aprotinin. After centrifuging at 10000 RPM at 4°C, the supernatant was designated as lysates. Protein concentration was measured using Pierce BCA kit. Samples (50 μg of protein) were resolved by SDS-PAGE and transferred to a nitrocellulose membrane, which was incubated in 5% milk in phosphate buffer saline (PBS)-0.3% Tween20 overnight at 4°C and then with primary antibodies in 2% milk in PBS-Tween buffer: anti-MuSK (1:1000, sc-33204, Santa Cruz Biotechnology), anti-LRP4 against the extracellular domain (1:1000, clone N207/27, UC Davis/NIH NeuroMab Facility) and antitubulin (1:2000,sc-23948, Santa Cruz Biotechnology). After washing, the membrane was incubated with PBS-Tween buffer containing HRP-conjugated goat antimouse and antirabbit IgG from Pierce (1:2000, PI-32230 [antimouse], PI-32260 [antirabbit]). The immunoreactive bands were exposed to autoradiography films, and the grayscale was quantified by ImageJ (NIH), as described before (18,58).
Immunoprecipitation
Immunoprecipitation (IP) experiments were described as previously in muscle (30). Briefly, muscle tissues were homogenized and lysed in IP buffer(150 mm NaCl, 2.5 mm EDTA, 50 mm Tris–HCl, pH, 7.4, 50 mm NaF, 2% SDS, 0.5% sodium deoxycholate, 20% glycerol, 0.1% sodium vanadate, 1% PMSF). Then, the lysis was centrifuged at max speed (14 000 rpm) for 15 min at 4°C. The supernatant was transferred to a new EP tube, 100 ~ 200 ug protein lysates were taken, appropriate 4× sample buffer was added and the mixture was boiled at 95°C for 5 ~ 10 min for the inputs. The rest of the supernatant was combined with 500 ~ 1000ul (about 500 ~ 1000ug protein) lysates, and 1–2 ug IP antibody was added overnight at 4°C with rotation. After that, 5 μl protein A/G agarose beads were added to the lysates (Pierce #20399) for 2–3 h at 4°C. Finally, the protein was pulled down by beads and added 20–30 ul appropriate 2× sample buffer and boiled at 95°C for 5 ~ 10 min. The supernatant was collected and evaluated by SDS-PAGE. (29,49).
Hematoxylin and eosin staining
Cryosections from fresh-frozen tissues were washed with PBS for 3 times and incubated in hematoxylin for 15 min and rinsed with deionized water followed by washing with tap water for at least 5 min to allow the stain to develop. Slides were then incubated in eosin for 3 min. The staining was dehydrated sequentially with 75, 95 and 100% ethanol for 2–5 min each. Slides were finally placed in xylene for 20 min and mounted with cytoseal. Images were collected with Olympus FSX-100 and image analysis was conducted using National Institutes of Health (NIH) Image J software.
Analysis of AChR clusters
The diaphragm, soleus or tibialis anterior muscle fibers were fixed in 4% PFA overnight and permeabilized for 2 h with 0.5% Triton X-100 in 3% BSA and 3% goat serum. They were then incubated with a mixture of rhodamine-conjugated a-bungarotoxin (R-BTX, 1:2000, Invitrogen), and antibodies against dystrophin (1:500, ab15277, Abcam), antibodies against neurofilament (NF) (1:1000, ab7795, Millipore) and synaptophysin (1:500, DAK-SYNAP, DAKO) at 4°C overnight. After washing with PBS, three times for 1 h each, muscle fibers were incubated with goat antimouse/antirabbit IgG conjugated with AlexaFluor-488 (1500, Invitrogen) overnight at 4°C. Images were collected with Olympus FSX-100 and image analysis was conducted using National Institutes of Health (NIH) Image J software (18,29).
qRT-PCR
Total RNA of mouse muscles was extracted using Trizol reagent (Sigma) and transcribed into cDNA templates using High-Capacity cDNA Reverse Transcription Kits (Sigma) following manufacture’s instruction. Quantitative PCRs were run in a StepOnePlusTM Real-Time Systems (ABI System) using PowerUpTM SYBR Green Master Mix (Thermo Scientific). The primers for specific genes are listed in Table 1.
Table 1.
Primer sequence used in the qRT-PCR
| Gene | Primer sequence |
|---|---|
| LRP4(Human) | F: 5′-GTG TGG CAG AAC CTT GAC AGT C-3′ |
| R: 5′-ACC GCT CTA ACT TGG CAT TCT CC-3′ | |
| MUSK(Human) | F: 5′-TTT GCT GTC CGT GCC AGA ATG C-3′ |
| R: 5′-GGC TTT GAG GAC GTC ATT GGT G-3′ | |
| DOK7(Human) | F: 5′-GCC ATC ATG CTG GGC TTT GAC A-3′ |
| R: 5′-AAC TTG GTG CCT GGA GCC ACT G-3′ | |
| RAPSYN(Human) | F: 5′-GGA CAA AGG TGC TGG AGA AGA G-3′ |
| R: 5′-TGT CGA TCT GGA CCA CAG CGA A-3′ | |
| CHRNG(Human) | F: 5′-CTG TCT TCC TCT TCC TTG TGG C-3′ |
| R: 5′-CGA CAA TGA GGA TGG TCA CCA C-3′ | |
| GAPDH(Human) | F: 5′-GTC TCC TCT GAC TTC AAC AGC G-3′ |
| R: 5′-ACC ACC CTG TTG CTG TAG CCA A-3′ | |
| Lrp4 | F: 5′-GTG TGG CAG AAC CTT GAC AGT C-3′ |
| R: 5′-TAC GGT CTG AGC CAT CCA TTC C-3′ | |
| MuSK | F: 5′-CTG AAG GCT GTG AGT CCA CTG T-3′ |
| R: 5′-TCC TTT ACC GCC AGG CAG TAC T-3′ | |
| Dok7 | F: 5′-TCA GCC TCA GAA GAG CGT GTT G-3′ |
| R: 5′-GCC TCA GAA GAG GAA CTG GAT AG-3′ | |
| Rapsyn | F: 5′-GTG GAT GAA GGT GCT GGA GAA G-3′ |
| R: 5′-CCG AGC AGT ATC AAT CTG GAC C-3′ | |
| AChRγ | F: 5′-CTT GTG GCT AAG AAG GTG CCT G-3′ |
| R: 5′-GCA AGG ACA CAT TGA GCA CGA C-3′ | |
| Utrophin | F: 5′-GCC ATC ATC TTG GTG AAT GCT CG-3′ |
| R: 5′-GGA TGA AGG GTC CTG ACC AAT C-3′ | |
| α-dystrobrevin | F: 5′-CGG CTT GAT GAA GAA CAC AGG C-3′ |
| R: 5′-CGA TGG TGA AGG AGA TGT CAG G-3′ | |
| α-DG | F: 5′-GTG GTT GGC ATT CCA GAC GGT A-3′ |
| R: 5′-CAG TGT AGC CAA GAC GGT AAG G-3′ | |
| NOS1 | F: 5′-ACC AGC ACC TTT GGC AAT GGA G-3′ |
| R: 5′-GAG ACG CTG TTG AAT CGG ACC T-3′ | |
| SNTA1 | F: 5′-CAG TTG GTG GAT GGC TGT CAT C-3′ |
| R: 5′-GTG AAG CCC TTG TCG ATG TGC A-3′ | |
| SNTB1 | F: 5′-AGC CTC TGT CAT CCC AGT CCT T-3′ |
| R: 5′-GTG TGC TTA GCA TCT GGC GAG T-3′ | |
| SGCA | F: 5′-ACT TCCGCGTTGACTGGTGCA-3′ |
| R: 5′-CAC CAA GGC ATC TGT CAG GAA G-3′ | |
| SGCB | F: 5′-GGC AAC TTA GCC ATC TGC GTG A-3′ |
| R: 5′-GTG GAA CTC CAT GCT ATC ACA CC-3′ | |
| SGCD | F: 5′-TGA GAC TGG AGT CCA AGG ATG G-3′ |
| R: 5′-CTC GAA GAC CTT CTG CCT CGT T-3′ | |
| SGCG | F: 5′-GTG ACA GTC AGT GCT CGC AAC T-3′ |
| R: 5′-GCA GAG AAC AGT GGC TTG CCA T-3′ | |
| LAMA2 | F: 5′-TGG AAG TAG CCG AAC CAG GAC A-3′ |
| R: 5′-CAC CTG GTT CAG AAC GAA GTC G-3′ | |
| GAPDH | F: 5′-GTG AAG GTC GGT GTG AAC GG-3′ |
| R: 5′-CAA GCT TCC CAT TCT CGG CCT-3′ |
Statistical analysis
Data were assumed to be normally distributed and equal variance between groups. Randomization was not performed. Datasets were statistically compared using a one- or two-way analysis of variance. Two-tailed, unpaired t-test was used to compare data between two groups. Unless otherwise indicated, data are expressed as mean or mean ± standard error of the mean (SEM). All analyses were performed double-blinded to genotype.
Conflict of Interest statement. The authors declare that they have no conflict of interest.
Contributor Information
Tiankun Hui, School of Life Science, Nanchang University, Nanchang, Jiangxi, China; Laboratory of Synaptic Development and Plasticity, Institute of Life Science, Nanchang University, Nanchang, Jiangxi, China.
Hongyang Jing, School of Life Science, Nanchang University, Nanchang, Jiangxi, China; Laboratory of Synaptic Development and Plasticity, Institute of Life Science, Nanchang University, Nanchang, Jiangxi, China.
Tian Zhou, School of Basic Medical Sciences, Nanchang University, Nanchang, Jiangxi, China.
Peng Chen, School of Life Science, Nanchang University, Nanchang, Jiangxi, China; Laboratory of Synaptic Development and Plasticity, Institute of Life Science, Nanchang University, Nanchang, Jiangxi, China.
Ziyang Liu, School of Life Science, Nanchang University, Nanchang, Jiangxi, China; Laboratory of Synaptic Development and Plasticity, Institute of Life Science, Nanchang University, Nanchang, Jiangxi, China.
Xia Dong, Laboratory of Synaptic Development and Plasticity, Institute of Life Science, Nanchang University, Nanchang, Jiangxi, China; School of Basic Medical Sciences, Nanchang University, Nanchang, Jiangxi, China.
Min Yan, Laboratory of Synaptic Development and Plasticity, Institute of Life Science, Nanchang University, Nanchang, Jiangxi, China; School of Basic Medical Sciences, Nanchang University, Nanchang, Jiangxi, China.
Dongyan Ren, School of Life Science, Nanchang University, Nanchang, Jiangxi, China; Laboratory of Synaptic Development and Plasticity, Institute of Life Science, Nanchang University, Nanchang, Jiangxi, China.
Suqi Zou, School of Life Science, Nanchang University, Nanchang, Jiangxi, China; Laboratory of Synaptic Development and Plasticity, Institute of Life Science, Nanchang University, Nanchang, Jiangxi, China.
Shunqi Wang, School of Life Science, Nanchang University, Nanchang, Jiangxi, China; Laboratory of Synaptic Development and Plasticity, Institute of Life Science, Nanchang University, Nanchang, Jiangxi, China.
Erkang Fei, School of Life Science, Nanchang University, Nanchang, Jiangxi, China; Laboratory of Synaptic Development and Plasticity, Institute of Life Science, Nanchang University, Nanchang, Jiangxi, China.
Daojun Hong, Department of Neurology, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, China.
Xinsheng Lai, School of Life Science, Nanchang University, Nanchang, Jiangxi, China; Laboratory of Synaptic Development and Plasticity, Institute of Life Science, Nanchang University, Nanchang, Jiangxi, China.
Funding
National Natural Science Foundation of China (31660268, 81601092 to X.L.; 31860268 to T.Z. and 31960176 to S.Z.); Natural Science Foundation of Jiangxi province (20181BAB215017).
References
- 1.Fairclough, R.J., Wood, M.J. and Davies, K.E. (2013) Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nat. Rev. Genet., 14, 373–378. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman, E.P., Brown, R.H., Jr. and Kunkel, L.M. (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell, 51, 919–928. [DOI] [PubMed] [Google Scholar]
- 3.Ervasti, J.M., Ohlendieck, K., Kahl, S.D., Gaver, M.G. and Campbell, K.P. (1990) Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature, 345, 315–319. [DOI] [PubMed] [Google Scholar]
- 4.Campbell, K.P. and Kahl, S.D. (1989) Association of dystrophin and an integral membrane glycoprotein. Nature, 338, 259–262. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu-Motohashi, Y., Komaki, H., Motohashi, N., Takeda, S., Yokota, T. and Aoki, Y. (2019) Restoring dystrophin expression in Duchenne muscular dystrophy: current status of therapeutic approaches. J. Pers. Med., 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimizu-Motohashi, Y., Miyatake, S., Komaki, H., Takeda, S. and Aoki, Y. (2016) Recent advances in innovative therapeutic approaches for Duchenne muscular dystrophy: from discovery to clinical trials. Am. J. Transl. Res., 8, 2471–2489. [PMC free article] [PubMed] [Google Scholar]
- 7.Barthelemy, F. and Wein, N. (2018) Personalized gene and cell therapy for Duchenne muscular dystrophy. Neuromuscul. Disord., 28, 803–824. [DOI] [PubMed] [Google Scholar]
- 8.Duan, D. (2018) Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol. Ther., 26, 2337–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aartsma-Rus, A., Fokkema, I., Verschuuren, J., Ginjaar, I., van Deutekom, J., van Ommen, G.J. and den Dunnen, J.T. (2009) Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat., 30, 293–299. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura, A. and Takeda, S. (2011) Exon-skipping therapy for Duchenne muscular dystrophy. Lancet, 378, 546–547. [DOI] [PubMed] [Google Scholar]
- 11.Wu, H., Xiong, W.C. and Mei, L. (2010) To build a synapse: signaling pathways in neuromuscular junction assembly. Development, 137, 1017–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tintignac, L.A., Brenner, H.R. and Ruegg, M.A. (2015) Mechanisms regulating neuromuscular junction development and function and causes of muscle wasting. Physiol. Rev., 95, 809–852. [DOI] [PubMed] [Google Scholar]
- 13.Li, L., Xiong, W.C. and Mei, L. (2018) Neuromuscular junction formation, aging, and disorders. Annu. Rev. Physiol., 80, 159–188. [DOI] [PubMed] [Google Scholar]
- 14.Hesser, B.A., Henschel, O. and Witzemann, V. (2006) Synapse disassembly and formation of new synapses in postnatal muscle upon conditional inactivation of MuSK. Mol. Cell. Neurosci., 31, 470–480. [DOI] [PubMed] [Google Scholar]
- 15.Kong, X.C., Barzaghi, P. and Ruegg, M.A. (2004) Inhibition of synapse assembly in mammalian muscle in vivo by RNA interference. EMBO Rep., 5, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eguchi, T., Tezuka, T., Miyoshi, S. and Yamanashi, Y. (2016) Postnatal knockdown of dok-7 gene expression in mice causes structural defects in neuromuscular synapses and myasthenic pathology. Genes Cells, 21, 670–676. [DOI] [PubMed] [Google Scholar]
- 17.Samuel, M.A., Valdez, G., Tapia, J.C., Lichtman, J.W. and Sanes, J.R. (2012) Agrin and synaptic laminin are required to maintain adult neuromuscular junctions. PLoS One, 7, e46663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barik, A., Lu, Y., Sathyamurthy, A., Bowman, A., Shen, C., Li, L., Xiong, W.C. and Mei, L. (2014) LRP4 is critical for neuromuscular junction maintenance. J. Neurosci., 34, 13892–13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohlendieck, K., Ervasti, J.M., Matsumura, K., Kahl, S.D., Leveille, C.J. and Campbell, K.P. (1991) Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron, 7, 499–508. [DOI] [PubMed] [Google Scholar]
- 20.Connolly, A.M., Keeling, R.M., Mehta, S., Pestronk, A. and Sanes, J.R. (2001) Three mouse models of muscular dystrophy: the natural history of strength and fatigue in dystrophin-, dystrophin/utrophin-, and laminin alpha 2-deficient mice. Neuromuscul. Disord., 11, 703–712. [DOI] [PubMed] [Google Scholar]
- 21.Theroux, M.C., Olivant, A. and Akins, R.E. (2008) C Histomorphology of neuromuscular junction in Duchenne muscular dystrophy. Paediatr. Anaesth., 18, 256–259. [DOI] [PubMed] [Google Scholar]
- 22.Ng, S.Y. and Ljubicic, V. (2020) Recent insights into neuromuscular junction biology in Duchenne muscular dystrophy: impacts, challenges, and opportunities. EBioMedicine, 61, 103032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Pijl, E.M., van Putten, M., Niks, E.H., Verschuuren, J.J., Aartsma-Rus, A. and Plomp, J.J. (2016) Characterization of neuromuscular synapse function abnormalities in multiple Duchenne muscular dystrophy mouse models. Eur. J. Neurosci., 43, 1623–1635. [DOI] [PubMed] [Google Scholar]
- 24.Grady, R.M., Zhou, H., Cunningham, J.M., Henry, M.D., Campbell, K.P. and Sanes, J.R. (2000) Maturation and maintenance of the neuromuscular synapse: genetic evidence for roles of the dystrophin--glycoprotein complex. Neuron, 25, 279–293. [DOI] [PubMed] [Google Scholar]
- 25.Banks, G.B., Chamberlain, J.S. and Froehner, S.C. (2009) Truncated dystrophins can influence neuromuscular synapse structure. Mol. Cell. Neurosci., 40, 433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyons, P.R. and Slater, C.R. (1991) Structure and function of the neuromuscular junction in young adult mdx mice. J. Neurocytol., 20, 969–981. [DOI] [PubMed] [Google Scholar]
- 27.Pratt, S.J.P., Shah, S.B., Ward, C.W., Kerr, J.P., Stains, J.P. and Lovering, R.M. (2015) Recovery of altered neuromuscular junction morphology and muscle function in mdx mice after injury. Cell. Mol. Life Sci., 72, 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pratt, S.J.P., Valencia, A.P., Le, G.K., Shah, S.B. and Lovering, R.M. (2015) Pre- and postsynaptic changes in the neuromuscular junction in dystrophic mice. Front. Physiol., 6, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen, C., Lu, Y., Zhang, B., Figueiredo, D., Bean, J., Jung, J., Wu, H., Barik, A., Yin, D.M., Xiong, W.C. and Mei, L. (2013) Antibodies against low-density lipoprotein receptor-related protein 4 induce myasthenia gravis. J. Clin. Invest., 123, 5190–5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao, K., Shen, C., Li, L., Wu, H., Xing, G., Dong, Z., Jing, H., Chen, W., Zhang, H., Tan, Z. et al. (2018) Sarcoglycan alpha mitigates neuromuscular junction decline in aged mice by stabilizing LRP4. J. Neurosci., 38, 8860–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao, K., Shen, C., Lu, Y., Huang, Z., Li, L., Rand, C.D., Pan, J., Sun, X.D., Tan, Z., Wang, H. et al. (2017) Muscle yap is a regulator of neuromuscular junction formation and regeneration. J. Neurosci., 37, 3465–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao, N., Zhao, K., Cao, Y., Ren, X., Jing, H., Xing, G., Xiong, W.C. and Mei, L. (2020) A role of Lamin A/C in preventing neuromuscular junction decline in mice. J. Neurosci., 40, 7203–7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weatherbee, S.D., Anderson, K.V. and Niswander, L.A. (2006) LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development, 133, 4993–5000. [DOI] [PubMed] [Google Scholar]
- 34.Xiong, L., Jung, J.U., Wu, H., Xia, W.F., Pan, J.X., Shen, C., Mei, L. and Xiong, W.C. (2015) Lrp 4 in osteoblasts suppresses bone formation and promotes osteoclastogenesis and bone resorption. Proc. Natl. Acad. Sci. USA, 112, 3487–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun, X.D., Li, L., Liu, F., Huang, Z.H., Bean, J.C., Jiao, H.F., Barik, A., Kim, S.M., Wu, H., Shen, C. et al. (2016) Lrp 4 in astrocytes modulates glutamatergic transmission. Nat. Neurosci., 19, 1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruegg, M.A. and Bixby, J.L. (1998) Agrin orchestrates synaptic differentiation at the vertebrate neuromuscular junction. Trends Neurosci., 21, 22–27. [DOI] [PubMed] [Google Scholar]
- 37.McMahan, U.J. (1990) The agrin hypothesis. Cold Spring Harb. Symp. Quant. Biol., 55, 407–418. [DOI] [PubMed] [Google Scholar]
- 38.Campanelli, J.T., Roberds, S.L., Campbell, K.P. and Scheller, R.H. (1994) A role for dystrophin-associated glycoproteins and utrophin in agrin-induced AChR clustering. Cell, 77, 663–674. [DOI] [PubMed] [Google Scholar]
- 39.Grady, R.M., Merlie, J.P. and Sanes, J.R. (1997) Subtle neuromuscular defects in utrophin-deficient mice. J. Cell Biol., 136, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan, M.C., Rowe, G.C., Raghuram, S., Patten, I.S., Farrell, C. and Arany, Z. (2014) Post-natal induction of PGC-1alpha protects against severe muscle dystrophy independently of utrophin. Skelet. Muscle, 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Handschin, C., Kobayashi, Y.M., Chin, S., Seale, P., Campbell, K.P. and Spiegelman, B.M. (2007) PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev., 21, 770–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pisani, C., Strimpakos, G., Gabanella, F., Di Certo, M.G., Onori, A., Severini, C., Luvisetto, S., Farioli-Vecchioli, S., Carrozzo, I., Esposito, A. et al. (2018) Utrophin up-regulation by artificial transcription factors induces muscle rescue and impacts the neuromuscular junction in mdx mice. Biochim. Biophys. Acta Mol. basis Dis., 1864, 1172–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trajanovska, S., Ban, J., Huang, J., Gregorevic, P., Morsch, M., Allen, D.G. and Phillips, W.D. (2019) Muscle specific kinase protects dystrophic mdx mouse muscles from eccentric contraction-induced loss of force-producing capacity. J. Physiol., 597, 4831–4850. [DOI] [PubMed] [Google Scholar]
- 44.Nelson, C.E., Hakim, C.H., Ousterout, D.G., Thakore, P.I., Moreb, E.A., Castellanos Rivera, R.M., Madhavan, S., Pan, X., Ran, F.A., Yan, W.X. et al. (2016) In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science, 351, 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Long, C., McAnally, J.R., Shelton, J.M., Mireault, A.A., Bassel-Duby, R. and Olson, E.N. (2014) Prevention of muscular dystrophy in mice by CRISPR/Cas 9-mediated editing of germline DNA. Science, 345, 1184–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tabebordbar, M., Zhu, K., Cheng, J.K.W., Chew, W.L., Widrick, J.J., Yan, W.X., Maesner, C., Wu, E.Y., Xiao, R., Ran, F.A. et al. (2016) In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science, 351, 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu, H., Lu, Y., Shen, C., Patel, N., Gan, L., Xiong, W.C. and Mei, L. (2012) Distinct roles of muscle and motoneuron LRP4 in neuromuscular junction formation. Neuron, 75, 94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi, H.Y., Liu, Y., Tennert, C., Sugiura, Y., Karakatsani, A., Kroger, S., Johnson, E.B., Hammer, R.E., Lin, W.C. and Herz, J. (2013) APP interacts with LRP4 and agrin to coordinate the development of the neuromuscular junction in mice. Elife, 2, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang, B., Luo, S., Wang, Q., Suzuki, T., Xiong, W.C. and Mei, L. (2008) LRP4 serves as a coreceptor of agrin. Neuron, 60, 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kishi, M., Kummer, T.T., Eglen, S.J. and Sanes, J.R. (2005) LL5 beta: a regulator of postsynaptic differentiation identified in a screen for synaptically enriched transcripts at the neuromuscular junction. J. Cell Biol., 169, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pratt, S.J.P., Shah, S.B., Ward, C.W., Inacio, M.P., Stains, J.P. and Lovering, R.M. (2013) Effects of in vivo injury on the neuromuscular junction in healthy and dystrophic muscles. J. Physiol., 591, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haddix, S.G., Lee, Y.I., Kornegay, J.N. and Thompson, W.J. (2018) Cycles of myofiber degeneration and regeneration lead to remodeling of the neuromuscular junction in two mammalian models of Duchenne muscular dystrophy. PLoS One, 13, e0205926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yumoto, N., Kim, N. and Burden, S.J. (2012) Lrp 4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature, 489, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martinez-Pena y Valenzuela, I., Mouslim, C., Pires-Oliveira, M., Adams, M.E., Froehner, S.C. and Akaaboune, M. (2011) Nicotinic acetylcholine receptor stability at the NMJ deficient in alpha-syntrophin in vivo. J. Neurosci., 31, 15586–15596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hara, Y., Balci-Hayta, B., Yoshida-Moriguchi, T., Kanagawa, M., Beltran-Valero de Bernabe, D., Gundesli, H., Willer, T., Satz, J.S., Crawford, R.W., Burden, S.J. et al. (2011) A dystroglycan mutation associated with limb-girdle muscular dystrophy. N. Engl. J. Med., 364, 939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grady, R.M., Akaaboune, M., Cohen, A.L., Maimone, M.M., Lichtman, J.W. and Sanes, J.R. (2003) Tyrosine-phosphorylated and nonphosphorylated isoforms of alpha-dystrobrevin: roles in skeletal muscle and its neuromuscular and myotendinous junctions. J. Cell Biol., 160, 741–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goddeeris, M.M., Wu, B., Venzke, D., Yoshida-Moriguchi, T., Saito, F., Matsumura, K., Moore, S.A. and Campbell, K.P. (2013) LARGE glycans on dystroglycan function as a tunable matrix scaffold to prevent dystrophy. Nature, 503, 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu, H., Barik, A., Lu, Y., Shen, C., Bowman, A., Li, L., Sathyamurthy, A., Lin, T.W., Xiong, W.C. and Mei, L. (2015) Slit 2 as a beta-catenin/Ctnnb 1-dependent retrograde signal for presynaptic differentiation. Elife, 4, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]