

Abstract
The antidiabetic drug pioglitazone is, to date, the most efficacious oral drug recommended off‐label for the treatment of nondiabetic or diabetic patients with biopsy‐proven nonalcoholic steatohepatitis (NASH). However, weight gain and edema side effects have limited its use for NASH. Pioglitazone is a mixture of two stereoisomers ((R)‐pioglitazone and (S)‐pioglitazone) that interconvert in vitro and in vivo. We aimed to characterize their individual pharmacology to develop a safer and potentially more potent drug for NASH. We stabilized the stereoisomers of pioglitazone with deuterium at the chiral center. Preclinical studies with deuterium‐stabilized (R)‐pioglitazone (PXL065) and (S)‐pioglitazone demonstrated that (R)‐pioglitazone retains the efficacy of pioglitazone in NASH, including reduced hepatic triglycerides, free fatty acids, cholesterol, steatosis, inflammation, hepatocyte enlargement, and fibrosis. Although both stereoisomers inhibit the mitochondrial pyruvate carrier, PXL065 shows limited to no peroxisome proliferator–activated receptor gamma (PPARγ) activity, whereas (S)‐pioglitazone appears responsible for the PPARγ activity and associated weight gain. Nonetheless, in preclinical models, both stereoisomers reduce plasma glucose and hepatic fibrosis to the same extent as pioglitazone, suggesting that these benefits may also be mediated by altered mitochondrial metabolism. In a phase 1a clinical study, we demonstrated safety and tolerability of single 7.5‐mg, 22.5‐mg, and 30‐mg doses of PXL065 as well as preferential exposure to the (R)‐stereoisomer in comparison to 45‐mg pioglitazone. Conclusion: PXL065 at a dose lower than 22.5 mg is predicted to exhibit efficacy for NASH equal to, or greater than, 45‐mg pioglitazone without the potentially detrimental weight gain and edema. The development of PXL065 for NASH represents a unique opportunity to leverage the therapeutic benefits of pioglitazone, while reducing or eliminating PPARγ‐related side effects.

Abbreviations
- CD
choline‐deficient
- D/H
deuterium/hydrogen
- FFA
free fatty acid
- IL
interleukin
- MCD
methionine/choline‐deficient
- MPC
mitochondrial pyruvate carrier
- PK
pharmacokinetic
- PPAR
peroxisome proliferator–activated receptor
- T2DM
type 2 diabetes mellitus
- TZD
thiazolidinedione
- NASH
nonalcoholic steatohepatitis
- pio
pioglitazone
- PPARγ
peroxisome proliferator‐activated receptor gamma
- TG
triglyceride
Thiazolidinediones (TZDs) are antidiabetic drugs that sensitize the body to insulin. Four TZDs (Supporting Fig. S1) have been approved for the treatment of type 2 diabetes mellitus (T2DM). These compounds were empirically discovered to be agonists of the peroxisome proliferator–activated receptor gamma (PPARγ), a ligand‐activated nuclear receptor that drives broad transcriptional programs associated with adipogenesis, lipid metabolism, innate immune function, and metabolic homeostasis.( 1 ) As such, the antidiabetic mechanism of action of TZDs has been attributed to activation of PPARγ.( 2 )
In contrast, emerging evidence suggests that TZDs exert many of their beneficial effects independently of PPAR activation.( 3 ) For instance, liver and skeletal muscle remain responsive to TZDs, despite tissue‐specific deletion of PPARγ,( 4 ) and the whole‐body insulin‐sensitizing effect of rosiglitazone persists following PPARγ deletion in mature adipocytes.( 5 ) Pharmacologic evidence also suggests that TZDs have nongenomic (i.e., PPAR‐independent) effects. They can acutely alter metabolic homeostasis on a timescale that is likely too rapid to be driven by broad gene‐expression changes,( 6 ) and in vitro and in vivo experiments have shown that rank‐order affinity for PPARγ does not always correlate with efficacy.( 3 )
Recent work shows that many PPARγ‐independent effects of TZDs could be attributed to inhibition of the mitochondrial pyruvate carrier (MPC), an inner mitochondrial membrane transporter responsible for the uptake of glucose‐derived pyruvate from the cytoplasm into the mitochondrial matrix. TZDs are acute, specific inhibitors of MPC activity at clinically relevant concentrations.( 7 ) Indeed, TZDs with reduced affinity to PPARγ still have an insulin‐sensitizing effect.( 8 ) In mice, liver‐specific MPC ablation maintains euglycemia following diet‐induced obesity,( 9, 10 ) and muscle‐specific MPC deletion improves whole‐body insulin sensitivity.( 11 ) Pioglitazone has also been shown to be a direct inhibitor of acyl‐CoA synthase 4, an enzyme that is specifically linked to metabolic pathways.( 12 ) Despite decades of use in humans, the repertoire of targets for pioglitazone and its exact mechanism of action is only now beginning to be unraveled.
To date, pioglitazone, the most extensively studied drug for nonalcoholic steatohepatitis (NASH), has demonstrated “resolution of NASH without worsening of fibrosis” in a phase 4 clinical trial.( 13 ) It is the only drug recommended for off‐label use to treat both nondiabetic and diabetic patients with biopsy‐proven NASH in the Practice Guidance published by the American Association for the Study of Liver Diseases( 14 ) and the European Association for the Study of the Liver.( 15 ) The use of pioglitazone for the treatment of NASH, however, has been limited due to its PPARγ‐related side effects, which include weight gain,( 16 ) edema,( 17 ) and bone fractures.( 18 )
All TZDs, including pioglitazone, are a mixture of stereoisomers and are characterized by the presence of a chiral center prone to rapid, nonenzymatic inversion of configuration (Supporting Fig. S1). This instability has prevented the full pharmacological characterization of the individual stereoisomers. For example, the (S)‐stereoisomer of rosiglitazone was identified as the most potent PPARγ agonist. However, (S)‐rosiglitazone rapidly equilibrated to create a 1:1 mixture of (R)‐rosiglitazone and (S)‐rosiglitazone, preventing further characterization of the stereoisomers.( 19 ) The glucose‐lowering and triglyceride‐lowering activity of the (R)‐stereoisomers and (S)‐stereoisomers of pioglitazone have been evaluated in rat, but the conclusion that they are equipotent was incorrect, as no consideration was given to the rapid in vivo interconversion of the stereoisomers.( 20 ) Interestingly, the anti‐inflammatory effects of pioglitazone were shown to be uniquely associated with the (R)‐stereoisomer in a rat model of chronic obstructive pulmonary disease.( 21 ) This was only achieved after stabilization of the stereoisomers in an acidic solution, followed by immediate intranasal dosing, which limited interconversion during the study. TZDs, however, are still developed and marketed as mixtures of stereoisomers, because their chemically unstable chiral center prevents characterization and development of the single, preferred stereoisomer.
We have previously reported that, by stabilizing the chiral center of drug‐like compounds with deuterium, we can reduce the rate of interconversion and characterize each of the stereoisomers( 22 ); herein, we report the preclinical characterization of the deuterium‐stabilized stereoisomers of pioglitazone, deuterated (R)‐pioglitazone (PXL065) and deuterated (S)‐pioglitazone (d‐S‐pio), and the initial clinical evaluation of PXL065 compared with pioglitazone. In preclinical models, we discovered that the pleiotropic pharmacological effects of pioglitazone can be differentiated between the two stereoisomers, and that PPARγ activity may not be critical for efficacy in models of T2DM or NASH. These results challenge the commonly accepted view that PPARγ activity is responsible for all of the beneficial effects ascribed to pioglitazone. In addition, activation of PPARγ has been linked to the major side effects of TZD treatment (i.e., weight gain, fluid retention, and bone loss),( 17, 18 ) while PPARγ‐sparing TZDs have been shown to not cause bone loss.( 23 ) In a phase 1a clinical trial, administration of PXL065 was shown to be safe and well‐tolerated and to result in increased relative exposure to the (R)‐stereoisomer compared with pioglitazone treatment. Therefore, these observations support the development of PXL065, a single, deuterium‐stabilized, non‐PPARγ‐active, stereoisomer of pioglitazone, as a safer and potentially more potent therapeutic than pioglitazone for NASH and other indications such as T2DM.
Materials and Methods
Synthesis of PXL065 and d‐S‐pio
Compounds were synthesized by deuterium/hydrogen (D/H) exchange from pioglitazone hydrochloride, followed by separation of the stereoisomers and formation of the salt, as detailed in the Supporting Information.
PPAR Agonism and Binding Assays
The PPARγ and PPARα binding affinity of PXL065 and d‐S‐pio were evaluated in agonist radioligand assays (Eurofins Discovery). The PPARα, δ, and γ agonist activities were evaluated in coactivator recruitment assays (Eurofins Discovery). Human recombinant PPARα, δ, or γ and coactivator were pre‐incubated at room temperature for 30 minutes in the presence of the test compound and a fluorescence acceptor. A more detailed description is provided in the Supporting Information.
Mitochondrial Functional Assays
Oxygen consumption was measured using an Agilent Seahorse XF96 and XFe96 (Santa Clara, CA). To measure respiration driven by pyruvate in intact HepG2 cells, cells were plated at 2.5 104 cells/well in Seahorse XF96 tissue culture plates for 2 days. Oligomycin (2 μM), FCCP (carbonyl cyanide‐p‐trifluoromethoxyphenylhydrazone; two sequential pulses of 400 nM), and rotenone (200 nM) with antimycin A (1 μM) were added acutely to the wells, and respiratory parameters were calculated as in Divakaruni et al.( 24 ) Pyruvate‐driven respiration was measured in Dulbecco’s modified Eagle’s medium (#D5030; Sigma‐Aldrich, St. Louis, MO) without glucose and supplemented with 5 mM pyruvate, 2 mM HEPES (4‐[2‐hydroxyethyl]‐1‐piperazine ethanesulfonic acid), and in the presence of 3 μM CB‐839 (glutaminase inhibitor) and 3 μM etomoxir (carnitine palmitoyltransferase Ia inhibitor). Respiration in permeabilized cells was measured as previously described.( 25 )
Animal Studies
All animal experiments were approved by the relevant committees in the institutions where the studies were performed. All animal experiments complied with the criteria outlined in the Guide for the Care and Use of Laboratory Animals by the National Academy of Sciences.
For the pharmacokinetic (PK) studies, a detailed description of the mouse, rat, and dog PK studies is provided in the Supporting Information.
For the efficacy studies, protocols and statistical methods for the db/db and ob/ob mouse models of T2DM, the choline‐deficient (CD) and methionine/choline‐deficient (MCD) diet models of NASH, as well as the mouse weight‐gain study are provided in the Supporting Information
Human Study
An institutional review board–approved open‐label, phase 1a, in‐human clinical trial of PXL065 was completed under protocol DRX‐065‐001, adhering to the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use guidelines for good clinical practice. The study was divided into two parts. In part 1, healthy subjects (6 per group, as 3 per gender per group) were randomly allocated to receive, after an overnight fast, either a single 22.5‐mg oral dose of PXL065, as active pharmaceutical ingredient in a capsule, or a single 45‐mg oral dose of Actos (pioglitazone; Takeda, Tokyo, Japan), as a commercially available tablet. After review of safety and tolerability and following the determination of comparative PK exposure, 12 more subjects (6 per group, as 3 per gender per group) were included in part 2 and received either a single 7.5‐mg dose of PXL065 or a single 30‐mg dose of PXL065. The primary objective was to evaluate the safety and tolerability of PXL065 by recording of adverse events and changes from baseline in hematology, clinical chemistry, urinalysis, electrocardiograms, physical examinations, and vital signs. The secondary objectives included the evaluation of the PK of Actos and PXL065. To be eligible, subjects were to be healthy based on pre‐identified inclusion and exclusion criteria (see Supporting Information) Subjects remained in the clinic for 36 hours following dose and returned as outpatients on days 4 and 7 for follow‐up assessments. PK samples (plasma) were collected before dose and at 13 time points following dose.
Bioanalysis and PK Analysis
Plasma samples from preclinical and clinical studies were processed and analyzed by chiral high‐performance liquid chromatography with tandem mass spectrometry, using validated methods and good laboratory practice, as appropriate. Normalized peak areas for the deuterated stereoisomers were corrected for interference from the isotopic peak of the corresponding protonated stereoisomer, and concentrations were calculated (more details can be found in the Supporting Information). Data were plotted in Excel 2013 (Microsoft Corp., Redmond, WA). Noncompartmental data analysis was performed using the PKSolver Excel add‐in( 26 ) for mouse, rat, and dog PK data, Phoenix WinNonlin (version 6.2; Pharsight Corp., Palo Alto, CA) for the ob/ob plasma PK data, and Excel for comparisons as well as a validated Phoenix WinNonlin for human data.
Results
PXL065 Modulates Mitochondrial Function With No Measurable PPARγ Agonist Activity
Stabilizing the chiral center of pioglitazone allowed the separation and characterization of the individual (R)‐enantiomers and (S)‐enantiomers (Supporting Fig. S1). First, we measured whether PXL065 and d‐S‐pio showed the same affinity for PPARγ as racemic pioglitazone. Using a coactivator recruitment assay, d‐S‐pio and pioglitazone showed similar PPARγ activity, while, remarkably, PXL065 exhibited negligible PPARγ activity (EC50 = 4.6, 3.2, and >100 µM for pioglitazone, d‐S‐pio, and PXL065, respectively; Fig. 1A). As verification that the two enantiomers of pioglitazone have starkly different affinity for PPARγ, a direct binding assay independently showed that PXL065 exhibited 25‐fold less binding affinity for PPARγ than d‐S‐pio (IC50 = 6.5 µM for PXL065 and 260 nM for d‐S‐pio; Supporting Fig. S2A). Neither pioglitazone, PXL065, nor d‐S‐pio showed binding to PPARα or agonist activity at PPARα or PPARδ (Supporting Fig. S2B‐D).
FIG. 1.
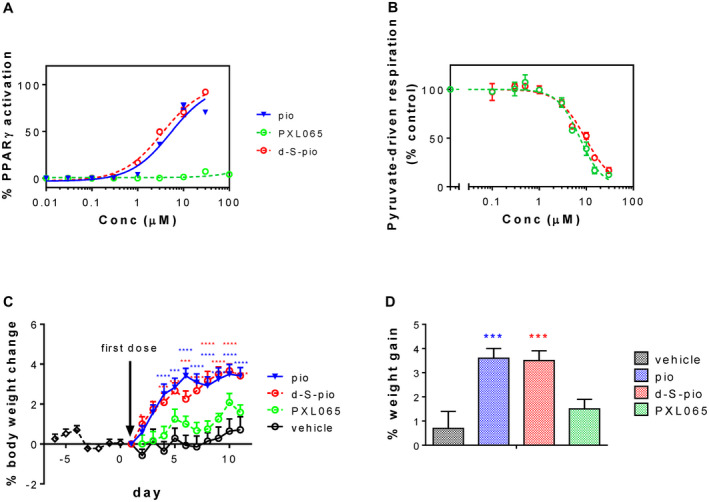
(A) PPARγ agonism of pioglitazone, PXL065, and d‐S‐pio (mean ± SEM). (B) Effect of pioglitazone, PXL065, and d‐S‐pio on pyruvate‐driven oxygen consumption (mean ± SD) in intact HepG2 cells: (pioglitazone [blue triangles, blue line], d‐S‐pio [red open circles, red dotted line], and PXL065 [green open circles, green dotted line]) and effect of pioglitazone, PXL065, and d‐S‐pio on body weight in C57BL/6J male mice dosed twice daily for 7 days (vehicle or 30, 15, and 15 mg/kg/day for pioglitazone, PXL065, and d‐S‐pio, respectively). (C,D) Body‐weight gain (mean ± SEM) versus time (C) and body weight on last day (mean + SEM) (D): vehicle, black; pioglitazone, blue; d‐S‐pio, red; and PXL065, green. Statistically different from vehicle group: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Abbreviation: pio, pioglitazone.
Unlike with PPARγ binding, PXL065 and d‐S‐pio were both shown to inhibit pyruvate‐driven respiration in HepG2 cells with single‐digit micromolar affinity (IC50s of 7.3 µM and 9.5 µM for PXL065 and d‐S‐pio, respectively; Fig. 1B). Qualitatively similar results were observed in C2C12 mouse myoblasts (IC50s of about 10 µM and 20 µM for PXL065 and d‐S‐pio, respectively), rat primary cortical neurons (IC50s of about 15 µM and 30 µM for PXL065 and d‐S‐pio, respectively), and permeabilized HepG2 cells (IC50s of about 3 µM for both compounds), demonstrating that this was not a cell type‐specific effect.
Relative Exposure to (R)‐Pioglitazone and (S)‐Pioglitazone Is Species‐Specific and Affected by Deuterium Substitution
Stabilization with deuterium at the chiral center of pioglitazone translated into significant changes in the relative exposure to the (R)‐stereoisomer and (S)‐stereoisomer in vivo, as observed after separately administering PXL065, d‐S‐pio, or pioglitazone. In all PK studies, the dose of pioglitazone (1:1 mixture of (R)‐pioglitazone and (S)‐pioglitazone) was twice the dose of PXL065 or d‐S‐pio, taking into account the 1:1 composition of stereoisomers in the solid form of pioglitazone. Deuterium stabilization of the stereoisomers leads to a strong in vivo stereoisomeric enrichment, but not to complete elimination of exposure, to the other stereoisomer, as over time and balanced by the elimination half‐life, some D/H exchange occurs, accompanied by inversion of configuration of the chiral center (PK parameters in Supporting Table S1).
In C57BL/6 mouse (Fig. 2A), administration of pioglitazone by oral gavage once daily for 5 days resulted in a dramatic stereoselective exposure to the (S)‐stereoisomer (R:S ratio of 0.25). Administration of PXL065 inverted that stereoselectivity and significantly increased the relative total exposure to (R)‐pioglitazone (sum of exposures to protonated and deuterated (R)‐pioglitazone), by 8‐fold (R:S ratio of 2). Conversely, administration of d‐S‐pio led to a 5‐fold increase in relative exposure to (S)‐pioglitazone (sum of exposure to protonated and deuterated (S)‐pioglitazone) with an R:S ratio of 0.056. No changes in elimination rates were observed for the enantiomers, consistent with the fact that the chiral center of pioglitazone is not involved in drug metabolism.
FIG. 2.
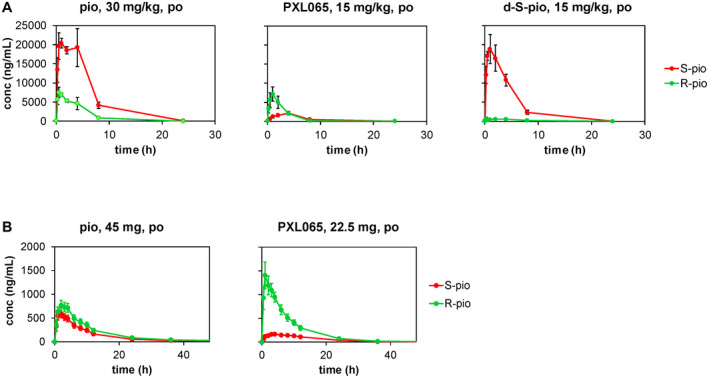
PK of pioglitazone, PXL065, and d‐S‐pio in mouse after once daily dosing for 5 days (mean ± SD) (A) and human after a single oral dose (mean ± SEM) (B): S‐pio, sum of protonated and deuterated (S)‐pioglitazone (red circles, red line); R‐pio, sum of protonated and deuterated (R)‐pioglitazone (green circles, green line).
In Sprague‐Dawley rat, repeated oral administration of pioglitazone once daily for 6 days resulted in stereoselective exposure to the (S)‐stereoisomer with an R:S ratio of 0.37 (Supporting Fig. S3A), which was increased 4‐fold in animals dosed with d‐S‐pio (R:S ratio of 0.099), while the relative exposure to (R)‐pioglitazone was increased 3‐fold in rats dosed with PXL065 (R:S ratio of 1.2). As in mouse, the elimination half‐lives of the stereoisomers were not affected by the compound administered.
In dog (Supporting Fig. S3B), a single oral dose of pioglitazone resulted in equivalent exposure to the two stereoisomers (R:S ratio of 1), whereas the relative exposure to (R)‐pioglitazone was increased 4‐fold after dosing PXL065 (R:S ratio of 4) and the relative exposure to (S)‐pioglitazone was increased 5‐fold after dosing d‐S‐pio (R:S ratio of 0.2). As in rodent, the elimination half‐lives were not affected. In dog, the relative exposure to human‐relevant pioglitazone metabolites M‐III and M‐IV is similar to that observed in human.( 20 ) After dosing pioglitazone, PXL065, or d‐S‐pio in dog, exposure to M‐IV was 8‐fold larger than exposure to M‐III and independent of compound administered (Supporting Table S1). However, absolute exposure to M‐III and M‐IV was 2‐fold lower in animals dosed with PXL065 or d‐S‐pio, as expected, because the dose of PXL065 or d‐S‐pio was half the dose of pioglitazone, showing that incorporation of deuterium did not affect metabolism.
In human (Fig. 2B and Supporting Table S2), administration of a single 45‐mg oral dose of Actos (pioglitazone) resulted in stereoselective exposure to (R)‐pioglitazone with an R:S ratio of 1.4. When a single dose of PXL065 (7.5, 22.5, or 30 mg; Fig. 2B and Supporting Fig. S4) was administered orally, the relative exposure to (R)‐pioglitazone increased by about 3‐fold to an R:S ratio of about 4.1. Exposure to (R)‐pioglitazone after dosing 22.5 mg PXL065 was 19% higher than in the 45‐mg Actos group, while the exposure to the (S)‐enantiomer was 42% lower. Interestingly, overall exposure to pioglitazone, as the sum of (R)‐pioglitazone and (S)‐pioglitazone, regardless of isotopic content, was similar after dosing 45 mg Actos or 22.5 mg PXL065. In contrast, as in dog, exposure to major metabolites M‐III and M‐IV was found to be lowered 2‐fold in subjects dosed with PXL065 22.5 mg versus Actos 45 mg. Of note, exposure to deuterated metabolites (d‐M‐III and d‐M‐IV) represented less than 10% of the exposure to total pioglitazone (i.e., sum of protonated and deuterated (R)‐pioglitazone, (S)‐pioglitazone, and metabolites M‐III and M‐IV).
PXL065, not d‐S‐pio, Is Responsible for Significantly Improving Hepatic Markers in Diet‐Induced Mouse Models of NASH
Although pioglitazone and PXL065 were studied in both the MCD and CD diet models, d‐S‐pio was only evaluated in the CD model. As previously observed in these models,( 27 ) both the MCD and CD diets caused weight loss. However, weight loss was significantly less severe with the CD diet. In mice on the MCD diet treated with pioglitazone, weight loss tended to be less than in vehicle‐treated or PXL065‐treated animals on the same diet (Supporting Fig. S5A). This is consistent with the PPARγ‐related weight gain observed with pioglitazone and other TZDs. PXL065 and pioglitazone significantly affected all markers of NASH progression in the two diet models (Table 1 and Fig. 3A,B), while d‐S‐pio was overall without effect. The only significant benefit of d‐S‐pio was an antifibrotic effect similar to that observed with PXL065 and pioglitazone (Supporting Fig. S5B). Interestingly, PXL065 appeared superior to pioglitazone for reducing hepatic steatosis and triglycerides (TGs) in the CD diet mice. PXL065, pioglitazone, and d‐S‐pio exhibited the same metabolic profile with a significant decrease in serum TGs and free fatty acids (FFAs) and an increase in adiponectin in the CD diet mice (Supporting Fig. S5C). A more detailed description is provided in the Supporting Information.
TABLE 1.
Qualitative Summary of the NASH‐Relevant Pharmacological Effects of Pioglitazone, PXL065, and d‐S‐pio in the Mouse Weight‐Gain Study and in the CD and MCD Diet Models of NASH
| Functional Parameters | Pio | PXL065 | d‐S‐Pio |
|---|---|---|---|
| ↓ Hepatic TGs | ✓ | ✓ | — |
| ↓ Hepatic FFAs | ✓ | ✓ | — |
| ↓ Hepatic cholesterol | ✓ | ✓ | — |
| ↓ Hepatic inflammation | ✓ | ✓ | — |
| ↓ Hepatic steatosis | ✓ | ✓ | — |
| ↓ Hepatic fibrosis | ✓ | ✓ | ✓ |
| ↑ Body‐weight gain | ✓ | — | ✓ |
| ↑ Fluid retention | ✓ | — | ✓ |
FIG. 3.
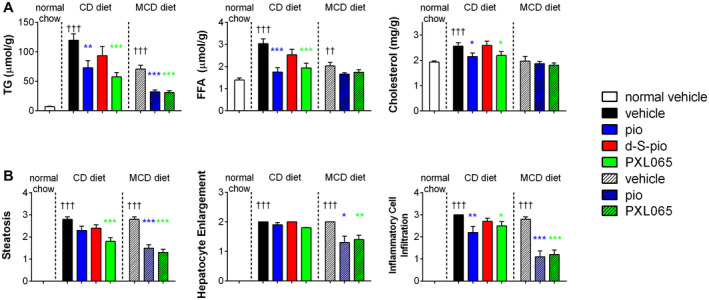
Pharmacological effect of pioglitazone, PXL065, and d‐S‐pio in CD diet and MCD diet mouse models of NASH: liver TGs, FFAs, and cholesterol (A) and liver histopathological measures of steatosis, enlarged hepatocytes, and inflammation (B): vehicle, black; pioglitazone, blue; d‐S‐pio, red; and PXL065, green (mean + SEM). Significantly different from normal chow group: †† P < 0.01 and ††† P < 0.001. Significantly different from vehicle group on the same diet: *P < 0.05, **P < 0.01, and ***P < 0.001.
d‐S‐pio, not PXL065, Causes Weight Gain
Only pioglitazone and d‐S‐pio significantly increased body weight over vehicle after 10 days of dosing in normal C57BL/6J male mice. In contrast, PXL065 did not increase body weight compared with vehicle (Fig. 1C,D). A trend was observed for increased fluid retention (measured as total body water) in animals administered d‐S‐pio or pioglitazone, but not PXL065 (Supporting Fig. S6).
PXL065, not d‐S‐pio, Significantly Reduces Inflammation in a db/db Mouse Model of T2DM
Inflammatory markers, serum amyloid A, and cytokines interleukin (IL) 1β and IL‐6, were significantly decreased only by pioglitazone and PXL065 (Fig. 4A), whereas d‐S‐pio had no effect. All three compounds statistically significantly reduced plasma glucose (Fig. 5A). Serum insulin was significantly decreased by pioglitazone, and a trend toward significant reduction was observed for d‐S‐pio (Fig. 5B). Similarly, serum adiponectin—a known marker of PPARγ activation—was significantly increased with pioglitazone and d‐S‐pio, while only a nonsignificant trend was seen with PXL065 (Fig. 4B), likely because of exposure to the PPARγ‐active (S)‐enantiomer. Serum TGs and FFAs were significantly decreased by all three compounds (Fig. 4C).
FIG. 4.
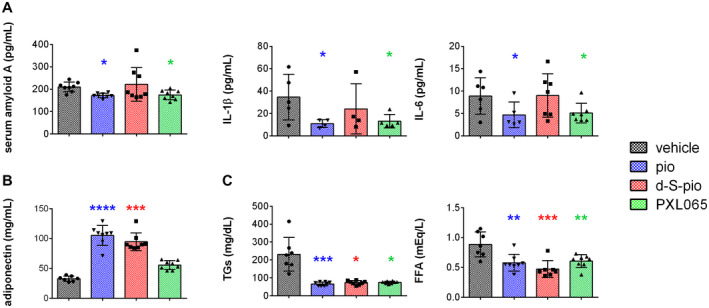
Pharmacological effect (mean ± SD) of pioglitazone, PXL065, and d‐S‐pio after 10 days of once‐daily dosing of vehicle, pioglitazone (30 mg/kg), PXL065 (20 mg/kg), or d‐S‐pio (20 mg/kg) by oral gavage in the db/db mouse model on inflammatory markers serum amyloid A and cytokines IL‐1β and IL‐6 (A), serum adiponectin (B), serum TGs, and FFAs (C): vehicle, black; pioglitazone, blue; d‐S‐pio, red; and PXL065, green. Significantly different from vehicle: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
FIG. 5.
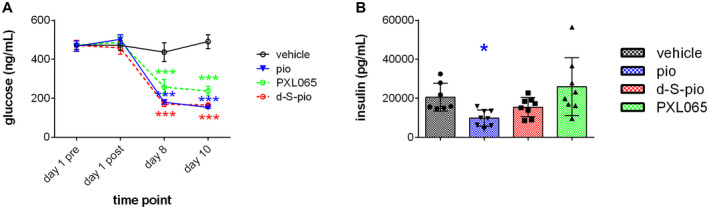
Pharmacological effect of once‐daily dosing for 10 days of pioglitazone (30 mg/kg), PXL065 (20 mg/kg), and d‐S‐pio (20 mg/kg) in the db/db mouse model on nonfasting blood glucose (mean ± SEM) (A) and insulin (mean ± SD) (B): vehicle, black; pioglitazone, blue; d‐S‐pio, red; and PXL065, green. Significantly different from vehicle: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
PXL065 Significantly Improves Markers of T2DM in the ob/ob Mouse
PXL065 dose‐dependently decreased basal glycemia and strongly decreased basal insulinemia, with a significant effect at the highest dose (30 mg/kg) (Supporting Fig. S7A,B). In contrast, the effects on basal glycemia of d‐S‐pio, and surprisingly pioglitazone, were not significant, although basal insulinemia was strongly and significantly decreased by both compounds. PXL065 dose‐dependently increased the plasma adiponectin level, with significance reached at 30 mg/kg. Interestingly, there appears to be a positive correlation of high molecular weight adiponectin increase with plasma exposure to the PPARγ‐active (S)‐enantiomer, while there is no correlation of the adiponectin increase with the exposure to the (R)‐enantiomer (Fig. 6A). An improvement of insulin secretion in response to glucose was also observed, as the insulinogenic index was increased after treatment with the three compounds, with the strongest and most significant effects observed after treatment with PXL065 (15 mg/kg and 30 mg/kg) (Supporting Fig. S7C). Both improvements in insulin action and insulin secretion in response to glucose likely contribute to the better glucose tolerance of ob/ob mice treated with the compounds (Fig. 6B). In addition, PXL065 treatment did not result in any significant body‐weight gain, except at the highest dose (30 mg/kg), compared to mice treated with vehicle. In contrast, d‐S‐pio (15 mg/kg) significantly increased body‐weight gain to the same extent as pioglitazone (30 mg/kg). Specifically, there appears to be a correlation between plasma exposure to the (S)‐enantiomer and body‐weight gain, while no such correlation is observed with the exposure to the (R)‐enantiomer (Fig. 6C). Finally, exposure to the enantiomers of pioglitazone in brain, liver, muscle, and fat tissue showed a larger tissue‐to‐plasma ratio for (R)‐pioglitazone than for (S)‐pioglitazone, regardless of the compound administered, indicating better tissue penetration of (R)‐pioglitazone (Supporting Fig. S8), in agreement with previously reported data on brain penetration of the stereoisomers of pioglitazone in the mouse.( 28 )
FIG. 6.
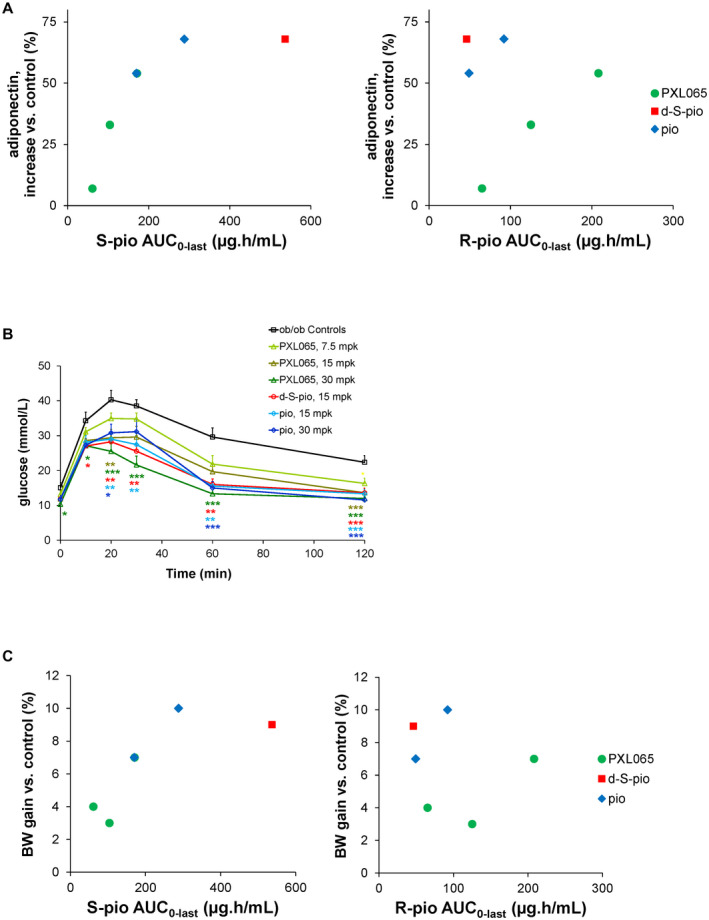
Pharmacological effect of pioglitazone (15 and 30 mg/kg), PXL065 (7.5, 15, and 30 mg/kg), and d‐S‐pio (15 mg/kg) after 4 weeks of once‐daily dosing by oral gavage in the ob/ob mouse model on plasma adiponectin (ratio high molecular weight to total) as percentage increase over ob/ob controls, as a function of exposure to S‐pio (sum of protonated and deuterated (S)‐pioglitazone, left) and R‐pio (sum of protonated and deuterated (R)‐pioglitazone, right) (A) and plasma glucose in an oral glucose tolerance test (B). Oral glucose tolerance test and analysis of variance with Dunnett’s post hoc test versus ob/ob controls: *P < 0.05, **P < 0.01, ***P < 0.001. (C) Body‐weight gain as percentage increase over ob/ob controls as function of exposure to S‐pio (left) and R‐pio (right). Abbreviations: AUC, area under the curve: and BW, body weight.
Discussion
The mechanism of action of pioglitazone and other TZDs has historically been assigned to their PPARγ agonist activity, given their high‐affinity PPARγ binding and their unequivocal effects on gene expression. However, PPARγ knockout models have shown that some pharmacological effects of TZDs are PPARγ‐independent.( 29, 30 ) PPARγ agonism has also been shown to increase hepatic steatosis (in rodents), thus potentially opposing any possible non‐PPARγ‐mediated beneficial effects of TZDs on lipid metabolism; the fact that liver‐specific PPARγ knockout mice are protected from this increase in steatosis supports this concept.( 31, 32 ) More recently, some pharmacological effects of TZDs have been linked to PPARγ‐independent mitochondrial and metabolic effects of these molecules, most notably inhibition of the MPC.( 7 )
Through deuterium stabilization, we were able to evaluate the pharmacology and pharmacokinetics of the stereoisomers of pioglitazone. In vivo, dosing of the stabilized stereoisomers was shown to affect the relative exposure to each stereoisomer. We observed a species‐dependent stereoselective exposure to the stereoisomers. When pioglitazone, a 1:1 mixture of stereoisomers, is administered, substantial (S)‐stereoselectivity is observed in rodents, while equivalent exposure to the stereoisomers and modest (R)‐stereoselectivity are seen in dog and human, respectively. Similar observations have already been made for other drugs,( 33 ) including several TZDs.( 34, 35, 36 ) These results highlight the problematic extrapolation of pharmacological effects of mixtures of stereoisomers from one species to another. Indeed, we have shown that the stereoisomers of pioglitazone have substantially different pharmacology with respect to PPARγ binding, activation, and classical related effects in vivo, such as weight gain, fluid retention, and increases in adiponectin. Specifically, the (S)‐stereoisomer is a PPARγ agonist, while the (R)‐stereoisomer has little or no activity at the same receptor, and both compounds are modulators of mitochondrial function. Therefore, the absolute exposure to each stereoisomer will affect the overall pharmacological effects of the mixture.
Dosing of the deuterium‐stabilized stereoisomers of pioglitazone offers a method to evaluate the pharmacological effects of each stereoisomer, by limiting exposure to the other stereoisomer. In preclinical species, dosing of the deuterium‐stabilized stereoisomers of pioglitazone at half the dose of the racemate resulted in a significant 3‐fold to 8‐fold enrichment in the relative exposure to the stereoisomer dosed. Interestingly, exposure to the dosed deuterium‐stabilized stereoisomer was the same as exposure to that stereoisomer when pioglitazone was administered at twice the dose. Similarly, elimination half‐life was not affected by the presence of deuterium.
In a db/db mouse model of T2DM, only PXL065 and pioglitazone decreased inflammatory markers. PXL065, d‐S‐pio, and pioglitazone had similar effects on blood glucose, plasma TGs and FFAs, while only d‐S‐pio and pioglitazone significantly increased adiponectin, a well‐known biomarker of PPARγ activity.( 37 ) Similarly, in an ob/ob mouse model of T2DM, PXL065 was the most potent at affecting markers of the disease, glycemia and insulinemia. Furthermore, plasma adiponectin and body‐weight gain were shown to be correlated with plasma exposure to the PPARγ active (S)‐enantiomer of pioglitazone. In a CD diet mouse model of NASH, the hepatic benefits of pioglitazone were further improved by PXL065, exhibiting a significant effect on hepatic TGs, FFAs, cholesterol, inflammation, and steatosis. In contrast, d‐S‐pio lacked any significant activity on these same endpoints. Liver fibrosis was affected to the same extent by both stereoisomers, which can be attributed to either PPARγ agonism and/or mitochondrial function modulation.( 38, 39 ) We confirmed these findings in an MCD diet–induced NASH model, in which PXL065 was equipotent to pioglitazone at reducing hepatic inflammation, hepatocyte enlargement, steatosis, and fibrosis.
These results demonstrate that the pleiotropic effects of pioglitazone are a composite of the distinct pharmacological activities of its stereoisomers. MPC inhibition by TZDs diminishes mitochondrial oxidation of glycolytic pyruvate, shifting reliance on fuels for oxidative phosphorylation to alternative substrates, including fatty acids and amino acids, while reducing de novo lipogenesis and gluconeogenesis.( 6, 7, 10, 11, 40 ) MPC inhibition may also acutely lead to increased cellular glucose uptake and AMPK (5'‐adenosine monophosphate–activated protein kinase) activation in skeletal muscle, contributing to insulin sensitization.( 6, 7 ) These shifts in metabolism are consistent with diminished disease pathogenesis in NASH/nonalcoholic fatty liver disease (NAFLD). In further support of this concept, recent work has shown that reduction in liver pyruvate kinase activity that decreases glycolytic pyruvate production is also protective from NASH/NAFLD in mice fed a high‐fat/high‐sucrose diet.( 41 ) PPARγ activation has been shown to result in not only fat relocalization to subcutaneous depots, leading to lipid depletion in serum and improved insulin sensitization through adiponectin up‐regulation,( 42, 43 ) but also to increase hepatic expression of genes related to fatty acid synthesis.( 32 ). In ob/ob mice, PPARγ activation by rosiglitazone was shown to lead to exacerbation of the fatty liver phenotype.( 31 ) The in vivo pharmacological effects of pioglitazone and other TZDs (in rodent) can, therefore, be explained as the result of the opposing effects of PPARγ activation and a shift in mitochondrial metabolism, the former increasing fat content in the liver while the latter decreases it. The balance between the two may be tilted one way or the other as a function of the relative potency of a TZD at its respective targets. This conclusion is supported by the comparison of rosiglitazone and pioglitazone,( 43, 44 ) where pioglitazone, a less potent PPARγ agonist with significant MPC inhibition potency when compared with its PPARγ agonism, has significantly better efficacy in rodent models with NASH and in human patients with NASH than rosiglitazone, a molecule with a substantially greater degree of PPARγ agonist potency relative to its intrinsic potency with respect to mitochondrial function modulatory activity.( 7, 45 )
In addition, unlike pioglitazone and the (S)‐stereoisomer, the (R)‐stereoisomer did not cause significant weight gain over the vehicle control in the mouse, confirming the known effect of PPARγ activation on body weight through mechanisms involving fluid retention and fat storage.( 2 ) In this study, we also observed a significant increase in total body water in mice administered pioglitazone, whereas a trend was observed with d‐S‐pio but not PXL065. Furthermore, as shown in the ob/ob model, the observed weight gain can be correlated to exposure to the potent PPARγ agonist (S)‐pioglitazone, while no such correlation exists between weight gain and exposure to the (R)‐enantiomer.
In humans, oral administration of a single dose of PXL065 up to 30 mg was safe and well tolerated. Exposure to major human metabolites M‐III and M‐IV (as a sum of protonated and deuterated molecular species) was 2‐fold lower after PXL065 dosing versus an equivalent dose of pioglitazone, indicating that metabolism is not affected by the deuterium introduction at the chiral center. In addition, elimination half‐lives of the deuterated metabolites were faster than those of the corresponding protonated forms, likely due to D/H exchange followed by elimination of the protonated forms, the elimination half‐lives of which were similar between PXL065 and pioglitazone dosing. The exposure to each of the deuterated metabolites as well as d‐S‐pio was less than 10% of the exposure to total pioglitazone (sum of the deuterated and protonated enantiomers of pioglitazone and major metabolites, M‐III and M‐IV). Absolute exposure to (R)‐pioglitazone, sum of protonated and deuterated isotopic species, was slightly increased after a single dose of 22.5 mg PXL065 versus 45 mg pioglitazone, whereas that of the (S)‐stereoisomer was more significantly decreased. Based on these clinical data and our PK modeling efforts (data not shown), we conclude that steady‐state dosing with a dose of PXL065 lower than 22.5 mg will result in exposure to the (R)‐stereoisomer comparable to that obtained with 45 mg pioglitazone. At that dose, exposure to the (S)‐stereoisomer should be similar to or lower than 7.5 mg pioglitazone, a dose that has been shown to not cause significant weight gain in humans.( 46, 47, 48 )
With stabilization of the stereoisomers, the PPARγ agonist activity of pioglitazone can be separated from the non‐PPARγ‐mediated mitochondrial and anti‐inflammatory effects. These results pave the way for the development of potentially safer TZDs, in which the side effects due to PPARγ activation are reduced and/or eliminated. These and earlier results from our group( 22 ) should prompt development of additional deuterium‐stabilized stereoisomers that comply with the FDA guidance on stereoisomers( 49 ) and lead to the advancement of improved therapeutics.
The clinical development of PXL065 for NASH is ongoing (phase 2; NCT04321343). Our hypothesis is that PXL065 will recapitulate the strong efficacy profile of pioglitazone with little or none of the adverse effects that are likely attributable to PPARγ, such as weight gain and edema. In addition, given the existence of extensive clinical and nonclinical data pertaining to pioglitazone as an approved product for diabetes, expedited development through the FDA abbreviated regulatory pathway, known as 505(b)(2), is being pursued.
Supporting information
Supporting Material
Acknowledgment
The authors thank Poxel colleagues for the useful discussions and review of the manuscript.
Supported by the National Institutes of Health (R01NS087611).
Potential conflict of interest: Dr. Van der Ploeg owns stock in Poxel. He owns stock in, consults for, and advises DeuteRx. Dr. Divakaruni owns stock in DeuteRx. Dr. Czarnik advises and owns stock in DeuteRx. Dr. Hallakou‐Bozec owns stock in and is employed by Poxel. Dr. Bolze owns stock in and is employed by Poxel. Dr. DeWitt owns stock in and is employed by DeuteRx. She owns stock in Poxel. Dr. Fouqueray owns stock in and is employed by Poxel. Dr. Jacques owns stock in and is employed by DeuteRx. He owns stock in Poxel. Dr. Murphy is employed by Cytokinetics. She owns stock in DeuteRx.
References
Author names in bold designate shared co‐first authorship.
- 1.Cariou B, Charbonnel B, Staels B. Thiazolidinediones and PPARγ agonists: time for a reassessment. Trends Endocrinol Metab 2012;23:205‐215. [DOI] [PubMed] [Google Scholar]
- 2.Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med 2013;19:557‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feinstein DL, Spagnolo A, Akar C, Weinberg G, Murphy P, Gavrilyuk V, et al. Receptor‐independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol 2005;70:177‐188. [DOI] [PubMed] [Google Scholar]
- 4.Norris AW, Chen L, Fisher SJ, Szanto I, Ristow M, Jozsi AC, et al. Muscle‐specific PPARγ‐deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J Clin Invest 2003;112:608‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang QA, Zhang F, Jiang L, Ye R, An YU, Shao M, et al. Peroxisome proliferator‐activated receptor γ and its role in adipocyte homeostasis and thiazolidinedione‐mediated insulin sensitization. Mol Cell Biol 2018;38:e00677‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.LeBrasseur NK, Kelly M, Tsao T‐S, Farmer SR, Saha AK, Ruderman NB, et al. Thiazolidinediones can rapidly activate AMP‐activated protein kinase in mammalian tissues. Am J Physiol Endocrinol Metab 2006;291:E175‐E181. [DOI] [PubMed] [Google Scholar]
- 7.Divakaruni AS, Wiley SE, Rogers GW, Andreyev AY, Petrosyan S, Loviscach M, et al. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc Natl Acad Sci U S A 2013;110:5422‐5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colca JR, VanderLugt JT, Adams WJ, Shashlo A, McDonald WG, Liang J, et al. Clinical proof‐of‐concept study with MSDC‐0160, a prototype mTOT‐modulating insulin sensitizer. Clin Pharmacol Ther 2013;93:352‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gray L, Sultana M, Rauckhorst A, Oonthonpan L, Tompkins S, Sharma A, et al. Hepatic mitochondrial pyruvate carrier 1 is required for efficient regulation of gluconeogenesis and whole‐body glucose homeostasis. Cell Metab 2015;22:669‐681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCommis K, Chen Z, Fu X, McDonald W, Colca J, Kletzien R, et al. Loss of mitochondrial pyruvate carrier 2 in the liver leads to defects in gluconeogenesis and compensation via pyruvate‐alanine cycling. Cell Metab 2015;22:682‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma A, Oonthonpan L, Sheldon RD, Rauckhorst AJ, Zhu Z, Tompkins SC, et al. Impaired skeletal muscle mitochondrial pyruvate uptake rewires glucose metabolism to drive whole‐body leanness. Elife 2019;8:e45873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim JH, Lewin TM, Coleman RA. Expression and characterization of recombinant rat Acyl‐CoA synthetases 1, 4, and 5. Selective inhibition by triacsin C and thiazolidinediones. J Biol Chem 2001;276:24667‐24673. [DOI] [PubMed] [Google Scholar]
- 13.Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz‐Lopez C, et al. Long‐term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus. Ann Intern Med 2016;165:305. [DOI] [PubMed] [Google Scholar]
- 14.Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328‐357. [DOI] [PubMed] [Google Scholar]
- 15.European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO) . EASL–EASD–EASO Clinical Practice Guidelines for the management of non‐alcoholic fatty liver disease. J Hepatol 2016;64:1388‐1402. [DOI] [PubMed] [Google Scholar]
- 16.Harrington WW, Britt CS, Wilson JG, Milliken NO, Binz JG, Lobe DC, et al. The effect of PPARalpha, PPARdelta, PPARgamma, and PPARpan agonists on body weight, body mass, and serum lipid profiles in diet‐induced obese AKR/J mice. PPAR Res 2007;2007:97125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guan YF, Hao C, Cha DR, Rao R, Lu W, Kohan DE, et al. Thiazolidinediones expand body fluid volume through PPARγ stimulation of ENaC‐mediated renal salt absorption. Nat Med 2005;11:861‐866. [DOI] [PubMed] [Google Scholar]
- 18.Lecka‐Czernik B. Bone loss in diabetes: use of antidiabetic thiazolidinediones and secondary osteoporosis. Curr Osteoporos Rep 2010;8:178‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parks DJ, Tomkinson NC, Villeneuve MS, Blanchard SG, Willson TM. Differential activity of rosiglitazone enantiomers at PPAR gamma. Bioorg Med Chem Lett 1998;8:3657‐3658. [DOI] [PubMed] [Google Scholar]
- 20.FDA . Drug Approval Package, Actos Tablets. 1999.
- 21.Finch H, Fox C, Sajad M. Respiratory Disease Treatment. Worldwide Patent Application WO2010015818, 2010;1‐45.
- 22.Dewitt S, Czarnik AW, Jacques V. Deuterium‐enabled chiral switching (DECS) yields chirally pure drugs from chemically interconverting racemates. ACS Appl Mater Interfaces 2020;11:1789‐1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukunaga T, Zou W, Rohatgi N, Colca JR, Teitelbaum SL. An insulin‐sensitizing thiazolidinedione, which minimally activates PPARγ, does not cause bone loss. J Bone Miner Res 2015;30:481‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Divakaruni AS, Paradyse A, Ferrick DA, Murphy AN, Jastroch M. Analysis and interpretation of microplate‐based oxygen consumption and pH data. In: Methods in Enzymology. Cambridge, MA: Academic Press; 2014:309‐354. [DOI] [PubMed] [Google Scholar]
- 25.Divakaruni AS, Rogers GW, Murphy AN. Measuring mitochondrial function in permeabilized cells using the seahorse XF analyzer or a clark‐type oxygen electrode. Curr Protoc Toxicol 2014;60:25.2.1‐16. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Huo M, Zhou J, Xie S. PKSolver: an add‐in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed 2010;99:306‐314. [DOI] [PubMed] [Google Scholar]
- 27.Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline‐deficient diet. J Lipid Res 2008;1068‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang KL, Pee HN, Yang S, Ho PC. Influence of drug transporters and stereoselectivity on the brain penetration of pioglitazone as a potential medicine against Alzheimer’s disease. Sci Rep 2015;5:9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Birnbaum Y, Long B, Qian J, Perez‐Polo JR, Ye Y. Pioglitazone limits myocardial infarct size, activates Akt, and upregulates cPLA2 and COX‐2 in a PPAR‐γ‐independent manner. Basic Res Cardiol 2011;106:431‐446. [DOI] [PubMed] [Google Scholar]
- 30.Miles PDG, Barak Y, He W, Evans RM, Olefsky JM. Improved insulin‐sensitivity in mice heterozygous for PPAR‐γ deficiency. J Clin Invest 2000;105:287‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsusue K, Haluzik M, Lambert G, Yim S‐H, Gavrilova O, Ward JM, et al. Liver‐specific disruption of PPARγ in leptin‐deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest 2003;111:737‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morán‐Salvador E, López‐Parra M, García‐Alonso V, Titos E, Martínez‐Clemente M, González‐Périz A, et al. Role for PPARγ in obesity‐induced hepatic steatosis as determined by hepatocyte‐ and macrophage‐specific conditional knockouts. FASEB J 2011;25:2538‐2550. [DOI] [PubMed] [Google Scholar]
- 33.Brocks DR. Drug disposition in three dimensions: an update on stereoselectivity in pharmacokinetics. Biopharm Drug Dispos 2006;27:387‐406. [DOI] [PubMed] [Google Scholar]
- 34.Drug Approval Package: Avandia (Rosiglitazone Maleate) NDA# 21‐071. https://www.accessdata.fda.gov/drugsatfda_docs/nda/99/21071_Avandia.cfm. Accessed June 5, 2020.
- 35.Izumi T, Tsuruta F, Ishizuka T, Nakamura K, Kothuma M, Takahashi M. Stereoselectivity in pharmacokinetics of rivoglitazone, a novel peroxisome proliferator‐activated receptor γ agonist, in rats and monkeys: model‐based pharmacokinetic analysis and in vitro‐in vivo extrapolation approach. J Pharm Sci 2013;102:3174‐3188. [DOI] [PubMed] [Google Scholar]
- 36.Shen Z, Bakhtiar R, Komuro M, Awano K, Taga F, Colletti A, et al. Enantiomer ratio of MK‐0767 in humans and nonclinical species. Rapid Commun Mass Spectrom 2005;19:1125‐1129. [DOI] [PubMed] [Google Scholar]
- 37.Combs TP, Wagner JA, Berger J, Doebber T, Wang W‐J, Zhang BB, et al. Induction of adipocyte complement‐related protein of 30 kilodaltons by PPARγ agonists: a potential mechanism of insulin sensitization. Endocrinology 2002;143:998‐1007. [DOI] [PubMed] [Google Scholar]
- 38.Caligiuri A, Bertolani C, Guerra CT, Aleffi S, Galastri S, Trappoliere M, et al. Adenosine monophosphate‐activated protein kinase modulates the activated phenotype of hepatic stellate cells. Hepatology 2007;47:668‐676. [DOI] [PubMed] [Google Scholar]
- 39.Lee HS, Shin H‐S, Choi J, Bae S‐J, Wee H‐J, Son T, et al. AMP‐activated protein kinase activator, HL156A reduces thioacetamide‐induced liver fibrosis in mice and inhibits the activation of cultured hepatic stellate cells and macrophages. Int J Oncol 2016;49:1407‐1414. [DOI] [PubMed] [Google Scholar]
- 40.Vacanti NM, Divakaruni AS, Green CR, Parker SJ, Henry RR, Ciaraldi TP, et al. Regulation of substrate utilization by the mitochondrial pyruvate carrier. Mol Cell 2014;56:425‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chella Krishnan K, Floyd RR, Sabir S, Jayasekera DW, Leon‐Mimila PV, Jones AE, et al. Liver pyruvate kinase promotes NAFLD/NASH in both mice and humans in a sex‐specific manner. Cell Mol Gastroenterol Hepatol 2021;11:389‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lutchman G, Promrat K, Kleiner DE, Heller T, Ghany MG, Yanovski JA, et al. Changes in serum adipokine levels during pioglitazone treatment for nonalcoholic steatohepatitis: relationship to histological improvement. Clin Gastroenterol Hepatol 2006;4:1048‐1052. [DOI] [PubMed] [Google Scholar]
- 43.Miyazaki Y, Mahankali A, Matsuda M, Mahankali S, Hardies J, Cusi K, et al. Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab 2002;87:2784‐2791. [DOI] [PubMed] [Google Scholar]
- 44.Djaouti L, Jourdan T, Demizieux L, Chevrot M, Gresti J, Vergès B, et al. Different effects of pioglitazone and rosiglitazone on lipid metabolism in mouse cultured liver explants. Diabetes Metab Res Rev 2010;26:297‐305. [DOI] [PubMed] [Google Scholar]
- 45.Ratziu V, Charlotte F, Bernhardt C, Giral P, Halbron M, LeNaour G, et al. Long‐term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the Fatty Liver Improvement by Rosiglitazone Therapy (FLIRT 2) extension trial. Hepatology 2010;51:445‐453. [DOI] [PubMed] [Google Scholar]
- 46.Satirapoj B, Watanakijthavonkul K, Supasyndh O. Safety and efficacy of low dose pioglitazone compared with standard dose pioglitazone in type 2 diabetes with chronic kidney disease: a randomized controlled trial. PLoS One 2018;13:e0206722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adachi H, Katsuyama H, Yanai H. The low dose (7.5 mg/day) pioglitazone is beneficial to the improvement in metabolic parameters without weight gain and an increase of risk for heart failure. Int J Cardiol 2017;227:247‐248. [DOI] [PubMed] [Google Scholar]
- 48.Rajagopalan S, Dutta P, Hota D, Bhansali A, Srinivasan A, Chakrabarti A. Effect of low dose pioglitazone on glycemic control and insulin resistance in type 2 diabetes: a randomized, double blind, clinical trial. Diabetes Res Clin Pract 2015;109:e32‐e35. [DOI] [PubMed] [Google Scholar]
- 49.Department of Health and Human Services; FDA . Guidance for the development of new stereoisomeric drugs. 1992. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm122883.htm. Accessed March 31, 2021.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Material