

Abstract
The serpin family A member 1 (SERPINA1) Z allele is present in approximately one in 25 individuals of European ancestry. Z allele homozygosity (Pi*ZZ) is the most common cause of alpha 1‐antitrypsin deficiency and is a proven risk factor for cirrhosis. We examined whether heterozygous Z allele (Pi*Z) carriers in United Kingdom (UK) Biobank, a population‐based cohort, are at increased risk of liver disease. We replicated findings in Massachusetts General Brigham Biobank, a hospital‐based cohort. We also examined variants associated with liver disease and assessed for gene–gene and gene–environment interactions. In UK Biobank, we identified 1,493 cases of cirrhosis, 12,603 Z allele heterozygotes, and 129 Z allele homozygotes among 312,671 unrelated white British participants. Heterozygous carriage of the Z allele was associated with cirrhosis compared to noncarriage (odds ratio [OR], 1.53; P = 1.1×10−04); homozygosity of the Z allele also increased the risk of cirrhosis (OR, 11.8; P = 1.8 × 10−09). The OR for cirrhosis of the Z allele was comparable to that of well‐established genetic variants, including patatin‐like phospholipase domain containing 3 (PNPLA3) I148M (OR, 1.48; P = 1.1 × 10−22) and transmembrane 6 superfamily member 2 (TM6SF2) E167K (OR, 1.34; P = 2.6 × 10−06). In heterozygotes compared to noncarriers, the Z allele was associated with higher alanine aminotransferase (ALT; P = = 4.6 × 10−46), aspartate aminotransferase (AST; P = 2.2 × 10−27), alkaline phosphatase (P = 3.3 × 10−43), gamma‐glutamyltransferase (P = 1.2 × 10−05), and total bilirubin (P = 6.4 × 10−06); Z allele homozygotes had even greater elevations in liver biochemistries. Body mass index (BMI) amplified the association of the Z allele for ALT (P interaction = 0.021) and AST (P interaction = 0.0040), suggesting a gene–environment interaction. Finally, we demonstrated genetic interactions between variants in PNPLA3, TM6SF2, and hydroxysteroid 17‐beta dehydrogenase 13 (HSD17B13); there was no evidence of epistasis between the Z allele and these variants. Conclusion: SERPINA1 Z allele heterozygosity is an important risk factor for liver disease; this risk is amplified by increasing BMI.

Abbreviations
- AAT
alpha 1‐antitrypsin
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BMI
body mass index
- FIB‐4
fibrosis‐4
- GGT
gamma‐glutamyltransferase
- HSD17B13
hydroxysteroid 17‐beta dehydrogenase 13
- ICD‐10
International Classification of Diseases, Tenth Revision
- LDL‐C
low‐density lipoprotein cholesterol
- Mass
Massachusetts
- OR
odds ratio
- Pi*Z
heterozygous for the SERPINA1 Z allele
- Pi*ZZ
homozygous for SERPINA1 Z allele
- PNPLA3
patatin‐like phospholipase domain containing 3
- SERPINA1
serpin family A member 1
- TM6SF2
transmembrane 6 superfamily member 2
- UK Biobank
United Kingdom Biobank
The serpin family A member 1 (SERPINA1) gene encodes the alpha 1‐antitrypsin (AAT) protein, an abundant circulating glycoprotein proteinase inhibitor synthesized primarily within hepatocytes.( 1, 2 ) The physiologic function of AAT is to bind and inactivate neutrophil elastase in the lung, protecting alveolar tissue from proteolytic degradation.( 3 ) AAT deficiency is a genetic disorder characterized by an increased risk for chronic obstructive pulmonary disease, emphysema, bronchiectasis, and chronic liver disease.( 1, 2 ) Point mutations in SERPINA1 lead to retention of mutant AAT in the liver and hepatocyte toxicity,( 4, 5 ) and the resulting lack of circulating AAT leads to chronic lung disease.( 3 )
The most common cause of AAT deficiency is homozygosity for the SERPINA1 Z allele (rs28929474).( 2 ) AAT deficiency is among the most common genetic diseases; the SERPINA1 Z allele is present in approximately one in 25 individuals of European ancestry, and one in 2,000 persons of European descent are homozygotes (Pi*ZZ).( 1, 2 ) The pathologic hallmark of Pi*ZZ‐related liver disease is the presence of intracytoplasmic deposition of insoluble AAT globules that are periodic acid‐Schiff positive, diastase‐resistant, and visualized with immunochemistry.( 4, 5 )
While the risk of chronic liver disease in patients homozygous for the SERPINA1 Z allele has been well studied,( 6, 7, 8, 9, 10 ) the risk of liver disease in heterozygous carriers of the SERPINA1 Z allele (Pi*Z) remains under investigation. Although initial reports found no association between Z allele heterozygotes and the risk of liver disease,( 11, 12, 13, 14 ) other case‐control studies have shown an effect of the Z allele on cystic fibrosis‐related liver disease,( 15 ) alcoholic and nonalcoholic fatty liver disease,( 16, 17, 18 ) portal hypertension,( 19 ) and liver stiffness and elevated serum transaminases.( 20 ) A limitation of these studies is that many have been carried out in relatively small sample sizes with a limited number of subjects with the Pi*Z genotype. More recently, genome‐wide association studies have demonstrated the association of the SERPINA1 Z allele with nonalcoholic liver disease, nonalcoholic cirrhosis, and alcoholic cirrhosis.( 21, 22, 23 ) However, these studies tend to employ an additive (per allele) genetic model where effect estimates are influenced by Z allele homozygotes and therefore do not specifically address risk in heterozygotes. Hence, we carried out the largest study to date to evaluate the association of Z allele heterozygosity (Pi*Z) and liver disease using International Classification of Diseases, Tenth Revision (ICD‐10) codes and biomarkers of liver injury. As the development of liver disease among subjects with Pi*ZZ is variable,( 10 ) we also assessed for gene–environment interactions that might amplify the phenotypic effect of sequence variation at the SERPINA1 Z allele. Finally, we investigated possible epistatic (nonadditive) interactions between established genetic variants associated with liver disease and the SERPINA1 Z allele. We performed our initial evaluation in the United Kingdom Biobank (UK Biobank) cohort, a population‐based study of 502,682 individuals that includes more than 18,000 Z allele heterozygotes. We replicated our findings in the Massachusetts (Mass) General Brigham Biobank cohort, a hospital‐based cohort of 43,534 genotyped individuals. We provide evidence that the SERPINA1 Z allele is among the most important genetic risk factors for liver injury and cirrhosis.
Participants and Methods
Population Stratification and Sample Quality Control
UK Biobank is a prospective population‐based cohort study with 502,682 persons, age 40‐69 years, for whom extensive baseline questionnaire data, physiologic measures, and biologic specimens have been obtained.( 24 ) Participants in the UK Biobank project provided written informed consent, and UK Biobank protocols are approved by the National Research Ethics Service. Analyses in this project were conducted under UK Biobank Resource Project 20915 and were approved by the Mass General Brigham Institutional Review Board. We used genotype data from the UK Biobank data set release version 2 and the Homo sapiens (human) genome assembly GRCh37 (hg19) human genome reference for all analyses in this study. To minimize variabilities due to population structure in our data set, we restricted the analysis to include individuals of self‐reported white British ancestry. We also excluded individuals with more genome‐wide heterozygosity than expected, an excess of missing genotype calls, putative sex chromosome aneuploidy, and more than 10 third‐degree relatives. To select unrelated individuals, we further removed at least 1 individual from each related pair with kinship coefficient >0.0625, giving preference to inclusion of patients with all‐cause cirrhosis by ICD‐10 codes. After quality control, 312,671 unrelated white British subjects were included in the analysis. We replicated our findings using Mass General Brigham Biobank, a large integrated database with clinical and genetic data from 43,534 individuals.( 25 ) To minimize variabilities due to population structure, we restricted the analysis of Mass General Brigham Biobank to include individuals of self‐reported white ancestry and removed at least 1 individual from each related pair with kinship coefficient >0.0885; 19,323 subjects with unrelated white ancestry were included in the analysis.
Single‐Nucleotide Polymorphism‐Based Genotyping
In the UK Biobank cohort, genotyping was performed using either the UK Biobank Lung Exome Variant Evaluation (UK BiLEVE) Axiom array or the UK Biobank Axiom array. We filtered out variants with minor allele frequency lower than 1%, variants with imputation quality lower than 0.5, and variants not included in the Haplotype Reference Consortium (HRC) imputation panel, as recommended by UK Biobank at the time of analysis. For Mass General Brigham Biobank, genotyping was performed using the Illumina MEGA array. Variants were imputed to the HRC using the Michigan Imputation Server. The SERPINA1 Z allele rs28929474(T) as well as patatin‐like phospholipase domain containing 3 (PNPLA3) I148M rs738409(G), transmembrane 6 superfamily member 2 (TM6SF2) E167K rs58542926(T), and the hydroxysteroid 17‐beta dehydrogenase 13 (HSD17B13) splice variant rs72613567(TA) were extracted using PLINK 2.0, and genotypes were coded using an additive model (0 for reference allele homozygotes, 1 for heterozygotes, and 2 for alternate allele homozygotes).
ICD‐10‐Based Liver Disease Phenotypes for Cirrhosis, Alcoholic Cirrhosis, and Fatty Liver Disease
In UK Biobank, ICD‐10 diagnosis codes were collated for each participant across all inpatient hospital records dating back to 1997 for England, 1998 for Wales, and 1981 for Scotland.( 26 ) In Mass General Brigham Biobank, participant samples were linked to the electronic medical record dating back to 1992, including physician diagnosis according to ICD‐10 codes.( 25 ) We defined an all‐cause cirrhosis phenotype by combining the following ICD‐10 codes: K702 (alcoholic fibrosis and sclerosis of liver), K703 (alcoholic cirrhosis of liver), K704 (alcoholic hepatic failure), K740 (hepatic fibrosis), K741 (hepatic sclerosis), K742 (hepatic fibrosis with hepatic sclerosis), K746 (other and unspecified cirrhosis of the liver), K766 (portal hypertension), I850 (bleeding esophageal varices), I859 (esophageal varices), K717 (toxic liver disease with fibrosis and cirrhosis of liver), K721 (chronic hepatic failure), K729 (hepatic failure, unspecified), and K767 (hepatorenal syndrome). We excluded cases of cirrhosis secondary to autoimmune indications, including primary biliary cholangitis, primary sclerosing cholangitis, and autoimmune hepatitis. We also investigated phenotypes for alcoholic cirrhosis and all‐cause fatty liver disease. For the alcoholic cirrhosis phenotype, we combined K702 (alcoholic fibrosis and sclerosis of liver), K703 (alcoholic cirrhosis of liver), and K704 (alcoholic hepatic failure). For the all‐cause fatty liver disease phenotype, we combined K700 (alcoholic fatty liver), K701 (alcoholic hepatitis), K709 (alcoholic liver disease, unspecified), K760 (fatty liver, not elsewhere classified), K758 (other specified inflammatory liver diseases), and K759 (inflammatory liver disease, unspecified).
Association Analysis for Binary Liver Disease Phenotypes and Quantitative Traits
R version 3.6.0 was used to perform association analyses for quantitative (circulating biomarkers of liver injury, lipids) and binary (ICD‐10 diagnosis) traits using linear and logistic regression, respectively. We adjusted for age, sex, body mass index (BMI), total number of medications taken by each participant, genotyping batch, and first 10 principal components of ancestry. Age refers to the age of the participant on the day they were enrolled in the Biobank project. To account for case‐control imbalance for binary liver disease phenotypes and lower allele frequencies, all logistic regression results were analyzed using the Firth penalized likelihood approach.( 27 ) We calculated per allele odds ratios (ORs) using the standard additive model. To estimate risk in heterozygotes, we calculated genotypic ORs (i.e., heterozygous versus wild type, excluding homozygotes); genotypic ORs for homozygotes (i.e., homozygous versus wild type, excluding heterozygotes) were also assessed. We log transformed alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), gamma‐glutamyltransferase (GGT), and total bilirubin, resulting in approximately normal distributions. A Bonferroni‐adjusted significance level of P < 0.0042 was used for the four genetic variants and three models tested.
Sensitivity Analysis for Alcohol Use
Given the known important role of alcohol intake on liver injury, we performed a sensitivity analysis exploring the association of genetic variants of interest with biomarkers of liver injury and cirrhosis, adding the self‐reported number of alcoholic drinks consumed as a covariate. We calculated the average units of alcohol consumed per week for each participant in UK Biobank, assuming 2 units (16 g) of pure alcohol in a pint of beer/cider; 1.5 units (12 g) in a glass of red wine, champagne, white wine, fortified wine, and “other” alcohol drinks; and 1 unit (8 g) in a measure of spirits. For participants who reported consuming alcohol monthly rather than weekly, we multiplied by 0.23 to convert monthly alcohol consumption to weekly.
Assessing for a SERPINA1 Z Allele × BMI Interaction
To evaluate the combined effects of the SERPINA1 Z allele and BMI on biomarkers of liver injury, we performed an interaction analysis using an additive model. The additive model included the main effects of the Z allele and BMI as well as an interaction term. BMI was entered as a continuous variable in all analyses. We also assessed the robustness of the interaction by excluding Z allele homozygotes in a sensitivity analysis. To depict the interaction between genotype and BMI visually, participants were divided into the following four categories of BMI: lean (≤25 kg/m2), overweight (25‐30 kg/m2), obese (30‐35 kg/m2), and very obese (>35 kg/m2).
Analysis of Genetic Interaction Between Variants Associated With Liver Injury
We evaluated the combined effects of two genetic variants of interest on biomarkers of liver injury by performing an association analysis using linear regression, modeling the main effects for each variant of interest, as well as a cross‐product interaction term, assuming an additive model. We also calculated an overall 4 degree‐of‐freedom test for interaction by modeling additive and dominance effects at each of the two loci as well as modeling four interaction terms and compared this with a null model with lack of epistasis implying that all interaction coefficients are 0.( 28 ) All models were adjusted for age, sex, BMI, total number of medications taken by each participant, genotyping batch, and first 10 principal components of ancestry. A Bonferroni‐adjusted significance level of P < 0.0083 was used for the six pairs of genetic variants tested.
Results
Study Population Characteristics, Evaluation of Cirrhosis PHENOTYPE
In UK Biobank, we identified 1,493 cases of cirrhosis, 12,603 SERPINA1 Z allele heterozygotes, and 129 SERPINA1 Z allele homozygotes in the 312,671 unrelated white British subjects included in our analysis. Patients with and without cirrhosis were compared (Table 1). Consistent with prior studies,( 29, 30 ) patients with cirrhosis had elevated ALT, AST, ALP, GGT, total bilirubin, direct bilirubin, and fibrosis‐4 (FIB‐4) scores and had lower albumin levels, platelet counts, total cholesterol, and direct low‐density lipoprotein cholesterol (LDL‐C) (P < 0.001 for all). Patients with cirrhosis were also older, were more likely to be men, had higher BMI and waist circumference, consumed more weekly alcohol among subjects reporting alcohol use, and took more medications (P < 0.001 for all). Age (OR, 1.04; P < 0.001), male sex (OR, 2.43; P < 0.001), BMI (OR, 1.07; P < 0.001), number of medications (OR, 1.2; P < 0.001), and weekly alcohol consumption (OR, 1.02; P < 0.001) were associated with cirrhosis by univariate analysis using logistic regression.
Table 1.
Characteristics of subjects with and without cirrhosis by ICD‐10 codes in UK Biobank
| No Cirrhosis (n = 311,178) | Cirrhosis (n = 1,493) | Total (N = 312,671) | P Value | |
|---|---|---|---|---|
| Age (years) | 56.85 (7.98) | 58.98 (7.10) | 56.86 (7.98) | <0.001 |
| Sex (male) | 144,170 (46%) | 1,011 (68%) | 145,181 (46%) | <0.001 |
| BMI (kg/m2) | 26.70 [24.12, 29.82] | 28.54 [25.43, 32.52] | 26.70 [24.12, 29.83] | <0.001 |
| Waist (cm) | 90.33 (13.39) | 99.09 (14.53) | 90.37 (13.41) | <0.001 |
| Number of Medications | 2.00 [0.00, 4.00] | 4.00 [2.00, 7.00] | 2.00 [0.00, 4.00] | <0.001 |
| ALT (U/L) | 20.16 [15.47, 27.31] | 29.97 [20.40, 48.06] | 20.18 [15.48, 27.37] | <0.001 |
| AST (U/L) | 24.40 [21.00, 28.80] | 36.30 [26.10, 57.00] | 24.40 [21.00, 28.80] | <0.001 |
| GGT (U/L) | 26.20 [18.50, 40.70] | 75.00 [35.80, 192.38] | 26.20 [18.50, 40.90] | <0.001 |
| ALP (U/L) | 80.30 [67.30, 95.60] | 96.93 [77.97, 126.90] | 80.30 [67.30, 95.70] | <0.001 |
| Total bilirubin (µmol/L) | 8.08 [6.45, 10.41] | 9.96 [7.49, 14.32] | 8.09 [6.45, 10.43] | <0.001 |
| Direct bilirubin (µmol/L) | 1.61 [1.30, 2.08] | 2.26 [1.64, 3.51] | 1.61 [1.30, 2.09] | <0.001 |
| Platelets (109 cells/L) | 247.80 [213.30, 286.50] | 204.30 [156.70, 260.00] | 247.60 [213.00, 286.30] | <0.001 |
| Albumin (g/L) | 45.26 (2.59) | 43.49 (3.88) | 45.26 (2.60) | <0.001 |
| FIB‐4 | 1.24 [0.98, 1.57] | 1.91 [1.33, 3.16] | 1.25 [0.98, 1.58] | <0.001 |
| Weekly alcohol use | 12.00 [6.00, 22.00] | 20.00 [9.00, 40.00] | 12.00 [6.00, 22.00] | <0.001 |
| LDL (mmol/L) | 3.57 (0.87) | 3.16 (0.92) | 3.57 (0.87) | <0.001 |
| HDL (mmol/L) | 1.46 (0.38) | 1.35 (0.45) | 1.45 (0.38) | <0.001 |
| Cholesterol (mmol/L) | 5.72 (1.14) | 5.15 (1.28) | 5.72 (1.14) | <0.001 |
Mean (SD) or number (%) are reported for normal variables and median [interquartile range] for non‐normal variables. Comparisons are made using the t test for normal variables and the Kruskal‐Wallis test for non‐normal variables. FIB‐4 is a noninvasive marker of hepatic fibrosis calculated by age × AST [U/L] / (PLT [109/L] × ALT1/2 [U/L]).( 30 ) Alcohol use is defined by average units of alcohol consumed per week among subjects reporting alcohol use.
Abbreviations: HDL, high‐density lipoprotein; PLT, platelet.
SERPINA1 Z Allele is Associated With Cirrhosis
We used UK Biobank to investigate the association of the SERPINA1 Z allele with broad categories of liver disease, including cirrhosis, alcoholic cirrhosis, and fatty liver disease. In an additive model, the SERPINA1 Z allele was associated with cirrhosis in an allele dose‐dependent manner (OR, 1.69; P = 2.3 × 10−07) (Fig. 1). The SERPINA1 Z allele was also associated with higher odds of cirrhosis in both heterozygotes versus noncarriers (OR, 1.53; P = 1.1 × 10−04) and homozygotes versus noncarriers (OR, 11.8; P = 1.8 × 10−09) (Fig. 1). The SERPINA1 Z allele was associated with alcoholic cirrhosis in a per allele additive model (OR, 1.89; P = 2.5 × 10−04), in heterozygotes versus noncarriers (OR, 1.83; P = 9.4 × 10−04), and in homozygotes versus noncarriers (OR, 9.21; P = 4.7 × 10−03) (Supporting Fig. S1). There was no association between the SERPINA1 Z allele and fatty liver disease (OR, 1.22; P = 0.17) (Supporting Fig. S2).
FIG. 1.
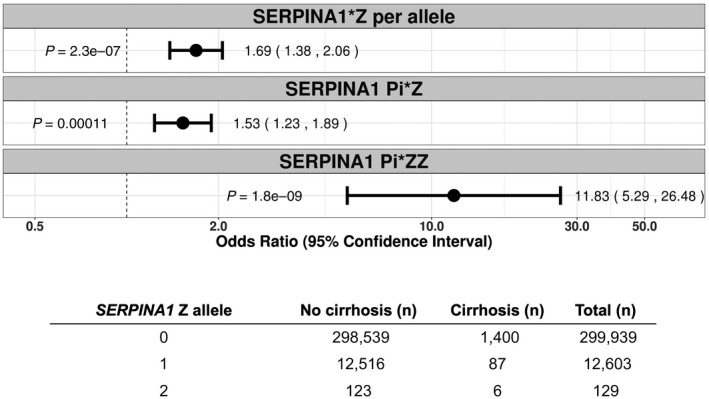
Association of SERPINA1 Z allele with cirrhosis phenotype by ICD‐10 diagnostic codes in UK Biobank, using the Firth penalized likelihood approach. ORs were calculated with the use of logistic regression, with adjustment for age, sex, BMI, total number of medications, genotyping batch, and first 10 principal components of ancestry. Subjects were coded 0 for reference allele homozygotes, 1 for heterozygotes, and 2 for alternate allele homozygotes. The first panel reflects an additive (per allele) model and the second and third panels reflect genotypic ORs for SERPINA1 Z allele heterozygotes (C/T) and homozygotes (T/T), respectively. The table reflects the number of subjects with and without cirrhosis for each genotype.
FIG. 2.
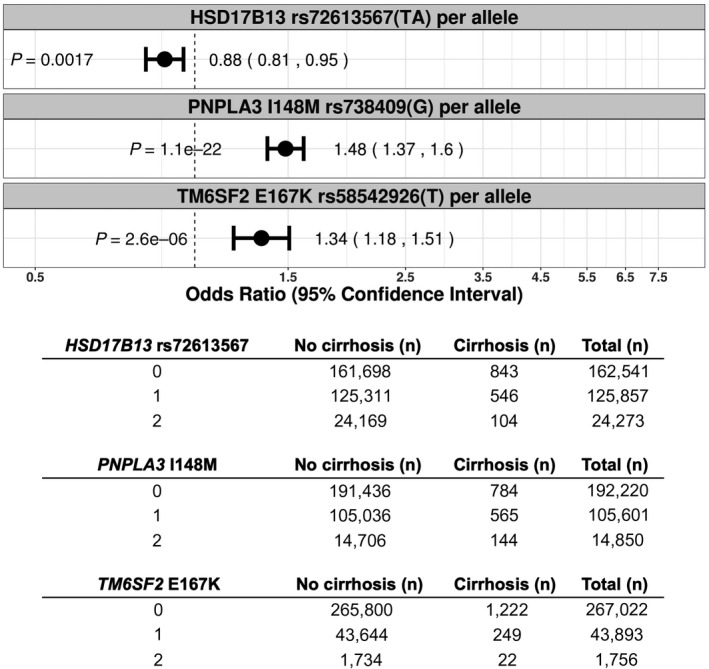
Association of PNPLA3 I148M, TM6SF2 E167K, and HSD17B13 rs72613567(TA) with cirrhosis by ICD‐10 diagnostic codes in the UK Biobank additive (per allele) model, using the Firth penalized likelihood approach. ORs were calculated with the use of logistic regression, with adjustment for age, sex, BMI, total number of medications, genotyping batch, and first 10 principal components of ancestry. Subjects were coded 0 for reference allele homozygotes, 1 for heterozygotes, and 2 for alternate allele homozygotes. The table reflects the number of subjects with and without cirrhosis for each genotype.
SERPINA1 Z Allele is Associated With Biomarkers of Liver Injury
Circulating liver enzymes are sensitive biomarkers of liver injury. We explored the association of the SERPINA1 Z allele with log‐transformed ALT, AST, ALP, GGT, and total bilirubin in the UK Biobank cohort. In an additive model, the SERPINA1 Z allele was associated with higher blood levels of ALT, AST, ALP, GGT and total bilirubin (Supporting Table S1). In heterozygotes versus noncarriers, the SERPINA1 Z allele was also associated with higher blood levels of ALT, AST, ALP, GGT, and total bilirubin (Table 2). Subjects homozygous for the Z allele had even greater elevations in circulating biomarkers of liver injury, including higher blood levels of ALT, AST, ALP, and GGT (Table 2).
Table 2.
Association of genetic variants with biomarkers of liver injury in SERPINA1 Z allele (Pi*Z) heterozygotes and homozygotes (Pi*ZZ) in UK Biobank
| Pi*Z heterozygotes | Measure (U/L) | Estimate | SE | Pr (>|t|) |
|---|---|---|---|---|
| log ALT | 5.43e−02 | 3.81e−03 | 4.64e−46 | |
| log AST | 2.67e−02 | 2.47e−03 | 2.23e−27 | |
| log ALP | 3.46e−02 | 2.51e−03 | 3.26e−43 | |
| log GGT | 2.31e−02 | 5.28e−03 | 1.21e−05 | |
| log total bilirubin | 1.56e−02 | 3.46e−03 | 6.35e−06 |
| Pi*ZZ homozygotes | Measure (U/L) | Estimate | SE | Pr (>|t|) |
|---|---|---|---|---|
| log ALT | 1.37e−01 | 3.75e−02 | 2.66e−04 | |
| log AST | 1.85e−01 | 2.43e−02 | 2.72e−14 | |
| log ALP | 6.03e−02 | 2.47e−02 | 1.45e−02 | |
| log GGT | 1.81e−01 | 5.22e−02 | 5.43e−04 | |
| log total bilirubin | 4.97e−02 | 3.41e−02 | 1.45e−01 |
Linear regression was performed adjusting for age, sex, BMI, number of medications, batch, and the first 10 principal components of ancestry.
Abbreviations: Pr (>|t|), P value for the proportion of the t distribution that is greater than the absolute value of the t statistic; SE, standard error.
Validation of Variants Previously Associated With Liver Disease
Common polymorphisms in PNPLA3 and TM6SF2 are important risk factors for liver disease,( 31, 32 ) and a splice variant in HSD17B13 has been associated with protection against liver disease.( 21 ) Consistent with prior studies that included an independent analysis in UK Biobank,( 22 ) we confirmed that PNPLA3 I148M and TM6SF2 E167K were associated with cirrhosis in an allele dose‐dependent manner and that HSD17B13 rs72613567(TA) was associated with protection from cirrhosis (Fig. 2). For PNPLA3 I148M, the additive OR for cirrhosis was 1.48 (P = 1.1 × 10−22) (Fig. 2); the OR for cirrhosis in heterozygotes versus noncarriers was 1.34 (P = 8.2 × 10−08) and in homozygotes versus noncarriers was 2.51 (P = 2.4 × 10−23) (Fig. 3). For TM6SF2 E167K, the additive OR for cirrhosis was 1.34 (P = 2.6 × 10−06) (Fig. 2); the OR for cirrhosis in heterozygotes versus noncarriers was 1.25 (P = 1.5 × 10−03) and in homozygotes versus noncarriers was 2.91 (P = 7.6 × 10−07) (Fig. 3). The PNPLA3 I148M allele and TM6SF2 E167K were both also associated with alcoholic cirrhosis (additive OR, 1.84; P = 2.0 × 10−18; and additive OR, 1.39; P = 2.9 × 10−03; respectively) (Supporting Fig. S3) and all‐cause fatty liver disease (additive OR, 1.43; P = 5.6 × 10−13; and additive OR, 1.22; P = 1.1 × 10−02; respectively) (Supporting Fig. S2). The splice variant in HSD17B13 rs72613567(TA) was protective against cirrhosis (additive OR, 0.88; P = 1.7 × 10−03) (Fig. 2) and fatty liver disease (additive OR, 0.85; P = 1.5 × 10−03) (Supporting Fig. S2).
FIG. 3.

Association of PNPLA3 I148M, and TM6SF2 E167K with cirrhosis phenotype by ICD‐10 diagnostic codes in the UK Biobank genotypic model, using the Firth penalized likelihood approach. Genotypic ORs are shown for PNPLA3 I148M heterozygotes (C/G) and homozygotes (G/G) as well as for TM6SF2 E167K heterozygotes (C/T) and homozygotes (T/T). ORs were calculated with the use of logistic regression, with adjustment for age, sex, BMI, total number of medications, genotyping batch, and first 10 principal components of ancestry.
Association of Known Genetic Variants With Biomarkers of Liver Injury
We explored the association of PNPLA3 I148M, TM6SF2 E167K, and HSD17B13 rs72613567(TA) with log‐transformed ALT, AST, ALP, GGT, and total bilirubin in UK Biobank. As expected, PNPLA3 I148M and TM6SF2 E167K were associated with elevated levels of ALT, AST, GGT and total bilirubin (Supporting Table S1). Both variants were associated with decreased ALP (Supporting Table S1). The HSD17B13 splice variant rs72613567(TA) was associated with decreased circulating levels of ALT, AST, GGT, and total bilirubin but was associated with increased levels of ALP (Supporting Table S1).
Association of Genetic Variants With Circulating Lipids
The PNPLA3 I148M and TM6SF2 E167K alleles that increase the risk of cirrhosis have been reported to decrease circulating LDL‐C and total cholesterol and decrease the risk of cardiovascular disease.( 32, 33, 34, 35 ) This raises the possibility that therapeutic strategies targeting PNPLA3, TM6SF2, or other variants associated with liver disease may adversely impact cardiovascular risk. We evaluated the association of PNPLA3 I148M, TM6SF2 E167K, HSD17B13 rs72613567(TA), and the SERPINA1 Z allele with total cholesterol and direct LDL‐C in UK Biobank (Supporting Table S2). Both PNPLA3 I148M and TM6SF2 E167K were associated with decreased cholesterol and direct LDL‐C. The splice variant in HSD17B13 that protects against cirrhosis was associated with increased total cholesterol but was not associated with direct LDL‐C. The SERPINA1 Z allele was not associated with either total cholesterol or LDL‐C.
BMI and the SERPINA1 Z Allele Interact to Increase the Risk of Liver Injury
To determine if the effect of the Pi*Z variant on hepatocellular injury is modified by BMI, we analyzed the relationship between the SERPINA1 genotype and ALT and AST after stratifying participants in UK Biobank into four categories based on BMI (Fig. 4). In all four BMI groups, the mean ALT or AST of Z allele heterozygotes was in between the levels of the reference allele homozygotes and Z allele homozygotes (Fig. 4). We performed linear regression analysis modeling the main effects for the Pi*Z variant and BMI as well as an interaction term (Z allele × BMI), assuming an additive model. We observed an interaction of BMI and the SERPINA1 Z allele in association with increased ALT (P interaction = 0.021) and AST (P interaction = 0.0040), using BMI as a continuous variable. Significant BMI and Z allele interaction was also observed in a sensitivity analysis excluding Z allele homozygotes (P interaction = 0.026 for ALT and P interaction = 0.0088 for AST).
FIG. 4.
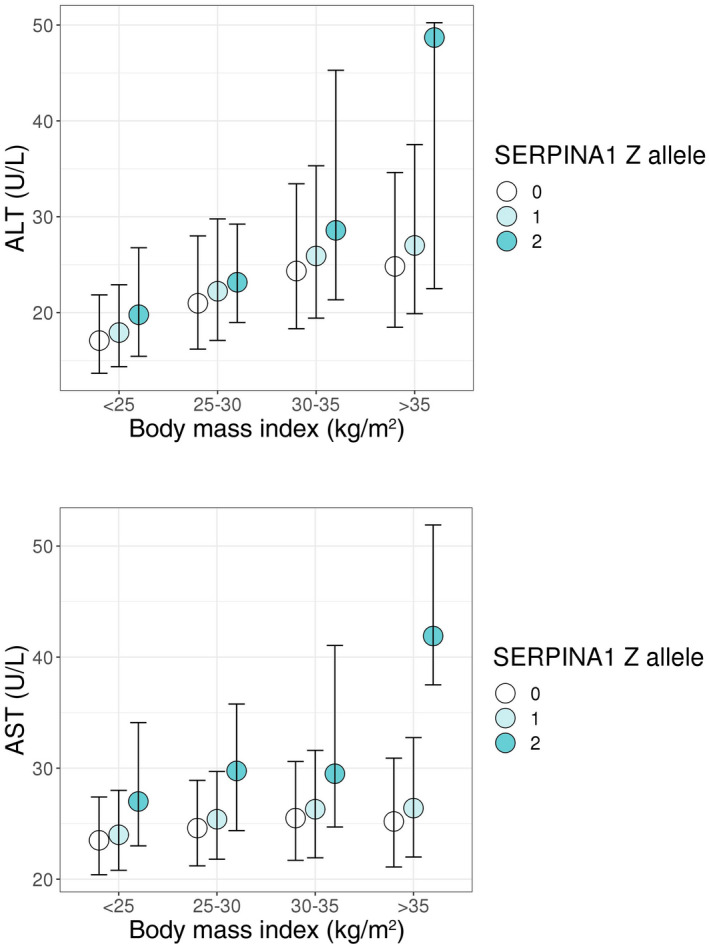
ALT and AST by BMI and SERPINA1 genotype in UK Biobank. Circles and bars depict medians and interquartile ranges, respectively. The ALT‐ and AST‐increasing effect of the SERPINA1 Z allele was amplified by increasing BMI (P for interaction SERPINA1 Z allele × BMI on ALT = 0.021; P for interaction SERPINA1 Z allele × BMI on AST = 0.0040). Significant interaction was also observed in a sensitivity analyses excluding Z allele homozygotes (P interaction = 0.026 for ALT; P interaction = 0.0088 for AST).
Genetic Interaction Between PNPLA3, TM6SF2, HSD17B13, and SERPINA1 Variants
We investigated possible epistatic interactions between genetic variants associated with liver disease in UK Biobank. By testing for interaction between the two variants in association with log‐transformed ALT and AST levels, we found that HSD17B13 rs72613567(TA) modified the risk of liver injury associated with PNPLA3 I148M. Each HSD17B13 rs72613567(TA) allele mitigated increases in ALT (P interaction = 2.0 × 10−10) and AST (P interaction = 5.9 × 10−14) associated with each PNPLA3 I148M allele (Supporting Table S3). The interactions remained significant when using an overall 4 degree‐of‐freedom test for genetic interaction,( 28 ) with P = 3.0 × 10−11 for interaction in association with ALT and P = 2.6 × 10−14 for interaction in association with AST (Supporting Table S4). We also found evidence of epistasis between PNPLA3 I148M and TM6SF2 E167K, such that each TM6SF2 E167K allele enhanced the increase in ALT (P interaction = 4.3 × 10−04) and AST (P interaction = 2.6 × 10−05) levels associated with each PNPLA3 I148M allele (Supporting Table S3). This interaction remained significant in an analysis using a 4 degree‐of‐freedom test, with P = 4.7 × 10−05 in association with ALT and P = 9.5 × 10−06 in association with AST (Supporting Table S4). There was no evidence of epistasis between the Z allele and other well‐established genetic risk factors for liver injury.
Table 3.
Characteristics of subjects with and without cirrhosis by ICD‐10 codes in Mass General Brigham Biobank
| No Cirrhosis (n = 18,394) | Cirrhosis (n = 929) | Total (N = 19,323) | P Value | |
|---|---|---|---|---|
| Age (years) | 63.00 [49.00, 73.00] | 65.00 [57.00, 73.00] | 63.00 [50.00, 73.00] | <0.001 |
| Sex (male) | 8,620 (47%) | 536 (58%) | 9,156 (47%) | <0.001 |
| BMI (kg/m2) | 26.58 [23.40, 30.55] | 28.25 [24.55, 32.79] | 26.61 [23.41, 30.66] | <0.001 |
| ALT (U/L) | 22.48 [17.00, 31.25] | 35.85 [24.91, 56.85] | 23.00 [17.05, 32.36] | <0.001 |
| AST (U/L) | 24.00 [19.91, 30.09] | 40.23 [28.36, 61.44] | 24.33 [20.00, 31.00] | <0.001 |
| ALP (U/L) | 73.00 [60.43, 89.44] | 95.39 [75.06, 131.14] | 74.00 [61.00, 91.00] | <0.001 |
| Platelets (109 cells/L) | 242.90 [205.00, 285.23] | 209.31 [159.74, 259.42] | 241.45 [203.00, 284.42] | <0.001 |
| Albumin (g/dL) | 4.26 [3.99, 4.47] | 3.93 [3.57, 4.25] | 4.25 [3.97, 4.47] | <0.001 |
| FIB‐4 | 1.29 [0.90, 1.81] | 2.17 [1.39, 3.57] | 1.32 [0.92, 1.87] | <0.001 |
| LDL (mg/dL) | 97.33 [77.00, 119.00] | 88.80 [70.52, 110.71] | 97.00 [76.50, 118.50] | <0.001 |
| HDL (mg/dL) | 53.00 [43.00, 65.76] | 46.00 [37.67, 57.33] | 52.67 [42.59, 65.17] | <0.001 |
| Cholesterol (mg/dL) | 183.17 [160.32, 206.99] | 174.00 [149.75, 196.29] | 182.50 [159.50, 206.36] | <0.001 |
Median [interquartile range] or number (%) are reported. Comparisons are made using the Kruskal‐Wallis test. FIB‐4 is a noninvasive marker of hepatic fibrosis calculated by age × AST [U/L] / (PLT [109/L] × ALT1/2 [U/L]).( 30 )
Abbreviations: HDL, high‐density lipoprotein; PLT, platelet.
Table 4.
Association of genetic variants with biomarkers of liver injury in the additive (per allele) model in Mass General Brigham Biobank
| Variant | Measure (U/L) | Estimate | SE | Pr (>|t|) |
|---|---|---|---|---|
| PNPLA3 I148M | log ALT | 3.69e−02 | 8.43e−03 | 1.21e−05 |
| log AST | 2.67e−02 | 6.19e−03 | 1.61e−05 | |
| log ALP | 4.13e−03 | 6.21e−03 | 5.06e−01 | |
| TM6SF2 E167K | log ALT | 2.75e−02 | 1.38e−02 | 4.63e−02 |
| log AST | 2.15e−02 | 1.01e−02 | 3.38e−02 | |
| log ALP | −1.51e−02 | 1.02e−02 | 1.38e−01 | |
| SERPINA1 Z allele | log ALT | 8.18e−02 | 2.70e−02 | 2.44e−03 |
| log AST | 3.34e−02 | 1.99e−02 | 9.23e−02 | |
| log ALP | 7.07e−02 | 1.99e−02 | 3.74e−04 |
Linear regression was performed adjusting for age, sex, BMI, batch, and the first 10 principal components of ancestry.
Abbreviations: Pr (>|t|), P value for the proportion of the t distribution that is greater than the absolute value of the t statistic; SE, standard error.
Sensitivity Analysis For Alcohol Consumption
Given the impact of alcohol intake on liver damage, we performed a sensitivity analysis in UK Biobank, analyzing the association of genetic variants of interest with biomarkers of liver injury and cirrhosis while accounting for weekly alcohol consumption. The results were consistent across all analyses, suggesting that PNPLA3 I148M, TM6SF2 E167K, HSD17B13 rs72613567(TA), and the SERPINA1 Z allele have an impact on liver disease independent of alcohol consumption (Supporting Table S5; Supporting Fig. S4).
Validation of Findings in an Independent Hospital‐Based Cohort
In the Mass General Brigham Biobank cohort, we identified 929 cases of cirrhosis, 615 SERPINA1 Z allele heterozygotes, and 13 SERPINA1 Z allele homozygotes in 19,323 unrelated white ancestry subjects. As expected, patients with all‐cause cirrhosis had elevated ALT, AST, ALP, and FIB‐4 scores and had lower albumin levels, platelet counts, total cholesterol, and LDL‐C (P < 0.001 for all) (Table 3). Patients with cirrhosis were also more likely to be men, were older, and had higher BMI (P < 0.001 for all). In this hospital‐based cohort, the proportion of individuals with cirrhosis was higher compared to UK Biobank (Table 3). Age (OR, 1.01; P < 0.001), male sex (OR, 1.55; P < 0.001), and BMI (OR, 1.04; P < 0.001) were associated with cirrhosis by univariate analysis using logistic regression. The SERPINA1 Z allele, PNPLA3 I148M, and TM6SF2 E167K were all associated with cirrhosis in an allele dose‐dependent manner. The OR for the SERPINA1 Z allele in association with cirrhosis was 1.64 (P = 3.5 × 10−02), compared to 1.48 (P = 2.0 × 10−06) for PNPLA3 I148M and 1.33 (P = 2.8 × 10−02) for TM6SF2 E167K (Fig. 5). SERPINA1 Z allele heterozygotes had a trend toward increased risk of cirrhosis (OR, 1.55; P = 8.3 × 10−02), while Pi*ZZ homozygous individuals had a significantly increased risk of cirrhosis (OR, 7.2; P = 5.0 × 10−02) (Supporting Fig. S5). PNPLA3 I148M and TM6SF2 E167K were associated with all‐cause fatty liver disease (additive OR, 1.25; P = 1.9 × 10−05; and additive OR, 1.26; P = 4.9 × 10−03; respectively), while the SERPINA1 Z allele was not associated with fatty liver disease (additive OR, 1.17; P = 0.35) (Supporting Fig. S6). The SERPINA1 Z allele was also associated with higher blood levels of log‐transformed ALT and ALP, and similar associations were demonstrated for PNPLA3 I148M and TM6SF2 E167K in association with ALT and AST (Table 4). Our study was not sufficiently powered to detect gene–environment interactions in the Mass General Brigham Biobank cohort.
FIG. 5.
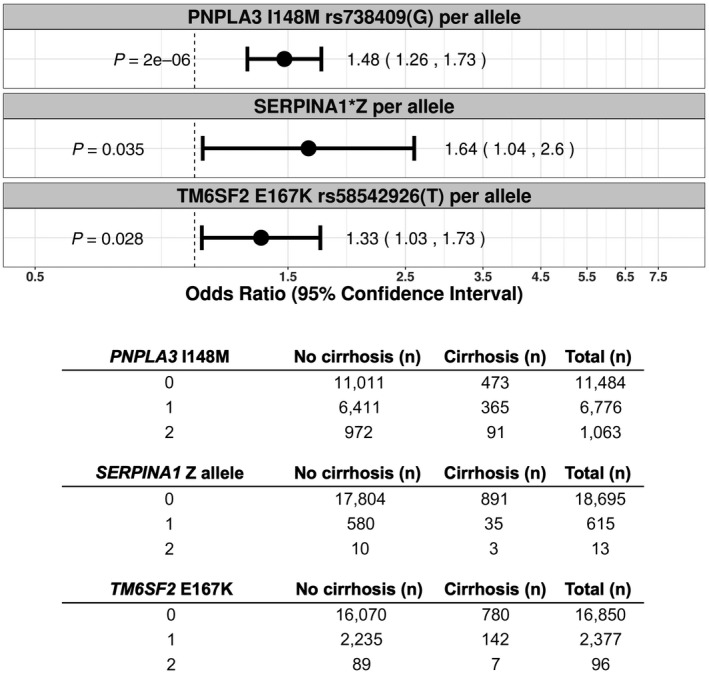
Association of PNPLA3 I148M, the SERPINA1 Z allele, and TM6SF2 E167K with cirrhosis by ICD‐10 diagnostic codes in the Mass General Brigham Biobank additive (per allele) model, using the Firth penalized likelihood approach. ORs were calculated with the use of logistic regression, with adjustment for age, sex, BMI, genotyping batch, and first 10 principal components of ancestry. Subjects were coded 0 for reference allele homozygotes, 1 for heterozygotes, and 2 for alternate allele homozygotes. The table reflects the number of subjects with and without cirrhosis for each genotype.
Discussion
In this study, we provide evidence from both a large population‐based cohort and a hospital‐based cohort that the SERPINA1 Z allele is associated with cirrhosis and biochemical tests of liver injury. We also validate several genetic variants previously associated with liver disease, including PNPLA3 I148M, TM6SF2 E167K, and HSD17B13 rs72613567(TA). Our findings suggest that the SERPINA1 Z allele is among the most important genetic risk factors for cirrhosis and liver injury in subjects of European ancestry. This study adds to a growing body of literature illustrating a liver disease burden in Z allele heterozygotes,( 16, 17, 18 ) including a recent large cross‐sectional analysis of the European Alpha‐1 Liver Cohort.( 20 ) Our analysis included 12,603 SERPINA1 Z allele heterozygotes from a population‐based cohort and 615 Z allele heterozygotes from a hospital‐based cohort, collectively representing the largest study associating the Z allele with liver disease outcomes. In contrast to prior case‐control studies in Z allele heterozygotes, a major strength of using UK Biobank is that participants were not recruited on the basis of having liver disease, enabling a population‐based assessment of the role of Z allele heterozygosity on liver phenotypes. Our study also complements prior genome‐wide association studies that have identified the SERPINA1 Z allele as a risk factor for liver disease. While these studies tend to employ an additive (per allele) genetic model,( 21, 22 ) our study provides the specific assessment of a heterozygous versus wild‐type model, where effect estimates are not influenced by the disease or biomarker status of the Z allele homozygotes. We demonstrate that Z allele heterozygotes have an intermediate liver phenotype compared to Pi*ZZ homozygous individuals. Our data also confirm the marked susceptibility of Pi*ZZ individuals to end‐stage liver disease.
As with the development of chronic lung disease,( 36, 37 ) the development of liver disease among Pi*ZZ homozygous or Pi*Z heterozygous subjects is variable,( 10 ) and other genetic and environmental risk factors may contribute to this variability. A recent study demonstrated that adiposity amplifies the risk of fatty liver disease conferred by multiple loci, including PNPLA3 I148M, TM6SF2 E167K, and glucokinase regulator (GCKR) P446L.( 38 ) We evaluated the combined effect of the SERPINA1 Z allele and BMI on biomarkers of liver injury and demonstrated an interaction of BMI with the SERPINA1 Z allele in association with ALT or AST in UK Biobank. This finding should be validated in an independent cohort. Potential mechanisms for the SERPINA1 Z allele and BMI interaction effect include obesity‐induced endoplasmic reticulum stress( 39 ) and autophagy dysregulation,( 40 ) which may aggravate proteotoxic stress from the accumulation of misfolded mutant AAT in the endoplasmic reticulum of hepatocytes.( 4, 5 ) Additional research will be required to further delineate the mechanism of the synergistic relationship between the SERPINA1 Z allele and BMI.
There are no pharmacologic interventions currently approved for liver disease associated with AAT deficiency, although several approaches have demonstrated preclinical proof‐of‐concept and have entered early stage clinical trials.( 2 ) Our data suggest that therapeutic silencing of the SERPINA1 Z allele( 41 ) may have benefit in patients with liver disease from homozygous AAT deficiency and also in heterozygous carriers of the Z allele. Whereas the PNPLA3 I148M and TM6SF2 E167K variants that increase the risk of cirrhosis have a protective effect on circulating lipids and reduce risk of coronary artery disease,( 32, 33, 34, 35 ) the lack of association of the SERPINA1 Z allele with total cholesterol or LDL‐C in UK Biobank suggests that therapeutic targeting of SERPINA1 may not necessarily lead to excess cardiovascular risk.
We hypothesized that genetic interactions may play an important role in disease susceptibility. We found that HSD17B13 rs72613567(TA) mitigated the risk of liver injury conferred by the PNPLA3 I148M variant; prior studies have demonstrated that the splice variant in HSD17B13 is associated with decreased PNPLA3 messenger RNA expression.( 21 ) We also found evidence of a genetic interaction between PNPLA3 I148M and TM6SF2 E167K in association with aminotransferase levels. PNPLA3 I148M and TM6SF2 E167K are both associated with reduced hepatic secretion of triglyceride‐rich lipoproteins.( 32, 34 ) There was no evidence of epistasis between the Z allele and other well‐established genetic risk factors for liver injury, suggesting that these known genetic variants act in an additive manner with the Z allele to increase risk for liver injury.
Collectively, these findings may have implications for individuals heterozygous for the SERPINA1 allele. In current clinical practice, measuring serum AAT levels is a cost‐effective method of identifying subjects who are Pi*ZZ homozygous and ruling out severe AAT deficiency.( 1, 2 ) The use of serum AAT to identify subjects who are Pi*Z heterozygous is more challenging as their AAT protein levels are often within the reference range.( 42 ) Moreover, the occurrence of insoluble AAT aggregates on liver biopsy is highly variable in subjects who are Pi*Z heterozygous.( 20 ) As such, individuals who are Pi*Z heterozygous may be clinically indistinguishable through conventional diagnostic approaches. Identification of the 2%‐4% of individuals of European ancestry that carry the Pi*Z allele may require genotyping or protein phenotyping in clinical practice.
Our results should be interpreted in the context of several important limitations. Further research will be needed to confirm these results across multiple ethnicities. Among liver disease cases analyzed in this study, the presence of hepatitis B or hepatitis C was not systematically assessed. ICD‐10 diagnostic codes are known to be imprecise in the context of clinical care; further studies on the impact of the SERPINA1 Z allele locus on biopsy‐confirmed liver disease are warranted. Future studies should also assess the incidence of liver‐related outcomes in SERPINA1 Z allele carriers using time‐to‐event or longitudinal analysis.
In conclusion, while severe AAT deficiency from SERPINA1 Z allele homozygosity (Pi*ZZ) is a proven genetic risk factor for developing cirrhosis, we provide evidence that Z allele heterozygotes also have a significantly increased risk of liver injury and cirrhosis and that this risk may increase synergistically in the setting of a higher BMI. We suggest that the SERPINA1 Z allele is among the most important genetic risk factors for liver injury and cirrhosis. We also provide preliminary evidence of genetic interactions between variants in PNPLA3, TM6SF2, and HSD17B13. Further studies may determine the relevance of these findings to patient risk stratification, disease prevention, and therapeutic intervention.
Supporting information
Supplementary Material
Acknowledgment
We thank all participants in UK Biobank and Mass General Brigham Biobank; their contribution to this study leads to advancing our understanding of liver disease. Ilana Usiskin is acknowledged for R version 3.6.0 expertise.
Supported by the Beth Israel Deaconess Medical Center Weener Family (Residency Research Small Grants Program to A.H.), National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (grants T32 HL007427 to M.M.; K01 HL129039 to D.Q.; R01 HL123915, R01 HL141826 to J.A.L.S.; P01 HL114501, R01 HL133135 to E.K.S; R01 HL137927 to E.K.S., M.H.C.; R01 HL147148 to E.K.S., B.D.H, M.H.C.; K08 HL136928 to B.D.H.; U01 HL089856, R01 HL135142 to B.D.H., M.H.C.), NIH National Institute of Diabetes and Digestive and Kidney Diseases (K08 DK113109 to S.V; K08 DK115883 to Z.G.J), American Association for the Study of Liver Diseases (Alan Hofmann Clinical and Translational Research Award to Z.G.J.), and American College of Gastroenterology (Clinical Research Award to Z.G.J.).
This research has been conducted using the United Kingdom Biobank Resource under Application Number 20915.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funding body has no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Potential conflict of interest: Dr. Hakim consults for Deep Track Capital, LP and advises Olix Pharmaceuticals, Inc. Dr. Silverman received grants from GlaxoSmithKline and Bayer. Dr. Cho consults for AstraZeneca, Illumina, and Genetech and received grants from Bayer and GSK. The other authors have nothing to report.
References
- 1.Silverman EK, Sandhaus RA. Clinical practice. Alpha1‐antitrypsin deficiency. N Engl J Med 2009;360:2749‐2757. [DOI] [PubMed] [Google Scholar]
- 2.Strnad P, McElvaney NG, Lomas DA. Alpha1‐antitrypsin deficiency. N Engl J Med 2020;382:1443‐1455. [DOI] [PubMed] [Google Scholar]
- 3.Ogushi F, Fells GA, Hubbard RC, Straus SD, Crystal RG. Z‐type alpha 1‐antitrypsin is less competent than M1‐type alpha 1‐antitrypsin as an inhibitor of neutrophil elastase. J Clin Invest 1987;80:1366‐1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1‐antitrypsin accumulation in the liver. Nature 1992;357:605‐607. [DOI] [PubMed] [Google Scholar]
- 5.Kuscuoglu D, Janciauskiene S, Hamesch K, Haybaeck J, Trautwein C, Strnad P. Liver ‐ master and servant of serum proteome. J Hepatol 2018;69:512‐524. [DOI] [PubMed] [Google Scholar]
- 6.Clark VC, Marek G, Liu C, Collinsworth A, Shuster J, Kurtz T, et al. Clinical and histologic features of adults with alpha‐1 antitrypsin deficiency in a non‐cirrhotic cohort. J Hepatol 2018;69:1357‐1364. [DOI] [PubMed] [Google Scholar]
- 7.Hamesch K, Mandorfer M, Pereira VM, Moeller LS, Pons M, Dolman GE, et al.; European Alpha1‐Liver Study Group . Liver fibrosis and metabolic alterations in adults with alpha‐1‐antitrypsin deficiency caused by the Pi*ZZ mutation. Gastroenterology 2019;157:705‐719.e18. [DOI] [PubMed] [Google Scholar]
- 8.Eriksson S, Carlson J, Velez R. Risk of cirrhosis and primary liver cancer in alpha1‐antitrypsin deficiency. N Engl J Med 1986;314:736‐739. [DOI] [PubMed] [Google Scholar]
- 9.Tanash HA, Nilsson PM, Nilsson JA, Piitulainen E. Clinical course and prognosis of never‐smokers with severe alpha‐1‐antitrypsin deficiency (PiZZ). Thorax 2008;63:1091‐1095. [DOI] [PubMed] [Google Scholar]
- 10.Tanash HA, Piitulainen E. Liver disease in adults with severe alpha‐1‐antitrypsin deficiency. J Gastroenterol 2019;54:541‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bell H, Schrumpf E, Fagerhol MK. Heterozygous MZ alpha‐1‐antitrypsin deficiency in adults with chronic liver disease. Scand J Gastroenterol 1990;25:788‐792. [DOI] [PubMed] [Google Scholar]
- 12.Vecchio FM, Fabiano A, Orsini G, Ragusa D, Massi G. Alpha‐1‐antitrypsin MZ phenotype and cryptogenic chronic liver disease in adults. Digestion 1983;27:100‐104. [DOI] [PubMed] [Google Scholar]
- 13.Theodoropoulos G, Fertakis A, Archimandritis A, Kapordelis C, Angelopoulos B. Alpha 1‐antitrypsin phenotypes in cirrhosis and hepatoma. Acta Hepatogastroenterol (Stuttg) 1976;23:114‐117. [PubMed] [Google Scholar]
- 14.Fisher RL, Taylor L, Sherlock S. Alpha‐1‐antitrypsin deficiency in liver disease: the extent of the problem. Gastroenterology 1976;71:646‐651. [PubMed] [Google Scholar]
- 15.Bartlett JR, Friedman KJ, Ling SC, Pace RG, Bell SC, Bourke B, et al.; Gene Modifier Study Group . Genetic modifiers of liver disease in cystic fibrosis. JAMA 2009;302:1076‐1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strnad P, Buch S, Hamesch K, Fischer J, Rosendahl J, Schmelz R, et al. Heterozygous carriage of the alpha1‐antitrypsin Pi*Z variant increases the risk to develop liver cirrhosis. Gut 2019;68:1099‐1107. [DOI] [PubMed] [Google Scholar]
- 17.Schaefer B, Mandorfer M, Viveiros A, Finkenstedt A, Ferenci P, Schneeberger S, et al. Heterozygosity for the alpha‐1‐antitrypsin Z allele in cirrhosis is associated with more advanced disease. Liver Transpl 2018;24:744‐751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graziadei IW, Joseph JJ, Wiesner RH, Therneau TM, Batts KP, Porayko MK. Increased risk of chronic liver failure in adults with heterozygous alpha1‐antitrypsin deficiency. Hepatology 1998;28:1058‐1063. [DOI] [PubMed] [Google Scholar]
- 19.Mandorfer M, Bucsics T, Hutya V, Schmid‐Scherzer K, Schaefer B, Zoller H, et al. Liver disease in adults with α1‐antitrypsin deficiency. United European Gastroenterol J 2018;6:710‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider CV, Hamesch K, Gross A, Mandorfer M, Moeller LS, Pereira V, et al.; European Alpha‐1 Liver Study Group . Liver phenotypes of European adults heterozygous or homozygous for Pi*Z variant of AAT (Pi*MZ vs Pi*ZZ genotype) and noncarriers. Gastroenterology 2020;159:534‐548.e11. [DOI] [PubMed] [Google Scholar]
- 21.Abul‐Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, et al. A protein‐truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med 2018;378:1096‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen VL, Chen Y, Du X, Handelman SK, Speliotes EK. Genetic variants that associate with cirrhosis have pleiotropic effects on human traits. Liver Int 2020;40:405‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valenti L. Uncovering the genetics of cirrhosis: new plots for the usual suspects. Liver Int 2020;40:281‐282. [DOI] [PubMed] [Google Scholar]
- 24.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 2015;12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gainer V, Cagan A, Castro V, Duey S, Ghosh B, Goodson A, et al. The Biobank Portal for Partners Personalized Medicine: a query tool for working with consented Biobank samples, genotypes, and phenotypes using i2b2. J Pers Med 2016;6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018;562:203‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heinze G, Schemper M. A solution to the problem of separation in logistic regression. Stat Med 2002;21:2409‐2419. [DOI] [PubMed] [Google Scholar]
- 28.Cordell HJ. Epistasis: what it means, what it doesn't mean, and statistical methods to detect it in humans. Hum Mol Genet 2002;11:2463‐2468. [DOI] [PubMed] [Google Scholar]
- 29.Privitera G, Spadaro L, Marchisello S, Fede G, Purrello F. Abnormalities of lipoprotein levels in liver cirrhosis: clinical relevance. Dig Dis Sci 2018;63:16‐26. [DOI] [PubMed] [Google Scholar]
- 30.Udell JA, Wang CS, Tinmouth J, FitzGerald JM, Ayas NT, Simel DL, et al. Does this patient with liver disease have cirrhosis? JAMA 2012;307:832‐842. [DOI] [PubMed] [Google Scholar]
- 31.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg‐Hansen A, et al. Exome‐wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grandone A, Cozzolino D, Marzuillo P, Cirillo G, Di Sessa A, Ruggiero L, et al. TM6SF2 Glu167Lys polymorphism is associated with low levels of LDL‐cholesterol and increased liver injury in obese children. Pediatr Obes 2016;11:115‐119. [DOI] [PubMed] [Google Scholar]
- 34.Pirazzi C, Adiels M, Burza MA, Mancina RM, Levin M, Ståhlman M, et al. Patatin‐like phospholipase domain‐containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J Hepatol 2012;57:1276‐1282. [DOI] [PubMed] [Google Scholar]
- 35.Liu DJ, Peloso GM, Yu H, Butterworth AS, Wang X, Mahajan A, et al.; Charge Diabetes Working Group; EPIC‐InterAct Consortium; EPIC‐CVD Consortium; GOLD Consortium; VA Million Veteran Program . Exome‐wide association study of plasma lipids in >300,000 individuals. Nat Genet 2017;49:1758‐1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeMeo DL, Sandhaus RA, Barker AF, Brantly ML, Eden E, McElvaney NG, et al. Determinants of airflow obstruction in severe alpha‐1‐antitrypsin deficiency. Thorax 2007;62:806‐813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silverman EK. Risk of lung disease in PI MZ heterozygotes. Current status and future research directions. Ann Am Thorac Soc 2016;13(Suppl. 4):S341‐S345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stender S, Kozlitina J, Nordestgaard BG, Tybjærg‐Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet 2017;49:842‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hummasti S, Hotamisligil GS. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ Res 2010;107:579‐591. [DOI] [PubMed] [Google Scholar]
- 40.Hidvegi T, Mukherjee A, Ewing M, Kemp C, Perlmutter DH. The role of autophagy in alpha‐1‐antitrypsin deficiency. Methods Enzymol 2011;499:33‐54. [DOI] [PubMed] [Google Scholar]
- 41.Wooddell CI, Blomenkamp K, Peterson RM, Subbotin VM, Schwabe C, Hamilton J, et al. Development of an RNAi therapeutic for alpha‐1‐antitrypsin liver disease. JCI Insight 2020;5:e135348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kok KF, Willems HL, Drenth JP. The cut‐off value of 100 mg/dl is insufficient to detect heterozygous alpha‐1 antitrypsin‐deficient liver disease patients. Liver Int 2010;30:491‐492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material