

Abstract
Aerobic alcohol oxidation catalyzed by transition-metal salts and aminoxyls are prominent examples of cooperative catalysis. Cu/aminoxyl catalysts have been studied previously and feature “integrated cooperativity”, in which CuII and the aminoxyl participate together to mediate alcohol oxidation. Here, we investigate a complementary Fe/aminoxyl catalyst system and provide evidence for “serial cooperativity”, involving a redox cascade in which alcohol is oxidized by in situ-generated oxoammonium species, which is directly detected in the catalytic reaction mixture by cyclic step chronoamperometry. The mechanistic difference between the Cu and Fe-based catalysts arises from the use iron(III) nitrate, which initiates a NOx-based redox cycle for oxidation of aminoxyl/hydroxylamine to oxoammonium. The different mechanisms for the Cu- and Fe-based catalyst systems are manifested in different alcohol oxidation chemoselectivity and functional group compatibility.
Graphical Abstarct
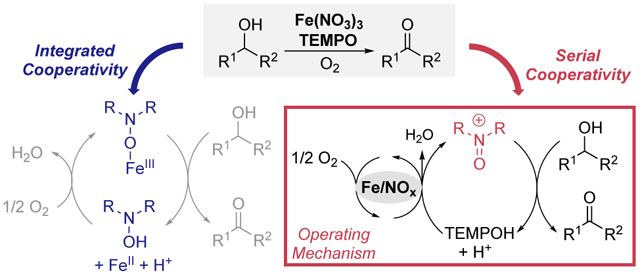
Aminoxyl radicals, such as TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl), are highly effective (co)catalysts for alcohol oxidation.1–7 They are used in the industrially important Anelli oxidation, which uses NaOCl as the oxidant,8,9 serve as effective electrocatalysts,7 and are key components in numerous aerobic alcohol oxidation catalysts.3–6 The Anelli and electrochemical oxidation methods are initiated by one-electron oxidation of TEMPO to TEMPO+, and the oxoammonium species promotes alcohol oxidation, generating TEMPOH as byproduct (Scheme 1). Catalytic turnover proceeds via two-electron oxidation of TEMPOH to TEMPO+ with NaOCl or at an electrode. Aerobic oxidation methods feature different mechanisms, depending on the cocatalyst used to promote oxidation of TEMPOH (Scheme 2A). Cu/aminoxyl cocatalyst systems have been developed for aerobic alcohol oxidation,4,5 and mechanistic studies reveal “integrated cooperativity” between CuII and an aminoxyl radical. Each component serves as a one-electron oxidant to support the two-electron oxidation reaction (Scheme 2B).10–13 Electrochemical studies confirm that the high-potential oxoammonium species is not formed in these reactions.14 A different class of aerobic alcohol oxidation catalysts employ NOx-based cocatalysts,6 derived from NaNO2, HNO3, or organic nitrites (Scheme 2A). Mechanistic studies show that these reactions feature a redox cascade involving “serial cooperativity”, in which O2 reacts with NO to generate NO2, and NO2 oxidizes TEMPOH to TEMPO+ (Scheme 2C).15,16 The final step closely resembles other TEMPO-catalyzed alcohol oxidation reactions that generate TEMPO+ as the reactive oxidant (e.g., using NaOCl or an electrode). Fe/TEMPO represents a third set of cocatalyst systems that show excellent aerobic alcohol oxidation reactivity.17–23 The mechanism of these reactions has not yet been characterized, but postulated pathways include both integrated and serial cooperativity.5,17,19–21,23 Stoichiometric alcohol oxidation reactions observed with FeCl3–TEMPO adducts are consistent with an “integrated” mechanism.24–26 On the other hand, nearly all Fe/TEMPO catalysts use iron nitrates or include another NOx source that could contribute to a “serial” pathway. In the present study, we probe the mechanism of Fe/TEMPO-catalyzed alcohol oxidation by operando electrochemical analysis, and the data provide evidence for serial cooperativity. This mechanism is further evident from reactions with substrates that reveal different chemoselectivity and synthetic scope relative to Cu/TEMPOcatalyzed alcohol oxidation.
Scheme 1.
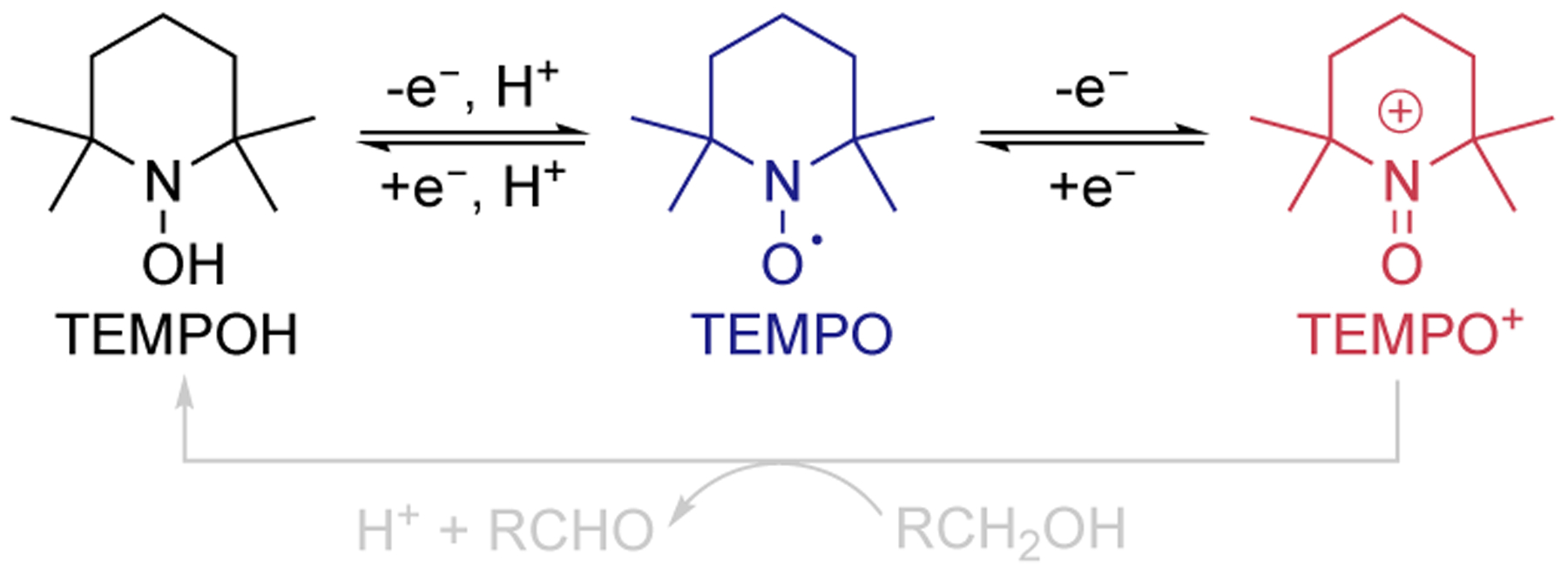
Aminoxyl-based redox reactions.
Scheme 2.
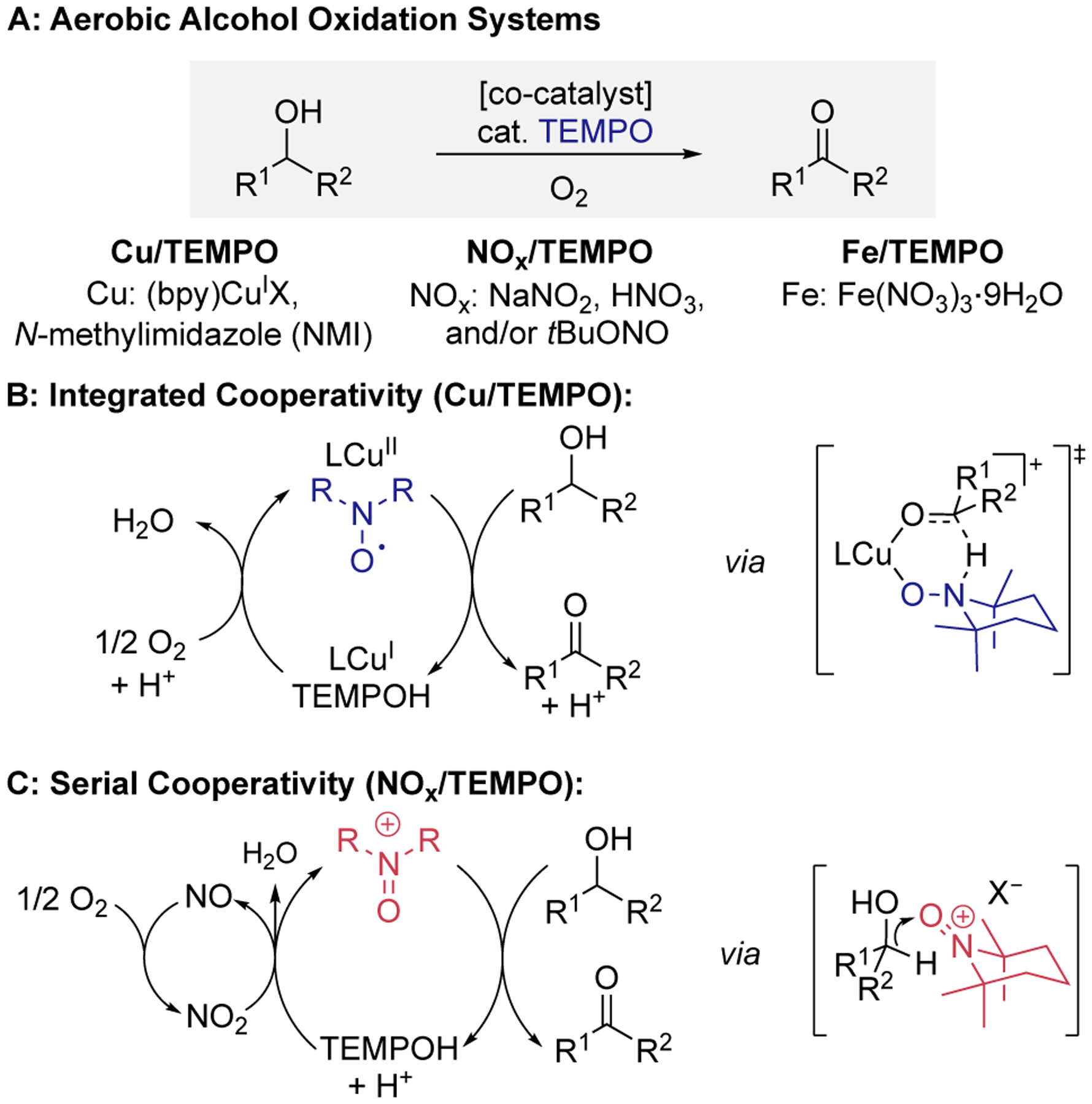
Aminoxyl co-catalyst systems for aerobic alcohol oxidation.
We have previously used cyclic step chronoamperometry (CSCA) to probe TEMPO-catalyzed alcohol oxidation reactions with NaOCl as the oxidant.27 This analytical technique detects redox active species in solution without influencing their bulk concentration. Use of a rotating disk electrode (RDE) avoids mass-transfer limitations at the electrode and provides real-time insights into the analytes in solution. The CSCA experiments were conducted by cycling the RDE between potentials above and below the TEMPO+/TEMPO redox couple (E1/2 = 0.24 V vs Fc+/0). When the RDE potential is above 0.24 V, positive current is detected from oxidation of TEMPO in solution and the magnitude of the current is proportional to [TEMPO]. When the RDE is below 0.24 V, negative current arises from reduction of TEMPO+ with a magnitude proportional to [TEMPO+].
Studies were initiated by analyzing TEMPO speciation in the presence of HCl and HNO3 in order to establish CSCA benchmarks for the investigation of Fe/TEMPO-catalyzed aerobic alcohol oxidation (Figure 1). The conditions were derived from a previously reported catalytic NOx/TEMPO method for alcohol oxidation.28 Addition of 1 equiv HCl to a solution of TEMPO initiates TEMPO disproportionation into a 1:1 mixture of TEMPO+ and TEMPOH2+.29–31 TEMPOH2+ is electrochemically inactive at these potentials and measured currents solely arise from TEMPO and TEMPO+. During a period of 12 min (t = 1–13 min), TEMPO disappearance and TEMPO+ appearance proceeds with the expected 2:1 stoichiometry (Figure 1B). Addition of HNO3 (1 equiv) at this point initiates oxidation of all of the TEMPO and TEMPOH2+ to TEMPO+, reflecting the TEMPOH/NOx/O2 portion of the mechanism in Scheme 2C.
Figure 1.
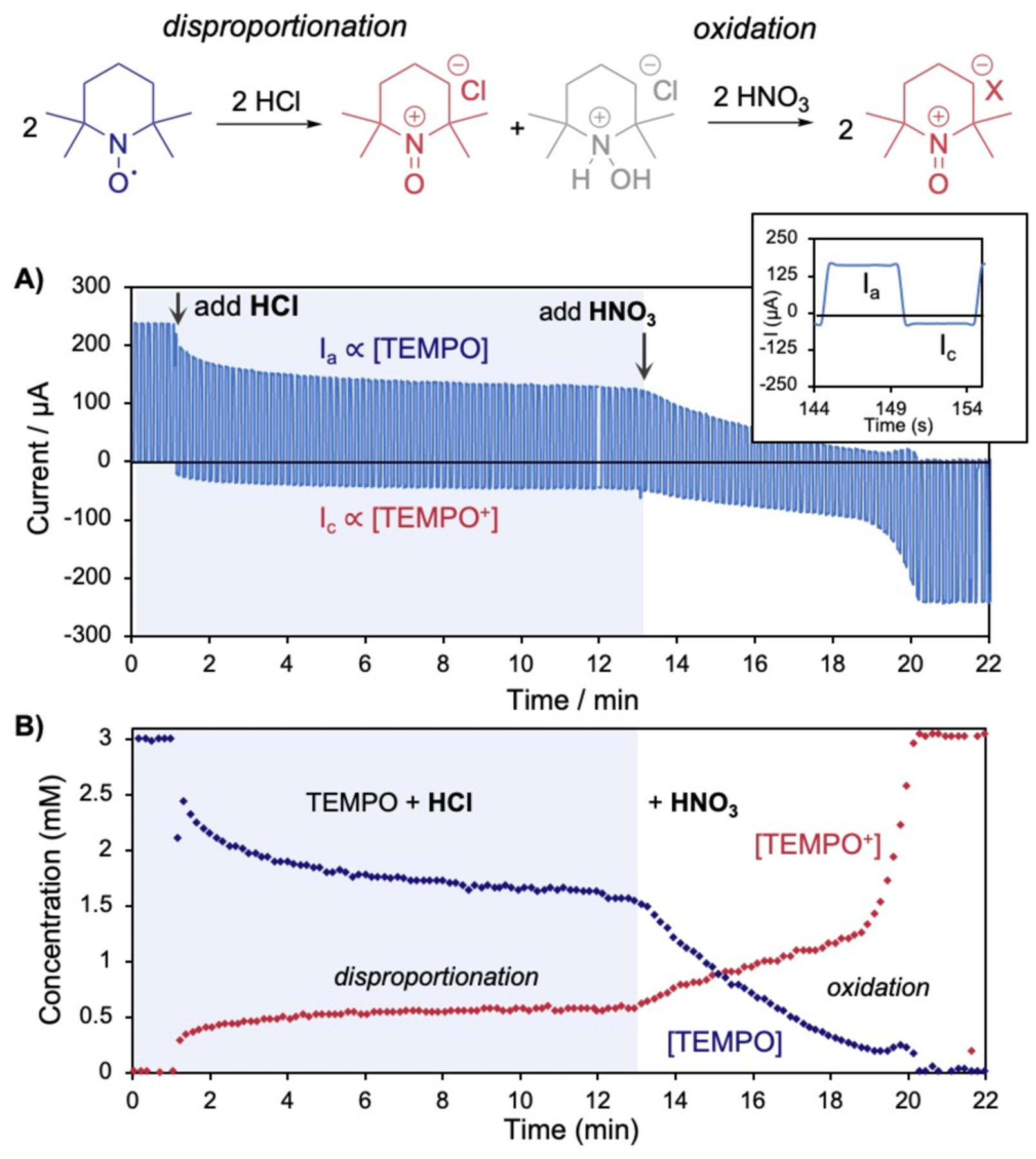
A) Cyclic step chronoamperometric (CSCA) analysis of TEMPO (3 mM), following sequential addition of 1 equiv HCl (at 1 min) and HNO3 (at 13 min) to the aerobic reaction mixture. Inset shows representative limiting anodic and cathodic currents for two CSCA steps. B) Concentration profiles of TEMPO and TEMPO+ based on the median Ic and Ia measured at −0.18 and 0.52 V vs. Fc0/+, respectively, for each potential step in (A). Conditions: 0.1 M NBu4PF6, CH3CN, 1000 RPM.
We then used CSCA to probe TEMPO speciation in the presence of Fe(NO3)3 under air in CH3CN (Figure 2). Fe(NO3)3 is electroactive and is reduced at a potential similar to TEMPO+. Nonetheless, it was possible to subtract the background current of Fe(NO3)3 in the absence of TEMPO (see Supporting Information for details; while this issue affects quantitative precision, the resulting data provide clear insights). An induction period is evident over the first 2 min of the reaction, during which time TEMPO disproportionation is evident from the 2:1 stoichiometry associated with TEMPO disappearance and TEMPO+ appearance). This phase is followed by a rapid oxidation of TEMPO species, resulting in depletion of TEMPO together with the formation of TEMPO+. These data show that, unlike the (bpy)Cu/NMI cocatalyst system,14, 32 Fe(NO3)3 is capable of generating TEMPO+ and implicate a serial cooperativity mechanism similar to Scheme 2C.
Figure 2.
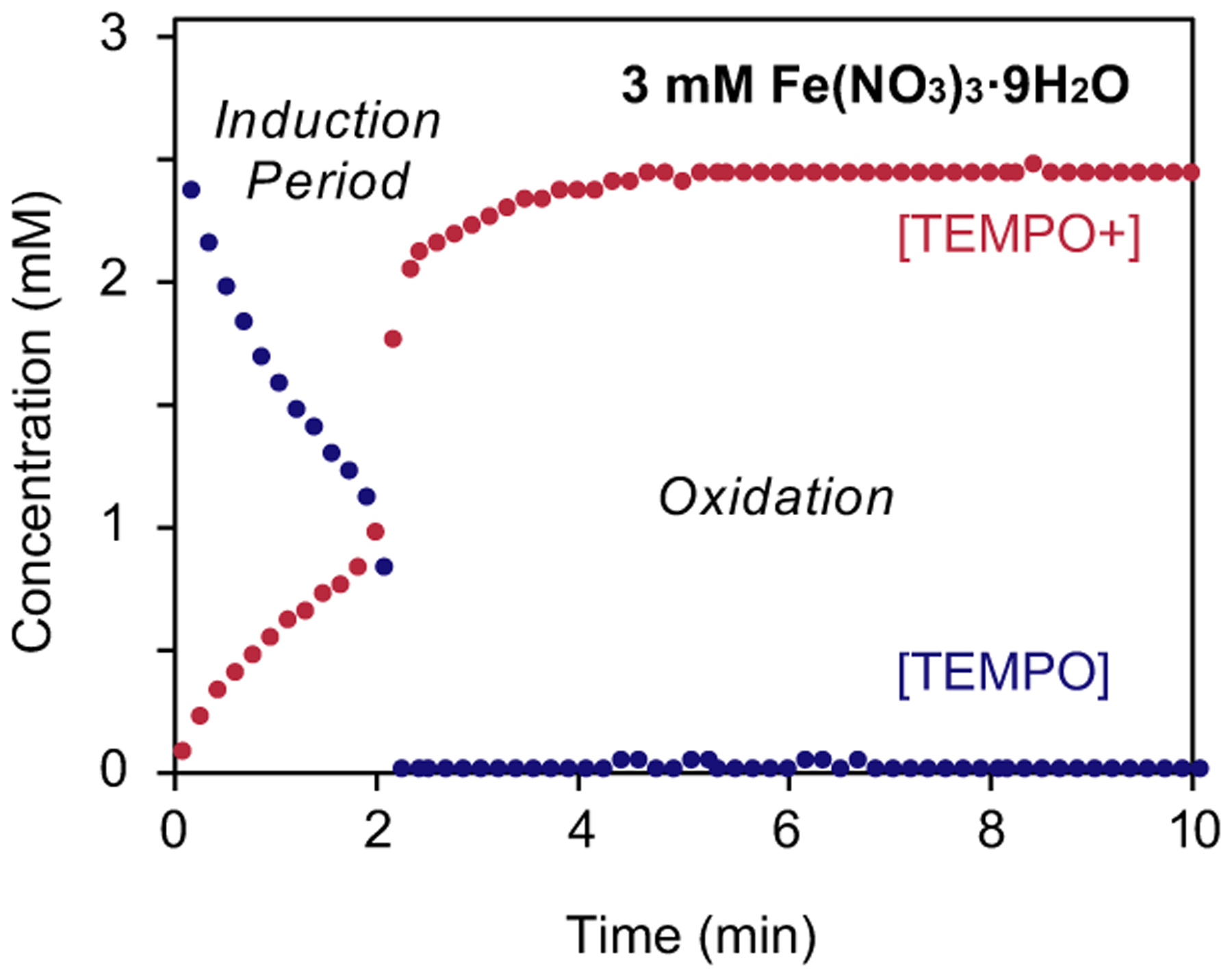
[TEMPO] (blue) and [TEMPO+] (red) obtained by using CSCA at a rotating disk electrode upon mixing CH3CN solutions of TEMPO and Fe(NO3)3•9H2O in the absence (A) and presence (B) of 1-phenylethanol (50 mM). Conditions: 3 mM [TEMPO] and [Fe], 0.1 M NBu4PF6 in CH3CN; Ic and Ia measured at −0.16 and 0.64 V vs. Fc0/+, respectively, 1000 RPM.
Additional experiments were conducted to gain additional insights into the origin of TEMPO disproportionation and redox behavior in the presence of Fe(NO3)3. Fe(OTf)3 and Al(OTf)3 represent M3+ Lewis acids, similar to Fe(NO3)3, and these species show very similar reactivity with TEMPO (Figures 3A and 3B). CSCA analysis reveals rapid formation of 0.5 equiv of TEMPO+, consistent with Lewis acid-promoted disproportionation of TEMPO. TEMPO disproportionation was independently confirmed by following the Al(OTf)3/TEMPO reaction by UV-visible spectroscopy (see Figure S6 in the Supporting Information).33 The anodic CSCA current in Figures 3A and 3B (blue traces) is assigned to “[TEMPO]” because the current at the anodic potential could include oxidation of species other than TEMPO (e.g., TEMPO-M adducts, M = FeIII, AlIII). Weaker Lewis acids, Zn(NO3)2 and Al(OTf)3/3 NBu4NO3, are less effective in promoting TEMPO disproportionation (Figures 3C and 3D). Negligible or slower formation of TEMPO+ is observed in these experiments.
Figure 3.
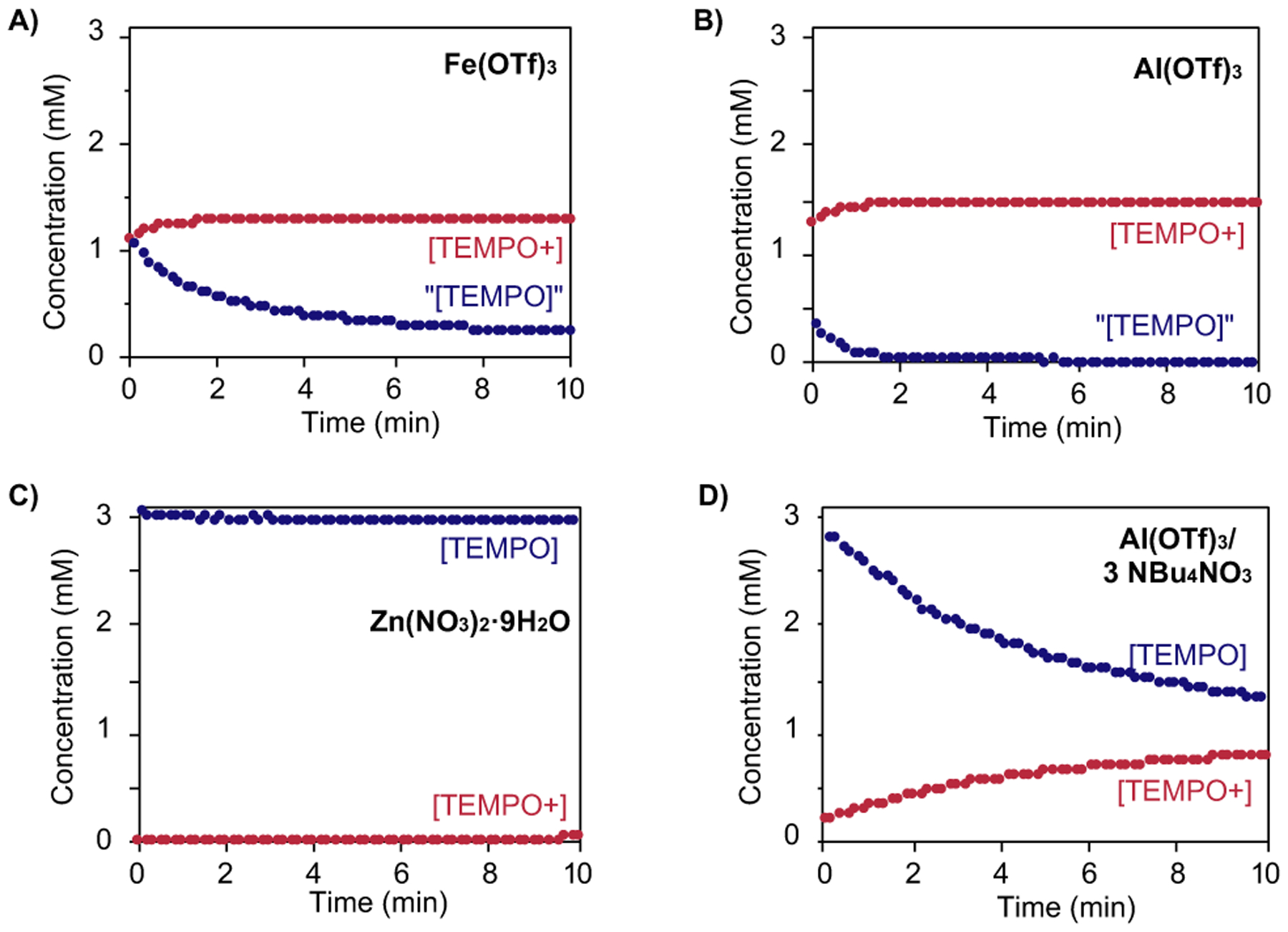
[TEMPO] (blue) and [TEMPO+] (red) obtained by using CSCA at a rotating disk electrode upon mixing CH3CN solutions of TEMPO and Fe(OTf)3, Al(OTf)3, Zn(NO)2, and Al(OTf)3/3 NBu4NO3. Conditions otherwise consistent with those in Figure 2.
The results in Figures 2 and 3 reveal that neither FeIII [e.g., in the form of Fe(OTf)3] nor nitrate salts alone can account for the TEMPO reactivity and cocatalytic role of Fe(NO3)3 in aerobic alcohol oxidation reactions. A rationale for the observations is that FeIII-promoted TEMPO disproportionation generates TEMPO anion, which will coordinate to FeIII and provide reducing equivalents needed to convert nitrate into catalytically relevant NOx species. The iron species likely play an active role in nitrate reduction34 and initiation of catalysis.
The results above show that Fe(NO3)3 and Brønsted-acid NOx-based catalyst systems feature similar mechanisms for aerobic alcohol oxidation, involving serial cooperativity (Scheme 2C). This pathway, which contrasts the integrated cooperativity of (bpy)Cu cocatalyst systems (Scheme 2B), has important synthetic implications. A series of substrate probes and kinetic comparisons were used to illuminate the similarities and differences among three prototypical catalyst systems for aerobic alcohol oxidation: Cu/TEMPO,35 Fe(NO3)3/TEMPO,20,21 and H+/NOx/TEMPO.36 These efforts prioritized mechanistic insights over synthetic optimization as the latter has been emphasized in previous reports. The diol substrates in Figures 4A an 4B feature an electronically activated 2° benzylic alcohol with a sterically less hindered 1° benzylic or aliphatic alcohol. In both cases, the Cu/TEMPO catalyst shows a preference for 1° alcohol oxidation. The Fe(NO3)3 and NaNO2/HNO3 cocatalyst systems exhibit similar reactivity, with little selectivity between 1°/2° benzylic alcohols (Figure 4A) but high selectivity for 2° benzylic over 1° aliphatic alcohol oxidation (Figure 4B). This selectivity contrasts that observed with the Cu/TEMPO catalyst system but matches that expected from oxoammonium-mediated alcohol oxidation under acidic conditions, aligning with the serial mechanistic pathway in Scheme 2C.37,38
Figure 4.
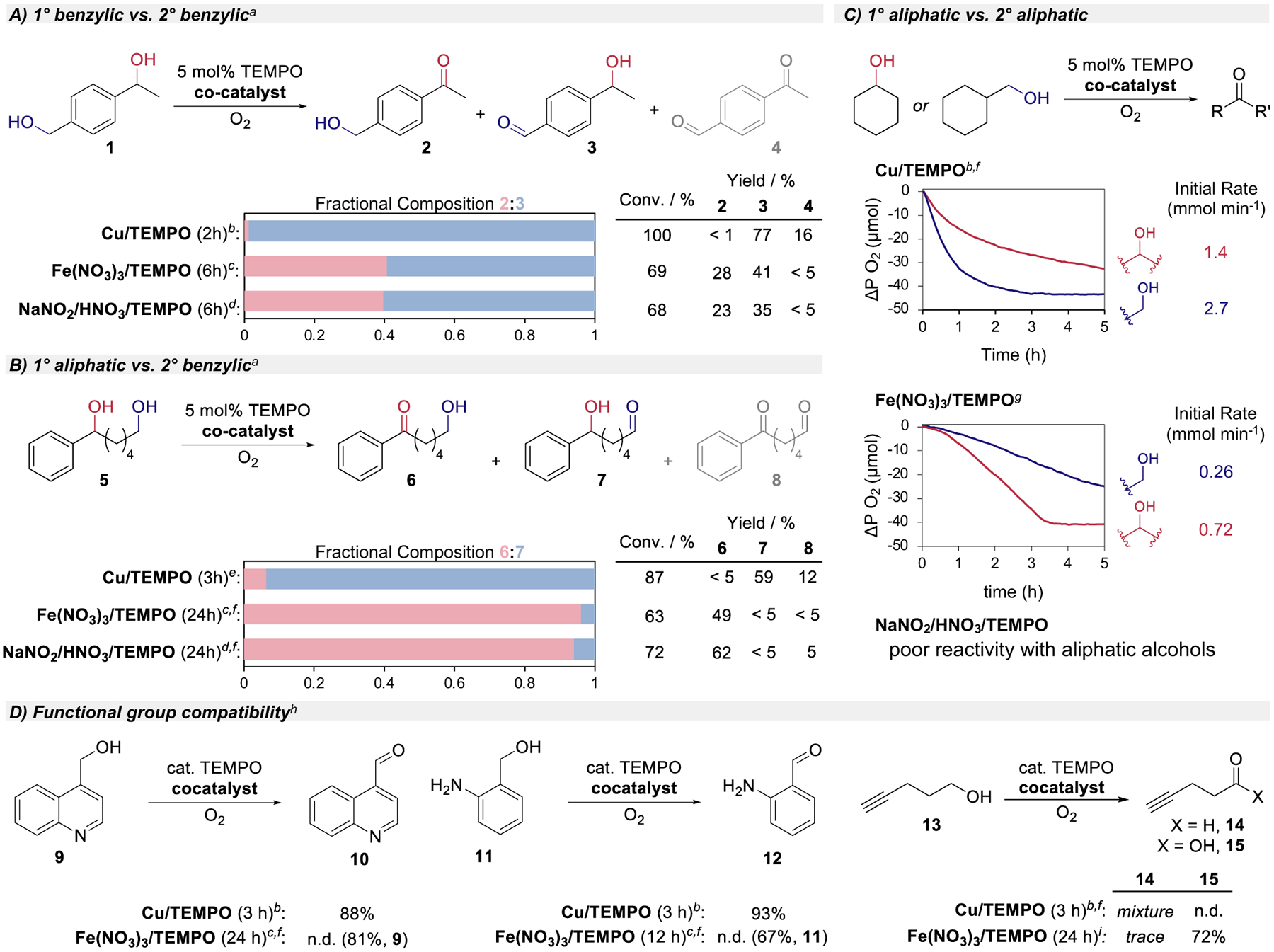
Comparison of chemoselectivity and functional compatibility with different aminoxyl cocatalyst systems for aerobic alcohol oxidation: A) intramolecular competition between 1° and 2° benzylic alcohols, B) intramolecular competition between 1° aliphatic and 2° benzylic alcohols, C) independent rate comparison of cyclohexanol and cyclohexanemethanol (for Cu/TEMPO, initial rates determined from the first 20 min; for Fe(NO3)3, rates determined from 40–60 min, i.e., after induction period), D) additional substrate probes. Conditions: rt, 5 mol% TEMPO unless noted otherwise. (A), (B), (C): 0.1 mmol substrate (0.1 M), (D): 1.0 mmol substrate (0.2 M). aRatios, yields determined by 1H NMR spectroscopy. b5 mol% [Cu(CH3CN)4]BF4, 5 mol% bpy, 10 mol% N-methylimidazole (NMI), CH3CN, air. c5 mol% Fe(NO3)3, CH3CN, air. d10 mol% NaNO2, 20 mol% HNO3, CH3CN, air. e5 mol% CuBr, 5 mol% bpy, 10 mol% NMI, CH3CN, air. f1 atm O2 rather than ambient air. g5 mol% Fe(NO3)3, 5 mol% KC1, 1,2-dichloroethane, 1 atm O2. hIsolated yields. i10 mol% TEMPO, 10 mol% Fe(NO3)3, 10 mol% KC1, 1,2-dichloroethane, 1 atm O2.
To compare 1° and 2° aliphatic alcohols, cyclohexanol and cyclohexylmethanol oxidation were monitored via gas-uptake methods (Figure 4C).39 Reaction time courses show that Cu/TEMPO oxidizes cyclohexylmethanol approximately two-fold faster than cyclohexanol. In contrast, Fe(NO3)3/TEMPO oxidizes cyclohexanol approximately three-fold faster than cyclohexylmethanol, following an induction period at the start of the reaction (attributed to in situ generation of NOx species as discussed above). The NaNO2/HNO3 system shows poor reactivity with aliphatic alcohols and kinetic data were not recorded. The latter observation highlights a benefit of the Fe(NO3)3-based catalyst system relative to other NOx-based catalysts that use a similar mechanism.
The Cu/TEMPO and Fe(NO3)3/TEMPO catalyst systems show complementary features with other substrates (Figure 4D). For example, Cu/TEMPO catalysts show good tolerance of basic and oxidatively sensitive functional groups.35,40–42 This feature is evident in oxidation of the quinoline- and aniline-containing substrates 9 and 11. In contrast, little reactivity for these substrates is observed with the Fe(NO3)3 catalyst system, probably arising from interference of the basic functional groups with the Lewis acidity of FeIII and/or generation of the NOx cocatalyst species. The Fe(NO3)3 catalyst system can be advantageous in other cases. Cu/TEMPO reacts poorly with 1° alcohol 13, which has a terminal alkyne; a mixture of unreacted starting material and products, including the aldehyde, are observed. In contrast, 13 shows good reactivity with the Fe(NO3)3/TEMPO catalyst system, affording the carboxylic acid 15 in good yield. This result highlights another complementary feature of Cu and Fe(NO3)3-based catalyst systems: Cu/TEMPO catalysts convert 1° alcohols to aldehydes and are poisoned by acidic functional groups, such as carboxylic acids. The Fe(NO3)3 system not only tolerates acidic groups but can also oxidize 1° alcohols to the carboxylic acid products.20
The substrate reactivity data in Figure 4 pairs with the mechanistic data in Figures 1–3 to support serial cooperativity between Fe(NO3)3 and TEMPO during catalytic aerobic alcohol oxidation (Scheme 2C). The combination of Fe3+ and nitrate represents an appealing NOx cocatalyst system capable of generating oxoammonium species with O2 (Scheme 3). We anticipate that aerobic alcohol oxidation catalysts that feature nitrate or other NOx components also take advantage of similar reactivity. Examples include recently reported Cu(NO3)2/TEMPO43 and Fe(NO3)3/bpy/TEMPO44 systems.45 The mechanism in Scheme 3 contrasts the integrated cooperativity mechanism of Cu/aminoxyl catalysts. These divergent modes of cooperativity are manifested by different chemoselectivity and synthetic scope for the catalytic reactions. The user-friendly and complementary synthetic features of both catalyst systems should contribute to broader adoption of aerobic alcohol oxidation methods in organic synthesis, including large scale applications.22,46–48
Scheme 3.
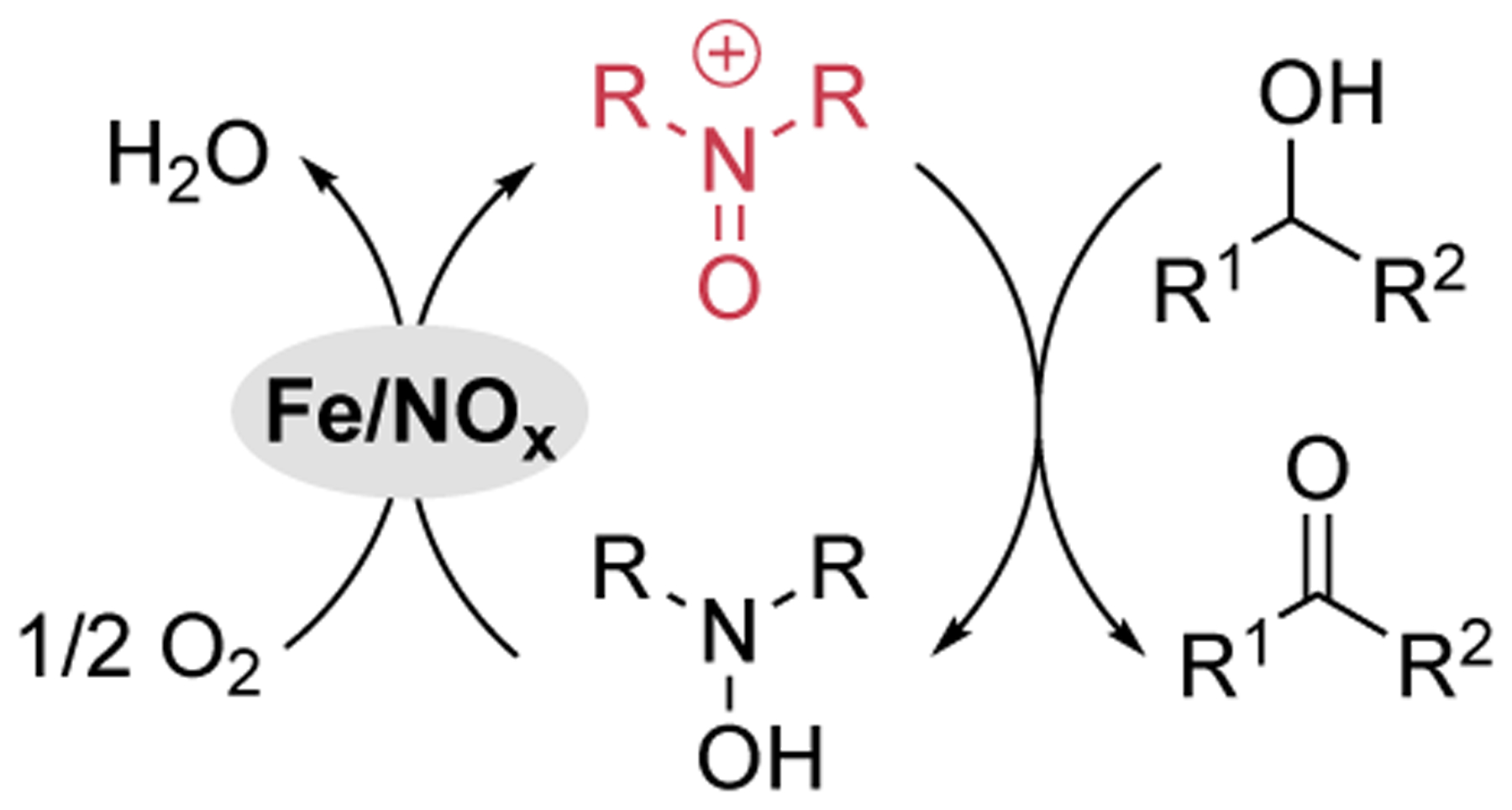
Simplified mechanism for Fe(NO3)3/aminoxylcatalyzed aerobic alcohol oxidation.
Supplementary Material
ACKNOWLEDGMENT
This work was funded by the Department of Energy (DE-FG02-05ER15690). J.E.N. acknowledges the NSF for a predoctoral fellowship (DGE-1747503). The spectrometers were purchased through NSF grant CHE-1048642 and by a generous gift from Paul J. and Margaret M. Bender, and the mass spectrometer was purchased through NIH grant NIH 1S10 OD020022-1.
Footnotes
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.1c05224
Experimental details, additional electrochemical data, and compound characterization data with NMR spectra (PDF).
The authors declare no competing financial interest.
REFERENCES
- 1.de Nooy AEJ; Besemer AC; van Bekkum H On the Use of Stable Organic Nitroxyl Radicals for the Oxidation of Primary and Secondary Alcohols. Synthesis 1996, 1153–1174. [Google Scholar]
- 2.Bobbitt JM; Brückner C; Merbouh N Oxoammonium- and Nitroxide-Catalyzed Oxidations of Alcohols. Org. React 2009, 74, 103–424. [Google Scholar]
- 3.Wertz S; Studer A Nitroxide-Catalyzed Transition Metal-Free Aerobic Oxidation Processes. Green Chem. 2013, 15, 3116–3134. [Google Scholar]
- 4.Ryland BL; Stahl SS Practical Aerobic Oxidations of Alcohols and Amines with Homogeneous Copper/TEMPO and Related Catalyst Systems. Angew. Chem. Int. Ed 2014, 53, 8824–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao Q; Dornan LM; Rogan L; Hughes NL; Muldoon MJ Aerobic Oxidation Catalysis with Stable Radicals. Chem. Commun 2014, 50, 4524–4543. [DOI] [PubMed] [Google Scholar]
- 6.Miles KC; Stahl SS Practical Aerobic Alcohol Oxidation with Cu/Nitroxyl and Nitroxyl/NOx Catalyst Systems. Aldrichimica Acta 2015, 48, 8–10. [Google Scholar]
- 7.Nutting JE; Rafiee M; Stahl SS Tetramethylpiperidine N-Oxyl (TEMPO), Phthalimide N-Oxyl (PINO), and Related N-Oxyl Species: Electrochemical Properties and Their Use in Electrocatalytic Reactions. Chem. Rev 2018, 118, 4834–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anelli PL; Biffi C; Montanari F; Quici S Fast and Selective Oxidation of Primary Alcohols to Aldehydes or to Carboxylic Acids and Secondary Alcohols to Ketones Mediated by Oxoammonium Salts under Two-Phase Conditions. J. Org. Chem 1987, 52, 2559–2562. [Google Scholar]
- 9.Caron S; Dugger RW; Gut Ruggeri S; Ragan JA; Ripin DHB Large-Scale Oxidations in the Pharmaceutical Industry. Chem. Rev 2006, 106, 2943–2989. [DOI] [PubMed] [Google Scholar]
- 10.Dijksman A; Arends IWCE; Sheldon RA Cu(II)-Nitroxyl Radicals as Catalytic Galactose Oxidase Mimics. Org. Biomol. Chem 2003, 1, 3232–3237. [DOI] [PubMed] [Google Scholar]
- 11.Belanzoni P; Michel C; Baerends EJ Cu(bipy)2+/TEMPOCatalyzed Oxidation of Alcohols: Radical or Nonradical Mechanism? Inorg. Chem 2011, 50, 11896–11904. [DOI] [PubMed] [Google Scholar]
- 12.Hoover JM; Ryland BL; Stahl SS Mechanism of Copper(I)/TEMPO-Catalyzed Aerobic Alcohol Oxidation. J. Am. Chem. Soc 2013, 135, 2357–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryland BL; McCann SD; Brunold TC; Stahl SS Mechanism of Alcohol Oxidation Mediated by Copper(II) and Nitroxyl Radicals. J. Am. Chem. Soc 2014, 136, 12166–12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Badalyan A; Stahl SS Cooperative Electrocatalytic Alcohol Oxidation with Electron-Proton-Transfer Mediators. Nature 2016, 535, 406–410. [DOI] [PubMed] [Google Scholar]
- 15.Goldstein S; Samuni A; Russo A Reaction of Cyclic Nitroxides with Nitrogen Dioxide: The Intermediacy of the Oxoammonium Cations. J. Am. Chem. Soc 2003, 125, 8364–8370. [DOI] [PubMed] [Google Scholar]
- 16.Gerken JB; Stahl SS High-Potential Electrocatalytic O2 Reduction with Nitroxyl/NOx Mediators: Implications for Fuel Cells and Aerobic Oxidation Catalysis. ACS Cent. Sci 2015, 1, 234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang N; Liu R; Chen J; Liang X NaNO2-Activated, Iron-TEMPO Catalyst System for Aerobic Alcohol Oxidation Under Mild Conditions. Chem. Commun 2005, 5322–5324. [DOI] [PubMed] [Google Scholar]
- 18.Yin W; Chu C; Lu Q; Tao J; Liang X; Liu R Iron Chloride/4-Acetamido-TEMPO/Sodium Nitrite-Catalyzed Aerobic Oxidation of Primary Alcohols to the Aldehydes. Adv. Synth. Catal 2010, 352, 113–118. [Google Scholar]
- 19.Ma S; Liu J; Li S; Chen B; Cheng J; Kuang J; Liu Y; Wan B; Wang Y; Ye J; Yu Q; Yan W; Yu S Development of a General and Practical Iron Nitrate/TEMPO-Catalyzed Aerobic Oxidation of Alcohols to Aldehydes/Ketones: Catalysis with Table Salt. Adv. Synth. Catal 2011, 353, 1005–1017. [Google Scholar]
- 20.Jiang X; Zhang J; Ma S Iron Catalysis for Room-Temperature Aerobic Oxidation of Alcohols to Carboxylic Acids. J. Am. Chem. Soc 2016, 138, 8344–8347. [DOI] [PubMed] [Google Scholar]
- 21.Wang L; Shang S; Li G; Ren L; Lv Y; Gao S Iron/ABNO-Catalyzed Aerobic Oxidation of Alcohols to Aldehydes and Ketones under Ambient Atmosphere. J. Org. Chem 2016, 81, 2189–2193. [DOI] [PubMed] [Google Scholar]
- 22.Jiang X; Liu J; Ma S Iron-Catalyzed Aerobic Oxidation of Alcohols: Lower Cost and Improved Selectivity. Org. Process Res. Dev 2019, 23, 825–835. [Google Scholar]
- 23.Hong M; Min J; Wu S; Cui H; Zhao Y; Li J; Wang S Metal Nitrate Catalysis for Selective Oxidation of 5-Hydroxymethylfurfural into 2,5-Diformylfuran under Oxygen Atmosphere. ACS Omega 2019, 4, 7054–7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scepaniak JJ; Wright AM; Lewis RA; Wu G; Hayton TW Tuning the Reactivity of TEMPO by Coordination to a Lewis Acid: Isolation and Reactivity of MCl3 (η1-TEMPO) (M = Fe, Al). J. Am. Chem. Soc 2012, 134, 19350–19353. [DOI] [PubMed] [Google Scholar]
- 25.Nguyen T-AD; Wright AM; Page JS; Wu G; Hayton TW Oxidation of Alcohols and Activated Alkanes with Lewis Acid-Activated TEMPO. Inorg. Chem 2014, 53, 11377–11387. [DOI] [PubMed] [Google Scholar]
- 26.See also:Bar-On P; Mohsen M; Zhang R; Feigin E; Chevion M; Samuni A Kinetics of Nitroxide Reaction with Iron(II). J. Am. Chem. Soc 1999, 121, 8070–8073. [Google Scholar]
- 27.Rafiee M; Miles KC; Stahl SS Electrocatalytic Alcohol Oxidation with TEMPO and Bicyclic Nitroxyl Derivatives: Driving Force Trumps Steric Effects. J. Am. Chem. Soc 2015, 137, 14751–14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The conditions resemble those used for aerobic alcohol oxidation with a TEMPO/HCl/HNO3 cocatalyst system: Rahimi A; Azarpira A; Kim H; Ralph J; Stahl SS Chemoselective Metal-Free Aerobic Alcohol Oxidation in Lignin. J. Am. Chem. Soc 2013, 135, 6415–6418.
- 29.Kishioka S.-y.; Ohsaka T; Tokuda K Electrochemical Studies of Acid-Promoted Disproportionation of Nitroxyl Radical. Electrochim. Acta 2003, 48, 1589–1594. [Google Scholar]
- 30.The pKa of HCl in CH3CN is estimated to be 10.6: Kütt A; Rodima T; Saame J; Raamat E; Mäemets V; Kaljurand I; Koppel IA; Garlyauskayte R. Yu.; Yagupolskii YL; Yagupolskii LM; Bernhardt E; Willner H; Leito I Equilibrium Acidities of Superacids. J. Org. Chem 2011, 76, 391–395.
- 31.Gerken JB; Pang YQ; Lauber MB; Stahl SS Structural Effects on the pH-Dependent Redox Properties of Organic Nitroxyls: Pourbaix Diagrams for TEMPO, ABNO, and Three TEMPO Analogs. J. Org. Chem 2018, 83, 7323–7330. [DOI] [PubMed] [Google Scholar]
- 32.Walroth RC; Miles KC; Lukens JT; MacMillan SM; Stahl SS; Lancaster KM Electronic Structural Analysis of Copper(II)–TEMPO/ABNO Complexes Provides Evidence for Copper(I)–Oxoammonium Character. J. Am. Chem. Soc 2017, 139, 1350713517. [DOI] [PubMed] [Google Scholar]
- 33.Wright AM; Page JS; Scepaniak JJ; Wu G; Hayton TW Divergent Reactivity of TEMPO with MBr3 (M = B, Al). Eur. J. Inorg. Chem 2013, 3817–3820. [Google Scholar]
- 34.Epstein IR; Kustin K; Warshaw LJ A Kinetics Study of the Oxidation of Iron(II) by Nitric Acid. J. Am. Chem. Soc 1980, 102, 3751–3758. [Google Scholar]
- 35.Hoover JM; Stahl SS Highly Practical Copper(I)/TEMPO Catalyst System for Chemoselective Aerobic Oxidation of Primary Alcohols. J. Am. Chem. Soc 2011, 133, 16901–16910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lauber MB; Stahl SS Efficient Aerobic Oxidation of Secondary Alcohols at Ambient Temperature with an ABNO/NOx Catalyst System. ACS Catal. 2013, 3, 2612–2616. [Google Scholar]
- 37.Golubev VA; Borislavskii VN; Aleksandrov AL Mechanism of Oxidation of Primary and Secondary Alcohols by Oxopiperidinium Salts. Russ. Chem. Bull 1977, 26, 1874–1881. [Google Scholar]
- 38.Bailey WF; Bobbitt JM; Wiberg KB Mechanism of the Oxidation of Alcohols by Oxoammonium Cations. J. Org. Chem 2007, 72, 4504–4509. [DOI] [PubMed] [Google Scholar]
- 39.Salazar CA; Thompson BJ; Knapp SMM; Myers SR; Stahl SS Multichannel Gas-Uptake/Evolution Reactor for Monitoring Liquid-Phase Chemical Reaction. Rev. Sci. Instrum 2021, 92, 044103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumpulainen ETT; Koskinen AMP Catalytic Activity Dependency on Catalyst Components in Aerobic Copper-TEMPO Oxidation. Chem. Eur. J 2009, 15, 10901–10911. [DOI] [PubMed] [Google Scholar]
- 41.Sasano Y; Nagasawa S; Yamazaki M; Shibuya M; Park J; Iwabuchi Y Highly Chemoselective Aerobic Oxidation of Amino Alcohols into Amino Carbonyl Compounds. Angew. Chem. Int. Ed 2014, 53, 3236–3240. [DOI] [PubMed] [Google Scholar]
- 42.Sasano Y; Kogure N; Nagasawa S; Kasabata K; Iwabuchi Y 2-Azaadamantane N-oxyl (AZADO)/Cu Catalysis Enables Chemoselective Aerobic Oxidation of Alcohols Containing Electron-Rich Divalent Sulfur Functionalities. Org. Lett 2018, 20, 6104–6107. [DOI] [PubMed] [Google Scholar]
- 43.Zhai D; Ma S Copper Catalysis for Highly Selective Aerobic Oxidation of Alcohols to Aldehydes/Ketones. Org. Chem. Front 2019, 6, 3101–3106. [Google Scholar]
- 44.Lagerblom K; Wrigstedt P; Keskiväli J; Parviainen A; Repo T Iron-Catalyzed Selective Aerobic Oxidation of Alcohols to Carbonyl and Carboxylic Compounds. Chem. Eur. J 2016, 81, 1160–1165. [DOI] [PubMed] [Google Scholar]
- 45.Preliminary studies provide support for this hypothesis, as elaborated in section 9 of the Supporting Information. For example, the Cu(NO3)2/TEMPO system exhibits selectivity more akin to Fe(NO3)3/TEMPO than Cu/bpy/TEMPO. The Fe(NO3)3/bpy/TEMPO system does not show the same 1°/2° alcohol selectivity as other NOx-based catalyst systems, but it should be noted that oxoammonium reagents can favor 1° over 2° alcohol oxidation under certain conditions (see ref. 2). These observations warrant further investigation, as they suggest NOx-based catalyst systems could switch product selectivity, even with the same active species.
- 46.Steves JE; Preger Y; Martinelli JR; Welch CJ; Root TW; Hawkins JM; Stahl SS Process Development of CuI/ABNO/NMI-Catalyzed Aerobic Alcohol Oxidation. Org. Process Res. Dev 2015, 19, 1548–1553 [Google Scholar]
- 47.Ortiz A; Soumeillant M; Savage SA; Strotman NA; Haley M; Benkovics T; Nye J; Xu Z; Tan Y; Ayers S; Gao Q; Kiau S Synthesis of HIV-Maturation Inhibitor BMS-955176 from Betulin by an Enabling Oxidation Strategy. J. Org. Chem 2017, 82, 4958–4963. [DOI] [PubMed] [Google Scholar]
- 48.Ochen A; Whitten R; Aylott HE; Ruffell K; Williams GD; Slater F; Roberts A; Evans P; Steves JE; Sanganee MJ Development of a Large-Scale Copper(I)/TEMPO-Catalyzed Aerobic Alcohol Oxidation for the Synthesis of LSD1 Inhibitor GSK2879552. Organometallics 2019, 38, 176–184. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.