

Abstract
Background:
The self-assembly of metabolic enzymes into filaments or foci highlights an intriguing mechanism for the regulation of metabolic activity. Recently, we identified the conserved polymerization of phosphoribosyl pyrophosphate synthetase (PRPS), which catalyzes the first step in purine nucleotide synthesis, in yeast and cultured mammalian cells. While previous work has revealed that loss of PRPS activity regulates retinal development in zebrafish, the extent to which PRPS filament formation affects tissue development remains unknown.
Results:
By generating novel alleles in the zebrafish PRPS paralogs, prps1a and prps1b, we gained new insight into the role of PRPS filaments during eye development. We found that mutations in prps1a alone are sufficient to generate abnormally small eyes along with defects in head size, pigmentation, and swim bladder inflation. Furthermore, a loss-of-function mutation that truncates the Prps1a protein resulted in the failure of PRPS filament assembly. Lastly, in mutants that fail to assemble PRPS filaments, we observed disorganization of the actin network in the lens fibers.
Conclusions:
The truncation of Prps1a blocked PRPS filament formation and resulted in a disorganized lens fiber actin network. Altogether, these findings highlight a potential role for PRPS filaments during lens fiber organization in zebrafish.
Keywords: metabolic enzyme filaments, retina, PRPS, lens fiber, actin
INTRODUCTION
One of the difficulties in studying inborn errors of metabolism has been that defects in different steps of a common pathway can lead to a wide variety of highly specific phenotypes. Historically, this has been interpreted as being due to specific cell types having a particular sensitivity to disruptions in a given pathway. However, over the past decade, metabolic enzymes across a variety of species have been found to assemble into higher order structures (i.e. filaments or foci) in the cytoplasm (An et al., 2008; Narayanaswamy et al., 2009; Noree et al., 2010; Shen et al., 2016). Interestingly, mutations in a subset of these assembling metabolic enzymes can result in various genetic disorders (Alli et al., 2008; Fitzmaurice et al., 2013; Sircar et al., 2012; Tong and Harwood, 2006). This has raised the possibility that the highly specific phenotypes associated with disrupting distinct steps in a common metabolic pathway might be connected to the absence of these metabolic structures.
Extensive genetic and biochemical studies have focused on establishing a connection between metabolic enzyme assembly and regulation of enzymatic activity. For instance, both E. coli and S. cerevisiae CTP synthetases form filaments in response to end product inhibition by CTP (Barry et al., 2014; Noree et al., 2014). In contrast, both mammalian acetyl-CoA carboxylase and phosphofructokinase polymerize in response to increased enzyme activity (Hunkeler et al., 2018; Webb et al., 2017). Furthermore, enzymes that act in consecutive steps of a pathway can co-assemble into large multi-enzyme clusters (An et al., 2008). For example, enzymes in the de novo purine biosynthesis pathway in cultured mammalian cells aggregate together in response to purine deprivation to promote substrate channeling and increase metabolic flux. Additionally, metabolic enzyme polymerization is a clinically relevant drug target. Mycophenolic acid, for example, is an immunosuppressant used in organ transplants that promotes IMPDH polymerization (Carcamo et al., 2011). Thus, formation of higher order structures can act as a mechanism to regulate flux through a particular metabolic pathway and can serve as a pharmaceutical target. However, there have been few attempts to link metabolic enzyme structures to inborn errors of metabolism. To date, the primary example of such a connection is that mutations in IMPDH associated with autosomal dominant retinitis pigmentosa have been shown to suppress IMPDH polymerization (Thomas et al., 2012).
Recently, we found that phosphoribosyl pyrophosphate (PRPP) synthetase (PRPS), which catalyzes the conversion of ribose-5-phosphate to PRPP, forms filaments in yeast, Drosophila oocytes, rat hippocampal neurons, and human fibroblasts (Noree et al., 2019). Similar to other self-assembling metabolic enzymes, PRPS assembly is linked to substrate availability and associated with inactive form of the PRPS in yeast (Noree et al., 2019). Additionally, the disease phenotypes associated with loss-of-function (LoF) and gain-of-function mutations in PRPS have long been perplexing and suggested that it would be an excellent candidate for investigating the role of enzyme polymerization in tissue development and function. For instance, LoF mutations in PRPP synthetase that reduce enzymatic activity cause sensorineural deafness, optic atrophy, ataxia, and, in severe cases, intellectual disability (Arts et al., 1993; Kim et al., 2007; Zoref et al., 1975). On the other hand, gain-of-function mutations that result in PRPS overexpression result in gout due to the overproduction of purines and increased PRPS enzymatic activity (Becker et al., 1982; Zoref et al., 1975). However, mutations that disrupt feedback inhibition of PRPP synthetase, which result in enhanced enzymatic activity, cause a combination of phenotypes reminiscent of what is seen in both gain- and loss-of-function scenarios: the sensorineural deafness of LoF mutants and the gout associated with increased enzyme activity (Ahmed et al., 1999; Becker et al., 1988, 1980; Zoref et al., 1975). The fact that “superactivity” and LoF mutations cause similar symptoms regardless of the differences in enzyme misregulation suggests that defects in PRPS polymerization could underlie these phenotypes.
Given the rarity of PRPS disorders, few animal models of PRPS LoF syndromes exist. In one example, Pei and colleagues found that, while zebrafish have two PRPS paralogs (prps1a and prps1b), loss of maternal and zygotic function of prps1a yielded embryos with smaller eyes and reduced hair cell number (Pei et al., 2016). These defects are consistent with the optic atrophy and hearing impairment observed in human patients. Additionally, these phenotypes were exacerbated in prps1a;prps1b double mutants, which also exhibited abnormal motor neuron development and hair cell innervation. These developmental perturbations were proposed to be due to cell cycle delays as a consequence of reduced nucleotide synthesis. In another example in zebrafish, morpholino knockdown of prps1a and prps1b resulted in sensorineural hearing loss as a result of reduced otic vesicles, otoliths, and inner ear hair cells (DeSmidt et al., 2019). While these genetic analyses of the PRPS paralogs suggested roles for PRPS activity during embryogenesis, they did not clarify whether PRPS filaments exist in zebrafish and whether alterations in PRPS polymerization would cause particular perturbations of tissue development.
In this report, we examine the relationship between PRPS polymerization and retinal development in zebrafish. We have found that PRPS forms filaments in a region adjacent to the retinal pigmented epithelium. To address the relationship between PRPS filaments and eye development, we generated two new LoF alleles in prps1a and one new LoF allele in prps1b. Loss of prps1a alone was sufficient to generate a small eye phenotype similar to that previously reported in prps1a;prps1b double mutants, whereas our prps1b mutants failed to exhibit any developmental abnormalities. In addition to a small eye phenotype, prps1a mutants displayed defects in head size, pigmentation, and swim bladder inflation not observed with previous prps1a or prps1b alleles (Pei et al., 2016). While both of our prps1a mutants resulted in smaller eyes, only the mutation predicted to truncate the Prps1a protein sequence failed to assemble PRPS filaments and also exhibited a disorganized actin network in the lens fibers. This observation represents a potential relationship between PRPS polymerization and actin network organization in the lens and suggests a possible developmental consequence of defects in metabolic enzyme polymerization. Taken together, these results highlight the first connection between PRPS filament formation and lens fiber development.
RESULTS
PRPS filaments assemble in the layer surrounding the retinal pigmented epithelium
Since previous studies of PRPS revealed its importance for eye development in zebrafish (Pei et al., 2016), we examined if PRPS assembled into filaments in the retina. Given that PRPS polymerization was identified using an antibody raised against the human ortholog of PRPS (hPRPS1) (Noree et al., 2019), we first performed BLAST analysis to compare hPRPS1 to both zebrafish paralogs, Prps1a and Prps1b. This analysis revealed that Prps1a and Prps1b have a ~92% and ~93% identity with hPRPS1 (Figure 1A), respectively, suggesting that our antibody would be likely to recognize these zebrafish PRPS proteins. Considering that prps1a and prps1b are both expressed in the retina from 36 hours post fertilization (hpf) onward (Pei et al., 2016), we chose to perform whole-mount immunofluorescence at 2 and 5 days post fertilization (dpf) to look for polymerization of PRPS in the eye. At 2 dpf, PRPS staining appeared diffuse in the cytoplasm of cells in the outer layer of the eye (Figure 1B). However, at 5 dpf, we observed robust filament formation of PRPS in the eyes of all embryos examined (Figure 1B). Despite this high frequency of detecting PRPS filaments, we found that the length and number of filaments varied among embryos, even among siblings (Figure 1C and 1D).
Figure 1. PRPS forms filaments in the retina at 5 dpf.

(A) Protein sequence alignment (UniProt) of human PRPP Synthetase (P60891) to zebrafish PRPP Synthetase paralogs Prps1a (Q4KME9) and Prps1b (Q08CA5). Human PRPS1 has ~96% identity to both zebrafish paralogs.
(B) Antibody raised against human PRPS1 recognizes filaments at 5 days post fertilization (dpf) in the zebrafish eye. Representative maximum intensity projections of confocal images depict left eyes at 2 and 5 dpf stained for PRPS; rostral is to the left and dorsal is to the top. We note that we have never detected filament-like staining in the wild-type eye using the goat anti-rabbit secondary antibody in the absence of the anti-PRPS antiserum. Inserts in bottom left corners show zoomed views of boxed areas. Scale bars: 50 μm. (n = 12 embryos for each stage)
(C) Graph plots the average length of the PRPS filaments observed in each eye examined at 5 dpf. Red line indicates the median value.
(D) Graph plots the number of PRPS filaments found in each eye examined at 5 dpf. Red line indicates the median value.
Since prior work had established that prps1a mutants and prps1a;prps1b double mutants exhibit decreased eye pigmentation (Pei et al., 2016), we investigated whether PRPS filaments localized to the retinal pigmented epithelium (RPE) layer by examining the relationship between PRPS staining and the localization of the RPE marker Zpr2 (Yazulla and Studholme, 2001). However, PRPS filaments were not evident in the RPE, but instead appeared to localize to an adjacent layer (Figure 2A). At this stage, this layer contains neural crest-derived iridophores and melanophores; later, the choroid and sclera will form in this region. Punctate PRPS staining was also observed in what appears to be the photoreceptor cell layer (PCL).
Figure 2. PRPS cytoplasmic filaments assemble in cells adjacent to the RPE.

(A) Single optical slice from a confocal z-stack shows a representative example of a 5 dpf eye stained for PRPS (green), Zpr2 (retinal pigmented epithelium (RPE) marker) (red), and DAPI (blue) and depicts PRPS filament formation in the cytoplasm of cells adjacent to the RPE layer. Rostral is to the left and dorsal is to the top. Inserts in bottom right corners show zoomed views of boxed areas. Scale bar: 40 μm for larger images, 3 μm for inserts (n = 6 embryos)
(B) Representative maximum intensity projections of confocal images of a single cell, dissected from retinal tissue and stained for PRPS (green) and DAPI (blue), depict cytoplasmic localization of PRPS filaments. Retinas were dissected, subjected to trypsin treatment, and plated before being fixed and immunostained. Arrow indicates PRPS filament. Scale bar: 10 μm. (n = 3 eye dissections; ~30% of the dissociated cells exhibited PRPS filaments)
In contrast to our observations in Drosophila, rat hippocampal neurons, and human fibroblasts (Noree et al., 2019), PRPS filaments in the zebrafish eye appeared to localize to the cytoplasm instead of to the nucleus (Figure 2A). We attempted to validate the cytoplasmic localization of PRPS filaments using cryosections, but we were unable to detect PRPS signal despite trying multiple antigen retrieval protocols. We therefore chose to dissect eyes at 5 dpf, followed by trypsinization to obtain cells from the outer layers of the retina. These dissociated cells were then plated, fixed, and immunostained for PRPS. Consistent with our other immunostaining, these cells displayed PRPS filaments in the cytoplasm (Figure 2B). We acknowledge that the dissection process could potentially alter the metabolic state of the dissociated cells; however, we think that it would be unlikely for our protocol to cause the disassembly of PRPS filaments, since nutrient starvation can trigger and/or maintain filament assembly in yeast (Noree et al., 2019). Altogether, the identification of PRPS filaments in a particular region of the zebrafish eye suggests the possibility that this territory has a distinct metabolic state and provides a foundation for examining the relationship between PRPS polymerization and eye development.
Loss-of-function of prps1a alone is sufficient to decrease eye size
In order to assess the role of PRPS filaments in eye development, we utilized CRISPR-Cas9 genome editing techniques to generate LoF mutants in both prps1a and prps1b. Two different guide RNAs (gRNAs) were used to target either exon 1 or exon 5 of the prps1a genomic locus, in addition to one gRNA that targets exon 2 in prps1b (Figure 3A). For prps1a, we identified two alleles that resulted in different changes in the predicted protein sequence. The prps1asd59 mutation caused a 1 bp deletion in exon 1 that introduced a frameshift and resulted in an early termination codon (Figure 3A). This mutation is predicted to truncate the Prps1a protein from 320 amino acids to 41 amino acids, thus eliminating nearly all of the protein, including residues required for oligomerization and enzymatic activity (Li et al., 2007). Our second prps1a allele, prps1asd60, had a 21 bp in-frame deletion in exon 5 resulting in a 7 amino acid deletion (196 aa – 202 aa) (Figure 3A and 3C). These deleted residues lie within a structural pocket involved in binding of PRPS substrates (ATP and ribose-5-phosphate), both of which are required for enzymatic activity (Li et al., 2007). For prps1b, our prps1bsd61 mutation introduced a 23 bp insertion that caused a frameshift and resulted in an early stop codon (Figure 3B and 3C). This mutation is predicted to truncate the Prps1b protein from 318 amino acids to 82 amino acids.
Figure 3. Generation and genotyping of prps1a and prps1b mutations.

(A) Schematic of gRNAs targeting genomic loci of prps1a along with changes in genomic and predicted protein sequences. Green arrows indicate gRNA target sites. Blue text depicts the sequence before the PAM recognition sequence (red). Premature stop codon is shown as an asterisk. Depicted protein sequences for Prps1a display amino acids 11 – 41 (prps1asd59) and 186 – 216 (prps1asd60).
(B) Schematic of gRNAs targeting genomic loci of prps1b along with changes in genomic and predicted protein sequences. Green arrows indicate gRNA target sites. Blue text depicts the sequence before the PAM recognition sequence (red). Premature stop codon is shown as an asterisk. Depicted protein sequence for Prps1b displays amino acids 61 – 84.
(C) Representative agarose gel images depict genotyping for prps1a and prps1b mutations. prps1asd60 and prps1bsd61 mutations can be identified by changes in PCR product size. The prsp1asd59 mutation eliminates a MboII restriction site in the PCR product. MT indicates homozygous mutants.
While heterozygotes for our prps1a and prps1b mutations showed no distinguishable phenotypes (data not shown), homozygotes for either prps1a mutation resulted in reduced retina size at both 2 and 5 dpf (Figure 4A and 4B). Reminiscent of LoF mutations in downstream purine biosynthesis enzymes gart and paics (Ng et al., 2009), both prps1a mutants also displayed decreased yellow xanthophore-derived pigmentation (Figure 4B). Additionally, both prps1a mutants exhibited small heads and reduced swim bladder inflation by 5 dpf (Figure 4B). Notably, only a few homozygous prps1a mutants (<5% of those examined) were able to recover from the swim bladder defect and reach adulthood. However, we were unable to breed any of these fish. Homozygotes for the prps1b mutation showed no evident morphological defects at either 2 or 5 dpf (Figure 4A and 4B). Furthermore, we found no additional or more severe phenotypes in prps1asd59;prps1bsd61 double homozygotes (Figure 4C).
Figure 4. Loss of prps1a, not prps1b, generates eye, pigmentation, head, and swim bladder phenotypes.

(A) Representative bright-field images of wild-type, prps1a mutant, and prps1b mutant embryos at 2 dpf. Reduced eye size is observed in both prps1a mutants, but not in prps1b mutants. (n = 16 embryos for each genotype)
(B) Representative bright-field images of wild-type, prps1a mutant, and prps1b mutant embryos at 5 dpf. Reduced eye and head size, reduced swim bladder inflation, and reduced xanthophore-derived yellow pigmentation are observed in prps1a mutants, but not in prps1b mutants. (n = 16 embryos for each genotype)
(C) Representative bright-field images of wild-type, prps1asd59 mutant, and prps1asd59; prps1bsd61 double mutant embryos at 2 dpf and 5 dpf. No additional phenotypes are observed in the double mutants compared to prps1asd59 mutants alone. (n = 12 embryos for each genotype and stage)
Our results suggest that loss of zygotic prps1a function alone is sufficient to generate severe developmental defects. While prior work from Pei and colleagues revealed that loss of zygotic prps1a function could result in slightly smaller eye size, they observed severe eye size defects only in either maternal-zygotic prps1a mutants or in prps1a;prps1b double mutants (Pei et al., 2016). In contrast, we found a dramatic reduction in eye size in zygotic homozygotes for both of our prps1a mutant alleles. Consistent with the notion that our prps1a alleles are particularly potent, defects in head size and swim bladder inflation were not reported in the previously studied prps1a mutants (Pei et al., 2016), but were observed in our prps1a mutants. We suspect that this discrepancy in potency between prps1a alleles relates to the fact that our prps1a alleles are predicted to alter the protein sequence, whereas the prps1ala015591 mutation (Pei et al., 2016) is the consequence of a retroviral insertion that reduces transcript levels of prps1a. Together, these results suggest a likely role for prps1a as the primary contributor to PRPS activity during embryonic development in zebrafish.
Truncation of prps1a blocks PRPS polymerization
Next, we sought to determine if any of our prps1 LoF alleles disrupted PRPS filament formation in the eye. We raised wild-type, prps1a mutant, and prps1b mutant embryos to 5 dpf and performed whole-mount immunofluorescence to examine PRPS polymerization. While PRPS filament assembly was relatively unaffected in prps1asd60 and prps1bsd61 mutants, we found that prps1asd59 mutants failed to assemble PRPS polymers (Figure 5A–5C). Interestingly, prps1asd60 mutants exhibited some PRPS signal that was not incorporated into discrete filaments (Figure 5A). While these mutants were still capable of forming filaments, the prps1asd60 mutation may reduce partitioning of monomeric PRPS into its filamentous form and affect its localization and patterning. Additionally, we note that the filaments in the prps1asd60 allele appear to be thinner than the filaments found in the wild-type prps1a and prps1bsd61 alleles. However, it’s unclear whether this change in morphology plays a relevant role in PRPS biology or retinal development.
Figure 5. Truncation of prps1a prevents PRPS filament formation.

(A) Representative maximum intensity projections of confocal images from wild-type, prps1a mutant, and prps1b mutant eyes at 5 dpf, stained for PRPS. Rostral is to the left and dorsal is to the top; zoomed views of boxed areas are displayed below each panel. Fractions indicate the number of embryos exhibiting PRPS filaments out of the number of embryos examined per genotype. Only prps1asd59 mutants failed to form PRPS filaments in the eye. Scale bar: 40 μm for larger images, 3 μm for magnified images.
(B) Graph plots the average length of the PRPS filaments observed in each wild-type, prps1a mutant, and prps1b mutant eye at 5 dpf. Red lines indicate median values. (ns = no statistically significant difference between data sets)
(C) Graph plots the number of PRPS filaments found in each wild-type, prps1a mutant, and prps1b mutant eye at 5 dpf. Red lines indicate median values. (ns = not significant)
These data argue that, while both prps1a alleles likely decrease enzymatic activity, as suggested by the nature of each mutation (Figure 3A) and the reduction of eye size in mutant embryos (Figure 4A and 4B), only a severe truncation of the Prps1a protein was able to disrupt PRPS polymerization. Together with the fact that the prps1asd59 mutation removes key residues required for both oligomerization and substrate-binding (Li et al., 2007), these results suggest that prps1asd59 is a strong loss-of-function mutation. In contrast, while enzymatic activity is likely to be reduced in prps1asd60 mutants (Li et al., 2007), this mutation does not remove residues required for oligomerization, suggesting that prps1asd60 is a less potent, hypomorphic allele. Since oligomerization regulates polymerization of other metabolic filaments (Lynch et al., 2017; Noree et al., 2014; Petrovska et al., 2014), it is reasonable to imagine that the difference in potential for oligomerization contributes to the presence of the PRPS polymerization defect in prps1asd59 mutants, but not in prps1asd60 mutants.
We were also intrigued to find that only prps1a seems to be required for filament formation, as indicated by the formation of PRPS filaments in prps1bsd61 mutants (Figure 5A–5C). This surprised us, especially considering the severity of the truncation of the Prps1b protein predicted to result from the prps1bsd61 mutation (Figure 3B). Given that both prps1a and prps1b are similarly expressed in the retina (Pei et al., 2016), both proteins should, in principle, be available to participate in PRPS filament formation. Yet, despite the similar sequences and expression patterns of prps1a and prps1b, it seems that Prps1b does not participate in polymerization, at least not in the absence of Prps1a, as in prps1asd59 mutants. Together, these results suggest that prps1a is the major contributor to PRPS filament assembly in the developing retina.
prps1a mutants that lack PRPS filaments exhibit disrupted lens fiber organization
Since both LoF and feedback-resistant mutations in hPRPS1 cause sensorineural deafness (Ahmed et al., 1999; Arts et al., 1993; Becker et al., 1988, 1980; Kim et al., 2007; Zoref et al., 1975), we sought to determine if defects in PRPS activity and/or polymerization could provide a link to hearing loss. Mutations in a variety of actin and actin-binding proteins disrupt actin network dynamics and organization and also disrupt auditory system function (Drummond et al., 2012). Although we did not observe PRPS filaments in hair cells of the inner ear or lateral line (data not shown), we explored whether changes in PRPS activity and/or filament formation disrupted the actin network in the developing retina. In wild-type embryos at 5 dpf, phalloidin staining revealed a robust, well-organized array of actin cables in the lens fibers at the center of the eye (Figure 6A). These actin cables appear to be arranged into regularly spaced radial and coaxial spokes; consistent with an organized network, we found that the angles between adjacent radial actin cables had similar values with a low standard deviation (Figure 6C–6E). In contrast, the actin array in lens fibers was disrupted in prps1asd59 embryos yielding irregularly positioned radial and coaxial spokes (Figure 6A). Notably, the angles between adjacent radial cables varied widely within each individual prps1asd59 mutant lens (Figure 6C–6E). Additionally, an accumulation of actin was present at the middle of the lens in prps1asd59 mutants (Figure 6A), and the lens diameter was smaller in prps1asd59 mutants than in wild-type embryos (average lens diameter: 111.1 ± 1.2 μm in wild-type, 94.7 ± 0.7 μm in prps1asd59 mutants; p<0.0001). Defects in actin network organization were observed only in prps1asd59 mutants: although prps1asd60 mutants also exhibited a small lens diameter (95.8 ± 0.7 μm, p<0.0001 in comparison to wild-type), their pattern of phalloidin staining appeared indistinguishable from that found in wild-type (Figure 6A, 6D, and 6E). Furthermore, both lens size (108.6 ± 1.7 μm) and actin network organization appeared normal in prps1bsd61 mutants (Figure 6A, 6D, and 6E). Together, our results suggest that loss of PRPS enzymatic activity alone is not sufficient to generate defects in lens fiber organization and that failure to assemble PRPS filaments contributes to this phenotype.
Figure 6. Loss of PRPS filaments results in lens fiber disorganization.

(A) Representative maximum intensity projections of confocal images from wild-type, prps1a mutant, and prps1b mutant eyes at 5 dpf, stained with PRPS and rhodamine phalloidin. Rostral is to the left and dorsal is to the top; zoomed views of boxed areas from the first column are displayed in the following columns. Fractions indicate the number of embryos exhibiting a regularly-spaced, lattice-like actin array in the lens out of the number of embryos examined per genotype. While both prps1a mutants displayed abnormal PRPS signal in the lens, only prps1asd59 mutants exhibited a disorganized actin array in the lens fibers. Scale bar: 40 μm for larger images, 20 μm for magnified images.
(B) Representative maximum intensity projections of confocal images from wild-type and prps1a mutant eyes at 2 dpf, stained with rhodamine phalloidin. Rostral is to the left and dorsal is to the top; zoomed views of boxed areas are displayed below each panel. Arrows indicate lens fibers. Fractions indicate the number of embryos exhibiting a honeycomb-like actin array in the lens out of the number of embryos examined per genotype. No defects were observed in prps1asd59 mutants at 2 dpf. Scale bar: 40 μm for larger images, 10 μm for magnified images.
(C) Graph plots the angles between adjacent radial actin cables in the lens fibers of one wild-type and one prps1asd59 eye at 5 dpf. Red lines indicate median values. Although the distribution of angle sizes is much broader in the prps1asd59 eye, there is not a statistically significant difference between these data sets. (ns = not significant)
(D) Graph plots the average angle size observed in each wild-type, prps1a mutant, and prps1b mutant eye at 5 dpf. Red lines indicate median values. Average angle size is similar for all genotypes. (ns = not significant)
(E) Graph plots the standard deviation of the actin cable angles measured in each wild-type, prps1a mutant, and prps1b mutant eye at 5 dpf. Red lines indicate median values. Wild-type, prps1asd60, and prps1bsd61 embryos displayed small standard deviations in angle size, whereas prps1asd59 embryos had much higher standard deviation values, indicating the relative disorganization of the actin cable array in prps1asd59 mutants. (**** signifies p<0.0001, ns = not significant)
The observed disorganization of the actin network in prps1asd59 mutants at 5 dpf seems to represent difficulties with maturation and/or maintenance of the lens fibers, rather than defects in initial lens fiber formation or morphogenesis, as the honeycomb-like actin array appears unaffected in prps1asd59 mutants at 2 dpf (Figure 6B). It is interesting to consider that this lens phenotype emerges by 5 dpf, temporally coincident with our observations of PRPS filament assembly in other portions of the eye (Figures 1B and 2A). Although we did not detect PRPS staining in the lens of wild-type embryos or prps1bsd61 mutants at 5 dpf (Figure 6A), we found aberrant PRPS localization in the lens of both prps1a mutants at this stage. Specifically, we detected diffuse PRPS signal in the lens of prps1asd59 mutants (Figure 6A; seen in 10/10 eyes examined), and we detected punctate PRPS staining in the lens of prps1asd60 mutants (Figure 6A; seen in 7/10 eyes examined). These abnormal patterns of PRPS localization in prps1a mutants suggest the possibility that PRPS filaments may form in the wild-type lens at a stage that we have not examined.
Cell polarity and retinal lamination are not affected in prps1asd59 mutants
Since mutations in genes that are required for retinal cell polarity and lamination, such as cdh2, prkci, and epb41l5 (Horne-Badovinac et al., 2001; Jensen and Westerfield, 2004; Masai et al., 2003), are known to cause a small eye phenotype in zebrafish, we next examined whether prps1 mutations had broader effects on the cytoskeleton throughout the eye. To test if cell polarity was disrupted in prps1asd59 mutants, we examined the localization of the tight junction marker ZO-1 in cryosections of eyes at 5 dpf. We found that ZO-1 appears to be localized to the outer limiting membrane in both wild-type and prps1asd59 mutant embryos (Figure 7A), arguing that the prps1asd59 mutation does not disrupt cell polarity (Krock and Perkins, 2014). Next, we examined if the prps1asd59 mutation disrupted retinal patterning. Phalloidin staining of both wild-type and prps1asd59 mutant embryos at 5 dpf revealed a similar retinal layer organization (Figure 7B). Thus, the lens fiber defects observed in prps1asd59 embryos do not appear to be accompanied by broader abnormalities in cell polarity or retinal lamination.
Figure 7. Cell polarity and retinal lamination are unaffected in prps1asd59 mutants.
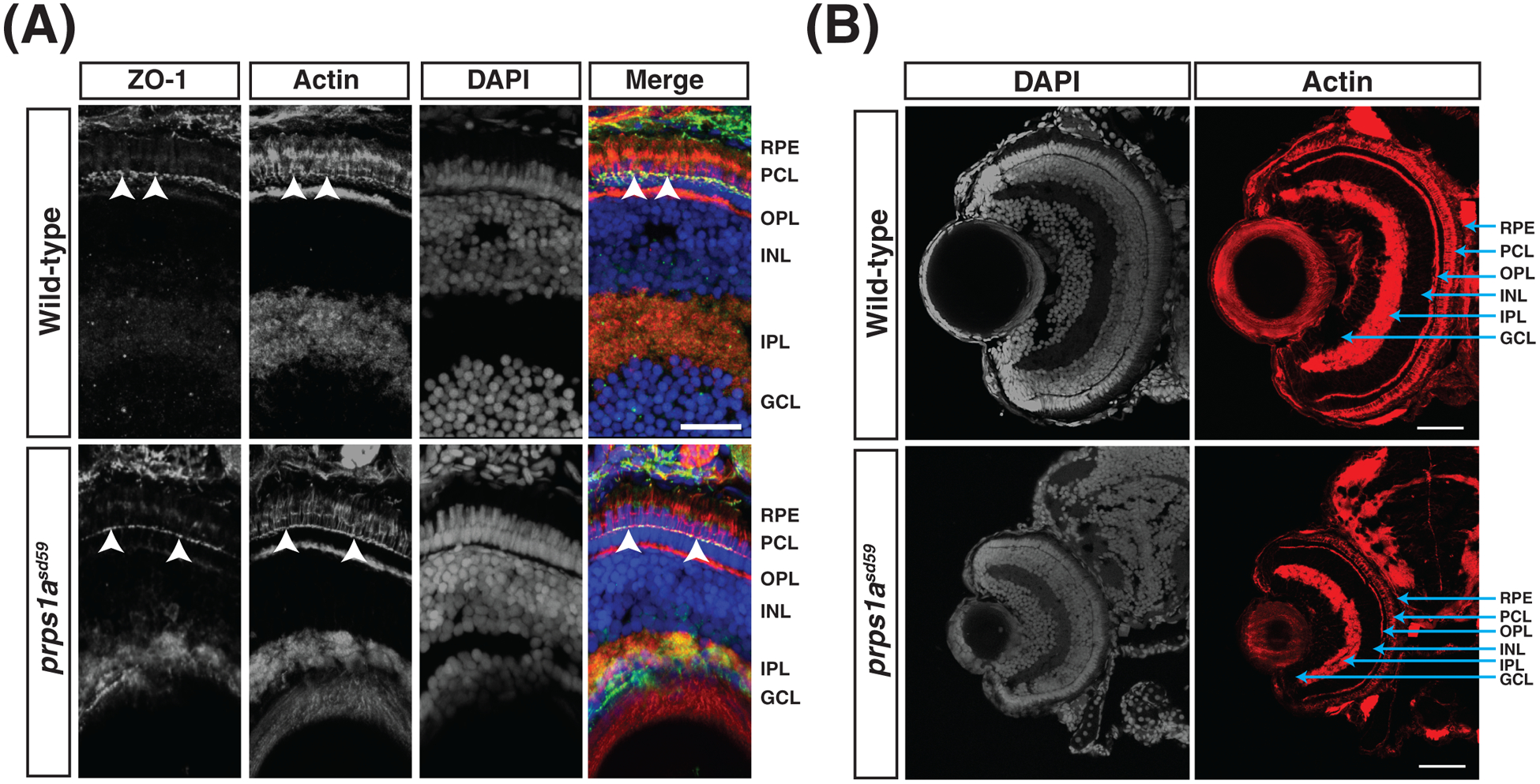
(A) Representative maximum intensity projections of confocal images of transverse cryosections from wild-type and prps1asd59 mutant embryos at 5 dpf, stained for ZO-1, actin, and DAPI. No change in ZO-1 localization is observed in prps1asd59 mutants compared to wild-type. Arrowheads indicate ZO-1 localization to the apical membrane of the photoreceptor cell layer. GCL, ganglion cell layer. IPL, inner plexiform layer. INL, inner nuclear layer. OPL, outer plexiform layer. PCL, photoreceptor cell layer. RPE, retinal pigmented epithelium. Scale bar: 20 μm. (n = 6 embryos for each genotype)
(B) Representative maximum intensity projections of confocal images of transverse cryosections from wild-type and prps1asd59 mutant embryos at 5 dpf, stained for actin and DAPI. Retinal patterning is unaffected in prps1asd59 mutants. Scale bar: 20 μm. (n = 6 embryos for each genotype)
DISCUSSION
Taken together, our studies provide several new insights into the role of PRPS in the developing zebrafish retina. Specifically, we find that disruption of prps1a alone results in a severe reduction in eye size, along with defects in head size, pigmentation, and swim bladder inflation. Furthermore, we find that an early truncation in the Prps1a protein prevents assembly of PRPS filaments in the eye. Interestingly, while both of our prps1a mutations cause small eye and small lens phenotypes, our two alleles differ in their effects on actin organization in the lens, with only the PRPS filament-deficient embryos displaying a disrupted actin network. Thus, our results suggest a potential connection between PRPS filament assembly and lens fiber organization.
Our finding that loss of PRPS activity resulted in a small eye phenotype is consistent with the prior analysis of another PRPS-deficient zebrafish model (Pei et al., 2016). However, it is interesting to note some distinctions between our two studies. Our data indicate a more potent function for zygotic prps1a than was suggested by the previous analysis: whereas the prior study saw a dramatic reduction in eye size only in maternal-zygotic prps1a mutants or in prps1a;prps1b double mutants, we found a comparably severe eye size defect in zygotic prps1a mutants. We suspect that this discrepancy is a consequence of the relative strength of the LoF alleles used in each study. Whereas the previously analyzed prps1ala015591 mutation is a retroviral insertion in the first intron that results in reduced mRNA levels, our prps1asd59 and prps1asd60 mutations are predicted to cause changes in the protein sequence that are likely to significantly diminish Prps1a activity. Consistent with the increased strength of our alleles, we found that prps1a mutants exhibited defects in head size, pigmentation, and swim bladder inflation that were not highlighted in the prior analysis of the prps1ala015591 allele. Since PRPP synthetase is a homohexomeric enzyme in which the active sites are positioned at the subunit interfaces, it is worthwhile to consider whether our prps1a mutations could exert a dominant effect on prps1b function. The prps1asd59 mutation removes the vast majority of the subunit contact sites; thus, it is unlikely that the strength of this allele reflects a dominant negative effect on prps1b. Furthermore, we note that prps1b function does not seem to be able to compensate for the loss of prps1a in our mutants, suggesting that prps1a is the primary paralog contributing to the aspects of zebrafish development that we have examined.
Interestingly, while both prps1asd59 and prps1asd60 mutants exhibited the same small eye and small lens phenotypes, only the mutation that disrupted filament formation, prps1asd59, caused defects in the actin network of the lens fibers. However, further analysis will be necessary to determine precisely when and how lens fiber development goes awry in prps1asd59 mutants. Initial steps in lens fiber formation and morphogenesis appear to proceed relatively normally in prps1asd59 mutants, in contrast to the lens fiber disruptions that are evident by 2 dpf when cell-extracellular matrix interactions are impeded, as in fn1 and itga5 mutants (Hayes et al., 2012). Instead, we suspect that the defects seen in prps1asd59 mutants at 5 dpf reflect an impact of PRPS function on the organization of the actin network during later steps of lens fiber maturation and maintenance, potentially related to the influences of vsp45 and psmd6, which are involved in endocytic trafficking and the ubiquitin proteasome system (Imai et al., 2010; Mochizuki et al., 2018), on lens fiber organization and differentiation. Furthermore, the observation of a lens defect only in prps1asd59 mutants, coincident with the absence of PRPS filaments, suggests the possibility that PRPS filament assembly, and not just PRPS enzymatic activity, is especially crucial during lens fiber development. Alternatively, we cannot rule out that other functions of PRPS, beyond its inherent enzymatic activity and filament assembly, are affected by the prps1asd59 allele and are particularly influential for the lens. For instance, it is possible that lens fiber disorganization is the consequence of a secondary metabolic effect in which altered metabolite levels lead to impaired organelle clearing during lens maturation (Bassnett and Beebe, 1992; Vihtelic et al., 2005; Wride, 2000). Consistent with this idea, mutations in the Drosophila ortholog of PRPS displayed defects in lysosome-mediated and autophagy processes that resulted in shortened lifespan and locomotive defects (Delos Santos et al., 2019). Future mechanistic studies will help to distinguish how PRPS filament assembly or metabolite levels affect lens fiber development. Finally, future studies will be necessary to determine whether the influence of PRPS on lens fiber organization reflects a cell-autonomous role of prps1a within the lens or a cell non-autonomous impact of its function elsewhere in the eye.
Co-assembly of metabolic enzymes into structures, such as the purinosome in purine biosynthesis (An et al., 2008), can be used to regulate flux through a pathway. If the PRPS filament acts a scaffold that regulates the activity of other metabolic enzymes, loss of the PRPS filament could lead to a more severe phenotype than loss of enzyme activity. This possibility has implications for our understanding of the wide array of PRPS-associated disease phenotypes. For example, optic atrophy is a common phenotype in PRPS-linked diseases (de Brouwer et al., 2010; Park et al., 2013). Missense variants of human PRPS1 have also been identified to cause retinal degeneration (Fiorentino et al., 2018). It is possible that the severity of the human disease phenotype might correlate with the impact of a mutation on PRPS filament formation as well as on PRPS enzymatic activity. This possibility might also help to explain how deafness and ataxia result from both LoF and feedback inhibition mutations in hPRPS1 (Ahmed et al., 1999; Arts et al., 1993; Becker et al., 1988, 1980; Kim et al., 2007; Zoref et al., 1975). Future studies examining whether the spectrum of human phenotypes and/or their severity is correlated with the effects of PRPS mutations on filament formation could test this intriguing hypothesis.
EXPERIMENTAL PROCEDURES
Zebrafish
Zebrafish (Danio rerio) were raised and maintained on a 14 h light/10 h dark cycle using standard protocols. The embryos used for these studies were obtained by natural spawning and raised at approximately 28°C with their developmental stage determined in days post-fertilization. All experiments followed Institutional Animal Care and Use Committee-approved protocols.
Generation of PRPS mutants and genotyping
Identification of target sites and generation of gRNAs were conducted according to a previously established protocol (Gagnon et al., 2014). In brief, an online webtool, CHOPCHOP, was used to identify target sites for CRISPR/Cas9-mediated mutagenesis. DNA templates for gRNAs were created using a cloning-independent protocol with oligonucleotides from IDT Technologies. gRNAs (prps1a exon 1: CGCATCCGGATCTGTCGCAGAAGATCGCGGACCG, prps1a exon 5: CATAAGGAGAGGAAGAAGGCCAATGAGGTGGAT, prps1b exon 2: ATCTGCCACTAGAGTCACTGCAGTCATCCCATGC) were transcribed using the MEGAshortscript T7 transcription kit and cleaned up according to the manufacturer’s protocol. gRNAs were stored at −20°C until use.
gRNAs were incubated with EnGen Spy Cas9 NLS protein (New England BioLabs) for 5 minutes at room temperature and placed on ice until ready for injection. All injections were performed in the wild-type strain AB. Injected embryos were raised to adulthood to generate potential founder fish (F0). F0 fish were outcrossed to create the F1 generation and germline transmission was determined by amplifying the genomic locus flanking the target loci via PCR using KOD Hot Start Polymerase. PCR products were purified and sequenced (Eton Sequencing) to identify secondary peaks in the resulting chromatogram. Next, adult F1 progeny were fin clipped to obtain genomic DNA. Fish were genotyped by performing TA cloning and sequencing to ensure clean sequencing reads. Heterozygous mutant fish were kept for further breeding. Both F2 and F3 embryos were used for all experiments.
For prps1asd59 mutants, the primer pair 5’-CTGTACGCCACCCCGACACTATAGG-3’ and 5’-GAGTCCGCTGCCTGTGAGACTCG-3’ amplified a 329 bp PCR product flanking the targeted gRNA site. While digestion with MboII creates 230 and 99 bp fragments in wild-type, the prps1asd59 allele has lost the MboII restriction site and remains uncut. In prps1asd60 mutants, the PCR product using the primer pair 5’-GTGCAGAGAAGTCTAAATGCATCT-3’ and 5’-CTGCATATCATGCATGTCCTCAC-3’ created a 258 bp band compared to the 279 bp size product in wild-type. In prps1bsd61 mutants, the PCR product using the primer pair 5’-TCCTGCAGTGTGGAGATTGG-3’ and 5’-TGGCTATAGACATTACCAGCACA-3’ created a 285 bp band compared to the 262 bp size product in wild-type. The change in PCR product size for prps1a and prps1b alleles could be visualized on a 3% TAE-agarose gel. Representative genotyping for each allele is shown in Figure 3C. Since prps1bsd61 mutants exhibited no morphological phenotypes, embryos were imaged prior to collecting genomic DNA for genotyping.
Wholemount immunofluorescence
Embryos were grown to indicated stages and fixed in 4% paraformaldehyde (PFA) in PBS at 4°C overnight. PFA was removed and embryos were washed 5 times in PBS for 5 minutes at room temperature. Embryos were then permeabilized in PBS-Triton 2% for 1.5 hours. After 5 PBS-0.5% Triton washes, embryos were blocked in 2% BSA in PBS-0.5% Triton for 1 hour. Rabbit anti-hPRPS1 polyclonal antiserum (Noree et al., 2019) was diluted 1:1000 in blocking solution and embryos were incubated overnight at 4°C. Following 5 PBS-0.5% Triton washes, embryos were incubated with 1:200 goat anti-rabbit Alexa Fluor 488 and 1:200 rhodamine phalloidin for 2 hours at room temperature in the dark. After washing to remove unbound secondary antibodies and phalloidin, embryos were stored in SlowFade Gold Antifade with DAPI prior to imaging at 4°C. 50% glycerol was added to embryos before they were mounted for imaging.
For Figure 2A, embryos were grown to indicated stages and fixed in 4% paraformaldehyde (PFA) in PBS at 4°C overnight. Following PBS washes, retinas were removed and the same immunostaining protocol mentioned above was performed. For rhodamine phalloidin staining of the lens (Figure 6A and 6B), the cornea was not removed prior to fixation or staining.
Dissection of eyes and immunostaining
Retina dissection and trypsinization followed a previously established protocol (Zou et al., 2008). In brief, eyes from embryos at 5 dpf were manually dissected and placed in DMEM media. Next, eyes were trypsinized in TrypLE Express for 30 minutes at room temperature. FBS was added to 10% to inhibit trypsin activity. Intact retinas were discarded and disassociated cells were pelleted for 2 minutes at 3000 rpm. Supernatant was removed and pellets were resuspended in PBS and added to poly-L-lysine coated dishes for 1 hour at room temperature. Adhered cells were then fixed in 4% PFA for 30 min at room temperature. Following 3 PBS washes, cells were permeabilized for 15 minutes in PBS-0.5% Triton and subsequently blocked in 2% BSA in PBS for 30 minutes. 1:1000 rabbit anti-hPRPS1 was added to blocking solution and incubated at 4°C overnight. Following 3 PBS washes, cells were incubated in 1:200 goat anti-rabbit Alexa Fluor 488 for 2 hours in the dark. After removing unbound secondary antibody, SlowFade Gold Antifade with DAPI was added, and cells were imaged immediately.
Histology
Embryos were grown to indicated stages in E3 containing PTU and fixed in 4% PFA in PBS at 4°C overnight. Following 3 PBS washes, embryos were dropped in 35% sucrose until they sunk to the bottom. 35% sucrose/PBS solution was removed by washing once with OCT freezing medium. Cryomolds were prepared by lining up embryos in OCT freezing medium and freezing on dry ice. Cryomolds were stored at −80°C until ready for use. Sections were cut at 12 μm thickness and transferred onto a microscope slide. Areas of interest were outlined with a hydrophobic PAP pen prior to rehydrating slides in PBS-0.1% Tween-20 for 5 minutes. Sections were blocked in 2% BSA in PBS-0.1% Tween-20 for 1 hour at room temperature followed by addition of primary antibodies in blocking solution (1:200 mouse anti-Zpr2 and 1:200 rabbit anti-ZO-1) overnight at 4°C. Following 3 PBS-0.1% Tween washes, sections were incubated in secondary solution (1:200 goat anti-rabbit Alexa Fluor 488, 1:200 sheep anti-mouse Alexa Fluor 546, or 1:200 rhodamine phalloidin) for 2 hours at room temperature in the dark. After 3 PBS-0.1% Tween washes, slides were dried and SlowFade Gold Antifade with DAPI was added before sealing with a coverslip and nail polish. Slides were stored at 4°C until ready for imaging.
Imaging and quantitative image analysis
Images were captured using a Zeiss Axioimager and Axiocam and were processed using Zeiss Axiovision and Adobe Creative Suite software. Confocal imaging was performed with a Leica SP5 confocal laser-scanning microscope and analyzed using Imaris software (Bitplane). For confocal images of whole-mount specimens, a 40X/1.10 water immersion objective was used to collect z-stacks composed of ~50 individual slices that were 2 μm thick. For confocal images of transverse cryosections, a 40X/1.10 water immersion objective was used to collect z-stacks composed of ~12 individual slices that were 0.5 μm thick.
For analysis of PRPS filament formation, maximum intensity projections of confocal reconstructions were imported into the Fiji image processing software package. A threshold was applied to subtract background fluorescence and identify PRPS filaments. Next, the “Analyze Particles” function was used to determine the number of filaments per retina, and the length of each filament was measured manually. Finally, Prism 8 (GraphPad) software was used to analyze and plot values.
For analysis of lens size and actin organization, maximum intensity projections of confocal reconstructions were imported into Fiji, and the lens region was segmented from the rest of the retina. The diameter of each lens was measured manually, and the angles between pairs of adjacent radial actin cables were measured using the angle tool, with the vertex of each angle located at the center of the lens. After measuring 20–30 angles per lens, Prism 8 software was used to calculate and plot the standard deviation for the set of angle values from each individual sample.
Replicates and statistical analysis
All assessments of morphology and immunofluorescence were replicated in at least two experiments with comparable results from independent crosses. For each experiment, the number of samples examined is reported in the associated figure legend. All statistical analysis was performed using either an unpaired Student’s t-test (Figure 6C and lens size comparisons) or one-way ANOVA (Figures 5B, 5C, 6D, and 6E) in Prism 8 software. Any p values greater than 0.05 were considered not significant.
ACKNOWLEDGMENTS
We thank Tami Sanchez for thoughtful zebrafish care and members of the Wilhelm and Yelon labs for valuable discussions. Work in the Wilhelm lab was supported by a grant from the HCIA program of HHMI and the James Wilhelm Memorial Fund. Kyle Begovich is a Howard Hughes Medical Institute Gilliam Fellow and was supported by NIH T32 GM007240. Work in the Yelon lab was supported by NIH R01 HL108599 and NIH R01 HL133166.
Grant support:
National Institutes of Health: R01 HL108599, R01 HL133166, T32 GM007240
HHMI: HCIA Program Grant
REFERENCES
- Ahmed M, Taylor W, Smith PR, Becker MA. 1999. Accelerated Transcription of PRPS1 in X- linked Overactivity of Normal Human Phosphoribosylpyrophosphate Synthetase. J Biol Chem 274: 7482–7488. [DOI] [PubMed] [Google Scholar]
- Alli N, Coetzee M, Louw V, Rensburg BV, Rossouw G, Thompson L, Pissard S, Thein SL. 2008. Sickle cell disease in a carrier with pyruvate kinase deficiency. Hematology 13: 369–372. [DOI] [PubMed] [Google Scholar]
- An S, Kumar R, Sheets ED, Benkovic SJ. 2008. Reversible Compartmentalization of de Novo Purine Biosynthetic Complexes in Living Cells. Science 320: 103–106. [DOI] [PubMed] [Google Scholar]
- Arts WFM, Loonen MCB, Sengers RCA, Slooff JL. 1993. X-linked ataxia, weakness, deafness, and loss of vision in early childhood with a fatal course. Annals of Neurology 33: 535–539. [DOI] [PubMed] [Google Scholar]
- Barry RM, Bitbol AF, Lorestani A, Charles EJ, Habrian CH, Hansen JM, Li HJ, Baldwin EP, Wingreen NS, Kollman JM, Gitai Z. 2014. Large-scale filament formation inhibits the activity of CTP synthetase. eLife 3: e03638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassnett S, Beebe DC. 1992. Coincident loss of mitochondria and nuclei during lens fiber cell differentiation. Dev Dyn 194: 85–93. [DOI] [PubMed] [Google Scholar]
- Becker MA, Losman MJ, Itkin P, Simkin PA. 1982. Gout with superactive phosphoribosylpyrophosphate synthetase due to increased enzyme catalytic rate. J Lab Clin Med 99: 495–511. [PubMed] [Google Scholar]
- Becker MA, Puig JG, Mateos FA, Jimenez ML, Kim M, Simmonds HA. 1988. Association of uric acid overproduction and sensorineural deafness. The American Journal of Medicine 85: 383–390. [DOI] [PubMed] [Google Scholar]
- Becker MA, Raivio KO, Bakay B, Adams WB, Nyhan WL. 1980. Variant human phosphoribosylpyrophosphate synthetase altered in regulatory and catalytic functions. J Clin Invest 65: 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carcamo WC, Satoh M, Kasahara H, Terada N, Hamazaki T, Chan JYF, Yao B, Tamayo S, Covini G, von Mühlen CA, Chan EKL. 2011. Induction of Cytoplasmic Rods and Rings Structures by Inhibition of the CTP and GTP Synthetic Pathway in Mammalian Cells. PLoS ONE 6: e29690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brouwer APM, van Bokhoven H, Nabuurs SB, Arts WF, Christodoulou J, Duley J. 2010. PRPS1 Mutations: Four Distinct Syndromes and Potential Treatment. Am J Hum Genet 86: 506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delos Santos K, Kim M, Yergeau C, Jean S, Moon NS. 2019. Pleiotropic role of Drosophila phosphoribosyl pyrophosphate synthetase in autophagy and lysosome homeostasis. PLoS Genet 15: e1008376. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- DeSmidt AA, Zou B, Grati M, Yan D, Mittal R, Yao Q, Richmond MT, Denyer S, Liu XZ, Lu Z. 2019. Zebrafish model for nonsyndromic X-linked sensorineural deafness, DFNX1. Anat Rec doi: 10.1002/ar.24115 [DOI] [PubMed] [Google Scholar]
- Drummond MC, Belyantseva IA, Friderici KH, Friedman TB. 2012. Actin in hair cells and hearing loss. Hear Res 288: 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentino A, Fujinami K, Arno G, Robson AG, Pontikos N, Armengol MA, Plagnol V, Hayashi T, Iwata T, Parker M, Fowler T, Rendon A, Gardner JC, Henderson RH, Cheetham ME, Webster AR, Michaelides M, Hardcastle AJ. 2018. Missense variants in the X-linked gene PRPS1 cause retinal degeneration in females. Human Mutation 39: 80–91. [DOI] [PubMed] [Google Scholar]
- Fitzmaurice AG, Rhodes SL, Lulla A, Murphy NP, Lam HA, O’Donnell KC, Barnhill L, Casida JE, Cockburn M, Sagasti A, Stahl MC, Maidment NT, Ritz B, Bronstein JM. 2013. Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proc Natl Acad Sci USA 110: 636–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon JA, Valen E, Thyme SB, Huang P, Ahkmetova L, Pauli A, Montague TG, Zimmerman S, Richter C, Schier AF. 2014. Efficient Mutagenesis by Cas9 Protein-Mediated Oligonucleotide Insertion and Large-Scale Assessment of Single-Guide RNAs. PLoS One 9: e98186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JM, Hartsock A, Clark BS, Napie HRL, Link BA, Gross JM. 2012. Integrin α5/fibronectin1 and focal adhesion kinase are required for lens fiber morphogenesis in zebrafish. Mol Biol Cell 23: 4725–4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne-Badovinac S, Lin D, Waldron S, Schwarz M, Mbamalu G, Pawson T, Jan YN, Stainier DYR, Abdelilah-Seyfried S. 2001. Positional cloning of heart and soul reveals multiple roles for PKCλ in zebrafish organogenesis. Current Biology 11: 1492–1502. [DOI] [PubMed] [Google Scholar]
- Hunkeler M, Hagmann A, Stuttfeld E, Chami M, Guri Y, Stahlberg H, Maier T. 2018. Structural basis for regulation of human acetyl-CoA carboxylase. Nature 558: 470–474. [DOI] [PubMed] [Google Scholar]
- Imai F, Yoshizawa A, Fujimori-Tonou N, Kawakami K, Masai I. 2010. The ubiquitin proteasome system is required for cell proliferation of the lens epithelium and for differentiation of lens fiber cells in zebrafish. Development 137: 3257–3268. [DOI] [PubMed] [Google Scholar]
- Jensen AM, Westerfield M. 2004. Zebrafish Mosaic Eyes Is a Novel FERM Protein Required for Retinal Lamination and Retinal Pigmented Epithelial Tight Junction Formation. Current Biology 14: 711–717. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Sohn KM, Shy ME, Krajewski KM, Hwang M, Park JH, Jang SY, Won HH, Choi BO, Hong SH, Kim BJ, Suh YL, Ki CS, Lee SY, Kim SH, Kim JW. 2007. Mutations in PRPS1, Which Encodes the Phosphoribosyl Pyrophosphate Synthetase Enzyme Critical for Nucleotide Biosynthesis, Cause Hereditary Peripheral Neuropathy with Hearing Loss and Optic Neuropathy (CMTX5). Am J Hum Genet 81: 552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krock BL, Perkins BD. 2014. The Par-PrkC Polarity Complex Is Required for Cilia Growth in Zebrafish Photoreceptors. PLoS One 9: e104661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Lu Y, Peng B, Ding J. 2007. Crystal structure of human phosphoribosylpyrophosphate synthetase 1 reveals a novel allosteric site. Biochem J 401: 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch EM, Hicks DR, Shepherd M, Endrizzi JA, Maker A, Hansen JM, Barry RM, Gitai Z, Baldwin EP, Kollman JM. 2017. Human CTP synthase filament structure reveals the active enzyme conformation. Nat. Struct Mol Biol 24: 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masai I, Lele Z, Yamaguchi M, Komori A, Nakata A, Nishiwaki Y, Wada H, Tanaka H, Nojima Y, Hammerschmidt M, Wilson SW, Okamato H. 2003. N-cadherin mediates retinal lamination, maintenance of forebrain compartments and patterning of retinal neurites. Development 130: 2479–2494. [DOI] [PubMed] [Google Scholar]
- Mochizuki T, Kojima Y, Nishiwaki Y, Harakuni T, Masai I. 2018. Endocytic trafficking factor VPS45 is essential for spatial regulation of lens fiber differentiation in zebrafish. Development 145: dev170282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanaswamy R, Levy M, Tsechansky M, Stovall GM, O’Connell JD, Mirrielees J, Ellington AD, Marcotte EM. 2009. Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc Natl Acad Sci USA 106: 10147–10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng A, Uribe RA, Yieh L, Nuckels R, Gross JM. 2009. Zebrafish mutations in gart and paics identify crucial roles for de novo purine synthesis in vertebrate pigmentation and ocular development. Development 136: 2601–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noree C, Monfort E, Shiau AK, Wilhelm JE. 2014. Common regulatory control of CTP synthase enzyme activity and filament formation. Mol Biol Cell 25: 2282–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noree C, Sato BK, Broyer RM, Wilhelm JE. 2010. Identification of novel filament-forming proteins in Saccharomyces cerevisiae and Drosophila melanogaster. J Cell Biol 190: 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noree C, Begovich K, Samilo D, Broyer R, Monfort E, Wilhelm JE. 2019. A quantitative screen for metabolic enzyme structures reveals patterns of assembly across the yeast metabolic network. Mol Biol Cell 30: 2721–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Hyun YS, Kim YJ, Nam SH, Kim S, Hong YB, Park JM, Chung KW, Choi BO. 2013. Exome Sequencing Reveals a Novel PRPS1 Mutation in a Family with CMTX5 without Optic Atrophy. J Clin Neurol 9: 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei W, Xu L, Varshney GK, Carrington B, Bishop K, Jones M, Huang SC, Idol J, Pretorius PR, Beirl A, Schimmenti LA, Kindt KS, Sood R, Burgess SM. 2016. Additive reductions in zebrafish PRPS1 activity result in a spectrum of deficiencies modeling several human PRPS1-associated diseases. Sci Rep 6: 29946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovska I, Nüske E, Munder MC, Kulasegaran G, Malinovska L, Kroschwald S, Richter D, Fahmy K, Gibson K, Verbavatz JM, Alberti S. Filament formation by metabolic enzymes is a specific adaptation to an advanced state of cellular starvation. eLife. 2014; 3: e02409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen QJ, Kassim H, Huang Y, Li H, Zhang J, Li G, Wang PY, Yan J, Ye F, Liu JL. 2016. Filamentation of Metabolic Enzymes in Saccharomyces cerevisiae. J Genet Genomics 43: 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sircar K, Huang H, Hu L, Cogdell D, Dhillon J, Tzelepi V, Efstathiou E, Koumakpayi IH, Saad F, Luo D, Bismar TA, Aparicio A, Troncoso P, Navone N, Zhang W. 2012. Integrative Molecular Profiling Reveals Asparagine Synthetase Is a Target in Castration-Resistant Prostate Cancer. Am J Pathol 180: 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas EC, Gunter JH, Webster JA, Schieber NL, Oorschot V, Parton RG, Whitehead JP. 2012. Different Characteristics and Nucleotide Binding Properties of Inosine Monophosphate Dehydrogenase (IMPDH) Isoforms. PLOS One 7: e51096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L, Harwood HJ. 2006. Acetyl-coenzyme A carboxylases: versatile targets for drug discovery. J Cell Biochem 99: 1476–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihtelic TS, Yamamoto Y, Springer SS, Jeffrey WR, Hyde DR. 2005. Lens opacity and photoreceptor degeneration in zebrafish lens opaque mutant. Dev Dyn 233: 52–65. [DOI] [PubMed] [Google Scholar]
- Webb BA, Dosey AM, Wittmann T, Kollman JM, Barber DL. 2017. The glycolytic enzyme phosphofructokinase-1 assembles into filaments. J Cell Biol 216: 2305–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wride MA. 2000. Minireview: apoptosis as seen through a lens. Apoptosis 5: 203–209. [DOI] [PubMed] [Google Scholar]
- Yazulla S, Studholme KM. 2001. Neurochemical anatomy of the zebrafish retina as determined by immunocytochemistry. J Neurocytol 592: 551–592. [DOI] [PubMed] [Google Scholar]
- Zoref E, De Vries A, Sperling O. 1975. Mutant feedback-resistant phosphoribosylpyrophosphate synthetase associated with purine overproduction and gout. Phosphoribosylpyrophosphate and purine metabolism in cultured fibroblasts. J Clin Invest 56: 1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Lathrop KL, Sun M, Wei X. 2008. Intact RPE maintained by Nok is essential for retinal epithelial polarity and cellular patterning in zebrafish. J Neurosci 28: 13684–13695. [DOI] [PMC free article] [PubMed] [Google Scholar]