

Abstract
Agents that inhibit the enzyme geranylgeranyl diphosphate synthase (GGDPS) have anti-cancer activity and our prior studies have investigated the structure-function relationship for a family of isoprenoid triazole bisphosphonates as GGDPS inhibitors. To further explore this structure-function relationship, a series of novel α-modified triazole phosphonates were prepared and evaluated for activity as GGDPS inhibitors in enzyme and cell-based assays. These studies revealed flexibility at the α position of the bisphosphonate derivatives with respect to being able to accommodate a variety of substituents without significantly affecting potency compared to the parent unsubstituted inhibitor. However, the monophosphonate derivatives lacked activity. These studies further our understanding of the structure-function relationship of the triazole-based GGDPS inhibitors and lay the foundation for future studies evaluating the impact of α-modifications on in vivo activity.
Keywords: bisphosphonate, geranylgeranyl diphosphate synthase, inhibition, isoprenoid biosynthesis, triazole, myeloma
Graphical Abstract
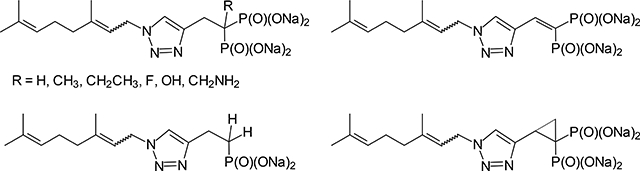
1. Introduction
The enzyme geranylgeranyl diphosphate synthase (GGDPS) is responsible for the production of the 20-carbon geranylgeranyl diphosphate which serves as the isoprenoid donor in protein geranylgeranylation reactions as well as a substrate for other isoprenoid biosynthetic pathway (IBP) enzymes.1 The protein geranylgeranylation reactions are mediated primarily by geranylgeranyl transferase (GGTase) I2–3 and II4, as well as the more recently identified GGTase III.5 Some of the key protein substrates for these enzymes include members of the Ras superfamily of small GTPases (e.g., the Rho proteins) which mediate cancer cell migration and metastasis,6 as well as the Rab GTPase family members, which play essential roles in regulating intracellular trafficking processes.7 We and others have been interested in developing GGDPS inhibitors as novel anti-cancer agents by virtue of their effects on disruption of protein geranylgeranylation.8–9 In particular, we have focused on the anti-myeloma effects of GGDPS inhibitors, as our work has demonstrated that disruption of Rab geranylgeranylation leads to impairment of intracellular monoclonal protein trafficking. In turn, this disruption in protein homeostasis results in induction of the unfolded protein response pathway and apoptosis.10–12
While our initial efforts focused on branched alkyl bisphosphonates,13–14 our more recent efforts have focused on developing families of monoalkyl triazole bisphosphonates with activity as GGDPS inhibitors.8, 15–21 In this latter family, the first to be identified was inhibitor 1 (Figure 1) which is a mixture of olefin isomers and exerts cellular activity at 0.5 μM in myeloma cells.15, 20 Subsequent structure-function work revealed that olefin stereochemistry and alkyl chain length significantly impact potency in vitro. When evaluating chain length, the homologue 2 was found to be the most potent analogue, with an IC50 of 45 nM against GGDPS and evidence of disruption of geranylgeranylation in cellular assays at concentrations as low as 30 nM.16, 20 Our studies also revealed differences in activity between E and Z isomers in these series and demonstrated synergistic activity amongst the two isomers comprising homologue 2.8, 15, 18 Notably, modification of the α-carbon had a further impact on activity with the α-methyl derivative (3) of compound 1 having a 5-fold increase in potency in cellular assays compared to the parent compound.20 In the case of the homologue 2, methylation at the α-position modestly affected cellular potency, but abrogated the impact of olefin stereochemistry on activity, rendering the two individual isomers to near equivalent potency.20 This was of particular significance, as from a drug development perspective, it is advantageous to have a compound which is a single isomer. An analogue of 1 that contained an ethyl substituent at the α-position (4) was found to have a five-fold decrease in potency in cellular assays, thus establishing a preliminary structure-function relationship with respect to the α-position and alkyl substituents.20
Figure 1.

Known triazole bisphosphonates that inhibit GGDPS.
Preclinical evaluation of several of our lead GGDPS inhibitors has revealed that these agents have metabolic stability, systemic biodistribution, prolonged t1/2 lives that allow once- or twice-weekly dosing, and confirmed the on-target effects with respect to disruption of protein geranylgeranylation.12, 22 Furthermore, we have established that these GGDPS inhibitors have in vivo anti-tumor activity in mouse models of myeloma and pancreatic adenocarcinoma.12, 23–24 Of note, our studies have revealed that the olefin stereochemistry as well as the α-carbon substituent have an impact on pharmacokinetic parameters, biodistribution profiles and toxicity profiles.12, 22
Given the promising drug-like characteristics of our lead inhibitors as well the structure-function relationship identified in both the in vitro and in vivo settings, we were interested in preparing additional derivatives with modifications at the α-position to gain further insight into the factors that determine activity. As an initial step, we have prepared seven novel analogues of compound 1 and evaluated their activity as potential GGDPS inhibitors. Compound 1 was chosen as the parent compound in these studies due to ease of synthesis with a more readily available azide as well as having previously established the relative activities of the corresponding α-methyl (3) and α-ethyl (4) derivatives.
2. Chemical Synthesis
Preparation of a family of α-modified triazole bisphosphonates could be approached via several different strategies, with two major variations based either on elaboration of an intact triazole or construction of an acetylene bearing the intact bisphosphonate followed by formation of the triazole. Generation of a bisphosphonic acid salt appropriate for bioassay would be accomplished as the final transformation in either strategy by hydrolysis of an appropriate bisphosphonate ester. One way to pursue the first option would be to take advantage of the acidity of the α position inherent to a geminal bisphosphonate tetraester. Introduction of the desired α substituent might be accomplished from the parent bisphosphonate ester through formation of the bisphosphonate anion as long as the substituent is amenable to a substitution reaction at the relatively hindered center. Several isoprenoid triazole bisphosphonates have been reported in our previous studies,8 while others could be prepared from a minimally substituted triazole. Alternatively, the triazole could be formed via a click reaction between an allylic azide and an acetylene bearing the complete bisphosphonate head group. This strategy would allow use of the relatively reactive propargyl bromide to assemble the α substituted bisphosphonate component from a substituted methylene bisphosphonate. Each route has inherent advantages. Related compounds with the same alkyl group and varied α substituents might be obtained through elaboration of the parent bisphosphonate. In contrast, variations in the alkyl tail with a single head group might be more efficiently accomplished through initial formation of the functionalized acetylene. Ultimately both strategies were explored to access different target compounds.
The initial efforts to elaborate an intact triazole are shown in Scheme 1. The triazole studied carried a mixed geranyl/neryl tail because it is readily available from geranyl (or neryl) azide25. After treatment of the parent bisphosphonate ester 5 with NaH, reaction with N-fluorobenzenesulfonimide (NFSI) allowed incorporation of an α-fluoro substituent in good yield.26–28 Hydrolysis of the phosphonate ester 6 was readily accomplished under standard conditions29 through treatment with TMSBr followed by reaction with NaOH. Even through the products were obtained as the expected mixture of olefin isomers in the isoprenoid chain, for both the ester (6) and the corresponding salt (7) the presence of fluorine was readily observed in the NMR spectra. Specifically, in the 31P NMR spectra, P-F coupling of ~70 Hz was observed in both cases.
Scheme 1.
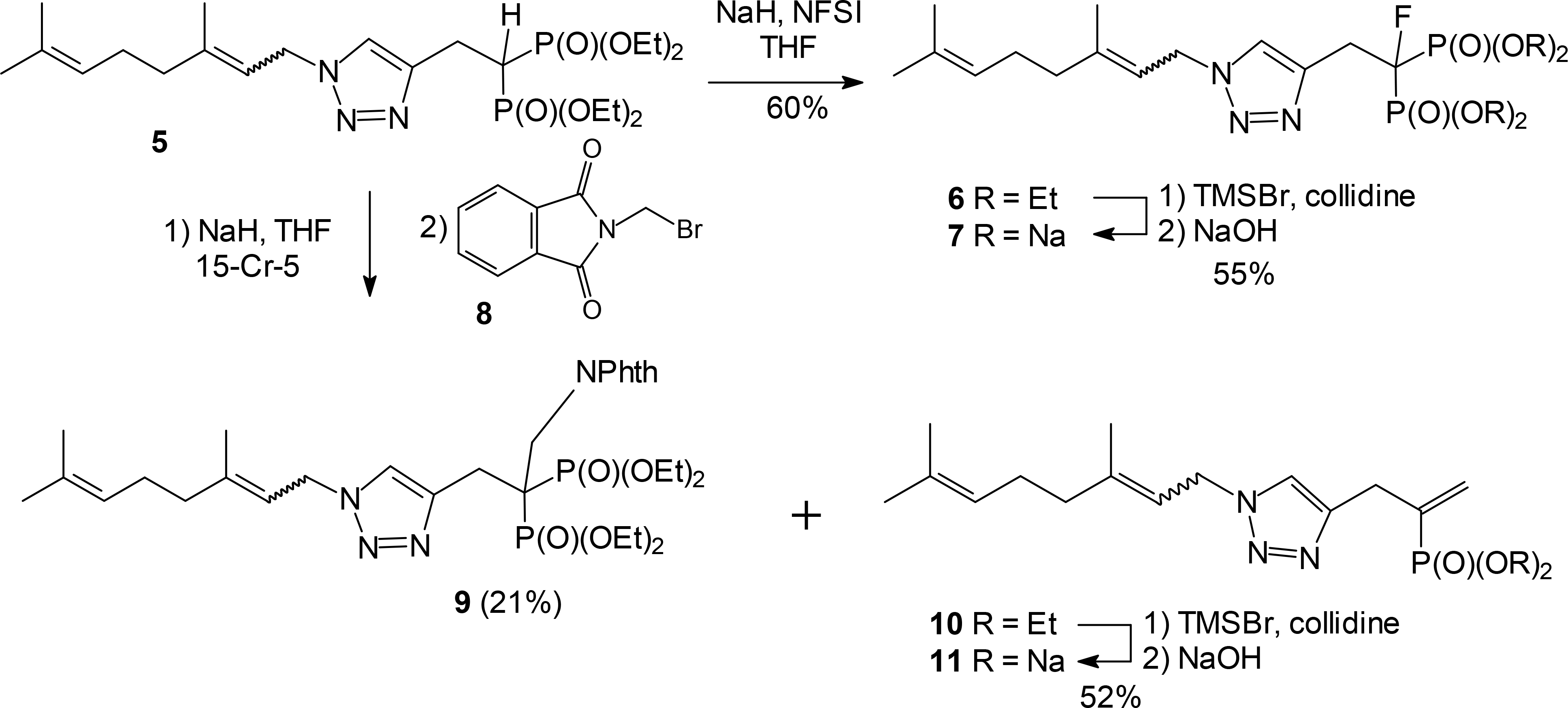
Synthesis of new triazole bisphosphonates via an α-anion.
Unfortunately, modification of bisphosphonate 5 through reaction with other electrophiles was not as straightforward. For example, treatment of the anion derived from bisphosphonate 5 with N-bromomethylphthalimide (8) gave varying amounts of the alkylated product 9 but the major product was identified as the terminal olefin 10. Formation of this olefin can be rationalized by nucleophilic attack at one of the phosphonates, followed by cleavage of the P-C bond and elimination of the phthalimide anion. However, even though this olefin was not targeted initially, it can be viewed as an α-modified product and after standard hydrolysis of the phosphonate esters the salt 11 was subjected to bioassays.
A more efficient synthesis of the α-aminomethylene compound 9 could be achieved by formation of the triazole after assembly of the substituted bisphosphonate. For this sequence (Scheme 2), a protected aminomethylene group was introduced through conjugate addition of phthalimide (12) to the unsaturated bisphosphonate 13. The resulting product 14 could be alkylated with propargyl bromide to give the intermediate acetylene 15 in good yield (80%). After cycloaddition with geranyl azide, the resulting triazole 9 was treated with hydrazine to generate the free amine 16. Final hydrolysis of the phosphonate esters under standard conditions gave the desired salt 17 in modest yield but sufficient quantity to allow bioassay.
Scheme 2.
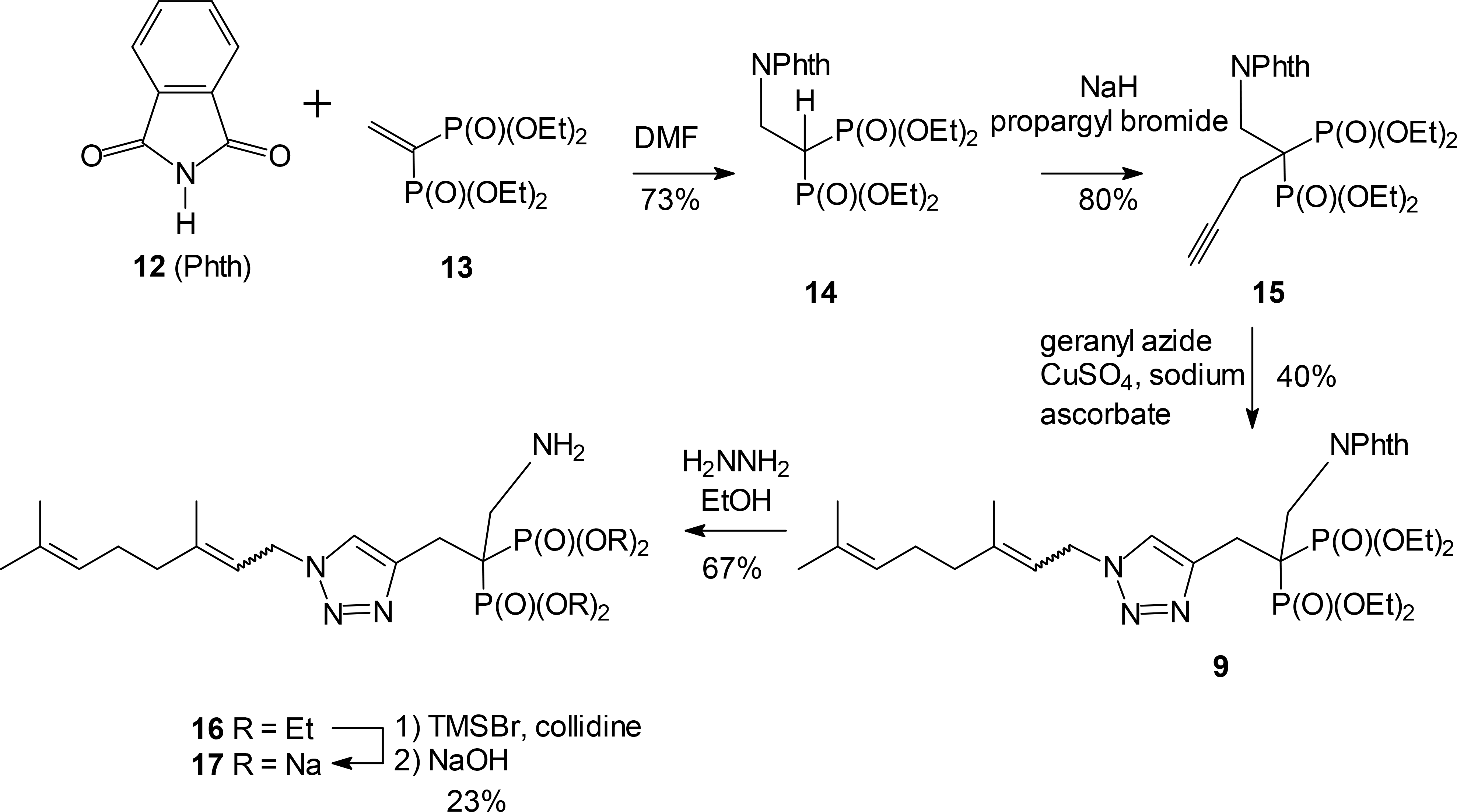
Late stage introduction of the triazole.
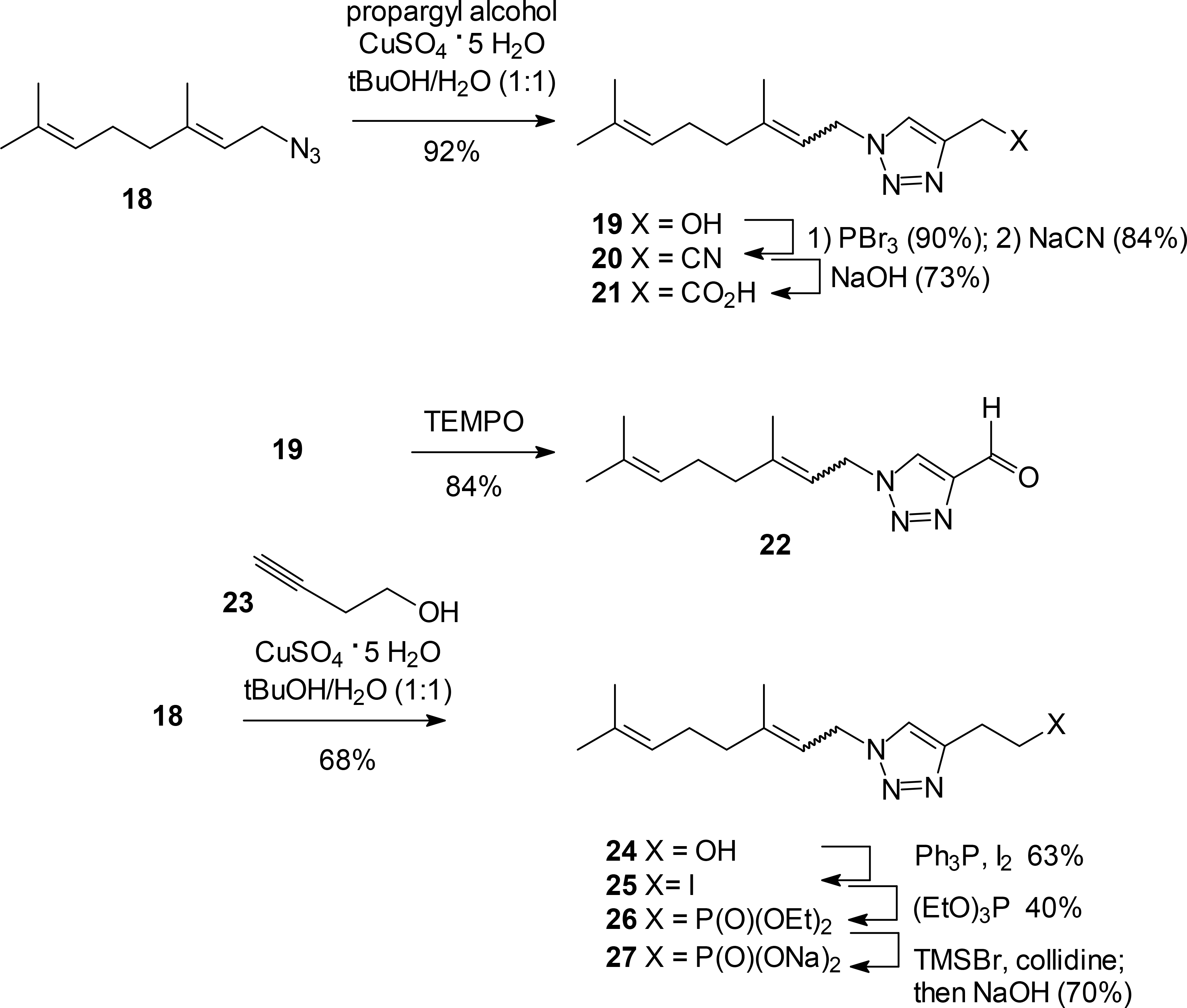
Other compounds that can be viewed as analogues to the triazole bisphosphonates were achieved by elaboration of simpler triazoles. For example, the triazole alcohol 19 has been reported as a single E-olefin isomer,30 although how that study avoided olefin isomerization through an allylic azide rearrangement25 is not clear. In our hands, cycloaddition of geranyl azide (18) with propargyl alcohol gave a mixture of the E and Z olefin isomers (19). Reaction of the alcohol 19 with PBr3 followed by treatment with NaCN gave the nitrile 20, and basic hydrolysis gave the corresponding carboxylic acid 21. Alternatively, the alcohol 19 undergoes smooth oxidation upon treatment with TEMPO to give the aldehyde 22. Geranyl azide (18) also undergoes smooth reaction with the C4 acetylene 23 to give the extended alcohol 24. This alcohol was converted to the corresponding iodide 25 upon treatment with iodine and triphenylphosphine, and then to the monophosphonate 26 through a classical Arbuzov reaction. Final hydrolysis of the phosphonate ethyl esters gave the monophosphonate 27, which can be viewed as the monophosphonate analogue to the bisphosphonate 1.
Both the triazole aldehyde 22 and the carboxylic acid 21 also could be employed to prepare other analogues to the lead triazole bisphosphonates. The Knoevenagel condensation31 of aldehyde 22 with tetraethyl methylenebisphosphonate gave the expected olefin 28 (Scheme 4). This olefin could be used directly to prepare the salt 29 for bioassay. Furthermore it readily undergoes reaction with a sulfur ylide to give the cyclopropyl compound 30.32 The cyclopropyl ring in compound 30 survives the mild conditions needed for hydrolysis of the phosphonate esters to give bisphosphonate 31.
Scheme 4.
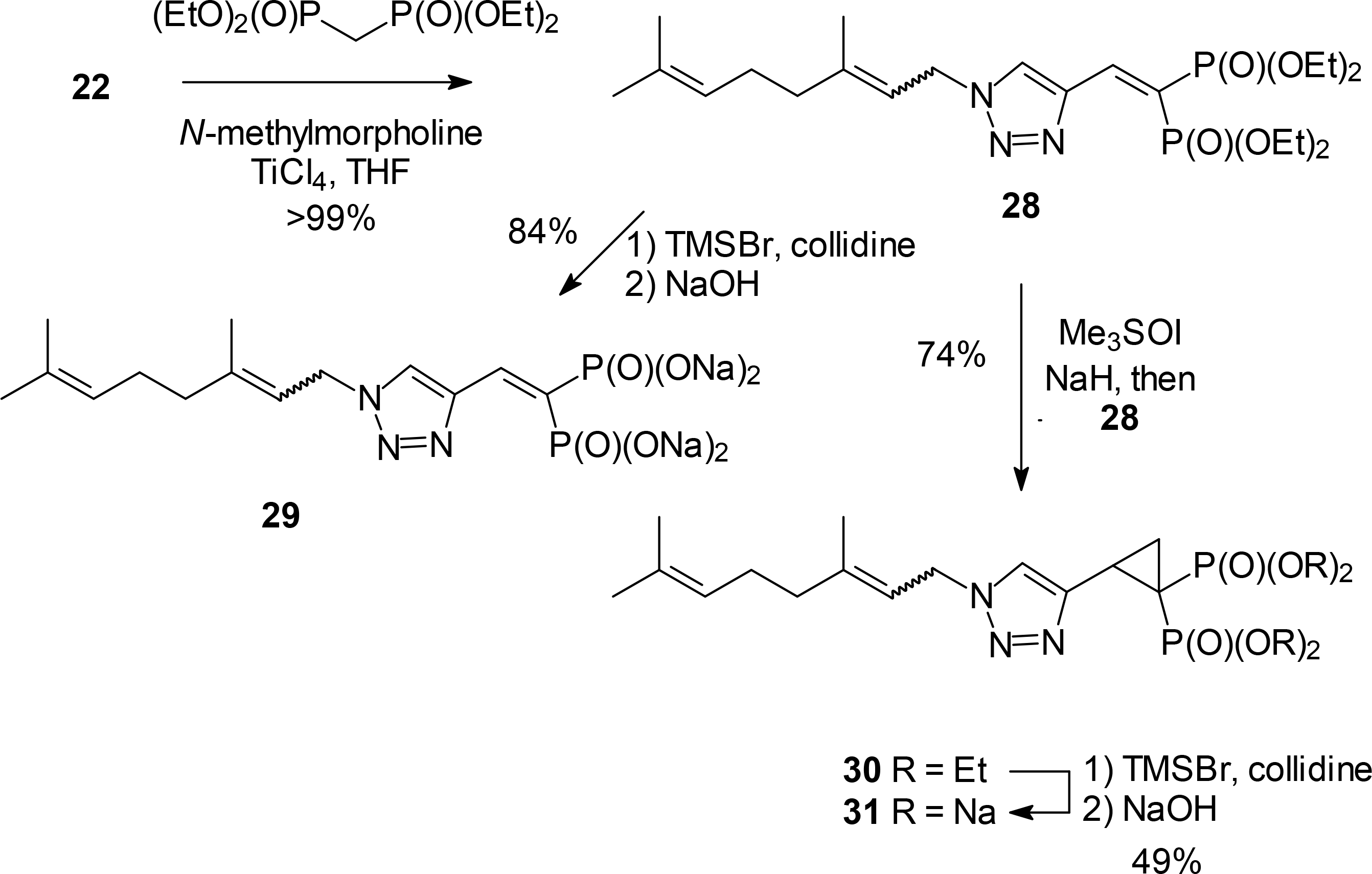
Access to bisphosphonates via a triazole aldehyde.
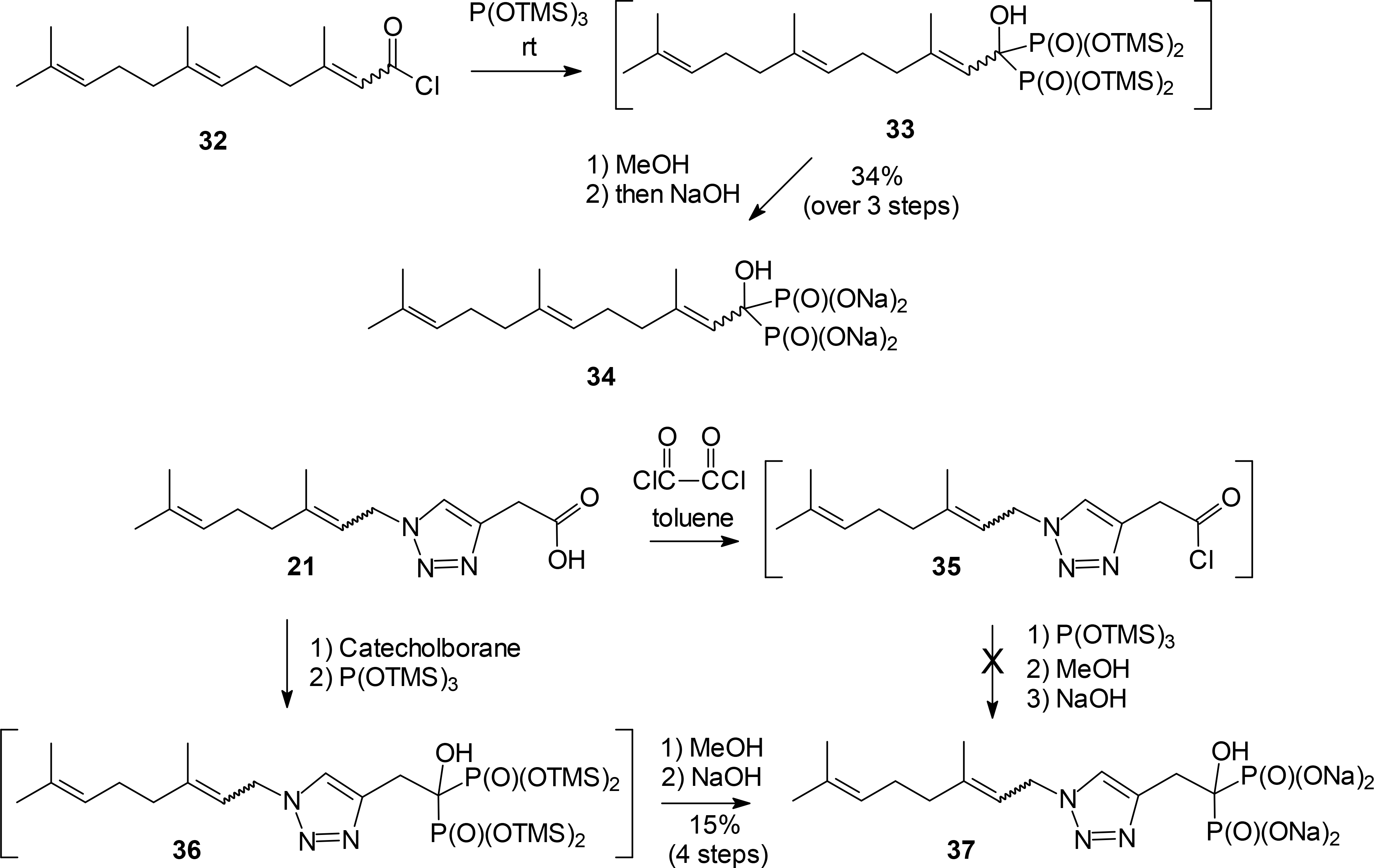
Finally, the triazole carboxylic acid 21 was viewed as a potential intermediate leading to an α-hydroxy bisphosphonate. Classically, preparation of α-hydroxy bisphosphonates has required rather harsh conditions,33–34 but LeCouvey and coworkers35–36 have developed a mild synthesis of α-hydroxy bisphosphonates through use of tris(trimethylsilyl) phosphite which avoids the known phosphonate-phosphate rearrangement.37
In our hands, this protocol worked smoothly with the known farnesoyl chloride (32)38–39 as a model carboxylic acid, giving the α-hydroxy bisphosphonate salt 34 from the presumed intermediate 33 in 34% yield over three steps. Unfortunately, attempts to apply the same conditions to the carboxylic acid 21 appeared to afford the acid chloride 35 but attempted conversion to the desired product 37 went unrewarded. In contrast, a method recently reported by Egorov and coworkers that involved activation of the carboxylic acid with catecholborane followed by reaction with tris(trimethylsilyl)phosphite gave the bisphosphonate intermediate 36.40 Hydrolysis of the TMS esters of the bisphosphonate 36 afforded the desired α-hydroxy bisphosphonate 37 in modest overall yield but in sufficient quantity to allow bioassays to proceed.
3. Biological Results
The seven novel triazole phosphonic acids were evaluated for activity as GGDPS inhibitors in an in vitro GGDPS enzyme assay as well as cellular assays in the human RPMI-8226 myeloma cell line. The cellular assays included analysis of intracellular light chain levels, which we have previously established as a biomarker for disruption of Rab geranylgeranylation.10 In addition, effects on Rap1a (a representative GGTase I substrate) geranylgeranylation and H-Ras (a representative FTase substrate) farnesylation were evaluated via immunoblot analysis. Of note, the antibody used for Rap1a is selective for unmodified Rap1a, thus under control conditions, no protein is detected, but under conditions in which cells are incubated with agents that disrupt geranylgeranylation, protein is detected. As a positive control, the HMG-CoA reductase inhibitor lovastatin was included in the cellular assays as treatment with this agent results in disruption of both farnesylation and geranylgeranylation as a consequence of depleting cells of both FPP and GGPP. Table 1 summarizes the results of these assays and includes as comparison, the previously reported non-substituted geranyl/neryl triazole bisphosphonate (1)15 as well as the α-methyl (3) and α-ethyl (4) derivatives20.
Table 1.
Summary of the bioassay results.
| Compound | GGDPS IC50 (μM) | Cellular LEC (μM)1 |
|---|---|---|
| 1 15 | 2.2 ± 0.6 | 0.5 |
| 3 20 | 1.3 ± 0.3 | 0.1 |
| 4 20 | 5.9 ± 1.3 | 2.5 |
| 7 | 3.0 ± 0.9 | 0.25 |
| 11 | >100 μM | No activity up to 100 μM |
| 17 | 2.0 ± 0.5 | 0.5 |
| 27 | >100 μM | No activity up to 100 μM |
| 29 | 2.7 ± 1.6 | 0.25 |
| 31 | 1.6 ± 0.7 | 0.5 |
| 37 | 1.4 ± 0.5 | 0.1 |
Cellular LEC (lowest effective concentration) is defined as the lowest concentration for which an unmodified Rap1a band is visible in the immunoblot and a statistically significant increase in intracellular lambda light chain is observed in the ELISA.
These studies demonstrated that all of the novel bisphosphonates have similar potency as GGDPS inhibitors in the GGDPS enzyme assay (1.4–3.0 μM range for IC50). The cellular potency varied somewhat more based on the substituent (0.1–0.5 μM range for LEC). The most potent of the derivatives was the α-hydroxy derivative 37 (LEC of 0.1 μM). As shown in Figure 2A–B, the α-fluoro derivative 7 was less potent than the α-hydroxy derivative 37 as evidenced by the immunoblot analysis of unmodified Rap1a and accumulation of intracellular light chain. Comparison of the vinyl (29) vs cyclopropyl (31) derivatives (Figure 2C–D), revealed that the vinyl derivative 29 was two-fold more potent in cellular assays than the cyclopropyl derivative 31. The aminomethylene compound 17 had very similar potency in cellular assays (Figure 3A–B) as the cyclopropyl derivative. Finally, the two novel monophosphonates (27 and 11) displayed no activity as GGDPS inhibitors in either the enzyme assay or in the cellular assays at concentrations up to 100 μM (Figure 3 C–D). None of the novel tested compounds showed evidence of disruption of farnesylation as evidenced by the H-Ras immunoblots (Figures 2–3).
Figure 2. Cellular activity of compounds 7, 37, 29 and 31 are consistent with GGDPS inhibition.
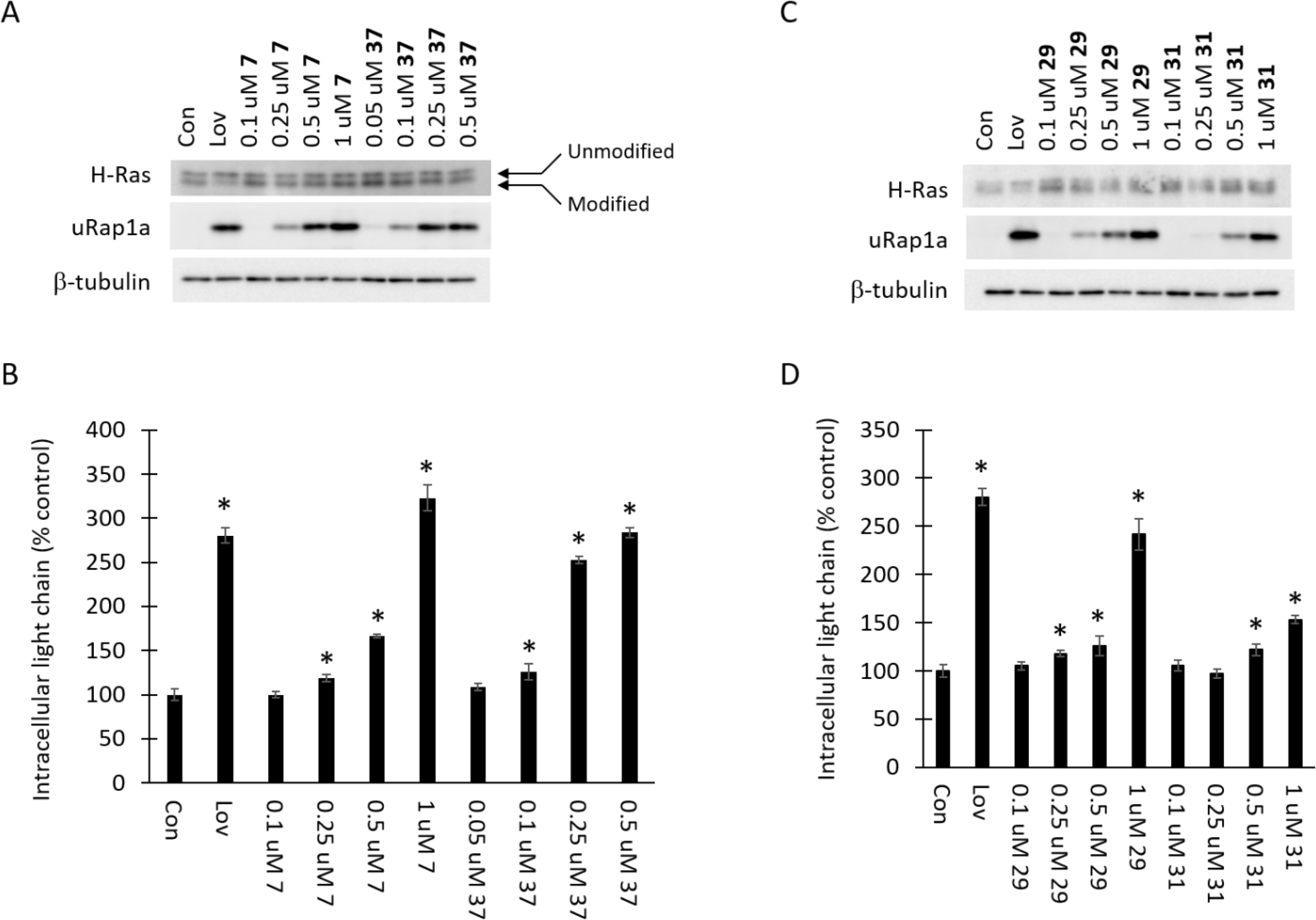
RPMI-8226 cells were incubated for 48 hours in the presence or absence of lovastatin (10 μM, Lov) or varying concentrations of compounds 7, 37, 29 or 31. A and C) Immunoblot analysis of H-Ras, unmodified Rap1a (uRap1a; antibody detects only unmodified protein) and β-tubulin (as a loading control) was performed. The gels are representative of three independent studies. B and D) Intracellular lambda light chain concentrations were determined via ELISA. Data are expressed as percentage of control (mean ± SD, n=3). The * denotes p<0.05 per t-test and compares treated cells to untreated control cells.
Figure 3. Cellular activity of compound 17 is consistent with GGDPS inhibition while monophosphonates 27 and 11 show no activity.
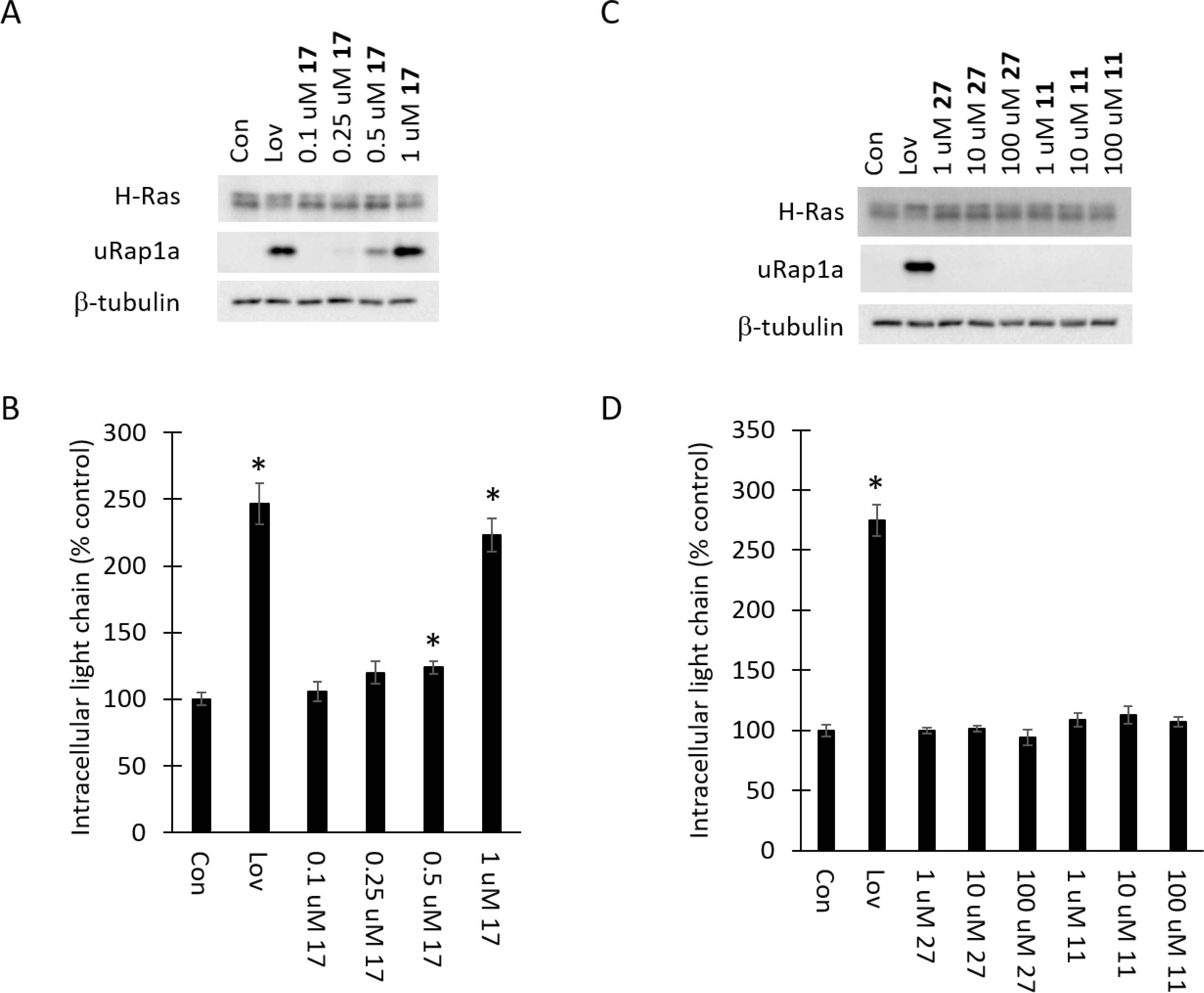
RPMI-8226 cells were incubated for 48 hours in the presence or absence of lovastatin (10 μM, Lov) or varying concentrations of compounds 17, 27 or 11. A and C) Immunoblot analysis of H-Ras, unmodified Rap1a (uRap1a; antibody detects only unmodified protein) and β-tubulin (as a loading control) was performed. The gels are representative of three independent studies. B and D) Intracellular lambda light chain concentrations were determined via ELISA. Data are expressed as percentage of control (mean ± SD, n=3). The * denotes p<0.05 per t-test and compares treated cells to untreated control cells.
Finally, the seven novel compounds were evaluated for their ability to induce cytotoxic effects in myeloma cells. As shown in Figure 4, all of the α-modified bisphosphonates (compounds 7, 37, 29, 31, 17) but neither of the monophosphonates (11, 27), induced cytotoxicity as determined via MTT assays. Consistent with the relative potency in the cellular assays which evaluated disruption of protein geranylgeranylation, the α-hydroxy derivative 37 was the most potent compound in the MTT assays.
Figure 4. The α-modified bisphosphonates, but not the monophosphonates, induce cytotoxicity in myeloma cells.
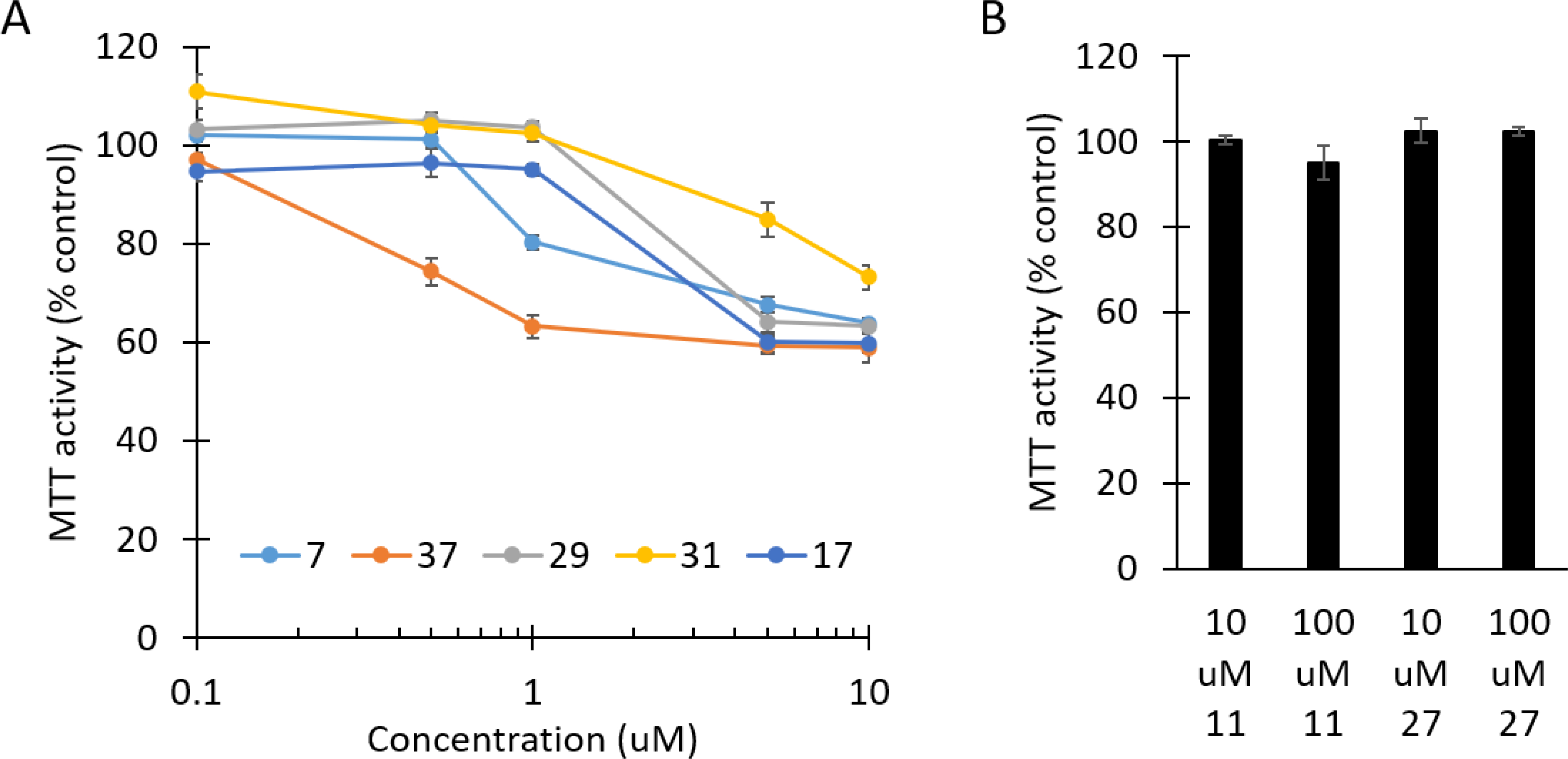
RPMI-8226 cells were incubated for 48 hours in the presence of the novel triazoles and MTT assays were performed. Data are expressed as percentage of control (mean ± standard deviation, n=4).
4. Discussion
We have prepared and assayed a series of novel isoprenoid triazole phosphonate derivatives as potential GGDPS inhibitors with a specific focus on modification at the α-carbon position. These studies demonstrate that there is flexibility at that position to accommodate a variety of substituents without significantly impairing potency compared to the parent unsubstituted compound. In particular we note that incorporation of a hydroxyl group at the α-carbon position (37) led to potency equivalent to the α-methyl derivative (3) and approximately five-fold enhanced potency (in cellular assays) relative to the parent unsubstituted compound 1. This is in contrast to an analogue in which the hydroxyl group was incorporated at the ω-position, which led to an approximately 20-fold decrease in potency (relative to 1).21 However, this ω-hydroxy derivative was of interest because of its ability to be linked to hyaluronic acid and enhance drug delivery,21 thus future studies will focus on determining whether the α-hydroxy derivative 37, as well as other potentially linkable derivatives such as the α-aminomethylene compound 17, can be conjugated to hyaluronic acid and alter not only uptake into myeloma cells but also in vivo biodistribution patterns.
Biological activity in the cell-based assays is dependent on several factors beyond binding to the target enzyme, including membrane permeability if cell entry occurs by passive diffusion or compatibility with an isoprenoid transporter if cell entry occurs via an active system. When compared to the lead compounds 3 and 4, the new alkyl derivatives (the olefin 29 and the cyclopropyl compound 31) vary little with respect to calculated LogP values and both show comparable activity. The fluoro compound 7 also shows comparable activity. The two new compounds with greatest cLogP values, the monophosphonates 11 and 27, show no activity up to 100 μM concentrations in the cell based assay, which suggests that further studies on monophosphonates are unlikely to be rewarding. Finally, the α-aminomethylene compound 17 and the α-hydroxy compound 37 have the smallest cLogP values and both show good cellular activity. Whether this is due to enhanced cell uptake, or factors such as increased acidity of the bisphosphonate or introduction of a group that allows tridentate complexation36, 41 within the active site, will have to be further studied.
Our prior studies have demonstrated that both olefin stereochemistry and chain length of the triazole bisphosphonate family of GGDPS inhibitors have significant effects on potency.8, 15–16, 18–19 In specific, we have found that the homogeranyl/homoneryl length is associated with the highest potency and that in most cases, the Z-isomers are more potent than the corresponding E-isomers. As the synthetic sequence needed to prepare homoneryl/homogeranyl derivatives are considerably longer than those needed to prepare the geranyl/neryl derivatives, and as the mixtures of isomers are more readily prepared than the individual isomers, our efforts at preparing novel α -modified derivatives focused on geranyl/neryl mixtures. Key next steps will be to pursue synthesis and biological evaluation of the homoneryl/homogeranyl derivatives of the most active compounds in this series (e.g., 37).
Legigan et al., recently reported a series of novel triazole bisphosphonates with alkyl or phenyl substituents at the C-4 or C-5 positions and a hydroxyl group at the α-carbon.36 In addition, a derivative which incorporated a propylene linker (instead of a methylene linker) between the bisphosphonate and triazole ring was prepared.36 These studies demonstrated significant reduction in potency (in MTT viability assays using solid tumor cell lines) for the C-5 series as well as the propylene linkage. As the reported bioassays did not include direct assessment of GGDPS inhibitory activity, it is not possible to draw any conclusions regarding relative potency of this series to the novel GGDPS inhibitors reported here. However, it may be of interest in the future to evaluate the impact of incorporating a propylene linker (between the bisphosphonate and the triazole) and determining whether this modification has an impact on the requirement to have a homoneryl length substituent at the C-4 position.
5. Experimental Procedures and Methods
5.1. General Experimental Procedures.
Diethyl ether (Et2O) and tetrahydrofuran (THF) both were freshly distilled from sodium and benzophenone. Acetonitrile, methylene chloride (CH2Cl2), pyridine, and triethylamine (Et3N) were freshly distilled from calcium hydride prior to use. All other solvents and reagents were purchased from commercial sources and used without further purification. All reactions in non-aqueous medium were conducted in flame-dried glassware under a positive pressure of argon or nitrogen. The NMR spectra were taken at 300, 400, or 500 MHz for 1H, and 75, 100, or 125 MHz for 13C, with internal standards of Si(CH3)4 (1H, δ 0.00) or CDCl3 (1H, δ 7.27; 13C, δ 77.2) for non–aqueous samples or D2O (1H, δ 4.80) for aqueous samples. The 31P chemical shifts were obtained at 121 or 162 MHz and reported in ppm relative to 85% H3PO4 (external standard). High resolution mass spectra were provided by the University of Iowa Mass Spectrometry Facility. Silica gel (60 Å, 0.040–0.063 mm) was used for flash chromatography.
5.2. (1-(Diethoxy-phosphoryl)-2-[1-(3,7-dimethyl-octa-2,6-dienyl)-1H-[1,2,3]triazol-4-yl]-1-fluoroethyl)-phosphonic acid diethyl ester (6).
To a suspension of NaH (80 mg, 60% dispersion in mineral oil) in THF (30 mL), a solution of bisphosphonate 5 (750 mg, 1.5 mmol) in THF (10 mL) was added slowly at 0 °C. The reaction mixture was stirred for 30 minutes while maintaining the temperature below 5 °C. A solution of N-fluorobenzenesulfonimide (570 mg, 1.8 mmol) in DMF (10 mL) was prepared separately and added to the reaction mixture. The reaction was allowed to warm to rt and stirred for 12 hours. The reaction mixture then was partitioned between EtOAc (50 mL) and H2O (40 mL). The organic layer was collected and the aqueous layer was extracted with EtOAc (30 mL times 3). The organic extracts were combined, dried (Na2SO4), and concentrated. The material obtained was subjected to silica columns (hexane: EtOH, 5:1) which afforded the desired compound 6 (450 mg, 60%): For the major isomer 1H NMR (400 MHz, CDCl3) δ 7.50 (s, 1H), 5.44 – 5.36 (m, 1H), 5.07–4.99 (m, 1H), 4.94 – 4.89 (m, 2H), 4.26 – 4.11 (m, 8H), 3.65 (q, JHP = 13 Hz, 2H), 2.19 – 2.06 (m, 4H), 1.74 (s 3H) 1.64 (s, 3H) 1.55 (s, 3H), 1.30 – 1.25 (td, J = 7.1, 2.9 Hz, 12H); 13C NMR (100 MHz, CDCl3) δ 142.8, 132.1, 123.4, 123.1, 118.1, 117.2, 64.2 (m, 2C), 64.0 (m, 2C), 47.8, 39.4, 30.0, 29.7, 26.1, 25.6, 23.4, 17.7, 16.4 – 16.3 (m, 4C); 31P NMR (162 MHz, CDCl3) 13.1 ppm (d, JPF = 74.0 Hz). HRMS (ES+, m/z) calcd for (M+H)+ C22H40FN3O6P2: 524.2455; found: 524.2462.
5.3. (2-[1-(3,7-Dimethyl-octa-2,6-dienyl)-1H-[1,2,3]triazol-4-yl]-1-fluoro-1-phosphono-ethyl)-phosphonic acid (7).
A solution of the ester 6 (104 mg, 0.20 mmol) in CH2Cl2 (3 mL) was cooled to 0 °C. Collidine (0.27 mL, 2 mmol) then was added slowly to the solution followed by the addition of TMSBr (0.32 mL, 2.4 mmol). The reaction was allowed to warm to rt gradually and stirred under argon while the progress of the reaction was monitored periodically by 31P NMR. After 12 hours, the solvent was removed under reduced pressure, and then 10 mL of toluene was added to the residue and the volatiles were removed in vacuo (repeated three times). A solution of 1N NaOH (2.2 mL) was added to the residue and it was stirred for 20 minutes at rt. The reaction mixture was transferred to a conical flask, the salt was precipitated by addition of anhydrous acetone and, after storage at 0 °C for 20 minutes, the reaction was filtered. The solid precipitate was dissolved in water and the aqueous solution was dried on a lyophilizer to afford the desired tetra-sodium salt 7 (50 mg, 55%). For the major isomer: 1H NMR (400 MHz, D2O) δ 7.75 (2s, 1H), 5.44 – 5.34 (m, 1H), 5.03 – 5.01 (m, 1H), 4.90 – 4.86 (m, 2H), 3.41 (dt, JHF = 26.1 Hz, JHP = 11.5 Hz, 2H), 2.14 – 1.67 (m, 4H), 1.62 – 1.45 (6s, 9H); 31P NMR (162 MHz, D2O) 12.2 ppm (d, JPF = 68.0 Hz). HRMS (ES−, m/z) calcd for (M−H)− C14H23FN3O6P2: 410.1045; found: 410.1035.
5.4. Compound 10.
To a stirred solution of bisphosphonate 5 (188 mg, 0.37 mmol) in THF (6 mL) at 0 °C were added NaH (27 mg, 0.67 mmol) and 15-crown-5 (12 μL, 0.07 mmol). After 30 minutes, N-(bromomethyl)phthalimide (152 mg, 0.63 mmol) was added. The reaction mixture was allowed to warm to rt and stirred overnight. The reaction was quenched by addition of NH4Cl (saturated) and extracted with ether (3 times). The combined organic layers were dried (MgSO4) and concentrated in vacuo. Final purification by flash chromatography (EtOAc to 10% EtOH in EtOAc) afforded two products, phosphonate 10 and bisphosphonate 9 (54% yield and 21%, respectively). For the major isomer of compound 10: 1H NMR (400 MHz, CDCl3) δ 7.37 (s, 1H), 6.13–6.07 (d, JHP = 22.4 Hz, 1H), 5.81–5.69 (d, JHP = 48.8 Hz, 1H), 5.39 (m, 1H), 5.04 (m, 1H), 4.91 (m, 2H), 4.05 (m, 4H), 3.66 (d, JHP = 12.0 Hz, 2H), 2.20–2.08 (m, 4H), 1.76 (s, 3H), 1.66 (s, 3H), 1.66 (s, 3H), 1.26 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 144.3 (m), 143.5, 137.4, 132.5, 132.1 (m), 123.8, 122.0, 117.5, 63.5 (d, JCP = 6.0H, 2C) 48.0, 39.5, 28.6 (d, JCP = 52.4 Hz), 26.8, 25.8, 23.5, 17.8, 16.3 (d, JCP = 6.0 Hz, 2C); 31P NMR (162 MHz, CDCl3) 18.2 ppm. HRMS (ESI) m/z calcd for C19H33N3O3P [M + H]+ 382.2254, found 382.2239.
5.5. Compound 11.
To a stirred solution of the phosphonate ester 10 (33 mg, 0.09 mmol) in CH2Cl2 (0.6 mL) at 0 °C, collidine (41 μL, 0.32 mmol) and TMSBr (47 μL, 0.36 mmol) were added dropwise in succession. The reaction was stirred overnight while it warmed to rt, and the solvent then was removed in vacuo. The resulting residue was diluted with toluene (2 mL) and concentrated in vacuo to remove any excess TMSBr (3 times). The residue was dissolved in CH2Cl2 (1 mL) and washed with water (1 mL). The organic layer was concentrated in vacuo. It then was treated with 0.3N NaOH (0.5 mL) and allowed to stir for 30 minutes at rt. After it was washed with CH2Cl2 (1 mL), the aqueous layer was concentrated in vacuo to provide the desired salt 11 as a yellow solid with a 52% yield (8.2 mg). For the major isomer: 1H NMR (400 MHz, D2O) δ 7.68 (s, 1H), 5.48–5.36 (m, 3H), 5.01 (m, 1H), 4.86 (m, 2H), 3.51 (d, JHP = 7.5 Hz, 2H), 2.13–2.09 (m, 4H), 1.79 (s, 3H), 1.69 (s, 3H), 1.52 (s, 3H); 13C NMR (100 MHz, D2O) δ 149.2 (m), 144.8, 134.7, 125.0, 124.9, 124.5, 120.4 (m), 118.0, 48.8, 39.4, 29.0 (d, JCP = 52.4 Hz), 26.3, 25.8, 17.9, 16.5; 31P NMR (162 MHz, D2O) 18.2 ppm. HRMS (ESI) m/z calcd for C15H23N3O3P [M − H]− 324.1472, found 324.1481.
5.6. Compound 14.
Through minor modifications of a literature procedure,42 the olefin 13 (1.3 mmol, 395 mg) was dissolved in DMF (5 mL), phthalmide (12, 218 mg, 1.48 mmol) was added, and the reaction was stirred at rt overnight. After water was added to the reaction mixture, it was extracted with ether (7 times). The combined organic extracts were dried (Mg2SO4) and filtered, and the filtrate was concentrated in vacuo. The final product 14 was obtained in a yield of 73% (424 mg) and used without further purification. Both the 1H and 13C NMR spectra were identical to published data.42
5.7. Compound 9.
To a stirred solution of bisphosphonate 14 (138 mg, 0.31 mmol) in THF (6 mL) at 0 °C were added NaH (23 mg, 0.58 mmol) and 15-crown-5 (13 μL, 0.06 mmol). After the reaction was stirred for 30 min, propargyl bromide (60 μL, 0.52 mmol) was added. The reaction mixture was allowed to warm to rt and stirred for 3 hours. The reaction mixture then was quenched by addition of NH4Cl (saturated) and extracted with ether (3 times). The combined organic layers were dried (MgSO4) and concentrated in vacuo. The final product 15 was obtained in 80% yield (120 mg) and used in the next reaction without further purification. dfw
To a stirred solution of acetylene 15 (120 mg, 0.25 mmol) and geranyl azide (67 mg, 3.7 mmol) in t-BuOH/H2O (4:1, 4 mL total volume), saturated CuSO4 (0.01 mL) and sodium ascorbate (15 mg, 0.08 mmol) were added in sequence. The resulting reaction mixture was stirred overnight at rt, and then the solvent was removed in vacuo. The resulting residue was dissolved in brine and extracted with EtOAc (3 times). The combined organic extracts were washed with 5% NH4OH, dried (Na2SO4) and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (EtOAc to 5% EtOH in EtOAc) afforded triazole 9 (60 mg, 40%). For the major isomer: 1H NMR (500 MHz, CDCl3) δ 7.89–7.85 (m, 3H), 7.74 (m, 2H), 5.42 (m, 1H), 5.11–5.02 (m, 1H), 4.93 (m, 2H), 4.40 (m, 2H), 4.16 (m, 4H), 3.99 (m, 2H), 3.84 (m, 2H), 3.72 (m, 2H), 2.20–2.06 (m, 4H), 1.79 (s, 3H), 1.66 (s, 3H), 1.58 (s, 3H), 1.23 (m, 6H), 1.07 (m, 6H); 13C NMR (126 MHz, CDCl3) δ 168.1 (2C), 142.7, 142.1 (m), 134.1 (2C), 132.5 (2C), 131.9, 124.5, 123.5, 123.1 (2C), 117.35, 64.0 (d, JCP= 7.6 Hz, 2C), 63.9 (d, JCP= 7.3 Hz, 2C), 47.8, 47.7 (t, JCP= 131.4 Hz), 39.4, 37.4, 32.1, 26.6, 26.2, 25.6, 17.7, 16.0 (d, JCP= 6.4 Hz, 2C), 15.9 (d, JCP= 6.6 Hz, 2C); 31P NMR (202 MHz, CDCl3) 22.2 ppm. HRMS (ESI) m/z calcd for C31H47N4O8P2 [M + H]+ 665.2859, found 665.2864.
5.8. Compound 16.
To a stirred solution of bisphosphonate 9 (60 mg, 0.09 mmol) in MeOH (1 mL), hydrazine hydrate (28 μL, 0.54 mmol) was added and the reaction was stirred overnight. The reaction mixture was filtered, and the filtrate was concentrated in vacuo. The residue was acidified to pH 2 by addition of 1M HCl and then was extracted with CH2Cl2 (3 times). The aqueous layer was made basic to pH 12 by addition of 1M NaOH and then was extracted with CH2Cl2 (3 times). The organic layer was dried (Na2SO4) and filtered, and the filtrate was concentrated in vacuo to obtain the final product amine 16 (32 mg, 67% yield) which was used in the next transformation without further purification. For the major isomer: 1H NMR (400 MHz, CDCl3) δ 7.48 (s, 1H), 5.38 (m, 1H), 5.03 (m, 1H), 4.89 (m, 2H), 4.12 (m, 8H), 3.33 (dd, JHP = 16, 12 Hz, 2H), 3.23 (t, JHP = 16 Hz, 2H), 2.11–2.04 (m, 4H), 1.76 (s, 3H), 1.66 (s, 3H), 1.57 (s, 3H), 1.27 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 143.0, 142.5 (m), 132.3, 123.6, 123.4, 117.2, 63.0 (m, 2C), 62.6 (m, 2C), 47.7, 43.0, 39.4, 26.2, 25.6, 25.3, 20.9, 17.7, 16.3 (m, 4C); 31P NMR (162 MHz, CDCl3) 25.5 ppm. HRMS (ESI) m/z calcd for C23H44N4O6P2 [M + H]+ 535.2809, found 535.2817.
5.9. Compound 17.
According to the procedure described for compound 11 the bisphosphonate ester 16 (30 mg, 0.06 mmol) in CH2Cl2 (0.6 mL) was treated with collidine (56 μL, 0.42 mmol) and TMSBr (97%, 61 μL, 0.50 mmol). After the reaction was stirred overnight, standard work-up and treatment with NaOH provided the desired salt 17 as a yellow solid (7 mg, 23%). For the major isomer: 1H NMR (400 MHz, D2O) δ 7.85 (s, 1H), 5.48–5.38 (m, 1H), 5.07 (m, 1H), 4.88 (m, 2H), 3.16 (t, JHP = 13.1 Hz, 2H), 2.98 (dd, JHP = 20.0, 13.5 Hz), 2.17–1.98 (m, 4H), 1.71 (s, 3H), 1.57 (s, 3H), 1.50 (s, 3H); 13C NMR (100 MHz, D2O) δ 146.0, 144.3, 134.7, 126.9, 124.9, 118.2, 48.9, 44.3, 39.5, 28.5, 26.4, 25.8, 22.9, 17.9, 16.5; 31P NMR (162 MHz, D2O) 22.7 ppm. HRMS (ESI) m/z calcd for C15H27N4O6P2 [M − H]− 421.1400, found 421.1418.
5.10. Compound 19.
To a stirred solution of propargyl alcohol (0.13 mL, 2.22 mmol) and geranyl azide (18, 597 mg, 3.3 mmol) in t-BuOH/H2O (1:1, 20 mL total volume), saturated CuSO4 (0.01 mL) and sodium ascorbate (132 mg, 0.66 mmol) were added in sequence. The resulting reaction mixture was stirred overnight at rt, and then the solvent was removed in vacuo. The resulting residue was dissolved in brine and extracted with EtOAc (3 times). The combined organic extracts were washed with 5% NH4OH, dried (Na2SO4) and filtered, and the filtrate was concentrated in vacuo. Final purification by column chromatography (1:1 hexane/ ethyl acetate to ethyl acetate) afforded triazole 19 (478 mg, 92%). The 1H NMR spectra shows a 2:1 mixture of E and Z olefin isomers. For the major isomer: 1H NMR (400 MHz, CDCl3) δ 7.48 (s, 1H), 5.40 (m, 1H), 5.10 (m, 1H), 4.92 (m, 2H), 4.75 (s, 2H), 3.30 (br s, 1H), 2.10–2.00 (m, 4H), 1.75 (s, 3H), 1.66 (s, 3H), 1.57 (s, 3H);13C NMR (100 MHz, CDCl3) δ 143.5, 132.2, 123.4, 123.1, 121.1, 117.8, 56.3, 47.9, 39.4, 26.1, 25.7, 23.5, 17.7.30
5.11. [1-(3,7-Dimethyl-octa-2,6-dienyl)-1H-[1,2,3]triazol-4-yl]-acetonitrile (20).
The alcohol 19 (1.6 g, 6.8 mmol) was dissolved in ether and the solution was cooled to 0 °C. After PBr3 (0.2 mL, 2.0 mmol) was added dropwise, the reaction was left to stir at 0 °C for 2 hr. Then the reaction mixture was poured into a separatory funnel filled with ice, and was extracted with ether (2 times). The combined ether layers were dried (MgSO4) and filtered through basic alumina, and then the filtrate was concentrated in vacuo. To a stirred mixture of the resulting bromide in DMF (25 mL), sodium cyanide (300 mg, 6.1 mmol) was added, and the mixture was heated to 40 °C and allowed to stir overnight. The reaction then was diluted with ether, washed with water (3 times), and then brine. The organic layer was dried (MgSO4) and filtered, and the filtrate was concentrated in vacuo. The resulting oil was purified by column chromatography (hexane to 2:1 hexane/ethyl acetate) to yield the nitrile 20 (880 mg, 84%) as a colorless oil. The 1H NMR spectrum showed a 2:1 mixture of Z and E olefin isomers. For the major isomer: 1H NMR (400 MHz, CDCl3) δ 7.54 (s, 1H), 5.40 (t, J = 7.0 Hz, 1H), 5.04 (m, 1H), 4.95 (m, 2H), 3.85 (s, 2H), 2.19–2.10 (m, 4H), 1.77 (s, 3H), 1.67 (s, 3H), 1.58 (s, 3H);13C NMR (100 MHz, CDCl3) δ 144.1, 137.3, 132.3, 123.3, 121.4, 117.2, 116.5, 48.2, 39.4, 26.1, 25.7, 23.4, 17.7, 15.5. HRMS (ESI) m/z calcd for C14H21N4 [M + H]+ 245.1766, found 245.1761.
5.12. [1-(3,7-Dimethyl-octa-2,6-dienyl)-1H-[1,2,3]triazol-4-yl]-acetic acid (21).
To a stirred solution of the nitrile 20 in ethanol (5 mL), 3 N NaOH (2 mL) was added and the solution was heated at reflux for two days. The reaction was diluted with water (10 mL) and extracted with ether (2 times). The aqueous layer was acidified to pH 2 by addition of concentrated HCl and then was extracted with ether (3 times). The combined ether layers were dried (MgSO4) and filtered, and the filtrate was concentrated in vacuo to obtain acid 21 (96 mg, 73%). The 1H NMR spectrum shows a 2:1 mixture of E and Z olefin isomers. For the major isomer: 1H NMR (400 MHz, MeOD) δ 7.80 (s, 1H), 5.50–5.46 (m,1H), 5.16–5.07 (m, 1H), 5.00 (m, 2H), 3.75 (s, 2H), 2.27–2.12 (m, 4H), 1.82 (s, 3H), 1.66 (s, 3H), 1.60 (s, 3H);13C NMR (100 MHz, MeOD) δ 173.5, 144.2, 132.8, 124.8, 124.6, 124.3, 118.7, 40.5, 32.0, 27.2, 25.9, 23.5, 17.8, 16.4. HRMS (ESI) m/z calcd for C14H22O2N3 [M + H]+ 264.1707, found 264.1698.
5.13. 1-[3,7-Dimethylocta-2,6-dien-1-yl]-1H-1,2,3-triazole-4-carbaldehyde (22).
In a procedure modified from the literature,43 triazole 19 (3.13 g, 13.3 mmol), acetonitrile (140 mL), and pH 7 buffer (6.5 mL) were stirred at 0 °C. Subsequently, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (210 mg, 1.33 mmol) and (diacetoxyiodo)benzene (5.14 g, 16 mmol) were added. After the reaction was stirred for 1 hour, the acetonitrile was removed under reduced pressure, and the resulting residue was diluted with ethyl acetate and water. The layers were separated, and the aqueous layer was extracted three times with ethyl acetate. The combined organic layers were dried (Na2SO4) and filtered, and the filtrate was concentrated under reduced pressure. The remaining residue was purified via flash chromatography to afford the title compound (22) as a yellow oil (2.62 g, 84% yield). For the major isomer: 1H NMR (400 MHz, CDCl3) δ 10.13 (s, 1H), 8.07 (s), 5.45 (t, J = 7.12 Hz, 1H), 5.03 (m, 3H), 2.14 (m, 4H), 1.80 (s), 1.69 (s, 3H), 1.60 (s, 3H); 13C (75.5 MHz, CDCl3) δ 185.0, 147.6, 145.0, 132.3, 124.4, 123.1, 115.7, 48.0, 39.2, 26.0, 25.5, 23.3, 17.6. HRMS (ESI) m/z calcd for C13H19N3ONa [M + Na]+ 256.1426, found 256.1418.
5.14. Compound 24.
To a stirred solution of 3-butyn-1-ol (23, 0.14 mL, 1.86 mmol) and geranyl azide (500 mg, 2.79 mmol) in t-BuOH/H2O (1:1, 20 mL total volume), saturated CuSO4 (0.05 mL) and sodium ascorbate (110 mg, 0.56 mmol) were added in sequence. The resulting reaction mixture was stirred overnight at rt, and then the solvent was removed in vacuo. The resulting residue was dissolved in brine and extracted with EtOAc (3 times). The combined organic extracts were washed with 5% NH4OH, dried (Na2SO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by column chromatography (1:1 hexane/ethyl acetate to ethyl acetate) afforded triazole 24 (317 mg, 68%). The 1H NMR spectra shows a 2:1 mixture of E and Z olefin isomers. For the major isomer: 1H NMR (400 MHz, CDCl3) δ 7.32 (s, 1H), 5.42 (t, J = 8.0 Hz, 1H), 5.06 (m, 1H), 4.93 (m, 2H), 3.95 (m, 2H), 2.95 (t, J = 6.8 Hz, 2H), 2.64 (br s, 1H), 2.20–2.11 (m, 4H), 1.80 (s, 3H), 1.68 (s, 3H), 1.59 (s, 3H);13C NMR (100 MHz, CDCl3) δ 143.0, 132.1, 123.9, 123.2, 117.9, 117.1, 66.3, 47.9, 39.4, 28.9, 26.1, 25.7, 17.6,16.4. HRMS (ESI) m/z calcd for C14H24N3O [M + H]+ 250.1914, found 250.1918.
5.15. Compound 25.
A solution of PPh3 (1.6 g, 6.02 mmol) and I2 (1.5 g, 6.02 mmol) in CH2Cl2 (40 mL) was stirred for 10 minutes at rt. Imidazole (0.68 g, 10.3 mmol) was added and the solution was stirred for another 10 minutes. The alcohol 24 (1.0 g, 4.0 mmol) in 5 mL of CH2Cl2 was added and the solution was stirred overnight. The reaction was diluted by addition of saturated sodium bisulfite and then was extracted with CH2Cl2 (2 times). The organic layers were combined, dried (Na2SO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by column chromatography (2:1 hexane/ ethyl acetate) afforded the iodide 25 (880 mg, 63%). For the major isomer: 1H NMR (400 MHz, CDCl3) δ 7.37 (s, 1H), 5.43 (m, 1H), 5.06 (m, 1H), 4.94 (m, 2H), 3.43 (m, 2H), 3.29 (m, 2H), 2.20–2.11 (m, 4H), 1.78 (s, 3H), 1.68 (s, 3H), 1.60 (s, 3H);13C NMR (100 MHz, CDCl3) δ 146.3, 143.3, 132.2, 123.5, 120.6, 117.1, 47.9, 39.5, 30.5, 26.2, 25.8, 17.8, 16.6, 4.19. HRMS (ESI) m/z calcd for C14H23N3I [M + H]+ 360.0931, found 360.0922.
5.16. Compound 26.
A solution of iodide 25 and triethyl phosphite in anhydrous toluene was heated at reflux for 7 days. After the solvent was removed in vacuo, purification by column chromatography (ethyl acetate to 3% EtOH in ethyl acetate) gave phosphonate 26 (315 mg, 40%). For the major isomer: 1H NMR (500 MHz, CDCl3) δ 7.45 (s, 1H), 5.44 (m, 1H), 5.08 (m, 1H), 4.94 (m, 2H), 4.11 (m, 4H), 3.02 (m, 2H), 2.25–2.09 (m, 6H), 1.81 (s, 3H), 1.70 (s, 3H), 1.61 (s, 3H), 1.32 (m, 6H); 13C NMR (126 MHz, CDCl3) δ 146.1, 142.4, 131.5, 123.1, 120.1, 116.9, 61.1 (d, JCP = 26.0 Hz, 2C), 47.3, 38.9, 31.5, 25.2, 25.3, 18.5, 17.2, 15.8 (m, 2C); 31P NMR (202 MHz, CDCl3) 29.9 ppm. HRMS (ESI) m/z calcd for C18H32N3O3P [M + H]+ 370.2254, found 370.2250.
5.17. Compound 27.
According to the procedure described for compound 11 the phosphonate ester 26 (50 mg, 0.135 mmol) in CH2Cl2 (0.6 mL) was treated with collidine (36 μL, 0.27 mmol) and TMSBr (97%, 39 μL, 0.30 mmol). After the reaction was stirred overnight standard work-up and treatment with NaOH provided the desired salt 27 as a white solid(34 mg, 70%). For the major isomer: 1H NMR (500 MHz, D2O) δ 7.77 (s, 1H), 5.46 (m, 1H), 5.13 (m, 1H), 4.99 (m, 2H), 2.91 (m, 2H), 2.28–2.14 (m, 4H), 1.84 (s, 3H), 1.77 (m, 2H), 1.74 (s, 3H). 1.62 (s, 3H); 13C NMR (126 MHz, D2O) δ 149.9, 144.2, 134.0, 123.8, 122.2, 117.2, 47.8, 38.5, 28.9, 25.4, 24.9, 20.6, 16.9, 15.6; 31P NMR (202 MHz, D2O) 21.3 ppm. HRMS (ESI) m/z calcd for C14H23N3O3P [M − H]− 312.1472, found 312.1480.
5.18. Diethyl [1-(diethoxyphosphoryl)-2-[1-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-1H-1,2,3-triazol-4-yl]ethyl]phosphonate (28).
To a Schlenk flask at 0 °C was added carbon tetrachloride (0.25 mL) and titanium tetrachloride (0.22 mL, 2 mmol). When THF (2 mL) was added dropwise as the mixture was stirred carefully, a yellow precipitate formed. Aldehyde 22 (233 mg, 1 mmol) then was added dropwise, followed by tetraethyl methylenebisphosphonate (288 mg, 1 mmol). After N-methylmorpholine (0.161 mL) in THF (0.3 mL) was added dropwise over the course of 1 h, the reaction mixture was stirred and allowed to warm to rt overnight. The reaction was quenched carefully by addition of aqueous saturated sodium bicarbonate solution and the THF was removed under reduced pressure. The resulting residue was diluted with ethyl acetate and water. The layers were separated, and the aqueous layer was extracted five times with ethyl acetate. The combined organic layers were dried (Na2SO4) and filtered, and then volatile materials were removed under reduced pressure. The resulting residue was purified via flash chromatography to afford the title compound as a yellow oil (28, 500 mg, 1 mmol, >99% yield). For the major isomer: 1H NMR (300 MHz, CDCl3) δ 8.82 (s, 1H), 8.33 (dd, J = 47.4, 29.2 Hz, 1H), 5.39 (t, J = 7.1 Hz, 1H), 5.02 (m, 1H), 4.95 (m, 2H), 4.10 (m, 8H), 2.10 (m, 4H), 1.75 (s, 3H), 1.63 (s, 3H), 1.54 (s, 3H), 1.32 (t, J = 7.1 Hz, 6H), 1.25 (t, J = 7.1 Hz, 6H); 13C (75.5 MHz, CDCl3) δ 149.6, 143.7, 142.4 (dd, JPC = 25.0, 10.2 Hz), 132.2, 127.7, 123.4, 117.6 (t, JPC = 169.4 Hz), 116.7, 62.8 (m, 4), 48.2, 39.5, 26.3, 25.7, 17.7, 16.7, 16.4 – 16.2 (m, 4C); 31P (121.5 MHz, CDCl3) 16.2 (d, J = 47.3 Hz, 1P), 12.4 (d, J = 47.4 Hz, 1P). HRMS (ESI) m/z calcd for C22H40N3O6P2 [M + H]+ 504.2392, found 504.2393.
5.19. Disodium [1-[bis(sodiooxy)phosphoryl]-2-[1-[3,7-dimethylocta-2,6-dien-1-yl]-1H-1,2,3-triazol-4-yl]ethenyl]phosphonate (29).
According to the procedure described for compound 7 bisphosphonate 36 (340 mg, 0.67 mmol, 1 eq), collidine (0.62 mL, 4.7 mmol, 7 eq), and TMSBr (0.71 mL, 5.36 mmol, 8 equivalents) in CH2Cl2 (7 mL) were allowed to react overnight. After a parallel work-up, final dissolution in water followed by lyophilization afforded compound 29 as a white solid (292 mg, 84%): For the major isomer 1H NMR (400 MHz, D2O) δ 8.78 (s, 1H), 7.48 (dd, J = 40.3, 26.3 Hz, 1H), 5.49 (m, 1H), 5.14 (m, 1H), 5.00 (m, 2H), 2.27 – 2.06 (m, 4H), 1.77 (s, 3H), 1.63 (s, 3H), 1.56 (s, 1H); 13C (100.6 MHz, D2O) δ 144.8 (m), 143.6, 140.9 (t, JCP = 137.6 Hz), 133.6, 131.7, 126.7, 123.8, 117.0, 48.1, 38.6, 25.5, 24.8, 17.0, 15.7; 31P (162 MHz, D2O) 15.8 (d, J = 42.9 Hz, 1P), 9.9 (d, J = 43.0 Hz, 1P). HRMS (ESI) m/z calcd for C14H22N3O6P2 [M − H]− 390.0984, found 390.0986.
5.20. Diethyl [1-(diethoxyphosphoryl)-2-(1-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-1H-1,2,3-triazol-4-yl)cyclopropyl]phosphonate (30).
To a suspension of sodium hydride (24 mg, 0.6 mmol) in anhydrous DMSO (0.3 mL) was added trimethylsulfoxonium bromide (132 mg, 0.6 mmol). The reaction was stirred for 1 hour, after which the vinyl bisphosphonate 28 (252 mg, 0.5 mmol) was added. The reaction was stirred for 4 hours and then quenched and diluted with brine. The aqueous layer was extracted seven times with ethyl acetate. The combined organic fractions were dried (Na2SO4) and filtered, and the filtrate was concentrated under reduced pressure. The resulting residue was purified by flash chromatography to afford compound 30 (192 mg, 74% yield). For the major isomer: 1H NMR (400 MHz, CDCl3) δ 7.49 (s, 1H), 5.38 (t, J = 6.7 Hz, 1H), 5.05 (m, 1H), 4.91 (m, 1H), 4.26 (m, 2H), 4.19 (p, J = 7.3 Hz, 2H), 4.05 (m, 2H), 3.73 (m, 2H), 3.26 (m, 1H), 2.22 – 1.97 (m, 5H), 1.85 (m, 1H), 1.76 (s, 3H), 1.67 (s, 3H), 1.58 (s, 3H), 1.37 (q, J = 7.4 Hz, 6H), 1.28 (t, J = 7.1 Hz, 3H), 1.08 (t, J = 7.1 Hz, 3H); 13C (100.6 MHz, CDCl3) δ 142.7, 142.6, 132.6, 123.4, 122.6, 118.1, 117.3, 63.1 (d, JPC = 6.6 Hz), 62.8 (d, JPC = 6.6 Hz), 62.5 (d, JPC = 6.6 Hz), 62.2 (d, JPC = 6.6), 47.9, 39.4, 26.1, 25.7, 23.4, 20.5 (dd, JPC = 4.4, 2.2), 19.1 (dd, JPC = 177.2, 167.6 Hz), 17.7, 16.5 – 16.0 (m 5C); 31P (162 MHz, CDCl3) 23.1 (d, J = 24.0 Hz, 1P), 21.1 (d, J = 24.0 Hz, 1P). HRMS (ESI) m/z calcd for C23H42N3O6P2 [M + H]+ 518.2549, found 518.2550.
5.21. Disodium (1-[bis(sodiooxy)phosphoryl]-2-(1-[3,7-dimethylocta-2,6-dien-1-yl]-1H-1,2,3-triazol-4-yl)cyclopropyl)phosphonate (31).
Following the general procedure for the preparation of compound 7, the title compound 31 was obtained from bisphosphonate 30 (65 mg, 0.13 mmol) as a white solid (30 mg, 0.061 mmol, 49%). For the major isomer: 1H NMR (400 MHz, D2O) δ 7.74 (s, 1H), 5.42 (t, J = 8.0 Hz, 7.2 Hz, 1H), 5.05 (m, 1H), 4.84 (m, 2H), 2.40 (m, 1H), 2.15 – 2.04 (m, 4H), 1.69 (s, 3H), 1.56 (s, 3H), 1.48 (s, 3H), 1.38 (m, 2H); 13C (100.6 MHz, D2O) δ 146.9 (m), 143.8, 133.7, 124.3, 123.9, 117.2, 47.8, 38.5, 25.4, 24.8, 22.4, 16.9, 16.7 (m, 2C), 15.6; 31P (162 MHz, D2O) 18.6 (br d, J = 27.0 Hz, 1P), 16.4 (d, J = 27.3, 1P). HRMS (ESI) m/z calcd for C15H26N3O6P2 [M − H]− 488.2087, found 488.2086.
5.22. [(2E,6E)-1-Hydroxy-3,7,11-trimethyl-1-phosphono-dodeca-2,6,10-trienyl]phosphonic acid tetrasodium salt (34).
To a flame dried and argon purged round bottom flask containing the acid chloride 32 (210 mg, 820 μmol) was added dropwise tris(trimethylsilyl)phosphite (550 μL, 1.60 mmol) and the reaction was allowed to stir at rt for 40 min. To the reaction was added anhydrous MeOH (5.0 mL) and the reaction was allowed to stir at rt for 12 h. To the reaction mixture was added 1 N NaOH (2.0 mL) and the reaction was allowed to stir for 80 min. After the solution was concentrated on a rotary evaporator, it was placed under high vacuum for 12 h. The resulting solid was dissolved in a minimum amount of water and the salt was precipitated by addition of 1:1 i-propyl alcohol:CH3CN (50 mL). The solid was collected by filtration to afford bisphosphonate 34 as a white solid (132 mg, 34% over 3 steps). For the major isomer: 1H NMR (500 MHz, D2O) δ 5.50 – 5.47 (m, 1H), 5.25 – 5.20 (m, 1H), 5.16 – 5.12 (m, 1H), 2.17 (s, 6H), 2.15 – 1.96 (m, 6H), 1.88 – 1.84 (m, 2H), 1.64 – 1.62 (m, 3H), 1.58 – 1.55 (m, 3H); 13C NMR (101 MHz, D2O) δ 139.2, 135.3, 134.6, 128.6, 123.4, 120.1, 79.1 (t, J = 129 Hz), 48.6, 42.4, 39.1, 31.8, 27.5, 24.1, 22.9, 22.7, 19.1, 17.0; 31P NMR (162 MHz, D2O) 18.0 ppm. HRMS (TOF ESI−) m/z calcd for C15H27O7P2 (M − H)− 381.1237, found 381.1227.
5.23. [2-[3,7-Dimethylocta-2,6-dienyl]triazol-4-yl]-1-hydroxy-1-phosphono-ethyl]phosphonic acid tetrasodium salt (37).
The title compound was prepared by extension of a literature procedure.40 To a flame dried and argon purged round bottom flask containing carboxylic acid 21 (237 mg, 900 μmol) was added catecholborane (1.0 M in THF, 2.79 mL, 2.79 mmol) and the reaction was allowed to stir at rt for 2 h. To the reaction was added dropwise tris(trimethylsilyl)phosphite (1.23 mL, 3.69 mmol) and the reaction was allowed to stir an additional 2 h. After the reaction was concentrated on high vacuum overnight, anhydrous MeOH (1 mL) was added and the reaction was allowed to stir for 5 min. The MeOH was removed under high vacuum and Et2O was added to the residue until a precipitate formed. The solid was filtered and washed with Et2O (1 mL). To the solid was added 1 N NaOH (5 mL) and the mixture was allowed to stir for 30 h at rt. The reaction was concentrated on a rotary evaporator to afford bisphosphonate 37 (65 mg, 15%). For the major isomer: 1H NMR (500 MHz, D2O) δ 7.77 (s, 1H), 6.77 (br d, 1H, disappears upon shaking with D2O), 5.46 – 5.33 (m, 1H), 5.03 (t, J = 7.2 Hz, 1H), 4.89 – 4.85 (m, 2H), 3.32 – 3.23 (m, 2H), 2.17 – 1.98 (m, 4H), 1.70 – 1.66 (m, 3H), 1.55 (s, 3H), 1.51 –1.45 (m, 3H); 13C NMR (126 MHz, D2O) δ 143.9, 143.0 (m), 133.9, 125.1, 123.7, 117.2, 73.2 (t, J = 134.9 Hz), 47.8, 38.6, 30.3, 25.6, 24.9, 16.9, 15.6; 31P NMR (202 MHz, D2O) 17.1 ppm. HRMS (TOF ESI−) m/z calcd for C14H24N3O7P2 (M − H)− 408.1090, found 408.1083.
5.24. GGDPS enzyme assay.
Recombinant GGDPS was kindly provided by Dr. Edward Snell (Hauptman-Woodward Medical Research Institute). Enzyme assays were performed as previously described.16 Compounds were tested in duplicate at multiple concentrations and three independent experiments were performed.
5.24. Immunoblot analysis.
RPMI-8226 cells (obtained from ATCC) were incubated with test compounds for 48 hrs. Whole cell lysate was obtained using RIPA buffer (0.15 M NaCl, 1% sodium deoxycholate, 0.1% SDS, 1% Triton (v/v) X-100, 0.05 M Tris HCl) containing protease and phosphatase inhibitors. Protein content was determined using the bicinchoninic acid (BCA) method (Pierce Chemical). Equivalent amounts of cell lysate were resolved by SDS-PAGE, transferred to polyvinylidene difluoride membrane, probed with the appropriate primary antibodies (anti-H-Ras (610001, BD Transduction Laboratories), anti-Rap1a (sc-373968, Santa Cruz Biotechnology), anti-β-tubulin (T-5201, Sigma)), and detected using HRP-linked secondary antibodies and Clarity ECL (for tubulin) or Millipore Immobilon ECL (for Ras and Rap1a) western blotting reagents per manufacturer’s protocols.
5.26. Lambda light chain ELISA.
Lysates generated for the immunoblot studies were utilized for intracellular lambda light chain quantification as determined by ELISA (Bethyl Laboratories).
5.27. MTT assay.
Cells were seeded (2.5 × 104 cells/100 μL per well) in 96-well flat-bottom plates and incubated with drugs for 48 hours. The MTT assay was performed as previously described.44 The absorbance for control cells treated with solvent only was defined as an MTT activity of 100%.
5.28. Statistical Analysis.
Two-tailed t-testing was used to calculate statistical significance. An α of 0.05 was set as the level of significance. Concentration response curves were analyzed via CompuSyn software (ComboSyn, Inc.,) to determine the IC50 values for the GGDPS enzyme assays.
Supplementary Material
Scheme 3.
Elaboration of intact triazoles.
Scheme 5.
Synthesis of α-hydroxy bisphosphonates from carboxylic acids.
Acknowledgements
Financial support from the Roy J. Carver Charitable Trust (01-224), the National Institutes of Health (R01 CA258621, P30 CA036727), and the American Society of Hematology is gratefully acknowledged. The Q-Exactive mass spectrometer used in this research was acquired through the National Science Foundation Major Research Instrumentation and the Chemical Instrumentation Programs (CHE-1919422).
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kuzuguchi T; Morita Y; Sagami I; Sagami H; Ogura K, Human geranylgeranyl diphosphate synthase. cDNA cloning and expression. J Biol Chem. 1999;274:5888–94. 10.1074/jbc.274.9.5888. [DOI] [PubMed] [Google Scholar]
- 2.Moomaw JF; Casey PJ, Mammalian protein geranylgeranyltransferase. Subunit composition and metal requirements. J Biol Chem. 1992;267:17438–43. [PubMed] [Google Scholar]
- 3.Seabra MC; Reiss Y; Casey PJ; Brown MS; Goldstein JL, Protein farnesyltransferase and geranylgeranyltransferase share a common alpha subunit. Cell. 1991;65:429–34. 10.1016/0092-8674(91)90460-g. [DOI] [PubMed] [Google Scholar]
- 4.Seabra MC; Goldstein JL; Südhof TC; Brown MS, Rab geranylgeranyl transferase. A multisubunit enzyme that prenylates GTP-binding proteins terminating in Cys-X-Cys or Cys-Cys. J Biol Chem. 1992;267:14497–503. [PubMed] [Google Scholar]
- 5.Kuchay S; Wang H; Marzio A; Jain K; Homer H; Fehrenbacher N; Philips MR; Zheng N; Pagano M, GGTase3 is a newly identified geranylgeranyltransferase targeting a ubiquitin ligase. Nat Struct Mol Biol. 2019;26:628–636. 10.1038/s41594-019-0249-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawson CD; Ridley AJ, Rho GTPase signaling complexes in cell migration and invasion. J Cell Biol. 2018;217:447–457. 10.1083/jcb.201612069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grosshans BL; Ortiz D; Novick P, Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci U S A. 2006;103:11821–7. 10.1073/pnas.0601617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haney SL; Wills VS; Wiemer DF; Holstein SA, Recent advances in the development of mammalian geranylgeranyl diphosphate synthase inhibitors. Molecules. 2017;22:886. https://doi.org/10.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lacbay CM; Waller DD; Park J; Gomez Palou M; Vincent F; Huang XF; Ta V; Berghuis AM; Sebag M; Tsantrizos YS, Unraveling the Prenylation-Cancer Paradox in Multiple Myeloma with Novel Geranylgeranyl Pyrophosphate Synthase (GGPPS) Inhibitors. J Med Chem. 2018;61:6904–6917. 10.1021/acs.jmedchem.8b00886. [DOI] [PubMed] [Google Scholar]
- 10.Holstein SA; Hohl RJ, Isoprenoid biosynthetic pathway inhibition disrupts monoclonal protein secretion and induces the unfolded protein response pathway in multiple myeloma cells. Leuk Res. 2011;35:551–9. 10.1016/j.leukres.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dykstra KM; Allen C; Born EJ; Tong H; Holstein SA, Mechanisms for autophagy modulation by isoprenoid biosynthetic pathway inhibitors in multiple myeloma cells. Oncotarget. 2015;6:41535–49. 10.18632/oncotarget.6365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haney SL; Chhonker YS; Varney ML; Talmon G; Smith LM; Murry DJ; Holstein SA, In Vivo Evaluation of Isoprenoid Triazole Bisphosphonate Inhibitors of Geranylgeranyl Diphosphate Synthase: Impact of Olefin Stereochemistry on Toxicity and Biodistribution. J Pharmacol Exp Ther. 2019;371:327–338. 10.1124/jpet.119.258624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shull LW; Wiemer AJ; Hohl RJ; Wiemer DF, Synthesis and biological activity of isoprenoid bisphosphonates. Bioorg Med Chem. 2006;14:4130–6. 10.1016/j.bmc.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 14.Wiemer AJ; Yu JS; Lamb KM; Hohl RJ; Wiemer DF, Mono- and dialkyl isoprenoid bisphosphonates as geranylgeranyl diphosphate synthase inhibitors. Bioorg Med Chem. 2008;16:390–9. 10.1016/j.bmc.2007.09.029. [DOI] [PubMed] [Google Scholar]
- 15.Zhou X; Ferree SD; Wills VS; Born EJ; Tong H; Wiemer DF; Holstein SA, Geranyl and neryl triazole bisphosphonates as inhibitors of geranylgeranyl diphosphate synthase. Bioorg Med Chem. 2014;22:2791–8. 10.1016/j.bmc.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wills VS; Allen C; Holstein SA; Wiemer DF, Potent triazole bisphosphonate inhibitor of geranylgeranyl diphosphate synthase. ACS Med Chem Lett. 2015;6:1195–8. 10.1021/acsmedchemlett.5b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matthiesen RA; Wills VS; Metzger JI; Holstein SA; Wiemer DF, Stereoselective synthesis of homoneryl and homogeranyl triazole bisphosphonates. J Org Chem. 2016;81:9438–9442. 10.1021/acs.joc.6b01693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allen C; Kortagere S; Tong H; Matthiesen RA; Metzger JI; Wiemer DF; Holstein SA, Olefin isomers of a triazole bisphosphonate synergistically inhibit geranylgeranyl diphosphate synthase. Mol Pharmacol. 2017;91:229–236. 10.1124/mol.116.107326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wills VS; Metzger JI; Allen C; Varney ML; Wiemer DF; Holstein SA, Bishomoisoprenoid triazole bisphosphonates as inhibitors of geranylgeranyl diphosphate synthase. Bioorg Med Chem. 2017;25:2437–2444. 10.1016/j.bmc.2017.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matthiesen RA; Varney ML; Xu PC; Rier AS; Wiemer DF; Holstein SA, alpha-Methylation enhances the potency of isoprenoid triazole bisphosphonates as geranylgeranyl diphosphate synthase inhibitors. Bioorg Med Chem. 2018;26:376–385. 10.1016/j.bmc.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhuiyan NH; Varney ML; Bhattacharya DS; Payne WM; Mohs AM; Holstein SA; Wiemer DF, omega-Hydroxy isoprenoid bisphosphonates as linkable GGDPS inhibitors. Bioorg Med Chem Lett. 2019;29:126633. 10.1016/j.bmcl.2019.126633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haney SL; Chhonker YS; Varney ML; Talmon G; Murry DJ; Holstein SA, Preclinical investigation of a potent geranylgeranyl diphosphate synthase inhibitor. Invest New Drugs. 2018;36:810–818. 10.1007/s10637-018-0571-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haney SL; Varney ML; Chhonker YS; Shin S; Mehla K; Crawford AJ; Smith HJ; Smith LM; Murry DJ; Hollingsworth MA; Holstein SA, Inhibition of geranylgeranyl diphosphate synthase is a novel therapeutic strategy for pancreatic ductal adenocarcinoma. Oncogene. 2019;38:5308–5320. 10.1038/s41388-019-0794-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haney SL; Varney ML; Chhonker Y; Talmon G; Smith LM; Murry DJ; Holstein SA, In vivo evaluation of combination therapy targeting the isoprenoid biosynthetic pathway. Pharmacol Res. 2021;167:105528. 10.1016/j.phrs.2021.105528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feldman AK; Colasson B; Sharpless KB; Fokin VV, The allylic azide rearrangement: achieving selectivity. J Am Chem Soc. 2005;127:13444–5. 10.1021/ja050622q. [DOI] [PubMed] [Google Scholar]
- 26.Coxon FP; Joachimiak L; Najumudeen AK; Breen G; Gmach J; Oetken-Lindholm C; Way R; Dunford JE; Abankwa D; Blazewska KM, Synthesis and characterization of novel phosphonocarboxylate inhibitors of RGGT. Eur J Med Chem. 2014;84:77–89. 10.1016/j.ejmech.2014.06.062. [DOI] [PubMed] [Google Scholar]
- 27.Marma MS; Xia Z; Stewart C; Coxon F; Dunford JE; Baron R; Kashemirov BA; Ebetino FH; Triffitt JT; Russell RG; McKenna CE, Synthesis and biological evaluation of alpha-halogenated bisphosphonate and phosphonocarboxylate analogues of risedronate. J Med Chem. 2007;50:5967–75. 10.1021/jm0702884. [DOI] [PubMed] [Google Scholar]
- 28.Marma MS; Khawli LA; Harutunian V; Kashemirov BA; McKenna CE, Synthesis of α-fluorinated phosphonoacetate derivatives using electrophilic fluorine reagents: Perchloryl fluoride versus 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) (Selectfluor®). J Flour Res. 2005;126:1467–1475. 10.1016/j.jfluchem.2005.04.002. [DOI] [Google Scholar]
- 29.McKenna CE; Higa MT; Cheung NH; McKenna M-C, The facile dealkylation of phosphonic acid dialkyl esters by bromotrimethylsilane. Tetrahedron Lett. 1977;18:155–158. [Google Scholar]
- 30.Dubey N; Sharma M; Kumar A; Sharma P, A click chemistry strategy to synthesize geraniol-coupled 1,4-disubstituted 1,2,3-triazoles and exploration of their microbicidal and antioxidant potential with molecular docking profile. Med Chem Res. 2015;24:2717–2731. 10.1007/s00044-015-1329-5. [DOI] [Google Scholar]
- 31.Lehnert W, Knoevenagel condensations with TiCl4 base. 4. Reactions of aldehydes and ketones with phosphonoacetic ester and methylenediphosphonates. Tetrahedron. 1974;30:301–305. 10.1016/s0040-4020(01)91461-9. [DOI] [Google Scholar]
- 32.Talele TT, The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules. J Med Chem. 2016;59:8712–8756. 10.1021/acs.jmedchem.6b00472. [DOI] [PubMed] [Google Scholar]
- 33.Ratrout SS; Al Sarabi A. e. M.; Sweidan KA, A One-Pot and Efficient Synthesis of Zoledronic Acid Starting from Tert-butyl Imidazol-1-yl Acetate. Pharm Chem J. 2015;48:835–839. 10.1007/s11094-015-1205-0. [DOI] [Google Scholar]
- 34.C KMC; Hudock MP; Zhang Y; Guo RT; Cao R; No JH; Liang PH; Ko TP; Chang TH; Chang SC; Song Y; Axelson J; Kumar A; Wang AH; Oldfield E, Inhibition of geranylgeranyl diphosphate synthase by bisphosphonates: a crystallographic and computational investigation. J Med Chem. 2008;51:5594–607. 10.1021/jm800325y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lecouvey M; Mallard I; Bailly T; Burgada R; Leroux Y, A mild and efficient one-pot synthesis of 1-hydroxymethylene-1,1-bisphosphonic acids. Preparation of new tripod ligands. Tetrahedron Lett. 2001;42:8475–8478. 10.1016/S0040-4039(01)01844-5. [DOI] [Google Scholar]
- 36.Legigan T; Migianu-Griffoni E; Redouane MA; Descamps A; Deschamp J; Gager O; Monteil M; Barbault F; Lecouvey M, Synthesis and preliminary anti-cancer evaluation of new triazole bisphosphonate-based isoprenoid biosynthesis inhibitors. Eur J Med Chem. 2021;214:113241. 10.1016/j.ejmech.2021.113241. [DOI] [PubMed] [Google Scholar]
- 37.Ruel R; Bouvier J-P; Young RN, Single-Step Preparation of 1-Hydroxybisphosphonates via Addition of Dialkyl Phosphite Potassium Anions to Acid Chlorides. J Org Chem. 1995;60:5209–5213. 10.1021/jo00121a044. [DOI] [Google Scholar]
- 38.Kulkarni YS; Snider BB, Intramolecular [2 + 2] cycloadditions of ketenes. 2. Synthesis of chrysanthenone, .beta.-pinene, .beta.-cis-bergamotene, and .beta.-trans-bergamotene. J Org Chem. 1985;50:2809–2810. 10.1021/jo00215a054. [DOI] [Google Scholar]
- 39.Kulkarni YS; Niwa M; Ron E; Snider BB, Synthesis of terpenes containing the bicyclo[3.1.1]heptane ring system by the intramolecular [2 + 2] cycloaddition reaction of vinylketenes with alkenes. Preparation of chrysanthenone, .beta.-pinene, .beta.-cis-bergamotene, .beta.-trans-bergamotene, .beta.-copaene, and .beta.-ylangene and lemnalol. J Org Chem. 1987;52:1568–1576. 10.1021/jo00384a035. [DOI] [Google Scholar]
- 40.Egorov M; Aoun S; Padrines M; Redini F; Heymann D; Lebreton J; Mathé-Allainmat M, A One-Pot Synthesis of 1-Hydroxy-1,1-bis(phosphonic acid)s Starting from the Corresponding Carboxylic Acids. Eur J Org Chem. 2011;2011:7148–7154. 10.1002/ejoc.201101094. [DOI] [Google Scholar]
- 41.Szajnman SH; Ravaschino EL; Docampo R; Rodriguez JB, Synthesis and biological evaluation of 1-amino-1,1-bisphosphonates derived from fatty acids against Trypanosoma cruzi targeting farnesyl pyrophosphate synthase. Bioorg Med Chem Lett. 2005;15:4685–90. 10.1016/j.bmcl.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 42.Lv M; Wang M; Lu K; Peng L; Zhao Y, An efficient synthesis of 2-Aminoethylidene-1,1-Bisphosphonates derivatives via Michael addition reaction. Phosphorus Sulfur. 2018;193:149–154. 10.1080/10426507.2017.1393421. [DOI] [Google Scholar]
- 43.Lodewyk MW; Lui VG; Tantillo DJ, Synthesis of (sulfonyl)methylphosphonate analogs of prenyl diphosphates. Tetrahedron Lett. 2010;51:170–173. 10.1016/j.tetlet.2009.10.119. [DOI] [Google Scholar]
- 44.Holstein SA; Hohl RJ, Interaction of cytosine arabinoside and lovastatin in human leukemia cells. Leuk Res. 2001;25:651–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.