

Abstract
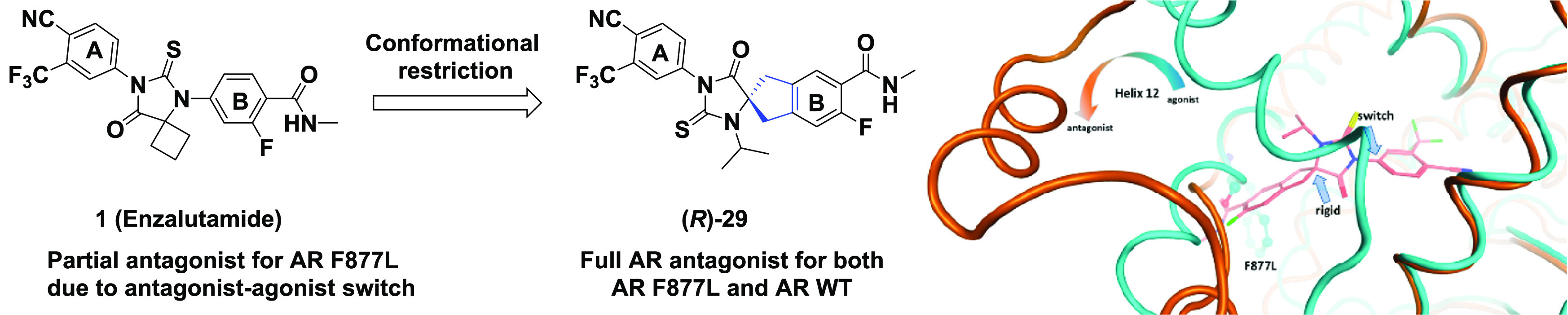
Androgen receptor (AR) transcriptional reactivation plays a key role in the development and progression of lethal castration-resistant prostate cancer (CRPC). Recurrent alterations in the AR enable persistent AR pathway signaling and drive resistance to the treatment of second-generation antiandrogens. AR F877L, a point mutation in the ligand binding domain of the AR, was identified in patients who acquired resistance to enzalutamide or apalutamide. In parallel to our previous structure–activity relationship (SAR) studies of compound 4 (JNJ-pan-AR) and clinical stage compound 5 (JNJ-63576253), we discovered additional AR antagonists that provide opportunities for future development. Here we report a highly potent series of spirocyclic thiohydantoins as AR antagonists for the treatment of the F877L mutant and wild-type CRPC.
Keywords: Androgen receptor, spirocyclic, thiohydantoin, resistance, apalutamide, LBD, prostate cancer
Molecular profiling studies have shown that recurrent genomic alterations in the master regulator androgen receptor (AR) and its pathway is a common feature that drives resistance to the second-generation AR-targeted therapies abiraterone acetate, enzalutamide (1, Figure 1), and apalutamide (3, Figure 2) for the treatment of advanced prostate cancer.1−5 AR transcriptional reactivation and persistent AR signaling are now understood as the central cores of resistance mechanisms in disease progression leading to lethal metastatic castration-resistant prostate cancer (mCRPC).6,7 Sustained AR signaling in CRPC tumors has been reported to be the result of numerous genomic aberrations including well-documented gene alteration, amplification, overexpression, splice variant isoform expression (AR-V7), ligand binding domain (LBD) point mutations, and glucocorticoid receptor (GR) bypass.1,8−12 Colloquially dubbed as “A Resilient Foe”, retargeting the AR by molecularly targeted therapy with a precision medicine approach still represents a major therapeutic opportunity for the treatment of mCRPC.13
Figure 1.

Chemical structures of enzalutamide (1) and darolutamide (2).
Figure 2.

Chemical structures of 3 (apalutamide ARN-509), 4 (JNJ-pan-AR), 5 (JNJ-63576253), and bioisosteric spirocyclic scaffolds 6, 7, and 8 proposed as antagonists of the AR WT and AR F877L. Compound class 8 was the focus of our lead optimization efforts.
The AR belongs to the steroid hormone group of nuclear receptors that includes the estrogen receptor (ER), GR, and progesterone receptor (PR), and it functions as a ligand-inducible transcription factor that is activated by the binding of androgens such as testosterone and dihydrotestosterone (DHT) to its LBD.14 Point mutations in the AR LBD have been associated with 10–20% of the resistance that converts antagonists into agonists upon ligand binding, driving the disease through the reactivation of AR signaling.15 For example, the missense mutation of phenylalanine to leucine at AR amino acid 877 (F877L, formerly known as F876L) was reported in 2013 to confer resistance to both enzalutamide and apalutamide, leading to an antagonist-to-agonist switch.10,11 AR F877L was found to occur spontaneously in cells after prolonged treatment with enzalutamide and apalutamide. AR F877L was also detected in the plasma circulating tumor DNA (ctDNA) for 3 of 29 progressing patients enrolled in the apalutamide Phase I clinical trial.15 Recently, the next-generation AR antagonist darolutamide (ODM-201, 2, Figure 1) was reported to overcome resistance mechanisms including AR F877L mutation and was subsequently approved for nonmetastatic castration-resistant prostate cancer (nmCRPC) by the U.S. Food and Drug Administration (FDA).16
Recently, we published our work on the characterization and optimization of compound 4 (JNJ-pan-AR). focusing on the substituents of ring “A” as well as ring “B” and its periphery, leading to the discovery of the clinical stage compound 5 (JNJ-63576253).17 In our putative AR homology model, the mechanism for the antagonist-to-agonist switch of enzalutamide (1) or apalutamide (3) in the AR F877L mutant tumors could be explained by comparing the open (“antagonistic”) and closed (“agonistic”) conformations, respectively, of Helix 12 located in the LBD.17−20
To complement our reported work on rings “A” and “B” of compound 4, we decided to expand our exploration of the hydantoin core by investigating additional spirocyclic compound classes such as scaffolds 6, 7, and 8 in an effort to preserve the key structural features of the pharmacophore, reduce the ligand conformational flexibility, and improve the suboptimal pharmacological parameters of compound 4. Here we report our work on the discovery of a series of spirocyclic thiohydantoins based on scaffold 8 that are highly potent antagonists of the AR WT and AR F877L (Figure 2).
Our early models of bicalutamide, enzalutamide (1), and apalutamide (3) suggested that reducing the flexibility within this class of ligands by conformational restriction would have an impact on the antagonist-to-agonist switch in AR F877L without the need for peripheral bulky substituents (Supplementary Section S-12).21,23,24 To test this hypothesis (Figure 3, Supplementary Section S-12), we designed rigidified scaffolds 6, 7, and 8 with a spirocyclic central core to add rotational restriction in thiohydantoin series (Figure 2). The concept of conformation restriction by the cyclization of an acyclic group via a bioisosteric spirocyclic ring has emerged as an effective approach in drug discovery in the past decade.22 Initially, we explored the structure–activity relationship (SAR) for compounds in classes 6 and 7, but unfortunately, either significant a loss of potency (6) or a surprising enhanced intrinsic agonism (7) rendered these compounds difficult to progress (data not shown). Thus we focused on scaffold 8 while simultaneously taking advantage of the insight gained from the development of 5 (JNJ-63576253).
Figure 3.
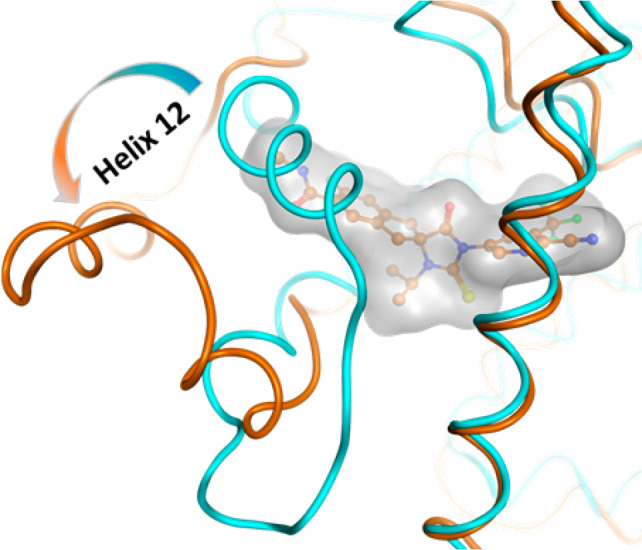
Manual docking model of (R)-28 (orange balls and sticks with gray surface) bound to a potentially antagonistic conformation (orange tubes) of the AR LBD. Superposition of a crystal structure of the AR in a putative agonistic conformation is shown in cyan tubes (PDB ID: 1T5Z).
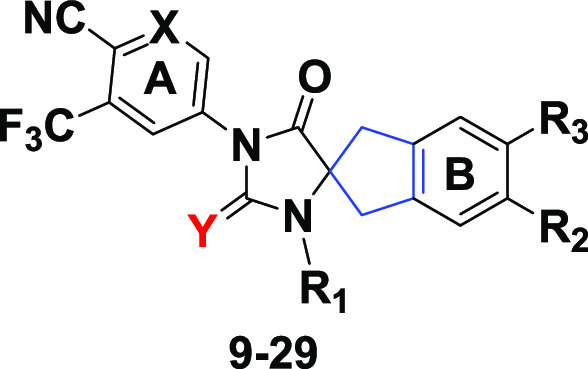
In contrast with our previous SAR studies, our exploration of scaffold 8 preserved the key pharmacophore elements of peripheral rings “A” and “B” in 1 (enzalutamide), 3 (apalutamide), and 4 (JNJ-pan-AR) to better understand the impact of the central spirocyclic core alteration, as shown in analogues 9–29 (Table 1). For example, compound 16 preserved two key features of 4: a piperidinyl group on ring “B” and an identical “A” ring. Similarly, compounds 14, 19, and (R)-29 kept the same “A” and “B” rings as 1.
Table 1. Chemical Structures of Compounds 9–29.

The general syntheses of analogues 9–16, 18–29, and metabolite 17 are outlined in Schemes 1–4. In Scheme 1, substituted 2-aminoindan-2-carboxylic ester 32 was prepared starting from 30 following literature procedures.25,26 Reductive alkylation reactions of 32 with an aldehyde such as isobutyraldehyde or a ketone such as acetone, cyclobutanone, cyclopentanone, or cyclohexanone in the presence of sodium triacetoxyborohydride provided N-alkylated 34, which was then cyclized to 36 by heating with isothiocyanate 35. The Pd-catalyzed Heck aminocarbonylation of 36 using W(CO)6 provided racemic analogues 18–22.27 The chiral supercritical fluid chromatography (SFC) separation of 18 and 19 led to the corresponding chirally pure (S)-26, (S)-27 and (R)-28, (R)-29. Because we were unable to obtain crystal structure of these molecules, the assignments of the absolute stereochemistry for 26–29 were based on vibrational circular dichroism (VCD) experiments (Supplementary Sections S-8 and S-9). Confirmation of these assignments by unambiguous asymmetric synthesis was beyond the scope of this program.
Scheme 1. Synthesis of Spirocyclic Thiohydantoin Analogues 18–22 and 26–29.

Reagents and conditions: (a) NBS, AIBN (cat), CCl4, 85 °C; (b) K2CO3, 18-crown-6, MeCN, rt; (c) aq. HCl, rt; (d) NaBH(OAc)3, DCE, rt; (e) TEA, THF/DMF, 80 °C; (f) MeNH2·HCl (or MeNHOMe·HCl), W(CO)6 (cat.), Pd(OAc)2 (cat.), Xantphos (cat.), TEA, 1,4-dioxane, 80 °C; (g) SFC chiral separation.
Scheme 4. Synthesis of Spirocyclic Thiohydantoin Analogues 23–25.

Reagents and conditions: (a) Zn(CN)2, Pd2dba3, PPh3, DMA, 120 °C; (b) aqueous conc. HCl, 135 °C; (c) TEA, THF/DMF, 80 °C; (d) NH4Cl, HBTU, DIPEA, DMAP (cat.), DMF, rt.
In Scheme 2, commercially available or synthetically readily accessible 37 (R2 = H or F) or synthetically readily accessible 32 (R2 = F) was protected with a Boc group to give 38.25,26 The N-methylation of 38 with iodomethane and sodium hydride followed by Pd-catalyzed Heck aminocarbonylation of 39 using W(CO)6 and the subsequent removal of the Boc group provided 41, which was cyclized with isothiocyanate 35 or isocyanate 42 to analogues 11–15. Analogues 11–15 could also be prepared by the Boc deprotection of 39 and elaboration following the methods previously described in Scheme 1. Compounds 9 and 10 were prepared from 38 by the procedures described in Scheme 2 but omitting N-methylation (step b) (Supplementary Section S-7).
Scheme 2. Synthesis of Spirocyclic Thiohydantoin or Hydantoin Analogues 11–15.

Reagents and conditions: (a) Boc2O, TEA, DCM, rt; (b) MeI, NaH, DMF, rt; (c) MeNHOMe·HCl, W(CO)6 (cat.), Pd(OAc)2 (cat.), Xantphos (cat.), DMAP, K3PO4, 1,4-dioxane, microwave, 120 °C; (d) TFA, DCM, rt; (e) TEA, THF, DMF, 80 °C.
In Scheme 3, phenol 44 was obtained by converting aryl bromide 39a to boronic ester 43 followed by oxidative cleavage. The subsequent removal of the Boc group and the cyclization of 45 with isothiocyanate 35 led to intermediate 17, which was then converted to analogue 16 by the Mitsunobu reaction and subsequent treatment with HCl. In Scheme 4, the palladium-catalyzed cyanation of aryl bromide 34 using Zn(CN)2 under mild conditions afforded nitrile 47, which was hydrolyzed to diacid 48 and subsequently cyclized to 49 without isolation by heating with isothiocyanate 35. By comparison with the route illustrated in Scheme 1, this route offered several advantages. The final step involving a flexible amide coupling reaction not only provided diversified “B”-ring carboxamide derivatives but also avoided potential contamination of the final product with palladium, in contrast with the Pd-catalyzed Heck aminocarbonylation used in the last step of Scheme 1.
Scheme 3. Synthesis of Spirocyclic Thiohydantoin Analogue 16 and Phenol Metabolite 17.

Reagents and conditions: (a) Pd(dppf)Cl2, KOAc, 1,4-dioxane, 80 °C; (b) oxone, acetone, rt; (c) TFA, DCM, rt; (d) TEA, THF, DMF, 80 °C; (e) DEAD, PPh3, THF, 60 °C; (f) HCl, 1,4-dioxane, rt.
To evaluate the SAR of these analogues for their AR antagonistic activities, ARE-luciferase reporter constructs were introduced into LNCaP prostate adenocarcinoma cancer cells that stably expressed either the WT AR (LNCaP AR cs) or the F877L mutant AR (LNCaP F877L) in a native AR setting.28 The analogues 9–16 and 18–29 all acted as full antagonists in both cell lines and inhibited luciferase transcription with IC50 values ranging from 50 to >30 000 nM. Compounds 1 (enzalutamide) and 4 (JNJ-pan-AR) were used as controls (Table 2). No agonism of either AR F877L or the AR WT was observed for spirocyclic compounds under the assay conditions.
Table 2. Transcriptional Reporter Assay Activity of 9–16 and 19–29 in LNCaP F877L (Mutant) and LNCaP AR cs (WT) Cells.
| compound | LNCaP F877La IC50 (nM)b | LNCaP WTa IC50 (nM)b |
|---|---|---|
| 1 | not fit | 117 ± 66 |
| 4 | 98 ± 63 | 191 ± 120 |
| 9 | >15000c | >15000d |
| 10 | 5012 ± 207 | 11220 ± 3826e |
| 11 | 676 ± 170 | 1380 ± 141 |
| 12 | 525 ± 322 | 646 ± 113 |
| 13 | 759 ± 81 | 1096 ± 7 |
| 14 | 339 ± 119 | 398 ± 52 |
| 15 | 1660 ± 1081 | >30000 |
| 16 | 178 ± 86 | 219 ± 116 |
| 18 | 145 ± 32 | 112 ± 28 |
| 19 | 93 ± 32 | 93 ± 30 |
| 20 | 102 ± 10 | 100 ± 20 |
| 21 | 123 ± 39 | 126 ± 8 |
| 22 | 257 ± 13 | 191 ± 11 |
| 23 | 170 ± 37 | 229 ± 25 |
| 24 | 132 ± 33 | 148 ± 39 |
| 25 | 174 ± 4 | 209 ± 6 |
| (S)-26 | 112 ± 116 | 178 ± 41 |
| (S)-27 | 151 ± 26 | 170 ± 19 |
| (R)-28 | 65 ± 47 | 52 ± 37 |
| (R)-29 | 78 ± 10 | 72 ± 23 |
LNCaP F877L and LNCaP WT reporter assays were repeated two times or more. All compounds except 1 were antagonists in AR F877L, and all were antagonists in the AR WT.
Max inhibition >90% for all compounds unless specifically mentioned.
Max inhibition 52%.
Max inhibition 35%.
Max inhibition 80%.
The unsubstituted (R1 = H) analogues 9 (F877L IC50 > 15 000 nM; WT IC50 > 15 000 nM) and 10 (F877L IC50 = 5012 nM; WT IC50 = 11 220 nM) were at least 10 times less potent compared with the corresponding N-methylated (R1 = Me) compounds 11 (F877L IC50 = 676 nM; WT IC50 = 1380 nM) and 12 (F877L IC50 = 525 nM; WT IC50 = 646 nM). However, the potencies of fluorinated (R2 = F) 13 (F877L IC50 = 759 nM; WT IC50 = 1039 nM) and 14 (F877L IC50 = 339 nM; WT IC50 = 398 nM) were almost equal to or slightly better than those of nonfluorinated (R2 = H) 11 and 12. By contrast, hydantoin 15 (F877L IC50 = 1660 nM; WT IC50 = 5129 nM) lost significant potency compared with the corresponding thiohydantoin 12 (F877L IC50 = 525 nM; WT IC50 = 646 nM), confirming the importance of the thiocarbonyl moiety. It was notable that analogue 16 (F877L IC50 = 178 nM; WT IC50 = 219 nM) was a full antagonist and was almost equally potent as benchmark 4 (F877L IC50 = 98 nM; WT IC50 = 191 nM); both compounds possess a piperidinyloxy substituent on ring “B”. A further potency increase was achieved in 18 (R1 = i-Pr, F877L IC50 = 145 nM; WT IC50 = 112 nM), 19 (R1 = i-Pr, F877L IC50 = 93 nM; WT IC50 = 93 nM), 20 (R1 = i-Bu, F877L IC50 = 102 nM; WT IC50 = 100 nM), and 21 (R1 = i-Bu, F877L IC50 = 123 nM; WT IC50 = 126 nM). An additional derivatization of R1 in 22–25 did not further enhance the potency. Nevertheless, the potencies of 18–21 were comparable to or slightly better than that of 4. Interestingly, chiral (R)-28 (F877L IC50 = 65 nM; WT IC50 = 52 nM) and (R)-29 (F877L IC50 = 78 nM; WT IC50 = 72 nM) were about two times more potent than the corresponding (S)-26 (F877L IC50 = 112 nM; WT IC50 = 178 nM) and (S)-27 (F877L IC50 = 151 nM; WT IC50 = 170 nM). The assignments of absolute stereochemistry for 26–29 were based on VCD experiments due to the unsuccessful efforts to obtain single crystals for X-ray studies and the inability to predict the potency difference in our homology model.29,30 Significantly, these analogues remained full antagonists in both AR F877L and the AR WT despite having the same “B” substituents as 1 (enzalutamide) or 3 (apalutamide), both of which are agonists in AR F877L. This observation suggests the intrinsic propensity of scaffold 8 for antagonism against both AR F877L and the AR WT as well as for selectivity over the GR (Figure 4).12
Figure 4.
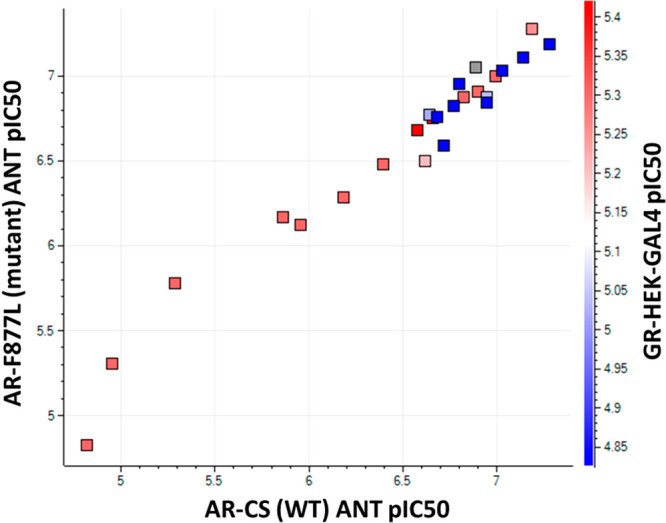
Correlation between AR F877L (y axis) and the AR WT (x axis) antagonistic pIC50 and the AR WT pIC50 in transporter assays along with the GR antagonistic pIC50 (red–blue color scale).
To correlate the AR antagonism with an antiproliferative effect in androgen-dependent tumor cell lines, 12, 16, 18–20, and 26–29 were also evaluated in a growth inhibition assay in the AR WT-dependent VCaP cells, again using 1 and 4 as comparators.31 Luciferase transcription inhibition appeared to translate into antiproliferative activity in VCaP cells, tracking well with LNCaP WT potency with IC50 values ranging from 10 to 2440 nM (Table 3). The potencies of 16, 18–20, (R)-28, and (R)-29 were comparable to or better than those of 1 and 4.
Table 3. Antiproliferative Activity of 12, 16, 18–20, and 26–29 in the VCaP Prostate Cancer Cell Line Compared with That of 1 and 4a.
| compound | 1 | 4 | 12 | 16 | 18 | 19 | 20 | (S)-26 | (S)-27 | (R)-28 | (R)-29 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| VCaP IC50 (nM) | 149 ± 30 | 92 ± 71 | 2440 ± 2010 | 40 ± 40 | 30 ± 20 | 50 ± 20 | 10 ± 10 | 680 ± 150 | 480 ± 50 | 40 ± 10 | 110 ± 30 |
VCaP cells (WT AR) were cultured in the presence of 30 pM R1881, and the extent of androgen-dependent proliferation was calculated. The VCaP antiproliferative assay was repeated two times or more.
In mouse single-dose pharmacokinetic (PK) studies, (R)-29, the more potent enantiomer of 19 (Table 1), displayed a higher area under the receiver operating characteristic (ROC) curve (AUC) (104 μg·h/mL) after oral dosing and lower clearance (CL 0.9 mL/min/kg) after IV dosing compared with the corresponding, less potent (S)-27 (AUC 50.4 μg·h/mL, CL 3.0 mL/min/kg) (Table 3). The same trend was observed by comparing (R)-28 and (S)-26 in terms of the exposure and clearance, indicating the subtle difference between (R)- and (S)-enantiomers in terms of the PK characteristics. The overall PK parameters of (R)-29 tracked well with its corresponding racemate, 19. All analogues displayed favorable PK parameters, with oral bioavailability ranging from 61 to >100% (Table 4).
Table 4. Human Liver Microsomal (HLM) T1/2, Permeability, and Mean Single-Dose PK Parameters of Compounds 16, 18, 19, and 26–29 in CD-1 Male Mice by PO and IV Dosing.
| compound | 16 | 18 | 19 | (S)-26 | (S)-27 | (R)-28 | (R)-29 |
|---|---|---|---|---|---|---|---|
| HLM_T1/2 (min)a | >180 | >180 | >180 | 118 | >180 | 135 | >180 |
| Papp A > B (+inh.) (cm/s × 10–6)c | 6.4 | 36.5 | 35.2 | 35.5 | 24.4 | 16.6 | 34.5 |
| PO dose (mg/kg) | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| AUC0-inf (μg·h/mL) | 15.2 | 27.0 | 107 | 39.1 | 50.4 | 99.4 | 104 |
| Cmax (μM) | 0.92 | 3.69 | 8.85 | 3.7 | 5.81 | 6.1 | 5.86 |
| IV dose (mg/kg) | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| CL (mL/min/kg) | 11.5 | 3.4 | 1.6 | 4.0 | 3.0 | 1.3 | 0.9 |
| T1/2 (h) | 16.7 | 19.5 | 14.8 | 6.9 | 5.2 | 15.8 | 10.3 |
| Vdss (L/kg) | 16.3 | 4.8 | 1.7 | 2.3 | 1.8 | 1.9 | 1.7 |
| F (%)b | 97 | 61 | 102 | 92 | 91 | 80 | 63 |
For HLM T1/2: high stability >180 min; 33 min < medium stability <180 min; low stability < 33 min.
Oral bioavailability.
Passive permeability was measured from the apical (A) to the basolateral side (B) of the MDCK-MDR1 cells in the presence of a P-glycoprotein (P-gp) inhibitor.
Compounds 16, 19, (S)-26, (S)-27, (R)-28, and (R)-29 were evaluated in rats for their inhibitory effect on the growth of androgen-sensitive organs (ASOs) under stimulation by testosterone propionate (TP) in the Hershberger assay to assess their in vivo antiandrogen activities against the WT AR (Figure 5). Treatment with compounds 16, (S)-26, and (R)-28 resulted in statistically significant reductions in ASOs versus the TP control at 30 mg/kg once daily oral dosing for 10 days (p > 0.0001; Figure 5), comparable to that of positive control enzalutamide (1) at 30 mg/kg. Importantly, compound 19 showed a similar reduction in ASOs at 5 mg/kg. In a separate study, treatment with compound (R)-29 resulted in statistically significant efficacy at 5 mg/kg once daily oral dosing for 10 days (p ≤ 0.0001; Figure 5) compared with that of positive-control flutamide at 3 mg/kg. In contrast, compound (S)-27 showed minimal effects on ASOs at 5 mg/kg (Figure 5), consistent with its less robust in vitro AR antagonistic potency (Tables 1 and 2) and lower in vivo PK exposure compared with (R)-29 (Table 4).
Figure 5.

Hershberger assay: Dosing effect of compounds 16 (30 mg/kg), 19 (5 mg/kg), (S)-26 (30 mg/kg), and (R)-28 (30 mg/kg) compared with control group 1 (enzalutamide, 30 mg/kg) and (R)-29 (5 mg/kg) and (S)-27 (5 mg/kg) compared with control group flutamide (3 mg/kg) on ASOs. The ASO development of seminal vesicles and coagulating glands (SVCG) and the ventral prostate (VP) is shown. The compound-dependent suppression of ASOs is significant for each compound tested (p ≤ 0.0001, t-test/Mann–Whitney). Data are the mean ± SD (n = 6).
We previously reported the identification of phenolic derivative 50 (Table 5) as a key metabolite of compound 4 (JNJ-pan-AR, Figure 2). In vitro experiments demonstrated that 50 could be bioactivated in glutathione (GSH) trapping experiments, as indicated in Table 5, by the formation of GSH adducts such as the putative catechol structure 51.17 It is notable that the structural features in the periphery of ring “B” in compound 16 closely resemble those of compound 4 (JNJ-pan-AR), thus raising the possibility of similar bioactivation.32 Like 4, phenolic 17 was indeed detected in a small amount (1.4%) in trapping experiments together with a trace amount of two GSH adducts. The small amount of 17 reflected the low turnover of 16 under the in vitro assay conditions. Furthermore, phenolic 17 was moderately metabolized in the same experiments to generate at least four GSH adducts in significant amounts, including the two observed for 16: UD+GSH-2H (10.7%) and UD+O+GSH-2H (30.1%). Presumably, one of the GSH adducts (UD+GSH-2H) was formed at the para-benzylic position of ring “B”. These results suggested that compound 16 remained vulnerable to the bioactivation liability, and the next round of SAR iteration should investigate bioisosteric “B” rings such as pyridyl to mitigate potential bioactivation risks. We previously used a similar approach in the optimization of compound 4, resulting in the successful identification of clinical stage compound 5 (JNJ-63576253).17 However, such additional B-ring modifications in the spiro series are beyond the scope of this work.
Table 5. Incubation of 16, 17, and 50 in Dog Liver Microsomes with GSH in the Absence or Presence of NADPHa.

| compound | 50 | 17 | 16 | |||
|---|---|---|---|---|---|---|
| NADPHb | – | + | – | + | – | + |
| UDc | 100.0% | 78.7% | 100.0% | 52.3% | 100.0% | 98.4% |
| UD+O | 9.2% | 2.0% | 0.19% | |||
| phenolic (50 or 17)d | NA | NA | NA | NA | 0.01% | 1.4% |
| UD+GSH-2H (a) | 10.7% | 0.002%d | ||||
| UD+GSH-2H (b) | 1.8% | |||||
| UD+O+GSH-2H | 12.1% | 30.1% | 0.01%d | |||
| UD+O+GSH | 3.1% | |||||
Final concentrations of the substrates (UD), dog liver microsomes, glutathione (GSH), and NADPH were 10 μM, 1 mg/mL, 5 mM, and 1 mM, respectively. The percent compositions of the parent drug and its metabolites were based on peak areas from 5 ppm accurate mass measurements with the assumption of equal positive electrospray ionizations.
–/+ means in the absence or in the presence of NADPH.
UD is defined as incubated substrate unchanged.
Phenolic refers to either metabolite 50 or 17.
GSH conjugates detected from incubation with 16 were from metabolite 17.
Next, we tested the efficacy of compounds (S)-26, (S)-27, (R)-28, and (R)-29 for their ability to suppress prostate tumor xenograft growth in vivo. Castrated male SHO mice bearing the established LNCaP AR F877L mutant-driven 3-D spheroid tumors were orally administered vehicle alone or compound (S)-26, (S)-27, (R)-28, or (R)-29 at 15 or 30 mg/kg once daily for 18 days. As shown in Figure 6, the continuous administration of compound (S)-26, (S)-27, (R)-28, or (R)-29 resulted in statistically significant tumor growth inhibition compared with the vehicle-treated control group: (S)-26 (30 mg/kg, 58%, p = 0.0002), (S)-27 (30 mg/kg, 32%, p = 0.0309), (R)-28 (30 mg/kg, 46%, p = 0.0152), and (R)-29 (15 mg/kg, 57%, p = 0.0008). Notably, compound (R)-29 had similar efficacy as compound (S)-26 but at half the dose (15 vs 30 mg/kg). This is likely a result of the improved in vitro potency of (R)-29 combined with the lower in vivo clearance and the higher exposure. A minimal impact on body weight loss was observed, indicating that all compounds were well tolerated. Together, these in vivo data provide evidence that spirocyclic AR antagonist compounds not only inhibited the growth of ASOs but also demonstrated statistically significant in vivo activity against the mutant AR-driven tumor growth in this LNCaP AR F877L xenograft model.
Figure 6.

Oral in vivo efficacy profile of (S)-26, (S)-27, (R)-28, and (R)-29 in castrated male SHO mice implanted with LNCaP F877L tumors cells. Tumors were measured twice weekly, and the results are presented as the average tumor volume, expressed as mm3 ± the standard error of mean (SEM) of each group.
In summary, we described the discovery of a new series of AR antagonists with a spirocyclic thiohydantoin core represented by scaffold 8. SAR studies revealed the intrinsic propensity of this bioisoteric replacement of the core ring for full antagonism against both the wild-type and the F877L mutant AR. Our findings highlighted the advantage of this conformationally restricted spirocyclic thiohydantoin core to avoid reliance on peripheral functional groups to impart full antagonism of both the wild-type and the mutant AR. We also described the favorable in vitro and in vivo pharmacological properties of (R)-29 as a full antagonist against AR F877L. This result stands in sharp contrast with the antagonist-to-agonist switch observed with 1 (enzalutamide), despite (R)-29 and 1 sharing almost identical structural features of peripheral rings “A” and “B”. Although our hypothesis on the impact of conformational restriction on the antagonist-to-agonist switch appears to be valid within the series derived from scaffold 8, we are unable to fully rationalize the mechanism of these antagonists at the molecular level. It will be useful to further validate our hypothesis using experimental approaches such as X-ray crystallography and nuclear magnetic resonance (NMR) in combination with molecular simulations. Nevertheless, the distinctive molecular architecture of scaffold 8 represents an appealing opportunity for the further exploration and development of next-generation AR pathway inhibitors.
Acknowledgments
We thank Kathryn Packman (Janssen R&D, Cambridge, MA) and Leopoldo Luistro (Janssen R&D, Spring House, PA) for their contributions to the in vivo Hershberger and LNCaP AR F877L xenograft model studies; Samantha Allen (Janssen R&D, Spring House, PA) and Lori Westover (Janssen R&D, Spring House, PA) for their contributions to the transcriptional reporter assays; Bing Huang (Wuxi AppTec, Shanghai, China) and Christopher Teleha (Janssen R&D, Spring House, PA) for their contributions to the preparation and chiral SFC separation of the compounds described herein; and Jordan Nafie (BioTools, Jupiter, FL), Rina Dukor (BioTools, Jupiter, FL), and Ann Vos (Janssen R&D, Beerse, Belgium) for VCD experiments.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00032.
Assay and protocols: transcriptional reporter, proliferation, Hershberger, tumor xenograft efficacy, GSH screen for phenolic metabolites, detailed chemistry, VCD experiments, SFC traces, and modeling (PDF)
Author Present Address
∥ I.H.: Cancer Research UK Newcastle Drug Discovery Unit, Newcastle University, Newcastle upon Tyne, United Kingdom.
Author Present Address
⊥ J.R.B.: F. Hoffmann-La Roche Ltd., Molecular Targeted Therapies (Oncology), Basel, Switzerland.
The authors declare no competing financial interest.
Supplementary Material
References
- Quigley D. A.; Dang H. X.; Zhao S. G.; Lloyd P.; Aggarwal R.; Alumkal J. J.; Foye A.; Kothari V.; Perry M. D.; Bailey A. M.; Playdle D.; Barnard T. J.; Zhang L.; Zhang J.; Youngren J. F.; Cieslik M. P.; Parolia A.; Beer T. M.; Thomas G.; Chi K. N.; Gleave M.; Lack N. A.; Zoubeidi A.; Reiter R. E.; Rettig M. B.; Witte O.; Ryan C. J.; Fong L.; Kim W.; Friedlander T.; Chou J.; Li H.; Das R.; Li H.; Moussavi-Baygi R.; Goodarzi H.; Gilbert L. A.; Lara P. N. Jr.; Evans C. P.; Goldstein T. C.; Stuart J. M.; Tomlins S. A.; Spratt D. E.; Cheetham R. K.; Cheng D. T.; Farh K.; Gehring J. S.; Hakenberg J.; Liao A.; Febbo P. G.; Shon J.; Sickler B.; Batzoglou S.; Knudsen K. E.; He H. H.; Huang J.; Wyatt A. W.; Dehm S. M.; Ashworth A.; Chinnaiyan A. M.; Maher C. A.; Small E. J.; Feng F. Y. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 2018, 174 (3), 758–769. 10.1016/j.cell.2018.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita K.; Nonomura N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Mens Health. 2019, 37 (3), 288–295. 10.5534/wjmh.180040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bono J. S.; Logothetis C. J.; Molina A.; Fizazi K.; North S.; Chu L.; Chi K. N.; Jones R. J.; Goodman O. B. Jr.; Saad F.; Staffurth J. N.; Mainwaring P.; Harland S.; Flaig T. W.; Hutson T. E.; Cheng T.; Patterson H.; Hainsworth J. D.; Ryan C. J.; Sternberg C. N.; Ellard S. L.; Fléchon A.; Saleh M.; Scholz M.; Efstathiou E.; Zivi A.; Bianchini D.; Loriot Y.; Chieffo N.; Kheoh T.; Haqq C. M.; Scher H. I. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364 (21), 1995–2005. 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher H. I.; Fizazi K.; Saad F.; Taplin M. E.; Sternberg C. N.; Miller K.; de Wit R.; Mulders P.; Chi K. N.; Shore N. D.; Armstrong A. J.; Flaig T. W.; Fléchon A.; Mainwaring P.; Fleming M.; Hainsworth J. D.; Hirmand M.; Selby B.; Seely L.; de Bono J. S. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367 (13), 1187–1197. 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- Smith M. R.; Saad F.; Chowdhury S.; Oudard S.; Hadaschik B. A.; Graff J. N.; Olmos D.; Mainwaring P. N.; Lee J. Y.; Uemura H.; Lopez-Gitlitz A.; Trudel G. C.; Espina B. M.; Shu Y.; Park Y. C.; Rackoff W. R.; Yu M. K.; Small E. J. Apalutamide treatment and metastasis-free survival in prostate cancer. N. Engl. J. Med. 2018, 378 (15), 1408–1418. 10.1056/NEJMoa1715546. [DOI] [PubMed] [Google Scholar]
- Schweizer M. T.; Yu E. Y. Persistent androgen receptor addiction in castration-resistant prostate cancer. J. Hematol. Oncol. 2015, 8, 128. 10.1186/s13045-015-0225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan S. R.; Ha G.; Hoff A. M.; Wala J. A.; Carrot-Zhang J.; Whelan C. W.; Haradhvala N. J.; Freeman S. S.; Reed S. C.; Rhoades J.; Polak P.; Cipicchio M.; Wankowicz S. A.; Wong A.; Kamath T.; Zhang Z.; Gydush G. J.; Rotem D.; et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell 2018, 174 (2), 433–447. 10.1016/j.cell.2018.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D.; Van Allen E. M.; Wu Y. M.; Schultz N.; Lonigro R. J.; Mosquera J. M.; Montgomery B.; Taplin M. E.; Pritchard C. C.; Attard G.; Beltran H.; Abida W.; Bradley R. K.; Vinson J.; Cao X.; Vats P.; Kunju L. P.; Hussain M.; Feng F. Y.; Tomlins S. A.; Cooney K. A.; Smith D. C.; Brennan C.; Siddiqui J.; Mehra R.; Chen Y.; Rathkopf D. E.; Morris M. J.; Solomon S. B.; Durack J. C.; Reuter V. E.; Gopalan A.; Gao J.; Loda M.; Lis R. T.; Bowden M.; Balk S. P.; Gaviola G.; Sougnez C.; Gupta M.; Yu E. Y.; Mostaghel E. A.; Cheng H. H.; Mulcahy H.; True L. D.; Plymate S. R.; Dvinge H.; Ferraldeschi R.; Flohr P.; Miranda S.; Zafeiriou Z.; Tunariu N.; Mateo J.; Perez-Lopez R.; Demichelis F.; Robinson B. D.; Schiffman M.; Nanus D. M.; Tagawa S. T.; Sigaras A.; Eng K. W.; Elemento O.; Sboner A.; Heath E. I.; Scher H. I.; Pienta K. J.; Kantoff P.; de Bono J. S.; Rubin M. A.; Nelson P. S.; Garraway L. A.; Sawyers C. L.; Chinnaiyan A. M. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161 (5), 1215–1228. 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis E. S.; Lu C.; Wang H.; Luber B.; Nakazawa M.; Roeser J. C.; Chen Y.; Mohammad T. A.; Chen Y.; Fedor H. L.; Lotan T. L.; Zheng Q.; De Marzo A. M.; Isaacs J. T.; Isaacs W. B.; Nadal R.; Paller C. J.; Denmeade S. R.; Carducci M. A.; Eisenberger M. A.; Luo J. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371 (11), 1028–1038. 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph J. D.; Lu N.; Qian J.; Sensintaffar J.; Shao G.; Brigham D.; Moon M.; Maneval E. C.; Chen I.; Darimont B.; Hager J. H. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discovery 2013, 3 (9), 1020–1029. 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- Korpal M.; Korn J. M.; Gao X.; Rakiec D. P.; Ruddy D. A.; Doshi S.; Yuan J.; Kovats S. G.; Kim S.; Cooke V. G.; Monahan J. E.; Stegmeier F.; Roberts T. M.; Sellers W. R.; Zhou W.; Zhu P. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discovery 2013, 3 (9), 1030–1043. 10.1158/2159-8290.CD-13-0142. [DOI] [PubMed] [Google Scholar]
- Arora V. K.; Schenkein E.; Murali R.; Subudhi S. K.; Wongvipat J.; Balbas M. D.; Shah N.; Cai L.; Efstathiou E.; Logothetis C.; Zheng D.; Sawyers C. L. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155 (6), 1309–1322. 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson P. S. Targeting the androgen receptor in prostate cancer--a resilient foe. N. Engl. J. Med. 2014, 371 (11), 1067–1069. 10.1056/NEJMe1409306. [DOI] [PubMed] [Google Scholar]
- Tan M. H.; Li J.; Xu H. E.; Melcher K.; Yong E. L. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36 (1), 3–23. 10.1038/aps.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathkopf D. E.; Smith M. R.; Ryan C. J.; Berry W. R.; Shore N. D.; Liu G.; Higano C. S.; Alumkal J. J.; Hauke R.; Tutrone R. F.; Saleh M.; Chow Maneval E.; Thomas S.; Ricci D. S.; Yu M. K.; de Boer C. J.; Trinh A.; Kheoh T.; Bandekar R.; Scher H. I.; Antonarakis E. S. Androgen receptor mutations in patients with castration-resistant prostate cancer treated with apalutamide. Ann. Oncol. 2017, 28 (9), 2264–2271. 10.1093/annonc/mdx283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moilanen A. M.; Riikonen R.; Oksala R.; Ravanti L.; Aho E.; Wohlfahrt G.; Nykänen P. S.; Törmäkangas O. P.; Palvimo J. J.; Kallio P. J. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci. Rep. 2015, 5, 12007. 10.1038/srep12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Connolly P. J.; Lim H. K.; Pande V.; Meerpoel L.; Teleha C.; Branch J. R.; Ondrus J.; Hickson I.; Bush T.; Luistro L.; Packman K.; Bischoff J. R.; Ibrahim S.; Parrett C.; Chong Y.; Gottardis M. M.; Bignan G. Discovery of JNJ-63576253: a clinical stage androgen receptor antagonist for F877L mutant and wild-type castration resistant prostate cancer (mCRPC). J. Med. Chem. 2021, 64 (2), 909–924. 10.1021/acs.jmedchem.0c01563. [DOI] [PubMed] [Google Scholar]
- Liu H.; Han R.; Li J.; Liu H.; Zheng L. Molecular mechanism of R-bicalutamide switching from androgen receptor antagonist to agonist induced by amino acid mutations using molecular dynamics simulations and free energy calculation. J. Comput.-Aided Mol. Des. 2016, 30 (12), 1189–1200. 10.1007/s10822-016-9992-2. [DOI] [PubMed] [Google Scholar]
- Liu H. L.; Zhong H. Y.; Song T. Q.; Li J. Z. A Molecular Modeling Study of the Hydroxyflutamide resistance mechanism induced by androgen receptor Mutations. Int. J. Mol. Sci. 2017, 18 (9), 1823 10.3390/ijms18091823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Wang L.; Tian J.; Li J.; Liu H. Molecular Dynamics Studies on the enzalutamide resistance mechanisms induced by androgen receptor mutations. J. Cell. Biochem. 2017, 118 (9), 2792–2801. 10.1002/jcb.25928. [DOI] [PubMed] [Google Scholar]
- Jung M. E.; Ouk S.; Yoo D.; Sawyers C. L.; Chen C.; Tran C.; Wongvipat J. Structure–activity relationship for thiohydantoin androgen receptor antagonists for castration-resistant prostate cancer (CRPC). J. Med. Chem. 2010, 53 (7), 2779–2796. 10.1021/jm901488g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soengas R. G.; Silva S. Spirocyclic nucleosides in medicinal chemistry: an overview. Mini-Rev. Med. Chem. 2012, 12 (14), 1485–1496. 10.2174/138955712803832681. [DOI] [PubMed] [Google Scholar]
- Balbas M. D.; Evans M. J.; Hosfield D. J.; Wongvipat J.; Arora V. K.; Watson P. A.; Chen Y.; Greene G. L.; Shen Y.; Sawyers C. L. Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife 2013, 2, e00499 10.7554/eLife.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A.; Wang Y.; Meng X.; Yang Y. Design, synthesis and biological evaluation of novel thiohydantoin derivatives as potent androgen receptor antagonists for the treatment of prostate cancer. Bioorg. Med. Chem. 2021, 31, 115953. 10.1016/j.bmc.2020.115953. [DOI] [PubMed] [Google Scholar]
- Murigi F. N.; Nichol G. S.; Mash E. A. Synthesis of the conformationally constrained tyrosine analogues, (R)- and (S)-5-hydroxy-2-aminoindan-2-carboxylic acids. J. Org. Chem. 2010, 75 (4), 1293–1296. 10.1021/jo902438s. [DOI] [PubMed] [Google Scholar]
- Kotha S.; Brahmachary E. Synthesis of indan-based unusual α-amino acid derivatives under phase-transfer catalysis conditions. J. Org. Chem. 2000, 65 (5), 1359–1365. 10.1021/jo991387v. [DOI] [PubMed] [Google Scholar]
- Wiȩckowska A.; Fransson R.; Odell L. R.; Larhed M. Microwave-assisted synthesis of Weinreb and MAP aryl amides via Pd-catalyzed Heck aminocarbonylation using Mo(CO)6 or W(CO)6. J. Org. Chem. 2011, 76 (3), 978–981. 10.1021/jo102151u. [DOI] [PubMed] [Google Scholar]
- Chen C. D.; Welsbie D. S.; Tran C.; Baek S. H.; Chen R.; Vessella R.; Rosenfeld M. G.; Sawyers C. L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10 (1), 33–39. 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Böselt L.; Sidler D.; Kittelmann T.; Stohner J.; Zindel D.; Wagner T.; Riniker S. Determination of Absolute Stereochemistry of Flexible Molecules Using a Vibrational circular dichroism spectra alignment algorithm. J. Chem. Inf. Model. 2019, 59 (5), 1826–1838. 10.1021/acs.jcim.8b00789. [DOI] [PubMed] [Google Scholar]
- Stephens P. J.; Devlin F. J.; Pan J. J. The determination of the absolute configurations of chiral molecules using vibrational circular dichroism (VCD) spectroscopy. Chirality 2008, 20 (5), 643–63. 10.1002/chir.20477. [DOI] [PubMed] [Google Scholar]
- Korenchuk S.; Lehr J. E.; Mclean L.; Lee Y. G.; Whitney S.; Vessella R.; Lin D. L.; Pienta K. J. VCaP, a cell-based model system of human prostate cancer. In Vivo 2001, 15 (2), 163–168. [PubMed] [Google Scholar]
- Naven R. T.; Swiss R.; Klug-McLeod J.; Will Y.; Greene N. The development of structure-activity relationships for mitochondrial dysfunction: uncoupling of oxidative phosphorylation. Toxicol. Sci. 2013, 131 (1), 271–278. 10.1093/toxsci/kfs279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.