

ABSTRACT
Antivirals targeting severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) could improve treatment of COVID-19. We evaluated the efficacy of clinically relevant hepatitis C virus (HCV) NS3 protease inhibitors (PIs) against SARS-CoV-2 and their interactions with remdesivir, the only direct-acting antiviral approved for COVID-19 treatment. HCV PIs showed differential potency in short-term treatment assays based on the detection of SARS-CoV-2 spike protein in Vero E6 cells. Linear PIs boceprevir, telaprevir, and narlaprevir had 50% effective concentrations (EC50) of ∼40 μM. Among the macrocyclic PIs, simeprevir had the highest (EC50, 15 μM) and glecaprevir the lowest (EC50, >178 μM) potency, with paritaprevir, grazoprevir, voxilaprevir, vaniprevir, danoprevir, and deldeprevir in between. Acyclic PIs asunaprevir and faldaprevir had EC50s of 72 and 23 μM, respectively. ACH-806, inhibiting the HCV NS4A protease cofactor, had an EC50 of 46 μM. Similar and slightly increased PI potencies were found in human hepatoma Huh7.5 cells and human lung carcinoma A549-hACE2 cells, respectively. Selectivity indexes based on antiviral and cell viability assays were highest for linear PIs. In short-term treatments, combination of macrocyclic but not linear PIs with remdesivir showed synergism in Vero E6 and A549-hACE2 cells. Longer-term treatment of infected Vero E6 and A549-hACE2 cells with 1-fold EC50 PI revealed minor differences in the barrier to SARS-CoV-2 escape. Viral suppression was achieved with 3- to 8-fold EC50 boceprevir or 1-fold EC50 simeprevir or grazoprevir, but not boceprevir, in combination with 0.4- to 0.8-fold EC50 remdesivir; these concentrations did not lead to viral suppression in single treatments. This study could inform the development and application of protease inhibitors for optimized antiviral treatments of COVID-19.
KEYWORDS: coronavirus, antiviral, repurposing, combination treatment, synergy, COVID-19
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a positive-sense single-stranded RNA virus of the Coronaviridae family, which emerged in humans in 2019 most likely originating from a bat-borne virus (1–3). SARS-CoV-2 causes coronavirus disease 2019 (COVID-19), a multisystemic disease with initial symptoms mostly localizing to the respiratory tract. At the end of April 2021, the COVID-19 pandemic had been responsible for >152 million infected, >3 million deaths, and an unknown number of individuals suffering from long-term health effects (4–8).
Repurposing of drugs approved for other medical indications is promoted as a time-saving approach to the identification of urgently needed treatments. At present, the only drug approved for treatment of COVID-19 that directly targets SARS-CoV-2 proteins is remdesivir, an inhibitor of the viral nonstructural protein (nsp) 12 polymerase, originally being an investigational broad-spectrum antiviral previously evaluated for treatment of chronic hepatitis C virus (HCV) infection and Ebola virus infection (9).
Another important target of antiviral drugs are viral proteases, which are essential for the cleavage of viral polyproteins into functional proteins (10–13). The coronavirus main protease (Mpro) or 3 chymotrypsin-like protease (3CLpro) is a cysteine protease corresponding to nsp5 and is highly conserved among coronaviruses. Mpro mediates 11 polyprotein cleavage events and is thus essential for viral replication (14–16). The second coronavirus protease, the papain-like protease (PLpro), is also a cysteine protease corresponding to the protease domain of nsp3 and is less conserved. PLpro mediates 3 cleavage events and has important roles for viral replication and regulation of host innate immunity (17, 18). Therefore, both SARS-CoV-2 proteases are considered potential drug targets (19, 20).
Hepatitis C virus is a positive-sense single-stranded RNA virus of the Flaviviridae family, which is classified into 8 major genotypes and various subtypes (21, 22). The main HCV protease, nonstructural protein 3 (NS3), is a chymotrypsin-like serine protease (23–25). Together with its essential cofactor, NS4A, it mediates 4 cleavage events of the polyprotein. Inhibitors of this protease are important components of recently developed, highly efficient HCV treatment regimens based on the combination of antivirals directly targeting HCV proteins (26, 27).
Initially developed HCV protease inhibitors (PIs) have a linear structure and include boceprevir and telaprevir, which were approved in 2011 in the United States and the European Union, as well as narlaprevir, approved in 2016 in Russia, for treatment of chronic HCV infection (see Fig. S1 and Table S1 in the supplemental material). Subsequently, PIs with a macrocyclic structure, including simeprevir, paritaprevir, grazoprevir, glecaprevir, voxilaprevir, vaniprevir, danoprevir, and deldeprevir, were developed. These macrocyclic PIs were approved between 2013 and 2019 in the United States, European Union, or China, with the exception of vaniprevir, only approved in Japan, and deldeprevir, which was never approved. Of the 2 acyclic PIs, asunaprevir and faldaprevir, asunaprevir was approved in Japan, Canada, and China, while faldaprevir was not approved. Several of the initially developed PIs were subsequently discontinued due to the development of more-efficient PIs with increased activity against the different HCV genotypes (Table S1). At present, clinical use in the United States, European Union, and China is focused on inhibitor combinations, including grazoprevir, glecaprevir, or voxilaprevir. Additionally, in China, inhibitor combinations, including paritaprevir, danoprevir, or asunaprevir are used in the clinic.
While an inhibitor of HCV NS4A (ACH-806) was tested in clinical phase 1 trials, development was halted due to reversible nephrotoxicity (28, 29).
In this study, we investigated in vitro efficacy of a panel of HCV PIs, including all clinically approved compounds and selected compounds tested in clinical studies, against SARS-CoV-2. These studies were carried out in African green monkey kidney Vero E6 cells, and results were verified in human Huh7.5 hepatoma and A549 lung carcinoma cells, the latter engineered to constitutively express the human SARS-CoV-2 entry receptor angiotensin-converting enzyme 2 (hACE2). All culture systems were previously demonstrated to be relevant for studies of antivirals targeting SARS-CoV-2 (30–36). We further evaluated the efficacy of an HCV NS4A protease cofactor inhibitor. In concentration-response antiviral assays, we determined the 50% effective concentrations (EC50s), 50% cytotoxic concentrations (CC50s), and selectivity indexes (SIs). Moreover, we evaluated interactions with remdesivir for selected linear and macrocyclic PI compounds. Finally, in longer-term assays, we evaluated selected PIs singly or in combination with remdesivir for their barrier to viral escape.
RESULTS
Differential potency of clinically relevant HCV protease and cofactor inhibitors against SARS-CoV-2 in vitro.
To determine the potency of a panel of HCV PIs and an HCV NS4A inhibitor against SARS-CoV-2, we developed a cell-based short-term antiviral treatment assay in 96-well plates, adapting an assay previously developed to determine potency of HCV PIs against HCV (29, 37–43). In this assay, replicate SARS-CoV-2-infected cultures were treated with different dilutions of inhibitors. Following incubation, cultures were subjected to immunostaining for SARS-CoV-2 spike protein and automated counting of single spike protein-positive cells (31, 44). Multiplicity of infection (MOI) and incubation time were chosen to avoid virus-induced cytopathogenic effects (CPEs) during the assay and to result in >1,000 single spike protein-positive cells in nontreated control cultures, as determined in pilot CPE and immunostaining assays (for details, see Materials and Methods). The potency of the inhibitors to reduce the number of spike protein-expressing cells compared to that of the nontreated controls was evaluated. Furthermore, cytotoxicity of inhibitors was determined by cell viability assays; in these assays, cell viability of >90% was confirmed for inhibitor concentrations used in treatment assays.
First, concentration-response studies were carried out in Vero E6 cells. Tested inhibitors were able to inhibit the virus with EC50 values in the micromolar range, with exception of glecaprevir, voxilaprevir, and deldeprevir, where EC50 values were not determined due to cytotoxicity of the drugs or antiviral activity of the diluent dimethyl sulfoxide (DMSO) at high drug concentrations (Fig. 1 and Table 1; see also Fig. S2). The linear PIs boceprevir, telaprevir, and narlaprevir showed comparable potencies, with EC50 values of ∼40 μM. Among the macrocyclic PIs, simeprevir showed the highest potency, with an EC50 of 15 μM. Furthermore, paritaprevir had an EC50 of 22 μM, while grazoprevir and vaniprevir had EC50 values of 42 and 51 μM, respectively. Finally, the EC50 was 87 μM for danoprevir. For the acyclic PIs, faldaprevir (EC50, 23 μM) was more potent than asunaprevir (EC50, 72 μM).
FIG 1.

Potency of a panel of HCV PIs and an HCV NS4A inhibitor against SARS-CoV-2 in Vero E6 cells. Vero E6 cells were seeded in 96-well plates and, the following day, infected with SARS-CoV-2 at an MOI of 0.002 followed by treatment with specified concentrations of the PIs boceprevir, telaprevir, narlaprevir, simeprevir, paritaprevir, grazoprevir, glecaprevir, voxilaprevir, vaniprevir, danoprevir, deldeprevir, asunaprevir, and faldaprevir as well as HCV NS4A inhibitor ACH-806, as described in Materials and Methods. After 46 to 50 h of incubation, SARS-CoV-2-infected cells were visualized by immunostaining for the SARS-CoV-2 spike protein and quantified by automated counting, as described in Materials and Methods. Data points (red dots) are means of counts from 7 replicate cultures ± standard errors of the means (SEMs) and represent percent residual infectivity, determined as percent SARS-CoV-2-positive cells relative to means of counts from 14 replicate infected nontreated control cultures. Sigmoidal concentration-response curves (red lines) were fitted and EC50 values were determined, as described in Materials and Methods. Cell viability data were obtained in replicate assays with noninfected cells using a colorimetric assay, as described in Materials and Methods. Data points (blue triangles) are means from 3 replicate cultures ± SEMs and represent percent cell viability relative to mean absorbance from 12 replicate nontreated control cultures. Sigmoidal concentration-response curves were fitted and CC50 values were determined as shown in Fig. S4 in the supplemental material. The red dotted lines represent the drug concentrations at which DMSO is expected to induce antiviral effects with reduction of residual infectivity to <70%, according to Fig. S2. The blue dotted lines represent the drug concentrations at which DMSO is expected to induce cytotoxicity with reduction of cell viability to <90%, according to Fig. S2.
TABLE 1.
Potency of a panel of HCV PIs and an HCV NS4A inhibitor against SARS-CoV-2 in vitro
| Inhibitor | EC50 (μM)a | CC50 (μM)b | SIc |
|---|---|---|---|
| Vero E6 cells | |||
| Boceprevir | 44 | >1,214 | >28 |
| Telaprevir | 40 | >432 | >11 |
| Narlaprevir | 37 | 269 | 7.3 |
| Simeprevir | 15 | 59 | 3.9 |
| Paritaprevir | 22 | 123 | 5.6 |
| Grazoprevir | 42 | 239 | 5.7 |
| Glecaprevir | >178 | >268 | ND |
| Voxilaprevir | >27 | 72 | <2.7 |
| Vaniprevir | 51 | 171 | 3.4 |
| Danoprevir | 87 | >243 | >2.8 |
| Deldeprevir | >20 | 56 | <2.8 |
| Asunaprevir | 72 | 263 | 3.7 |
| Faldaprevir | 23 | 146 | 6.3 |
| ACH-806 | 46 | >429 | >9.3 |
| Huh7.5 cells | |||
| Boceprevir | 42 | 701 | 17 |
| Simeprevir | 14 | 33 | 2.4 |
| Grazoprevir | 20 | 133 | 6.7 |
| A549-hACE2 cells | |||
| Boceprevir | 20 | >1,213 | >61 |
| Simeprevir | 9 | 56 | 6.2 |
| Grazoprevir | 26 | 125 | 4.8 |
| Glecaprevir | >94 | >268 | ND |
| Voxilaprevir | 10 | 81 | 8.1 |
EC50, 50% effective concentration, determined in antiviral treatment assays as described in Materials and Methods. For voxilaprevir and deldeprevir in Vero E6 cells, >50% residual infectivity was observed at the highest noncytotoxic concentrations; for glecaprevir in both Vero E6 cells and A549-hACE2 cells, the highest applied concentration was limited due to antiviral effects of the diluent DMSO; thus, for these PIs, no precise EC50 was determined. EC50 values are also included in Fig. 1 to 3.
CC50, 50% cytotoxic concentration, determined in cell viability assays as described in Materials and Methods. For boceprevir, telaprevir, glecaprevir, danoprevir, and ACH-806 in Vero E6 cells and for boceprevir and glecaprevir in A549-hACE2 cells, >50% cell viability was observed at the highest concentrations tested; thus, no precise CC50 was determined. Tested concentrations were those at which DMSO was not expected to reduce cell viability to <90% (see Fig. S2 in the supplemental material).
SI, selectivity index, determined as CC50/EC50 based on results from antiviral treatment assays and cell viability assays. ND, not determined.
To validate the immunostaining-based treatment assay, concentration-response assays quantifying CPE were carried out in Vero E6 cells for selected PIs. In this assay, replicate SARS-CoV-2-infected cultures were treated with different dilutions of inhibitors. MOI and incubation time were chosen to induce relatively strong virus-induced CPEs in nontreated control cultures upon termination of the assay (30% to 50% cell viability). In treated cultures, the potency of the inhibitors to inhibit virus-induced CPE compared to that in the nontreated control cultures was evaluated. In these assays, the tested PIs, boceprevir, simeprevir, grazoprevir, glecaprevir, and voxilaprevir, showed similar potency and EC50 values as in the immunostaining-based assays (see Fig. S3).
To confirm the potency of PIs in human cells, selected PIs were studied in human Huh7.5 hepatoma cells. In these assays, the tested PIs, boceprevir, simeprevir, and grazoprevir, had similar concentration-response curves and EC50 values as in Vero E6 cells (Fig. 2, Table 1).
FIG 2.
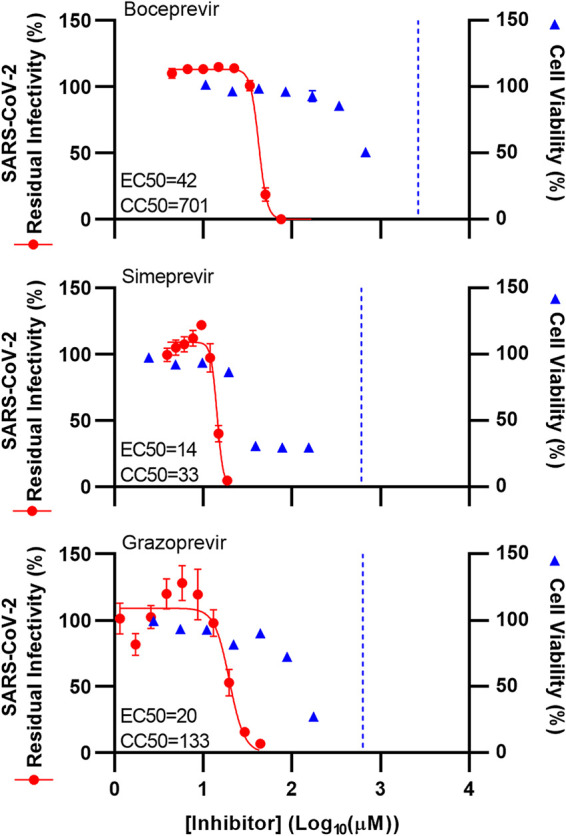
Potency of selected HCV PIs against SARS-CoV-2 was confirmed in Huh7.5 cells. Huh7.5 cells were seeded in 96-well plates and, the following day, infected with SARS-CoV-2 at an MOI of 0.02 followed by treatment with specified concentrations of the PIs boceprevir, simeprevir, and grazoprevir, as described in Materials and Methods. After 70 to 74 h of incubation, SARS-CoV-2-infected cells were visualized by immunostaining for the SARS-CoV-2 spike protein and quantified by automated counting, as described in Materials and Methods. Data points (red dots) are means from 7 replicates ± SEMs and represent percent residual infectivity, determined as percent SARS-CoV-2-positive cells relative to means of counts from 14 replicate infected nontreated control cultures. Sigmoidal concentration-response curves (red lines) were fitted and EC50 values were determined, as described in Materials and Methods. Cell viability data were obtained in replicate assays with noninfected cells using a colorimetric assay as described in Materials and Methods. Data points (blue triangles) are means from 3 replicate cultures ± SEMs and represent percent cell viability relative to mean absorbance from 12 nontreated controls. Sigmoidal concentration-response curves were fitted and CC50 values were determined, as shown in Fig. S5. The blue dotted line represents the drug concentrations at which DMSO is expected to induce cytotoxicity with reduction of cell viability to <90%, according to Fig. S2; DMSO did not induce antiviral effects in the tested concentration ranges (Fig. S2).
Finally, we confirmed differential potency of selected PIs in human A549 lung carcinoma cells transduced to express the SARS-CoV-2 entry receptor hACE2. In these A549-hACE2 cells, inhibitors showed slightly increased potency. Thus, boceprevir, simeprevir, and grazoprevir showed an ∼2-fold decreased EC50 compared to the EC50 in Vero E6 cells. In line with this observation and in contrast to that in Vero E6 cells, the EC50 value for voxilaprevir was determined in A549-hACE2 cells (10 μM). Similarly to Vero E6 cells, no EC50 was determined for glecaprevir in A549-hACE2 cells due to antiviral activity of DMSO at high inhibitor concentrations (Fig. 3).
FIG 3.
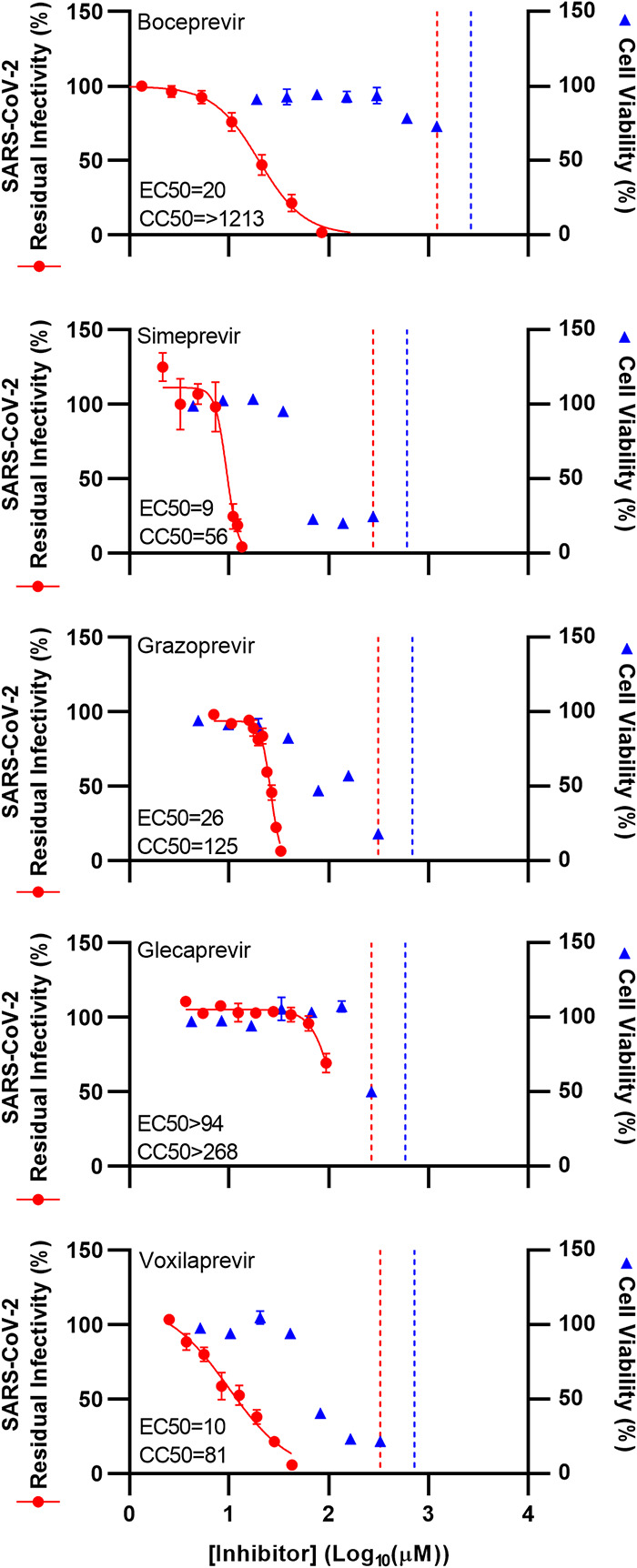
Potency of selected HCV PIs against SARS-CoV-2 was confirmed in A549-hACE2 cells. A549-hACE2 cells were seeded in 96-well plates and, the following day, infected with SARS-CoV-2 at an MOI of 0.003 followed by treatment with specified concentrations of the PIs boceprevir, simeprevir, grazoprevir, glecaprevir, and voxilaprevir, as described in Materials and Methods. After 46 to 50 h of incubation, SARS-CoV-2-infected cells were visualized by immunostaining for the SARS-CoV-2 spike protein and quantified by automated counting, as described in Materials and Methods. Data points (red dots) are means from 7 replicates ± SEMs and represent percent residual infectivity, determined as percent SARS-CoV-2-positive cells relative to means of counts from 14 replicate infected nontreated control cultures. Sigmoidal concentration-response curves (red lines) were fitted and EC50 values were determined, as described in Materials and Methods. Cell viability data were obtained in replicate assays with noninfected cells using a colorimetric assay as described in Materials and Methods. Data points (blue triangles) are means from 3 replicate cultures ± SEMs and represent percent cell viability relative to mean absorbance from 12 nontreated controls. Sigmoidal concentration-response curves were fitted and CC50 values were determined, as shown in Fig. S6. The red dotted lines represent the drug concentrations at which DMSO is expected to induce antiviral effects with reduction of residual infectivity to <70%, according to Fig. S2. The blue dotted lines represent the drug concentrations at which DMSO is expected to induce cytotoxicity with reduction of cell viability to <90%, according to Fig. S2.
All inhibitors were diluted in DMSO. At the DMSO dilutions used for generation of specific data, no antiviral effect was observed in Vero E6, Huh7.5, or A549-hACE2 cells (Fig. 1 to 3 and Fig. S2).
Cell viability assays were carried out for all studied drugs to determine their level of in vitro cytotoxicity and CC50 values. In these assays, drug concentrations were used at which no DMSO-induced cytotoxicity was observed (Fig. 1 to 3 and Fig. S2). In Vero E6 cells, the linear PIs showed the lowest cytotoxicity, with all CC50 values >200 μM (>1,214, >432, and 269 μM for boceprevir, telaprevir, and narlaprevir, respectively) (Fig. 1, Table 1, and Fig. S4). Among the macrocyclic inhibitors, grazoprevir, glecaprevir, and danoprevir showed the lowest cytotoxicity, with CC50s >200 μM. Paritaprevir and vaniprevir showed intermediate cytotoxicity, with CC50s between 100 and 200 μM, while simeprevir, voxilaprevir, and deldeprevir showed the highest cytotoxicity, with CC50s between 50 and 100 μM. Cell viability assays carried out in Huh7.5 and in A549-hACE2 cells for selected PIs showed similar results (Fig. 2 and 3, Table 1, and Fig. S5 and S6).
Based on these assays, in Vero E6 cells, the linear inhibitors had the highest selectivity indexes (SI = CC50/EC50), with SIs of >28 for boceprevir, >11 for telaprevir, and 7.3 for narlaprevir (Table 1). Of the macrocyclic inhibitors, paritaprevir and grazoprevir had the highest SIs (5.6 and 5.7, respectively), while simeprevir and vaniprevir had slightly lower SIs of 3.9 and 3.4, respectively. For glecaprevir, voxilaprevir, danoprevir, and deldeprevir, SIs could not be determined. For the acyclic inhibitors, SI values were 6.3 for faldaprevir and 3.7 for asunaprevir. Finally, SI values calculated based on assays in Huh7.5 and in A549-hACE2 cells were comparable to those based on assays in Vero E6 cells (Table 1).
For the HCV NS4A inhibitor ACH-806, in Vero E6 cells, the EC50 was 46 μM, CC50 was >429 μM, and SI was >9.3 (Fig. 1, Table 1, and Fig. S4).
HCV PIs showed differential interactions with remdesivir in short-term treatment assays.
To study interactions between selected PIs and remdesivir, 96-well-based short-term immunostaining-based drug interaction assays were carried out. SARS-CoV-2-infected Vero E6 or A549-hACE2 cell cultures were treated with selected PIs singly, with remdesivir singly, or with a combination of PIs and remdesivir. Inhibitor dilution series were chosen based on determined EC50 values (Table 1; Fig. S7). Inhibitors were only used at concentrations where no cytotoxicity or antiviral effects of DMSO were observed (see Fig. S2, S8, and S9). For each inhibitor pair to be evaluated, 7 to 10 treatment conditions were chosen. Each treatment condition was defined by a given concentration of PI applied singly, a given concentration of remdesivir applied singly, and a combination of these same concentrations of a PI and remdesivir (see Table S2).
For each treatment condition, the residual infectivity of the culture receiving combination treatment was compared to the residual infectivity of the cultures receiving the corresponding single treatments. A graphical representation of the data is shown in Fig. 4. All treatment conditions are detailed and the corresponding residual infectivity values are shown in Table S2.
FIG 4.
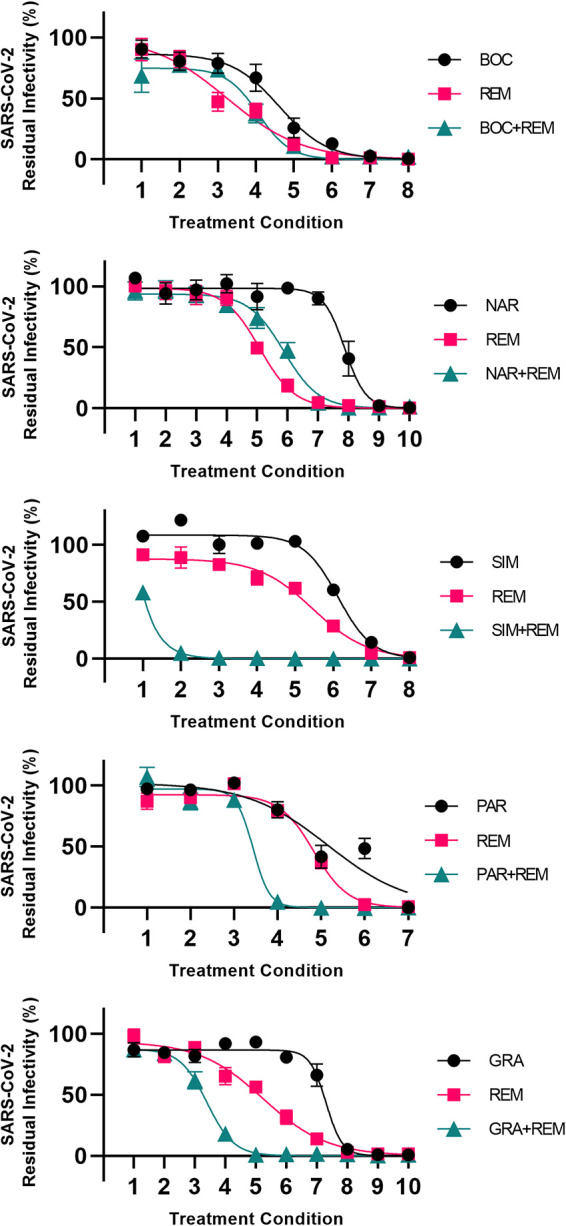
Analysis of interactions of selected HCV PIs with remdesivir in Vero E6 cells. Vero E6 cells seeded in 96-well plates were infected the following day with SARS-CoV-2 at an MOI of 0.002 followed by treatment with serial dilutions of the linear PI boceprevir (BOC) or narlaprevir (NAR), the macrocyclic PI simeprevir (SIM), paritaprevir (PAR), or grazoprevir (GRA), polymerase inhibitor remdesivir (REM), or a combination of these PIs and remdesivir, as described in Materials and Methods. After 46 to 50 h of incubation, SARS-CoV-2-infected cells were visualized by immunostaining for the SARS-CoV-2 spike protein and quantified by automated counting, as described in Materials and Methods. For each inhibitor pair to be evaluated, 7 to 10 treatment conditions were used (indicated on x axes). Each treatment condition was defined by a given concentration of PI applied singly, a given concentration of remdesivir applied singly, and a combination of these same concentrations of PIs and remdesivir, as specified in Table S2, resulting in 3 data points per treatment condition. Data points are means from 6 or 7 replicates ± SEMs and represent percent residual infectivity, determined as percent SARS-CoV-2-positive cells relative to means of counts from infected nontreated control cultures. Sigmoidal concentration-response curves were fitted as described in Materials and Methods. The tested inhibitor concentrations did not impact cell viability (Fig. S8). DMSO did not induce antiviral effects in the tested concentration ranges (Fig. S2).
For the linear inhibitors boceprevir and narlaprevir, under almost all treatment conditions, the effect of the combination did not exceed the effect of the most efficient single inhibitor, remdesivir (Fig. 4). For example, under treatment condition 3, 17.4 μM boceprevir resulted in 79% residual infectivity and 1.7 μM remdesivir resulted in 47% residual infectivity. Combination treatment with 17.4 μM boceprevir and 1.7 μM remdesivir resulted in 74% residual infectivity. Thus, treatment with remdesivir alone proved equally or more efficient than a combination of a linear PI with remdesivir.
In contrast, for the macrocyclic inhibitors simeprevir, paritaprevir, and grazoprevir, under various treatment conditions, the effect of the combination exceeded the effect of both single inhibitors (Fig. 4). For example, under treatment condition 3, 11.1 μM simeprevir resulted in 100% residual infectivity and 0.6 μM remdesivir resulted in 83% residual infectivity. However, combination treatment with 11.1 μM simeprevir and 0.6 μM remdesivir resulted in 1% residual infectivity. Thus, treatment with the combination showed an added effect compared to that with the corresponding single drug treatments.
We confirmed the above-described differential interactions of linear versus macrocyclic PIs with remdesivir in human A549-hACE2 cells by carrying out similar assays with the selected PIs boceprevir, simeprevir, and grazoprevir (Fig. 5 and Table S2).
FIG 5.
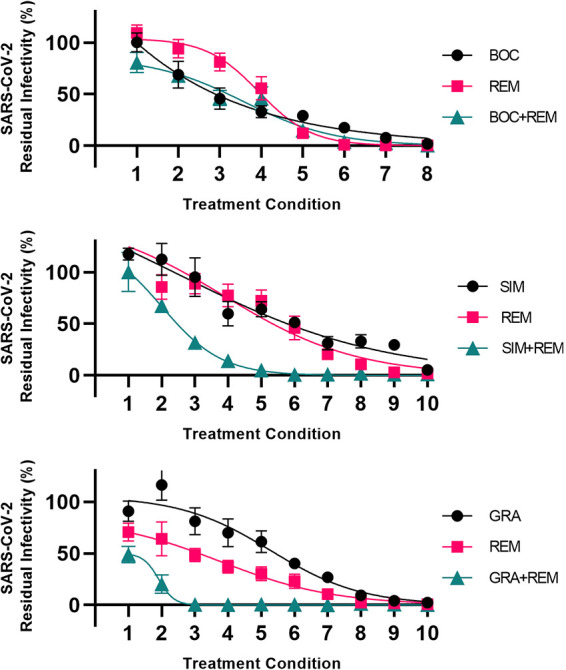
Analysis of interactions of selected HCV PIs with remdesivir in A549-hACE2 cells. A549-hACE2 cells seeded in 96-well plates were infected the following day with SARS-CoV-2 at an MOI of 0.003 followed by treatment with serial dilutions of the linear PI boceprevir (BOC), the macrocyclic PI simeprevir (SIM) or grazoprevir (GRA), polymerase inhibitor remdesivir (REM), or a combination of these PIs and remdesivir, as described in Materials and Methods. After 46 to 50 h of incubation, SARS-CoV-2-infected cells were visualized by immunostaining for the SARS-CoV-2 spike protein and quantified by automated counting, as described in Materials and Methods. For each inhibitor pair to be evaluated, 8 to 10 treatment conditions were used (indicated on x axes). Each treatment condition was defined by a given concentration of PI applied singly, a given concentration of remdesivir applied singly, and a combination of these same concentrations of PIs and remdesivir, as specified in Table S2, resulting in 3 data points per treatment condition. Data points are means from 7 replicates ± SEMs and represent percent residual infectivity, determined as percent SARS-CoV-2-positive cells relative to means of counts from infected nontreated control cultures. Sigmoidal concentration-response curves were fitted as described in Materials and Methods. DMSO was kept constant in all cultures. The tested inhibitor concentrations did not impair cell viability (Fig. S8). DMSO did not induce antiviral effects in the tested concentration ranges (Fig. S2).
To further define the nature of the interactions between the tested PIs and remdesivir, the above-described data sets were analyzed using the method of Chou and Talalay using CompuSyn software (45, 46). This analysis revealed primarily antagonistic interactions between remdesivir and the linear PIs boceprevir and narlaprevir. In contrast, primarily synergistic interactions were observed between remdesivir and the macrocyclic PIs paritaprevir and grazoprevir (see supplemental text, Fig. S10 and S11, and Table S3 and S4).
CompuSyn software provided a suboptimal fit for the simeprevir plus remdesivir data sets, most probably due to the steep slope of the simeprevir concentration-response curve. Therefore, an alternative software, SynergyFinder 2.0, was applied to analyze alternative data sets generated as required for this analysis and as described in the supplemental text. This analysis showed overall synergistic interactions between simeprevir and remdesivir in Vero E6 and A549-hACE2 cells (supplemental text and Fig. S12 and S13).
HCV PIs showed small differences in the barrier to escape of SARS-CoV-2.
To investigate their barriers to escape, all PIs for which an EC50 was determined were used for longer-term treatment of SARS-CoV-2-infected Vero E6 cells in culture flasks at the highest possible equipotent concentration (1-fold EC50) according to predicted cytotoxicity (Fig. 1, Table 1, and Fig. S4). In the nontreated control cultures, the infection spread to 50% of culture cells on day 1 and to 90% of culture cells on day 3 postinfection, as estimated by immunostaining for the SARS-CoV-2 spike protein (Fig. 6). Following day 3, typically massive virus-induced cell death was observed in these control cultures. For all PI-treated cultures (Fig. 6), initial viral suppression was observed with 10% to 30% of infected culture cells on day 1 postinfection and treatment initiation. On day 3, only cultures treated with narlaprevir, grazoprevir, vaniprevir, asunaprevir, and faldaprevir showed viral suppression, with infections of 10% to 50% of culture cells, while in cultures treated with boceprevir, telaprevir, simeprevir, paritaprevir, and danoprevir, 90% of culture cells were infected. On day 5, virus spread to 90% of culture cells in grazoprevir-treated cultures, while cultures treated with vaniprevir and asunaprevir were closed due to massive cell death, assumed to be due to PI-induced cytotoxicity, possibly enhanced by SARS-CoV-2 infection. On day 7, in narlaprevir- and faldaprevir-treated cultures, 60% of culture cells were infected; these cultures were closed on day 9 due to massive cell death.
FIG 6.

Comparison of barriers to escape for HCV PIs at equipotent concentrations in Vero E6 cells. Vero E6 cells seeded the previous day in T25 flasks were infected with SARS-CoV-2 at an MOI of 0.00002 followed by treatment with 1-fold EC50 of boceprevir, telaprevir, narlaprevir, simeprevir, paritaprevir, grazoprevir, vaniprevir, danoprevir, asunaprevir, and faldaprevir, which were administered immediately after infection and subsequently at the indicated time points postinfection when cells were split, as described in Materials and Methods. (Left) Percentages of SARS-CoV-2-infected cells on the specified days postinfection were determined by anti-spike protein immunostaining of replicate cultures derived following cell splitting and treatment. (Middle) SARS-CoV-2 RNA titers determined in cell culture supernatants as genome copies per milliliter on the specified days postinfection were determined by RT-qPCR assays. The black line indicates the lower limit of quantification (LLOQ). In the left and middle panels, to facilitate comparisons, bars are color coded according to the day postinfection, and blue and red dotted lines were inserted to highlight day 1 and 3 values of the nontreated culture, respectively. (Right) Replicate cultures were derived following cell splitting and treatment, immunostained for the SARS-CoV-2 spike protein (green) and counterstained with Hoechst dye (blue), and images were acquired, as described in Materials and Methods. Cultures summarized in this figure are derived from different experimental setups, each including an infected nontreated control culture, which showed viral spread comparable to that in the depicted representative culture. *, culture was terminated, or infection data not recorded, due to virus-induced cell death; **, culture was terminated due to drug-induced cytotoxicity, possibly enhanced by viral infection.
Viral spread kinetics monitored by immunostaining were confirmed by the determination of SARS-CoV-2 RNA titers in cell culture supernatants using a reverse transcription-quantitative PCR (RT-qPCR) assay (Fig. 6). In the nontreated control culture, the number of SARS-CoV-2 genome copies increased by 4 orders of magnitude from 5.5 × 106/ml on day 1 to 9.7 × 1010/ml on day 3 postinfection. Compared to these values, all treatments resulted in a small decrease in genome copies on days 1 and 3; however, none of the treatments was able to prevent viral spread as monitored by determination of viral RNA titers.
To confirm the PI barrier to escape in human cells, similar longer-term treatments were carried out in A549-hACE2 cells. Compared to the nontreated culture with SARS-CoV-2 RNA titers of 3.4 × 1010 and 2.5 × 1010 genome copies/ml on days 3 and 5, respectively, treatment with 1-fold EC50 of boceprevir, simeprevir, and grazoprevir had no to little effect on viral spread as monitored by determination of viral RNA titers (Fig. 7, left). While A549-hACE2 cultures were also followed by immunostaining, in contrast to that for Vero E6 cells, percent infection was difficult to estimate following day 1 postinfection, when a higher percentage of culture cells had become infected.
FIG 7.

Comparison of barriers to escape for HCV PIs in A549-hACE2 cells. A549-hACE2 cells seeded the previous day in T25 flasks were infected with SARS-CoV-2 at an MOI of 0.0005, followed by treatment with indicated concentrations of specified inhibitors administered immediately after infection and subsequently at the listed time points when cells were split, as described in Materials and Methods. BOC, boceprevir; SIM, simeprevir; GRA, grazoprevir; REM, remdesivir. Upon splitting of cells, cell culture supernatant was harvested and subjected to RT-qPCR for determination of SARS-CoV-2 RNA titers determined as genome copies per milliliter. The black lines indicate the LLOQs. To facilitate comparisons, bars are color coded according to the day postinfection, and blue and red dotted lines were inserted to highlight day 1 and 3 values of the nontreated culture, respectively. Cultures summarized in this figure are derived from different experimental setups, each including an infected nontreated control culture, which showed viral spread comparable to that in the depicted representative culture. (Left) Treatment with 1-fold EC50 boceprevir, simeprevir, or grazoprevir. (Middle) Treatment with 1-, 2-, 3-, 4-, 5-, and 8-fold EC50 boceprevir. *, culture was terminated due to virus- or drug-induced cytotoxicity; #, culture was maintained for a total of 13 days without indication of infection (RNA titers were around the LLOQ and no observation of single SARS-CoV-2 spike protein-positive cells). (Right) Treatment with 0.8-fold EC50 remdesivir, 1-fold EC50 boceprevir, 1-fold EC50 simeprevir, or 1-fold EC50 grazoprevir singly or with a combination of remdesivir with either of the PIs.
In conclusion, 1-fold EC50 of the tested PIs did not suppress SARS-CoV-2 in vitro.
Boceprevir had the potential to completely suppress viral infection in vitro.
As suboptimal viral suppression was observed under treatment with 1-fold EC50, we chose the PI with the highest SI to enable longer-term treatment at higher-fold EC50 concentrations. Vero E6 cells infected with SARS-CoV-2 were treated with 1-, 1.5-, 2-, 2.5, 3-, and 5-fold EC50 of boceprevir (Fig. 8). Treatment with 1- and 1.5-fold EC50 boceprevir only had a minor impact on viral spread on day 1 postinfection and treatment initiation, while 90% of culture cells became infected on day 3, as observed for nontreated control cells. Also, in cultures treated with 2- and 2.5-fold EC50, 90% of culture cells became infected on day 5. In contrast, treatment with 3- and 5-fold EC50 resulted in sustained viral suppression with no evidence of infected cells in the culture treated with 3-fold EC50 from day 3 and in the culture treated with 5-fold EC50 from day 1 during follow-up periods of 9 and 17 days, respectively. In addition, from cultures treated with 3- and 5-fold EC50 on day 5 and day 3, respectively, replicate cultures receiving no treatment going forward were derived, which did not show any infected cells during a follow-up period of 10 days, suggesting that the infection was cured under these treatments.
FIG 8.

Boceprevir was capable of completely suppressing SARS-CoV-2 in Vero E6 cells. Vero E6 cells seeded the previous day in T25 flasks were infected with SARS-CoV-2 at an MOI of 0.00002 followed by treatment with 1-, 1.5-, 2-, 2.5-, 3-, and 5-fold EC50 boceprevir, which was administered immediately after infection and subsequently at the indicated time points when cells were split, as described in Materials and Methods. (Left) Percentages of SARS-CoV-2-infected cells on the specified days postinfection were determined by anti-spike protein immunostaining of replicate cultures derived following cell splitting and treatment. (Middle) SARS-CoV-2 RNA titers determined in cell culture supernatants as genome copies per milliliter on the specified days postinfection were determined by RT-qPCR assays. The black line indicates the LLOQ. In the left and middle panels, to facilitate comparisons, bars are color coded according to the day postinfection, and blue and red dotted lines were inserted to highlight day 1 and 3 values of the nontreated culture, respectively. (Right) Replicate cultures were derived following cell splitting and treatment, immunostained for the SARS-CoV-2 spike protein (green) and counterstained with Hoechst dye (blue), and images were acquired, as described in Materials and Methods. Cultures summarized in this figure are derived from different experimental setups, each including an infected nontreated control culture, which showed viral spread comparable to that in the depicted representative culture. *, culture was terminated, or infection data not recorded, due to virus-induced cell death; #, culture was maintained for a total of 17 days without indication of infection (no observation of single SARS-CoV-2 spike protein-positive cells and RNA titers were around the LLOQ).
Viral spread kinetics monitored by immunostainings were confirmed by the determination of SARS-CoV-2 RNA titers in cell culture supernatants (Fig. 8). In cultures treated with 1- to 2.5-fold EC50 boceprevir, viral RNA titers increased during the experiment; however, compared to the nontreated control culture, this increase was decelerated in a concentration-dependent manner, and overall peak titers were lower. In contrast, in cultures treated with 3- and 5-fold EC50 boceprevir, viral RNA titers decreased to values around the lower limit of quantification (LLOQ) during the experiment (Fig. 8). In follow-up derived replicate cultures receiving no treatment, viral RNA titers were around the LLOQ.
To further confirm elimination of SARS-CoV-2 from cultures treated with 3- and 5-fold EC50 boceprevir, we carried out a 96-well based infectivity assay monitoring presence of infectious virus in all cell culture supernatants from the derived replicate cultures by inoculation of Vero E6 cell indicator cultures. Derived replicate cultures were chosen for this analysis, as they were not treated with antivirals, which might inhibit infection of indicator cultures. Using this assay, we did not find evidence of any SARS-CoV-2-infected cells in the indicator cultures.
To investigate the potential of boceprevir to suppress SARS-CoV-2 infection in human cells, SARS-CoV-2-infected A549-hACE2 cells were treated with 1-, 2-, 3-, 5-, and 8-fold EC50 boceprevir (Fig. 7, middle). Virus RNA titrations revealed that 1- to 5-fold EC50 boceprevir only had a minor impact on viral spread, while 8-fold EC50 boceprevir resulted in viral suppression, with viral titers decreasing to values around the LLOQ during an observation period of 13 days. In addition, from the culture treated with 8-fold EC50 on day 7, a replicate culture receiving no treatment was derived, from which viral RNA titers were around the LLOQ during a period of 7 days.
Thus, 3- to 8-fold EC50 boceprevir was able to suppress SARS-CoV-2 in vitro.
Simeprevir or grazoprevir in combination with remdesivir completely suppressed viral infection in vitro.
To further study and confirm the interactions of PIs with remdesivir, three PIs with apparent differential interactions with remdesivir were selected for longer-term treatment of SARS-CoV-2-infected cells. Vero E6 cells infected with SARS-CoV-2 were treated with the PIs boceprevir, simeprevir, or grazoprevir singly, remdesivir singly, or either PI in combination with remdesivir, using 1-fold EC50 PI and 0.4-fold EC50 remdesivir. Inhibitor concentrations were selected to confer suboptimal effects in single treatments in order to rule out viral suppression in these treatments. Equipotent concentrations of PIs were used based on data shown in Fig. 1 and 6 and Table 1. For remdesivir, potency was evaluated based on concentration-response curves obtained from data shown in Fig. 4 and Fig. S7 and, in addition, based on pilot longer-term treatment assays. Treatment with remdesivir, boceprevir, simeprevir, or grazoprevir singly, as well as treatment with boceprevir plus remdesivir, had no or only a minor impact on viral spread on day 1 postinfection and treatment initiation, while 70% to 90% of culture cells became infected on day 3, as observed for nontreated control cells (Fig. 9). In contrast, in cultures treated with simeprevir plus remdesivir or grazoprevir plus remdesivir, complete and sustained viral suppression was achieved with no evidence of infection from day 1 during a follow-up period of 15 days. In addition, to confirm complete viral suppression, from these cultures, replicate cultures receiving no treatment were derived on day 5. Replicate cultures derived from cultures treated with simeprevir plus remdesivir and with grazoprevir plus remdesivir did not show any infected cells during a follow-up period of 19 and 14 days, respectively.
FIG 9.

At equipotent concentrations, simeprevir and grazoprevir but not boceprevir synergized with remdesivir to completely suppress viral infection in Vero E6 cells. Vero E6 cells seeded the previous day in T25 flasks were infected with SARS-CoV-2 at an MOI of 0.00002 followed by treatment with 0.4-fold EC50 remdesivir (REM), 1-fold EC50 boceprevir (BOC), 1-fold EC50 simeprevir (SIM), or 1-fold EC50 grazoprevir (GRA) singly or with a combination of remdesivir with either of the PIs, which were administered immediately after infection and subsequently at the indicated time points when cells were split, as described in Materials and Methods. (Left) Percentages of SARS-CoV-2-infected cells on the specified days postinfection were determined by anti-spike protein immunostaining of replicate cultures derived following cell splitting and treatment. (Middle) SARS-CoV-2 RNA titers determined in cell culture supernatants as genome copies per milliliter on the specified days postinfection were determined by RT-qPCR assays. The black lines indicate the LLOQs. In the left and middle panels, to facilitate comparisons, bars are color coded according to the day postinfection, and blue and red dotted lines were inserted to highlight day 1 and 3 values of the nontreated culture, respectively. (Right) Replicate cultures were derived following cell splitting and treatment, immunostained for the SARS-CoV-2 spike protein (green) and counterstained with Hoechst dye (blue), and images were acquired, as described in Materials and Methods. Cultures summarized in this figure are derived from two different experimental setups (REM/BOC/SIM and REM/GRA experiments), each including the respective depicted nontreated control culture. *, culture was terminated, or infection data not recorded, due to virus-induced cell death; #, culture was maintained for a total of 15 days without indication of infection (no observation of single SARS-CoV-2 spike protein-positive cells and RNA titers were around the LLOQ).
Determination of SARS-CoV-2 RNA titers in cell culture supernatants confirmed viral spread kinetics monitored by immunostainings (Fig. 9). Treatment with single inhibitors and boceprevir plus remdesivir had no or a minor impact on RNA titers compared to that in the nontreated control culture (Fig. 9). However, under treatment with simeprevir plus remdesivir or with grazoprevir plus remdesivir, RNA titers decreased to values around the LLOQ during the experiment. In derived replicate cultures receiving no treatment, viral RNA titers were around the LLOQ.
Elimination of SARS-CoV-2 from cultures treated with simeprevir plus remdesivir was confirmed by an infectivity assay monitoring the presence of infectious virus in all cell culture supernatants from the derived replicate cultures not receiving treatment.
To verify interactions of boceprevir, simeprevir, or grazoprevir with remdesivir in human cells, SARS-CoV-2-infected A549-hACE2 cells were treated with 1-fold EC50 boceprevir, simeprevir, or grazoprevir singly, 0.8-fold remdesivir singly, or either PI in combination with remdesivir (Fig. 7, right). Virus RNA titrations revealed that treatment with single inhibitors or boceprevir plus remdesivir only had a minor impact on viral spread, while treatment with simeprevir plus remdesivir or grazoprevir plus remdesivir resulted in viral suppression with viral RNA titers around the LLOQ.
Thus, combination of simeprevir or grazoprevir with remdesivir was able to suppress SARS-CoV-2 in vitro.
DISCUSSION
In the present study, we provided a head-to-head comparison of the efficacy of a panel of clinically relevant HCV PIs, including all PIs approved for the treatment of chronic hepatitis C, against SARS-CoV-2 in cell-based assays. In short-term antiviral assays, PIs showed differential potencies, with EC50 values between 15 μM (simeprevir) and >178 μM (glecaprevir) in Vero E6 cells and 9 μM (simeprevir) and >94 μM (glecaprevir) in A549-hACE2 cells. Detailed short-term synergy studies in both cell types using a PI subpanel revealed PI structure-dependent interactions with remdesivir, with linear inhibitors showing mostly antagonism and macrocyclic inhibitors showing mostly synergism. In longer-term Vero E6 and A549-hACE2 cell culture assays, at relatively low equipotent concentrations, PIs showed small differences regarding the barrier to viral escape. For boceprevir, a relatively high SI facilitated treatment with higher concentrations, revealing its potential to completely suppress viral infection. Furthermore, combination of simeprevir or grazoprevir with remdesivir suppressed viral infection at relatively low inhibitor concentrations, shown to be subtherapeutic in single treatments.
Even though cell lines do not entirely recapitulate in vivo conditions, they provide robust models for preclinical antiviral activity studies. Our findings are strengthened by the fact that similar results were obtained in three different cell lines, including two human cell lines, of which one was derived from human lung. Future studies to evaluate the efficacy of HCV PIs on SARS-CoV-2 in primary cells, organoids, animals, or humans would be of interest.
The sequence homology between different SARS-CoV-2 isolates is high. In comparison to the Wuhan reference isolate (GenBank accession number NC_045512), the isolate used in this study harbored six consensus amino acid changes (T85I in nsp2, P323L in nsp12, E309K and D614G in S, Q57H in ORF3a, and R209I in N); however, there were no changes in Mpro or PLpro, the proposed main targets of the studied HCV PI (31).
EC50s against SARS-CoV-2 were in the micromolar range, with the lowest EC50s (9 and 15 μM for simeprevir in A549-hACE2 and Vero E6 cells, respectively) approaching the EC50 of remdesivir (0.1 and 2.5 μM in A549-hACE2 and Vero E6 cells, respectively); EC50s of remdesivir were in line with previously reported results (30–32, 35, 36). However, EC50s of PIs against SARS-CoV-2 were higher than the EC50s against HCV. Initially developed HCV PIs such as boceprevir and simeprevir were roughly 10- to 1,000-fold less potent, while optimized HCV PIs such as grazoprevir, glecaprevir, and voxilaprevir were roughly 1,000- to 100,000-fold less potent against SARS-CoV-2 than against different HCV isolates (29, 38–43, 47–49). This suggested that optimization of inhibition of the HCV protease counteracted broader activity against proteases of other RNA viruses, such as SARS-CoV-2.
Boceprevir showed the highest SIs (>61 and >28 in A549-hACE2 and Vero E6 cells, respectively), while simeprevir showed the lowest SIs (6.2 and 3.9 in A549-hACE2 and Vero E6 cells, respectively). Of note, some clinically relevant drugs such as digoxin have low therapeutic breadth, with SIs as low as 2 (50), and HCV PIs have been proven safe in clinical practice. To estimate the clinical potential of inhibitors, comparisons of their EC50 with clinically achievable plasma and tissue concentrations are more relevant than comparison with in vitro CC50 values. For most HCV PIs, peak plasma concentrations (Cmax) were significantly lower than the determined EC50 values (see Table S5 in the supplemental material). The most favorable Cmax/EC50 ratio was found for simeprevir (Cmax/EC50 of ∼1), followed by faldaprevir (Cmax/EC50 of ∼0.2) as well as boceprevir, telaprevir, and vaniprevir (Cmax/EC50 of ∼0.1) based on the EC50s obtained in Vero E6 cells. Furthermore, plasma concentrations of free and biologically active compounds are expected to be lower than reported Cmax values due to the high plasma protein binding of HCV PIs. Regarding tissue concentrations, for HCV PIs, mostly liver concentrations were reported and were 20- to 280-fold higher than plasma concentrations (43, 51–53). In rats, following a single oral administration of simeprevir, concentrations in the intestine, which is permissive to SARS-CoV-2 infection (54), were up to 128-fold higher than in the plasma, while concentrations in other tissues were roughly equal to plasma concentrations (55). Therefore, it would be relevant to determine HCV PI lung concentrations in humans following multiple doses in steady state (56). For remdesivir showing high in vitro efficacy, clinical efficacy might in part be limited by poor distribution to the lungs (31, 32, 56). Poor lung distribution following systemic application of inhibitors might be overcome by improved formulations allowing topic application, such as inhalable formulations.
Inhibitor efficacy can be improved by combination treatments with synergistic and thus drug-saving effects as reported here for the combination of remdesivir with the macrocyclic PIs simeprevir, paritaprevir, or grazoprevir in Vero E6 cells and with simeprevir or grazoprevir in A549-hACE2 cells. Synergism was recently suggested for the combination of remdesivir with simeprevir (35, 36) or grazoprevir (36) in short-term assays in Vero E6 cells (35, 36) and human embryonic kidney (HEK293T) cells (36). Our extensive results in Vero E6 and A549-hACE2 cells using short-term drug interaction assays demonstrated that the mode of interaction between PI and remdesivir depended on the PI structure. Thus, combination of remdesivir with the linear PIs boceprevir and narlaprevir showed mostly antagonism, while combination with macrocyclic PIs showed mostly synergism. In contrast to the previous studies (35, 36), we confirmed these PI structure-dependent interactions with remdesivir in longer-term treatment assays in Vero E6 and A549-hACE2 cells, where the combination of remdesivir with simeprevir or grazoprevir, but not with boceprevir, resulted in added efficacy. This structure dependence might be explained by differences in viral targets. While the investigated HCV PIs were suggested to target Mpro (30, 33–36, 57–77), simeprevir was suggested to also target the SARS-CoV-2 polymerase (35), and simeprevir, grazoprevir, and paritaprevir were suggested to also target PLpro (36, 62). It should be noted that additional alternative viral targets, including nsp13 (helicase), nsp14 (exonuclease and methyltransferase), nsp15 (endoribonuclease), nsp16 (2′-O-ribose methyltransferase), as well as structural proteins N (capsid) and spike, were suggested for paritaprevir, grazoprevir, and simeprevir by modeling studies (62, 74, 78–82). Future detailed molecular studies are required to fully define the viral targets of different HCV PIs.
HCV PIs were designed and optimized to bind the HCV NS3 protease. In modeling studies, structural similarity between the HCV NS3 protease and SARS-CoV-2 Mpro, including their active sites, was reported (36, 57, 68, 77), despite a lack of overall sequence conservation. While both viral proteases are chymotrypsin-like proteases, the HCV NS3 protease has a larger and more shallow binding groove (34). The HCV NS3 protease and PLpro do not show structural similarity (36).
While carrying out and revising this study, several research articles addressing a potential effect of HCV PIs on SARS-CoV-2 were published. Using in silico modeling approaches, more than 20 studies predicted binding of different linear and macrocyclic HCV PIs to SARS-CoV-2 Mpro (34, 36, 57–77). Fewer studies predicted binding of macrocyclic HCV PIs to PLpro (36, 62). Furthermore, crystal structures of boceprevir, narlaprevir, and telaprevir bound to Mpro were solved (34, 77, 83–85). In addition, reports were published on inhibition of Mpro by linear and macrocyclic HCV PIs (33–36, 83, 84) and of PLpro by macrocyclic and acyclic HCV PIs (36).
Recently, five groups demonstrated the efficacy of different HCV PIs in Vero E6 cells (30, 33–36). Most EC50s reported in these studies were in the same range as those reported here; the slightly higher EC50 values observed in our study are most likely caused by differences in experimental assay conditions. EC50s in human lung cells were only reported for simeprevir (EC50, 1 μM) (35) and voxilaprevir (>10 μM), also using the A549-hACE2 cell line (30). The similar PI EC50 values in monkey Vero E6 cells, human Huh7.5 cells, and human A549-hACE2 cells reported in our study validate Vero E6 cells for the study of HCV PI efficacy against SARS-CoV-2. Furthermore, the short-term treatment assay based on the quantification of SARS-CoV-2 spike protein-expressing cells was validated here by an assay measuring virus-induced CPE.
Finally, we report antiviral activity of the HCV NS4A inhibitor ACH-806. Future studies are required to define the SARS-CoV-2 target of this compound and to investigate its potential to inform design of SARS-CoV-2 inhibitors.
In conclusion, we here provide a head-to-head comparison of the efficacy of a panel of clinically relevant HCV PIs against SARS-CoV-2, including detailed studies of interaction with remdesivir using different cell lines. Of the currently, clinically widely used HCV PIs, grazoprevir and voxilaprevir showed activity against SARS-CoV-2 in lung cells. For clinical use of HCV PIs with higher potency and higher plasma concentrations, such as simeprevir, production would need to be reinitiated, as was the case for remdesivir. While HCV PIs showed relatively low Cmax/EC50 ratios assuming systemic administration, it remains possible that active concentrations will be attainable in relevant tissues. Furthermore, treatment efficacy might be increased by topical administration of improved formulations and by combination with remdesivir. As this study demonstrated structure-dependent differential interaction of HCV PIs with remdesivir, novel PIs should be tested for interaction with remdesivir if combination treatment is considered. While clinical studies would be needed to investigate if HCV PIs studied here could have direct clinical relevance, this study identifies compounds that could assist the development of further optimized PI molecules for future COVID-19 treatment regimes.
MATERIALS AND METHODS
Cell cultivation.
All cells were maintained at 37°C and 5% CO2. African green monkey kidney Vero E6 cells (gift from J. Dubuisson) and human Huh7.5 hepatoma cells (86) were maintained in Dulbecco’s modified Eagle medium (Invitrogen, Paisley, UK) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Sigma, St. Louis, MO, USA), 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco/Invitrogen Corp., Carlsbad, CA, USA). A549-hACE2 cells (InvivoGen, Toulouse, France) were maintained in Dulbecco’s modified Eagle medium nutrient mixture F-12 (Gibco, Paisley, UK) supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.5 μg/ml puromycin (InvivoGen). Cells were split every 2 to 3 days with trypsin (Sigma) to maintain a subconfluent monolayer.
Virus isolate.
The corona virus isolate SARS-CoV-2/human/Denmark/DK-AHH1/2020 was derived from a swab sample from a Danish patient that was passaged in Vero E6 cells. For the experiments presented here, we used a sequence-confirmed 2nd viral passage stock with an infectivity titer of 5.5 log10 50% tissue culture infective dose (TCID50)/ml (31).
Inhibitors.
All inhibitors were purchased from Acme Bioscience (Palo Alto, CA, USA) and dissolved in DMSO (Sigma, St. Louis, MO, USA).
Short-term concentration-response antiviral treatment assays for evaluation of inhibitor potency.
Ninety-six-well-based short-term antiviral treatment assays in Vero E6 cells, Huh7.5 cells, and A549-hACE2 cells were developed based on assays previously established for the determination of the potency of HCV PIs against HCV (29, 37–43). Vero E6 and A549-hACE2 cells were seeded at 10,000 cells per well, and Huh7.5 cells were seeded at 9,000 cells per well in 96-well flat-bottom plates (Thermo Fisher Scientific, Roskilde, Denmark). The following day, for Vero E6 cells, medium was changed to 50 μl fresh medium and cells were inoculated with SARS-CoV-2/human/Denmark/DK-AHH1/2020 at an MOI of 0.002 by adding 50 μl virus stock diluted in medium to each well. Huh7.5 cells were inoculated at an MOI of 0.02, and A549-hACE2 cells were inoculated at an MOI of 0.003 (based on the infectivity titer determined in Vero E6 cells) by exchanging the medium with 50 μl virus stock diluted in medium. Following a 1-h incubation at 37°C and 5% CO2, infected cells were treated with a dilution series of inhibitors by adding 50 μl medium with inhibitor, resulting in the specified concentrations. Alternatively, cells were treated with a dilution series of DMSO alone serving as a control for the antiviral activity of DMSO. All concentrations of inhibitor were tested in 7 replicates; 14 infected and nontreated and 12 noninfected and nontreated replicates were included in each assay. For A549-hACE2 cells, the concentration of DMSO was kept constant in all cultures. Cells were subjected to immunostaining for the SARS-CoV-2 spike protein and evaluated as described below after incubation for 46 to 50 h for Vero E6 and A549-hACE2 cells or for 70 to 74 h for Huh7.5 cells.
Additionally, 96-well based concentration-response CPE assays were carried out in Vero E6 cells. Vero E6 cells were seeded at 10,000 cells per well in 96-well flat-bottom plates (Thermo Fisher Scientific). The following day, cells were inoculated with SARS-CoV-2/human/Denmark/DK-AHH1/2020 at an MOI of 0.01 by exchanging the medium with 50 μl virus stock diluted in medium. After a 1-h incubation, infected cells were treated with a dilution series of inhibitor by adding 50 μl medium with inhibitor, resulting in the specified concentrations. All concentrations of inhibitor were tested in 4 replicates; 8 infected and nontreated and 16 noninfected and nontreated replicates were included in each assay. The concentration of DMSO was kept constant in all cultures. After 70 to 74 h of incubation at 37°C and 5% CO2, CPE was evaluated using viral ToxGlo assay (Promega, Madison, WI, USA) according to the manufacturer’s guidelines. Relative light units (RLU) from infected and treated wells were related to the mean RLU from the 16 noninfected control wells. Data points are given as means from 4 replicates with standard errors of the means (SEMs). Sigmoidal concentration-response curves were fitted and EC50 values were calculated using GraphPad Prism 8.0.0 applying the formula Y = Bottom + ((Top − Bottom)/(1 + 10(Log10EC50-X)*HillSlope)).
Immunostaining and evaluation of 96-well plates.
Cells were fixed and virus was inactivated by submersion into methanol (J.T.Baker, Gliwice, Poland) for 20 min at room temperature. For immunostaining for the SARS-CoV-2 spike protein, plates were washed 2 times with phosphate buffered saline (PBS; Sigma, Gillingham, UK) containing 0.1% Tween 20 (Sigma, St Louis, MO) (PBS-Tween). Then, endogenous peroxidase activity was blocked by adding H2O2 and incubating for 10 min followed by 2 more washes with PBS-Tween and blocking with PBS containing 1% bovine serum albumin (Roche, Mannheim, Germany) and 0.2% skim milk (Easis, Aarhus, Denmark) (PBSK) for 30 min. Next, plates were emptied and incubated with primary antibody, SARS-CoV-2 spike chimeric monoclonal antibody (number [no.] 40150-D004; Sino Biological, Beijing, China), diluted 1:5,000 in PBSK for 2 h at room temperature. Then, plates were washed 2 times with PBS-Tween and incubated for 1 h at room temperature with secondary antibody, F(ab′)2-goat anti-human IgG-Fc cross-adsorbed secondary antibody conjugated with horseradish peroxidase (HRP) (no. A24476; Invitrogen, Carlsbad, CA, USA) or goat F(ab′)2 anti-human IgG-Fc (HRP), preadsorbed (no. 98595; Abcam, Cambridge, UK), diluted 1:2,000 in PBSK. Finally, plates were washed 2 times with PBS-Tween, and SARS-CoV-2 spike protein-positive cells were stained using diaminobenzidine (DAB) substrate BrightDAB kit (no. BS04-110; Immunologic, Duiven, Netherlands) according to the manufacturer’s guidelines. Plates were evaluated by automated counting of single SARS-CoV-2 spike protein-positive cells using an ImmunoSpot series 5 UV analyzer (CTL Europe GmbH, Bonn, Germany) (87). The mean of counts from noninfected and nontreated wells, which was usually <50, was subtracted from counts of infected wells. Counts from infected and treated wells were related to the mean count of the 14-replicate infected nontreated wells to calculate percent residual infectivity; mean counts of infected nontreated wells were 3,000 to 4,000 for Vero E6 cells, 1,000 to 2,000 for Huh7.5 cells, and 2,000 to 3,000 for A549-hACE2 cells. Data points are given as means from 7 replicates with SEMs. Sigmoidal concentration-response curves were fitted and EC50 values calculated as described previously using GraphPad Prism 8.0.0 with a bottom constraint of 0 applying the formula Y = Top/(1 + 10[Log10 EC50 − X] × Hill slope) (43, 88). Representative images from concentration-response antiviral treatment assays are shown by Zhou et al. (44).
Short-term concentration-response antiviral treatment assays for analysis of interactions of PIs and remdesivir in Vero E6 and A549-hACE2 cells.
Interactions of selected PIs in combination with remdesivir for inhibition of SARS-CoV-2 were investigated based on protocols previously established for HCV (38). The experimental design was similar to that of the concentration-response antiviral treatment assays described above. In brief, Vero E6 cells or A549-hACE2 cells were seeded at 10,000 cells per well in 96-well flat-bottom plates, medium was changed to 50 μl fresh medium, and cells were inoculated with SARS-CoV-2/human/Denmark/DK-AHH1/2020 at an MOI of 0.002 (Vero E6 cells) or an MOI of 0.003 (A549-hACE2 cells) by adding 50 μl virus stock diluted in medium to each well. Following a 1-h incubation at 37°C and 5% CO2, infected cells were treated with a dilution series of inhibitors by adding 50 μl medium with inhibitor resulting in the specified concentrations. Regarding inhibitor treatment, dilution series of selected PIs singly, remdesivir singly or a combination of PIs and remdesivir were used that were based on EC50 values against SARS-CoV-2. Thus, for inhibitors and combinations of inhibitors, 1.15- to 2-fold dilution series with at least 7 different dilutions were applied spanning the respective EC50 values, aiming at achieving residual infectivity between 0% and 100%. For combination treatments, the same PI and remdesivir concentrations as used in single treatments were applied with a fixed ratio, except for the simeprevir plus remdesivir data set, where a nonconstant ratio was used. All treatment conditions were tested in 6 or 7 replicates, including 21 to 70 infected and nontreated replicates per experiment (with at least 7 replicates per experimental plate) and 12 noninfected and nontreated replicates per experimental plate. In experiments with A549-hACE2 cells, the concentration of DMSO was kept constant in all cultures. After 46 to 50 h of incubation, infected cells were visualized by immunostaining for the SARS-CoV-2 spike protein, and plates were evaluated by automated counting of single SARS-CoV-2 spike protein-positive cells, as described above.
Concentration-response cell viability assays.
To evaluate cytotoxic effects of the inhibitors and DMSO, cell viability was monitored using the CellTiter 96 Aqueous one solution cell proliferation assay (Promega, Madison, WI, USA). Vero E6, Huh7.5, and A549-hACE2 cells were seeded in 96-well flat-bottom plates at 10,000, 9,000, and 10,000 cells per well, respectively, and the following day were treated with dilution series of inhibitors or combinations of inhibitors by adding 100 μl of medium containing inhibitors at the specified concentrations or DMSO alone at the specified dilutions. After 46 to 50 h for Vero E6 and A549-hACE2 cells and after 70 to 74 h for Vero E6 and Huh7.5 cells, cell viability was evaluated according to the manufacturer’s guidelines. Vero E6 cells were tested with different incubation times to control for assays with 48 versus 72 h of incubation. In brief, 20 μl CellTiter 96 aqueous one solution reagent was added to each well, and plates were then incubated for 1 to 3 h at 37°C and 5% CO2. After incubation, for each well, absorbance at 492 nm was recorded by use of a FLUOstar OPTIMA 96-well plate reader (BMG, LABTECH, Offenburg, Germany). Each inhibitor concentration was tested in 2 to 4 replicate wells, and each experimental plate included 12 replicate nontreated control wells. Absorbance values from treated wells were related to the mean absorbance of the nontreated wells to estimate percent cell viability. Data points are given as means from 2 to 4 replicates with SEMs. Sigmoidal concentration-response curves were fitted and 50% cytotoxic concentration (CC50) values were calculated using GraphPad Prism 8.0.0, with a bottom constraint of 0 applying the formula Y = Top/(1 + 10[Log10 EC50 − X] × Hill slope).
Viral cytopathogenic effect assay for determination of MOI for short-term treatment assays.
To select a suitable MOI for short-term treatment assays not resulting in virus-induced CPE (immunostaining-based assay) or relatively strong CPE (CPE-based assay) during the assays, viral CPE assays were carried out. Cells were plated on 96-well flat-bottom plates as described for short-term treatment assays. The following day, cells were infected at different MOIs with SARS-CoV-2/human/Denmark/DK-AHH1/2020, using 100 μl per well with 4 replicates per MOI. Following the incubation times used in the short-term treatment assays, CPE was evaluated using the viral ToxGlo assay (Promega) according to the manufacturer’s guidelines. Relative light units (RLU) from infected cultures were related to the mean RLU from noninfected control cultures to detect CPE.
Longer-term antiviral treatment assays in SARS-CoV-2-infected Vero E6 and A549-hACE2 cells.
Cells were seeded at 106 cells per flask in T25 flasks (Nunc) and, the following day, infected at an MOI of 0.00002 (Vero E6 cells) or an MOI of 0.0005 (A549-hACE2 cells) with SARS-CoV-2/human/Denmark/DK-AHH1/2020. Cells were treated with specified-fold EC50s of inhibitors on the day of infection by adding inhibitors together with the virus and again on day 1 postinfection. The cells were split and treated every 2 days with the specified concentrations of inhibitors, and the percentage of infected culture cells was evaluated by immunostaining for the SARS-CoV-2 spike protein and immunofluorescence imaging, as described below. In experiments with A549-hACE2 cells, the concentration of DMSO was kept constant in all cultures. Upon cell splitting, culture supernatants were harvested and stored at −80°C. Selected culture supernatants were used to determine viral RNA titers by RT-qPCR and to evaluate the presence of infectious virus by using an infectivity assay. For each experiment, a nontreated infected culture was included serving as a positive control for infection. Cultures were closed when massive cell death occurred, induced by viral infection and/or inhibitor treatment. Cell death was monitored with a light microscope.
Immunostaining and immunofluorescence imaging for evaluation of longer-term Vero E6 and A549-hACE2 cell cultures.
In longer-term SARS-CoV-2-infected and PI-treated cultures, following cell splitting and treatment, replicate cell cultures were seeded in 8-well chamber slides (Thermo Fisher Scientific, Rochester, NY, USA). The next day, cells were fixed, and virus was inactivated by submersion into methanol for 20 min. Chamber slides were washed twice with PBS-Tween and then incubated with primary antibody, SARS-CoV-2 spike chimeric monoclonal antibody (no. 40150-D004; Sino Biological, Beijing, China), diluted 1:1,000 in PBSK for 2 h at room temperature. Following 2 washes with PBS-Tween, chamber slides were incubated with secondary antibody, Alexa-Fluor 488 goat anti-human IgG(H+L) (no. A-11013; Invitrogen, Paisley, UK), diluted 1:500 and Hoechst 33342 (Invitrogen) diluted 1:1,000 in PBS-Tween for 20 min at room temperature. The percentage of SARS-CoV-2 spike protein-positive cells was evaluated by fluorescence microscopy (Axio Vert.A1; Zeiss, Jena, Germany), assigning the following designations: 0% infected cells (no cells infected), single infected cells, and 10% to 90% infected cells (in steps of 10%). The images were acquired with ZEN 3.0 software.
RT-qPCR assay for determination of SARS-CoV-2 RNA titers for evaluation of longer-term Vero E6 and A549-hACE2 cell cultures.
For longer-term cultures, upon cell splitting and treatment, supernatant was harvested and stored at −80°C. Supernatant was mixed 1:3 with TRIzol LS (Life Technologies), and RNA was extracted with chloroform (Sigma) using 5PRIME phase gel lock heavy tubes (Quantabio). RNA was purified using an RNA clean and concentrator-5 kit (ZYMO Research) according to the manufacturer’s guidelines, and RNA was eluted in nuclease-free water (Ambion). qPCRs were carried out using the TaqMan fast virus 1-step master mix (Thermo Fischer) with previously described primers and probes (89): E_Sarbeco F (5′-ACAGGTACGTTAATAGTTAATAGCGT-3′), E_Sarbeco_R (5′-ATATTGCAGCAGTACGCACACA-3′), and E_Sarbeco_P (6-carboxyfluorescein [FAM]-5′-ACACTAGCCATCCTTACTGCGCTTCG-3′-black hole quencher 1 [BHQ1]). Primers were used at 400 nM and probe was used at 200 nM together with 2.5 μl purified RNA. Cycling conditions were as follows: for reverse transcription, 10 min at 55°C, followed by 3 min at 95°C and 45 cycles of 95°C for 15 s and 58°C for 30 s using the LightCycler 96 System (Roche). For every assay, a negative control and RNA standards ranging from 10 to 105 RNA copies per μl (Twist Bioscience) were included. RNA titers (genome copies/milliliter) were calculated by interpolation of cycle threshold values of the standard curve generated using the standard panel and the LightCycler software. The LLOQ of the assay was calculated as mean of RNA titers in supernatants derived from noninfected control cultures plus 3 standard deviations.
Infectivity assay for evaluation of presence of infectious virus in supernatants from longer-term Vero E6 cell cultures.
Vero E6 indicator cell cultures were seeded at 10,000 cells per well in 96-well flat-bottom plates. The following day, the medium was exchanged with 100 μl of cell culture supernatants diluted 1:5 in cell culture medium. Culture supernatants were harvested every 2 to 3 days from replicate cultures not receiving treatment derived from longer-term Vero E6 cell cultures. Longer-term Vero E6 cell cultures treated with 3- or 5-fold EC50 boceprevir (Fig. 8) or 1-fold EC50 simeprevir in combination with 0.4-fold EC50 remdesivir (Fig. 9) were investigated. For each supernatant, 4 replicate indicator cultures were inoculated. Twelve cultures inoculated with SARS-CoV-2/human/Denmark/DK-AHH1/2020 at an MOI of 0.01 served as a positive control for infection, and 12 noninfected cultures served as negative controls. After 46 to 50 h of incubation at 37°C and 5% CO2, cells were subjected to immunostaining for the SARS-CoV-2 spike protein, and the number of single infected cells was evaluated by automated counting as described above.
ACKNOWLEDGMENTS
This work was supported by Ph.D. stipends from the Candys Foundation (K.A.G., A.O., L.V.P., J.B., and J.M.G.) and the China Scholarship Council (Y.Z., J.M.G.) and by grants from the Novo Nordisk Foundation (J.B.), the Weimann Foundation (U.F.), the Region H Foundation (J.B. and J.M.G.), the Amager and Hvidovre Hospital Research Foundation (C.R.D.H. and J.M.G.), the Independent Research Fund Denmark (J.B.), the Læge Sofus Carl Emil Friis og Hustru Olga Doris Friis' Foundation (J.M.G.), and the Danish Agency for Science and Higher Education (S.R., J.B., and J.M.G.).
We thank Bjarne Ørskov Lindhardt (Copenhagen University Hospital, Hvidovre) and Carsten Geisler (University of Copenhagen) for support. We thank Line Abildgaard Ryberg for discussion on structural interactions of Mpro and PI, as well as Pia Pedersen, Lotte Mikkelsen, and Anna-Louise Sørensen (Copenhagen University Hospital, Hvidovre) for general laboratory assistance. We also thank Jean Dubuisson and Sandrine Belouzard for providing Vero E6 cells and Charles Rice for providing Huh7.5 cells.
S.R., J.B., and J.M.G. conceived this project. K.A.G., Y.Z., A.G., S.R., and J.M.G. designed the experiments. K.A.G., Y.Z., C.R.D.H., A.G., and A.O. carried out the experiments. K.A.G., Y.Z. C.R.D.H. A.G., A.O., R.C., and J.M.G. analyzed and interpreted the data. K.A.G., Y.Z., A.O., R.C., L.V.P., U.F., S.F., T.K.H.S., and S.R. contributed to isolation and characterization of SARS-CoV-2/human/Denmark/DK-AHH1/2020 in vitro and established experimental systems. K.A.G., Y.Z., and J.M.G. prepared an initial manuscript draft. All authors contributed to and discussed the manuscript. J.M.G. supervised the study.
We declare no conflict of interest.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Zhou P, Lou Yang X, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang R, Di Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W. 2020. A novel coronavirus from patients with pneumonia in China. N Engl J Med 382:727–733. 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, Zhang YZ. 2020. A new coronavirus associated with human respiratory disease in China. Nature 579:265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie J, Tong Z, Guan X, Du B, Qiu H. 2020. Clinical characteristics of patients who died of coronavirus disease 2019 in China. JAMA Netw Open 3:e205619. 10.1001/jamanetworkopen.2020.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guan W, Ni Z, Hu Y, Liang W, Ou C, He J, Liu L, Shan H, Lei C, Hui DSC, Du B, Li L, Zeng G, Yuen KY, Chen R, Tang C, Wang T, Chen P, Xiang J, Li S, Wang JL, Liang Z, Peng Y, Wei L, Liu Y, Hu YH, Peng P, Wang JM, Liu J, Chen Z, Li G, Zheng Z, Qiu S, Luo J, Ye C, Zhu S, Zhong N, China Medical Treatment Expert Group for Covid-19. 2020. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382:1708–1720. 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Wang Y, Chen Y, Qin Q. 2020. Unique epidemiological and clinical features of the emerging 2019 novel coronavirus pneumonia (COVID-19) implicate special control measures. J Med Virol 92:568–576. 10.1002/jmv.25748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.World Health Organization. 2020. WHO coronavirus disease (COVID-19). https://covid19.who.int/.
- 8.Johns Hopkins University. 2020. COVID-19 dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). https://coronavirus.jhu.edu/map.html.
- 9.Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, Hohmann E, Chu HY, Luetkemeyer A, Kline S, Lopez de Castilla D, Finberg RW, Dierberg K, Tapson V, Hsieh L, Patterson TF, Paredes R, Sweeney DA, Short WR, Touloumi G, Lye DC, Ohmagari N, Oh M, Ruiz-Palacios GM, Benfield T, Fätkenheuer G, Kortepeter MG, Atmar RL, Creech CB, Lundgren J, Babiker AG, Pett S, Neaton JD, Burgess TH, Bonnett T, Green M, Makowski M, Osinusi A, Nayak S, Lane HC, ACTT-1 Study Group Members. 2020. Remdesivir for the Treatment of Covid-19—Final Report. N Engl J Med 383:1813–1826. 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agbowuro AA, Huston WM, Gamble AB, Tyndall JDA. 2018. Proteases and protease inhibitors in infectious diseases. Med Res Rev 38:1295–1331. 10.1002/med.21475. [DOI] [PubMed] [Google Scholar]
- 11.Scheel TKH, Rice CM. 2013. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med 19:837–849. 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartenschlager R, Lohmann V, Penin F. 2013. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat Rev Microbiol 11:482–496. 10.1038/nrmicro3046. [DOI] [PubMed] [Google Scholar]
- 13.Cannalire R, Cerchia C, Beccari AR, Di Leva FS, Summa V. 13November2020. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. J Med Chem 10.1021/acs.jmedchem.0c01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R. 2020. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368:409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y, Yu J, Wang L, Yang K, Liu F, Jiang R, Yang X, You T, Liu X, Yang X, Bai F, Liu H, Liu X, Guddat LW, Xu W, Xiao G, Qin C, Shi Z, Jiang H, Rao Z, Yang H. 2020. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582:289–293. 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 16.Mengist HM, Fan X, Jin T. 2020. Designing of improved drugs for COVID-19: crystal structure of SARS-CoV-2 main protease Mpro. Signal Transduct Target Ther 5:67. 10.1038/s41392-020-0178-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, Schulz L, Widera M, Mehdipour AR, Tascher G, Geurink PP, Wilhelm A, van der Heden van Noort GJ, Ovaa H, Müller S, Knobeloch KP, Rajalingam K, Schulman BA, Cinatl J, Hummer G, Ciesek S, Dikic I. 2020. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587:657–662. 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Báez-Santos YM, St John SE, Mesecar AD. 2015. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res 115:21–38. 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akaji K, Konno H. 2020. Design and evaluation of anti-SARS-coronavirus agents based on molecular interactions with the viral protease. Molecules 25:3920. 10.3390/molecules25173920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Osipiuk J, Azizi SA, Dvorkin S, Endres M, Jedrzejczak R, Jones KA, Kang S, Kathayat RS, Kim Y, Lisnyak VG, Maki SL, Nicolaescu V, Taylor CA, Tesar C, Zhang YA, Zhou Z, Randall G, Michalska K, Snyder SA, Dickinson BC, Joachimiak A. 2021. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat Commun 12:743. 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Simmonds P. 2014. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology 59:318–327. 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borgia SM, Hedskog C, Parhy B, Hyland RH, Stamm LM, Brainard DM, Subramanian MG, McHutchison JG, Mo H, Svarovskaia E, Shafran SD. 2018. Identification of a novel hepatitis C virus genotype from Punjab, India: expanding classification of hepatitis C virus into 8 genotypes. J Infect Dis 218:1722–1729. 10.1093/infdis/jiy401. [DOI] [PubMed] [Google Scholar]
- 23.Kim JL, Morgenstern KA, Lin C, Fox T, Dwyer MD, Landro JA, Chambers SP, Markland W, Lepre CA, O’Malley ET, Harbeson SL, Rice CM, Murcko MA, Caron PR, Thomson JA. 1996. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 87:343–355. 10.1016/S0092-8674(00)81351-3. [DOI] [PubMed] [Google Scholar]
- 24.Hahm B, Han DS, Back SH, Song OK, Cho MJ, Kim CJ, Shimotohno K, Jang SK. 1995. NS3-4A of hepatitis C virus is a chymotrypsin-like protease. J Virol 69:2534–2539. 10.1128/JVI.69.4.2534-2539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morikawa K, Lange CM, Gouttenoire J, Meylan E, Brass V, Penin F, Moradpour D. 2011. Nonstructural protein 3-4A: the Swiss army knife of hepatitis C virus. J Viral Hepat 18:305–315. 10.1111/j.1365-2893.2011.01451.x. [DOI] [PubMed] [Google Scholar]
- 26.Pawlotsky JM. 2016. Hepatitis C virus resistance to direct-acting antiviral drugs in interferon-free regimens. Gastroenterology 151:70–86. 10.1053/j.gastro.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 27.Li DK, Chung RT. 2019. Overview of direct-acting antiviral drugs and drug resistance of hepatitis C virus. Methods Mol Biol 1911:3–32. 10.1007/978-1-4939-8976-8_1. [DOI] [PubMed] [Google Scholar]
- 28.Pottage JC, Lawitz E, Mazur D, Wyles D, Vargas H, Ghalib R, Gugliotti R, Donohue M, Robison H. 2007. Short-term antiviral activity and safety of ACH-806(GS-9132), an NS4A antagonist, in HCV genotype 1 infected individuals. J Hepatol 46:294–295. 10.1016/S0168-8278(07)62381-2. [DOI] [Google Scholar]
- 29.Gottwein JM, Jensen SB, Serre SBN, Ghanem L, Scheel TKH, Jensen TB, Krarup H, Uzcategui N, Mikkelsen LS, Bukh J. 2013. Adapted J6/JFH1-based hepatitis C virus recombinants with genotype-specific NS4A show similar efficacies against lead protease inhibitors, alpha interferon, and a putative NS4A inhibitor. Antimicrob Agents Chemother 57:6034–6049. 10.1128/AAC.01176-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie X, Muruato AE, Zhang X, Lokugamage KG, Fontes-Garfias CR, Zou J, Liu J, Ren P, Balakrishnan M, Cihlar T, Tseng C-TK, Makino S, Menachery VD, Bilello JP, Shi P-Y. 2020. A nanoluciferase SARS-CoV-2 for rapid neutralization testing and screening of anti-infective drugs for COVID-19. Nat Commun 11:5214. 10.1038/s41467-020-19055-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramirez S, Fernandez-Antunez C, Galli A, Underwood A, Pham LV, Ryberg LA, Feng S, Pedersen MS, Mikkelsen LS, Belouzard S, Dubuisson J, Sølund C, Weis N, Gottwein JM, Fahnøe U, Bukh J. 2021. Overcoming culture restriction for SARS-CoV-2 in human cells facilitates the screening of compounds inhibiting viral replication. Antimicrob Agents Chemother 65:e00097–21. 10.1128/AAC.00097-21. [DOI] [PMC free article] [PubMed]
- 32.Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G. 2020. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 30:269–271. 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma C, Sacco MD, Hurst B, Townsend JA, Hu Y, Szeto T, Zhang X, Tarbet B, Marty MT, Chen Y, Wang J. 2020. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res 30:678–692. 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu L, Ye F, Feng Y, Yu F, Wang Q, Wu Y, Zhao C, Sun H, Huang B, Niu P, Song H, Shi Y, Li X, Tan W, Qi J, Gao GF. 2020. Both boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat Commun 11:4417. 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lo HS, Hui KPY, Lai H-M, He X, Khan KS, Kaur S, Huang J, Li Z, Chan AKN, Cheung HH-Y, Ng K-CC, Ho JCW, Chen YW, Ma B, Cheung PM-H, Shin D, Wang K, Lee M-H, Selisko B, Eydoux C, Guillemot J-C, Canard B, Wu K-P, Liang P-H, Dikic I, Zuo Z, Chan FKL, Hui DSC, Mok VCT, Wong K-B, Mok CKP, Ko H, Aik WS, Chan MCW, Ng W-L. 2021. Simeprevir potently suppresses SARS-CoV-2 replication and synergizes with remdesivir. ACS Cent Sci 7:792–802. 10.1021/acscentsci.0c01186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bafna K, White K, Harish B, Rosales R, Ramelot TA, Acton TB, Moreno E, Kehrer T, Miorin L, Royer CA, García-Sastre A, Krug RM, Montelione GT. 2021. Hepatitis C virus drugs that inhibit the SARS-CoV-2 papain-like protease synergize with remdesivir to suppress viral replication in cell culture. Cell Rep 35:109133. 10.1016/j.celrep.2021.109133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li YP, Ramirez S, Humes D, Jensen SB, Gottwein JM, Bukh J. 2014. Differential sensitivity of 5′UTR-NS5A recombinants of hepatitis C virus genotypes 1–6 to protease and NS5A inhibitors. Gastroenterology 146:812–821. 10.1053/j.gastro.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 38.Gottwein JM, Jensen SB, Li YP, Ghanem L, Scheel TKH, Serre SBN, Mikkelsen L, Bukh J. 2013. Combination treatment with hepatitis C virus protease and NS5A inhibitors is effective against recombinant genotype 1a, 2a, and 3a viruses. Antimicrob Agents Chemother 57:1291–1303. 10.1128/AAC.02164-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jensen SB, Humes DG, Ramirez S, Li Y, Bukh J, Gottwein JM, 2015. Substitutions at NS3 residue 155, 156, or 168 of hepatitis C virus genotypes 2 to 6 induce complex patterns of protease inhibitor resistance. Antimicrob Agents Chemother 59:7426–7436. 10.1128/AAC.01953-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Serre SBN, Jensen SB, Ghanem L, Humes DG, Ramirez S, Li Y-P, Krarup H, Bukh J, Gottwein JM. 2016. Hepatitis C virus genotype 1 to 6 protease inhibitor escape variants: in vitro selection, fitness, and resistance patterns in the context of the infectious viral life cycle. Antimicrob Agents Chemother 60:3563–3578. 10.1128/AAC.02929-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pham LV, Jensen SB, Fahnøe U, Pedersen MS, Tang Q, Ghanem L, Ramirez S, Humes D, Serre SBN, Schønning K, Bukh J, Gottwein JM. 2019. HCV genotype 1–6 NS3 residue 80 substitutions impact protease inhibitor activity and promote viral escape. J Hepatol 70:388–397. 10.1016/j.jhep.2018.10.031. [DOI] [PubMed] [Google Scholar]
- 42.Jensen SB, Fahnøe U, Pham LV, Serre S, Tang Q, Ghanem L, Pedersen MS, Ramirez S, Humes D, Pihl AF, Filskov J, Sølund CS, Dietz J, Fourati S, Pawlotsky J, Sarrazin C, Weis N, Schønning K, Krarup H, Bukh J, Gottwein JM. 2019. Evolutionary pathways to persistence of highly fit and resistant hepatitis C virus protease inhibitor escape variants. Hepatology 70:771–787. 10.1002/hep.30647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gottwein JM, Scheel TKH, Jensen TB, Ghanem L, Bukh J. 2011. Differential efficacy of protease inhibitors against HCV genotypes 2a, 3a, 5a, and 6a NS3/4A protease recombinant viruses. Gastroenterology 141:1067–1079. 10.1053/j.gastro.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Y, Glimore K, Ramirez S, Settles E, Gammeltoft KA, Pham LV, Fahnøe U, Feng S, Offersgaard A, Trimpert T, Bukh J, Osterrieder K, Gottwein JM, Seeberger PH. In vitro efficacy of artemisinin-based treatments against SARS-CoV-2. Sci Rep, in press. [DOI] [PMC free article] [PubMed]
- 45.Chou T-C, Martin N. 2007. CompuSyn software for drug combinations and for general dose effect analysis, and user’s guide. ComboSyn, Inc., Paramus, NJ. [Google Scholar]
- 46.Chou TC, Talalay P. 1984. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22:27–55. 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 47.Ramirez S, Li YP, Jensen SB, Pedersen J, Gottwein JM, Bukh J. 2014. Highly efficient infectious cell culture of three hepatitis C virus genotype 2b strains and sensitivity to lead protease, nonstructural protein 5A, and polymerase inhibitors. Hepatology 59:395–407. 10.1002/hep.26660. [DOI] [PubMed] [Google Scholar]
- 48.Imhof I, Simmonds P. 2010. Development of an intergenotypic hepatitis C virus (HCV) cell culture method to assess antiviral susceptibilities and resistance development of HCV NS3 protease genes from HCV genotypes 1 to 6. J Virol 84:4597–4610. 10.1128/JVI.02698-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Imhof I, Simmonds P. 2011. Genotype differences in susceptibility and resistance development of hepatitis C virus to protease inhibitors telaprevir (VX-950) and danoprevir (ITMN-191). Hepatology 53:1090–1099. 10.1002/hep.24172. [DOI] [PubMed] [Google Scholar]
- 50.Becker DE. 2007. Drug therapy in dental practice: general principles. Part 2 - pharmacodynamic considerations. Anesth Prog 54:19–24. 10.2344/0003-3006(2007)54[19:DTIDPG]2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramsden D, Tweedie DJ, Chan TS, Taub ME, Li Y. 2014. Bridging in vitro and in vivo metabolism and transport of faldaprevir in human using a novel cocultured human hepatocyte system, HepatoPac. Drug Metab Dispos 42:394–406. 10.1124/dmd.113.055897. [DOI] [PubMed] [Google Scholar]
- 52.Wright DH, Caro L, Cerra M, Panorchan P, Du L, Anderson M, Potthoff A, Nachbar RB, Wagner J, Manns MP, Talal AH. 2015. Liver-to-plasma vaniprevir (MK-7009) concentration ratios in HCV-infected patients. Antivir Ther 20:843–848. 10.3851/IMP2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Center for Drug Evaluation and Research. 2009. Clinical pharmacology and biopharmaceuticals review(s). Application number 205123Orig1s000. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/205123Orig1s000ClinPharmR.pdf.
- 54.Wölfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Müller MA, Niemeyer D, Jones TC, Vollmar P, Rothe C, Hoelscher M, Bleicker T, Brünink S, Schneider J, Ehmann R, Zwirglmaier K, Drosten C, Wendtner C. 2020. Virological assessment of hospitalized patients with COVID-2019. Nature 581:465–469. 10.1038/s41586-020-2196-x. [DOI] [PubMed] [Google Scholar]
- 55.Lin TI, Lenz O, Fanning G, Verbinnen T, Delouvroy F, Scholliers A, Vermeiren K, Rosenquist Â, Edlund M, Samuelsson B, Vrang L, De Kock H, Wigerinck P, Raboisson P, Simmen K. 2009. In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor. Antimicrob Agents Chemother 53:1377–1385. 10.1128/AAC.01058-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Chen L. 2020. Tissue distributions of antiviral drugs affect their capabilities of reducing viral loads in COVID-19 treatment. Eur J Pharmacol 889:173634. 10.1016/j.ejphar.2020.173634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eleftheriou P, Amanatidou D, Petrou A, Geronikaki A. 2020. In silico evaluation of the effectivity of approved protease inhibitors against the main protease of the novel SARS-CoV-2 virus. Molecules 25:2529. 10.3390/molecules25112529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khan RJ, Jha RK, Amera GM, Jain M, Singh E, Pathak A, Singh RP, Muthukumaran J, Singh AK. 2020. Targeting SARS-CoV-2: a systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. J Biomol Struct Dyn 39:2679–2692. 10.1080/07391102.2020.1753577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rahman MM, Saha T, Islam KJ, Suman RH, Biswas S, Rahat EU, Hossen MR, Islam R, Hossain MN, Mamun A, Al Khan M, Ali MA, Halim MA. 21July2020. Virtual screening, molecular dynamics and structure–activity relationship studies to identify potent approved drugs for Covid-19 treatment. J Biomol Struct Dyn 10.1080/07391102.2020.1794974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Talluri S. 2021. Molecular Docking and Virtual Screening based prediction of drugs for COVID-19. Comb Chem High Throughput Screen 24:716–728. 10.2174/1386207323666200814132149. [DOI] [PubMed] [Google Scholar]
- 61.Iftikhar H, Ali HN, Farooq S, Naveed H, Shahzad-Ul-Hussan S. 2020. Identification of potential inhibitors of three key enzymes of SARS-CoV2 using computational approach. Comput Biol Med 122:103848. 10.1016/j.compbiomed.2020.103848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Manikyam JH, Joshi SK. 2020. Whole genome analysis and targeted drug discovery using computational methods and high throughput screening tools for emerged novel coronavirus (2019-nCoV). J Pharm Drug Res 3:341–361. [PMC free article] [PubMed] [Google Scholar]
- 63.Chtita S, Belhassan A, Aouidate A, Belaidi S, Bouachrine M, Lakhlifi T. 2020. Discovery of potent SARS-CoV-2 inhibitors from approved antiviral drugs via docking and virtual screening. Comb Chem High Throughput Screen 24:441–454. 10.2174/1386207323999200730205447. [DOI] [PubMed] [Google Scholar]
- 64.Bahadur Gurung A, Ajmal Ali M, Lee J, Abul Farah M, Mashay Al-Anazi K. 2020. Structure-based virtual screening of phytochemicals and repurposing of FDA approved antiviral drugs unravels lead molecules as potential inhibitors of coronavirus 3C-like protease enzyme. J King Saud Univ Sci 32:2845–2853. 10.1016/j.jksus.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shamsi A, Mohammad T, Anwar S, AlAjmi MF, Hussain A, Md Tabish R, Islam A, Md Imtaiyaz H. 2020. Glecaprevir and maraviroc are high-affinity inhibitors of SARS-CoV-2 main protease: possible implication in COVID-19 therapy. Biosci Rep 40:BSR20201256. 10.1042/BSR20201256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ghahremanpour MM, Tirado-Rives J, Deshmukh M, Ippolito JA, Zhang CH, De Vaca IC, Liosi ME, Anderson KS, Jorgensen WL. 2020. Identification of 14 known drugs as inhibitors of the main protease of SARS-CoV-2. ACS Med Chem Lett 11:2526–2533. 10.1021/acsmedchemlett.0c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abhithaj J, Francis D, Sharanya CS, Arun KG, Sadasivan C, Variyar EJ. 2 September 2020. Repurposing simeprevir, calpain inhibitor IV and a cathepsin F inhibitor against SARS-CoV-2 and insights into their interactions with Mpro. J Biomol Struct Dyn 10.1080/07391102.2020.1813200. [DOI] [PubMed] [Google Scholar]
- 68.Calligari P, Bobone S, Ricci G, Bocedi A. 2020. Molecular investigation of SARS–COV-2 proteins and their interactions with antiviral drugs. Viruses 12:445. 10.3390/v12040445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hosseini FS, Amanlou M. 2020. Anti-HCV and anti-malaria agent, potential candidates to repurpose for coronavirus infection: virtual screening, molecular docking, and molecular dynamics simulation study. Life Sci 258:118205. 10.1016/j.lfs.2020.118205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lima de Oliveira MD, Teixeira de Oliveira KM. 5May2021. Comparative docking of SARS-CoV-2 receptors antagonists from repurposing drugs. ChemRxiv 10.26434/chemrxiv.12044538. [DOI] [Google Scholar]
- 71.Elzupir AO. 2020. Inhibition of SARS-CoV-2 main protease 3CLpro by means of α-ketoamide and pyridone-containing pharmaceuticals using in silico molecular docking. J Mol Struct 1222:128878. 10.1016/j.molstruc.2020.128878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ahmed SA, Abdelrheem DA, El-Mageed HRA, Mohamed HS, Rahman AA, Elsayed KNM, Ahmed SA. 2020. Destabilizing the structural integrity of COVID-19 by caulerpin and its derivatives along with some antiviral drugs: an in silico approaches for a combination therapy. Struct Chem 31:2391–2412. 10.1007/s11224-020-01586-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alamri MA, Tahir Ul Qamar M, Mirza MU, Bhadane R, Alqahtani SM, Muneer I, Froeyen M, Salo-Ahen OMH. 24 June 2020. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CLpro. J Biomol Struct Dyn 10.1080/07391102.2020.1782768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ahmed S, Mahtarin R, Ahmed SS, Akter S, Islam MS, Al Mamun A, Islam R, Hossain MN, Ali MA, Sultana MUC, Parves MR, Ullah MO, Halim MA. 28 July 2020. Investigating the binding affinity, interaction, and structure-activity-relationship of 76 prescription antiviral drugs targeting RdRp and Mpro of SARS-CoV-2. J Biomol Struct Dyn 10.1080/07391102.2020.1796804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fatoki TH, Ibraheem O, Ogunyemi IO, Akinmoladun AC, Ugboko HU, Adeseko CJ, Awofisayo OA, Olusegun SJ, Enibukun JM. 20 July 2020. Network analysis, sequence and structure dynamics of key proteins of coronavirus and human host, and molecular docking of selected phytochemicals of nine medicinal plants. J Biomol Struct Dyn 10.1080/07391102.2020.1794971. [DOI] [PubMed] [Google Scholar]
- 76.Keretsu S, Bhujbal SP, Cho SJ. 2020. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci Rep 10:17716. 10.1038/s41598-020-74468-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bafna K, Krug RM, Montelione G. 21April2020. Structural similarity of SARS-CoV2 Mpro and HCV NS3/4A proteases suggests new approaches for identifying existing drugs useful as COVID-19 therapeutics. ChemRxiv 10.26434/chemrxiv.12153615.V1. [DOI] [Google Scholar]
- 78.Gurung AB. 2020. In silico structure modelling of SARS-CoV-2 Nsp13 helicase and Nsp14 and repurposing of FDA approved antiviral drugs as dual inhibitors. Gene Rep 21:100860. 10.1016/j.genrep.2020.100860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bhowmik D, Nandi R, Jagadeesan R, Kumar N, Prakash A, Kumar D. 2020. Identification of potential inhibitors against SARS-CoV-2 by targeting proteins responsible for envelope formation and virion assembly using docking based virtual screening, and pharmacokinetics approaches. Infect Genet Evol 84:104451. 10.1016/j.meegid.2020.104451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Khan RJ, Jha RK, Singh E, Jain M, Amera GM, Singh RP, Muthukumaran J, Singh AK. 4 September 2020. Identification of promising antiviral drug candidates against non-structural protein 15 (NSP15) from SARS-CoV-2: an in silico assisted drug-repurposing study. J Biomol Struct Dyn 10.1080/07391102.2020.1814870. [DOI] [PubMed] [Google Scholar]
- 81.Kadioglu O, Saeed M, Johannes Greten H, Efferth T. 2021. Identification of novel compounds against three targets of SARS CoV-2 coronavirus by combined virtual screening and supervised machine learning. Comput Biol Med 133:104359. 10.1016/j.compbiomed.2021.104359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Trezza A, Iovinelli D, Santucci A, Prischi F, Spiga O. 2020. An integrated drug repurposing strategy for the rapid identification of potential SARS-CoV-2 viral inhibitors. Sci Rep 10:13866. 10.1038/s41598-020-70863-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kneller DW, Galanie S, Phillips G, O'Neill HM, Coates L, Kovalevsky A. 2020. Malleability of the SARS-CoV-2 3CL Mpro active-site cavity facilitates binding of clinical antivirals. Structure 28:1313.e3–1320.e3. 10.1016/j.str.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Anson B, Chapman M, Lendy E, Pshenychnyi S, D’Aquila R, Satchell K, Mesecar A. 1May. 2020. Broad-spectrum inhibition of coronavirus main and papain-like proteases by HCV drugs. Res Sq 10.21203/rs.3.rs-26344/v1. [DOI] [Google Scholar]
- 85.Kneller DW, Phillips G, O’Neill HM, Jedrzejczak R, Stols L, Langan P, Joachimiak A, Coates L, Kovalevsky A. 2020. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat Commun 11:3202. 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blight KJ, Mckeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76:13001–13014. 10.1128/jvi.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gottwein JM, Scheel TKH, Callendret B, Li Y-PP, Eccleston HB, Engle RE, Govindarajan S, Satterfield W, Purcell RH, Walker CM, Bukh J. 2010. Novel infectious cDNA clones of hepatitis C virus genotype 3a (strain S52) and 4a (strain ED43): genetic analyses and in vivo pathogenesis studies. J Virol 84:5277–5293. 10.1128/JVI.02667-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Scheel TKHH, Gottwein JM, Mikkelsen LS, Jensen TB, Bukh J. 2011. Recombinant HCV variants with NS5A from genotypes 1–7 have different sensitivities to an NS5A inhibitor but not interferon-α. Gastroenterology 140:1032–1042. 10.1053/j.gastro.2010.11.036. [DOI] [PubMed] [Google Scholar]
- 89.Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DKW, Bleicker T, Brünink S, Schneider J, Schmidt ML, Mulders DGJC, Haagmans BL, Van Der Veer B, Van Den Brink S, Wijsman L, Goderski G, Romette JL, Ellis J, Zambon M, Peiris M, Goossens H, Reusken C, Koopmans MPG, Drosten C. 2020. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 25:2000045. 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download AAC.02680-20-s0001.pdf, PDF file, 2.1 MB (2.1MB, pdf)