

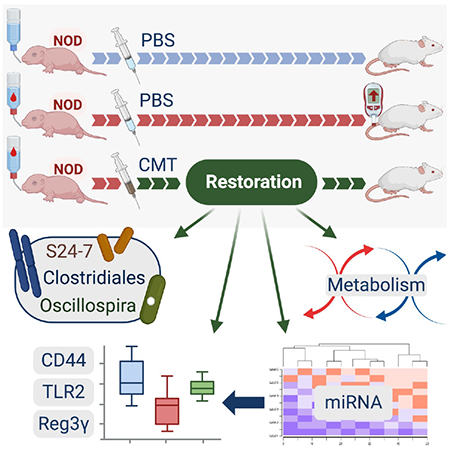
SUMMARY
Early-life antibiotic exposure perturbs the intestinal microbiota and accelerates type 1 diabetes (T1D) development in the NOD mouse model. Here we found that maternal cecal microbiota transfer (CMT) to NOD mice after early-life antibiotic perturbation largely rescued the induced T1D enhancement. Restoration of the intestinal microbiome was significant and persistent, remediating the antibiotic-depleted diversity, relative abundance of particular taxa, and metabolic pathways. CMT also protected against perturbed metabolites and normalized innate and adaptive immune effectors. CMT restored major patterns of ileal microRNA and histone regulation of gene expression. Further experiments suggest a gut microbiota-regulated T1D protection mechanism centered on Reg3γ, in an innate intestinal immune network involving CD44, TLR2, and Reg3γ. This regulation affects downstream immunological tone, which may lead to protection against tissue-specific T1D injury.
Graphical Abstract
In brief
Using a mouse model where early-life antibiotics enhances T1D, Zhang et al. show that subsequent maternal cecal microbiota transfer reduces illness. The restorative effects on intestinal microbiome and metabolism, ileal wall gene expression and regulation, and innate and adaptive immune effectors suggest a gut microbiota-regulated T1D protective mechanism.
INTRODUCTION
Infancy is a critical period for establishing a healthy gut microbiota and facilitating individual immune development (Gensollen et al., 2016; Gollwitzer and Marsland, 2015; Pennisi, 2016). Alteration of gut microbiota composition in infancy may change age-associated immunity and organ-specific inflammation, increasing risk of immune-mediated diseases (Amenyogbe et al., 2017; Aversa et al., 2020; Kemppainen et al., 2017; Mullaney et al., 2019).
Type 1 diabetes (T1D) is the most common autoimmune disease in childhood with pancreatic insulin-producing β cells destroyed by autoreactive T cells and other effectors (Atkinson and Eisenbarth, 2001; Wilson et al., 1998). The triggers and intermediary molecular mechanisms of T1D remain unclear, but innate immunity may play key roles in the initial pathogenetic steps (Atkinson and Chervonsky, 2012; Pino et al., 2010; Wen et al., 2008). In infants, both the gut microbiota (Dominguez-Bello et al., 2011) and the immune system are developing (Olin et al., 2018). Antibiotic exposure could potentially change the microbial interplay with immune system development, and experimental models in the NOD (non-obese diabetic) mouse indicate altered susceptibility to T1D development (Brown et al., 2016; Candon et al., 2015; Hu et al., 2017; Livanos et al., 2016; Zhang et al., 2018). Our recent studies showed that early-life antibiotic-exposure induced gut microbiota perturbation, interfering with adaptive immune effectors, altering ileal gene maturational patterns and incidence of T1D onset, especially in male NOD mice (Livanos et al., 2016; Zhang et al., 2018).
Faced with massive disruption of the intestinal microbiota due to antibiotic exposures and other perturbations, clinicians have developed fecal microbiota transplantation (FMT), a restorative procedure to return to eubiosis and control illness (Kelly et al., 2015; Malikowski et al., 2017). This technique has been successful in permitting the recovery of patients with Clostridioides difficile colitis (van Nood et al., 2013) and is being investigated for efficacy in numerous clinical conditions (Bowman et al., 2015; Kellermayer, 2019; Pathak et al., 2013).
Since the antibiotic-perturbed microbiota T1D model that we have developed in NOD mice also involves substantial dysbiosis (Livanos et al., 2016; Zhang et al., 2018), we now ask whether we can restore the microbiota and return the enhanced disease risk to baseline. The underlying questions are to determine which microbiota might be optimal for restoration and to assess the intermediate steps affected by the perturbation and restored by the transplantation. This experimental approach establishes the feasibility of the process and identifies key microbiota members, metagenomic pathways, metabolites, host genes and signaling mechanisms that can be harnessed for future focused interventions. These discoveries provide insights into analogous human pathophysiology.
RESULTS
Cecal microbiota transfer normalizes post-antibiotic gut microbiome compositions
We first evaluated whether specific microbiota transfer can restore changes in the intestinal microbiota and intestinal innate immunity induced by an early-life antibiotic course in NOD mice (Figure 1A). To reflect microbial populations relevant to the maturation of the recipient pups, we prepared cecal microbiota pools from dams or pups from four different donor types: i) pups on their fifth day of life (Pup-P5); ii) dams on pup day of life 5 (Mom-P5); iii) dams at parturition (Mom-P0); and iv) dams at parturition, who received an antibiotic (tylosin) course during pregnancy to disrupt their microbiome (Mom-T-P0). These inocula contained ~107 copies of 16S rRNA genes and varied in their α-diversity with the Mom-P0 inoculum showing the highest values (Table S1). Community structure (β-diversity) using unweighted UniFrac, showed the dam pool inocula clustering together, whereas the pup pool inoculum was distinct (data not shown). We then performed cecal microbiota transfer (CMT) via gavage to recipient mice at P15 that had received the standard antibiotic treatment (1PAT, with tylosin) from P5-P10 and evaluated the gut microbiota and the intestinal innate responses at two early timepoints after the CMT or control transfers (Figure 1). Pups exposed to 1P followed by the control gavage showed markedly diminished α-diversity, persisting to P42 (Figure 1BC). Donor pool i CMT(Pup-P5) did not significantly change 1PAT cecal microbiota α-diversities for either timepoint (Figure 1B). Donor pool ii CMT(Mom-P5) significantly increased 1P cecal microbiota α-diversities at P42 but not at P28 (Figure 1B). Donor pool iii CMT(Mom-p0) significantly increased recipient cecal microbiota α-diversities at both P28 and P42 (Figure 1B). Compared to donor pool iii CMT(Mom-p0), donor pool iv CMT(Mom-T-P0) did not significantly increase cecal microbiota α-diversities at P28 (Figure 1B).
Figure 1. Study design, microbiome and ileal gene expression analysis of STR experiment.
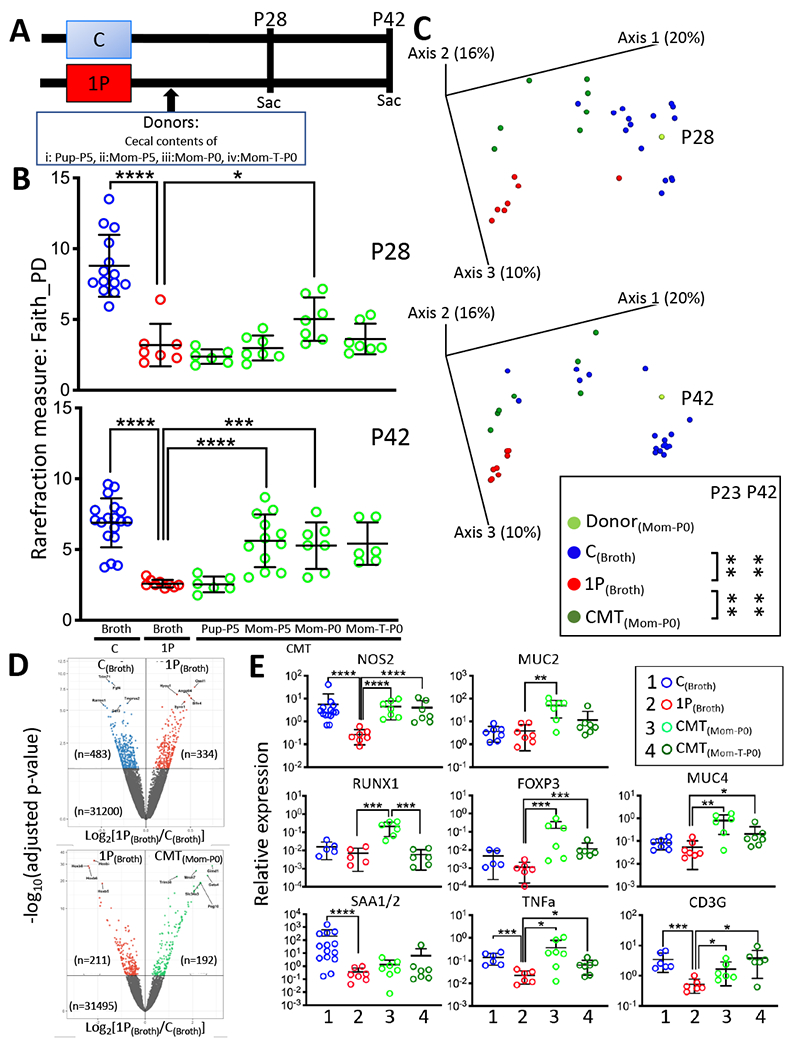
A) Study design. Pregnant NOD/ShiLtJ mice were randomized into six groups. Pups in five groups received atylosin course (1PAT; 1P) from P5-P10 and then were gavaged at P15 with broth alone (1P with broth), or with one of four pools of donor cecal contents in broth (i: CMT containing Pup-P5, ii: Mom-P5, iii: Mom-P0 and iv: Mom-T-P0 cecal contents). One pup group did not receive 1P (Control; C), and mice were gavaged in parallel at P15 with broth alone (C with broth). Mice (n=6-18/group) were followed and sacrificed at P28 and P42 to assess effects of the gavages on the microbiota and on ileal gene expression. B) α-diversity of cecal microbiome at P28 and P42, with rarefaction at 3000 sequences/sample, in mice receiving the different gavages. mean±SD shown, *p<0.05; ***p<0.001; ****p<0.0001; Mann-Whitney U test. C) β-diversity of cecal microbiome in mice receiving the different gavages, as measured by unweighted UniFrac analysis. Groups are: C(Broth), control pups receiving blank broth: 1P(Broth), 1P pups receiving blank broth; CMT(Mom-P0), 1P pups receiving CMT from normal dams at day of birth. **p=0.0017; one-way-ANOVA with Tukey’s HSD post-test for multiple comparisons. D) Volcano plot of RNAseq-based differential ileal gene expression at P42 between C and 1P mice receiving broth alone (upper panel), and between 1P mice receiving Mom-P0 (CMT) and 1P mice receiving broth alone (1P) (lower panel) (n=3 mice/group). In the upper panel, the 10 genes most differentiated by 1P(Broth) are Trim71, Fgf4, Tmprss2, Rarres1, Gdf3 (repressed), and Oasl1, Hyou1, Angptl4, Slfn4, Syvn1 (induced). In the lower panel, the 10 genes most differentiated by CMT(Mom-P0) are Hoxb5-8 (repressed), and Gimd1, Mroh7, Trim38, Gata4, Slc34a3, Peg10 (induced). E) RT-qPCR-based P28 ileal gene expression of Nos2, Muc2, Muc4, Runxl, Foxp3, CD3g, Saa1/2 and TNFα. Group 1: C(Broth), Group 2: 1P(Broth), Group 3: CMT(Mom-P0), Group 4: CMT(Mom-T-P0), 1P pups receiving CMT from dams on the day of birth who were exposed during pregnancy to tylosin. Mean±SD shown; n=6-14 mice/group; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; Mann-Whitney Utest. Related to Figure S1D, Tables S1,S2.
The 1PAT mice that received the Mom-P0 CMT or control broth had significantly different microbial compositions from each other (Figure 1C). The other maternal inocula (Mom-P5 and Mom-T-P0, but not the Pup-P5) yielded similar results (data not shown). Compared to control mice without antibiotic exposure, 1PAT depleted particular Firmicutes (including genera Oscillospira, Ruminococcus) and enriched genus Akkermansia in the cecal microbiota from early timepoints (Figure S1A), consistent with our prior observation (Zhang et al., 2018). Based on these Short-Term Restoration (STR) analyses, we concluded that Mom-P0 CMT best restored early life α-diversity, β-diversity and specific taxa of interest, making it the optimal transfer material among those studied for long-term restoration (LTR) experiments.
Maternal CMT normalized ileal gene expression and metabolic pathways regulating intestinal immune markers in antibiotic-treated mice
We next asked whether the partially restored microbiota affected gene expression in ileum. 1PAT significantly affected P42 ileal expression of 817 of the ~30,000 detected genes, downregulating 483 and upregulating 334 (Figure 1D upper panel); Mom-P0 CMT affected expression of 403 genes compared to 1PAT, downregulating 211, upregulating 192; Figure 1D lower panel); KEGG analysis showed genes related to retinol, cysteine, methionine, pyruvate, and sulfur metabolism as well as cytochromes and steroid hormone biosynthesis (Figure S1C). The genes most repressed by CMT compared to 1PAT included a group of Antp homeobox family transcription factor genes (Hoxb5, Hoxb6, Hoxb7 and Hoxb8) and Ifi202b (autoimmune-development associated interferon activated gene), and those most induced included Gata4 (β-cell transcription factor), Trim 38 (inhibitor of TLR3/4 mediated inflammation) and Ighv1-47 (immunoglobulin heavy chain gene) (Figure 1D, Table S2). In total, these findings indicate that Mom-P0 CMT changed host ileal gene expression including genes important in early-life immunological development.
We next evaluated the effects of Mom-P0 CMT on specific ileal innate and adaptive immune early marker signature genes that we previously identified 1PAT-perturbed (Zhang et al., 2018) (Figure 1E). Ileal expression of NOS2 (innate immune-inducible nitric oxide synthase), downregulated by 1PAT, was partially restored by Mom-P0 CMT. Similar trends were seen for MUC2 and MUC4 (genes central for mucin synthesis), TNFα, RUNX1 (early-life transcription factor) and two of its downstream genes (FOXP3, CD3G) critically involved in adaptive immunity (Figure 1E). Serum amyloid A1 (SAA1) was significantly downregulated by 1P at P28 in the ileum, but Mom-P0 CMT had little effect (Figure 1E). CMT with antibiotic-perturbed maternal microbiota (Mom-T-P0) had effects similar to Mom-P0 CMT, but generally to a lesser extent. Collectively, findings from the STR experiment provided evidence that Mom-P0 CMT significantly restored both microbiota population structure and host responses, suggesting the utility of an experiment to assess whether the 1PAT-enhanced T1D could be blocked.
Maternal CMT largely rescued the antibiotic-induced T1D enhancement
To assess whether the Mom-P0 CMT protected NOD mice against antibiotic-enhanced T1D, we used male NOD mice which showed stronger antibiotic effects than females (Livanos et al., 2016; Zhang et al., 2018). In the LTR experiment, male mice received 1PAT (or not as C, Control) at P5-10 and then were gavaged with Mom-P0 CMT or with blank medium (PBS) on P13-17 and then were monitored for 30 weeks for T1D development (Figure 2A). By the study end, control (C) mice were significantly more often TID-free (50%) than were the 1P mice (15%) (Figure 2B), consistent with our prior studies (Livanos et al., 2016; Zhang et al., 2018). Mom-P0 CMT restored week 30 survival (45%) similar to that observed in C mice (50%) (Figure 2B). Moreover, according to the Kaplan-Meier analyses and weighted log-rank tests, there were significant differences in disease-free rates across the three experimental groups (p=0.005), especially during the middle and late time points. Disease-free rates for the three groups were similar at the beginning phase (week 0 to ~20), but the rate for group 1P dropped during the middle phase compared to those of C (p=0.009) and CMT (p=0.045) (Figure 2B). Thus, we now confirmed our model of T1D enhancement with a single early-life antibiotic course and showed the ability to significantly reverse the phenotype. Since the interval between the early-life antibiotic exposure (ending on P10) and gavages (ending on P17), and T1D onset (earliest at P85, median at P180) represents a long latency period, we focused on early time points to better understand the initial mechanisms in T1D pathogenesis.
Figure 2. Effect of CMT on T1D development and early-life fecal microbiome in a longterm restoration (LTR) experiment.
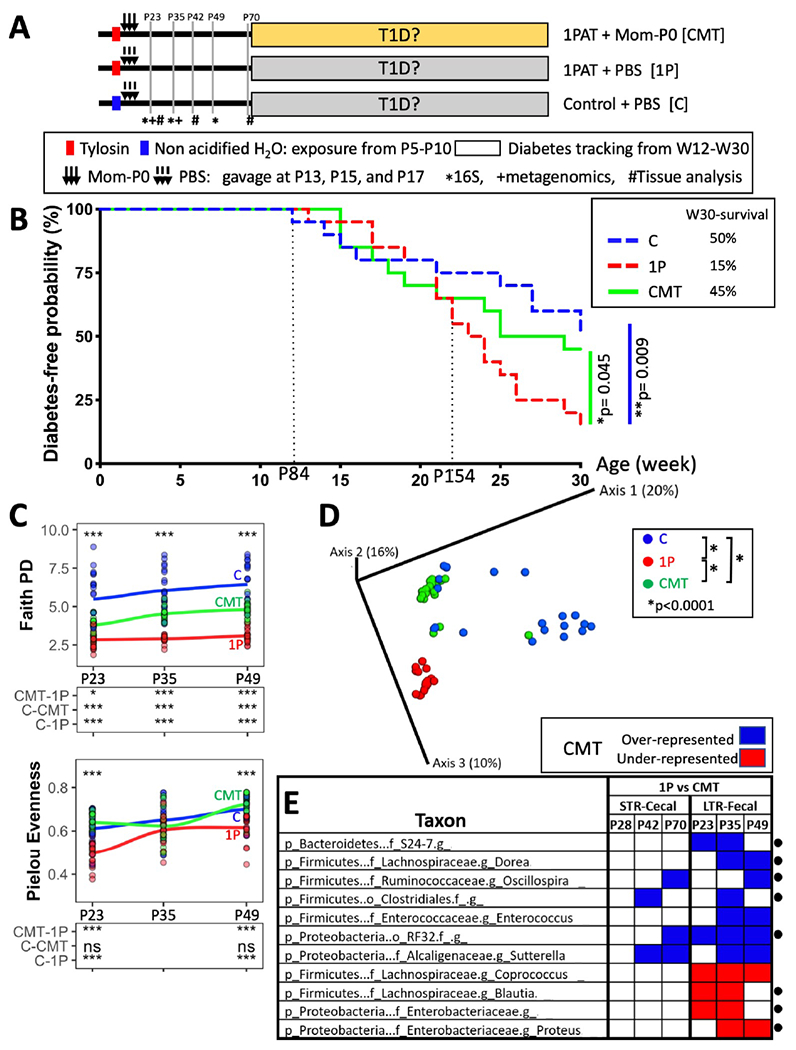
A) Design of LTR experiment. Pregnant NOD/ShiLtJ male mice were randomized into three groups: Control mice gavaged with PBS at P13-17 (group C); antibiotic-treated (1P) mice receiving either PBS (group 1P) or gavages of Mom-P0 cecal contents (group CMT). Mice (n=20/group) were tested weekly for diabetes by blood glucose measurement from weeks 11-30. In addition, to provide specimens for the intermediate analyses of mechanism, 72 mice were sacrificed earlier [12 each in the C, 1P, and CMT groups at P23, and six in each group at P42 (n=18) and at P70 (n=18)]. Symbols indicate times at which the fecal microbiome was analyzed by *16S rRNA sequencing, and +shotgun metagenomic sequencing, or a subset was #sacrificed for examining ileal and cecal tissues and contents, and metabolomics. B) Kaplan-Meier analysis of T1D incidence in mice (n=20/group). The first T1D case occurred at P84; after P154, the 3 groups were clearly differentiated in T1D outcomes. Statistical significance was determined by the Gρ,γ weighted log rank test (Fleming and Harrington, 1991). Post-hoc pairwise comparisons were adjusted by a Benjamini-Hochberg procedure. C) α-diversity overtime measuring Faith’s PD and Pielou evenness. Upper panel: ***p<0.001, one-way ANOVA; Lower panel: *p<0.01, ***p<0.001, ns p>0.05, one-way ANOVA with Tukey’s HSD post-test. D) β-diversity, as determined by unweighted UniFrac analysis of the fecal microbiota of the three groups. Inter-group UniFrac distances were all significant (p<0.0001), one-way-ANOVA with Tukey’s HSD post-test. E) Early-life taxa significantly under-(red) or over-(blue) represented in either the STR or LTR experiments in CMT compared to 1P mice, according to MaAsLin2 analysis. The abundance of taxa marked with • were significantly different in 1P compared to C and also were restored by CMT (also see Figure S1A). Analysis identified a set of genera (from family S24-7, orders Clostridiales and RF32 and genera Dorea and Oscillospira) significantly decreased by 1P but increased by CMT, and also identified a set of genera (from family Enterobacteriaceae, and genera Blautia and Proteus) significantly increased by 1P but decreased by CMT. “g” indicates unclassified genus below the family or Order levels. Related to Figures S1,S2.
CMT significantly restored fecal microbiome community characteristics toward baseline
Next, we assessed the effects of the maternal CMT on the pup microbiome. Community richness, measured by Faith’s PD, was markedly diminished by 1PAT compared to C mice, but was intermediate in the CMT mice and community richness increased faster for C and CMT from P23 to P49 than for 1P mice (Figure 2C upper panel). Community evenness (Pielou score) also was markedly reduced in the 1P mice, and P23 to P49 trends differed for each group (Figure 2C lower panel); however, CMT restored community evenness to the control level. Community structure (β-diversity) significantly differed between C and 1P mice, and CMT led to an intermediate group, significantly distinct from 1P mice and C mice (Figure 2D). Estimating the total number of bacterial cells in P23 fecal samples using universal bacterial qPCR showed that 1PAT significantly decreased total bacterial abundance, but CMT was restorative (Figure S1B left panel). All these metrics indicate that CMT restored antibiotic-perturbed global bacterial populations toward normal. We also noted substantial 16S count heterogeneity in C mice, with low numbers associated with early (<week22) T1D development (Figure S1B right panel).
Effect of post-antibiotic CMT on specific taxa
We next assessed the specific taxa restored by CMT. Across the 267 fecal and cecal samples obtained at early timepoints in the Mom-P0 CMT STR and LTR experiments, we identified 194 individual taxa at the genus level. Applying the MaAsLin2 algorithm to the pre-T1D STR cecal samples and LTR fecal samples, we observed 1PAT differentially selected taxa (Figure S1A) with significantly differential taxa after CMT (Figure 2E). Bacteroidales family S24-7, dominant in the NOD mouse gut, were depleted in 1P mice (Figure S1A) and were restored by CMT (Figures 2E,3B). Firmicutes unclassified species from genera Oscillospira and Dorea, and unclassified species from orders Clostridiales and RF32, were under-represented in 1P mice, respectively, and restored by CMT (Figures 2E,S1A,S2D). Among them, Oscillospira relative abundances in C mice that developed T1D early (<week22) were significantly lower at all three early timepoints than those developing T1D late (>week28) or never (Figure 3A right panel), and unclassified species within Order Clostridiales at P35 and P49 was also negatively associated with T1D-development (Figure S2A), independently in all three treatment groups (Figure S2B). In contrast, Blautia producta, and two unclassified species from Enterobacteriaceae (a Proteus species and an unclassified genus) were enriched in 1P compared to C, consistent with prior findings (Zhang et al., 2018), and reduced by CMT (Figures 2E,S1C,S2D). We also identified unclassified species from families Erysipelotrichaceae and genera Lactobacillus depleted by 1PAT but significantly restored by CMT, while Akkermansia muciniphila was enriched by 1PAT but partially repressed by CMT (Figure S2D). Comparing taxa abundances across groups, 1PAT significantly decreased the Lachnospiraceae/Enterococcus ratio from that in C, while CMT was significantly restorative (Figure S2C). In total, these analytical methods identified taxa associated or not with protection against T1D development.
Figure 3. CMT effects on specific bacterial taxa and on bacterial metabolic pathways.
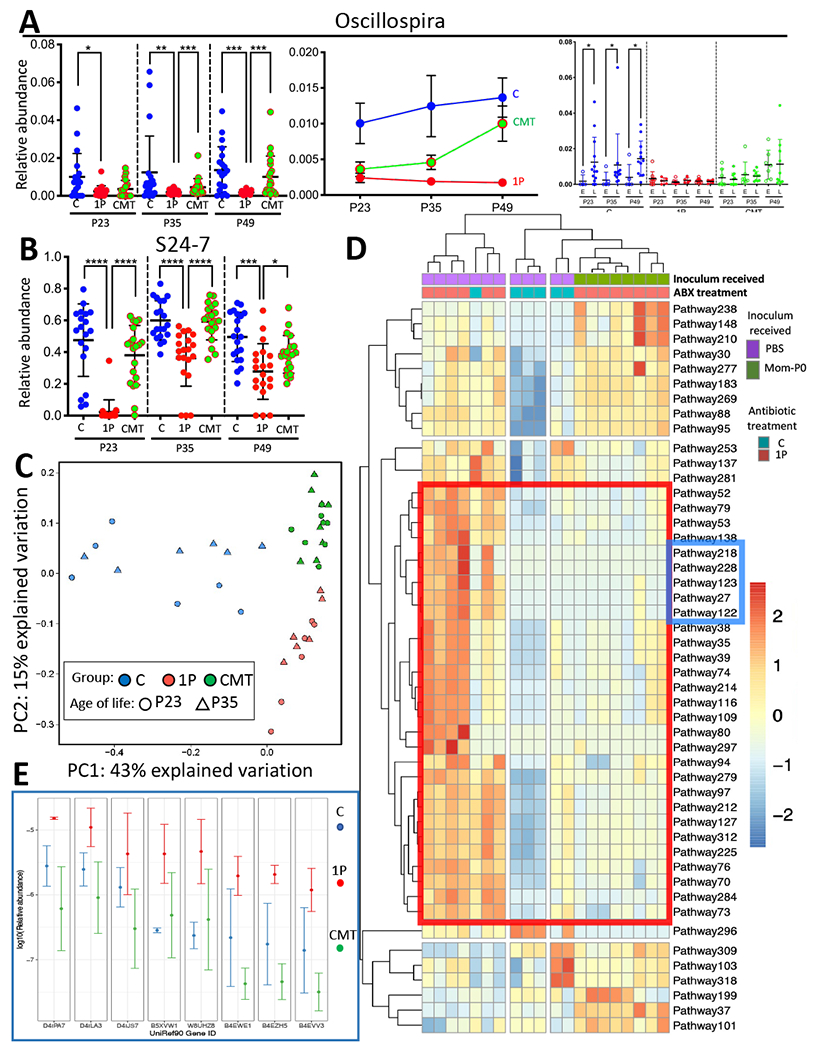
A)Oscillospira genus relative abundance with respect to the average microbe in groups (C, 1P and CMT) at P23, P35 and P49 (left panel), changing with time (middle panel); and in the mice that developed T1D early (E) or late or never (L) within each treatment group (right panel). B) S24-7 family relative abundance in groups (C, 1P and CMT), at P23, P35 and P49; Analysis was performed by QIIME2 based on serial fecal samples from 19~20 mice/group. Mean±SEM shown; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; Mann-Whitney Utest. C) Metagenomic (MetaCyc) pathway distribution (Caspi et al., 2018) at P23 and P35 in C (blue, n=6), or 1P (red, n=6), or CMT mice (green, n=8) visualized by principal component analysis; D) Unsupervised clustering of 48 fecal microbial pathways with significant differences at P23. A group of 29 pathways, which were restored to control levels by CMT, is highlighted within the red box [see Table S2]. A subgroup of 5 pathways with the greatest consistency between CMT and C is highlighted within the blue box, including three fatty acid salvage, and two fatty acid β-oxidation pathways [also see Figures S1D,S1E]. E) Comparison of the relative abundance of the most abundant genes in the fatty acid metabolism-related pathways most restored by CMT. Of the 5 pathways, the eight genes with highest relative abundance (D4ILA3-B4EVV3) encode long-chain acyl-CoA synthetases and ligases (E.C.6.2.1.3; n=5) or 3-ketoacyl-CoA thiolases (E.C.2.3.1.16; n=3) and long-chain fatty acid CoA ligases (n=2). Mean±SD shown from determinations in 5.2±1.7 individual mice/group [also see Figures S1D,S1E]. Related to Figures S1,S2,S3, Tables S3,S4.
CMT partially restores gut microbial metabolic pathways
To determine whether the altered microbiota following CMT differed in its metabolic functions, we used shotgun sequencing to examine post-gavage fecal metagenomes. Based on Bray-Curtis analysis of 325 metabolic pathways identified at P23 and P35, 1PAT globally affected metagenomic composition, whereas CMT was at least partially restorative (Figure 3C). The P23 and P35 results clustered together within each treatment group, indicating the stability of the findings. CMT restored 29 (71%) of the 41 pathways that differentiated the 1P and C mice (Figure 3D, Table S3), as shown in the heatmap red box (from pathway 52 to pathway 73, and Table S3). The restoration of gut microbial metabolic pathways to baseline levels provides intermediate mechanisms for the restoration of the baseline T1D phenotype (Figliuolo et al., 2017; Hosomi et al., 2019; Tiso and Schechter, 2015). A subcluster of 5 pathways (Blue box; Pathways 218-122) show the greatest consistency between C and CMT, in terms of the strength and uniformity of the effect, and all relate to fatty acid β-oxidation (Figures 3D,S1E). Of the 5 pathways in this cluster, three (PWY-7094 (fatty acid salvage), PWY-5136 (fatty acid β-oxidation 2) and FAO-PWY fatty acid β-oxidation 1), show high abundance of eight genes in the PAT mice (Figures 3E,S1D), that is significantly greater than in the C or CMT groups. Thus, CMT restored the abundances of these genes in the metagenome after 1P to the control levels. Each of these genes encodes long-chain acyl-CoA synthetases (AMP-forming), whereas most of the other 35 genes that constitute these pathways were present in very low abundance. Overexpression of long-chain acyl-CoA synthetases, leading to production of toxic lipids (e.g. peroxides) can be damaging to host cells (Chiu et al., 2001). Fatty acid β-oxidation also creates common intermediates (such as AMP, NADH/NADPH), with TCA cycle end products of acetyl-CoA and ATP. Thus, antibiotic exposure affecting the products of the most abundant differential genes significantly increased by PAT but restored by CMT impacts fatty acid metabolism by regulating the addition of acyl CoA to a 2,3,4-saturated fatty acid and releasing acetyl-CoA from a 2,3,4-saturated 3-oxoacyl-CoA (Figure S1E). Conjugated linoleic acids produced by lactobacilli are anti-inflammatory (Chiu et al., 2001), but their removal through β-oxidation in PAT mice would decrease their availability to check inflammation. The ileal mitochondria concentration was significantly diminished in the PAT mice immediately following the antibiotic exposure (Figure S3F), supporting the hypothesis of immediate metabolic damage to the intestinal wall with the antibiotic treatment.
CMT restored host gut and blood metabolites
We next asked whether the CMT-altered microbiome affected host metabolic phenotypes. Untargeted metabolomic analysis of the P23 cecal contents showed 41 metabolites with ≥2-fold significant differences between C and 1P mice (Table S5). Five metabolites (cholic acid, L-pipecolic acid, β-homoproline, cytidine, and 1-Methyladenosine) fell significantly with 1PAT and were restored by CMT, and four metabolites (dipeptide valine-tyrosine, glycitein, o-acetyl-L-serine and hexanedioic acid, bis(2-ethylhexyl) ester) rose significantly with 1PAT and were restored by CMT (Figure 4A upper panel). In targeted analyses, we evaluated several key microbial products including cecal short chain fatty acids (SCFAs) and plasma trimethylamine N-oxide (TMAO)-associated metabolites. Among six measured SCFAs, only cecal lactate was significantly increased in 1P mice but restored by CMT (Figure 4A lower panels), whereas trends for cecal propionate and serum TMAO were inverse. Collectively, these studies define metabolic products in the host perturbed by 1PAT and restored by CMT.
Figure 4. CMT effects on early life metabolism and ileal gene expression.
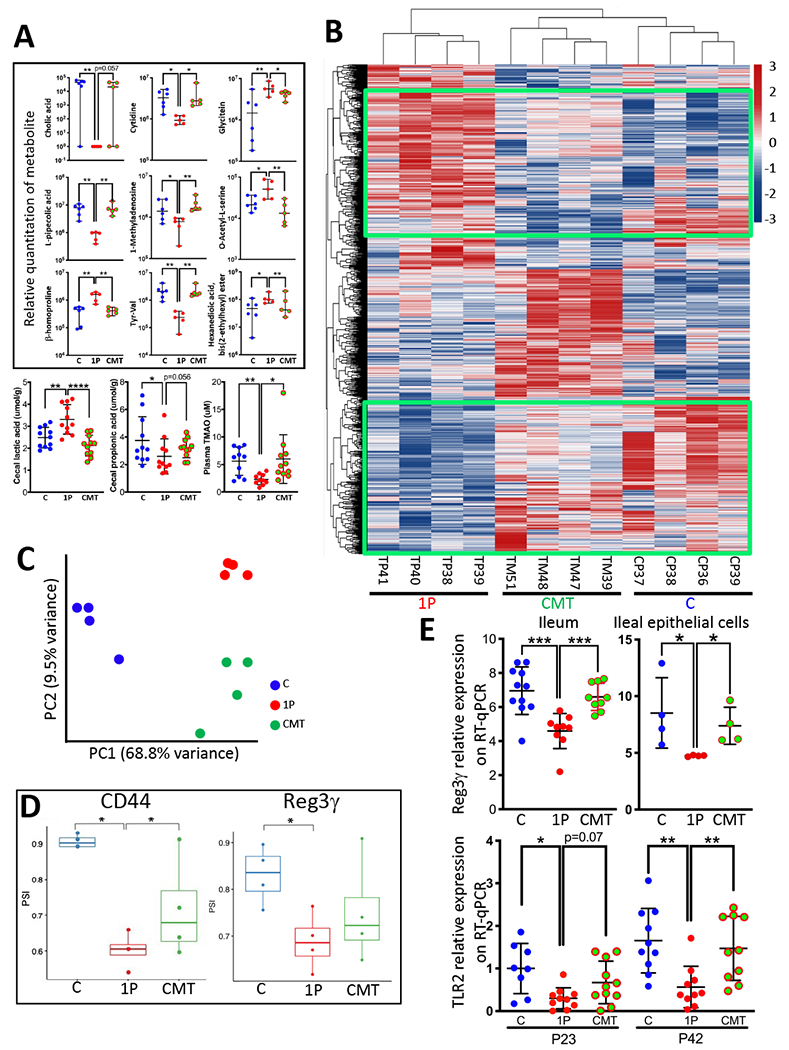
A) CMT rescues early life metabolism. Upper panels: Relative quantitation of 9 cecal metabolites, identified by untargeted metabolomics, with significant differences at P23 between the C and 1P, with restoration by CMT (n=5-6 mice/group, median±range shown). Lower panels: Quantitation of 3 targeted metabolites at P23 (n=10-11 mice/group, mean±SD shown). Cecal propionic acid and lactic acid, and plasma TMAO. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; Mann-Whitney Utest. B) Heat map showing unsupervised hierarchical clustering of the P23 ileal genes with the most highly differentiated expression across the three groups (C, 1P, CMT, n=4 mice/group) using the Seq-N-Slide sequencing data analysis pipeline (https://github.com/igordot/sns). The green boxes highlight the specific genes that were restored in the CMT group. C) Principal Component Analysis of global ileal RNA percentage spliced-in index (PSI) distribution in the three groups: C (n=4; blue); 1P (n=4; red); CMT (n=4; green). D) Differential splicing rates for a 207-nt exon of CD44 (ID: ENSMUSE00000429028) (Left) and a 4-nucleotide retained intron of Reg3γ (between ID: ENSMUSE00000194370 and ID: ENSMUSE00001055347) (Right) in the three groups. PSI in C, 1P, CMT groups (n=4 mice/group, median±range shown). *p <0.05; Welch’s t-test. E) Expression of indicator genes, determined by specific RT-qPCRs. Top: Reg3γ expression in whole ileum (n=9-11 mice/group) and in ileal epithelial cells (n=4 independent cell collection/group) at P23. Bottom: TLR2 expression in P23 and P42 ileum (n=8-11 mice/group). Mean±SD shown; *p<0.05; **p<0.01; ***p<0.001; Mann-Whitney U test. Related to Figures S3,S4,S5,S6, Table S5,S7.
CMT restored host ileal gene expression
To more directly understand the mechanisms by which CMT reduced 1PAT-induced T1D development, we examined P23 ileal gene expression profiles by RNAseq. Unsupervised hierarchical clustering analysis indicated that gene profiles of all 1P mice formed a separate cluster from the C mice, as expected (Zhang et al., 2018). However, the CMT mice clustered with the C mice rather than with the 1P mice (Figure 4B), providing evidence at a global level for CMT restoration of gene expression. Next, compared with the 1P mice, GO term enrichment pathway analysis indicated that CMT decreased expression of genes related to B-cells, immunoglobulins, complement activation, phagocytosis and increased expression of genes related to extracellular space functions and stress responses (Figure S4A). Recent studies provide evidence that exon-splicing differences, histone modification and microRNA expression all affect gene expression as well as host cell phenotypes (Gosline et al., 2016; Karlic et al., 2010; Koch, 2017; Miao et al., 2014). As such, we explored each of these avenues to better understand the mechanisms at the microbial-host cell interface.
Restoration of abnormal exon-splicing profiles of key genes
Since abnormal RNA splicing of host genes also may have pathophysiologic significance (Garcia-Blanco et al., 2004; Li et al., 2019), which may be affected by the gut microbiota (Hasler et al., 2017), we examined the percent spliced in (PSI) index of exons based on the RNAseq studies. This analysis identified 192 differentially spliced exons (Figure S5A), which globally and significantly differentiated between groups (Figure 4C). By PCoA, the CMT are more similar to the C on PC1, but are more similar to the 1P on PC2, consistent with partial restoration (Figure 4C). We identified a group of exons whose alternative splicing patterns differ significantly between the C and 1P mice, but are similar between C and CMT (Figure S5A bottom box).
We then focused on two identified genes, CD44 and Reg3γ with alternative splicing patterns differentiated by antibiotic exposure (Figure 4D). CD44 encodes a cell surface glycoprotein involved in cell-cell interaction, responses to bacterial infection (Senbanjo and Chellaiah, 2017), and TLR regulation (Pure and Cuff, 2001), suppressing TLR2-mediated inflammation (Kawana et al., 2008). The CD44 splicing pattern involves a 207-bp exon (ID: ENSMUSE00000429028), which is significantly lower in 1P mice, while partially but significantly restored by CMT (Figure 4D left panel). Consistent with this finding, ileal TLR2 gene expression at early timepoints was significantly decreased by 1PAT but restored by CMT (Figure 4B lower panel), providing evidence in this model that CD44 regulates TLR2 gene expression through alternative splicing (Qadri et al., 2018). Reg3γ encodes a protein with multipotent activities including antibacterial activity in the intestinal lumen (Mukherjee and Hooper, 2015; Shin and Seeley, 2019) and islet regeneration in the pancreas (Xia et al., 2016). We now identified a significant Reg3γ modification with a 4-nucleotide retained intron that was significantly selected by the 1PAT exposure and partially reversed by the CMT restoration (Figure 4D right panel). Intron retention enhances gene regulatory complexity (Schmitz et al., 2017), which has been found to be associated with developmental and complex diseases, including cancers (Dvinge and Bradley, 2015) and Alzheimer’s disease (Adusumalli et al., 2019). These studies provide further evidence that CD44 and Reg3γ are critical mechanistic intermediates between the altered microbiome in 1P and its restoration with CMT, affecting T1D pathogenesis.
CMT modulated ileal histone post-translational modification
Having found that 1PAT changed histone post-translational modification (PTM) states (Zhang et al., 2018), we now evaluated early-life PTM states in ileum and liver to assess whether CMT restored baseline chromatin regulation. Among the 55 PTM states detected, all three treatments led to distinct patterns at P23 and P42 in ileum and liver (Figure S5B). At P42, for histone variant H3.3, present at active transcription areas, CMT led to decreased K27ac and increased K27 methylation (K27me3K36un and K27me2K36un) (Figure 5A). Since K27 methylation is associated with repressed gene expression, and K27 acetylation with active transcription, our findings indicate that CMT leads to repression of genes otherwise upregulated by the antibiotic-perturbed microbiota. Since K27me3 methylation state is under the regulation of histone demethylase Jmjd3 which is inducible in a NFkB-dependent manner by TLR2 agonists (De Santa et al., 2007), the CMT-increased K27me3 methylation is consistent with our observation that CMT increased TLR2 expression which reduces the TLR2 receptor occupancy ratio by the TLR2 agonist.
Figure 5. CMT effects on early-life ileal histone modification and microRNA profiles.
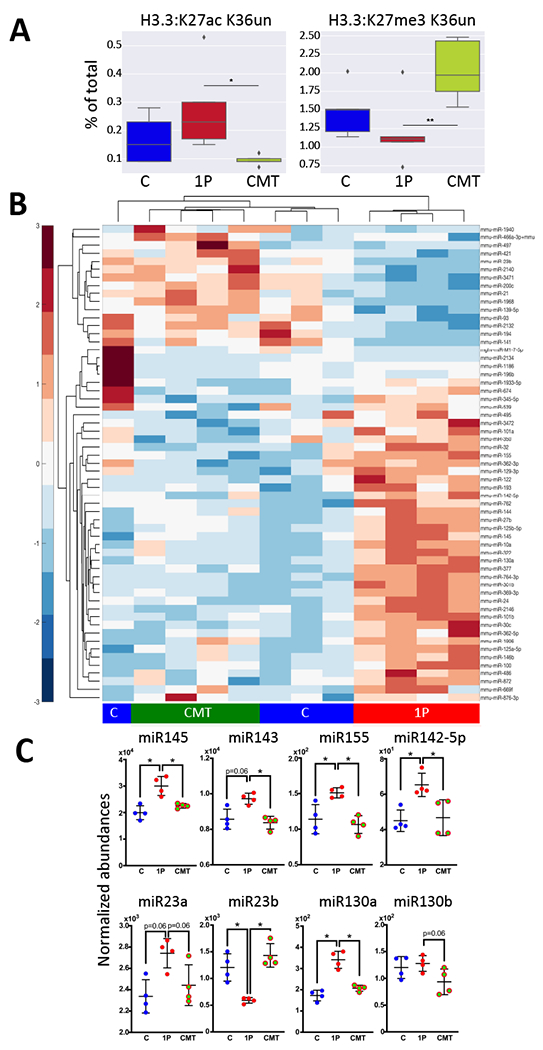
A) CMT rescues long-term histone acetylation and methylation at specific sites at P42. High-abundance, representative peptides [Left: Histone H3.3: K27ac K36un acetylation and Right: Histone H3.3: K27me3 K36un] are shown as a percent of the peptide family total. Medians and IQR are shown. Whiskers extend to points within 1.5 interquartile ranges of the lower and upper bounds, and points outside this range are graphed as individual points. n=5 mice/group, *p<0.05, **p<0.01, One-way ANOVA with Tukey’s HSD post-test. B) Heat map showing unsupervised hierarchical clustering of the 46 most abundant and significantly differential miRNAs by NanoString in the C, 1P and CMT groups (n=4 mice/group) at P23. C) Normalized abundance of eight miRNAs (miR145, miR143, miR155, miR142-5p, miR23a/b & miR130a/b) that regulate CD44, TLR2, and Reg3γ. n=4 mice/group, mean±SD shown, *p<0.05; **p<0.01; Mann-Whitney U test. Related to Figures S4,S5, Table S6.
CMT restored host ileal microRNA expression profiles
Next we examined the expression of all 599 known murine miRNAs in ileal samples from the same P23 mice used for the ileal RNAseq studies. Unsupervised hierarchical clustering of the 46 highest abundance miRNAs indicated that the profiles of the mice receiving CMT clustered with the C rather than with the 1P mice (Figure 5B), paralleling the result of the RNAseq experiment (Figure 4B), and indicating that CMT restored ileal miRNA expression at a global level. We identified a set of specific ileal miRNAs reported to regulate genes affected by 1PAT including CD44, TLR2 and Reg3γ, which were differentiated by 1PAT but restored or partially restored by CMT (Table S6, Figure 4E,5C). miR145 regulating CD44 (Pagliuca et al., 2013), miR143 regulating-CD44 (Pagliuca et al., 2013; Yang et al., 2016) and TLR2 (Guo et al., 2013), and miR155 (Bayraktar et al., 2019) regulating-TLR2 were increased by 1P compared to C, but restored by CMT (Figure 5C). Reg3γ-regulating miR23b (McKenna et al., 2010) was decreased by 1P and restored by cecal microbiota transfer (Figure 5C).
CMT effects on gene expression in isolated epithelial cells
Thus far, the studies of gene expression were based on analysis of full thickness distal ileal tissues. As such, we next sought to explore the interaction of the perturbed and restored microbiota at the more precise interface of isolated ileal epithelial cells. We repeated the exposure of male mice to antibiotics (1P), or not (C), with CMT restoration, exactly as in Figure 2A, and following mouse sacrifice and isolation of purified ileal epithelial cells (Figure S6A), gene expression was compared across the experimental groups by Nanostring assay of 547 immune-associated genes. 1PAT exposure changed the gene expression profile of NOD ileal epithelial cells and CMT altered the profile from that of the 1PAT treatment at a global level (Figure S6BC). 1PAT significantly differentiated 66 genes (Figure S6C, Table S7) and CMT restored 31 of the 46 downregulated and 14 of the 20 upregulated. CMT restored 1PAT-differentiated Reg3γ expression in ileal tissue in the intact mouse (Figure 4E upper left panel) and in the purified IECs (Figure 4E upper right panel). As expected, these studies indicate that ileal epithelial cells participate in the transduction of the altered and restored microbiome signals that ultimately lead to the observed T1D phenotypes.
CMT-restored network of microbial-host intestinal gene expression interaction
Since the effects of 1PAT on the interaction between microbiome and the host were global and extensive, we conducted network analysis to identify the key linkages (Figure 6A). Key taxa in the network were family S24-7, genera Oscillospira and Lactobacillus, and Clostridiales, which were depleted by 1PAT but restored by CMT, and taxa from genera Akkermansia, Blautia, Enterococcus, with the inverse relationships. Key host ileal pathways were involved in immunity (CD molecules, cell adhesion molecules, lysosome and peptidases), metabolism (PPAR signaling, Fat digestion and absorption, cholesterol metabolism) and cellular processing (transcription factors, messenger RNA biogenesis, splicesome, exosome, histone modification proteins, membrane trafficking, phosphorus-containing group transfer and ubiquitination). The effects of the early life antibiotic treatment were widespread; this analysis identifies central mechanisms downstream of the perturbations.
Figure 6. Multi-omic analysis of microbe-host gene expression interactions and verification of key targets in the network.
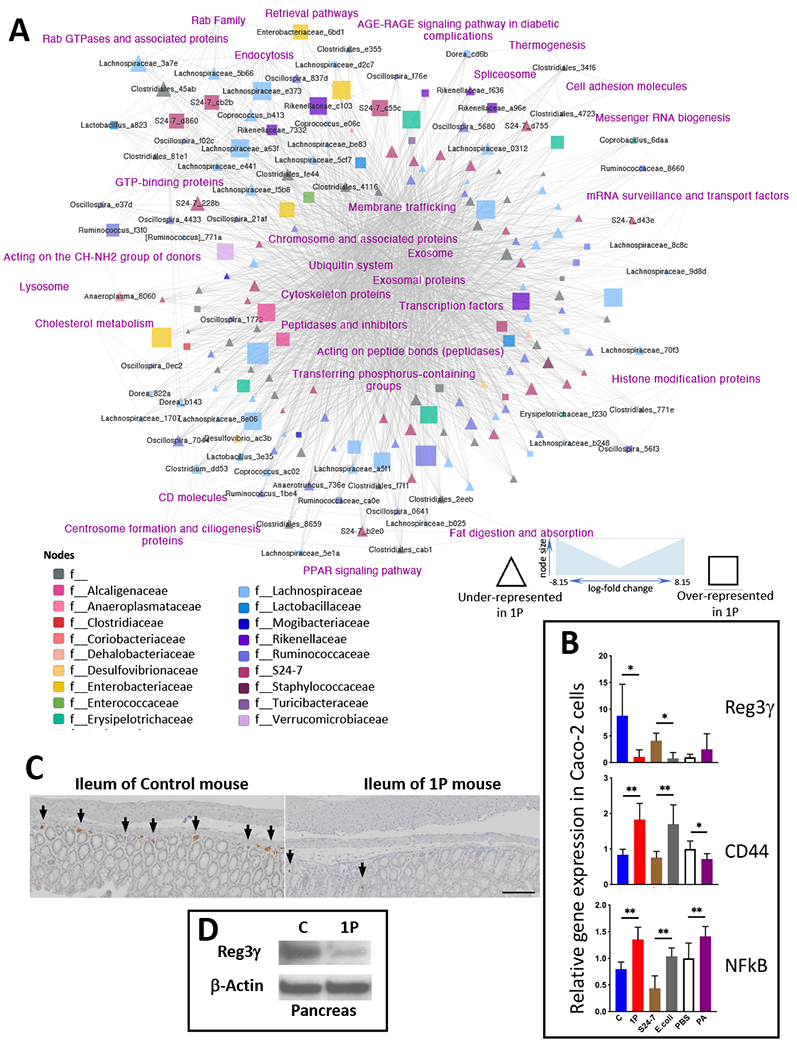
A) Multi-omic analysis of microbe-host gene expression interactions. A total 1336 host pathways of C, 1P and CMT mice in the previous (Zhang et al., 2018) and current study identified by RNAseq and 188 fecal taxa of these mice identified by 16S sequencing were applied to a co-occurrence model using MMvec that outputs rankings (Morton et al., 2019). The network was visualized using Cytoscape, with taxa colored by Family and node size scaled to mean relative abundance of the OTU. Differential abundance was calculated using Songbird (Morton et al., 2019), comparing the three groups. We grouped taxa based on the over- or under-representation in 1P. B) Effects of intestinal luminal contents on expression of key host genes in intestinal epithelial cells in culture. To extend our observations and to verify targets identified in the multi-omic analysis, human Caco-2 intestinal epithelial cells were co-cultured for 8 h with cell-free supernatants from cecal contents of C and 1P mice at P23, or with bacterial cells of S24-7 strain MH8C or E. coli strain K12, or with propionic acid (PA), or PBS as a blank control (PBS). The cells were collected, RNA extracted and RT-qPCR performed to evaluate expression of 3 targeted host genes, Reg3γ and CD44, identified in the NOD mouse model as potential differential players in immune-regulatory pathways, as well as NFkB as a master regulator of inflammatory responses. mean±SD shown; n=6 independent culture/group; *p<0.05; **p<0.01; Mann-Whitney U test between each pair of stimuli. C) Immunohistochemical staining of Reg3γ in ileal tissue in NOD mice at P23. Formalin-fixed and paraffin-embedded ileal tissues of C (left) and 1P (right) mice were stained with rabbit anti-Reg3γ-specific primary antibody, goat HRP α-rabbit secondary antibody and visualized with 3,3’-diaminobenzidine and with hematoxylin as counter-stain. Arrowheads illustrate some of the cells expressing Reg3γ. Scale bar is 100μm. D) Immunoblot analysis of 1P effects on pancreatic Reg3γ in NOD mice at P23. Total protein extracts from pancreatic tissue lysates were subjected to SDS-PAGE for immunoblotting with rabbit anti-REG3G to compare Reg3γ protein levels and for immunoblotting with mouse anti-Actin as a loading control.
Effects of intestinal luminal constituents on expression of targeted host genes in intestinal epithelial cells
Our global model identified cell surface molecules and transcription factors as elements of the differential gene regulation related to antibiotic exposure. We next examined in vitro the expression of three genes, Reg3γ (a C-type lectin and anti-microbial peptide), CD44 (a cell-surface glycoprotein) to represent epithelial cell surface molecules, and NFkB, a master regulator of inflammation, to represent transcription factors. To extend our observation of the differential regulatory effects of intestinal epithelial Reg3γ gene expression in NOD mice (Figure 4E), human intestinal epithelial Caco-2 cells were co-cultured with constituents from mouse cecal contents. First, we examined cell-free supernatants from the intestinal contents of C mice as our standard, and compared with those from 1P mice. We also studied bacterial cells from a single strain from the S24-7 family that is markedly diminished in 1P mice vs. an individual E. coli strain, representative of the Enterobacteriaceae which are increased in 1P mice (Figures 2E,3B). Finally, we compared a 1PAT-decreased metabolite, the SCFA propionic acid (PA) (Figure 4A) to PBS as control. Reg3γ expression was significantly (8-fold) diminished in the presence of cecal contents from 1P mice than those from C mice. Similarly, expression was significantly (9-fold) lower in the presence of the E. coli cells than with the commensal S24-7 bacterial cells (that are increased and decreased in 1P mice, respectively) and trended to be upregulated in the presence of propionic acid (PA) compared to PBS (Figure 6B, top panel). Taken together with our prior findings in the NOD mice, these results indicate that the balance between the particular commensal bacterial taxa (e.g. the dominant S24-7) and the opportunists (e.g. the Enterobacteriaceae) are involved in the regulation of expression of the key host gene Reg3γ in intestinal epithelial cells and that are perturbed by the antibiotic exposure. Next, we examined CD44, since there were significant differences in alternative splicing in NOD mice (Figure 4D). Our results (Figure 6B middle panel), indicate that CD44 expression in Caco-2 cells also is regulated; significantly higher in the presence of cecal contents from 1P than from C mice, significantly higher in the presence of E. coli cells than S24-7 family cells and significantly lower in the presence of PA than PBS alone, results that are consistently inverse to those for Reg3γ. Finally, we also examined Caco-2 cell expression of NFkB (Figure 6B bottom panel). Expression was significantly higher in the presence of cecal contents from 1P than from C, significantly higher in the presence of E. coli cells than S24-7 bacterial cells, paralleling that for CD44; in this assay, PA had the opposite effect. In total, these findings confirm the identity of important microbes and their products that affect the gene regulatory environment in the intestinal epithelium and suggest that the balance between them may be detected by Reg3γ which transduces the signals more deeply.
We then extended our observations to assess the involvement of Reg3γ, identified from gene expression studies as a host product whose early age (P23) expression was consistently reduced by 1PAT and restored by CMT (Figure 4E). Immunohistochemical observations further show that antibiotic exposure decreased Reg3γ expression at the protein level (Figure 6C). Immunoblot-based analysis also showed that in 1P mice, there were decreases in pancreatic Reg3γ at P23 (Figure 6D). These findings further support our model that antibiotic exposure with its consequent changes to the composition of the intestinal microbiota has effects on pathways that go through Reg3γ, affecting propensity for T1D (Figures 7,S7).
Figure 7. Summary of mechanisms perturbed by 1PAT and restored by CMT.
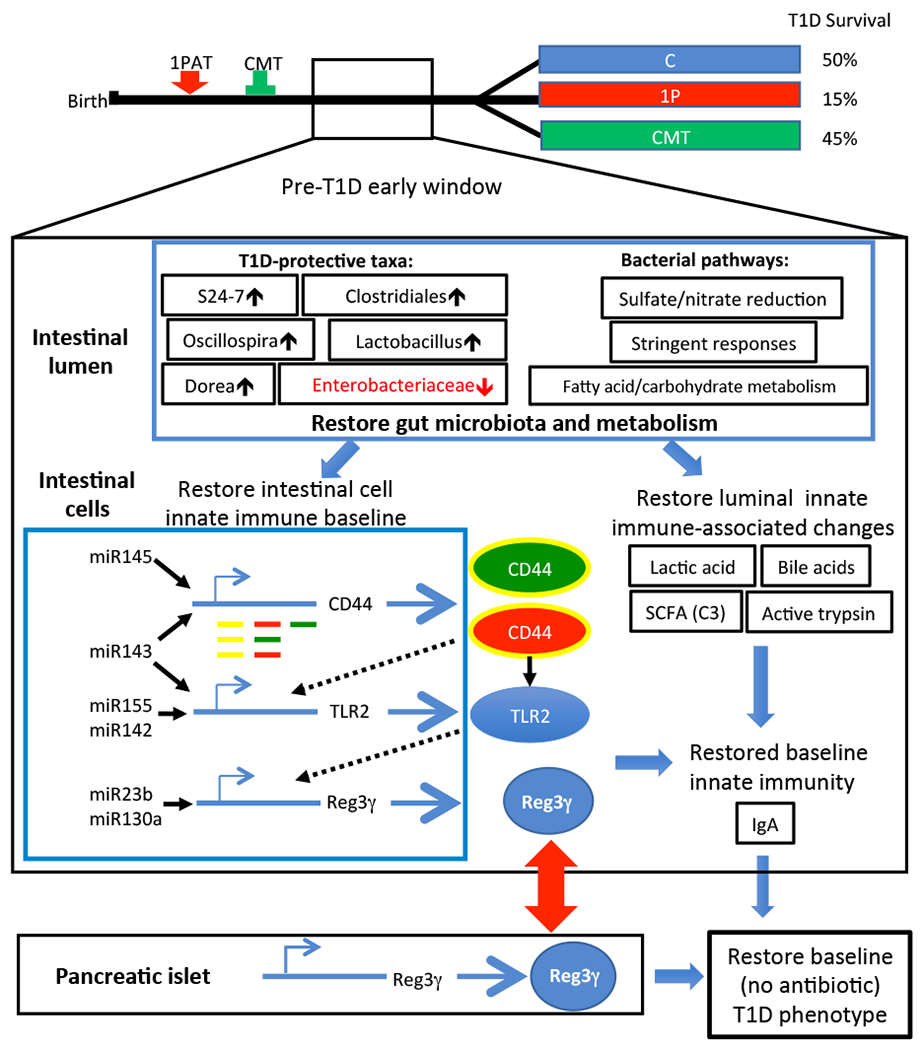
Changes in the microbiome affecting the balance of commensals and opportunists (e.g. Enterobacteriaceae), and thus in the intestinal lumen and their metabolites, led to alterations in intestinal cell gene regulation affecting innate immune effectors. The altered balance of these effectors shapes the milieu relevant to adaptive immunity that is injurious to the pancreatic islets. Related to Figure S7.
DISCUSSION
Prior studies in both humans and mice show that antibiotics given early in life, at doses that are therapeutic for treating bacterial infections, disrupt the developing microbiome (Langdon et al., 2016; Nobel et al., 2015), often with substantial pathophysiological consequences (Aversa et al., 2020; Ortqvist et al., 2019; Stark et al., 2019). An important question is whether restoration is possible. We addressed this question in a well-established murine model of antibiotic-enhanced development of T1D (Livanos et al., 2016; Zhang et al., 2018) and now provide evidence that restoration of both microbiota and health status can be achieved.
Our preliminary studies indicated that microbiota from mothers on the day of birth (Mom-P0) had the most favorable characteristics of those tested for partially normalizing the microbiota of the pups that had been altered beginning at P5. This is consistent with the evidence that at birth, mothers pass a comprehensive microbiota to their offspring, with subsequent selection and succession of dominant species (Kimura et al., 2020; Mueller et al., 2015). Alternative sources and exact timing for optimal transplantation must be examined in future studies, but inocula of unperturbed maternal (Mom-P0) microbiota may be important candidates. Consistent with this point, a recent study of restoration following Cesarean section in human children showed that inocula from late pregnancy conferred partial restoration of the infant’s microbiota (Korpela et al., 2020).
The intermediate species richness phenotype produced by CMT suggests incomplete engraftment of a robust microbial community potentially influenced by residual antibiotic effects. However, CMT mice had comparable community evenness to control, suggesting that while species richness was reduced, the communities assembled into structures resembling the unperturbed communities. Oscillospira could be one of the T1D-protective intestinal taxa and its depletion may be used as an early biomarker to predict T1D onset. Oscillospira, a currently unculturable and not well-characterized genus (Konikoff and Gophna, 2016), also may be important in human health, with abundance associated with leanness (Goodrich et al., 2014; Tims et al., 2013) and fecal bile acid levels (David et al., 2014), and inversely related to inflammatory diseases (Walters et al., 2014). Consistent with these reports, we observed that Oscillospira relative abundance and cecal bile acids, as mediators regulating lipid and glucose metabolism (Ferrell and Chiang, 2019) and T cell homeostasis (Song et al., 2020), were decreased by antibiotic perturbation and restored by unperturbed maternal microbiota transfer. Analysis across the experiments shows greater consistency in the microbes depleted by antibiotics and restored by transfer than in the ones enhanced by antibiotics; such findings are consistent with a model of antibiotic-induced loss of keystone species, and replacement by opportunists, that may vary between individuals. If correct, that phenomenon provides further rationale for early replacement approaches.
With partial restoration of the microbial populations, consistent restoration of metagenome and metabolites were observed, improving confidence in the methodologies, and suggesting mechanisms for abrogating the disruption-enhanced T1D phenotype. We identified a short list of relevant metagenomic metabolic pathways and metabolites significantly perturbed by antibiotics and recovered with restoration. Intestinal cysteine and methionine reduce oxidative stress and increase goblet cell and crypt cell proliferation (Ruth and Field, 2013). Steroid signaling pathways via NF-kappa B regulation suppress immune and inflammatory responses (McKay and Cidlowski, 1999). Retinoic acid affects mucosal immune responses by promoting dendritic cell CD103-expression, enhancing Foxp3+ inducible regulatory T cell differentiation and inducing gut-homing T cell specificities (Oliveira et al., 2018). Since bacterial bile acid metabolites are essential for maintaining intestinal RORγ+ regulatory T cell homeostasis (Song et al., 2020), cholic acid, reduced by antibiotic exposure and restored by CMT, provides a potential intermediary for the altered immune phenotypes (Fiorucci et al., 2018; He et al., 2018).
We showed that the microbiota degrades trypsin and antibiotics suppress this activity with restoration by CMT (Figure S3). That trypsin damages the colonic epithelium, mediating tight junction degradation and intestinal permeability, and activates signaling pathways through protease-activated receptors (PARs) (Cenac et al., 2003; Midtvedt et al., 2013; Van Spaendonk et al., 2017; Vergnolle, 2016), provides another mechanism through which a perturbed microbiota can lead to enhanced auto-immunity.
In the ileum, we observed substantial alterations in gene expression at a global level due to the altered microbiota, and identified a subset of genes with altered expression, consistent with prior reports (Livanos et al., 2016; Zhang et al., 2018), but now in which CMT restored normal phenotypes (Figures 6,7), including genes related to innate immunity (e.g. Saal, Muc2, Reg3γ). The CMT-induced restoration also led to a global shift away from immune activation (Figure 7), emphasizing the importance of the microbiota-intestinal wall interface in regulating the T1D phenotype (Daft and Lorenz, 2015; Hu et al., 2015). Studies of isolated epithelial cells indicate the critical position of the ileal epithelium in the transduction of luminal signals to downstream immunological effector cells (Birchenough et al., 2016; Haber et al., 2017; Pott and Hornef, 2012).
Considering factors upstream of ileal gene expression were affected in opposite directions by antibiotic exposure and CMT, we showed: i) altered expression of ileal miRNAs. Affected miRNAs include regulators of innate and adaptive immunity (e.g. miR143 and miR155, which target CD44 and TLR2, respectively (O’Neill et al., 2011; Wen et al., 2013; Yang et al., 2016); ii) altered gene splicing, as illustrated by the shifts in CD44 (Prochazka et al., 2014). iii) altered intestinal histone modification epithelium, as we reported (Zhang et al., 2018). In particular, reciprocal changes in variant histone H3.3, deposited in areas of active transcription, and in canonical histone H3 occurred with antibiotic exposure and restoration, consistent with heightened and restored gene expression, respectively (Zhao and Garcia, 2015). The opposing patterns between histone H3.3:K27 acetylation and H3.3:K27me3 methylation in response to CMT affect Th17 differentiation through the key transcription factor RORγT (Cribbs et al., 2020).
By identifying factors perturbed by antibiotics and restored by CMT, we propose that CMT rescues T1D through a pathway involving gut commensal bacteria S24-7 and host Reg3γ as key elements (Figure 7). Moreover, we narrow the model to understand the most active elements (Figure S7) that now can be approached experimentally to understand the essentiality of their role. Although our studies in cell culture point toward mechanisms involving specific microbes and specific host effects, such as involving Reg3γ, our ability to test those hypotheses is limited by the lack of further in vivo experiments. Nevertheless, these studies emphasize the centrality of early life events in the intestine for determining systemic effects (e.g. immunopathology in the pancreas) that may occur well into adult life. The availability of a functional restoration (CMT) permits more focused explorations of T1D pathogenesis, the broader biological issue of the crucial interface between early life microbiota and host immunological and metabolic development, and points to approaches for remediation.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information requests should be directed to and will be fulfilled by the lead contact, Martin J. Blaser at martin.blaser@cabm.rutgers.edu.
Materials availability
This study did not generate new unique reagents
Data and code availability
RNA-Seq data that support the findings of this study have been deposited in the ArrayExpress database (https://www.ebi.ac.uk) with accession code E-MTAB-9981 (RNA-seq of ileum at P42 in STR) and E-MTAB-9982 (RNA-Seq of ileum at P23 in LTR). Ileal NanoString data have been deposited in the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession numbers GSE163887 (Ileal epithelial cells immune gene profile at P23) and GSE163888 (ileal miRNA profile at P23). 16S rRNA data have been deposited in QIITA with the identifier 13529 (https://qiita.ucsd.edu/study/description/13529). Shotgun metagenomics data have been deposited in the European Nucleotide Archive (ENA) (https://www.ebi.ac.uk/metagenomics/) under accession number, PRJEB42282.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
NOD/ShiLtJ mice (6 weeks old) were purchased from Jackson Laboratory (Bar Harbor ME), and bred in an SPF vivarium at the New York University Langone Medical Center (NYUMC) Skirball animal facility and at Rutgers University School for Public Health animal facility. All animal procedures were approved by the NYUMC Institutional Animal Care and Use Committee (IACUC protocol no. 160623) and by the Rutgers University Institutional Animal Care and Use Committee (IACUC protocols no. 201900017 and 201900032). Mice were housed in individual ventilated cages (IVC) with no more than 5 mice per cage in SPH vivarium room with room temperature as stable as 22-24°C, the relative humidity 45-60%, and 12-h light-dark cycle. The dams and their litters were randomly assigned to control (C) or 1PAT antibiotic (1P) groups. At postnatal (P) day 23, the pups were weaned and housed to separate males and females with 2-5 mice per cage. All mice received acidified drinking water supplied by the facility routinely except for the periods when some litters were receiving antibiotic treatment. A therapeutic dose of the macrolide tylosin tartrate (Sigma-Aldrich, Billerica MA) was given to 1P pups in their non-acidified drinking water at 333 mg/L on P5-P10, as described (Zhang et al., 2018).
Purification and analysis of isolated intestinal epithelial cells
Mouse small intestinal epithelial cells were isolated based on established protocols (Gracz et al., 2012; Graves et al., 2014) with further modifications. P23 male NOD mice were subjected to C, 1P, and CMT treatments, as above, and at sacrifice, ~12 cm of the terminal ileum was removed, fat and Peyer’s patches excised, the tissue opened longitudinally and cut into 1-cm pieces, and incubated on ice in 20 mL of DMEM (Corning, Tewksbury MA) supplemented with 10% FCS (Gibco, Grand Island NY). After a brief vortex, each sample was incubated in 20 mL DMEM with 10% FCS and 1 mM DTT (Sigma) at 37°C for 15 min, with shaking at 180 rpm. The sample was filtered through 70 μM filter mesh, and the filtrate containing IECs were centrifuged at 300 g and stored on ice. Remaining tissue was transferred to 20 mL PBS with 15 mM EDTA, briefly vortexed, then incubated at 37°C for 10 min with shaking at 180 rpm. The supernatant was filtered through 70 μM mesh, combined with IECs from the previous step, and re-suspended in 5 mL DMEM with 0.02 mg/mL DNase I (Sigma) for 10 min at room temperature. Cells were filtered a final time, re-suspended in DMEM with 10% FCS, and viable cells counted by trypan blue exclusion. After cell counting, IECs from three mice in the same treatment group were pooled for each of the three IEC treatments pools. IECs were stained using a LIVE/DEAD viability dye, anti-CD45-FITC, and anti-EpCAM-APC (BioLegend, San Diego CA), according to the manufacturer’s instructions, and subjected to fluorescence-activated cell sorting using a 100 μM nozzle to collect live IECs using a Coulter MoFlo™ XDP at Rutgers University Ernest Mario School of Pharmacy Cytometry and Cell Sorting Laboratory. The collected cells subjected to TRIzol reagent (ThermoFisher Scientific, Carlsbad, CA) directly without further culture for RNA extraction.
Cell line
Human intestinal cell line Caco-2 (ATCC, Manassas VA) were cultured and maintained in surface-treated sterile tissue flasks or 6-well plates (ThermoFisher Scientific) in Eagle’s Minimum Essential medium (MEM) (Corning, Tewksbury MA) with 10% fetal calf serum (FCS) (Corning) and 1x Penicillin-streptomycin (Gibco) in a humidified incubator at 37°C with 5% CO2 air atmosphere. Cells were split and media were changed every 72 hours.
Microbe strains
S24-7 family MH8C strain (S24-7) (gift from the Carolina Tropini Lab) was cultured on TSA with sheep blood agar medium (Thermo Scientific, Waltham MA) for 72 h at 37°C in an anaerobic Chamber (Coy Lab Products, Grass Lake MI) under an atmosphere of 90% N2, 5% CO2, and 5% H2. E. coli K12 strain was cultured on LB liquid medium for 16 h at 37°C with shaking (160 rpm) under aerobic conditions.
METHOD DETAILS
Microbiota transfer
To prepare donor cecal materials for the short-term restoration (STR) experiment, we bred 12 pairs of NOD mice in 3 groups with 4 pairs for each group. For group 1, dams and pups did not receive any treatment and were sacrificed at pup day of life 5 (P5). Cecal contents from the dams were collected separately, and pooled as Mom-P5. Cecal contents from the 24 pups of the 4 litters were collected and pooled as Pup-P5. For group 2, dams did not receive any treatment and were sacrificed on the day of birth, and cecal contents were collected and pooled as Mom-P0. For group 3, dams received a 5-day 1PAT tylosin course (333 mg/L) during pregnancy (E13-E18), and were sacrificed on the day of birth, and cecal contents were collected and pooled as Mom-T-P0. During collection, cecal tissues were fully opened and cecal contents were immediately released into pre-reduced anaerobic dental transport media (Anaerobe Systems, Morgan Hill CA), mixed well, and frozen at −80°C. To create the final pools, the cecal materials were thawed in an anaerobic chamber, pooled, thoroughly mixed and, diluted in ice-cold pre-reduced broth medium with glycerol. After settling to precipitate large particulates, the liquid suspensions were transferred, mixed thoroughly and aliquoted in 1 mL volumes at OD600 (1.0~1.5), frozen at −80°C until use. On the day of use, 50 μL of each suspension was thawed, and transferred via gavage at P15 to the 1P pups. Broth medium/glycerol alone was used in the control groups. For the long-term restoration (LTR) experiment, cecal materials were collected from 20 NOD dams at the day of pup birth, and pooled together as Mom-P0. Pre-reduced PBS (phosphate buffered saline) in anaerobic conditions was used for resuspension and dilution, and used in control groups. By oral gavage, 50, 75, and 100 μL of the suspension were transferred to 1P pups at P13, P15, and P17, respectively, representing the cecal microbiota transfer group (CMT).
Diabetes monitoring
Diabetes monitoring of NOD male mice (n=20) was performed weekly beginning at week 11 of age and continued to week 30, as described (Wen et al., 2008; Zhang et al., 2018). Diabetes progression evaluation and detecting significant differences between treatments was performed by Kaplan-Meier analysis (Kaplan and Meier, 1958) and the Log-rank (Mantel-Cox) test (Harrington and Fleming, 1982), respectively. Considering that the proportional hazards assumption may not hold throughout the study and there may be delayed effects on disease risk, the Gρ,γ weighted log rank test (Fleming and Harrington, 1991) was adopted, and higher weight was assigned to the middle time points with both parameters set to 1. Post-hoc pairwise comparisons were adjusted by a Benjamini-Hochberg procedure.
Collection of fecal and tissue samples
Fresh fecal collection from individual NOD male mice was performed weekly after weaning as described (Zhang et al, 2018). Briefly, each mouse was placed in an empty clean beaker for 2–3 minutes to allow them to defecate normally to obtain 4 pellets, which were frozen at −80°C for later analysis. At mouse sacrifice, the distal ileum (1/3 of the section) was collected; after removing contents, tissue was kept in RNAlater (Qiagen, Valencia CA). Cecum samples with contents were divided into two sections, one directly frozen at −80°C for 16S rRNA and metabolic analyses and the other was added to pre-reduced anaerobic dental transport media (Anaerobe Systems) and frozen at −80°C for bacterial isolation. After removal of colonic contents, colonic tissues were collected into RNAlater. Blood samples were collected from cardiac puncture and plasma samples were prepared and frozen at −80°C for metabolite analyses. From mice sacrificed at P70, the pancreas was fixed in freshly prepared modified Bouin’s fixative (Leiter, 2001), paraffin-embedded, sectioned, stained, and scored, as described (Livanos et al, 2016; Zhang et al., 2018) with modification by using methyl green as counterstain (Forestier et al., 2007).
16S rRNA assessments of microbiota and community analysis
Microbiome assessment was performed as described (Zhang et al., 2018). Briefly, microbiota DNA was extracted from fecal samples (1 pellet) and cecal samples using the DNeasy PowerSoil-HTP 96 Well Soil DNA Isolation Kit (Qiagen). An amplicon library of the bacterial 16S rRNA V4 regions was obtained with barcoded fusion primers and triplicate PCR, and combined with each DNA sample at equal concentrations, as described (Zhang et al., 2018). The library was sequenced with the Ilumina MiSeq platform using 2×150 bp paired-end format (Illumina, San Diego CA) at the New York University Langone Genome Technology Center. QIIME 2.0 was used as the amplicon read processing pipeline as described (https://qiime2.org/) (Bolyen et al., 2019). Briefly, demultiplexed paired-end reads from MiSeq were trimmed, denoised and joined with DADA2 (Callahan et al., 2016). A phylogenetic tree was created by FastTree (Price et al., 2010) after the multiple sequence alignment and masking of highly variable positions using MAFFT (Katoh and Standley, 2013). Taxonomy was assigned using a pre-trained Naïve Bayes classifier (Bokulich et al., 2018) based on a pre-created Greengene 13_8 99% identity OTUs (DeSantis et al., 2006). Microbial α-diversity and β-diversity were generated using QIIME2.0. α-diversity was measured using phylogenetic diversity (Faith’s PD) (Faith, 1992), Shannon index, and observed amplicon sequence variant (ASV) and analyzed with GraphPad Prism 8.0 (GraphPad Software, La Jolla CA). Longitudinal analysis of α-diversity was performed in R (R Core Team, 2017). Each timepoint was tested individually using a one-way ANOVA, individual comparisons between all groups were calculated for significant timepoints (p<0.05) post hoc using Tukey’s HSD post-test. Figures were made using ggplot2 (Wickham, 2009). Unweighted UniFrac was used for β-diversity analysis (Lozupone and Knight, 2005). Assessment of significantly different taxa with respect to the average microbe was performed using the ANCOM program (Mandal et al., 2015) within QIIME2.0 (Bolyen et al., 2019). Assessment of longitudinal changes of microbes binned at species-level taxonomy was performed in R (R Core Team, 2017) using the tool MaAsLin2 (Mallick et al., 2021) with default parameters. In brief, taxa were normalized using total sum scaling (TSS) and log-transformed. Normalized taxa were modeled with fixed effect of treatment group and random effects of timepoint and mouse ID. Significant taxa (FDR-corrected p-value <0.05), were further tested for significance between groups at each timepoint. Total 16S copy number of fecal micrbiome was also evaluated by qPCR using 1 μL fecal microbime DNA per fecal pellet with bacterial universal primer pairs 338F and 518R (Muyzer et al., 1993) in a LightCycler 480 system (Roche, Branchburg NJ) and QuantiTect SYBR® Green PCR Kit (Qiagen). For group mean comparisons, the Mann-Whitney U test was performed with p-value <0.05 indicating significance.
Microbiome assessment by shotgun metagenomic sequencing
A total 36 fecal samples from 18 male NOD mice, including 6 each from C, 1P and CMT mice, were examined at 3 and 5 weeks in the LTR experiment. Whole genome shotgun sequencing was performed as described (Zhang et al., 2018). Briefly, Genomic DNA (5 ng) from each sample was extracted with QIAamp Powerfecal DNA Kit (QIAgen), and library preparation and subsequent whole genome sequencing (WGS) was performed using the Illumina HiSeq 2500 platform with 4 flow cell lanes as 150-bp paired-end reads, at NYU Langone’s Genome Technology Center. A total of 377 Gb raw sequence data was obtained from sequencing center for the 36 samples, averaging ~31.5 million pairs of PE150 reads per sample (standard deviation=4.4 million). Kneaddata v0.6.1 (https://huttenhower.sph.harvard.edu/kneaddata) was used for quality filtering with a sliding window of 3, minimum quality score of 20, and minimum sequencing length of 100 bases. Both paired and orphan reads, which passed the initial filtering were further decontaminated using genomes of host (Mus musculus) and PhiX (an Illumina sequencing control). After these rigorous quality control steps, ~263 Gb data, (average of 70.1% of raw reads (range: 56.0-84.0%)), were considered high-quality and further analyzed. Quality filtered reads from each sample were then passed to HuMAnN2 v0.11.1 (Franzosa et al., 2018), for gene function and pathway analysis with default parameters. Abundance of genes or pathways were renormalized to relative abundance using the HuMAnN2 utility script. Principal component analysis (PCA) calculated using Bray-Curtis measurements. Differentially abundant pathways were identified using MaAsLin2 (Mallick et al., 2021) with FDR correction. Features with corrected q-value <0.1 were identified as significant. A heatmap of significant pathways at each time point were generated with pheatmap R-package, rows and columns arranged based on hierarchical clustering of arcsin-square-root transformed relative abundance in accordance with MaAsLin2. MaAsLin2-identified significant pathways were further examined using tools provided by the MetaCyc metabolic pathway database (Caspi et al., 2018) to screen for those found in bacteria.
High resolution GC-MS/MS analysis of cecal short chain fatty acids (SCFAs)
From the LTR experiment, 33 P23 cecal content samples, including 11 each from C, 1P, or CMT mice, underwent SCFA analysis as described (Lieber et al., 2019). Briefly, cecal contents were aliquoted, weighed and hydrated with water solution of NaOH (5 mM). After 40 min vortex-mixing in the cold and centrifugation, 50 μL of supernatant was placed in a GC vial containing NaOH (5 mM in water) and isotopically labeled internal standards (IS; 5 μL). Solution of 2-Butanol/Pyridine (3:2; v/v) (50 μL) and isobutyl chloroformate (10 μL) was added, the mixture vortexed and sonicated, the derivatized acid were extracted with hexane (50 μL) and the hexane layer transferred into another vial for the GC-MS analysis. The quantitation of acetic acid, butyric acid, isovaleric acid, lactic acid, propionic acid, and succinic acid was performed using isotope dilution GC-MS/MS by using MRM mode. The absolute quantity of each SCFA was determined using calibration curves measured for each analyte. Samples were analyzed by using the Thermo TSQ-Evo triple quadrupole in tandem with the Trace 1310 gas chromatograph (ThermoFisher Scientific). Chromatographic separation was achieved by using an HP-5MS fused-silica capillary column (30 m × 0.250 mm × 0.25 μm; Agilent Technologies, Santa Clara, CA, USA) coated with 5% phenymethyl siloxane. Each extract (1 μL) was injected in split mode (10:1). Helium as carrier gas flow was 1 ml/min. The GC oven temperature program was as follows. The initial temperature of 40 °C was held for 2 min after injection before it was increased up to 50°C at 3°C/min, followed by increase to 110°C at 5°C/min, then 250°C at 30°C/min and 310°C at 70°C/min, and then held at 310°C for 3 min. Argon was used as collision gas. The injector, transfer line, and ion source temperature were set at 260, 290, and 230 °C, respectively. The mass spectrometer was tuned to an electron impact ionization energy of 70 eV in the MRM mode with the following parent to daughter ion transitions: m/z 61.0 → 43.0 for acetic acid, m/z 63.0 → 45.0 for [13C2]-acetic acid, m/z 61.0 → 43.0 m/z 71.0 → 41.0 for butyric acid, m/z 78.1 → 46.1 for D7-butyric acid, m/z 85.1 → 57.1 for isovaleric acid, m/z 87.1 → 59.1 for D2-isovaleric acid, m/z 135.1 → 45.1 for lactic acid, m/z 138.1 → 48.0 for D3-lactic acid, m/z 75.1 → 57.0 for propionic acid, m/z 77.1 → 59.0 for D2-propionic acid, m/z 101.1 → 55.0 for succinic acid, and m/z 105.1 → 57.0 for D6-succinic acid.
High resolution GC-TOF-MS analysis of serum TMAO associated metabolites
From the same mice as above, P23 serum samples underwent TMAO analysis, using stable isotope dilution HPLC with online electrospray ionization tandem mass spectrometry on an API 365 triple quadrupole mass spectrometer (Applied Biosystems, Foster City CA) with upgraded source (Ionics, Bolton, Ontario) interfaced with a Cohesive HPFC (Franklin, MA), as described (Schugar et al., 2017). TMAO-trimethyl-d9 (d9-TMAO) and TMA-d9 (d9-TMA) were used as internal standards, and TMAO and TMA standards and internal standards were spiked into control sera to prepare calibration curves. Stable isotope dilution liquid chromatography with tandem mass spectrometry (LC/MS-MS) was used to quantify circulating trimethylamine N-oxidc (TMAO) in positive ion multiple reaction monitoring (MRM) mode using characteristic parent to daughter ion transitions: m/z 76.00 → 59.10 for TMAO and m/z 85.00 → 66.25 for D9-TMAO as described previously described with slight modifications (Wang et al., 2011). Briefly, proteins from plasma samples (10 μL) were precipitated with ice cold methanol containing D9-TMAO as internal standard. After vortexing and centrifugation, supernatants were injected on a Luna silica column (2.0 × 150 mm, 5 μm; Phenomenex, Torrance, CA, USA) at a flow rate of 0.35 ml/min composed of solvent A, 0.1% propionic acid in water, and solvent B, 0.1% acetic acid in methanol, using a Shimadzu Nexera Ultra High Performance Liquid Chromatograph system interfaced with Shimadzu 8050 Triple Quadrupole Mass Spectrometer (Shimadzu, Kyoto, Japan). Liquid chromatography gradient starting from 100% A over 1 min, then to 10% B over 4 min, then to 50% B over 4 min, and then to 100% B over 0.5 min, followed by 100% B and A washing for 3 min was used to resolve analytes. Spectra were continuously acquired after the initial 2 min.
LC-MS global metabolomics of mouse cecal samples
From the LTR experiment, 16 P23 samples (6 C, 5 1P, and 5 CMT) were subjected to LCMS analysis to detect and identify putative metabolite structure and assess their relative quantities, at the NYU Metabolomic core, as described (Jones et al., 2014). A MilliporeTM ZIC (1.0 ×l50 mm, 3.5 μm), an LC column, was coupled to a Dionex Ultimate 3000TM system at 25°C for the gradient elution. With a 1 μL injection volume, the flow rate was 75 μL/min with 10 mM ammonium formate in water (A), or acetonitrile (B). The gradient profile was: 85-85%B (0-5 min), 85-77.5%B (5-5.1 min), 77.5-77.5%B (5.1-7 min), 77.5-70%B (7-8.5 min), 70-70%B (8.5-14 min), 70-5%B (14-15min), 5-5%B (15-17 min), 5-85%B (17-18 min), 85-85%B (18-24 min). MS analyses coupled the LC system to a Thermo Q Exactive HFTM mass spectrometer operating in heated electrospray ionization mode (HESI). Method duration was 20 min with a polarity switching data-dependent Top 5 method for both positive and negative modes. Spray voltage for both positive and negative modes was 3.5kV and capillary temperature was set to 320°C with a sheath gas rate of 35, aux gas of 10, and max spray current of 100 μA. The full MS scan for both polarities utilized 120,000 resolution with an AGC target of 3e6 and a maximum IT of 100 ms, with scan range from 110-1000 m/z. Tandem MS spectra for both positive and negative mode used a resolution of 15,000, AGC target of 1e5, maximum IT of 50 ms, isolation window of 0.4 m/z, isolation offset of 0.1 m/z, fixed first mass of 50 m/z, and 3-way multiplexed normalized collision energies (nCE) of 10, 35, 80. The minimum AGC target was 1e4 with an intensity threshold of 2e5. All data were acquired in profile mode.
Measurement of fecal IgA
Mouse fecal samples were resuspended in PBS at a concentration of 50 mg/mL by extensive vortexing, allowed to stand for 20 min on ice, and centrifuged at 16,000 g for 10 min to collect supernatant, as described (Haneberg et al., 1994; Ruiz et al., 2017). Each supernatant was assessed for IgA using the Mouse IgA ELISA Kit (Bethyl, Montgomery TX) with suitable dilutions, according to the manufacturer’s instructions, and absorbance was measured at 450 nm using the SpectraMax iD3 Multi-Mode Microplate Reader (Molecular Devices, San Jose CA).
Measurement of fecal and cecal trypsin activity
Mouse cecal or fecal content trypsin activity were quantitated based on colorimetric-based assay (Norin et al., 1986) (Abcam, Cambridge UK) following the manufacturer’s instructions. Briefly, 10 mg mouse cecal or fecal content samples were diluted in 100 μL trypsin assay buffer and homogenized. Homogenized samples were centrifuged for 5 min at 4°C, at −22,000 g to remove insoluble materials and supernatants collected. Absorbance was measured immediately at 405 nm using Dynex Mrx Revelation micro-plate reader (Chantilly VA). The values of active trypsin of each sample were obtained by the standard curves generated in the same assay according to the kit protocol. Using an in-lab trypsin assay kit, fecal samples (5mg) were homogenized in 100 μL 0.1M Tris buffer (pH 8.2), and supernatant (50 μL) of each sample homogenate was incubated with 1ul of chymotrypsin inhibitor (TPCK, Sigma-Aldrich) at 25 C for 10 min, then 10 μL 0.003 M BAPNA (N-benzoyl-DL-argine-4 nitroanilide hydrochloride) was added to each sample well. After incubation at 25 °C for 10 min, 405 nm absorbance was measured using SpectraMax iD3 Multi-Mode Microplate Reader (Molecular Devices, San Jose CA) and compared with standard curves from titration series of bovine pancreatic trypsin (Sigma-Aldrich).
RNA-Seq
Total RNA of the ileal tissues was extracted from mouse tissues using the PureLink™ RNA Mini Kit (Life Technologies, Eugene OR), and contaminating genomic DNA removed by treatment with DNase I (Qiagen). Total RNA quality and quantity were determined using the NanoDrop ND-1000 UV-Vis Spectrophotometer (NanoDrop Technologies, Inc., Wilmington DE), and Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara CA). RNA-Seq was performed using the Illumina Hiseq 250 at the New York University Langone Genome Technology Center, as described (Ruiz et al., 2017). Reads were aligned to the mouse GENCODE GRCm38.p5 (M14 release) genome with STAR v2.5.2b (Dobin et al., 2013), and were mapped against annotated genes with the read summarization program FeatureCounts (Liao et al., 2014). Differential expression analysis and KEGG pathways visualization was performed using DESeq2 in the R-package (Love et al., 2014). Unsupervised hierarchical clustering was generated using the Seq-N-Slide sequencing data analysis pipeline (https://github.com/igordot/sns).
NanoString analysis for immune genes and microRNA
For NanoString analysis, total RNA of ileum tissue and total RNA of FACS obtained small intestinal epithelial cells was extracted with QIAgen miRNeasy mini Kit (QIAgen) according to manufacturer’s instructions. The expression profile of total 547 immune genes for each RNA samples was evaluated using the nCounter XT mouse immunology kit with XT_PGX_MmV1_Immunology Code Set (NanoString Technologies, Seattle WA) at Rutgers Cancer Institute of New Jersey Immune Monitoring Core Facility. Counts were first filtered away no detected genes based on internal negative controls, and normalized according to manufacturer’s instructions, and Spearman’s rank correlation Principal component analysis were performed based on 414 expression-detected genes using statistical software XLSTAT data analytics package (Addinsoft, New York NY), and heatmap was generated based on the normalized counts of 65 significantly differentiated immune genes between each other groups (p<0.05) using statistical software Heatmapper (Babicki et al., 2016). The expression profile of 599 mouse miRNAs for each RNA sample was evaluated using the nCounter Mouse v1.5 miRNA Expression Assay kit (NanoString Technologies) at the New York University Langone Genome Technology Core. Counts were normalized according to manufacturer’s instructions, Heat maps were generated using the pheatmap package in R (Kolde and Vilo, 2015).
RT-qPCR for host target gene expression
To evaluate specific gene expression in mouse ileum, colon, and ileum epithelial cells, cDNA was synthesized from total RNA samples above with the Verso cDNA Synthesis kit (Thermo Scientific, Waltham MA) according to the manufacturer’s instructions. qPCR was run in a LightCycler 480 system (Roche, Branchburg NJ) using 10 ng synthesized cDNA, target gene-specific primer pairs based on Primerbank (Wang and Seed, 2003) (Table S8) and QuantiTect SYBR® Green PCR Kit (Qiagen). Target mRNA was normalized to 18S rRNA or housekeeping gene HPRT as an internal control in each sample. For group mean comparisons, the Mann-Whitney U test was performed with p-value <0.05 indicating significance.
Analysis of alternative splicing patterns
To investigate how the alternative splicing patterns in host cells are affected by PAT and CMT, we used the percent spliced-in index (PSI) (Venables et al., 2008) to quantify the relative abundance of the alternative exons, and compared the PSIs of the same exons in the three experimental groups. The PSI of an exon is defined based on the ratio between RNAseq reads including or excluding the exon:
where IR and ER denote the normalized RNA-seq read counts including and excluding the exon, respectively. These two terms are defined as
We performed transcriptome-wide quantification of PSI values following an established protocol (Schafer et al., 2015). The reads were mapped to the reference genome GRCm38 using the aligner STAR version 2.5.2a. The candidate alternative exons were identified from the GENCODE transcriptome annotation M10 (Frankish et al., 2019).To identify the exons with differential PSIs in the three groups, we first filtered the exons having constant PSIs or <5 mapped reads. Then, we performed ANOVA to identify the statistically significant exons with different mean PSIs among the three groups and Welch’s t-test to compare between each two groups among the three groups. We also performed a principal component analysis (PCA) to investigate the relative similarity of the RNAseq samples based on the PSIs of the identified exons.
Nano-liquid chromatography and electrospray ionization tandem mass spectrometry of global histone PTMs
Histone extracts were prepared from flash-frozen post-mortem ileum and liver from P23 and P42 mice (n=5 biological replicates per treatment and timepoint) and prepared for analysis by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) as described (Krautkramer et al., 2016). For each sample, 0.3 μg of derivatized histone peptides was injected onto Thermo EASY-nLC 1200 with a reverse-phase column packed in-house (Reprosil-Pur C18-AQ 3 μM particles C18 (Dr. Maisch GmbH, Ammerbuch, Germany), 150 mm x 0.075 mm inner diameter (silica tubing from New Objective, Woburn MA)). Mobile phase consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). Samples were resolved over the following gradient: 2-30% B over 35 min, 30-80% B over 20 min, followed by 10 min at 80% B and equilibration to 2% B between each injection. Data were acquired in DIA mode on a Thermo Q Exactive HF-X operating in positive polarity. Full MS1 scans were obtained at resolution 60,000; AGC target 1x106; and maximum fill time 25ms, and a scan range of 390-910 m/z. DIA scans were obtained with the following settings: 10m/z non-overlapping isolation windows; resolution 30,000; AGC target 1x106; maximum fill time 60 ms; loop count 15; and stepped normalized collision energy at 30, 33, and 35. Raw DIA data were imported into Skyline (MacLean et al., 2010; Pino et al., 2020). Daily v. 20.1.1.158 and matched to an in-house spectral library and quantified as described (Krautkramer et al., 2016; Krautkramer et al., 2015). Data are reported as a percentage of the peptide family total, where a peptide family is defined as a group of peptides spanning the same residues within a histone protein, (e.g.: Histone H3 residues 18-26 (KQLATKAAR), which contains lysine residues 18 and 23, is a peptide family comprised of the following members: K18ac+K23un, K18un+K23ac, K18ac+K23ac, K18me1+K23un, K18un+K23me1, and K18un+K23un). Isobaric and co-eluting peptides were not deconvoluted and are denoted accordingly (a single abundance value for K18ac K23un and K18un K23ac is labeled as K18ac/K23ac and histone H4 peptides containing 1-3 acetylated lysines are denoted as H4 1ac, H4 2ac, and H4 3ac). Exported peak values from Skyline were normalized within peptide families to the total area prior to calculation of fold changes and statistical analyses. Statistically significant differences were defined by p<0.05 by one-way ANOVA with a Tukey’s HSD post-test.
Multi-omic analysis
Using MMvec (Morton et al., 2019), we aimed to detect co-occurence associations between taxa in the gut microbiome and host intestinal gene expression profiles. To bolster the number of samples with paired profiling available, we combined samples from the current study with our previous study (Zhang et al., 2018). To mitigate batch effects, we subset to only taxa and genes observed in both studies. First, we used DEICODE to examine if there were batch effects between the two studies. DEICODE robust Aitchison PCA was used to examine differences in β-diversity using compositionally robust methods (Bolyen et al., 2019; Martino et al., 2019; Vazquez-Baeza et al., 2017; Vazquez-Baeza et al., 2013). Then a total of 1336 host intestinal pathways in male NOD mice from the C, 1P and CMT in the current and previous studies (Zhang et al., 2018) identified by RNAseq and 188 fecal taxa of these mice as identified by 16S sequencing were applied into a co-occurrence model which outputs rankings using a neural network (Morton et al., 2019). For each taxon, the top 10 pathways according to rank were selected as edges in the network. The assembled network was visualized using a force-directed layout in Cytoscape (Shannon et al., 2003).
Assessment of mitochondrial DNA relative abundance with qPCR
Total DNA with nuclear DNA and mitochondria DNA was extracted from quick-frozen P12 NOD mouse ileum tissue samples using the DNeasy Blood & Tissue Kit (Qiagen, Valencia CA). To evaluate mitochondria relative abundance in cells, mitochondria DNA (MtDNA) was quantified by qPCR using mouse mtDNA-specific primer pairs as described (Malik et al., 2016), and 18S nuclear DNA quantified by qPCR using mouse 18S specific primer pairs (Kuchipudi et al., 2012) to provide a cell nuclear reference. The ratio of MtDNA/18S nuclear DNA of control group and antibiotic group (n=12 samples per group) was used to calculate the differential significance of relative mitochondrial abundances between control and antibiotic-treated mice.
Co-culture assay
Fresh Caco-2 cells were seeded with high density to 6-well tissue culture plates (Corning) in MEM medium with 10% FBS without antibiotics (2 ml per well), cultured to >90% confluence, then co-cultured with each of following materials per treatment [200 μl of 0.22 μm-filtered cell-free supernatant components from PBS (10 ml)-suspended pooled cecal contents (500 mg) of C or 1P NOD mice at P23; 200 μl of freshly cultured and PBS-prewashed bacterial cells (2.5×108 cells/ml) of S24-7 family bacterial MH8C strain (S24-7) (gift from the Carolina Tropini Lab) or E. coli K12 strain (E. coli), or 200 ul of propionic acid (PA) (10 mM in PBS) or PBS as reference control (PBS)] for 8 h at 37C in 5% CO2 conditions. The attached cells were washed with ice-cold PBS and subjected to TRIzol reagent (Qiagen). RNA were extracted, cDNA were synthesized, RT-qPCR were performed as above to evaluate expression of the host genes, CD44, REG3G, and NFkB genes using primer pairs for Homo sapiens gene sequences in Primerbank (Wang and Seed, 2003) and using 18S rRNA as reference using Homo sapiens 18S primer pairs (Schmittgen and Zakrajsek, 2000) (Table S8).
Immunoblot analysis
Allprotect Tissue Reagent (Qiagen)-stabilized mouse tissues of control and mouse were lysed by sonication in RIPA lysis and extraction buffer (Thermo Scientific Pierce, Rockford IL) with protease inhibitor cocktail (Thermo Scientific). Then lysates was separated by SDS-PAGE (Expedeon Inc. San Diego CA), transferred to PVDF membrane (Thermo Scientific). Membranes were blocked in TPBT with 3% BSA for 1 h at room temperature and incubated with primary antibodies, rabbit anti-Reg3γ (Abcam, ab198216) or mouse anti-β-Actin (Sigma, A2228) according to instructions of the manufacturer. Immunodetection of targets were performed using horseradish peroxidase–conjugated anti-rabbit or anti-mouse polyvalent sheep immunoglobulin secondary antibodies and using chemiluminescence reagents, SuperSignal West Femto Chemiluminescent Substrate (Thermo Scientific) or Amersham ECL Western Blotting Detection Reagents (GE Healthcare, Piscataway NJ) in accordance with the manufacturers’ instructions.
Immunohistochemistry
Mouse ileum tissue were collected during, fixed in 10% neutral buffered formalin for 24 h, and processed into paraffin blocks according to standard procedures by the Rutgers Cancer Institute of New Jersey Biospecimen Repository and Histopathology Service Shared Resource. The Immunohistochemistry was performed on the Discovery XT immunostainer. The DAB kit was automatically dispensed according to the general protocol. Rabbit anti-Reg3γ (Abcam, ab198216) was used as primary, and secondary antibodies were manually dispensed accordingly. Counterstain for the procedures was Hematoxylin. Specimens were digitized at 20x at Rutgers Cancer Institute of New Jersey Biomedical Informatics shared resource using an Olympus VS 120 whole slide scanner (Olympus Corporation of the Americas, Center Valley, PA). Image analysis protocol was custom developed on Visiopharm image analysis platform (Visiopharm A/S, Hoersholm, Denmark) to identify specifically stained cells/particles and compute its area burden on affected tissue.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis were done by using GraphPad Prism 8.0 (GraphPad Software), QIIME2.0 (Bolyen et al., 2019), and R package (R Core Team, 2017). Mann-Whitney U test was performed using a 95% confidence interval to determine difference among different groups, for quantifying microbiome α-diversity Faith’s PD (Figure1B), relative expression of genes (Figure1E, 4D), relative abundances of specific taxa (Figure 3AB), metabolites (Figure 4A), and normalized abundance of miRNA (Figure 5C) with “n” representing mouse number, and for quantifying co-culture cell gene expression (Figure 6B) with “n” representing independent cell cultures. Statistical details were indicated in the Figure legends. Welch’s t-test was performed using a 95% confidence interval to determine difference among different groups for quantifying differential splicing rates of exons (Figure 4D), with “n” representing mouse number. Statistical details were indicated in the Figure legend. One-way-ANOVA for multiple comparisons were performed to determine difference among different mouse groups for α-diversity overtime measuring Faith’s PD evenness (Figure 2C upper panel). One-way-ANOVA with Tukey’s HSD post-test for correction for multiple comparisons were performed to determine difference among different mouse groups for β-diversity analysis of cecal and fecal microbiome (Figure 1C, 2D), α-diversity over time measuring Pielou evenness (Figure 2C lower panel), histone acetylation and methylation (Figure 5A), with “n” representing mouse number. Statistical details were indicated in the Figure legends. For Kaplan-Meier analysis of T1D incidence (Figure 2B), statistical analysis was performed by the Gρ,γ weighted log rank test, and post-hoc pairwise comparisons were adjusted by a Benjamini-Hochberg procedure. For different taxa analysis (Figure 2E), the ANCOM program (Mandal et al., 2015) within QIIME2.0 (Bolyen et al., 2019) was used. For Differentially abundant pathways analysis (Figure 3D), MaAsLin2 (Mallick et al., 2021) with FDR correction was used with features corrected q-value <0.1 were identified as significant. Principle component analysis (PCA) were calculated using Bray-Curtis measurements for metagenomics pathway distribution (Figure 3C) and global RNA PSI distribution (Figure 4C). Statistical details were indicated in the Figure legends and Star Methods. DEICODE robust Aitchison PCA using compositionally robust methods (Bolyen et al., 2019; Martino et al., 2019; Vazquez-Baeza et al., 2017; Vazquez-Baeza et al., 2013) for multi-omic analysis (Figure 6A). Statistical details were indicated in the Figure legends and Star Methods.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Reg3γ | Abcam | Cat # ab198216 |
| anti-β-Actin | Sigma | Cat # A2228 |
| anti-CD45-FITC | ThermoFisher Scientific | Cat # 11-9459-42 |
| anti-EpCAM-APC | ThermoFisher Scientific | Cat # 17-5791-80 |
| anti-CD16/CD32 | ThermoFisher Scientific | Cat # MA5-18012 |
| Bacterial strains | ||
| S24-7 | C. Tropini Lab | Strain MH8C |
| E. coli | Blaser Lab | Strain K12 |
| Biological samples | ||
| Fecal samples (mouse) | This study | N/A |
| Cecal content samples (mouse) | This study | N/A |
| Pancreatic tissue (mouse) | This study | N/A |
| Ileal tissue (mouse) | This study | N/A |
| Colonic tissue (mouse) | This study | N/A |
| Liver tissue (mouse) | This study | N/A |
| Serum (mouse) | This study | N/A |
| Ileal epithelial cells (mouse) | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Tylosin tartrate | Sigma-Aldrich | Cat # T6271 |
| Critical commercial assays | ||
| DNeasy PowerSoil-HTP 96 Well Soil DNA Isolation Kit | Qiagen | Cat # 12955-4 |
| QIAamp Powerfecal DNA Kit | Qiagen | Cat # 51804 |
| Qiaquick PCR purification kit | Qiagen | Cat # 28104 |
| miRNeasy mini Kit | Qiagen | Cat # 1038703 |
| QuantiTect SYBR® Green PCR Kit | Qiagen | Cat # 204145 |
| Quant-iT PicoGreen dsDNA assay kit | Life Technologies | Cat # P11496 |
| PureLink™ RNA Mini Kit | Life Technologies | Cat # 12183025 |
| Verso cDNA Synthesis kit | ThermoFisher Scientific | Cat # AB1453B |
| TruSeq RNA Sample Preparation Kit v2 | Illumina | Cat # RS-122-2001 |
| nCounter Mouse v1.5 miRNA Expression Assay kit | NanoString Technologies | NS_M_MIR_V1.5 |
| nCounter XT mouse immunology kit | NanoString Technologies | XT_PGX_MmV1 |
| Deposited data | ||
| RNA-Seq data | This study | ArrayExpress database: accession codes E-MTAB-9981 & E-MTAB-9982 |
| NanoString data | This study | NCBI Gene Expression Omnibus: accession codes GSE163887 & GSE163888 |
| 16S rRNA data | This study | QIITA: identifier 13529 |
| Shotgun metagenomics data | This study | European Nucleotide Archive (ENA): accession number PRJEB42282 |
| Mouse genome | The Ensembl Project |
http://useast.ensembl.org/Mus_musculus/Info/Index GRCm38.p5 |
| Human genome | The Ensembl Project |
http://useast.ensembl.org/Homo_sapiens/Info/Index GRCh38.p13 |
| Silva | The SILVA rRNA database project |
https://www.arb-silva.de/ Release of December 2019 RRID:SCR_006423 |
| GENCODE transcriptome annotation | The Gencode Project |
https://www.gencodegenes.org/mouse Release M10 (GRCm38.p4) RRID:SCR_014966 |
| Experimental models: Organisms | ||
| Mouse | Jackson Laboratories | NOD/ShiLtJ RRID:IMSR_JAX:001976 |
| Experimental models: Cell lines | ||
| Caco-2 | ATCC | HTB-37™ RRID:CVCL_0025 |
| Oligonucleotides | ||
| Bacterial Universal 16S primer pair 338F/518R in Table S8 | Muyzer et al., 1993 | NA |
| MtDNA (Mus musculus) primer pair in Table S8 | Malik et al., 2016 | NA |
| 18S (Mus musculus) primer pair in Table S8 | Livanos et al., 2016 | NA |
| 18S (Homo sapiens) primer pair in Table S8 | Schmittgen and Zakrajsek, 2000 | NA |
| Primers pairs targeting different genes in Table S8 | Primerbank Wang and Seed, 2003 |
https://pga.mgh.harvard.edu/primerbank/ |
| Software and algorithms | ||
| STAR | Dobin et al., 2013 | v2.5.2b RRID:SCR_004463 |
| featureCounts | Liao et al., 2014 |
http://subread.sourceforge.net/ v1.6.3 |
| DESeq2 package | Love et al., 2014 |
https://bioconductor.org/packages/release/bioc/html/DESeq2.html v1.22.2 RRID:SCR_015687 |
| Seq-N-Slide | The Ryoo Lab | https://github.com/igordot/sns |
| QIIME2 | Bolyen et al., 2019 |
https://qiime2.org/ v2020.8 |
| MAFFT | Katoh and Standley, 2013 |
https://mafft.cbrc.jp/alignment/software/ v7 RRID:SCR_011811 |
| DADA2 | Callahan et al., 2016 |
https://benjjneb.github.io/dada2/ v1.16 |
| ANCOM | Mandal et al., 2015 |
https://github.com/sidhujyatha/ANCOM v2.0 |
| MaAsLin2 | Mallick et al. 2021 | https://huttenhower.sph.harvard.edu/maaslin/ |
| Kneaddata | The Huttenhower Lab |
https://huttenhower.sph.harvard.edu/kneaddata/ v0.6.1 |
| HuMAnN2 | Franzosa et al., 2018 |
https://huttenhower.sph.harvard.edu/humann2/ v0.11.1 |
| Ingenuity Pathway Analysis (Qiagen IPA) | Qiagen |
https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ v01-14 RRID:SCR_008653 |
| Cytoscape | National Institute of General Medical Sciences |
https://cytoscape.org/ v3.7.1 RRID:SCR_003032 |
| MMvec | Morton et al., 2019 |
https://github.com/biocore/mmvec v1.0 |
| Songbird | Morton et al., 2019 |
https://github.com/biocore/songbird v1.0.4 |
| GraphPad Prism | GraphPad Software Inc. | V8 RRID:SCR_002798 |
Highlights:
Cecal microbiota transfer (CMT) rescued antibiotic-induced T1D enhancement in NOD mice
CMT restored potential T1D-protective bacterial taxa
CMT restored global patterns of gene expression and modifications in the ileum
The study reveals a regulatory network of innate immunity sensing intestinal signals
ACKNOWLEDGEMENTS
We thank Tadasu Lizumi, Menghan Liu, Kun Qian, and Drew Jones of New York University Medical Center, Anthony R. Williamson & Marcus Rauch of Janssen Prevention Center London, Gang Fang of Mount Sinai Medical Center, Carolina Tropini laboratory of University of British Columbia, NYU Langone Health Genome Technology Center & Metabolomics Core Resource, Rutgers Cancer Institute Histopathology core facility & Comprehensive Genomics Shared Resource, and Rutgers University Ernest Mario School of Pharmacy Cytometry & Cell Sorting Laboratory assistance with these studies. These studies were supported by Janssen Labs London (15-A0-00-00-0039-29-01), NIH Grants [R01GM128955-01, P01HL14783, 5R01DK110014, R01DK120679], the TransAtlantic Networks of Excellence Program (33.17CVD01) from the Fondation Leducq, the C&D Research Fund, the Emch Fund for Microbial Diversity, and an anonymous donor.
DECLARATION OF INTERESTS
Z.W. & S.L.H. report being named as co-inventors on pending and issued patents held by the Cleveland Clinic relating to cardiovascular diagnostics and therapeutics, and being eligible to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics or therapeutics from Cleveland HeartLab, Quest Diagnostics and Procter & Gamble. S.L.H. also reports, being a paid consultant for Procter & Gamble, having received research funds from Procter & Gamble, and Roche Diagnostics, and being eligible to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics or therapeutics from Cleveland HeartLab, Quest Diagnostics and Procter & Gamble. The other authors report that they have no relationships relevant to the contents of this paper to disclose.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
INCLUSION AND DIVERSITY STATEMENT
One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper self-identifies as living with a disability.
REFERENCES
- Adusumalli S, Ngian ZK, Lin WQ, Benoukraf T, and Ong CT (2019). Increased intron retention is a post-transcriptional signature associated with progressive aging and Alzheimer’s disease. Aging Cell 18, e12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amenyogbe N, Kollmann TR, and Ben-Othman R (2017). Early-Life Host-Microbiome Interphase: The Key Frontier for Immune Development. Front Pediatr 5, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson MA, and Chervonsky A (2012). Does the gut microbiota have a role in type 1 diabetes? Early evidence from humans and animal models of the disease. Diabetologia 55, 2868–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson MA, and Eisenbarth GS (2001). Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet 358, 221–229. [DOI] [PubMed] [Google Scholar]
- Aversa Z, Atkinson EJ, Schafer MJ, Theiler RN, Rocca WA, Blaser MJ, and LeBrasseur NK (2020). Association of Infant Antibiotic Exposure With Childhood Health Outcomes. Mayo Clin Proc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babicki S, Arndt D, Marcu A, Liang Y, Grant JR, Maciejewski A, and Wishart DS (2016). Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res 44, W147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayraktar R, Bertilaccio MTS, and Calin GA (2019). The Interaction Between Two Worlds: MicroRNAs and Toll-Like Receptors. Front Immunol 10, 1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchenough GM, Nystrom EE, Johansson ME, and Hansson GC (2016). A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 352, 1535–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, and Gregory Caporaso J (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 6, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37, 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman KA, Broussard EK, and Surawicz CM (2015). Fecal microbiota transplantation: current clinical efficacy and future prospects. Clin Exp Gastroenterol 8, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K, Godovannyi A, Ma C, Zhang Y, Ahmadi-Vand Z, Dai C, Gorzelak MA, Chan Y, Chan JM, Lochner A, et al. (2016). Prolonged antibiotic treatment induces a diabetogenic intestinal microbiome that accelerates diabetes in NOD mice. ISME J 10, 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, and Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candon S, Perez-Arroyo A, Marquet C, Valette F, Foray AP, Pelletier B, Milani C, Ventura M, Bach JF, and Chatenoud L (2015). Antibiotics in early life alter the gut microbiome and increase disease incidence in a spontaneous mouse model of autoimmune insulin-dependent diabetes. PLoS One 10, e0125448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R, Billington R, Fulcher CA, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Midford PE, Ong Q, Ong WK, et al. (2018). The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res 46, D633–D639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenac N, Garcia-Villar R, Ferrier L, Larauche M, Vergnolle N, Bunnett NW, Coelho AM, Fioramonti J, and Bueno L (2003). Proteinase-activated receptor-2-induced colonic inflammation in mice: possible involvement of afferent neurons, nitric oxide, and paracellular permeability. J Immunol 170, 4296–4300. [DOI] [PubMed] [Google Scholar]
- Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, and Schaffer JE (2001). A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest 107, 813–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs AP, Terlecki-Zaniewicz S, Philpott M, Baardman J, Ahern D, Lindow M, Obad S, Oerum H, Sampey B, Mander PK, et al. (2020). Histone H3K27me3 demethylases regulate human Th17 cell development and effector functions by impacting on metabolism. Proc Natl Acad Sci U S A 117, 6056–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daft JG, and Lorenz RG (2015). Role of the gastrointestinal ecosystem in the development of type 1 diabetes. Pediatr Diabetes 16, 407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, and Natoli G (2007). The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 130, 1083–1094. [DOI] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, and Andersen GL (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72, 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Bello MG, Blaser MJ, Ley RE, and Knight R (2011). Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology 140, 1713–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, and Bradley RK (2015). Widespread intron retention diversifies most cancer transcriptomes. Genome Med 7, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith D (1992). Conservation evaluation and phylogenetic diversity. Biological Conservation 61, 1–10. [Google Scholar]
- Ferrell JM, and Chiang JYL (2019). Understanding Bile Acid Signaling in Diabetes: From Pathophysiology to Therapeutic Targets. Diabetes Metab J 43, 257–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figliuolo VR, Dos Santos LM, Abalo A, Nanini H, Santos A, Brittes NM, Bernardazzi C, de Souza HSP, Vieira LQ, Coutinho-Silva R, et al. (2017). Sulfate-reducing bacteria stimulate gut immune responses and contribute to inflammation in experimental colitis. Life Sci 189, 29–38. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Biagioli M, Zampella A, and Distrutti E (2018). Bile Acids Activated Receptors Regulate Innate Immunity. Front Immunol 9, 1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming T, and Harrington D (1991). Counting Processes and Survival Analysis (John Wiley & Sons; ). [Google Scholar]
- Forestier C, Takaki T, Molano A, Im JS, Baine I, Jerud ES, Illarionov P, Ndonye R, Howell AR, Santamaria P, et al. (2007). Improved outcomes in NOD mice treated with a novel Th2 cytokine-biasing NKT cell activator. J Immunol 178, 1415–1425. [DOI] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47, D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, et al. (2018). Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods 15, 962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Blanco MA, Baraniak AP, and Lasda EL (2004). Alternative splicing in disease and therapy. Nat Biotechnol 22, 535–546. [DOI] [PubMed] [Google Scholar]
- Gensollen T, Iyer SS, Kasper DL, and Blumberg RS (2016). How colonization by microbiota in early life shapes the immune system. Science 352, 539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollwitzer ES, and Marsland BJ (2015). Impact of Early-Life Exposures on Immune Maturation and Susceptibility to Disease. Trends Immunol 36, 684–696. [DOI] [PubMed] [Google Scholar]
- Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, et al. (2014). Human genetics shape the gut microbiome. Cell 159, 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosline SJ, Gurtan AM, JnBaptiste CK, Bosson A, Milani P, Dalin S, Matthews BJ, Yap YS, Sharp PA, and Fraenkel E (2016). Elucidating MicroRNA Regulatory Networks Using Transcriptional, Post-transcriptional, and Histone Modification Measurements. Cell Rep 14, 310–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracz AD, Puthoff BJ, and Magness ST (2012). Identification, isolation, and culture of intestinal epithelial stem cells from murine intestine. Methods Mol Biol 879, 89–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves CL, Harden SW, LaPato M, Nelson M, Amador B, Sorenson H, Frazier CJ, and Wallet SM (2014). A method for high purity intestinal epithelial cell culture from adult human and murine tissues for the investigation of innate immune function. J Immunol Methods 414, 20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Chen Y, Hu X, Qian G, Ge S, and Zhang J (2013). The regulation of Toll-like receptor 2 by miR-143 suppresses the invasion and migration of a subset of human colorectal carcinoma cells. Mol Cancer 12, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, Burgin G, Delorey TM, Howitt MR, Katz Y, et al. (2017). A single-cell survey of the small intestinal epithelium. Nature 551, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haneberg B, Kendall D, Amerongen HM, Apter FM, Kraehenbuhl JP, and Neutra MR (1994). Induction of specific immunoglobulin A in the small intestine, colon-rectum, and vagina measured by a new method for collection of secretions from local mucosal surfaces. Infect Immun 62, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington D, and Fleming T (1982). A class of rank test procedures for censored survival data. Biometrika 69, 553–566. [Google Scholar]
- Hasler R, Sheibani-Tezerji R, Sinha A, Barann M, Rehman A, Esser D, Aden K, Knecht C, Brandt B, Nikolaus S, et al. (2017). Uncoupling of mucosal gene regulation, mRNA splicing and adherent microbiota signatures in inflammatory bowel disease. Gut 66, 2087–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Wu C, Li P, Li N, Zhang D, Zhu Q, Ren W, and Peng Y (2018). Functions and Signaling Pathways of Amino Acids in Intestinal Inflammation. Biomed Res Int 2018, 9171905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosomi K, Kiyono H, and Kunisawa J (2019). Fatty acid metabolism in the host and commensal bacteria for the control of intestinal immune responses and diseases. Gut Microbes, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Peng J, Tai N, Hu C, Zhang X, Wong FS, and Wen L (2015). Maternal Antibiotic Treatment Protects Offspring from Diabetes Development in Nonobese Diabetic Mice by Generation of Tolerogenic APCs. J Immunol 195, 4176–4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Wong FS, and Wen L (2017). Antibiotics, gut microbiota, environment in early life and type 1 diabetes. Pharmacol Res 119, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DR, Wu Z, Chauhan D, Anderson KC, and Peng J (2014). A nano ultra-performance liquid chromatography-high resolution mass spectrometry approach for global metabolomic profiling and case study on drug-resistant multiple myeloma. Anal Chem 86, 3667–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan E, and Meier P (1958). Nonparametric estimation from incomplete observations. J Am Statist Assoc 53, 457–481. [Google Scholar]
- Karlic R, Chung HR, Lasserre J, Vlahovicek K, and Vingron M (2010). Histone modification levels are predictive for gene expression. Proc Natl Acad Sci U S A 107, 2926–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, and Standley DM (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawana H, Karaki H, Higashi M, Miyazaki M, Hilberg F, Kitagawa M, and Harigaya K (2008). CD44 suppresses TLR-mediated inflammation. J Immunol 180, 4235–4245. [DOI] [PubMed] [Google Scholar]
- Kellermayer R (2019). Fecal microbiota transplantation: great potential with many challenges. Transl Gastroenterol Hepatol 4, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly CR, Kahn S, Kashyap P, Laine L, Rubin D, Atreja A, Moore T, and Wu G (2015). Update on Fecal Microbiota Transplantation 2015: Indications, Methodologies, Mechanisms, and Outlook. Gastroenterology 149, 223–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemppainen KM, Vehik K, Lynch KF, Larsson HE, Canepa RJ, Simell V, Koletzko S, Liu E, Simell OG, Toppari J, et al. (2017). Association between early-life antibiotic use and the risk of islet or celiac disease autoimmunity. JAMA Pediatr 171, 1217–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I, Miyamoto J, Ohue-Kitano R, Watanabe K, Yamada T, Onuki M, Aoki R, Isobe Y, Kashihara D, Inoue D, et al. (2020). Maternal gut microbiota in pregnancy influences offspring metabolic phenotype in mice. Science 367. [DOI] [PubMed] [Google Scholar]
- Koch L (2017). Alternative splicing: A thermometer controlling gene expression. Nat Rev Genet 18, 515. [DOI] [PubMed] [Google Scholar]
- Kolde R, and Vilo J (2015). GOsummaries: an R package for visual functional annotation of experimental data. F1000Res 4, 574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konikoff T, and Gophna U (2016). Oscillospira: a Central, Enigmatic Component of the Human Gut Microbiota. Trends Microbiol 24, 523–524. [DOI] [PubMed] [Google Scholar]
- Korpela K, Helve O, Kolho KL, Saisto T, Skogberg K, Dikareva E, Stefanovic V, Salonen A, Andersson S, and de Vos WM (2020). Maternal Fecal Microbiota Transplantation in Cesarean-Born Infants Rapidly Restores Normal Gut Microbial Development: A Proof-of-Concept Study. Cell 183, 324–334 e325. [DOI] [PubMed] [Google Scholar]
- Krautkramer KA, Kreznar JH, Romano KA, Vivas EI, Barrett-Wilt GA, Rabaglia ME, Keller MP, Attie AD, Rey FE, and Denu JM (2016). Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol Cell 64, 982–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krautkramer KA, Reiter L, Denu JM, and Dowell JA (2015). Quantification of SAHA-dependent changes in histone modifications using data-independent acquisition mass spectrometry. J Proteome Res 14, 3252–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchipudi SV, Tellabati M, Nelli RK, White GA, Perez BB, Sebastian S, Slomka MJ, Brookes SM, Brown IH, Dunham SP, et al. (2012). 18S rRNA is a reliable normalisation gene for real time PCR based on influenza virus infected cells. Virol J 9, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langdon A, Crook N, and Dantas G (2016). The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med 8, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiter EH (2001). The NOD mouse: a model for insulin-dependent diabetes mellitus. Curr Protoc Immunol Chapter 15, Unit 15 19. [DOI] [PubMed] [Google Scholar]
- Li WV, Li S, Tong X, Deng L, Shi H, and Li JJ (2019). AIDE: annotation-assisted isoform discovery with high precision. Genome Res 29, 2056–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lieber AD, Beier UH, Xiao H, Wilkins BJ, Jiao J, Li XS, Schugar RC, Strauch CM, Wang Z, Brown JM, et al. (2019). Loss of HDAC6 alters gut microbiota and worsens obesity. FASEB J 33, 1098–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livanos AE, Greiner TU, Vangay P, Pathmasiri W, Stewart D, McRitchie S, Li H, Chung J, Sohn J, Kim S, et al. (2016). Antibiotic-mediated gut microbiome perturbation accelerates development of type 1 diabetes in mice. Nat Microbiol 1, 16140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, and Knight R (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71, 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik AN, Czajka A, and Cunningham P (2016). Accurate quantification of mouse mitochondrial DNA without co-amplification of nuclear mitochondrial insertion sequences. Mitochondrion 29, 59–64. [DOI] [PubMed] [Google Scholar]
- Malikowski T, Khanna S, and Pardi DS (2017). Fecal microbiota transplantation for gastrointestinal disorders. Curr Opin Gastroenterol 33, 8–13. [DOI] [PubMed] [Google Scholar]
- Mallick H, Rahnavard P, McIver LJ, Ma S, Zhang Y, Nguyen LH, Tickle TL, Weingart G, Ren B, Schwager EH, et al. (2021). Multivariable Association Discovery in Population-scale Meta-omics Studies. bioRxiv URL https://huttenhower.sph.harvard.edu/maaslin/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal S, Van Treuren W, White RA, Eggesbo M, Knight R, and Peddada SD (2015). Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis 26, 27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino C, Morton JT, Marotz CA, Thompson LR, Tripathi A, Knight R, and Zengler K (2019). A Novel Sparse Compositional Technique Reveals Microbial Perturbations. mSystems 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay LI, and Cidlowski JA (1999). Molecular control of immune/inflammatory responses: interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr Rev 20, 435–459. [DOI] [PubMed] [Google Scholar]
- McKenna LB, Schug J, Vourekas A, McKenna JB, Bramswig NC, Friedman JR, and Kaestner KH (2010). MicroRNAs control intestinal epithelial differentiation, architecture, and barrier function. Gastroenterology 139, 1654–1664, 1664, e1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao F, Chen Z, Genuth S, Paterson A, Zhang L, Wu X, Li SM, Cleary P, Riggs A, Harlan DM, et al. (2014). Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes 63, 1748–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midtvedt T, Zabarovsky E, Norin E, Bark J, Gizatullin R, Kashuba V, Ljungqvist O, Zabarovska V, Mollby R, and Ernberg I (2013). Increase of faecal tryptic activity relates to changes in the intestinal microbiome: analysis of Crohn’s disease with a multidisciplinary platform. PLoS One 8, e66074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton JT, Aksenov AA, Nothias LF, Foulds JR, Quinn RA, Badri MH, Swenson TL, Van Goethem MW, Northen TR, Vazquez-Baeza Y, et al. (2019). Learning representations of microbe-metabolite interactions. Nat Methods 16, 1306–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller NT, Bakacs E, Combellick J, Grigoryan Z, and Dominguez-Bello MG (2015). The infant microbiome development: mom matters. Trends Mol Med 21, 109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, and Hooper LV (2015). Antimicrobial defense of the intestine. Immunity 42, 28–39. [DOI] [PubMed] [Google Scholar]
- Mullaney JA, Stephens JE, Geeling BE, and Hamilton-Williams EE (2019). Early-life exposure to gut microbiota from disease-protected mice does not impact disease outcome in type 1 diabetes susceptible NOD mice. Immunol Cell Biol 97, 97–103. [DOI] [PubMed] [Google Scholar]
- Muyzer G, de Waal EC, and Uitterlinden AG (1993). Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59, 695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobel YR, Cox LM, Kirigin FF, Bokulich NA, Yamanishi S, Teitler I, Chung J, Sohn J, Barber CM, Goldfarb DS, et al. (2015). Metabolic and metagenomic outcomes from early-life pulsed antibiotic treatment. Nat Commun 6, 7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norin KE, Gustafsson B, and Midtvedt T (1986). Strain differences in faecal tryptic activity of germ-free and conventional rats. Laboratory animals 20, 67–69. [DOI] [PubMed] [Google Scholar]
- O’Neill LA, Sheedy FJ, and McCoy CE (2011). MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol 11, 163–175. [DOI] [PubMed] [Google Scholar]
- Olin A, Henckel E, Chen Y, Lakshmikanth T, Pou C, Mikes J, Gustafsson A, Bernhardsson AK, Zhang C, Bohlin K, et al. (2018). Stereotypic Immune System Development in Newborn Children. Cell 174, 1277–1292 e1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira LM, Teixeira FME, and Sato MN (2018). Impact of Retinoic Acid on Immune Cells and Inflammatory Diseases. Mediators Inflamm 2018, 3067126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortqvist AK, Lundholm C, Halfvarson J, Ludvigsson JF, and Almqvist C (2019). Fetal and early life antibiotics exposure and very early onset inflammatory bowel disease: a population-based study. Gut 68, 218–225. [DOI] [PubMed] [Google Scholar]
- Pagliuca A, Valvo C, Fabrizi E, di Martino S, Biffoni M, Runci D, Forte S, De Maria R, and Ricci-Vitiani L (2013). Analysis of the combined action of miR-143 and miR-145 on oncogenic pathways in colorectal cancer cells reveals a coordinate program of gene repression. Oncogene 32, 4806–4813. [DOI] [PubMed] [Google Scholar]
- Pathak R, Enuh HA, Patel A, and Wickremesinghe P (2013). Treatment of relapsing Clostridium difficile infection using fecal microbiota transplantation. Clin Exp Gastroenterol 7, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi E (2016). Microbiome. The right gut microbes help infants grow. Science 351, 802. [DOI] [PubMed] [Google Scholar]
- Pino LK, Searle BC, Bollinger JG, Nunn B, MacLean B, and MacCoss MJ (2020). The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom Rev 39, 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pino SC, Kruger AJ, and Bortell R (2010). The role of innate immune pathways in type 1 diabetes pathogenesis. Curr Opin Endocrinol Diabetes Obes 17, 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pott J, and Hornef M (2012). Innate immune signalling at the intestinal epithelium in homeostasis and disease. EMBO Rep 13, 684–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP (2010). FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochazka L, Tesarik R, and Turanek J (2014). Regulation of alternative splicing of CD44 in cancer. Cell Signal 26, 2234–2239. [DOI] [PubMed] [Google Scholar]
- Pure E, and Cuff CA (2001). A crucial role for CD44 in inflammation. Trends Mol Med 7, 213–221. [DOI] [PubMed] [Google Scholar]
- Qadri M, Almadani S, Jay GD, and Elsaid KA (2018). Role of CD44 in Regulating TLR2 Activation of Human Macrophages and Downstream Expression of Proinflammatory Cytokines. J Immunol 200, 758–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2017). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: URL https://wwwR-projectorg/. [Google Scholar]
- Ruiz VE, Battaglia T, Kurtz ZD, Bijnens L, Ou A, Engstrand I, Zheng X, Iizumi T, Mullins BJ, Muller CL, et al. (2017). A single early-in-life macrolide course has lasting effects on murine microbial network topology and immunity. Nat Commun 8, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruth MR, and Field CJ (2013). The immune modifying effects of amino acids on gut-associated lymphoid tissue. J Anim Sci Biotechnol 4, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer S, Miao K, Benson CC, Heinig M, Cook SA, and Hubner N (2015). Alternative Splicing Signatures in RNA-seq Data: Percent Spliced in (PSI). Curr Protoc Hum Genet 87, 11 16 11–11 16 14. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, and Zakrajsek BA (2000). Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys Methods 46, 69–81. [DOI] [PubMed] [Google Scholar]
- Schmitz U, Pinello N, Jia F, Alasmari S, Ritchie W, Keightley MC, Shini S, Lieschke GJ, Wong JJ, and Rasko JEJ (2017). Intron retention enhances gene regulatory complexity in vertebrates. Genome Biol 18, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schugar RC, Shih DM, Warrier M, Helsley RN, Burrows A, Ferguson D, Brown AL, Gromovsky AD, Heine M, Chatterjee A, et al. (2017). The TMAO-Producing Enzyme Flavin-Containing Monooxygenase 3 Regulates Obesity and the Beiging of White Adipose Tissue. Cell Rep 19, 2451–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senbanjo LT, and Chellaiah MA (2017). CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front Cell Dev Biol 5, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, and Seeley RJ (2019). Reg3 Proteins as Gut Hormones? Endocrinology 160, 1506–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W, Geva-Zatorsky N, Jupp R, Mathis D, Benoist C, et al. (2020). Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature 577, 410–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark CM, Susi A, Emerick J, and Nylund CM (2019). Antibiotic and acid-suppression medications during early childhood are associated with obesity. Gut 68, 62–69. [DOI] [PubMed] [Google Scholar]
- Tims S, Derom C, Jonkers DM, Vlietinck R, Saris WH, Kleerebezem M, de Vos WM, and Zoetendal EG (2013). Microbiota conservation and BMI signatures in adult monozygotic twins. ISME J 7, 707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiso M, and Schechter AN (2015). Nitrate reduction to nitrite, nitric oxide and ammonia by gut bacteria under physiological conditions. PLoS One 10, e0119712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, et al. (2013). Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med 368, 407–415. [DOI] [PubMed] [Google Scholar]
- Van Spaendonk H, Ceuleers H, Witters L, Patteet E, Joossens J, Augustyns K, Lambeir AM, De Meester I, De Man JG, and De Winter BY (2017). Regulation of intestinal permeability: The role of proteases. World J Gastroenterol 23, 2106–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Baeza Y, Gonzalez A, Smarr L, McDonald D, Morton JT, Navas-Molina JA, and Knight R (2017). Bringing the Dynamic Microbiome to Life with Animations. Cell Host Microbe 21, 7–10. [DOI] [PubMed] [Google Scholar]
- Vazquez-Baeza Y, Pirrung M, Gonzalez A, and Knight R (2013). EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables JP, Klinck R, Bramard A, Inkel L, Dufresne-Martin G, Koh C, Gervais-Bird J, Lapointe E, Froehlich U, Durand M, et al. (2008). Identification of alternative splicing markers for breast cancer. Cancer Res 68, 9525–9531. [DOI] [PubMed] [Google Scholar]
- Vergnolle N (2016). Protease inhibition as new therapeutic strategy for GI diseases. Gut 65, 1215–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters WA, Xu Z, and Knight R (2014). Meta-analyses of human gut microbes associated with obesity and IBD. FEBS Lett 588, 4223–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, and Seed B (2003). A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res 31, e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. (2011). Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, et al. (2008). Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 455, 1109–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Xu L, Chen X, Xu W, Yin Z, Gao X, and Xiong S (2013). Autoantibody induction by DNA-containing immune complexes requires HMGB1 with the TLR2/microRNA-155 pathway. J Immunol 190, 5411–5422. [DOI] [PubMed] [Google Scholar]
- Wickham H (2009). ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- Wilson SB, Kent SC, Patton KT, Orban T, Jackson RA, Exley M, Porcelli S, Schatz DA, Atkinson MA, Balk SP, et al. (1998). Extreme Th1 bias of invariant Valpha24JalphaQ T cells in type 1 diabetes. Nature 391, 177–181. [DOI] [PubMed] [Google Scholar]
- Xia F, Cao H, Du J, Liu X, Liu Y, and Xiang M (2016). Reg3g overexpression promotes beta cell regeneration and induces immune tolerance in nonobese-diabetic mouse model. J Leukoc Biol 99, 1131–1140. [DOI] [PubMed] [Google Scholar]
- Yang Z, Chen D, Nie J, Zhou S, Wang J, Tang Q, and Yang X (2016). MicroRNA143 targets CD44 to inhibit breast cancer progression and stem cell-like properties. Mol Med Rep 13, 5193–5199. [DOI] [PubMed] [Google Scholar]
- Zhang XS, Li J, Krautkramer KA, Badri M, Battaglia T, Borbet TC, Koh H, Ng S, Sibley RA, Li Y, et al. (2018). Antibiotic-induced acceleration of type 1 diabetes alters maturation of innate intestinal immunity. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, and Garcia BA (2015). Comprehensive catalog of currently documented histone modifications. Cold Spring Harb Perspect Biol 7, a025064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq data that support the findings of this study have been deposited in the ArrayExpress database (https://www.ebi.ac.uk) with accession code E-MTAB-9981 (RNA-seq of ileum at P42 in STR) and E-MTAB-9982 (RNA-Seq of ileum at P23 in LTR). Ileal NanoString data have been deposited in the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession numbers GSE163887 (Ileal epithelial cells immune gene profile at P23) and GSE163888 (ileal miRNA profile at P23). 16S rRNA data have been deposited in QIITA with the identifier 13529 (https://qiita.ucsd.edu/study/description/13529). Shotgun metagenomics data have been deposited in the European Nucleotide Archive (ENA) (https://www.ebi.ac.uk/metagenomics/) under accession number, PRJEB42282.