

Abstract
Correction of mis-splicing events is a growing therapeutic approach for neurological diseases such as spinal muscular atrophy or neuronal ceroid lipofuscinosis 7, which are caused by splicing-affecting mutations. Mis-spliced effector genes that do not harbour mutations are also good candidate therapeutic targets in diseases with more complex aetiologies such as cancer, autism, muscular dystrophies or neurodegenerative diseases. Next-generation RNA sequencing (RNA-seq) has boosted investigation of global mis-splicing in diseased tissue to identify such key pathogenic mis-spliced genes. Nevertheless, while analysis of tumour or dystrophic muscle biopsies can be informative on early stage pathogenic mis-splicing, for neurodegenerative diseases, these analyses are intrinsically hampered by neuronal loss and neuroinflammation in post-mortem brains. To infer splicing alterations relevant to Huntington’s disease pathogenesis, here we performed intersect-RNA-seq analyses of human post-mortem striatal tissue and of an early symptomatic mouse model in which neuronal loss and gliosis are not yet present. Together with a human/mouse parallel motif scan analysis, this approach allowed us to identify the shared mis-splicing signature triggered by the Huntington’s disease-causing mutation in both species and to infer upstream deregulated splicing factors. Moreover, we identified a plethora of downstream neurodegeneration-linked mis-spliced effector genes that—together with the deregulated splicing factors—become new possible therapeutic targets. In summary, here we report pathogenic global mis-splicing in Huntington’s disease striatum captured by our new intersect-RNA-seq approach that can be readily applied to other neurodegenerative diseases for which bona fide animal models are available.
Keywords: RNA-sequencing (RNA-seq), Huntington’s disease, splicing, RNA-binding proteins (RBP), splicing factors
Elorza et al. reveal a mis-splicing signature in Huntington’s disease patients, devoid of human post-mortem related artefacts by intersecting data with an early symptomatic mouse model. They identify five families of altered splicing factors and six key pathogenic genes, unveiling new targets for therapeutic intervention.
Introduction
Alternative splicing of pre-mRNA is the differential processing of introns and exons to generate multiple transcript isoforms from individual genes, thereby increasing molecular diversity. However, when it is not properly executed, alternative splicing leads to mis-splicing, which may result in proteins with altered function and stability.1 A number of splicing factors and other RNA-binding proteins (RBPs) are responsible for proper regulation of alternative splicing,2 and growing evidence has implicated mis-splicing in a range of pathologies such as cancer,3 muscular dystrophies,4 autism5,6 and neurodegenerative diseases7 such as Alzheimer’s disease,8,9 amyotrophic lateral sclerosis10,11 and Huntington’s disease.12-15 Next-generation RNA sequencing (RNA-seq) has boosted investigation of global mis-splicing in diseased tissue. Nevertheless, while analysis of tumour or dystrophic muscle biopsies can be informative on early stage pathogenic mis-splicing, RNA-seq studies of neurodegenerative diseases using post-mortem brains are confounded by the dramatically altered cellular composition in end-state disease tissue due to neuronal loss and increased gliosis, the latter besides leading to a chronic inflammatory status. These caveats make it difficult to identify the causative molecular alterations that may serve as bases for therapeutic approaches.
Huntington’s disease is a devastating neurological disorder characterized by prominent motor symptoms and marked atrophy of the nucleus striatum.16 Huntington’s disease is caused by a polyglutamine (polyQ)-encoding CAG repeat expansion in the huntingtin (HTT) gene.17 Similar pathogenic CAG mutations in different genes cause multiple dominant spinocerebellar ataxias (SCAs), including SCA-1, -2, -3, -6, -7 and -1718 and there is evidence of toxicity being mediated by both the expanded CAG-containing mRNAs and the polyQ-containing proteins.19,20 The proteins that interact with expanded CAG mRNA include splicing factors such as MBNL1,21 U2AF222 and SRSF6,15,23 the latter being also sequestered into the characteristic polyQ inclusion bodies found in Huntington’s disease brains.12 All this has led to the proposal that splicing alterations may, at least in part, underlie Huntington’s disease.12–15 In fact, hypothesis-driven studies have identified two mis-splicing events in the aetiology of Huntington’s disease. Namely, a retained intron in the HTT gene giving rise to a highly toxic exon 1-encoded N-terminal truncated form of mutant Htt,14 and elevated inclusion of tau (MAPT) exon 10, leading to a pathogenic increase in tau isoforms with four tubulin binding repeats (4R-tau).12 However, analysis of global mis-splicing in striatum of patients with Huntington’s disease is missing due to the limitations imposed by the altered cellular composition in post-mortem samples discussed above.
To investigate mis-splicing patterns potentially relevant to the onset of Huntington’s disease, here we performed at par RNA-seq analyses of striatum of Huntington’s disease patients and of a highly validated Huntington’s disease transgenic mouse model at an early disease stage, in which motor symptoms are due to circuit dysfunction prior to appearance of neuronal loss and gliosis. Intersection of both analyses yielded a shared mis-splicing signature triggered by the Huntington’s disease-causing mutation in striatum of both species (Fig. 1). Characterization of this signature allowed us to infer a network of upstream splicing factors deregulated in both species and to identify a plethora of downstream neurodegeneration-linked effector genes whose aberrant splicing led us to corroborate their decreased protein levels in Huntington’s disease striatum.
Figure 1.

Workflow of the technical approach. Steps for human/mouse intersect-RNA-seq analysis of neurodegenerative disease-specific mis-splicing and identification of underlying altered splicing factors (SF) and pathogenic effectors. AS = alternative splicing; HD = Huntington’s disease; WB = western blot.
Materials and methods
Human brain tissue samples
Brain specimens used in this study from striatum of Huntington’s disease patients and controls were provided by Institute of Neuropathology Brain Bank (HUB-ICO-IDIBELL, Hospitalet de Llobregat, Spain), the Neurological Tissue Bank of the IDIBAPS Biobank (Barcelona, Spain), the Banco de Tejidos Fundación Cien (BT-CIEN, Madrid, Spain) and the Netherlands Brain Bank (Amsterdam, The Netherlands). Written informed consent for brain removal after death for diagnostic and research purposes was obtained from brain donors and/or next of kin. Procedures, information and consent forms have been approved by the Bioethics Subcommittee of Consejo Superior de Investigaciones Científicas (CSIC, Madrid, Spain). The post-mortem interval in tissue processing was between 05:00 and 07:45 for RNA-seq analysis and between 05:00 and 23:30 for western blot analyses. Neuropathological examination in the Huntington’s disease cases revealed a diagnosis of Huntington’s disease grade 3–4 following Vonsattel’s criteria.24 The characteristics of the donors for the samples used in the RNA-seq analysis are provided in Supplementary Table 1.
Mice
Different previously reported mouse models were used: R6/1 transgenic mice for the human exon-1-Htt gene25 in B6CBAF1 background, and heterozygous knock-in of an expanded CAG repeat in exon 1 of the huntingtin gene, zQ175 mice26 in C57BL/6J background. All mice were bred and housed at the Centro de Biología Molecular Severo Ochoa animal facility. Mean CAG repeat length of R6/1 mice in our colony was determined as in Mangiarini et al.25 and corresponds to ∼185 repeats. Mice were housed four per cage with food and water available ad libitum and maintained in a temperature-controlled environment on a 12/12-h light-dark cycle with light onset at 08:00. Animal housing and maintenance protocols followed the guidelines of Council of Europe Convention ETS123. Animal experiments were performed under protocols (PROEX293/15) approved by the Centro de Biología Molecular Severo Ochoa Institutional Animal Care and Utilization Committee (Comité de Ética de Experimentación Animal del CBM, CEEA-CBM), Madrid, Spain.
Rotarod test
Motor coordination was assessed with an accelerating rotarod apparatus (Ugo Basile). Mice were trained during two consecutive days, the first day: four trials at fixed 4 rpm for 1-min each, the second day: four trials for 2 min (the first minute at 4 rpm and the second minute at 8 rpm). On the third day rotarod was set to accelerate from 4 to 40 rpm over 5 min and mice were tested in four trials. The latency to fall from the rotarod was measured as a mean of the four accelerating trials.
Tissue preparation for staining
Wild-type and R6/1 mice euthanasia was performed using CO2. Brains were quickly extracted and the left hemisphere immersed in 4% paraformaldehyde overnight. After profuse washing in PBS, hemispheres were immersed in sucrose 30% in PBS for at least 72 h and then included in OCT (Optimum Cutting Temperature compound, Tissue-Tek, Sakura Finetek Europe, ref. 4583), frozen and stored at −80°C until use. Mouse sagittal and coronal sections (30-µm thick) were sequentially cut on a cryostat (Thermo Scientific), collected and stored free floating in glycol-containing solution (30% glycerol, 30% ethylene glycol in 0.02 M phosphate buffer) at −20°C.
Immunohistochemistry and immunofluorescence
Sections were first washed in PBS and then immersed in 0.3% H2O2 in phosphate-buffered saline (PBS) for 45 min to quench endogenous peroxidase activity. After PBS washes, sections were immersed for 1 h in blocking solution [PBS containing 0.5% foetal bovine serum (FBS), 0.3% TritonTM X-100 and 1% bovine serum albumin (BSA)] and incubated overnight at 4°C with anti-DARPP32 (1:5000, Chemicon, AB1656), anti-IBA1 (1:500, Wako, 019-19741), anti-HNRNPC (1:1000, Abcam, ab10294) or anti-TIA1 (1:1000, Abcam, ab40693) diluted in blocking solution. After washing, brain sections were incubated first with biotinylated goat anti-rabbit or anti-mouse secondary antibody and then with avidin-biotin complex using the Elite Vectastain kit (Vector Laboratories, PK-6101 and PK-6102). Chromogen reactions were performed with diaminobenzidine (SIGMAFASTTM DAB, Sigma, D4293) for 10 min. Mouse sections were mounted on glass slides and coverslipped with Mowiol® (Calbiochem, Cat. 475904). Images were captured using an Olympus BX41 microscope with an Olympus camera DP-70 (Olympus Denmark A/S).
For immunofluorescence, mouse sections were washed in PBS and pretreated with 0.1% TritonTM X-100 for 30 min, 1 M glycine for 15 min and blocking solution (1% BSA, 0.3% FBS and 0.1% TritonTM X-100) for 1 h. Sections were then incubated overnight at 4°C with rabbit anti-TIA1 (1:300) and mouse anti-huntingtin (1:300, EM48 Millipore, MAB5374) in blocking solution. After washing in PBS, sections were incubated with anti-rabbit DyLightTM 488 (1:500, Vector Laboratories, DI-1088) and anti-mouse Alexa 555 (1:500, Thermo Fisher, A-31570) for 1 h. Nuclei were counterstained with DAPI (1:4000, Calbiochem). Finally, sections were mounted on glass slides, coverslipped with Mowiol® (Calbiochem, 475904) and maintained at 4°C. Images were acquired with a laser confocal A1R system coupled to the invert Eclipse Ti-E microscope (Nikon). Images were processed using ImageJ 1.47v.
Striatal volumetry
Coronal sections (30-µm thick) were cut on a cryostat and every sixth section was counterstained with toluidine blue pH 4.0 (1 g/l Toluidine Blue, Sigma, 198161 in 0.8 M glacial acetic acid). Digital images were captured at ×2.5 magnification (Canon EOS 450 D digital camera) and striatal areas from 19–22 sections for each animal were calculated by using ImageJ software.27 Considering a separation of 180 μm between each section, total structure volume in each mouse was calculated.
Stereology
Coronal sections (30-μm thick) counterstained with toluidine blue pH 4.0 [1 g/l Toluidine Blue (Sigma, 198161), 0.8 M glacial acetic acid] from the volumetric analysis were used. Sections containing striatum were selected and the 15 most central sections were analysed. One randomly selected 60 µm × 60 µm optical dissector at ×60 magnification with an Olympus BX41 microscope with an Olympus camera DP-70 (Olympus Denmark A/S) was analysed in each section. Total neuronal cell number per dissector was assessed by a researcher blind to genotype. Striatal neuronal cell density was calculated and compared for wild-type (n = 3) and R6/1 mice (n = 3).
RNA sequencing
Total RNA was isolated using the Maxwell® 16 LEV simplyRNA Tissue Kit (Promega, AS1280) and quantified by Qubit® RNA BR Assay kit (Thermo Fisher Scientific) and the RNA integrity number (RIN) was estimated by using RNA 6000 Nano Bioanalyzer 2100 Assay (Agilent). The RNA-seq libraries were prepared with KAPA Stranded mRNA-Seq Illumina® Platforms Kit (Roche) following the manufacturer’s recommendations. Briefly, 500 ng of total RNA was used as the input material, the poly-A fraction was enriched with oligo-dT magnetic beads and the mRNA was fragmented. The strand specificity was achieved during the second strand synthesis performed in the presence of dUTP instead of dTTP. The blunt-ended double stranded cDNA was 3′ adenylated and Illumina platform compatible adaptors with unique dual indexes and unique molecular identifiers (Integrated DNA Technologies) were ligated. The ligation product was enriched with 15 PCR cycles and the final library was validated on an Agilent 2100 Bioanalyzer with the DNA 7500 assay. The libraries were sequenced on HiSeq 2500 (Illumina, Inc) with a read length of 2 × 101 bp using HiSeq 4000 SBS kit in a fraction of a HiSeq 4000 PE Cluster kit sequencing flow cell lane generating 55–102 million paired-end reads per sample. Image analysis, base calling and quality scoring of the run were processed using the manufacturer’s software Real Time Analysis (RTA 2.7.7).
RNA-sequencing analysis
Differentially expressed genes between the biological groups were analysed using vast-tools v2.1.0 compare_expr with default parameters28 and Salmon software29 (version 1.2.1 and Ensembl versions GRCh38.100 and GRCm38.100 for the alignment of fastq files with salmon command quant), followed by R packages tximport30 and DESeq231 (Supplementary Tables 2 and 3). To verify correct clustering according to genotype, hierarchical clustering of corrected reads (per mappability) per kilobase million (cRPKM) expression values obtained with vast-tools was performed using ‘hclust’ and ‘heatmap.2’ R packages over significantly changed genes in Huntington’s disease versus control and R6/1 versus wild-type mice (Benjamini-Hochberg correction method). Cell-type marker genes of neurons, astrocytes, oligodendrocytes and microglia as described previously32,33 (Supplementary Table 4) and those of specific striatal cell types: direct pathway spiny neurons (dSPNs), indirect pathway spiny neurons (iSPNs), astrocytes and microglia described in Merienne et al.,34 were analysed by calculating for each cell type the geometric mean of their cRPKM values (vast-tools analysis) or normalized counts (Salmon analysis), respectively, excluding those genes with null values in at least one sample. Data were plotted and analysed using GraphPad software (La Jolla, CA, USA).
To identify differential alternative splicing events between the two sample groups per species we used three complementary software:
(i) vast-tools v1.1.0, using human (hg19; vastdb.hsa.13.11.15) or mouse (mm9; vastdb.mmu.13.11.15) junction libraries for the align and combine modules.28 Differential splicing analysis was done using the module compare with default parameters (|ΔPSI| ≥ 15 and a minimum ΔPSI of 5 in the same direction among all replicates; PSI, per cent spliced in).
(ii) rMATS, v3.0.8,35 utilizing TopHat228 to align fastq reads against the GRCh38.p2 (dec. 2014) and GRCm38.p3 (jan. 2012) genomes together with a custom splice-junction library. For differential splicing analyses, default parameters were used (FDR < 5%).
(iii) SANJUAN v1.0-beta, which detects de novo splicing junctions and alternative splicing events (https://github.com/ppapasaikas/SANJUAN; accessed November 2016). For differential splicing analyses, medium confidence level of constraints on differential splicing junctions was used. The combined result of all tools is shown in Supplementary Tables 5 and 6. The relative contribution of each tool to the detection of mis-spliced genes is shown in Supplementary Fig. 4B and Supplementary Tables 7–12.
The entire RNA-seq dataset will be available at the European Nucleotide Archive (ENA) database with accession number PRJEB44140.
Enrichment analysis of Gene Ontology terms
Enrichment analysis of genes with differential upregulated expression only in Huntington’s disease samples was performed using DAVID Bioinformatics Resources 6.8, Biological Process annotation36 corrected for multiple testing with Benjamini-Hochberg false discovery rate (FDR) (adjusted P-value < 0.05). Genes downregulated both in human and mouse were analysed with Enrichr (http://amp.pharm.mssm.edu/Enrichr/; accessed October 2020) and significantly enriched biological processes and molecular functions were identified by Fisher’s exact test with adjusted P-value < 0.05.
Ingenuity pathway analysis analysis
Genes mis-spliced in both human and mouse were annotated to diseases and functions using Ingenuity Pathway Analysis (IPA) software (http://www.ingenuity.com). The analysis was performed in October 2019 and the results were filtered for neurological diseases related functions.
Semiquantitative reverse transcription-PCR validation
Selected events were evaluated with semiquantitative reverse transcription (RT)-PCR. Total RNA (500 ng) was reverse-transcribed using Invitrogen SuperScript® IV reverse transcriptase and cDNA (50 ng) was amplified with gene/exon-specific primers. PCR products were resolved on 2% high-resolution MetaPhorTM agarose gels (Lonza).
Primers
Primers used in the study are as follows: Ccdc88c Forward 5′-GGATTGGAGCCAAAGCCCTAG-3′, Reverse 5′-CTGAGGTTGAAAAGGTCCGGG-3′; Kctd17 Forward 5′-GGTGAACATCGGCTCCTCCTA-3′, Reverse 5′-GGGTGCCTCTGGCTTGTAATAG-3′; Synj1 Forward 5′-GCTGAGGGTGAAGAGGAGTGA-3′, Reverse 5′-GCTGATGGCATCTCGGATGTT-3′; Vps13c Forward 5′-CCCAGACTCTAGAGCCCAAGA-3′, Reverse 5′-GCTTGAGGGGAAGGCTGATTAC-3′; Trpm7 Forward 5′-TGCCGAATTGAAGAAGCCCTT-3′, Reverse 5′-TGTGAATGTCTGTAATTTTCACTTGA-3′; Slc9a5 Forward 5′-TCCAGCTTCTGTGACGTATGC-3′, Reverse 5′-AGTATTGCTTTTCTGGGAACACC-3′.
Western blot
Samples from human brain were stored at −80°C and ground with a mortar in a frozen environment with liquid nitrogen to prevent thawing of the samples, resulting in tissue powder. Mouse brains were quickly dissected on an ice-cold plate and the different structures stored at −80°C. Human and mouse protein extracts were prepared by homogenizing brain structures in ice-cold extraction buffer [20 mM HEPES pH 7.4, 100 mM NaCl, 20 mM NaF, 1% TritonTM X-100, 1 mM sodium orthovanadate, 1 μM okadaic acid, 5 mM sodium pyrophosphate, 30 mM β-glycerophosphate, 5 mM EDTA, protease inhibitors (Complete, Roche, Cat. No 11697498001)]. Homogenates were centrifuged at 15 000 rpm for 15 min at 4°C. The resulting supernatant was collected, and protein content determined by Quick StartTM Bradford Protein Assay (Bio-Rad, 500-0203). Twenty micrograms of total protein were electrophoresed on a 10% SDS-polyacrylamide gel, transferred to a nitrocellulose blotting membrane (Amersham Protran 0.45 μm, GE Healthcare Life Sciences, 10600002) and blocked in TBS-T (150 mM NaCl, 20 mM Tris-HCl, pH 7.5, 0.1% Tween 20) supplemented with 5% non-fat dried milk. Membranes were incubated overnight at 4°C with the primary antibody in TBS-T supplemented with 5% non-fat dried milk, washed with TBS-T and next incubated with HRP-conjugated anti-mouse IgG (1:2000, DAKO, P0447) or anti-rabbit IgG (1:2000, DAKO, P0448) and developed using the ECL detection kit (PerkinElmer, NEL105001EA).
Antibodies
Rabbit anti-CCDC88C (1:500, Sigma, HPA005832), rabbit anti-KCTD17 (1:500, Thermo Fisher, PA5-72101), mouse anti-SYNJ1 (1:500, Thermo Fisher, MA3-936), rabbit anti-TRPM7 (1:1000, Novus Biologicals, NBP2-20739), rabbit anti-SLC9A5 (1:500, Abcam, ab191528), rabbit anti-TIA1 (1:1000, Abcam, ab40693), rabbit anti-U2AF2 (1:500, Santa Cruz, sc-53942), mouse anti-RBFOX1 (1:2000, Merck Millipore, MABE985) for mouse samples, mouse anti-RBFOX1 (1:1000, antibodies-online, ABIN1580418) for human samples, mouse anti-RBFOX2 (1:500, Abcam, ab57154), rabbit anti-RBFOX3 (1:1000, Merck Millipore, MAB377), rabbit anti-ELAVL4 (1:500, Abcam, ab96474), rabbit anti-ELAVL2 (1:500, Abcam, ab96471), rabbit anti-ELAVL1 (1:500, Abcam, ab200342), rabbit anti-HNRNPC (1:500, Abcam, ab10294), rabbit anti-ELAVL3 (1:500, Abcam, ab129254), rabbit anti-PTBP1 (1:1000, Abcam, ab5642), rabbit anti-PTBP2 (1:2000, Merck Millipore, ABE431), rabbit anti-SMC1 (1:1000, Bethyl, A300-055A), mouse anti-β-ACTIN (1:25000, Sigma, A2228), rabbit anti-VINCULIN (1:20000, DAKO, P0448).
Motif enrichment analysis
Seventy-three orthologous exons with differential splicing in both human and mouse were evaluated in parallel for RBP motif enrichment. As a control set, we selected 914 conserved alternative splicing exons with no detectable changes (|ΔPSI| < 5) in the RNA-seq experiments of both species. RBP motifs deposited in the CIS-BP database (http://cisbp-rna.ccbr.utoronto.ca/index.php; accessed July 2016) were scanned separately in exons, 250 intronic bases upstream the exon (3′ splicing signal—3’ss, excluding 20 bp within the 3′ splice site) and 250 intronic bases downstream the exon (5′ splicing signal—5’ss, excluding 6 bp within the 5′ splice site). Motif occurrences (log-odd ≥6 for the respective RBP Position Weight Matrix) for each RBP were counted for regulated and control exons, respectively. A motif can be counted zero or once in a given sequence. Motif enrichment for each RBP in regulated alternative splicing exons compared to the control set was evaluated using Fisher’s exact test. To correct for multiple testing, Benjamini-Hochberg FDR was used for adjustment of P-values. Enrichment was considered significant when the adjusted P < 0.01.
Data analysis
Statistical analysis was performed with SPSS 21.0 (SPSS® Statistic IBM®). Data are represented as mean ± standard error of the mean (SEM). The normality of the data was analysed by Shapiro-Wilk. For two-group comparison, two-tailed Student’s t-test was performed (data with normal distribution) or Mann-Whitney U-test (data with non-normal distribution) was performed. A critical value for significance of P < 0.05 was used throughout the study. Benjamini-Hochberg correction was applied for multiple testing in RNA-seq analysis and motif enrichment analysis, in the latter enrichment was considered significant when the adjusted P < 0.01.
Data availability
Data are available from the corresponding author upon reasonable request.
Results
Human-mouse intersect-RNA-seq captures a mis-splicing signature affecting movement disorder genes
Given the fully penetrant and dominant nature of the Huntington’s disease-causing mutation, transgenic animal models that truly recapitulate symptoms and neuropathology have been generated. In particular, mice of the R6/1 model express a transgene driven by the murine Htt promoter that encodes the highly toxic exon 1-encoded N-terminal form of Htt with a CAG expansion resulting in a robust, yet slowly-progressing, motor phenotype.25 At the age of 3.5 months, R6/1 mice in our colony already showed a clear motor coordination deficit (P = 5.0 × 10−3; Student's t-test; Supplementary Fig. 1A), but no significant striatal atrophy or neuropathology, as evidenced by volumetric analysis of the striatum together with stereological neuronal count and immunostaining with markers of gliosis (Supplementary Fig. 1BandC). Given the lack of gliosis and neuronal loss at this stage, we reasoned that a transcriptomic comparison of striatum from this mouse model and from human patients with Huntington’s disease could reveal shared Huntington’s disease-specific pathogenic mis-splicing signatures triggered by the Huntington’s disease mutation in both species, devoid of secondary alteration-related artefacts. Therefore, we performed at par RNA-seq analyses of post-mortem striatum of Huntington’s disease patients (Vonsattel’s grade 3–4) and matching control subjects (n = 3), and of striatum of 3.5-month-old R6/1 mice, together with matching controls (n = 3). Between 55 and 102 million reads (101-bp, pair-ended) were generated per sample (Fig. 2A). Prior to the alternative splicing analysis, we performed an initial characterization of differentially expressed genes (DEGs). As expected, analysis of the R6/1 RNA-seq samples revealed similarities with previous studies performed on striatum of R6/1 mice,37 particularly those performed in young pre-symptomatic mice (Supplementary Fig. 2A and Supplementary Table 3). In agreement with the motor symptoms already observed in the 3.5-month-old R6/1 mice, we found decreased levels of specific marker genes related to the function of medium sized spiny neurons such as Darpp32, Drd1 and Drd2 (Supplementary Fig. 2B), which are known to decrease early in the course of R6/1 and R6/2 mouse phenotype.38–41 Accordingly, when we analysed batteries of molecular marker genes of specific striatal cell types,34 we found a slight decrease in the global expression of markers of the spiny projection neurons of the striatonigral direct pathway (dSPN) and striatopallidal indirect pathway (iSPN) (Supplementary Fig. 2C). However, in line with the lack of overt neuronal loss and gliosis in 3.5-month-old R6/1 mice (Supplementary Fig. 1BandC), we found no significant alterations of a set of pan-neuronal and glial marker genes32,33 (Supplementary Table 4) that would be expected to be altered in the event of generalized neuronal loss and gliosis (Fig. 2B). In contrast, as expected from the marked neuronal loss and gliosis of human Huntington’s disease post-mortem striatum,24 we observed a strong decrease in the relative expression of pan-neuronal marker genes (P = 8.0 × 10−5; Student's t-test) and an increase in glial marker genes, particularly those of astrocytes (P = 0.018; Student’s t-test) and microglia (P = 4.2 × 10−3; Student’s t-test) (Fig. 2B and Supplementary Fig. 2C). Comparison of the human and mouse Huntington’s disease-associated DEG signatures revealed that the number of up- or downregulated genes was much higher in human samples (Supplementary Fig. 3A, B andSupplementary Table 2), most likely reflecting the altered cellular composition and chronic neuroinflammation. In fact, upregulated genes show enrichment for inflammation-related genes (Supplementary Fig. 3B). This may also explain why, in good agreement with earlier human/mouse comparisons based on gene chip analyses,42 the overlap was much stronger for the downregulated genes (Supplementary Fig. 3A). Also in good agreement with previous literature,42,43 shared downregulated genes show an enrichment in synaptic transmission-related gene ontology categories (Supplementary Fig. 3C).
Figure 2.
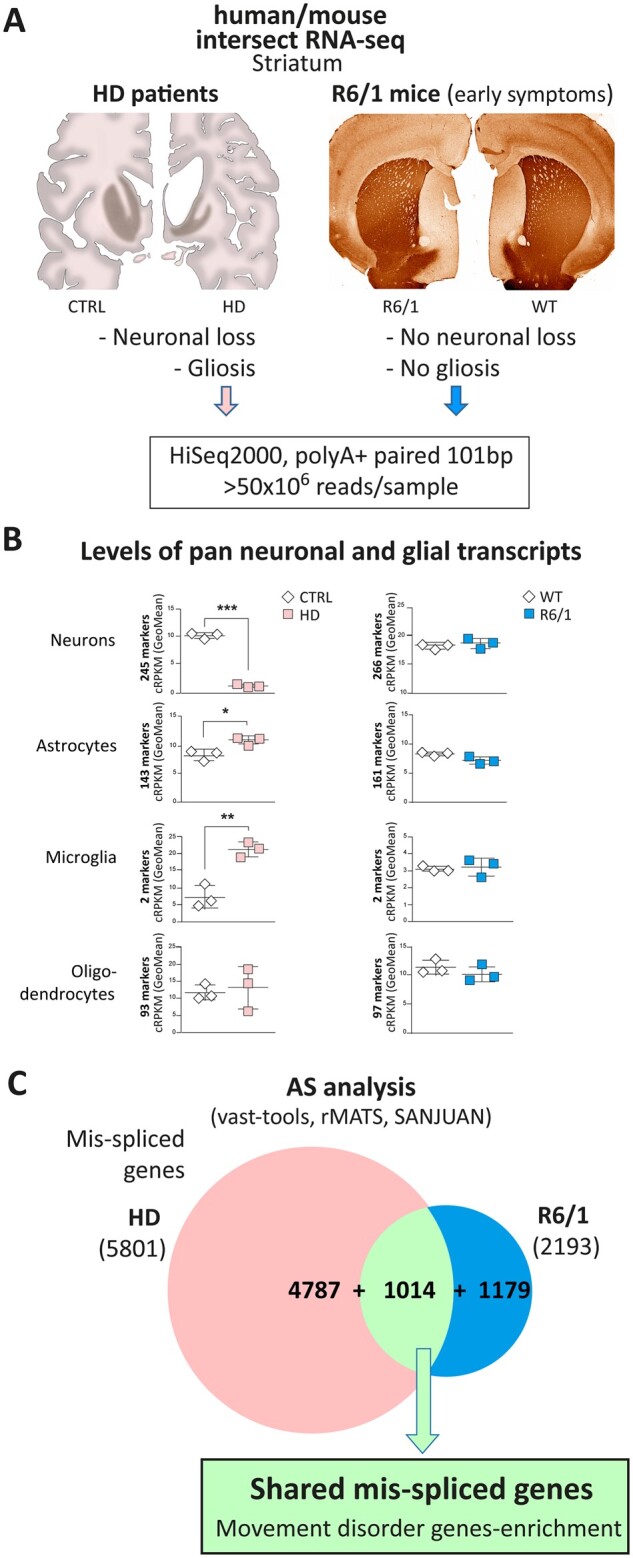
Intersect-RNA-seq analysis of striatum of patients with Huntington’s disease and early symptomatic R6/1 mice. (A) For at par RNA-seq analysis, polyA+ RNA was prepared from post-mortem striatum of patients with Huntington’s disease (Vonsattel’s grade 3–4) and matching control subjects (n = 3), and from striatum of 3.5-month-old R6/1 mice, together with matching controls (n = 3). The schematic drawings on the left depict coronal sections of human control and Huntington’s disease patient brain showing striatal atrophy and micrographs on the right show representative images of DARPP32 immunostaining of non-atrophied striatum in coronal sections of 3.5-month-old R6/1 mice as compared to wild-type mice. (B) Analysis of levels of cell-type-specific transcripts in human (left, n = 3) and mouse (right, n = 3) striatal RNA-seq samples using geometric mean of the cRPKM values of cell type marker genes of neurons, astrocytes, microglia and oligodendrocytes (Student’s t-test; *P < 0.05, **P < 0.01, ***P < 0.001). (C) Splicing was analysed with vast-tools, rMATs and SANJUAN software and the Venn diagram shows the number of mis-spliced genes in Huntington’s disease patients (5801) and R6/1 mice (2193) with respect to controls, as well as the intersect of 1,014 mis-spliced genes common to both species (P = 3.22 × 10−139, two-sided Fisher’s exact test using only one-to-one orthologues with sufficient read coverage in both species as background; n = 12 882). AS = alternative splicing; WT = wild-type.
Next, we compared alternative splicing between Huntington’s disease and control samples in both species using three complementary software packages (vast-tools, rMATS and SANJUAN; see ‘Materials and methods’ section for details). Combining the results obtained from each tool produced a list of 5801 genes with at least one differentially spliced alternative splicing event in Huntington’s disease patients respect to control subjects (Fig. 2C and Supplementary Table 5). Since isoform usage differs widely across cell types44,45 and is regulated upon inflammation,46 a substantial fraction of the observed alternative splicing changes are expected to be due to the altered neuronal/glia cellular composition and/or to the pro-inflammatory signals in post-mortem Huntington’s disease tissue. In line with this, the number of differentially spliced genes in R6/1 mice respect to controls was much smaller (2193 genes; Fig. 2C and Supplementary Table 6). Remarkably, a total of 1014 one-to-one orthologues were differentially spliced in both human and mouse (P = 3.22 × 10−139, two-sided Fisher’s exact test; Fig. 2C and Supplementary Tables 5, 6 and 13). This set of genes is expected to reflect alternative splicing alterations that are mainly due to the toxicity of the Huntington’s disease mutation independently of altered cellular content-associated artefacts in the human samples. Accordingly, this shared mis-splicing signature represents an important fraction of all mis-spliced genes in the mouse model (46.2%) but only a minority of those observed in human tissue (17.5%). Interestingly, we could further corroborate that we eliminated inflammation-related noise by intersecting with early symptomatic R6/1 mice, as a recent study revealed the interleukin-6-dependent gene expression of R6 mice47 and this is overrepresented among the genes whose mis-splicing develops in R6/1 mice after the age of 3.5 months (SupplementaryFig.4A).
Remarkably, Gene Ontology analysis (Supplementary Table 15) revealed that the genes in the shared mis-splicing signature were strongly enriched for terms related to basal ganglia and movement disorders (including Huntington’s disease), this being even more pronounced in the restricted list of the 372 mis-spliced genes that are coincidentally detected with a given alternative splicing-analysis tool in both species (Supplementary Table 16 and Supplementary Fig. 4B). This enrichment in basal ganglia- and movement disorder-related genes suggests that these shared alternative splicing alterations in striatum are a plausible contributor to Huntington’s disease pathogenesis. Finally, to test whether this shared mis-splicing signature was neuronal- or glial-specific, or even restricted to certain neuronal subtypes, we used available striatal cell-type-specific transcriptomic databases.34,41 However, we found no significantly biased distributions across cell types (Supplementary Fig. 4CandD).
Mis-splicing of neurodegeneration-linked genes correlates with reduced protein levels
To explore the pathogenic relevance of mis-spliced genes, we next focused on a subset of manually curated genes with differential alternative splicing in both species and whose mutations cause monogenic forms of neurodegeneration in humans: CCDC88C (Coiled-coil domain containing 88 C, linked to SCA40, OMIM #616053), KCTD17 [potassium channel tetramerization domain containing 17, linked to myoclonic dystonia 26 (DYT26) OMIM #616398], SYNJ1 [synaptojanin 1, linked to early onset Parkinson disease 20 (PARK20), OMIM #615530], VPS13C [vacuolar protein sorting 13 homolog C, linked to early onset Parkinson disease 23 (PARK23), OMIM #616840], TRPM7 [transient receptor potential cation channel subfamily M member 7, linked to amyotrophic lateral sclerosis-parkinsonism/dementia complex (ALSP/DC), OMIM #105500] and SLC9A5 [solute carrier family 9 member A5, also known as Na(+)/H(+) exchanger 5, which has been suggested to be linked to episodic kinesigenic dyskinesia 2 (EKD2), as it maps to the 16q13-q22.1 candidate region48] (Fig. 3A). Using VastDB,28 we identified the orthologous alternative splicing events that were differentially spliced in both species, and which corresponded to skipped exons in all cases except for TRPM7, which had a differentially retained intron (Fig. 3B). All these mis-spliced events detected by RNA-seq were validated by RT-PCR assays using striatal mRNA from an independent set of R6/1 and wild-type mice (n = 5; Fig. 3C).
Figure 3.

Mis-splicing of neurodegeneration-causing genes. (A) The OMIM database was used to select, among the genes that are mis-spliced in both human and mouse striatum, genes whose mutations in humans cause monogenic forms of neurodegeneration. (B) Using VastDB,28 the orthologous alternative spliced events differentially processed in both species were identified and the PSI (per cent spliced in) values of the mis-spliced events in each gene are shown in three wild-type versus three R6/1 samples (according to the RNA-seq vast-tools analysis). (C) RT-PCR assay and quantification of mis-spliced events in striatal RNA from an independent set of wild-type and R6/1 mice (n = 5). (Student’s t-test; *P < 0.05, **P < 0.01). Data represent mean ± SEM.
Next, we tested whether the splicing alterations observed in striatum of R6/1 mice in these neurodegeneration-associated genes correlated with changes in their protein levels. Western blot analyses revealed a significant decrease in protein levels for all of them except for VPS13C, which also showed a trend of reduced protein levels respect to the control samples (Fig. 4). Since both recessive mutations and decreased protein levels may result in decreased protein function, these data suggest that the observed global mis-splicing in striatum of patients with Huntington’s disease and mice may have a pathogenic effect by diminishing protein levels of multiple neurodegeneration-linked genes.
Figure 4.
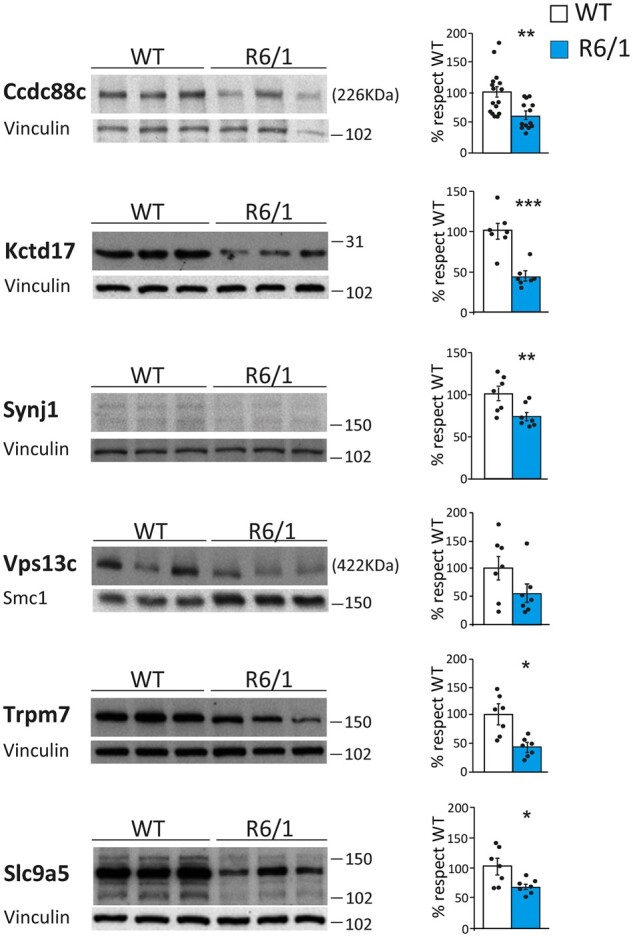
Decreased protein levels of neurodegeneration-causing genes in striatum of R6/1 mice. Protein levels in striatum of wild-type (WT) and R6/1 mice and quantifications normalized to vinculin (n = 7–16). (Student’s t-test; *P < 0.05, **P < 0.01, ***P < 0.001). Data represent mean ± SEM.
Parallel RBP motif enrichment analysis reveals splicing factors altered in Huntington’s disease striatum
We next looked for potential RBPs that may be responsible for the shared mis-splicing Huntington’s disease signature. For this purpose, we searched for known binding motifs for RBPs49 inside and around orthologous exons that were mis-regulated in both species. We reasoned that motifs associated with relevant RBPs would be similarly enriched in both exons sets. Since vast-tools directly provides exon-based homology information between the two species, we focused on 73 orthologous alternative exons detected by this software as mis-spliced in both human and mouse (|ΔPSI| ≥ 15) (Supplementary Table 17). As a control gene set, we used 914 alternatively spliced exons whose usage was not altered in Huntington’s disease and R6/1 samples respect to controls (|ΔPSI| < 5) (Fig. 5A). Motifs associated with six RBP families showed significant enrichments in the equivalent positions in both species: binding motifs of TIA1, U2AF2, HNRNPC and PTBP were enriched in the upstream intronic sequences and of RBFOX and ELAVL in downstream intronic sequences (Fig. 5B and Table 1).
Figure 5.
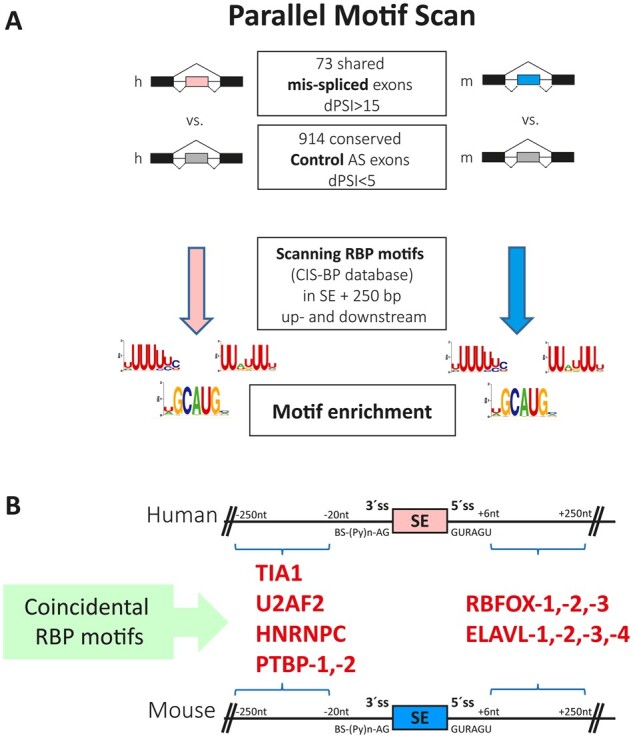
Human-mouse parallel RBP motif scan analysis in mis-regulated alternatively spliced events identifies enrichment of several splicing factor binding motifs. (A) Schematic representation of scan RBP motif analysis performed in the 73 differentially included alternative exons detected with vast-tools and conserved in human (h) and mouse (m). (B) Significantly enriched binding sites of splicing factors in 250 intronic base pairs adjacent to differentially skipped exons in Huntington’s disease and R6/1 samples. SE = skipped exons; ss = splice site.
Table 1.
Significantly enriched binding sites of splicing factors in 250 intronic base pairs adjacent to differentially skipped exons in Huntington’s disease and R6/1 samples
| Splicing factor | Binding motif |
Human
|
Mice
|
||
|---|---|---|---|---|---|
| P-value | Adjusted P-value | P-value | Adjusted P-value | ||
| 3′ splicing signal | |||||
| TIA1 | UUUUUBK | 1.607 × 10−6 | 1.141 × 10−4 | 2.002 × 10−6 | 1.261 × 10−4 |
| U2AF2 | UUUUUYC | 2.657 × 10−7 | 1.913 × 10−5 | 5.070 × 10−6 | 3.143 × 10−4 |
| HNRNPC | HUUUUUK | 1.713 × 10−5 | 1.182 × 10−3 | 3.516 × 10−5 | 2.075 × 10−3 |
| PTBP-1,-2,-3 | HYUUUYU | 1.139 × 10−8 | 8.427 × 10−7 | 2.009 × 10−12 | 1.306 × 10−10 |
| 5′ splicing signal | |||||
| RBFOX-1,-2,-3 | WGCAUG | 9.688 × 10−7 | 7.072 × 10−5 | 1.405 × 10−4 | 8.851 × 10−3 |
| ELAVL-1,-2,-3,-4 | UUDUUUU | 8.126 × 10−8 | 6.094 × 10−6 | 1.698 × 10−6 | 1.087 × 10−4 |
Western blot analysis of these RBPs in striatum of R6/1 and control mice revealed a significant decrease in protein levels for nine different members belonging to five of six identified RBP families. Specifically, we found decreased levels for TIA1 (71%, P = 9.2 × 10−4; Student’s t-test), U2AF2 (26%, P = 2.2 × 10−3; Student’s t-test), RBFOX1 (28% P = 0.016; Student’s t-test), RBFOX2 (46%, P = 8.5 × 10−3; Student’s t-test), RBFOX3 (60%, P = 1.3 × 10−5; Student’s t-test), ELAVL4 (76%, P = 1.5 × 10−4; Student’s t-test), ELAVL2 (55%, P = 2.3 × 10−5; Student’s t-test), ELAVL1 (30%, P = 2.9 × 10−4; Student’s t-test) and HNRNPC (66%, P = 1.9 × 10−3; Student’s t-test) (Fig. 6A). In contrast, the protein levels of ELAVL3 did not show any change in R6/1 striatum, neither did the two paralogues of the PTBP family, which are preferentially expressed in astrocytes (https://www.proteinatlas.org/) (Supplementary Fig. 5). To gain mechanistic insight into why these splicing factors show decreased protein levels, we first checked in our R6/1 RNA-seq data whether their transcript levels were also diminished. However, we found unaltered levels for most of them, except for Rbfox1, which showed significantly decreased transcript levels, and Hnrnpc, which counterintuitively showed increased levels (Fig. 6B). We also checked whether any of the altered splicing factors was particularly expressed in neuronal and/or glial cell-types of the striatum based on available datasets.34,41 According to laser capture followed by RNA-seq technology,34Rbfox1 mRNA is significantly enriched in spiny neurons, both globally (dSPN+iSPN) and also particularly in iSPN cells (logFC = 0.57, FDR = 0.042), while Elavl4 mRNA is enriched in microglial cells (logFC = 0.74, FDR = 0.091). According to TRAP analysis,41Rbfox3 mRNA is also significantly enriched in spiny neurons. We then performed immunostainings to test whether the splicing factors showing altered protein levels in R6/1 mice are being sequestered into the characteristic polyQ-containing inclusion bodies of Huntington’s disease, as it has been previously reported for SRSF6.12 Compared to wild-type mice, only HNRNPC and TIA1 showed and altered immunostaining pattern in striatum of R6/1 mice. This consisted of a more pronounced punctate pattern resembling that of RNA-foci (Fig. 6C). Moreover, TIA1 and polyQ exhibited a modest co-localization in inclusion-bearing neurons, which, in turn, seem to have diminished number of TIA1-positive RNA foci-like puncta (Fig. 6D). In summary, these results show that most splicing factors predicted by the parallel scan motif analysis show decreased protein levels in striatum of R6/1 mice but that, with the exception of RBFOX1, this cannot be attributed to decreased transcript levels or sequestration into inclusion bodies.
Figure 6.

Decreased protein levels of splicing factors in striatum of Huntington’s disease mouse model. (A) Protein levels in striatum of wild-type and R6/1 mice and quantifications normalized with β-actin (n = 7-20) (Student’s t-test; *P < 0.05, **P < 0.01, ***P < 0.001). Data represent mean ± SEM. (B) Messenger RNA levels of Rbfox1 and Hnrnpc in three wild-type versus three R6/1 samples, according to the RNA-seq Salmon DEG-analysis. (C) HNRNPC and TIA1 immunohistochemistry staining in striatum of R6/1 and wild-type mice. (D) Double immunofluorescence with TIA1 (green) and HTT (EM48, red) antibodies in striatum of R6/1 mice. Empty arrows show TIA1 punctate pattern in the absence of mutant HTT (mHtt) inclusions. Filled arrows show discrete co-localization of TIA1 and mHtt fluorescence in inclusion bodies. Nuclei were counterstained with DAPI (blue). (E) Protein levels in striatum of wild-type and zQ175 mice and quantifications normalized with β-actin (n = 6). (Student’s t-test; *P < 0.05). Data represent mean ± SEM.
Finally, to infer which of the decreased splicing factors are more likely to play an early role in Huntington’s disease pathogenesis, we analysed their protein levels in the zQ175 mouse model of prodromal Huntington’s disease. These mice are a heterozygous knock-in Huntington’s disease model with the CAG expansion in the endogenous Htt gene to resemble the human Huntington’s disease mutation better, but it does not develop an overt motor phenotype within the maximal (∼2.5 years) lifespan of a mouse and it is thus considered a model of pre-manifest Huntington’s disease.26 We found that RBFOX1 protein levels are decreased in striatum of zQ175 mice at the early age of 6 months and decreased protein levels or a strong tendency is also observed for RBFOX2, RBFOX3 and TIA1 in 20-month-old zQ175 mice. These results suggest that these splicing factors might be among the earliest ones to decrease in the human condition (Fig. 6E).
Validation of altered splicing factors and pathogenic downstream effectors in patient striatum
Finally, we assessed whether the protein-level alterations observed in R6/1 striatum for both splicing factors and neurodegenerative-associated genes reflected matching alterations in the striatum of Huntington’s disease patients, as expected from our intersect-RNA-seq analysis. Regarding the splicing factors, we found decreased protein levels for TIA1 (91%, P = 0.036; Student’s t-test), U2AF2 (65%, P = 0.045; Student’s t-test), RBFOX1 (76%, P = 5.3 × 10−4; Student’s t-test), RBFOX2 (46%, P = 4.6 × 10−3; Student’s t-test), RBFOX3 (96%, P = 3.3 × 10−3; Student’s t-test) and ELAVL2 (62%, P = 1.8 × 10−4; Student’s t-test). A similar trend for decreased protein levels (53%, P = 0.17; Student’s t-test) was also observed for HNRNPC (Fig. 7A). For the subset of neurodegeneration-linked mis-spliced genes, we observed a sharp decrease in protein levels for almost all of them (Fig. 7B). More precisely, for CCDC88C we found a 90% decrease in a low molecular weight form (P = 3.2 × 10−5; Student’s t-test) and a trend for decrease in the canonical 228 kDa form (60%, P = 0.081; Student’s t-test), for KCTD17 we observed a 58% decrease (P = 1.7 × 10−3; Student’s t-test) in the monomeric (36 kDa) form and a 59% decrease (P = 0.048; Student’s t-test) in the pentameric form (data not shown), and we also observed decreased levels of SYNJ1 (56%, P = 0.021; Student’s t-test), TRPM7 (82%, P = 6.0 × 10−3; Student’s t-test), VPS13C (69%, P = 0.012; Student’s t-test) and SLC9A5 (83%, P = 1.0 × 10−3; Student’s t-test) (Fig. 7B). Altogether, these results indicate that the mis-splicing Huntington’s disease signature and the candidate splicing factors identified by our intersect-RNA-seq and parallel motif scan analyses show high validation rates also at the protein level in striatum of patients with Huntington’s disease.
Figure 7.

Alteration of splicing factors and neurodegeneration-causing genes protein levels in striatum of patients with Huntington’s disease. (A) Representative western blots of protein levels of splicing factors in striatum of patients with Huntington’s disease and controls, and quantification normalized with vinculin (n = 7–15). (Student’s t-test; *P < 0.05, **P < 0.01, ***P < 0.001). Data represent mean ± SEM. (B) Localization of binding motifs of splicing factors enriched in the proximity of the mis-spliced events in each pathogenic gene and their protein levels in control and Huntington’s disease striatum. Histograms show quantification of protein levels normalized with vinculin (n = 7–17). (Student’s t-test; *P < 0.05, **P < 0.01, ***P < 0.001). Data represent mean ± SEM. WT = wild-type.
Discussion
Here we report a novel approach to identify potentially pathological signatures of mis-splicing in Huntington’s disease patients overcoming the artefacts associated with altered cell type composition and neuroinflammation in human post-mortem brain tissues. We performed at par RNA-seq analysis of striatal tissue from a mouse model of Huntington’s disease at an early symptomatic stage and from human post-mortem Huntington’s disease brain, whose intersection revealed a shared Huntington’s disease-specific mis-splicing signature affecting 1014 one-to-one orthologous genes. These included a subset of genes that had been previously linked to neurodegenerative movement disorders and that we showed to have reduced protein levels in the mouse model as well as in Huntington’s disease patients. In addition, by human-mouse parallel complementary motif searches on common mis-spliced events, we inferred a network of candidate upstream splicing factors with reduced protein levels in both species.
Post-mortem Huntington’s disease cerebral cortex shows a less marked atrophy than striatum, which is the brain region primarily affected in the disease, and RNA-seq has been previously performed on post-mortem Huntington’s disease BA4 motor cortex to analyse splicing.13 Nevertheless, it is well documented that the cortex also degenerates early in the course of disease,50 and therefore is likely affected by the mentioned caveats associated to neurodegeneration and neuroinflammation. In fact, comparison with our study revealed that 46% of the genes with alternative splicing alterations detected by Lin and co-workers13 in Huntington’s disease cortex (222 of 478) are also detected as mis-spliced in our RNA-seq analysis of Huntington’s disease striatum. However, only 36 of these genes are also altered in striatum of early symptomatic mice. Therefore, their alternative splicing analysis detected only 3.6% of the genes in the shared mis-splicing signature detected by our intersect RNA-seq analysis which, as we also demonstrate here, faithfully pinpointed upstream deregulated splicing factors and decreased protein levels of downstream mis-spliced pathogenic effectors. Furthermore, in their set of differential alternative splicing events, they detected enrichment of motifs of 15 splicing factors. However, only PTBP1—which is not expressed in neurons (https://www.proteinatlas.org/; accessed June 2020)—matches those detected in our analysis. Besides, PTBP1 protein levels are not altered neither in human nor mouse Huntington’s disease striatum, unlike the other five families of splicing factors that we detected through our parallel motif scan. Moreover, of the nine candidate splicing factors that show altered mRNA levels in the work of Lin et al.,13 the majority also change in human striatum according to our RNA-seq data, but only one is altered in striatum of early symptomatic mice. Overall, these observations point to neurodegeneration-associated confounding factors and highlight that, by refining mis-splicing analysis by intersecting results from human patients and an early mouse model, neurodegeneration- and neuroinflammation-associated artefacts are cleared out in our study.
Regarding specific genes whose mis-splicing detected through RT-PCR had previously been related to Huntington’s disease pathogenesis (e.g. MAPT,12CREB1,15TAF1,50 and HTT itself14), in our RNA-seq analysis we confirmed: the increased inclusion of MAPT exon 10, a CREB1 intron retention, and the altered usage of the alternative 3′ splicing signal of TAF1 exon 5 that favours the long (+63 nt) version of this exon. However, concerning HTT mis-splicing, there is a technical limitation to study 5′ sequences in very long transcripts such as that of HTT. This is because of a bias in favour of 3′ sequences in poly(A)+ RNA-seq analyses like the one we have performed here. Consequently, despite the considerable depth of our sequencing, we did not obtain reads with enough mapping to the first exons and introns of HTT to perform a quantitative analysis concerning the previously reported increased retention of the first intron of HTT.
Eight additional neurological disorders including the spinal and bulbar muscular atrophy (SBMA) and several SCAs are caused by expanded CAG repeats in coding sequences of different genes that give rise to expanded polyQ containing proteins.18 Given the similarity in the causing mutations, the molecular mechanisms in Huntington’s disease pathogenesis—including splicing alterations— are likely contributors to the other CAG repeat polyglutaminopathies. Most of these diseases show maximal affectation in cerebellum and the analysis of global mis-splicing in post-mortem cerebellar patient tissue for these diseases poses the same challenges as those addressed here for Huntington’s disease striatum. Since there are also good animal models for these triplet repeat disorders,52 at par early mouse model RNA-seq to identify the intersecting mis-splicing signature and parallel motif scans to infer associated splicing regulators will be useful to determine whether similar alternative splicing-related pathogenic mechanisms underlie the different CAG trinucleotide repeat disorders. Similarly, the human-mouse intersect-RNA-seq approach might be applied to any neurodegenerative disease for which animal models with construct validity are available and for which alterations in RNA processing are suspected as a pathogenic mechanism. These include the amyotrophic lateral sclerosis/frontotemporal dementia disease continuum (ALS/FTD),53 particularly the forms associated to mutations in genes encoding RBPs (TDP43 and FUS) or affecting formation of RNA foci (C9orf72)54 for which bona fide animal models exist.1
Pathological expanded CAG-containing mRNAs adopt a hairpin conformation55 and some of the splicing factors found decreased here are likely to be affected by direct and aberrant interaction with expanded CAG mRNA. This is in fact the case of U2AF215,22 and HNRNPC.15 Both U2AF2 and HNRNPC bind to U-rich motifs, as also do TIA1 and the ELAVL family, all identified here as mis-regulated in human Huntington’s disease patients and the mouse model. Since they compete for similar target mRNAs and may be regulated by common interactors, it is conceivable that deregulation of TIA1 and ELAVL might be secondary to U2AF2 and HNRNPC alterations driven by their interaction with expanded CAG repeats. The RBFOX family is the only one altered in our study that does not bind U-rich motifs, as it binds the GCAUG consensus.56 However, RBFOX splicing factors also act through a multiprotein complex called LASR (Large Assembly of Splicing Regulators) that includes HNRNPC57 and it is conceivable that sequestration of HNRNPC by expanded CAG-HTT exon1 RNA15 might subsequently affect RBFOX binding and stability.
Correction of mis-splicing events with antisense oligonucleotide drugs is a growing therapeutic approach for neurological diseases such as spinal muscular atrophy58 or neuronal ceroid lipofuscinosis 7,59 which are caused by splicing-affecting mutations. Here we identify at least six mis-spliced key pathogenic genes (CCDC88C, KCTD17, SYNJ1, VPS13C, TRPM7 and SLC9A5) that become good candidate therapeutic targets, as their alternative splicing is susceptible to be modified with similar antisense oligonucleotide drugs able to favour the normal ratio of alternative splicing generated mRNA isoforms. However, additional preclinical research needs to be conducted in order to explore if these mis-splicing events—and the concomitant decreased protein levels—significantly contribute to the Huntington’s disease mouse phenotype, either individually or in combination. This could be performed through mouse genetics and/or in vivo delivery of splicing-modifying oligonucleotides to identify the best candidates for potential clinical developments. Apart from correcting specific mis-splicing events, a more comprehensive but technically challenging therapeutic strategy would be the correction of the decreased protein levels of the splicing factors that may be acting upstream of the mis-splicing signature. In this regard, Rbfox1 and Rbfox3 are neuronal genes early downregulated at the transcriptional level in the striatum of Huntington’s disease knock-in mice.40,41 Since knock-in mice are models of prodromal Huntington’s disease, decreased expression of Rbfox splicing factors might be one of the earliest alterations underlying the mis-splicing signature that we report here. Accordingly, correcting the expression of Rbfox genes might have a higher impact in normalizing alternative splicing than that of other splicing factors that probably get deregulated later in the course of disease progression.
In summary, by intersect-RNA-seq analysis of at par-processed early mouse model and human post-mortem brain tissue, we have identified a Huntington’s disease-specific mis-splicing signature that allowed us to infer altered upstream splicing factors and downstream pathogenic effectors that can become new therapeutic targets for Huntington’s disease. Moreover, this approach can be applied to investigate the potential relevance of altered alternative splicing in other neurodegenerative diseases.
Supplementary Material
Acknowledgements
We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI). Human tissue was obtained from Institute of Neuropathology (HUB-ICO-IDIBELL) Brain Bank, the Neurological Tissue Bank of the IDIBAPS Biobank, the Banco de Tejidos Fundación CIEN, and the Netherlands Brain Bank. We thank Juan Valcárcel for scientific advice. This work was partially supported by the computing facilities of Extremadura Research Centre for Advanced Technologies (CETA-CIEMAT), funded by the European Regional Development Fund (ERDF). CETA-CIEMAT belongs to CIEMAT and the Government of Spain. The data submission to a public repository was performed by the Genomics and NGS Core Facility at the Centro de Biología Molecular Severo Ochoa (CBMSO, CSIC-UAM) which is part of the CEI UAM+CSIC, Madrid, Spain - http://www.cbm.uam.es/genomica. We also thank excellent technical assistance by Miriam Lucas and by the following core facilities: CBMSO-Genomics & Massive Sequencing, CBMSO-Animal Facility and CBMSO-Optical & confocal microscopy. We also thank Álvaro Lucas for help with illustrations.
Funding
This work was supported by CIBERNED-ISCIII collaborative grants PI2015-2/06-3 and PI2018/06-1; by grants: SAF2015-65371-R (MINECO/AEI/FEDER, UE); RTI2018-096322-B-I00 (MCIU/AEI/FEDER, UE) from Spanish Ministry of Economy and Competitiveness/Ministry of Science, Innovation and Universities (MINECO/MICINN) to J.J.L. and BFU2017-89201-P (MINECO/AEI/FEDER, UE) to M.I.; PI18/00263 from the Instituto de Salud Carlos III (Ministry of Economy, Industry and Competitiveness) co-funded by the European Regional Development Fund- to R.G.-E.; institutional grant from Fundación Ramón Areces to CBMSO; Fundación BBVA to J.J.L.; and by the European Research Council under the European Union's Horizon 2020 research and innovation program ERC-StG-LS2-637591 to M.I.
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.
Glossary
- RBP
RNA binding protein
References
- 1.Montes M, Sanford BL, Comiskey DF, Chandler DS.. RNA splicing and disease: Animal models to therapies. Trends Genet. 2019;35(1):68–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ule J, Blencowe BJ.. Alternative splicing regulatory networks: Functions, mechanisms, and evolution. Mol Cell. 2019;76(2):329–345. [DOI] [PubMed] [Google Scholar]
- 3.Srebrow A, Kornblihtt AR.. The connection between splicing and cancer. J Cell Sci. 2006;119(Pt 13):2635–2641. [DOI] [PubMed] [Google Scholar]
- 4.Scotti MM, Swanson MS.. RNA mis-splicing in disease. Nat Rev Genet. 2016;17(1):19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Irimia M, Weatheritt RJ, Ellis JD, et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell. 2014;159(7):1511–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parras A, Anta H, Santos-Galindo M, et al. Autism-like phenotype and risk gene mRNA deadenylation by CPEB4 mis-splicing. Nature. 2018;560(7719):441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nik S, Bowman TV.. Splicing and neurodegeneration: Insights and mechanisms. Wiley Interdiscip Rev RNA. 2019;10(4):e1532. [DOI] [PubMed] [Google Scholar]
- 8.Hsieh YC, Guo C, Yalamanchili HK, et al. Tau-mediated disruption of the spliceosome triggers cryptic RNA splicing and neurodegeneration in Alzheimer's disease. Cell Rep. 2019;29(2):301–316.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raj T, Li YI, Wong G, et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer's disease susceptibility. Nat Genet. 2018;50(11):1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fratta P, Sivakumar P, Humphrey J, et al. Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J. 2018;37(11):e98684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luisier R, Tyzack GE, Hall CE, et al. Intron retention and nuclear loss of SFPQ are molecular hallmarks of ALS. Nat Commun. 2018;9(1):2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernandez-Nogales M, Cabrera JR, Santos-Galindo M, et al. Huntington's disease is a four-repeat tauopathy with tau nuclear rods. Nat Med. 2014;20(8):881–885. [DOI] [PubMed] [Google Scholar]
- 13.Lin L, Park JW, Ramachandran S, et al. Transcriptome sequencing reveals aberrant alternative splicing in Huntington's disease. Hum Mol Genet. 2016;25(16):3454–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sathasivam K, Neueder A, Gipson TA, et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci U S A. 2013;110(6):2366–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schilling J, Broemer M, Atanassov I, et al. Deregulated splicing is a major mechanism of RNA-induced toxicity in Huntington's disease. J Mol Biol. 2019;431(9):1869–1877. [DOI] [PubMed] [Google Scholar]
- 16.Walker FO.Huntington's disease. Lancet. 2007;369(9557):218–228. [DOI] [PubMed] [Google Scholar]
- 17.Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72(6):971–983. [DOI] [PubMed] [Google Scholar]
- 18.Orr HT, Zoghbi HY.. Trinucleotide repeat disorders. Ann Rev Neurosci. 2007;30:575–621. [DOI] [PubMed] [Google Scholar]
- 19.Li LB, Yu Z, Teng X, Bonini NM.. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature. 2008;453(7198):1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shieh SY, Bonini NM.. Genes and pathways affected by CAG-repeat RNA-based toxicity in Drosophila. Hum Mol Genet. 2011;20(24):4810–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mykowska A, Sobczak K, Wojciechowska M, Kozlowski P, Krzyzosiak WJ.. CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res. 2011;39(20):8938–8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsoi H, Lau CK, Lau KF, Chan HY.. Perturbation of U2AF65/NXF1-mediated RNA nuclear export enhances RNA toxicity in polyQ diseases. Hum Mol Genet. 2011;20(19):3787–3797. [DOI] [PubMed] [Google Scholar]
- 23.Neueder A, Dumas AA, Benjamin AC, Bates GP.. Regulatory mechanisms of incomplete huntingtin mRNA splicing. Nat Commun. 2018;9(1):3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP. Jr. Neuropathological classification of Huntington's disease. J Neuropathol Exper Neurol. 1985;44(6):559–577. [DOI] [PubMed] [Google Scholar]
- 25.Mangiarini L, Sathasivam K, Seller M, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87(3):493–506. [DOI] [PubMed] [Google Scholar]
- 26.Heikkinen T, Lehtimaki K, Vartiainen N, et al. Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock-in mouse model of Huntington's disease. PLoS One. 2012;7(12):e50717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider CA, Rasband WS, Eliceiri KW.. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tapial J, Ha KCH, Sterne-Weiler T, et al. An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms. Genome Res. 2017;27(10):1759–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C.. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14(4):417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soneson C, Love MI, Robinson MD.. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Love MI, Huber W, Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cahoy JD, Emery B, Kaushal A, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J Neurosci. 2008;28(1):264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuhn A, Thu D, Waldvogel HJ, Faull RL, Luthi-Carter R.. Population-specific expression analysis (PSEA) reveals molecular changes in diseased brain. Nat Methods. 2011;8(11):945–947. [DOI] [PubMed] [Google Scholar]
- 34.Merienne N, Meunier C, Schneider A, et al. Cell-type-specific gene expression profiling in adult mouse brain reveals normal and disease-state signatures. Cell Rep. 2019;26(9):2477–2493.e9. [DOI] [PubMed] [Google Scholar]
- 35.Shen S, Park JW, Huang J, et al. MATS: A Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res. 2012;40(8):e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang da W, Sherman BT, Lempicki RA.. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. [DOI] [PubMed] [Google Scholar]
- 37.Yildirim F, Ng CW, Kappes V, et al. Early epigenomic and transcriptional changes reveal Elk-1 transcription factor as a therapeutic target in Huntington's disease. Proceedings of the National Academy of Sciences of the United States of America. 2019;116(49):24840–24851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Achour M, Le Gras S, Keime C, et al. Neuronal identity genes regulated by super-enhancers are preferentially down-regulated in the striatum of Huntington's disease mice. Hum Mol Genet. 2015;24(12):3481–3496. [DOI] [PubMed] [Google Scholar]
- 39.Desplats PA, Kass KE, Gilmartin T, et al. Selective deficits in the expression of striatal-enriched mRNAs in Huntington's disease. J Neurochem. 2006;96(3):743–757. [DOI] [PubMed] [Google Scholar]
- 40.Langfelder P, Cantle JP, Chatzopoulou D, et al. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci. 2016;19(4):623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee H, Fenster RJ, Pineda SS, et al. Cell type-specific transcriptomics reveals that mutant huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. Neuron. 2020;107(5):891–908.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuhn A, Goldstein DR, Hodges A, et al. Mutant huntingtin's effects on striatal gene expression in mice recapitulate changes observed in human Huntington's disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet. 2007;16(15):1845–1861. [DOI] [PubMed] [Google Scholar]
- 43.Luthi-Carter R, Strand A, Peters NL, et al. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet. 2000;9(9):1259–1271. [DOI] [PubMed] [Google Scholar]
- 44.Ellis JD, Barrios-Rodiles M, Colak R, et al. Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol Cell. 2012;46(6):884–892. [DOI] [PubMed] [Google Scholar]
- 45.Heinzen EL, Ge D, Cronin KD, et al. Tissue-specific genetic control of splicing: Implications for the study of complex traits. PLoS Biol. 2008;6(12):e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ortis F, Naamane N, Flamez D, et al. Cytokines interleukin-1beta and tumor necrosis factor-alpha regulate different transcriptional and alternative splicing networks in primary beta-cells. Diabetes. 2010;59(2):358–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wertz MH, Pineda SS, Lee H, Kulicke R, Kellis M, Heiman M.. Interleukin-6 deficiency exacerbates Huntington's disease model phenotypes. Mol Neurodegener. 2020;15(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spacey SD, Szczygielski BI, McRory JE, Wali GM, Wood NW, Snutch TP.. Mutation analysis of the sodium/hydrogen exchanger gene (NHE5) in familial paroxysmal kinesigenic dyskinesia. J Neural Transm (Vienna). 2002;109(9):1189–1194. [DOI] [PubMed] [Google Scholar]
- 49.Ray D, Kazan H, Chan ET, et al. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat Biotechnol. 2009;27(7):667–670. [DOI] [PubMed] [Google Scholar]
- 50.Rosas HD, Liu AK, Hersch S, et al. Regional and progressive thinning of the cortical ribbon in Huntington's disease. Neurology. 2002;58(5):695–701. [DOI] [PubMed] [Google Scholar]
- 51.Hernandez IH, Cabrera JR, Santos-Galindo M, et al. Pathogenic SREK1 decrease in Huntington's disease lowers TAF1 mimicking X-linked dystonia parkinsonism. Brain. 2020;143(7):2207–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bates GP, Gonitel R.. Mouse models of triplet repeat diseases. Mol Biotechnol. 2006;32(2):147–158. [DOI] [PubMed] [Google Scholar]
- 53.Ling SC, Polymenidou M, Cleveland DW.. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thomas M, Alegre-Abarrategui J, Wade-Martins R.. RNA dysfunction and aggrephagy at the centre of an amyotrophic lateral sclerosis/frontotemporal dementia disease continuum. Brain. 2013;136(Pt 5):1345–1360. [DOI] [PubMed] [Google Scholar]
- 55.Sobczak K, de Mezer M, Michlewski G, Krol J, Krzyzosiak WJ.. RNA structure of trinucleotide repeats associated with human neurological diseases. Nucleic Acids Res. 2003;31(19):5469–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuroyanagi H.Fox-1 family of RNA-binding proteins. Cell Mol Life Sci. 2009;66(24):3895–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ying Y, Wang XJ, Vuong CK, Lin CH, Damianov A, Black DL.. Splicing activation by Rbfox requires self-aggregation through its tyrosine-rich domain. Cell. 2017;170(2):312–323.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus Sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378(7):625–635. [DOI] [PubMed] [Google Scholar]
- 59.Kim J, Hu C, Moufawad El Achkar C, et al. Patient-customized oligonucleotide therapy for a rare genetic disease. N Engl J Med. 2019;381(17):1644–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the corresponding author upon reasonable request.