

Abstract
Neuroimaging genomic studies of autism spectrum disorder and schizophrenia have mainly adopted a ‘top-down’ approach, beginning with the behavioural diagnosis, and moving down to intermediate brain phenotypes and underlying genetic factors. Advances in imaging and genomics have been successfully applied to increasingly large case-control studies. As opposed to diagnostic-first approaches, the bottom-up strategy begins at the level of molecular factors enabling the study of mechanisms related to biological risk, irrespective of diagnoses or clinical manifestations. The latter strategy has emerged from questions raised by top-down studies: why are mutations and brain phenotypes over-represented in individuals with a psychiatric diagnosis? Are they related to core symptoms of the disease or to comorbidities? Why are mutations and brain phenotypes associated with several psychiatric diagnoses? Do they impact a single dimension contributing to all diagnoses? In this review, we aimed at summarizing imaging genomic findings in autism and schizophrenia as well as neuropsychiatric variants associated with these conditions. Top-down studies of autism and schizophrenia identified patterns of neuroimaging alterations with small effect-sizes and an extreme polygenic architecture. Genomic variants and neuroimaging patterns are shared across diagnostic categories suggesting pleiotropic mechanisms at the molecular and brain network levels. Although the field is gaining traction; characterizing increasingly reproducible results, it is unlikely that top-down approaches alone will be able to disentangle mechanisms involved in autism or schizophrenia. In stark contrast with top-down approaches, bottom-up studies showed that the effect-sizes of high-risk neuropsychiatric mutations are equally large for neuroimaging and behavioural traits. Low specificity has been perplexing with studies showing that broad classes of genomic variants affect a similar range of behavioural and cognitive dimensions, which may be consistent with the highly polygenic architecture of psychiatric conditions. The surprisingly discordant effect sizes observed between genetic and diagnostic first approaches underscore the necessity to decompose the heterogeneity hindering case-control studies in idiopathic conditions. We propose a systematic investigation across a broad spectrum of neuropsychiatric variants to identify putative latent dimensions underlying idiopathic conditions. Gene expression data on temporal, spatial and cell type organization in the brain have also considerable potential for parsing the mechanisms contributing to these dimensions’ phenotypes. While large neuroimaging genomic datasets are now available in unselected populations, there is an urgent need for data on individuals with a range of psychiatric symptoms and high-risk genomic variants. Such efforts together with more standardized methods will improve mechanistically informed predictive modelling for diagnosis and clinical outcomes.
Keywords: autism, schizophrenia, copy number variants, neuroimaging
Moreau et al. review advances and roadblocks in neuroimaging genomic studies of autism and schizophrenia. They propose combining diagnosis-first (‘top-down’) and gene-first (‘bottom-up’) strategies to parse out the contribution of genes and biological processes to dimensions involved in these psychiatric conditions.
Introduction: clinical diversity in autism and schizophrenia
Evolving boundaries
The nature and definition of autism spectrum disorder (ASD) and schizophrenia have been highly debated for decades. Classifications evolved over time, merging and splitting clinical manifestations. The broadening of diagnostic criteria together with improved clinical awareness has resulted in an increase of ASD prevalence in the past decades, reaching estimates of 1 in 59.1 In contrast, the schizophrenia population prevalence of ∼1% has remained relatively stable.2 Clinical diversity in schizophrenia was already reported by Bleuler, who described schizophrenia as a ‘group of schizophrenia(s)’ suggesting that this was a disorder with many possible clinical manifestations. Autism was introduced as a term in 1911 as one of four ‘types of impairment in SZ with affectivity, association, and ambivalence’.3 Autism was later described by Kanner4 and Asperger,5 to refer to a dimension of schizoid disorders. By the 1970s, researchers had clearly defined autism and childhood schizophrenia as separate conditions.6
The introduction of positive and negative symptoms in the 1980s helped to delineate subgroups of schizophrenia-like manifestations and therefore subgroups of patients. Negative symptoms in schizophrenia (such as social avoidance and emotional flatness) are also partially found in autism where they may be referred to as impairments in communication and motivation.7 Patients with either ASD or schizophrenia present difficulties in interpreting social cues associated with eye gaze, as well as deficits in theory of mind tasks.8 Schizophrenia is now defined as a severe mental illness involving disordered thought and perception, with a characteristic onset in late adolescence or early adulthood.9
To help distinguish both conditions, a ‘trumping rule’ accompanied autism in the DSM-III: autism should not be diagnosed in the presence of delusions, hallucinations, and incoherence. Today (DSM-V), spectrum terminology in ASD unifies three previously separate (DSM-IV) diagnoses: autistic’s disorder, Asperger’s disorder, and pervasive developmental disorder-not otherwise specified (PDD-NOS). Childhood-onset schizophrenia is now a recognized subtype of schizophrenia, defined by an onset before the age of 13 years. Approximately 30% of children and adolescents with childhood-onset schizophrenia also have ASD.10-12
It has been suggested that ASD and schizophrenia are extreme representations of symptomatic dimensions that extend into the normal range,13,14 but these putative dimensions have not yet been identified. Measures of autistic-like traits have been developed (e.g. the Social Responsiveness Scale) to examine subthreshold autistic features in other psychiatric conditions (such as schizophrenia) and non-psychiatric populations.15 Measures of social communication performed in the general population are genetically correlated with both ASD (during middle childhood) and schizophrenia (later adolescence).16 These approaches are in line with dimensional models such as the National Institute of Mental Health’s Research Domain Criteria Project (RDoC).13
Comorbidities are major pitfalls in top-down studies
Psychiatric comorbidities, which are common in neuropsychiatric disorders, present major caveats for any diagnosis-first studies. When a major diagnosis is assigned to an individual, it will guide treatment and enrolment in future research projects, often ignoring comorbidities. Neuroimaging and genetics findings may relate to core features of the diagnosis of interest or the spectrum of accompanying comorbidities.
Indeed, over a third of patients with ASD meet criteria for other conditions such as obsessive-compulsive disorder (OCD), anxiety, mood disorders, intellectual disability, attention deficit hyperactivity disorder (ADHD), or epilepsy.17,18 Although 15–25% of youth with ADHD meet the criteria for ASD, and 50–70% of those with ASD present comorbid ADHD,19 diagnostic criteria for ADHD and ASD did not allow their simultaneous diagnosis until the latest revision of the DSM-V.20 Intellectual disability, classified as an ASD specifier in the DSM-V, is likewise observed in ∼35% of individuals with ASD and can confound diagnostic instruments.21,22 A study of comorbidity within mental disorders in 5.9 million Danish individuals showed that a prior diagnosis of schizophrenia increased the risk of additional developmental disorders (including autism and intellectual disability, hazard ratio > 15), substance use, as well as personality and behavioural disorders (hazard ratio > 10).23 A prior diagnosis of developmental disorders increased the risk for intellectual disability (hazard ratio = 50), organic and behavioural disorders (hazard ratio > 15), and schizophrenia (hazard ratio = 8).
Comorbidities are also sex-dependent.24 For example, adult females with ASD are more likely to be diagnosed with comorbid OCD, mood, or eating disorders, rather than ASD, thereby underestimating the rate of ASD in young females.
Lessons learned from top-down studies
Reproducible neuroimaging findings in autism spectrum disorder and schizophrenia are limited
The most consistent structural MRI finding in ASD is, on average, a higher total brain volume (Fig. 1). This is mainly reported before 24 months,33,38,46,47 but is also observed in older individuals with autism (+0.25 Cohen’s d).39 Although debated,48 lower volumes of the cerebellum and corpus callosum and increased CSF volume were also recurrently reported in ASD compared to controls.38,49 Inconsistent findings have been reported for the hippocampus, amygdala, thalamus and basal ganglia.38 Such heterogeneity and small effect sizes (Cohen’s d < 0.3; Fig. 2) underscore the necessity for large samples allowing subtyping strategies.53
Figure 1.

Genomic variants and neuroimaging alterations associated with ASD and schizophrenia. Top: Common and rare genetic variants (in green and blue, respectively) associated with ASD (left) or schizophrenia (SZ, right).25–30Top middle: Genomic variants associated with both conditions and genetic correlation between ASD and schizophrenia.26Bottom: Structural and resting-state functional MRI (in blue and green, respectively) intermediate brain phenotypes associated with ASD (right) or schizophrenia (left). Results were reported based on meta-analyses or from the largest study to date.31–42Bottom middle: Shared anatomical and functional alterations associated with ASD and schizophrenia.43–45 BP = breakpoint; CT = cortical thickness; d = dorsal; Del = deletion; Dup = duplication; FC = functional connectivity; FPN = frontoparietal network; SA = surface area; SN = salience network; v = ventral; vol = volume.
Figure 2.

Effect size across three psychiatric conditions and CNVs. Distributions of Cohen’s d are represented for case-control studies in ASD, schizophrenia (SZ), ADHD and CNVs using three modalities: cortical thickness (A, D and G, from Park et al.50 and Modenato et al.51); surface area (B, E and H, from Moreau et al.44 and Modenato et al.51) and Functional connectivity (C, F and I, from Moreau et al.44,52). The same Cohen’s d distributions are presented for two large (22q11.2 and 16p11.2), one moderate (1q21.1) and one small effect size (15q11.2) deletion and duplication (D–I) from Modenato et al.51 and Moreau et al.52 For cortical thickness, surface area, and functional connectivity, CNVs show a much larger effect size at the global (mean shift) and regional level (spread of the Cohen’s d distribution) compared with psychiatric conditions.
To improve reproducibility, the Enhancing Neuro Imaging Genetics through Meta-Analysis (ENIGMA) consortium increased sample size by aggregating data from 49 scanning sites. This effort identified smaller volumes of the pallidum, putamen, amygdala, and nucleus accumbens with small effect sizes (0.13 Cohen’s d). Cortical thickness was higher in the frontal cortex and smaller in the temporal cortex (0.21 Cohen’s d).39 Subsequent studies of cortical morphometry in ASD40 reported higher mean cortical thickness (Cohen’s d = 0.22) compared to controls, in particular in the inferior frontal and prefrontal cortex, in the superior temporal, postcentral and posterior cingulate gyri, and the precuneus (Cohen’s d < 0.32). Superior temporal gyrus and inferior frontal sulcus cortical thickness were negatively correlated with age and full-scale intelligence quotient (FSIQ) in the ASD group. These two large studies provided convergent results, but authors also noted inconsistencies (cortical thickness decreases in the ENIGMA study39), which in part have been reconciled by adjusting the stringency of quality checking (e.g. motion) across both studies. Asymmetry in ASD has also been under scrutiny. An ENIGMA study of 54 datasets reported cortical thickness asymmetries involving mainly the superior frontal gyrus (Cohen’s d = 0.13), the medial frontal, orbitofrontal, inferior temporal, and cingulate regions, that were reduced in ASD compared to controls.54
Likewise, functional connectivity has been investigated in ASD. Resting-state functional MRI is particularly appropriate to study psychiatric paediatric population because it enables data acquisition on functional connectivity without patient participation (contrary to task-based functional MRI) and limits excessive motion during scanning. Several analytical methods applied to a large aggregate dataset showed a widespread decrease of connectivity in ASD compared to controls across all datasets.55,56 Underconnectivity was predominantly observed in the default mode network (DMN; Fig. 3 and Table 1), the salience, the visual, and the auditory networks. Thalamocortical overconnectivity (in particular, between the thalamus and the sensorimotor network) is also a finding replicated in most studies.36,55,60 Many other findings are inconsistent across sites41 and may reflect differences in ascertainment and mechanistic heterogeneity in ASD.61 These include reduced long-range connectivity, increased short-range connectivity, and decreased homotopic connectivity.41 Furthermore, functional connectivity is a field that lacks standardization and many analytical strategies are used by different groups (e.g. whether to perform global signal regression is an ongoing debate in the field and creates discrepancies across publications).62,63 As an example, the largest resting-state functional MRI study in ASD identified across four datasets reproducible patterns of underconnectivity within sensorimotor networks and overconnectivity within the frontoparietal networks (Fig. 3 and Table 1) across datasets (0.2–0.6 Cohen’s d).64 However, these results were not found by previous studies, likely due to different analytical strategies.
Figure 3.
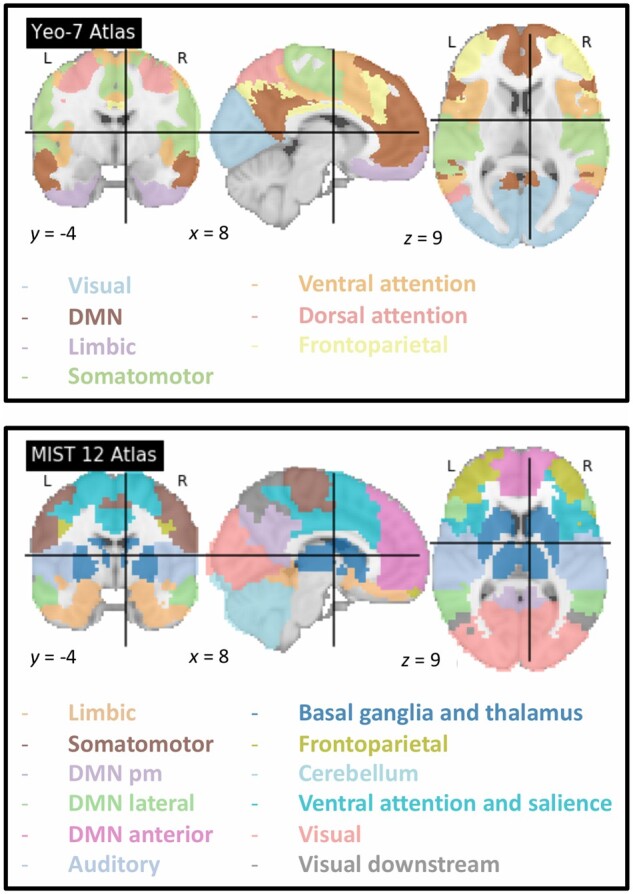
Correspondence between brain regions and functional networks. What constitutes a core functional network is not clear, and no universal taxonomy has been adopted yet.57 Networks have been defined at several levels of resolution including the commonly used 7-network parcellation (top right58) compared to the 12-network MIST parcellation (bottom right59) (https://simexp.github.io/multiscale_dashboard/index.html). See also Table 1.
Table 1.
Regions involved in the main functional networks
| Networks | Seed regions included |
|---|---|
| Salience network | Anterior insula, anterior cingulate |
| Frontoparietal = central executive network | Dorsolateral prefrontal cortex, posterior parietal cortex |
| Auditory network | Superior temporal gyrus, posterior insula dorsal, auditory region |
| Somatomotor network | Ventrolateral, dorsolateral, medial motor regions (precentral gyrus) |
| Sensorimotor network | Somatomotor and somatosensory networks (postcentral gyrus) |
| Limbic network | Amygdala, hippocampus, fusiform gyrus, posterior insula sulcus, temporal pole, inferior temporal gyrus and orbitofrontal cortex |
| Basal ganglia and thalamus | Caudate nucleus, putamen, thalamus |
| DMN | Ventromedian prefrontal, posterior cingulate cortices, precuneus, temporal medial lobe |
| Cerebellar network | Cerebellum |
| Ventral attention network | Right temporal-parietal junction and right ventral frontal cortex |
| Dorsal attention network | Intraparietal sulci and frontal eye fields |
| Visual network | Visual regions |
See also Fig. 3.
The ‘gradient’ analysis of human functional networks provides an additional coordinate system.65 It has been studied in the general population, and more recently in ASD. In normative/typically developing studies, this framework identifies a smooth transition along a gradient from unimodal areas of function (sensory, auditory, motor, visual) to higher-order transmodal areas (e.g. DMN). Studies showed that both extremes of the rostrocaudal gradient were decreased in ASD.66 Further analyses revealed cortical surface area decreases in ASD specifically within transmodal medial prefrontal and posterior cingulate regions.66
Results have been less conflicted in schizophrenia. Although both conditions are associated with small effects, those detected in schizophrenia are typically 2–3-fold larger than in ASD (Fig. 2). This difference, which is puzzling as ASD and schizophrenia have similar severities and prevalence, may suggest a lower level of neuroanatomical heterogeneity in schizophrenia compared to autism. A large meta-analysis reported a global grey matter reduction that was mainly driven by the dorsomedial and orbitofrontal cortex, as well as the medial temporal, insula, thalamic, and striatal area.31 The ENIGMA consortium (2028 schizophrenia and 2540 controls) reported smaller hippocampus (Cohen’s d = −0.46), amygdala (Cohen’s d = −0.31), thalamus (Cohen’s d = −0.31), nucleus accumbens (Cohen’s d = −0.25), and larger pallidum (Cohen’s d = 0.21) and lateral ventricle volumes (Cohen’s d = 0.37).34 A follow-up study (4474 schizophrenia and 5098 controls) examined cortical thickness and surface area35 showing a decrease in the total surface area driven by frontal and temporal lobe regions (Cohen’s d = −0.25). A widespread decrease in cortical thickness (Cohen’s d = −0.52) was also reported. Adjusting for mean cortical thickness showed thinner cortex in fusiform, parahippocampal, and inferior temporal gyri, and thicker cortex in the precuneus, and superior parietal cortex (Cohen’s d = 0.25). CT differences were greater in the group of individuals treated with antipsychotic medication and were correlated with illness duration.35 Of note, treatment may play a larger role in neuroimaging studies in schizophrenia compared to ASD due to the lower frequency of medication in the latter group.
Functional imaging studies in schizophrenia show reduced mean connectivity but in the absence of large functional MRI datasets in schizophrenia, results should be interpreted with caution.32,67 This is predominantly observed within the DMN, ventral attention, frontoparietal, and somatomotor networks (Fig. 3 and Table 1).68 In contrast, the thalamus has been reported as overconnected with the somatomotor network and the middle temporal gyrus (correlated with positive symptoms) and underconnected with cerebellar regions (correlated with delusions and bizarre behaviour).37,69 Cerebellar (Crus-I, lobule IX and lobule X) overconnectivity has been also reported with the salience and sensorimotor networks.42
An ongoing debate is whether to consider resting-state as a collection of individual states that may be captured using dynamic connectivity. Studies showed that functional networks are dominated by contributions from common organizational principles and conjunction of individual features.70 Therefore, disease-related effects that are state-dependent might appear as highly heterogeneous because of limited temporal sampling.
Earlier top-down studies were vastly underpowered to report effects in ASD and schizophrenia (e.g. analyses of the corpus callosum volume in ASD48), but larger studies are now yielding more reproducible findings. Small effect sizes reported in both schizophrenia and ASD might be an indicator of significant heterogeneity (Fig. 2). Several factors such as medication exposure (e.g. antipsychotic medications might modulate the functional MRI signal71,72) and the stage of the disease could confound these findings. There are likely subgroups associated with different patterns of brain alterations, possibly cancelling each other out in idiopathic cohorts. Examples of such effects are 16p11.2 deletions and duplications that equally increase autism risk but are associated with mirror effects on neuroimaging traits such as the insula volume.73 The subgroups and dimensions nested within conditions have however remained elusive. Furthermore, many of the alterations described above have been observed across several conditions. A core set of vulnerable brain regions and networks may be present across psychiatric diagnoses.
The polygenic architecture of autism spectrum disorder and schizophrenia
Twin studies estimate the genetic contribution to ASD and schizophrenia around 73–93% and 79%, respectively.74–76 Heritability estimates are, however, based on models [phenotype (P) = G(genetic) + E(environment)] that do not take into account the interaction between G and E. These estimated values may, therefore, be inflated by mechanisms such as assortative matting or dynastic effects.77
Most of the genetic contribution is due to common variants. Although the contribution of rare mutations to the total population liability is modest (5%), they contribute substantially to individual risk78 and occur mostly de novo. They are identified in 20% of individuals with ASD79,80 and have important implications for carriers. Among these rare variants, copy number variants (CNVs) are routinely screened in the clinic using chromosomal microarray analysis. Sixteen recurrent CNVs have been associated with ASD (Fig. 1).30,81 However, studies of non-recurrent CNVs estimate that any 1 megabase deletion or duplication including coding elements increases autism risk (albeit mildly) with a mean odds ratio (OR) of 1.6 and 1.2, respectively.82
Large effect size CNVs, such as the 16p11.2 deletion, are identified in 7–14% of patients with ASD.21,81 Rare large effect-size SNVs are identified in 13–15% of individuals with ASD.83 Exome sequencing studies have identified 102 genes conferring high risk for ASD, intellectual disability, and related neurodevelopmental conditions.84,85 These large risk ASD genes were enriched in the genome-wide association study (GWAS) signal of schizophrenia and educational attainment, as well as gene ontology terms including gene neuronal regulation and neuronal communication.85
For schizophrenia, large risk variants have been harder to identify in comparison with ASD.86 Early candidate gene studies identified rare putative large risk schizophrenia genes (e.g. COMT, DISC1, DTNBP1, and NRG1), but they were not subsequently replicated.87 Burden analyses show that de novo variants distributed across many coding genes are over-represented in schizophrenia.88 However, few genes have been robustly identified as large effect-size risk factors for schizophrenia (i.e. SETD1A, NRXN1).89,90 Eight CNVs have been formally associated with schizophrenia with OR ranging from 2 to 309,91 and eight additional CNVs met criteria for suggestive association (Fig. 1).27,92 However, burden analyses have demonstrated that many more CNVs increase risk for schizophrenia.27
The common-allele model posits that the psychiatric condition results from the cumulative effect of multiple common alleles with small effects. The yield of GWAS studies has significantly increased with sample size. The latest studies in ASD and schizophrenia identified five single nucleotide polymorphisms (SNPs)25 and 145 SNPs,29 respectively. However, predictive models suggest that as sample sizes increase, the rate of future common variant discoveries for ASD will be between those for schizophrenia and major depression.93
Neuroimaging and genomic dimensions across diagnostic boundaries
Neuroimaging traits and genetic factors specific to a psychiatric diagnosis have yet to be identified. The field has, however, progressed in characterizing neural substrates and genomic variants common across disorders.
In one of the first large transdiagnostic efforts, anterior cingulate area and anterior insula were among the top regions to demonstrate shared anatomical alterations across schizophrenia, bipolar disorder, major depression, addiction, OCD, and anxiety.43,94 Shared alterations were the highest between psychotic disorders and minimum with anxiety and OCD. A neuroanatomical investigation of ASD, schizophrenia, and ADHD has suggested that shared dimensions may arise through alterations in functional networks responsible for processing complex cognitive traits. Patterns associated with ASD and ADHD were distributed within the DMN, while ADHD and schizophrenia patterns were preferentially observed in the ventral attentional network (Fig. 3 and Table 1).50 The remaining components of the ASD and schizophrenia alteration profiles were distributed within the frontoparietal and limbic networks. Interestingly, thickness and surface alterations were observed within the same network, but not necessarily with the same directionality across conditions. Identifying overlap between these three conditions was difficult possibly because of the small neuroimaging effect size in ASD and ADHD, and the lower correlation between schizophrenia and these two earlier onset conditions.
Deficits in the social communication questionnaire measured in individuals with ASD, ADHD, and OCD were associated with a decrease in the right insula cortical thickness and the ventral striatum volume.95 Larger amygdala and hippocampus volumes were associated with higher scores on the ‘Reading the Mind in the Eyes Test’.95
At the functional level, studies showed that underconnectivity in the medial prefrontal cortex, anterior and posterior cingulate cortex, as well as the precuneus, were altered along a psychosis spectrum (i.e. bipolar disorder and schizophrenia).67,96,97 A large meta-analysis45 across eight psychiatric disorders identified shared alterations in network connectivity predominantly in the ventral salience and the frontoparietal networks, and the DMN. An underconnectivity pattern was identified between the DMN and the ventral salience network and between the frontoparietal and the salience networks. An overconnectivity pattern was found between the DMN and frontoparietal network and between the DMN and salience network.
Overall, these studies suggest that neuropsychiatric disorders may be related to similar hubs of vulnerability including the anterior cingulate cortex, the DMN, the frontoparietal network (especially prefrontal regions), and the insular cortex. Although these findings should be interpreted with caution, recurrent involvement of these brain areas could be due to their complex functions such as social cognition and executive functions,94 in line with the RDoC and p-factor. Neuroimaging dimensional reduction such as the gradient approach98 may help position psychiatric conditions along general dimensions.
Genetic correlations between psychiatric conditions are well-replicated findings and are much higher than what has been observed for neurological conditions.25,26,99,100 A recent study showed that among 146 genome-wide significant SNPs reported in ASD, ADHD, schizophrenia, bipolar disorder, major depression, anorexia nervosa, OCD, and Tourette syndrome, 109 (75%) showed association with two or more conditions and 23 with four or more neuropsychiatric disorders. These 23 SNPs were located within genes expressed in the brain from the second trimester. Modelling the joint genetic architecture of these eight conditions identified three groups of neuropsychiatric disorders: compulsive, mood, and psychotic, as well as early-onset conditions. These results suggest pleiotropic mechanisms as well as genetic dimensions spanning diagnoses.26,101
Similar observations have been reported for rare variants. Twenty nine pathogenic CNVs were shared across ASD and schizophrenia, including recurrent CNVs at 12 loci (such as 1q21.1, 3q29, 15q11.2, 16p11.2, 16p13.11, 17p12, 22q11.2).102 Gene set analyses pointed towards a substantial overlap of biological pathways involved in both disorders. Identified mechanisms included synapse/neuron projection, cell adhesion/junction, MAPK signalling, transcription/gene expression regulation, and the actin cytoskeleton. Shared mechanisms have been also investigated using gene expression data. Analyses of post-mortem cortex samples revealed shared gene-expression profiles between ASD and schizophrenia, as well as bipolar disorder and schizophrenia. Shared differential expression profiles involved downregulation of neuronal and synaptic signalling pathways with a gradient of transcriptomic severity showing the largest changes in ASD compared with schizophrenia or bipolar disorder.103
Overall, genetic factors appear to converge early on at the transcriptional level, which may in part explain phenotypic and neuroimaging traits shared across psychiatric conditions.
Bottom-up approach: large effect genetic variants to dissect mechanisms in psychiatry
The relevance of conducting bottom-up studies emerged from the questions raised by genetic discoveries of top-down studies. First, why are mutations overrepresented in individuals with a psychiatric diagnosis, and are they related to core symptoms of the disease or to comorbidities? Second, why are mutations associated with several diagnoses (pleiotropy), and do they impact a single dimension contributing to all diagnoses?
By contrast to the top-down approach, the bottom-up recruitment based on the presence of a genetic risk factor for neuropsychiatric disorders (Fig. 4), allows for the investigation of pathways related to a particular biological risk for psychiatry irrespective of any clinical phenotype. The statistical power required to conduct bottom-up studies limits this approach to genetic variants with large enough effect size and population frequency. Clinical routine investigation using whole-genome chromosomal microarrays revealed that CNVs are present in 10–15% of children with neuropsychiatric disorders.104 Many recurrent neuropsychiatric CNVs have large effect sizes (∼1 Cohen’s d; Fig. 2) on cognitive and neuroimaging traits and are natural candidates to conduct genetic first studies.
Figure 4.
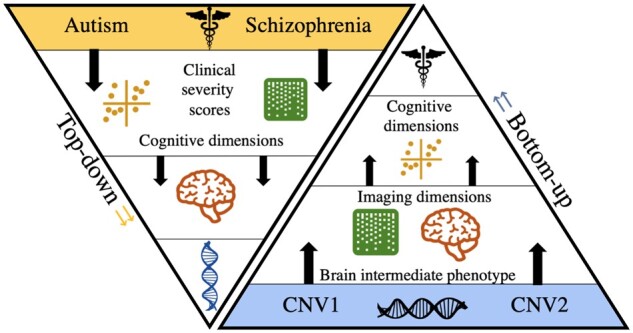
Top-down versus bottom-up approaches. The genetic-first, bottom-up approach (right) can build models/signatures from a lower level in the hierarchy (e.g. intermediate brain phenotype), and then asks questions about how such low-level models can explain observations higher up in the hierarchy (clinical manifestations).
Deep phenotyping one mutation at a time
Recurrent CNVs at the 16p11.2 and 22q11.2 loci are among the most frequent high-risk mutations associated with ASD and schizophrenia. Deletions and duplications between breakpoints 4 and 5 on chromosome 16p11.2 were first linked to ASD in 2008.105 Carriers of the duplication have a higher risk of developing schizophrenia (OR = 9.4).27,30 Both 16p11.2 deletions and duplications have been enriched in a broad spectrum of other conditions including ADHD and intellectual disability.106 Genetic first studies have estimated effect sizes of −1.5 Cohen’s d on IQ, and −1.4 Cohen’s d on phonological memory for deletions.107,108 A smaller decrease in IQ is associated with duplications (−0.8 Cohen’s d). Both CNVs do also affect social responsiveness (−3 Cohen’s d), as well as gross and fine motor skills.73,109 Mirror anthropometric phenotype has been reported with deletions mainly associated with obesity and macrocephaly and duplications associated with underweight, and microcephaly. Again, effects are large ranging from 0.8 to 1 Cohen’s d.107,108,110,111 Neuroimaging analyses reported negative gene-dosage effects on total brain volume, total grey and white matter with again similar large effect sizes.112 Once effects on global volumes are taken into account, a mirror negative gene dosage effect was observed for the insula volume.73 Other altered regions were only observed in deletions: transverse temporal gyrus, the calcarine cortex (Cohen’s d > 1), superior and middle temporal gyrus (Cohen’s d < −1) or in duplications: caudate and hippocampus (control> duplication, −0.5 > Cohen’s d > −1).73 At the functional level, a negative gene dosage effect on global connectivity was identified. After accounting for global signal, regional alterations in deletion included a thalamic-sensorial overconnectivity, impairment of frontoparietal network with temporoparietal regions, and strong disturbance of the posterior insula, the presupplementary motor cortex, and the basal ganglia (beta values from −0.8 to 1.4 z-scores).44,113 Duplications had a smaller effect on connectivity and mostly involved the amygdala-hippocampus complex, the cerebellum, and the basal ganglia.
Deletion at the 22q11.2 locus is the largest risk factor for schizophrenia (OR = 68) and up to 30% of adolescents and adults will develop psychosis.27,114,115 Children with 22q11.2 deletion have also a high risk of developing ASD (OR = 32),30 ADHD, and anxiety disorders.116,117 Duplications are less severe and are inherited in 70% of the cases (compared to deletions which are de novo in over 90% of individuals). While studies suggested a protective effect for schizophrenia118 (OR = 0.15),27 the duplication has been associated with a wide range of phenotypes, including ASD, psychomotor development, speech delay, and cognitive deficits.118 Ascertainment bias remains an issue in the study of genomic disorders which are often recruited in the clinic. Although this is particularly true for smaller effect size variants but may also apply to a lesser degree to 22q11.2.119
ENIGMA 22q11.2 deletion T-weighted studies reported a global decrease in surface area and an increase in mean cortical thickness (Cohen’s d: surface area = 1, cortical thickness = 0.6), particularly in temporal and cingulate cortices.120 Subcortical analyses showed decreased volumes and abnormal shape of the thalamus, putamen, hippocampus, and amygdala volumes (Cohen’s d = −0.9), as well as a greater lateral ventricle volume.121 The duplication shows an opposing pattern for mean cortical thickness, intracranial volume,122 and the hippocampus volume. Functional MRI studies have shown mirror effects at the global connectivity level. Underconnectivity between DMN, limbic, and frontoparietal networks is observed in deletions compared to control.44,123 Studies replicated a thalamocortical overconnectivity involving somatomotor regions and underconnectivity involving default mode network. The opposite effect was observed for the hippocampus in regard to somatomotor and frontoparietal network connectivity.44,124
However, it remains unclear if rare variants such as 16p11.2 and 22q11.2 represent mechanistic exceptions or if they may delineate dimensions that are generalized to idiopathic ASD and schizophrenia. This has been investigated at the functional connectivity level. The 16p11.2 deletion connectivity signature showed similarities with individuals diagnosed as either idiopathic schizophrenia or ASD and was associated with higher cognitive and behavioural impairments. Connectivity similarities were driven by the thalamus, the basal ganglia, and the cingulate areas. The 22q11.2 deletion connectivity profile showed similarities with individuals with idiopathic schizophrenia, ASD, and to a lesser extent with ADHD in particular through the thalamus, temporal pole, putamen, and the posterior insula.44 The thalamus and somatomotor regions played a critical role in dysconnectivity observed across both deletions and idiopathic psychiatric conditions.
Studies have sought to identify major genes driving phenotypic effects in CNV carriers to understand cellular mechanisms that give rise to the risk conferred by these variants. The 16p11.2 chromosomal region contains 29 unique genes and none of them has been formally linked to the 16p11.2 clinical phenotype.125 However, a smaller critical region of five genes, which includes TAOK2 and KCTD13, has been identified. Animal studies on TAOK2 reported dosage-dependent effects including changes in brain size and neural connectivity.126 Loss of TAOK2 activity was related to a reduction in RhoA activation, suggesting that this pathway is a mediator of TAOK2-dependent synaptic development. Of note, TAOK2 is interacting with KCTD13 in the RhoA signalling pathway, and with MAPK3.84 The overexpression of the human KCTD13 gene in zebrafish embryos induces a decrease in head size whereas deletion of the zebrafish orthologue yields a macrocephalic phenotype,127 but follow-up studies did not replicate KCTD13 findings.125
Among the 50 genes within the 22q11.2 locus,114COMT, TBX1, SEPT5, and DGCR8 were studied as putative critical drivers of the phenotype but results remain inconsistent.128 Importantly, an excess of de novo loss of function mutations has not been reported in any of the genes within the 16p11.2 and 22q11.2 regions. Overall, these studies show that association evidence for a CNV does not automatically imply that a single or even few genes are driving the effects.129
Common and specific effects of genomic variants on intermediate brain phenotypes
Single variant approaches reported in the previous chapter can only be applied to a few recurrent pathogenic variants frequent enough to establish a case-control study design. Thus, the effects of most rare deleterious variants remain undocumented. Single variant studies are therefore at odds with the extreme polygenicity of schizophrenia and ASD highlighted by GWAS discussed in the top-down approach. Several studies have suggested that every megabase of the genome contains common variation associated with increased risk for schizophrenia. This infinitesimal model also referred to as omnigenic applies to ASD and evidence shows that any megabase deletion including coding genes increases the risk for this condition.82 In this context, two non-exclusive hypotheses could be pursued: (i) an infinite number of disease-associated variants map onto an infinite number of neuroimaging patterns; and (i) variants converge on a parsimonious set of large scale network alterations. The first hypothesis alone appears improbable because ASD and schizophrenia case-control neuroimaging studies would have otherwise obtained no result.
In the effort to characterize specific and shared effects of CNVs on neuroimaging outcomes, a first cross-genetic study clustered neuroanatomical alterations across 26 different genetic mouse models of autism (including 16p11.2 CNVs, MECP2, NRXN1, and FMR1).130 Regional differences (relative to total brain volume) were heterogeneous but some regions were recurrently affected across models including the temporoparietal area, the cerebellar cortex, the frontal lobe, the hypothalamus, and the striatum. The authors clustered anatomical alterations and identified three distinct subgroups driven respectively by the limbic system, white matter structures/basal ganglia/thalamus, and cerebellar regions. Knockout mouse models from this study seemed to recapitulate the heterogeneity seen with the imaging findings in autism patients.
Similar studies in humans have been extremely difficult to implement because of the lack of data on individuals with genomic variants. Recent access to neuroimaging genetic data in the UK Biobank enabled the study of 12 schizophrenia-associated CNVs in the general population (n = 49 unaffected CNV carriers with schizophrenia, including 16p11.2, 22q11.2, NRXN1, 15q11.2, and 1q21.1 CNVs).131 The thalamus, the hippocampus, and the nucleus accumbens showed decreased volumes in CNV schizophrenia carriers. Thalamic and hippocampal volumes appeared to mediate effects on cognitive performances. A functional resting-state study of 502 carriers of eight neuropsychiatric CNVs (22q11.2, 16p11.2, 15q11.2, and 1q21.1 CNVs) showed that deletions and duplications had strong effects on connectivity. The level of brain dysfunction was also associated with the known levels of risk conferred by mutations. Connectivity signatures of 16p11.2 and 22q11.2 deletions showed similarities across several networks involving the frontoparietal, DMN, ventral attentional, and somatomotor networks.44 Dysconnectivity profiles across eight CNVs and idiopathic ASD, schizophrenia, and ADHD were summarized by three latent components involving the thalamus, the temporal pole, the anterior cingulate, and the ventromedial prefrontal cortex. The level of similarity between CNVs and idiopathic conditions was associated with mutation severity and was driven by the thalamus, and the posterior cingulate cortex, previously identified as hubs in transdiagnostic psychiatric studies (cf. ‘Lessons learned from top-down studies’ section). Beyond categorical diagnoses, CNV connectivity signatures were correlated with measures of autism severity and IQ.44
The extreme polygenicity of ASD and schizophrenia suggests that broad groups of rare and common variants share cognitive effects and neuroimaging patterns. A weighted linear model was developed to estimate the effect of CNVs on IQ using scores of intolerance to protein loss of function in a dataset of 24 000 individuals from unselected and psychiatric cohorts with cognitive assessments.132–134 These models could predict the effect size of any CNV with 80% accuracy. Deletions of >50% of the coding genome negatively impacted IQ, and this is consistent with infinitesimal/omnigenic models. The same linear weighted model using scores of intolerance to protein loss-of-function was used to explain functional connectivity across CNVs at 18 genomic loci in 502 carriers and 4427 non-carrier individuals. Deletions measured with scores of intolerance to probability of being loss-of-function intolerant (pLI) were associated with a general connectivity signature involving the thalamus, the anterior cingulate cortex, and the somatomotor network. This general deletion signature was correlated with lower general intelligence and higher autism severity scores in unselected, ASD, and ADHD cohorts.
A similar approach showed that schizophrenia relative-risk was correlated with diminished performance on at least one cognitive test. This approach was also applied to 21 carriers of either 22q11.2, 15q11.2, 1q21.1, 16p11.2, and 17q12 CNVs and 15 non-carriers showing that macro and microstructural properties of the cingulum bundles were associated with schizophrenia relative-risk.135
Bottom-up approaches have also been conducted in the general population using the aggregated genetic effect of common variants (polygenic risk scores). Polygenic risk scores use a set of trait-related SNPs that may not achieve significance at the individual level but collectively may explain a portion of the trait variance.136 Negative associations were observed in the general population between schizophrenia-polygenic risk score and mean cortical thickness, insular lobe137–139 and, frontotemporal cortical thickness as well as left hippocampus volume.140 This demonstrated again that some neuroanatomical alterations are shared between individuals at risk for schizophrenia and diagnosed with schizophrenia. Of note, studies (Generation R) of polygenic risk scores for ASD, schizophrenia, ADHD, bipolar disorder, and major depression, did not yield any results.141 This may be due to the fact that as opposed to bottom-up studies of single mutations, polygenic risk scores are likely to be mechanistically heterogeneous, diluting the neuroimaging signal. Computing polygenic scores informed by biological and brain processes (e.g. genes highly expressed in sensory-motor regions) has considerable potential to parse out the contribution of specific pathways to alterations of brain architecture (Fig. 5).
Figure 5.
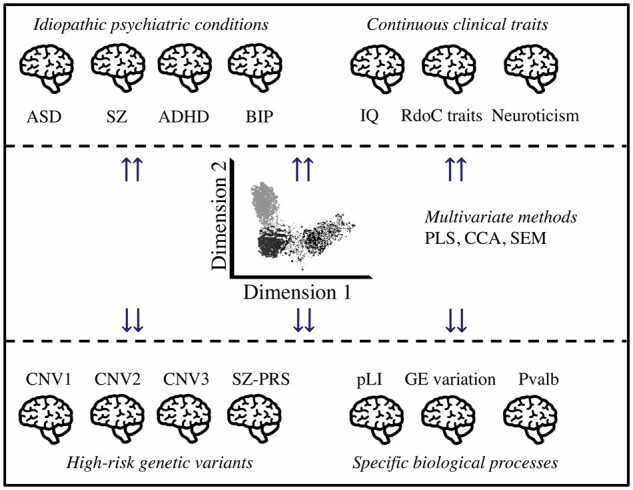
Integrating top-down and bottom-up strategies in neuroimaging genomics. We propose a systematic investigation of a broad spectrum of neuropsychiatric variants to identify dimensions underlying idiopathic conditions. Multiscale and multimodal studies using multivariate approaches would allow for the identification of latent brain dimensions that best explain the relationships between genomic variants, biological processes, psychiatric conditions, and cognitive traits. Neuroimaging proxies of specific biological processes are identified through bottom-up approaches using individuals who carry mutations in genes involved in defined gene sets (akin to a polygenic score). Computing polygenic scores informed by biological and brain processes (e.g. genes highly expressed in sensory-motor regions) has considerable potential to parse out the contribution of specific pathways to alterations of brain architecture. Multivariate approaches such as canonical correlation analysis or structural equation modelling142 will allow investigating the relationship between genomic variants, neuroimaging features, psychiatric conditions, and behavioural traits. BIP = bipolar disorder; CCA = canonical correlation analysis; GE = gene expression components143; IQ = intelligence quotient; pLI = probability of being loss-of-function intolerant; PLS = partial least square regression; PRS-SZ = polygenic risk score for schizophrenia; Pvalb = parvalbumin143,144; SEM = structural equation modelling; SZ = schizophrenia. See also Table 1.
Larger GWAS studies will improve the amount of variance explained by polygenic risk scores and will increase the relevance of bottom-up neuroimaging genetics studies using common variants.137–139 Such scores can capture individual-level variation and will be particularly appropriate for future predictive models.145
Future directions: linking microscale and macroscale observations
Gene expression data from the brain at the developmental, spatial and cell type levels provides highly granular information to annotate the brain function genetic variants at the micro- and macroscale levels (Fig. 5). A major hypothesis is that patterns of gene expression will allow us to understand the relationship between mutations and their effects on brain architecture and behaviour. Work from the Allen Institute suggests that a set of genes constitutes the core transcriptional machinery of the human brain.146 Thirty-two modules of co-expressed genes were identified—based on their spatial patterns of expression—highlighting a genome-wide redundancy. They were enriched for specific cell types, intracellular components, and associated with neurodevelopmental and degenerative conditions.146 These modules recapitulate large-scale gradients of brain organization.98 This canonical transcriptional organization of the genome (the default gene network146) is also highly correlated with the brain’s functional network architecture, such as with the default mode network and the principal gradient of macroscale cortical organization.65,143
Genomic variants in genes with a similar cortical organization or temporal pattern may lead to a shared set of brain alterations at the structural and functional levels. In other words, patterns of gene expression may predict patterns of neuroimaging alterations in carriers of CNVs and other genomic variants. Recently spatial patterns of cortical anatomy changes in individuals with 22q11.2 deletions, as well as aneuploidies (sex chromosomes and Down syndrome), were found to be correlated with cortical spatial expression of genes within the 22q11.2, X and Y chromosomes.147 The same observations have been reported at the functional connectivity level, by testing the association between connectivity-signatures of 22q11.2 and 16p11.2 deletion profiles and the brain expression patterns of genes encompassed in these genomic loci.44 However, it appears that these relationships are not specific. For example, the spatial brain expression pattern of 1834 genes (genome-wide false discovery rate) were correlated with the 22q11.2 functional brain connectivity profile. Indeed, many genes share similar spatial and temporal expression patterns, which may potentially explain the polygenic architecture of brain organization and psychiatric condition as well as the shared variance of cortical alterations across psychiatric disorders.148
The cytoarchitecture of the human brain may also help understand the link between genomic variants, their associated brain alterations and psychiatric conditions. For example, genes preferentially expressed in oligodendrocytes show a cortical distribution in their expression that is positively correlated with intracortical myelination measured by magnetization transfer.149 Brain alterations caused by CNVs and sex chromosome aneuploidies have also been associated with gene expression distributed along gradients of cell types.147 A similar approach has also linked cell types to patterns of brain alterations associated with ASD, ADHD, bipolar disorder, schizophrenia, OCD, and major depression. This ‘virtual histology’ approach reveals that the cortical expression patterns of pyramidal, microglia, astrocyte genes were correlated with cortical thickness alteration maps of eight psychiatric conditions.148
Although temporal expression during brain development is a dimension that is likely to impact brain architecture, it has not yet been associated with MRI alteration observed in carriers of CNVs and genomic variants. These exciting attempts to bridge macro- and micro-scale observations are initiating fruitful collaborations between genomics, neurobiology, computational and evolutionary neuroscience.
Functional dimensions disturbed across psychiatric conditions may also be distributed through these modules of co-expression and functional gradients.150 Such properties might be related to emerging properties of the genome and the recent evolution of the human brain.151,152
Conclusion: what have we learned and what are the next steps?
Early neuroimaging genomic studies in psychiatry were plagued by small sample sizes and inappropriate candidate gene strategies. Studies of psychiatric disorders were performed on the assumption of relative specificity (Box 1). With access to larger datasets in the past years, both top-down and bottom-up neuroimaging-genomics studies have gained traction with increased reproducibility of nature and effect-size of the alterations. The effect sizes of rare variants on neuroimaging endophenotypes are concordant with effects previously measured for the same variants on brain structure, cognitive and behavioural traits.73,133 This is in striking contrast with the effect sizes observed for functional connectivity and brain structure in schizophrenia, ASD, and ADHD, which are 3–5-fold lower (Fig. 2).35,39
This surprising discordance of effect-sizes observed between bottom-up (rare variants) and top-down studies (idiopathic conditions) underscore the necessity to dissect results from case-control studies conducted in idiopathic conditions with results from large-effect size rare variants. We propose a genetically-informed stratification by systematically investigating a broad spectrum of neuropsychiatric variants. This should allow for the identification of latent dimensions in idiopathic conditions.
The shared neuroimaging dimensions identified across psychiatric conditions are in line with the genetic correlation demonstrated between the same conditions as well as pleiotropic effects of genomic variants (Fig. 1). Findings also suggest a staggering diversity of brain endophenotypes across different genomic variants and idiopathic psychiatric conditions. Therefore, the time has not yet arrived to draw firm conclusions about the nature of the potential neuroimaging convergence (or lack thereof), across genetic risk and psychiatric conditions.
The neuroimaging field is increasingly moving towards harmonization using systematic analytical methods, atlas, and data structure58,153,154 as well as reporting standards including effect-sizes and un-thresholded beta map (Poldrack Nature). Large efforts have been in building platforms to associate imaging modalities and genetic data.155–158
Only a few datasets currently allow neuroimaging genomic studies (Fig. 6): UKBB161 and ABCD173 are large population cohorts with great potential to study genomic variation and neuroimaging phenotypes, but they include few individuals with mental illnesses and behavioural deficits. EU-AIMS is among the few psychiatric cohorts integrating genomics, neuroimaging and cognitive data, in ∼250 individuals with autism.172 Given our assumptions on the mechanistic heterogeneity in ASD, one would expect that a neuroimaging genomic dataset of several thousand individuals with autism would be required to provide the power to investigate brain-molecular dimensions. Of note, there are currently no neuroimaging genomic cohorts in schizophrenia that are available to the community. The ENIGMA consortium160 has also been instrumental in moving the field and has provided well-powered meta-analytic studies.
Figure 6.

Historical timeline in neuroimaging genetics. Many advances in neuroimaging genomics have been made by large-scale initiatives and cohort studies, such as the Autism Brain Imaging Data Exchange (ABIDE),55 the Psychiatric Genomics Consortium (PGC),159 the Enhancing Neuro Imaging Genetics through Meta-Analysis (ENIGMA) Consortium160 and the UK Biobank.161 These collaborative initiatives, among others, facilitate advances in psychiatry by providing large brain imaging and genomics datasets to the research community worldwide. Human Genome Project (HGP)162; Neuroimaging Tools and Resources Collaboratory (NITRC platform); Psychiatric Genomics Consortium (PGC)159; Alzheimer’s Disease Neuroimaging Initiative (ADNI)163; 1000 Genomes164; ADHD-200165; Open fMRI166; Human Brain Project (HBP)167; ENIGMA Consortium = Enhancing Neuro Imaging Genetics through Meta-Analysis (ENIGMA Consortium)160; = Autism Brain Imaging Data Exchange (ABIDE-1)55; NeuroVault168; UK Biobank161; HCP = Human Connectome Project (HCP)169; PING = Pediatric Imaging; Neurocognition; and Genetics (PING)170; SchizConnect171; EU-Aims172; Adolescent Brain Cognitive Development (ABCD).173
Neuroimaging genetic studies investigating large effect size mutations are lagging behind those focused on common variation. Closing this gap will require investing in new large scale cohorts with exome/genome sequencing data collected in individuals with a broad spectrum of psychiatric conditions. Cohorts with such data include UKBB and EU-AIMS. Alternative strategies include gene cohorts ascertaining individuals with previously identified large effect size neuropsychiatric variants such as ENIGMA-CNV, ENIGMA 22q11.2, and Quebec 1000 families. These efforts should provide significant power to associate brain mechanisms to genomic variants, molecular mechanisms, and mental illnesses. They will likely improve predictive modelling at the individual level and guide the development of mechanistically informed predictive tests with clinical utility.
Acknowledgements
We thank M. T. Park for providing Cohen’s d data for ASD, schizophrenia, and ADHD from his paper.50 We used them in Fig. 2.
Funding
This research was supported by the Brain Canada Multi investigator research initiative (MIRI), funds from the Institute of Data Valorization (IVADO). S.J. is supported by the Canadian Institute of Health Research CIHR_400528 and the The Institute of Data Valorization (IVADO) through the Canada First Research Excellence Fund, Healthy Brains for Healthy Lives through the Canada First Research Excellence Fund. P.B. is a fellow (‘Chercheur boursier Junior 2’) of the ‘Fonds de recherche du Québec—Santé’.
Competing interests
P.M.T. is supported in part by a grant from Biogen, Inc. (Boston, USA) for research unrelated to this manuscript.
Glossary
- ADHD
attention deficit hyperactivity disorder
- ASD
autism spectrum disorder
- CNV
copy number variant
- DMN
default mode network
- OCD
obsessive-compulsive disorder
- SNP
single nucleotide polymorphism
References
- 1.Baio J, Wiggins L, Christensen DL, et al. Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveill Summ. 2018;67:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rødgaard E-M, Jensen K, Vergnes J-N, Soulières I, Mottron L.. Temporal changes in effect sizes of studies comparing individuals with and without autism: A meta-analysis. JAMA Psychiatry. 2019;76(11):1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bleuler E.Dementia praecox oder Gruppe der Schizophrenien. Deuticke; 1911. [Google Scholar]
- 4.Kanner L.Autistic disturbances of affective contact. Nervous Child. 1943;2:217–250. [PubMed] [Google Scholar]
- 5.Asperger H.Die “Autistischen Psychopathen” im Kindesalter. Archiv Für Psychiatrie Und Nervenkrankheiten. 1944;117(1):76–136. [Google Scholar]
- 6.Chisholm K, Lin A, Abu-Akel A, Wood SJ.. The association between autism and schizophrenia spectrum disorders: A review of eight alternate models of co-occurrence. Neurosci Biobehav Rev. 2015;55:173–183. [DOI] [PubMed] [Google Scholar]
- 7.Craddock N, Owen MJ.. The Kraepelinian dichotomy - going, going… but still not gone. Br J Psychiatry. 2010;196(2):92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sasson NJ, Pinkham AE, Weittenhiller LP, Faso DJ, Simpson C.. Context effects on facial affect recognition in schizophrenia and autism: Behavioral and eye-tracking evidence. Schizophr Bull. 2016;42(3):675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bearden CE, Forsyth JK.. The many roads to psychosis: Recent advances in understanding risk and mechanisms. F1000Res. 2018;7:1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canitano R, Pallagrosi M.. Autism spectrum disorders and schizophrenia spectrum disorders: Excitation/inhibition imbalance and developmental trajectories. Front Psychiatry. 2017;8:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Driver DI, Gogtay N, Rapoport JL.. Childhood onset schizophrenia and early onset schizophrenia spectrum disorders. Child Adolesc Psychiatr Clin N Am. 2013;22(4):539–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rapoport J, Chavez A, Greenstein D, Addington A, Gogtay N.. Autism spectrum disorders and childhood-onset schizophrenia: Clinical and biological contributions to a relation revisited. J Am Acad Child Adolesc Psychiatry. 2009;48(1):10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Insel T, Cuthbert B, Garvey M, et al. Research domain criteria (RDoC): Toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010;167(7):748–751. [DOI] [PubMed] [Google Scholar]
- 14.Owen MJ, O’Donovan MC.. Schizophrenia and the neurodevelopmental continuum: Evidence from genomics. World Psychiatry. 2017;16(3):227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kincaid DL, Doris M, Shannon C, Mulholland C.. What is the prevalence of autism spectrum disorder and ASD traits in psychosis? A systematic review. Psychiatry Res. 2017;250:99–105. [DOI] [PubMed] [Google Scholar]
- 16.St Pourcain B, Robinson EB, Anttila V, et al. ; iPSYCH-SSI-Broad Autism Group. ASD and schizophrenia show distinct developmental profiles in common genetic overlap with population-based social communication difficulties. Mol Psychiatry. 2018;23(2):263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacob S, Wolff JJ, Steinbach MS, Doyle CB, Kumar V, Elison JT.. Neurodevelopmental heterogeneity and computational approaches for understanding autism. Transl Psychiatry. 2019;9(1):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joshi G, Petty C, Wozniak J, et al. The heavy burden of psychiatric comorbidity in youth with autism spectrum disorders: A large comparative study of a psychiatrically referred population. J Autism Dev Disord. 2010;40(11):1361–1370. [DOI] [PubMed] [Google Scholar]
- 19.Antshel KM, Zhang-James Y, Wagner KE, Ledesma A, Faraone SV.. An update on the comorbidity of ADHD and ASD: A focus on clinical management. Expert Rev Neurother. 2016;16(3):279–293. [DOI] [PubMed] [Google Scholar]
- 20.Ramtekkar UP.DSM-5 changes in attention deficit hyperactivity disorder and autism spectrum disorder: Implications for comorbid sleep issues. Children. 2017;4(8):62–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geschwind DH, State MW.. Gene hunting in autism spectrum disorder: On the path to precision medicine. Lancet Neurol. 2015;14(11):1109–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Volkmar FR, McPartland JC.. From Kanner to DSM-5: Autism as an evolving diagnostic concept. Annu Rev Clin Psychol. 2014;10:193–212. [DOI] [PubMed] [Google Scholar]
- 23.Plana-Ripoll O, Pedersen CB, Holtz Y, et al. Exploring comorbidity within mental disorders among a Danish National Population. JAMA Psychiatry. 2019;76(3):259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loomes R, Hull L, Mandy WPL.. What is the male-to-female ratio in autism spectrum disorder? A systematic review and meta-analysis. J Am Acad Child Adolesc Psychiatry. 2017;56(6):466–474. [DOI] [PubMed] [Google Scholar]
- 25.Grove J, Ripke S, Als TD, et al. ; 23andMe Research Team. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 2019;51(3):431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee PH, Anttila V, Won H, et al. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell. 2019;179(7):1469–1482.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marshall CR, Howrigan DP, Merico D, et al. ; CNV and Schizophrenia Working Groups of the Psychiatric Genomics Consortium. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 2017;49(1):27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moreno-De-Luca D, Moreno-De-Luca A, Cubells JF, Sanders SJ.. Cross-disorder comparison of four neuropsychiatric CNV loci. Curr Genet Med Rep. 2014;2(3):151–161. [Google Scholar]
- 29.Pardiñas AF, Holmans P, Pocklington AJ, et al. ; CRESTAR Consortium. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet. 2018;50(3):381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanders SJ, Sahin M, Hostyk J, et al. A framework for the investigation of rare genetic disorders in neuropsychiatry. Nat Med. 2019;25(10):1477–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fornito A, Yücel M, Patti J, Wood SJ, Pantelis C.. Mapping grey matter reductions in schizophrenia: An anatomical likelihood estimation analysis of voxel-based morphometry studies. Schizophr Res. 2009;108(1-3):104–113. [DOI] [PubMed] [Google Scholar]
- 32.Fornito A, Zalesky A, Pantelis C, Bullmore ET.. Schizophrenia, neuroimaging and connectomics. Neuroimage. 2012;62(4):2296–2314. [DOI] [PubMed] [Google Scholar]
- 33.Hazlett HC, Poe MD, Gerig G, et al. Early brain overgrowth in autism associated with an increase in cortical surface area before age 2 years. Arch Gen Psychiatry. 2011;68(5):467–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Erp TGM, Hibar DP, Rasmussen JM, et al. Subcortical brain volume abnormalities in 2028 individuals with schizophrenia and 2540 healthy controls via the ENIGMA consortium. Mol Psychiatry. 2016;21(4):547–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Erp TGM, Walton E, Hibar DP, et al. ; Karolinska Schizophrenia Project. Cortical brain abnormalities in 4474 individuals with schizophrenia and 5098 control subjects via the enhancing neuro imaging genetics through meta analysis (ENIGMA) consortium. Biol Psychiatry. 2018;84(9):644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woodward ND, Giraldo-Chica M, Rogers B, Cascio CJ.. Thalamocortical dysconnectivity in autism spectrum disorder: An analysis of the Autism Brain Imaging Data Exchange. Biol Psychiatry Cogn Neurosci Neuroimaging. 2017;2(1):76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferri J, Ford JM, Roach BJ, et al. Resting-state thalamic dysconnectivity in schizophrenia and relationships with symptoms. Psychol Med. 2018;48(15):2492–2499. [DOI] [PubMed] [Google Scholar]
- 38.Pagnozzi AM, Conti E, Calderoni S, Fripp J, Rose SE.. A systematic review of structural MRI biomarkers in autism spectrum disorder: A machine learning perspective. Int J Dev Neurosci. 2018;71:68–82. [DOI] [PubMed] [Google Scholar]
- 39.van Rooij D, Anagnostou E, Arango C, et al. Cortical and subcortical brain morphometry differences between patients with autism spectrum disorder and healthy individuals across the lifespan: Results from the ENIGMA ASD working group. Am J Psychiatry. 2018;175(4):359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bedford SA, Park MTM, Devenyi GA, et al. ; MRC AIMS Consortium. Large-scale analyses of the relationship between sex, age and intelligence quotient heterogeneity and cortical morphometry in autism spectrum disorder. Mol Psychiatry. 2020;25(3):614–628. [DOI] [PubMed] [Google Scholar]
- 41.King JB, Prigge MBD, King CK, et al. Generalizability and reproducibility of functional connectivity in autism. Mol Autism. 2019;10:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim D-J, Moussa-Tooks AB, Bolbecker AR, et al. Cerebellar-cortical dysconnectivity in resting-state associated with sensorimotor tasks in schizophrenia [Internet]. Hum Brain Mapp. 2020;41(11):3119–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goodkind M, Eickhoff SB, Oathes DJ, et al. Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry. 2015;72(4):305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moreau CA, Urchs SGW, Kuldeep K, et al. Mutations associated with neuropsychiatric conditions delineate functional brain connectivity dimensions contributing to autism and schizophrenia. Nat Commun. 2020;11(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sha Z, Wager TD, Mechelli A, He Y.. Common dysfunction of large-scale neurocognitive networks across psychiatric disorders. Biol Psychiatry. 2019;85(5):379–388. [DOI] [PubMed] [Google Scholar]
- 46.Courchesne E, Karns CM, Davis HR, et al. Unusual brain growth patterns in early life in patients with autistic disorder: An MRI study. Neurology. 2001;57(2):245–254. [DOI] [PubMed] [Google Scholar]
- 47.Li D, Karnath H-O, Xu X.. Candidate biomarkers in children with autism spectrum disorder: A review of MRI studies. Neurosci Bull. 2017;33(2):219–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lefebvre A, Beggiato A, Bourgeron T, Toro R.. Neuroanatomical diversity of corpus callosum and brain volume in autism: Meta-analysis, analysis of the autism brain imaging data exchange project, and simulation. Biol Psychiatry. 2015;78(2):126–134. [DOI] [PubMed] [Google Scholar]
- 49.Valenti M, Pino MC, Mazza M, Panzarino G, Di Paolantonio C, Verrotti A.. Abnormal structural and functional connectivity of the corpus callosum in autism spectrum disorders: A review. Rev J Autism Dev Dis. 2020;7(1):46–62. [Google Scholar]
- 50.Park MTM, Raznahan A, Shaw P, Gogtay N, Lerch JP, Chakravarty MM.. Neuroanatomical phenotypes in mental illness: Identifying convergent and divergent cortical phenotypes across autism, ADHD and schizophrenia. J Psychiatry Neurosci. 2018;43(3):201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Modenato C, Kumar K, Moreau C, et al. Neuropsychiatric copy number variants exert shared effects on human brain structure. medRxiv. [Preprint] doi:10.1101/2020.04.15.20056531 [Google Scholar]
- 52.Moreau C, Huguet G, Urchs S, et al. The general impact of haploinsufficiency on brain connectivity underlies the pleiotropic effect of neuropsychiatric CNVs. medRxiv. [Preprint] doi:10.1101/2020.03.18.20038505
- 53.Abraham A, Milham MP, Di Martino A, et al. Deriving reproducible biomarkers from multi-site resting-state data: An Autism-based example. Neuroimage. 2017;147:736–745. [DOI] [PubMed] [Google Scholar]
- 54.Postema MC, van Rooij D, Anagnostou E, et al. Altered structural brain asymmetry in autism spectrum disorder in a study of 54 datasets. Nat Commun. 2019;10(1):4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Di Martino A, Yan C-G, Li Q, et al. The autism brain imaging data exchange: Towards a large-scale evaluation of the intrinsic brain architecture in autism. Mol Psychiatry. 2014;19(6):659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lau WKW, Leung M-K, Lau BWM.. Resting-state abnormalities in autism spectrum disorders: A meta-analysis. Sci Rep. 2019;9(1):3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uddin LQ, Yeo BTT, Spreng RN.. Towards a universal taxonomy of macro-scale functional human brain networks. Brain Topogr. 2019;32(6):926–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yeo BTT, Krienen FM, Sepulcre J, et al. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J Neurophysiol. 2011;106(3):1125–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Urchs S, Armoza J, Moreau C, et al. MIST: A multi-resolution parcellation of functional brain networks. MNI Open Res. 2019;1:3. [Google Scholar]
- 60.Tomasi D, Volkow ND.. Reduced local and increased long-range functional connectivity of the thalamus in autism spectrum disorder. Cereb Cortex. 2019;29(2):573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He Y, Byrge L, Kennedy DP.. Nonreplication of functional connectivity differences in autism spectrum disorder across multiple sites and denoising strategies. Hum Brain Mapp. 2020;41(5):1334–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li J, Bolt T, Bzdok D, et al. Topography and behavioral relevance of the global signal in the human brain. Sci Rep. 2019;9(1):14286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murphy K, Fox MD.. Towards a consensus regarding global signal regression for resting state functional connectivity MRI. Neuroimage. 2017;154:169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Holiga Š, Hipp JF, Chatham CH, et al. Patients with autism spectrum disorders display reproducible functional connectivity alterations. Sci Transl Med. 2019;11(481):eaat9223. [DOI] [PubMed] [Google Scholar]
- 65.Margulies DS, Ghosh SS, Goulas A, et al. Situating the default-mode network along a principal gradient of macroscale cortical organization. Proc Natl Acad Sci U S A. 2016;113(44):12574–12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hong S-J, de Wael RV, Bethlehem RAI, et al. Atypical functional connectome hierarchy in autism. Nat Commun. 2019;10(1):1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fornito A, Bullmore ET.. Reconciling abnormalities of brain network structure and function in schizophrenia. Curr Opin Neurobiol. 2015;30:44–50. [DOI] [PubMed] [Google Scholar]
- 68.Dong D, Wang Y, Chang X, Luo C, Yao D.. Dysfunction of large-scale brain networks in schizophrenia: A meta-analysis of resting-state functional connectivity. Schizophr Bull. 2018;44(1):168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Giraldo-Chica M, Woodward ND.. Review of thalamocortical resting-state fMRI studies in schizophrenia. Schizophr Res. 2017;180:58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gratton C, Laumann TO, Nielsen AN, et al. Functional brain networks are dominated by stable group and individual factors, not cognitive or daily variation. Neuron. 2018;98(2):439–452.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McCabe C, Mishor Z.. Antidepressant medications reduce subcortical-cortical resting-state functional connectivity in healthy volunteers. Neuroimage. 2011;57(4):1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Y, Tang W, Fan X, et al. Resting-state functional connectivity changes within the default mode network and the salience network after antipsychotic treatment in early-phase schizophrenia. Neuropsychiatr Dis Treat. 2017;13:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martin-Brevet S, Rodríguez-Herreros B, Nielsen JA, et al. Quantifying the effects of 16p11.2 copy number variants on brain structure: A multisite genetic-first study. Biol Psychiatry. 2018;84(4):253–264. [DOI] [PubMed] [Google Scholar]
- 74.Bai D, Yip BHK, Windham GC, et al. Association of genetic and environmental factors with autism in a 5-country cohort. JAMA Psychiatry. 2019;76(10):1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hilker R, Helenius D, Fagerlund B, et al. Heritability of schizophrenia and schizophrenia spectrum based on the Nationwide Danish Twin Register. Biol Psychiatry. 2018;83(6):492–498. [DOI] [PubMed] [Google Scholar]
- 76.Taylor MJ, Rosenqvist MA, Larsson H, et al. Etiology of autism spectrum disorders and autistic traits over time. JAMA Psychiatry. 2020;77(9):936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morris TT, Davies NM, Hemani G, Smith GD.. Population phenomena inflate genetic associations of complex social traits. Sci Adv. 2020;6(16):eaay0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gaugler T, Klei L, Sanders SJ, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46(8):881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Iossifov I, Ronemus M, Levy D, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74(2):285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pinto D, Delaby E, Merico D, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94(5):677–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moreno-De-Luca D, Sanders SJ, Willsey AJ, et al. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol Psychiatry. 2013;18(10):1090–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Douard E, Zeribi A, Schramm C, et al. Effect sizes of deletions and duplications on autism risk across the genome. Am J Psychiatry. 2021;178(1):87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sanders SJ, Murtha MT, Gupta AR, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485(7397):237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Coe BP, Stessman HAF, Sulovari A, et al. Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat Genet. 2019;51(1):106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Satterstrom FK, Kosmicki JA, Wang J, et al. ; iPSYCH-Broad Consortium. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 2020;180(3):568–584.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Howrigan DP, Rose SA, Samocha KE, et al. Exome sequencing in schizophrenia-affected parent-offspring trios reveals risk conferred by protein-coding de novo mutations. Nat Neurosci. 2020;23(2):185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Farrell MS, Werge T, Sklar P, et al. Evaluating historical candidate genes for schizophrenia. Mol Psychiatry. 2015;20(5):555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fromer M, Pocklington AJ, Kavanagh DH, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506(7487):179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Singh T, Kurki MI, Curtis D, et al. ; Swedish Schizophrenia Study. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci. 2016;19(4):571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Takata A, Xu B, Ionita-Laza I, Roos JL, Gogos JA, Karayiorgou M.. Loss-of-function variants in schizophrenia risk and SETD1A as a candidate susceptibility gene. Neuron. 2014;82(4):773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Walsh T, McClellan JM, McCarthy SE, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320(5875):539–543. [DOI] [PubMed] [Google Scholar]
- 92.International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nishino J, Ochi H, Kochi Y, Tsunoda T, Matsui S.. Sample size for successful genome-wide association study of major depressive disorder. Front Genet. 2018;9:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Downar J, Blumberger DM, Daskalakis ZJ.. The neural crossroads of psychiatric illness: An emerging target for brain stimulation. Trends Cogn Sci. 2016;20(2):107–120. [DOI] [PubMed] [Google Scholar]
- 95.Baribeau DA, Dupuis A, Paton TA, et al. Structural neuroimaging correlates of social deficits are similar in autism spectrum disorder and attention-deficit/hyperactivity disorder: Analysis from the POND Network. Transl Psychiatry. 2019;9(1):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Meda SA, Ruaño G, Windemuth A, et al. Multivariate analysis reveals genetic associations of the resting default mode network in psychotic bipolar disorder and schizophrenia. Proc Natl Acad Sci U S A. 2014;111(19):E2066–E2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ongür D, Lundy M, Greenhouse I, et al. Default mode network abnormalities in bipolar disorder and schizophrenia. Psychiatry Res. 2010;183(1):59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huntenburg JM, Bazin P-L, Margulies DS.. Large-scale gradients in human cortical organization. Trends Cogn Sci. 2018;22(1):21–31. [DOI] [PubMed] [Google Scholar]
- 99.Brainstorm Consortium. Analysis of shared heritability in common disorders of the brain. Science. 2018;360(6395):eaap8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cross-Disorder Group of the Psychiatric Genomics Consortium, Lee SH, Ripke S, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45(9):984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marshall M.The hidden links between mental disorders. Nature. 2020;581(7806):19–21. [DOI] [PubMed] [Google Scholar]
- 102.Kushima I, Aleksic B, Nakatochi M, et al. Comparative analyses of copy-number variation in autism spectrum disorder and schizophrenia reveal etiological overlap and biological insights. Cell Rep. 2018;24(11):2838–2856. [DOI] [PubMed] [Google Scholar]
- 103.Gandal MJ, Haney JR, Parikshak NN, et al. ; CommonMind Consortium. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science. 2018;359(6376):693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011;13(7):680–685. [DOI] [PubMed] [Google Scholar]
- 105.Weiss LA, Shen Y, Korn JM, et al. ; Autism Consortium. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358(7):667–675. [DOI] [PubMed] [Google Scholar]
- 106.Niarchou M, Chawner SJRA, Doherty JL, et al. Psychiatric disorders in children with 16p11.2 deletion and duplication. Transl Psychiatry. 2019;9(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.D’Angelo D, Lebon S, Chen Q, et al. ; for the Cardiff University Experiences of Children With Copy Number Variants (ECHO) Study, the 16p11.2 European Consortium, and the Simons Variation in Individuals Project (VIP) Consortium. Defining the effect of the 16p11.2 duplication on cognition, behavior, and medical comorbidities. JAMA Psychiatry. 2016;73(1):20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Moreno-De-Luca A, Evans DW, Boomer KB, et al. The role of parental cognitive, behavioral, and motor profiles in clinical variability in individuals with chromosome 16p11.2 deletions. JAMA Psychiatry. 2015;72(2):119–126. [DOI] [PubMed] [Google Scholar]
- 109.Hippolyte L, Maillard AM, Rodriguez-Herreros B, et al. ; 16p11.2 European Consortium, Simons Variation in Individuals Project Consortium. The number of genomic copies at the 16p11.2 locus modulates language, verbal memory, and inhibition. Biol Psychiatry. 2016;80(2):129–139. [DOI] [PubMed] [Google Scholar]
- 110.Jacquemont S, Reymond A, Zufferey F, et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature. 2011;478(7367):97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zufferey F, Sherr EH, Beckmann ND, et al. ; 16p11.2 European Consortium. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J Med Genet. 2012;49(10):660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Maillard AM, Ruef A, Pizzagalli F, et al. ; 16p11.2 European Consortium. The 16p11.2 locus modulates brain structures common to autism, schizophrenia and obesity. Mol Psychiatry. 2015;20(1):140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bertero A, Liska A, Pagani M, et al. Autism-associated 16p11.2 microdeletion impairs prefrontal functional connectivity in mouse and human. Brain. 2018;141(7):2055–2065. [DOI] [PubMed] [Google Scholar]
- 114.Jonas RK, Montojo CA, Bearden CE.. The 22q11.2 deletion syndrome as a window into complex neuropsychiatric disorders over the lifespan. Biol Psychiatry. 2014;75(5):351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Karayiorgou M, Morris MA, Morrow B, et al. Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc Natl Acad Sci U S A. 1995;92(17):7612–7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Philip N, Bassett A.. Cognitive, behavioural and psychiatric phenotype in 22q11.2 deletion syndrome. Behav Genet. 2011;41(3):403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schneider M, Debbané M, Bassett AS, et al. ; for the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: Results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am J Psychiatry. 2014;171(6):627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rees E, Kirov G, Sanders A, et al. ; Wellcome Trust Case Control Consortium. Evidence that duplications of 22q11.2 protect against schizophrenia. Mol Psychiatry. 2014;19(1):37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Olsen L, Sparsø T, Weinsheimer SM, et al. Prevalence of rearrangements in the 22q11.2 region and population-based risk of neuropsychiatric and developmental disorders in a Danish population: A case-cohort study. Lancet Psychiatry. 2018;5(7):573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sun D, Ching CRK, Lin A, et al. Large-scale mapping of cortical alterations in 22q11.2 deletion syndrome: Convergence with idiopathic psychosis and effects of deletion size. Mol Psychiatry. 2020;25(8):1822–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ching CRK, Gutman BA, Sun D, et al. Mapping subcortical brain alterations in 22q11.2 deletion syndrome: Effects of deletion size and convergence with idiopathic neuropsychiatric illness. Am J Psychiatry. 2020;177(7):589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lin A, Ching CRK, Vajdi A, et al. Mapping 22q11.2 gene dosage effects on brain morphometry. J Neurosci. 2017;37(26):6183–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mattiaccio LM, Coman IL, Thompson CA, Fremont WP, Antshel KM, Kates WR.. Frontal dysconnectivity in 22q11.2 deletion syndrome: An atlas-based functional connectivity analysis. Behav Brain Funct. 2018;14(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schleifer C, Lin A, Kushan L, et al. Dissociable disruptions in thalamic and hippocampal resting-state functional connectivity in youth with 22q11.2 deletions. J Neurosci. 2019;39(7):1301–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Escamilla CO, Filonova I, Walker AK, et al. Kctd13 deletion reduces synaptic transmission via increased RhoA. Nature. 2017;551(7679):227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Richter M, Murtaza N, Scharrenberg R, et al. Altered TAOK2 activity causes autism-related neurodevelopmental and cognitive abnormalities through RhoA signaling. Mol Psychiatry. 2019;24(9):1329–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Golzio C, Willer J, Talkowski ME, et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485(7398):363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hiroi N, Takahashi T, Hishimoto A, Izumi T, Boku S, Hiramoto T.. Copy number variation at 22q11.2: From rare variants to common mechanisms of developmental neuropsychiatric disorders. Mol Psychiatry. 2013;18(11):1153–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Motahari Z, Moody SA, Maynard TM, LaMantia A-S.. In the line-up: Deleted genes associated with DiGeorge/22q11.2 deletion syndrome: Are they all suspects? J Neurodev Disord. 2019;11(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ellegood J, Anagnostou E, Babineau BA, et al. Clustering autism: Using neuroanatomical differences in 26 mouse models to gain insight into the heterogeneity. Mol Psychiatry. 2015;20(1):118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Warland A, Kendall KM, Rees E, Kirov G, Caseras X.. Schizophrenia-associated genomic copy number variants and subcortical brain volumes in the UK Biobank. Mol Psychiatry. 2020;25(4):854–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Huguet G, Schramm C, Douard E,. et al. Genome-wide analysis of gene dosage in 24,092 individuals estimates that 10,000 genes modulate cognitive ability. Mol Psychiatry. Published online 7 January 2021. doi:10.1038/s41380-020-00985-z [DOI] [PMC free article] [PubMed]
- 133.Huguet G, Schramm C, Douard E, et al. ; for the IMAGEN Consortium. Measuring and estimating the effect sizes of copy number variants on general intelligence in community-based samples. JAMA Psychiatry. 2018;75(5):447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lek M, Karczewski KJ, Minikel EV, et al. ; Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Drakesmith M, Parker GD, Smith J, et al. Genetic risk for schizophrenia and developmental delay is associated with shape and microstructure of midline white-matter structures. Transl Psychiatry. 2019;9(1):102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shen L, Thompson PM.. Brain imaging genomics: Integrated analysis and machine learning. Proc IEEE. 2020;135:108(1):125–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Neilson E, Shen X, Cox SR, et al. Impact of polygenic risk for schizophrenia on cortical structure in UK biobank. Biol Psychiatry. 2019;86(7):536–544. [DOI] [PubMed] [Google Scholar]
- 138.Reus LM, Shen X, Gibson J, et al. Association of polygenic risk for major psychiatric illness with subcortical volumes and white matter integrity in UK Biobank. Sci Rep. 2017;7:42140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Van der Auwera S, Wittfeld K, Homuth G, Teumer A, Hegenscheid K, Grabe HJ.. No association between polygenic risk for schizophrenia and brain volume in the general population. Biol Psychiatry. 2015;78(11):e41–e42. [DOI] [PubMed] [Google Scholar]
- 140.Alnæs D, Kaufmann T, van der Meer D, et al. ; Karolinska Schizophrenia Project Consortium. Brain heterogeneity in schizophrenia and its association with polygenic risk. JAMA Psychiatry. 2019;76(7):739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Alemany S, Jansen PR, Muetzel RL, et al. Common polygenic variations for psychiatric disorders and cognition in relation to brain morphology in the general pediatric population. J Am Acad Child Adolesc Psychiatry. 2019;58(6):600–607. [DOI] [PubMed] [Google Scholar]
- 142.Grotzinger AD, Rhemtulla M, de Vlaming R, et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat Hum Behav. 2019;3(5):513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Burt JB, Demirtaş M, Eckner WJ, et al. Hierarchy of transcriptomic specialization across human cortex captured by structural neuroimaging topography. Nat Neurosci. 2018;21(9):1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Müller E, Munn B, Hearne LJ, et al. Core and matrix thalamic sub-populations relate to spatio-temporal cortical connectivity gradients. Neuroimage. 2020;222:117224. [DOI] [PubMed] [Google Scholar]
- 145.Lewis CM, Vassos E.. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020;12(1):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hawrylycz M, Miller JA, Menon V, et al. Canonical genetic signatures of the adult human brain. Nat Neurosci. 2015;18(12):1832–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Seidlitz J, Nadig A, Liu S, et al. Transcriptomic and cellular decoding of regional brain vulnerability to neurogenetic disorders. Nat Commun. 2020;11(1):3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Writing Committee for the Attention-Deficit/Hyperactivity Disorder; Autism Spectrum Disorder; Bipolar Disorder; Virtual Histology of Cortical Thickness and Shared Neurobiology in 6 Psychiatric Disorders. JAMA Psychiatry. 2021;78(1):47–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Whitaker KJ, Vértes PE, Romero-Garcia R, et al. ; NSPN Consortium. Adolescence is associated with genomically patterned consolidation of the hubs of the human brain connectome. Proc Natl Acad Sci U S A. 2016;113(32):9105–9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kebets V, Holmes AJ, Orban C, et al. Somatosensory-motor dysconnectivity spans multiple transdiagnostic dimensions of psychopathology. Biol Psychiatry. 2019;86(10):779–791. [DOI] [PubMed] [Google Scholar]
- 151.Reardon PK, Seidlitz J, Vandekar S, et al. Normative brain size variation and brain shape diversity in humans. Science. 2018;360(6394):1222–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.van den Heuvel MP, Scholtens LH, de Lange SC, et al. Evolutionary modifications in human brain connectivity associated with schizophrenia. Brain. 2019;142(12):3991–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Esteban O, Markiewicz CJ, Blair RW, et al. fMRIPrep: A robust preprocessing pipeline for functional MRI. Nat Methods. 2019;16(1):111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pedregosa F, Varoquaux G, Gramfort A.. Scikit-learn: Machine learning in Python. J Mach Learn Res. 2011;12(85):2825−2830. [Google Scholar]
- 155.Crawford KL, Neu SC, Toga AW.. The image and data archive at the laboratory of neuro imaging. Neuroimage. 2016;124(Pt B):1080–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Das S, Zijdenbos A, Vins D, Harlap J, Evans A.. LORIS: A web-based data management system for multi-center studies. Front Neuroinform. 2011;5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gorgolewski KJ, Auer T, Calhoun VD, et al. The brain imaging data structure, a format for organizing and describing outputs of neuroimaging experiments. Sci Data. 2016;3:160044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Moreau CA, Jean-Louis M, Blair R, et al. The genetics-BIDS extension: Easing the search for genetic data associated with human brain imaging. Gigascience. 2020;9(10):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sullivan PF, Agrawal A, Bulik CM, et al. ; for the Psychiatric Genomics Consortium. Psychiatric genomics: An update and an agenda. Am J Psychiatry. 2018;175(1):15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Thompson PM, Stein JL, Medland SE, et al. ; Alzheimer’s Disease Neuroimaging Initiative, EPIGEN Consortium, IMAGEN Consortium, Saguenay Youth Study (SYS) Group. The ENIGMA Consortium: Large-scale collaborative analyses of neuroimaging and genetic data. Brain Imaging Behav. 2014;8(2):153–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sudlow C, Gallacher J, Allen N, et al. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3):e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.McPherson JD, Marra M, Hillier L, et al. A physical map of the human genome. Nature. 2001;409:934–941. [DOI] [PubMed] [Google Scholar]
- 163.Weiner MW, Aisen PS, Jack CR Jr, et al. ; Alzheimer's Disease Neuroimaging Initiative. The Alzheimer’s disease neuroimaging initiative: progress report and future plans. Alzheimers Dement. 2010;6(3):202–211.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.1000 Genomes Project Consortium, Abecasis GR, Altshuler D, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.HD-200 Consortium. The ADHD-200 Consortium: A model to advance the translational potential of neuroimaging in clinical neuroscience. Front Syst Neurosci. 2012;6:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Poldrack RA, Barch DM, Mitchell JP, et al. Toward open sharing of task-based fMRI data: The OpenfMRI project. Front Neuroinform. 2013;7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Amunts K, Ebell C, Muller J, Telefont M, Knoll A, Lippert T.. The human brain project: Creating a European research infrastructure to decode the human brain. Neuron. 2016;92(3):574–581. [DOI] [PubMed] [Google Scholar]
- 168.Gorgolewski KJ, Varoquaux G, Rivera G, et al. NeuroVault.org: A web-based repository for collecting and sharing unthresholded statistical maps of the human brain. Front Neuroinform. 2015;9:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Glasser MF, Smith SM, Marcus DS, et al. The Human Connectome Project’s neuroimaging approach. Nat Neurosci. 2016;19(9):1175–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jernigan TL, Brown TT, Hagler DJ Jr, et al. ; Pediatric Imaging, Neurocognition and Genetics Study. The Pediatric Imaging, Neurocognition, and Genetics (PING) Data Repository. Neuroimage. 2016;124(Pt B):1149–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kogan A, Alpert K, Ambite JL, Marcus DS, Wang L.. Northwestern University schizophrenia data sharing for SchizConnect: A longitudinal dataset for large-scale integration. Neuroimage. 2016;124(Pt B):1196–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Loth E, Charman T, Mason L, et al. The EU-AIMS Longitudinal European Autism Project (LEAP): Design and methodologies to identify and validate stratification biomarkers for autism spectrum disorders. Mol Autism. 2017;8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Casey BJ, Cannonier T, Conley MI, et al. ; ABCD Imaging Acquisition Workgroup. The Adolescent Brain Cognitive Development (ABCD) study: Imaging acquisition across 21 sites. Dev Cogn Neurosci. 2018;32:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]