

Abstract
The ageing brain is vulnerable to a wide array of neuropathologies. Prior work estimated that the three most studied of these, Alzheimer’s disease, infarcts, and Lewy bodies, account for ∼40% of the variation in late life cognitive decline. However, that estimate did not incorporate many other diseases that are now recognized as potent drivers of cognitive decline [e.g. limbic predominant age-related TDP-43 encephalopathy (LATE-NC), hippocampal sclerosis, other cerebrovascular conditions]. We examined the degree to which person-specific cognitive decline in old age is driven by a wide array of neuropathologies. Deceased participants (n = 1164) from two longitudinal clinical-pathological studies, the Rush Memory and Aging Project and Religious Orders Study, completed up to 24 annual evaluations including 17 cognitive performance tests and underwent brain autopsy. Neuropathological examinations provided 11 pathological indices, including markers of Alzheimer’s disease, non- Alzheimer’s disease neurodegenerative diseases (i.e. LATE-NC, hippocampal sclerosis, Lewy bodies), and cerebrovascular conditions (i.e. macroscopic infarcts, microinfarcts, cerebral amyloid angiopathy, atherosclerosis, and arteriolosclerosis). Mixed effects models examined the linear relation of pathological indices with global cognitive decline, and random change point models examined the relation of the pathological indices with the onset of terminal decline and rates of preterminal and terminal decline. Cognition declined an average of about 0.10 unit per year (estimate = −0.101, SE = 0.003, P < 0.001) with considerable heterogeneity in rates of decline (variance estimate for the person-specific slope of decline was 0.0094, P < 0.001). When considered separately, 10 of 11 pathological indices were associated with faster decline and accounted for between 2% and 34% of the variation in decline, respectively. When considered simultaneously, the 11 pathological indices together accounted for 43% of the variation in decline; Alzheimer’s disease-related indices accounted for 30–36% of the variation, non-Alzheimer’s disease neurodegenerative indices 4–10%, and cerebrovascular indices 3–8%. Finally, the 11 pathological indices combined accounted for less than a third of the variation in the onset of terminal decline (28%) and rates of preterminal (32%) and terminal decline (19%). Although age-related neuropathologies account for a large proportion of the variation in late life cognitive decline, considerable variation remains unexplained even after considering a wide array of neuropathologies. These findings highlight the complexity of cognitive ageing and have important implications for the ongoing effort to develop effective therapeutics and identify novel treatment targets.
Keywords: cognitive ageing, neuropathology, Alzheimer's disease, vascular, dementia
Boyle et al. examined the degree to which person-specific cognitive decline in old age is driven by an array of neuropathologies. Surprisingly, the neuropathologies together explained only 43% of the decline, suggesting that other factors are at play and warrant investigation as potential therapeutic targets.
Introduction
Prevention of late life cognitive decline is a top priority in ageing research, but efforts to mitigate cognitive decline continue to show limited success. This is in large part because cognitive ageing is a complex and heterogeneous phenomenon affected by a wide array of neuropathologies. That is, although it was long thought that late life cognitive decline was primarily the result of Alzheimer’s disease, it is now clear that the ageing brain is vulnerable to a variety of cognition impairing neuropathologies, including non-Alzheimer’s disease neurodegenerative diseases and cerebrovascular conditions.1-5 Neuropathologies can accumulate years or even decades prior to cognitive impairment, but their prevalence increases dramatically with advancing age, with one or more pathologies evident in many individuals over the age of 60 and nearly all over 80.1,6,7 Moreover, distinct diseases contribute to particular manifestations of dementia, but age-related neuropathologies also accumulate in individuals without cognitive impairment and can differentially impact cognition depending on their combination, severity and pattern of accumulation.7–11 We previously reported that the three most studied neuropathologies, Alzheimer’s disease, infarcts, and Lewy bodies, account for ∼40% of the person-specific variation in late life cognitive decline.12 However, that estimate did not incorporate many other diseases that have since emerged as potent drivers of cognitive decline [e.g. limbic predominant age-related TDP-43 encephalopathy (LATE-NC), hippocampal sclerosis, other vascular conditions].3,5,13 Further, we now have a much larger sample size and longer follow-up, which provide additional power. Understanding the extent to which the fuller complement of age-related neuropathologies account for cognitive decline is essential to inform research priorities and facilitate effective therapeutic interventions.
In this study, we examined the degree to which person-specific cognitive decline in old age is driven by a wide array of neuropathologies, including three Alzheimer’s disease indices, three non-Alzheimer’s disease neurodegenerative diseases, and five cerebrovascular indices. Participants were more than 1100 deceased persons from two longitudinal clinical-pathological studies, the Rush Memory and Aging Project and Religious Orders Study.14 They completed detailed annual cognitive assessments for up to 24 years (mean = 8.7) and underwent brain autopsy. Neuropathological examinations provided 11 pathological indices (i.e. global Alzheimer’s disease, amyloid-β, PHFtau tangles, LATE-NC, hippocampal sclerosis, Lewy bodies, macroscopic infarcts, microinfarcts, cerebral amyloid angiopathy, atherosclerosis and arteriolosclerosis). Linear mixed effects models first examined the relation of pathological indices with the annual rate of global cognitive decline, and random change point models then examined the relation of the pathological indices with the onset of terminal decline and rates of preterminal and terminal cognitive decline.
Materials and methods
Participants
Data came from deceased older subjects from two ongoing clinical-pathological cohort studies, the Religious Orders Study (ROS), which began in 1994, and the Rush Memory and Aging Project (MAP), which began in 1997.14 ROS participants are older Catholic nuns, priests, and monks from across the USA, and MAP participants are community-based older individuals from the greater Chicago area. The studies share essentially identical designs and operations. Participants enrol without known dementia and undergo detailed annual clinical evaluations and brain autopsy. All participants signed an informed consent form and Anatomical Gift Act for organ donation and an Institutional Review Board of Rush University Medical Center approved each study.
Eligibility for these analyses required at least two cognitive evaluations and complete data on all 11 pathological indices. At the time of these analyses (16 December 2019), 3614 participants had completed the baseline evaluation. We excluded 142 who died before the first follow-up cognitive evaluation and 185 who were enrolled less than 1 year. This left 3287 individuals and follow-up cognitive data were available for 3274 (99.6%). Of those, 1747 died and complete neuropathological data were available for 1344 (76.9%). We further excluded 180 who had other pathological diagnoses (e.g. brain tumour). Thus, there were a total of 1164 individuals in the main analytic group.
Clinical evaluation and cognitive assessment
Annual clinical evaluations include detailed neurological examinations, cognitive assessments, and medical history interviews.14,15 Cognitive function is assessed annually using a battery of 19 cognitive performance tests. Two tests (Mini-Mental State Examination, Complex Ideational Material) are used only for diagnostic purposes. The remaining 17 tests are used to create a composite measure of global cognitive function; tests include: immediate and delayed recall of the East Boston Story and Logical Memory Story A; Word List Memory, Word List Recall, Word List Recognition; Symbol Digit Modalities Test; Number Comparison; Boston Naming Test; Verbal Fluency; Word Reading; Digit Span Forward; Digit Span Backward; Digit Ordering; Judgment of Line Orientation; Standard Progressive Matrices. To generate the composite score, we converted raw scores on each test to z-scores, using the baseline mean and standard deviation (SD) from the combined parent studies, and then averaged the z-scores together. The use of a composite score minimizes floor and ceiling effects and other sources of random variability and is optimal for studies examining change in cognition over many years.12,14,16,17
Neuropathological assessment
Details of the neuropathological assessment and creation of summary measures of 11 neuropathologies, including markers of Alzheimer’s disease (i.e. global Alzheimer’s disease pathology, amyloid-β, PHFtau tangles), non-Alzheimer’s disease neurodegenerative diseases (i.e. LATE-NC, hippocampal sclerosis, Lewy bodies), and cerebrovascular conditions (i.e. macroscopic infarcts, microinfarcts, cerebral amyloid angiopathy, atherosclerosis, arteriolosclerosis) are provided in the Supplementary material.
Post-mortem MRI
In supporting analyses, we examined whether a metric of tissue integrity derived from post-mortem MRI, the transverse relaxation rate constant R2, might serve as a relatively independent pathological marker associated with late life cognitive decline. MRI procedures have been described previously.17 For the R2 measure, we identified groups of voxels whose R2 values trended together in different R2 characteristics within those voxels. To this end, we adapted FSL’s ‘melodic’ tool to carry out independent component analysis (ICA)1 on smoothed versions (half-width = 1.25 mm) of the normalized R2 maps. We derived 30 ICs as a trade-off between dimensionality reduction and spatial specificity of the resultant IC maps. This number of ICs captured most of the variance in the R2 images and is in line with several prior brain imaging studies that used a range of imaging modalities. The mixing matrix returned by the ICA tool contains the 30 IC weights or loadings for each specimen, which reflect the influence of each IC in the composition of each individual R2 map. We used these IC values in analyses.
Statistical analysis
We first used linear mixed effects models to characterize the linear rates of cognitive decline, examine the relation of pathology with cognitive decline, and determine the contribution of the pathological indices to the reduction of between-subject variation in cognitive decline. The outcome of interest was the longitudinal global cognition score. In a linear mixed effects model, each individual cognitive trajectory is assumed to follow a mean linear path of change in cognition function plus a random intercept and a random slope, which reflect the subject-specific level of cognition and the subject-specific rate of change in cognition. The variance of the random slope captures the between-subject variation in cognitive decline. We began with an unadjusted model with only the term for time, defined as the time in years before death, and then added in terms for the pathological indices and their interactions with time in a series of models. Each pathology × time interaction characterizes the difference from the mean slopes of cognitive decline due to the particular pathological index. Consequently, the reduction of the random slope variance from the unadjusted model to pathology-adjusted model reflects the between-subject variation in cognitive decline explained by the corresponding pathology. We first analysed the effect of each pathological index, and then included simultaneously the six pathological indices considered previously. However, as we have several additional pathological indices currently available, the fully adjusted core model included 11 pathological indices. We also conducted two sensitivity analyses: (i) we excluded persons whose cognition improved over time; and (ii) we repeated the fully adjusted model using two sub-composites of cognition based on tests that require vision and those that do not (Supplementary material) to examine whether vision altered the associations between the pathological indices and cognitive decline. Finally, we examined the three classes of pathology as groupings (i.e. Alzheimer’s disease pathologies, non-Alzheimer’s disease neurodegenerative pathologies, and cerebrovascular diseases) and varied the order of entry of the three pathology groups into the model to determine the range of variances explained by each class of pathology.
Next, because cognitive decline accelerates in the years just prior to death (i.e. terminal decline),18 we further applied random change point models to determine when the rate of cognitive decline accelerated prior to death and characterize rates of cognitive decline before and after the change point. The random change point models included four basic components: the change point, preterminal slope, terminal slope, and intercept proximate to death. The change point indicates the onset of terminal decline, and the preterminal and terminal slopes indicate the rate of change before and after its onset; the intercept reflects the level of cognition proximate to death. Each component was parameterized as a linear function of variables of interest, including the pathological indices. The term for each pathological index on the change point estimates the association of that pathological index with the onset of terminal decline. The term for each pathological index in the preterminal slope estimates the association of that pathological index with the rate of preterminal decline. The term for each pathological index in the terminal slope estimates the association of that pathological index with the rate of decline during the terminal period. The pathological indices, if significant, account for a proportion of the total variances in random change point, and preterminal and terminal slopes, and the reduction of the random slope variance reflects the between-subject variation in cognitive decline explained by pathological indices. Model estimation was done with a Bayesian Monte Carlo Markov Chain approach implemented in OpenBUGS software.
Data availability
The data that support the findings of this study are available upon request via the Rush Alzheimer’s Disease Center Resource Sharing Hub (www.radc.rush.edu).
Results
Descriptive characteristics of participants
At the time of these analyses, 1164 participants had died with two or more cognitive evaluations and had complete autopsy data. They had an average of 8.4 years of follow up (SD = 5.0, range = 1–23), an average age at death of 90.0 (SD = 6.4, range = 65.9–108.3), and 806 (69.2%) were females. Additional descriptive data are provided in Table 1. At autopsy, 1160 (99.7%) persons had Alzheimer’s disease pathology (global Alzheimer’s disease pathology > 0); more specifically, 1025 (88.1%) patients were positive for amyloid-β, and 1163 (99.9%) were positive for PHFtau tangles; additional neuropathological data are provided in Table 1. Table 2 shows the intercorrelations among pathology variables and demographics.
Table 1.
Descriptive‘ characteristics of the cohort
| Variable | Mean (SD) or n (%) |
|---|---|
| Age at death, years | 90.0 (6.4) |
| Education, years | 16.2 (3.6) |
| Female | 806 (69.2%) |
| Follow-up years | 8.4 (5.0) |
| Baseline global cognition | −0.11 (0.61) |
| Proximate to death global cognition | −0.75 (1.21) |
| Global Alzheimer’s disease pathology | 0.79 (0.63) |
| Amyloid-β, square-root transformed | 1.67 (1.13) |
| PHFtau tangles, square-root transformed | 1.73 (1.35) |
| Neocortical Lewy bodiesa | 159 (13.7%) |
| LATE-NC (4 stages) | |
| 0 (None) | 549 (47.2%) |
| 1 (Amygdala) | 216 (18.6%) |
| 2 (Amygdala + Limbic) | 124 (10.7%) |
| 3 (Amygdala + Limbic + Neocortical) | 275 (23.6%) |
| Hippocampal sclerosisa | 117 (10.1%) |
| Gross infarcts, chronica | 420 (36.1%) |
| Microscopic infarcts, chronica | 352 (30.2%) |
| Atherosclerosis | |
| 0 (none) | 221 (19.0%) |
| 1 (mild) | 570 (49.0%) |
| 2 (moderate) | 294 (25.3%) |
| 3 (severe) | 79 (6.8%) |
| Arteriolosclerosis | |
| 0 (none) | 376 (32.3%) |
| 1 (mild) | 431 (37.0%) |
| 2 (moderate) | 274 (23.5%) |
| 3 (severe) | 83 (7.1%) |
| Cerebral amyloid angiopathy | |
| 0 (None) | 244 (21.0%) |
| 1 (Mild) | 490 (42.1%) |
| 2 (Moderate) | 282 (24.2%) |
| 3 (Severe) | 148 (12.7%) |
| Braak Score | |
| 0 | 9 (<1%) |
| 1,2 | 159 (14%) |
| 3,4 | 643 (55%) |
| 5,6 | 353 (30%) |
| CERAD Score | |
| None | 239 (20.5%) |
| Sparse | 94 (8%) |
| Moderate | 412 (35%) |
| Frequent | 419 (36%) |
| Thal phase | |
| 0 | 74 (8%) |
| 1,2 | 167 (18%) |
| 3 | 267 (29%) |
| 4,5 | 406 (44%) |
Lewy bodies present.
Table 2.
Intercorrelations‘ among the pathological indices
| Variable | Global Alzheimer’s disease | Amyloid-β | PHFtau tangles | LB | LATE-NC | HS | Gross infarcts | Micro-infarcts | Athero- sclerosis | Arteriolo- sclerosis | CAA |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Global Alzheimer’s disease | 0.77a | 0.73a | 0.07b | 0.22a | 0.08b | −0.003 | −0.02 | 0.007 | 0.03 | 0.39a | |
| Amyloid-β | 0.53a | 0.06b | 0.19a | 0.08b | 0.03 | 0.01 | −0.04 | 0.03 | 0.36a | ||
| PHFtau tangles | 0.10a | 0.3a | 0.11a | 0.03 | −0.002 | 0.04 | 0.08b | 0.32a | |||
| LB | 0.07b | 0.05c | −0.02 | −0.006 | −0.07b | −0.05c | −0.03 | ||||
| LATE-NC | 0.35a | 0.05c | 0.04 | 0.07b | 0.09b | 0.14a | |||||
| HS | 0.04 | 0.04 | 0.02 | 0.06c | 0.06b | ||||||
| Gross infarcts | 0.21a | 0.25a | 0.19a | 0.007 | |||||||
| Micro-infarcts | 0.08b | 0.09b | 0.003 | ||||||||
| Atherosclerosis | 0.34a | −0.004 | |||||||||
| Arteriolosclerosis | 0.03 |
CAA = cerebral amyloid-β angiopathy; HS = hippocampal sclerosis; LB = neocortical Lewy bodies.
P < 0.001.
P < 0.05.
Trend, P < 0.10, based on Spearman correlations.
Heterogeneity of age-related cognitive decline
We first used linear mixed effects models to estimate the mean trajectory of cognitive decline and the variability of linear decline across individuals. In the initial unadjusted model, global cognitive function declined a mean of about 0.1 standard units per year (estimate = −0.101, SE = 0.003, P < 0.001; Table 3); the variance of the subject-specific slope of decline was 0.0094 (P < 0.001; Table 4). Figure 1 shows a spaghetti plot of the observed longitudinal trajectories for a random sample (n = 50) of participants (Fig. 1, left), and the model-derived mean trajectory of cognitive decline (Fig. 1, right) superimposed on the estimated individual trajectories for those participants. Considerable heterogeneity is evident, with some individuals improving, some remaining stable, some declining moderately (close to mean trajectory), and some declining rapidly.
Table 3.
Association of‘ each pathological index with rate of cognitive decline
|
Separate models
|
Multivariate models
|
|||||
|---|---|---|---|---|---|---|
|
Six pathologies
|
Eleven pathologies
|
|||||
| Estimate (SE) | P-value | Estimate (SE) | P-value | Estimate (SE) | P-value | |
| Global Alzheimer’s disease pathology | −0.080 (0.005) | <0.001 | −0.020 (0.008) | 0.013 | −0.021 (0.008) | 0.009 |
| Amyloid-β | −0.030 (0.003) | <0.001 | −0.002 (0.003) | 0.596 | −0.001 (0.003) | 0.730 |
| PHFtau tangles | −0.043 (0.002) | <0.001 | −0.034 (0.003) | <0.001 | −0.032 (0.003) | <0.001 |
| Neocortical Lewy bodies | −0.059 (0.009) | <0.001 | −0.045 (0.008) | <0.001 | −0.047 (0.008) | <0.001 |
| LATE-NC (4 stages) | −0.021 (0.003) | <0.001 | – | – | −0.003 (0.002) | 0.278 |
| Hippocampal sclerosis | −0.064 (0.010) | <0.001 | – | – | −0.035 (0.009) | <0.001 |
| Gross infarcts, chronic | −0.022 (0.007) | <0.001 | −0.024 (0.006) | <0.001 | −0.015 (0.006) | 0.010 |
| Microscopic infarcts, chronic | −0.003 (0.007) | 0.627 | −0.0005 (0.006) | 0.938 | 0.002 (0.006) | 0.686 |
| Atherosclerosis | −0.020 (0.004) | <0.001 | – | – | −0.015 (0.003) | <0.001 |
| Arteriolosclerosis | −0.015 (0.003) | <0.001 | – | – | −0.006 (0.003) | 0.073 |
| Cerebral amyloid angiopathy | −0.023 (0.003) | <0.001 | – | – | −0.002 (0.003) | 0.444 |
Table 4.
Variances explained‘ by each of the pathological indices and combinations thereof
| Predictor |
Global cognition decline
|
||
|---|---|---|---|
| Total variance | Reduction | % Total variance explained | |
| Reference model | 0.0094 | – | – |
| Separate models | |||
| Global Alzheimer’s disease pathology | 0.0069 | 0.0025 | 27% |
| Amyloid-β | 0.0082 | 0.0012 | 13% |
| PHFtau tangles | 0.0061 | 0.0033 | 35% |
| Neocortical Lewy bodies | 0.0090 | 0.0004 | 4% |
| LATE-NC (4 stages) | 0.0088 | 0.0006 | 7% |
| Hippocampal sclerosis | 0.0091 | 0.0003 | 3% |
| Gross infarcts, chronic | 0.0093 | 0.0001 | 1% |
| Microscopic infarcts, chronic | 0.0094 | 0.0000 | 0% |
| Atherosclerosis | 0.0092 | 0.0002 | 3% |
| Arteriolosclerosis | 0.0093 | 0.0001 | 2% |
| Cerebral amyloid angiopathy | 0.0090 | 0.0004 | 5% |
| Multivariate models | |||
| Six pathologiesa | 0.0057 | 0.0037 | 40% |
| All 11 pathologies | 0.0054 | 0.0040 | 43% |
Includes global‘ Alzheimer’s disease pathology, amyloid-β, PHFtau tangles, neocortical Lewy bodies, gross infarcts, and microscopic infarcts.
Figure 1.

Spaghetti plot of individual‘ trajectories from a random sample of subjects (n = 50, left) and mean slope of cognitive decline superimposed on their estimated individual slopes (model-derived slopes, right).
Relation of age-related neuropathologies to cognitive decline
In separate analyses, we first examined the relation of each pathological index with the rate of cognitive decline. Global Alzheimer’s disease pathology was associated with a faster rate of cognitive decline (estimate = −0.080, SE = 0.005, P < 0.001), and alone explained 27% of the between-subject variability in decline. Amyloid-β (estimate = −0.030, SE = 0.003, P < 0.001) and PHFtau tangles (estimate = −0.043, SE = 0.002, P < 0.001) were also associated with a faster rate of decline and explained 13% and 35% of the between-subject variance, respectively. Similarly, other neurodegenerative pathologies were associated with a faster rate of cognitive decline but accounted for less of the between-subject variance (neocortical Lewy bodies estimate = −0.102, SE = 0.014, P < 0.001, 8% variance explained; LATE-NC estimate = −0.021, SE = 0.003, P < 0.001, 7% variance explained; hippocampal sclerosis estimate = −0.064, SE = 0.010, P < 0.001, 3% variance explained). Finally, with the exception of microinfarcts, all vascular indices were associated with faster decline but accounted for the least between-subject variance (atherosclerosis estimate = −0.020, SE = 0.004, P < 0.001, 3% variance explained; arteriolosclerosis estimate = −0.015, SE = 0.003, P < 0.001, 2% variance explained; gross infarcts estimate = −0.022, SE = 0.007, P < 0.001, 1% variance explained; cerebral amyloid-β angiopathy estimate = −0.023, SE = 0.003, P < 0.001, 5% variance explained). See Tables 3 and 4 for details.
Next, we examined the relation of multiple pathological indices simultaneously. We first considered the six pathologies examined previously (i.e. global Alzheimer’s disease pathology, amyloid-β, PHFtau tangles, gross infarcts, microinfarcts, and neocortical Lewy bodies) for comparison with our prior work as we are now using a much larger sample with considerably longer follow-up data. Together, the six pathological indices accounted for 40% of the variation in cognitive decline, which is almost exactly the same as previously reported (41%). Then, we included all 11 pathologies simultaneously (Table 3). Together, the 11 pathological indices accounted for 43% of the variation in cognitive decline.
In sensitivity analyses, because neuropathologies are associated with cognitive decline rather than improvement, we conducted an analysis in which we excluded participants who improved and repeated the core analysis only among decliners. The core findings were essentially unchanged (data not shown). Further, because vision impairment is common among older subjects and can affect cognitive performance, we examined the association of neuropathologies with cognitive decline separately for cognitive tests that require vision and those that do not. The results largely remained unchanged; the only exception was gross infarcts, which were only significantly associated with decline in tests that did not require vision (Supplementary material).
Contribution of different classes of neuropathologies to cognitive decline
To facilitate an understanding of the degree to which different classes of neuropathologies contribute to cognitive decline, we grouped the pathological indices into three subgroups: (i) Alzheimer’s disease-related neurodegenerative pathology, i.e. amyloid-β, PHFtau tangles, and global Alzheimer’s disease pathology; (ii) other neurodegenerative pathologies, i.e. neocortical Lewy bodies, LATE-NC, and hippocampal sclerosis; and (iii) cerebrovascular pathologies, i.e. atherosclerosis, arteriolosclerosis, gross infarcts, microinfarcts and cerebral amyloid angiopathy. Figure 2 is based on the fully adjusted model and shows the additive effects of the three classes on the rate of cognitive decline.
Figure 2.
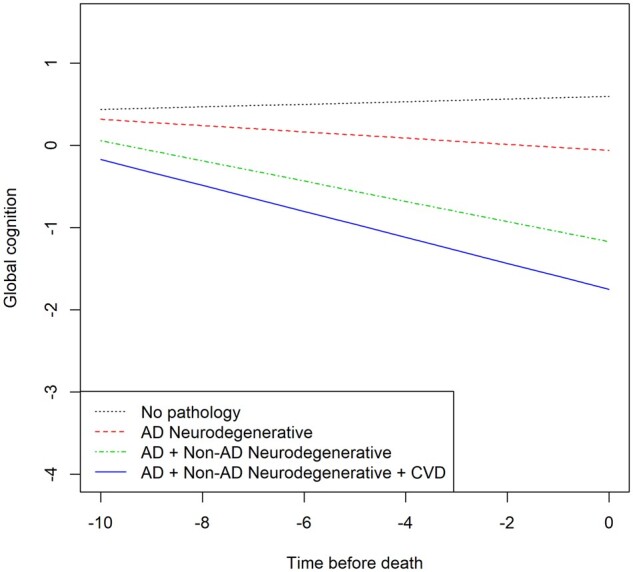
Contributions of‘ combinations of the pathological indices to cognitive decline (model derived slopes). Alzheimer’s disease (AD) = neurodegenerative pathologies related to Alzheimer’s disease, i.e. global Alzheimer’s disease pathology, amyloid-β and PHFtau tangles; CVD = cerebrovascular diseases, i.e. gross infarcts, microscopic infarcts, atherosclerosis, arteriolosclerosis, cerebral amyloid-β angiopathy; non-Alzheimer’s disease (non-AD) = neurodegenerative pathologies, i.e. Lewy bodies, hippocampal sclerosis and LATE-NC.
We further investigated the variances explained by each of the three pathology classes, and the additional variance reduction by adding one subgroup to the others; thus, we determined the range of variances accounted for by each subgroup. Results showed that Alzheimer’s disease-related neurodegenerative pathologies explained 30–36% of the between-subjects variation, non-Alzheimer’s disease neurodegenerative pathologies explained 4–10%, and cerebrovascular disease indices explained 3–8%, depending on the order of entry (data not shown). Figure 3 summarizes the cumulative influence of the pathological indices on the between-subject variation in cognitive decline, as well as the unexplained variation.
Figure 3.

Variation in cognitive‘ decline explained by the pathological indices (dark grey) and the residual, unexplained variation in cognitive decline (light grey) derived from fully adjusted models. Alzheimer’s disease (AD) = neurodegenerative pathologies related to Alzheimer’s disease, i.e. global Alzheimer’s disease pathology, amyloid-β and PHFtau tangles; CVD = cerebrovascular indices, i.e. gross infarcts, microscopic infarcts, atherosclerosis, arteriolosclerosis, cerebral amyloid-β angiopathy; non-Alzheimer’s disease (non-AD) = neurodegenerative pathologies, i.e. Lewy bodies, hippocampal sclerosis and LATE-NC.
Relation of pathological indices with preterminal and terminal cognitive decline
Cognitive decline accelerates in the years just before death and recent work suggests that much of late life cognitive decline is in fact terminal.15 To capture terminal change, we used random change point models to characterize the onset of terminal decline and rates of preterminal and terminal decline and then examine the contribution of the pathological indices to the different components of the cognitive trajectory. For robustness of model estimation, these analyses were done in a subset of individuals (n = 933) with at least five cognitive assessments; the mean length of follow-up was 9.9 years (SD = 4.5, range = 4–23). In the initial analysis, the onset of terminal cognitive decline (i.e. the change point) began 3.6 years prior to death [95% credible interval (CI) = −3.79 to −3.42]. The rate of decline in the preterminal period was 0.04 per year (95% CI = −0.047 to −0.037), and this increased 8-fold in the terminal period to −0.32 (95% CI = −0.346 to −0.301). We then simultaneously examined the six pathological indices considered previously and again the findings were very consistent with our prior findings. In total, the six pathological indices accounted for 22.5% of the variation in the onset of terminal decline, 28.6% of the variation in preterminal decline, and 17.3% of the variation in terminal decline. Finally, we examined simultaneously all 11 pathological indices (Table 5). Together, the 11 pathological indices accounted for 28.0% of the variation in the onset of terminal decline, 32.3% of preterminal, and 18.6% of terminal decline. Supplementary Fig. 1 illustrates the additive effect of the different classes of neuropathologies on the various components of the cognitive trajectory.
Table 5.
Association of the‘ pathological indices with the onset of accelerated decline as well as rates of preterminal and terminal cognitive decline
| Model term | Index | Estimate | SD |
95% CI
|
|
|---|---|---|---|---|---|
| Lower | Upper | ||||
| No pathology | Intercept | 0.453 | 0.100 | 0.258 | 0.651 |
| Preterminal slope | 0.011 | 0.005 | 0.001 | 0.022 | |
| Terminal slope | −0.170 | 0.029 | −0.227 | −0.115 | |
| Change point | −1.769 | 0.234 | −2.237 | −1.322 | |
| Global Alzheimer’s disease pathology | Intercept | −0.352 | 0.115 | −0.578 | −0.127 |
| Preterminal slope | −0.006 | 0.007 | −0.020 | 0.007 | |
| Terminal slope | −0.033 | 0.028 | −0.088 | 0.021 | |
| Change point | −0.409 | 0.236 | −0.868 | 0.045 | |
| Amyloid-β | Intercept | 0.009 | 0.046 | −0.082 | 0.100 |
| Preterminal slope | −0.003 | 0.003 | −0.008 | 0.003 | |
| Terminal slope | −0.001 | 0.012 | −0.024 | 0.022 | |
| Change point | −0.002 | 0.096 | −0.190 | 0.190 | |
| PHFtau tangles | Intercept | −0.488 | 0.045 | −0.575 | −0.399 |
| Preterminal slope | −0.017 | 0.003 | −0.022 | −0.011 | |
| Terminal slope | −0.029 | 0.011 | −0.050 | −0.008 | |
| Change point | −0.462 | 0.091 | −0.642 | −0.285 | |
| Neocortical Lewy bodies | Intercept | −0.697 | 0.114 | −0.921 | −0.471 |
| Preterminal slope | −0.016 | 0.006 | −0.028 | −0.004 | |
| Terminal slope | −0.147 | 0.028 | −0.203 | −0.092 | |
| Change point | −0.306 | 0.220 | −0.741 | 0.132 | |
| LATE-NC | Intercept | −0.049 | 0.034 | −0.114 | 0.018 |
| Preterminal slope | −0.002 | 0.002 | −0.005 | 0.002 | |
| Terminal slope | −0.006 | 0.008 | −0.022 | 0.010 | |
| Change point | −0.067 | 0.070 | −0.208 | 0.071 | |
| Hippocampal sclerosis | Intercept | −0.665 | 0.127 | −0.909 | −0.416 |
| Preterminal slope | −0.028 | 0.007 | −0.043 | −0.014 | |
| Terminal slope | 0.013 | 0.029 | −0.043 | 0.070 | |
| Change point | −1.238 | 0.257 | −1.746 | −0.734 | |
| Atherosclerosis | Intercept | −0.175 | 0.049 | −0.271 | −0.079 |
| Preterminal slope | −0.005 | 0.003 | −0.011 | 0.000 | |
| Terminal slope | −0.006 | 0.012 | −0.031 | 0.019 | |
| Change point | −0.166 | 0.105 | −0.371 | 0.040 | |
| Arteriolosclerosis | Intercept | −0.077 | 0.044 | −0.162 | 0.008 |
| Preterminal slope | −0.001 | 0.003 | −0.006 | 0.004 | |
| Terminal slope | −0.025 | 0.011 | −0.047 | −0.004 | |
| Change point | 0.087 | 0.092 | −0.094 | 0.266 | |
| Gross infarcts | Intercept | −0.295 | 0.081 | −0.459 | −0.137 |
| Preterminal slope | −0.011 | 0.005 | −0.020 | −0.002 | |
| Terminal slope | −0.034 | 0.021 | −0.075 | 0.006 | |
| Change point | −0.135 | 0.172 | −0.478 | 0.199 | |
| Microscopic infarcts | Intercept | −0.154 | 0.081 | −0.310 | 0.006 |
| Preterminal slope | −0.002 | 0.005 | −0.011 | 0.007 | |
| Terminal slope | 0.006 | 0.021 | −0.034 | 0.046 | |
| Change point | −0.278 | 0.173 | −0.617 | 0.060 | |
| Cerebral amyloid-β angiopathy | Intercept | −0.074 | 0.043 | −0.158 | 0.010 |
| Preterminal slope | 0.004 | 0.002 | −0.001 | 0.009 | |
| Terminal slope | −0.006 | 0.011 | −0.027 | 0.014 | |
| Change point | −0.166 | 0.090 | −0.343 | 0.010 | |
Contribution of a novel brain imaging marker to cognitive decline
The above findings suggest that the full complement of neuropathological indices studied here account for less than half of the variation in late life cognitive decline. Thus, other (as yet unknown or unmeasured) pathological and resilience factors must be at play. Because we previously showed in a relatively small sample that post-mortem neuroimaging markers (i.e. R2) may be independently related to cognitive decline,14 we analysed data from a subsample of 680 individuals (mean length of follow-up was 9.2 years, SD = 5.1, range = 1–23) in whom post-mortem R2 data were available. In this analysis, we repeated the core model (with all 11 pathological indices) but with additional terms for 30 post-mortem R2 imaging indices and their interactions with time. In this analysis, 21 components were associated with the rate of decline (12 remained significant after Bonferroni correction; data not shown). The 11 pathologies explained 44% of the between-subjects variance in decline, almost the same as in the core analysis (43%). Further, the 30 R2 components combined explained an additional 4%, bringing the total variance explained to 48% in the subsample (Supplementary Fig. 2).
Discussion
We previously reported that pathological indices of Alzheimer’s disease, infarcts, and Lewy bodies accounted for ∼40% of the person-specific variation in late life cognitive decline.12 However, it is now clear that several other age-related neuropathologies also are important drivers of late life cognitive decline.3–5,8,9,11,18–23 Here, we examined the degree to which a much wider array of neuropathologies (i.e. 11 pathological indices, including markers of Alzheimer’s disease, non-Alzheimer’s disease neurodegenerative diseases, and several cerebrovascular conditions) account for person-specific variation in cognitive decline. To our surprise, the full complement of pathological indices accounted for only 43% of the variation in decline; Alzheimer’s disease-related indices accounted for between 30% and 36%, non-Alzheimer’s disease neurodegenerative indices 4–10%, and cerebrovascular indices 3–8%. Further, the 11 pathological indices together accounted for less than a third of the variation in the onset of terminal decline (28.0%) and rates of preterminal (32.3%) and terminal (18.6%) decline. These findings confirm that the neuropathologies that are the main targets of therapeutic intervention are important determinants of cognitive decline but they are not the whole story. More than half of the decline remains unexplained. These findings have important implications for the development of therapeutic agents and suggest that a focus on other pathological and resilience factors is needed to identify novel treatment targets.
First, these findings highlight the tremendous complexity of late life cognitive decline. In this study, 10 of the 11 neuropathological indices studied were associated with an increased rate of cognitive decline. Alzheimer’s disease-related pathological indices were by far the most potent, accounting for around a third of the variability in decline overall. Non-Alzheimer’s disease neurodegenerative diseases and cerebrovascular conditions were less potent, accounting for less than ∼10% overall. These findings, together with prior work showing that co-existing pathologies may act in concert to impair cognition, magnify some of the challenges inherent in developing therapeutics. For example, cerebrovascular or and other comorbid neurodegenerative diseases can impair cognition independent of Alzheimer’s disease pathology and lower the threshold for dementia, particularly early in the pathophysiological course of Alzheimer’s disease.7,8,10,20–23 The role of vascular and other pathologies as initiators, catalysts, or additive contributors to neurodegeneration is complex and may vary depending on when the lesions develop.10 Moreover, to complicate matters further, the impact of any given pathology at a person-specific level varies depending on the specific combination of other neuropathologies present.11 Thus, it is clear that disease-modifying therapies for Alzheimer’s disease in particular are urgently needed, but with so many other diseases present and in varying combinations, some cognitive decline is still likely even with an effective Alzheimer’s disease treatment. Further, more than half of the variation in decline remained unexplained after considering the full complement of the neuropathological indices studied here. Thus, other pathological factors (unknown or not yet quantified) must be at play.
Indeed, here, the neuroimaging metric of R2 accounted for an additional 4% of the variation in decline above and beyond neuropathology. Neuroimaging may reveal pathology that is not fully evident via traditional histopathological evaluation or provide insight into where to look for new pathologies, but this work is still emerging.17,20 For example, a recent study reported an association of white matter hyperintensities with memory decline.22,24 Neuroimaging techniques can quantify additional vascular indices such as microbleeds and enlarged perivascular spaces, as well as other brain changes, and future work leveraging contemporary imaging techniques is needed to complement standard pathological approaches to elucidate the full spectrum of pathological factors that contribute to cognitive decline in old age.
Second, these findings suggest an urgent need to identify novel therapeutic targets, particularly resilience factors.24 That is, the residual (unexplained) variation observed here in part reflects the reality that cognitive ageing involves a delicate balance between neuropathology and resilience factors. Numerous risk factors for cognitive decline and dementia have been identified, including psychological (e.g. purpose in life), experiential (e.g. cognitive and physical activity), and genomic risk factors, as well as structural elements of reserve (e.g. presynaptic proteins).25–29 Importantly, many such factors provide protection independent of the known neuropathologies, and some (e.g. purpose in life, social networks) interact with pathology to provide a buffer against cognitive decline even in the face of accumulating neuropathology.28,29 It is very likely that other behavioural, genomic, and structural resilience factors exist yet have not been identified, as relatively few studies have the behavioural, cognitive, genetic and neuropathological data needed to determine how risk factors work in the face of accumulating neuropathology. The present findings suggest that an increased focus on resilience may yield novel therapeutic targets. Critically, interventions that increase resilience may be amenable to broad application and possibly can be applied in middle age or even earlier in the life course, before pathology takes its hold on the brain. Thus, such interventions may help stave off the effects of accumulating neuropathology and could confer greater benefit than interventions targeting specific disease mechanisms. Indeed, in recent years, we have identified several novel genes and proteins associated with cognitive decline separate from brain pathologies.24,25,27
Third, these findings indicate a need for a greater focus on the factors associated with terminal cognitive decline. Approximately 70% of the cognitive loss older adults experience is due to terminal decline, a phase that overlaps considerably with dementia, yet very little is known about the factors that drive terminal decline.30 In this study, the 11 pathological indices combined accounted for 28% of the variation in the onset of terminal decline, 32.3% of preterminal decline, and only 18.6% of terminal decline. These findings build on prior work showing that age-related neuropathologies exert their strongest effects in the initial stages of cognitive decline, before the terminal phase begins.12 Therefore, other factors must cause the rapid acceleration in cognitive decline during the last years of life. We suspect that declining physical health, medical comorbidities, frailty and associated changes in self-care play a role, and may even interact with pathology to impact cognition during the terminal phase. For example, one study reported that frailty interacts with Alzheimer’s disease pathology to impact cognition; specifically, older adults who were more frail were more likely both to have Alzheimer’s disease pathology and for it to be expressed as dementia compared to those with less frailty.31 We are not aware of studies that have examined such associations in light of terminal decline, however. Future work that incorporates terminal decline explicitly and seeks to understand both its basis and factors that mitigate it are essential for reducing the burden of late life cognitive decline.
This study has strengths and weaknesses, some of which may contribute to our unexpected finding that, even after considering a wide spectrum of cognition impairing neuropathologies, more than half of the variation in late life decline remains unexplained. First, although the pathological indices studied here are currently the gold standard markers of common age-related diseases, they nonetheless are markers of complex diseases that cause a cascade of changes in the brain, not all of which are readily quantifiable. Second, we did not systematically examine frontotemporal lobar degeneration (FTLD) and some other pathological indices that may be important determinants of cognitive decline [e.g. ageing-related tau astrogliopathy (ARTAG), inflammation], although the distinction between LATE-NC and FTLD is still being determined.32–39 We are in the process of collecting data on ARTAG and other new indices and will examine their contributions in future work as sufficient data accrue. Third, we did not examine asymmetry in pathology findings or focus explicitly on strategic versus non-strategic infarcts. Finally, we did not examine interactions among neuropathologies on cognition, as how best to do so is not straightforward in the context of this study and thus far we have limited evidence of such effects. However, we acknowledge that there may be mechanistic interactions that result in neurodegeneration or other tissue injury and this is an area of active interest. Despite these limitations, these data provided a solid foundation for examining the degree to which the array of neuropathologies that are the primary focus of ageing research drives late life cognitive decline. Our findings underscore the complexity of cognitive ageing and offer a new perspective on the considerable challenges that lie ahead in regard to the development of effective and novel therapeutics.
Funding
Funding for this study came from the National Institute on Aging (R01AG34374, R01AG17917, R0133678, P30AG10161, R01AG15819, R01AG067482) and the Illinois Department of Public Health.
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.
Supplementary Material
Glossary
- LATE-NC
limbic predominant age-related TDP-43 encephalopathy
References
- 1.White LR, Edland SD, Hemmy LS, et al. Neuropathologic comorbidity and cognitive impairment in the Nun and Honolulu-Asia aging studies. Neurology. 2016;86:1000–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kawas CH, Kim RC, Sonnen JA, Bullain SS, Trieu T, Corrada MM.. Multiple pathologies are common and related to dementia in the oldest-old: The 90+ study. Neurology. 2015;85(6):535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA.. Relation of cerebral vessel disease to Alzheimer's disease dementia and cognitive function in elderly people: A cross-sectional study. Lancet Neurol. 2016;15:934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brayne C, Richardson K, Matthews FE, et al. ; Cambridge City Over-75s Cohort Cc75c Study Neuropathology Collaboration. Neuropathological correlates of dementia in over-80-year old brain donors from the population-based Cambridge city over-75s cohort (CC75C) study. J Alzheimers Dis. 2009;18:645–658. [DOI] [PubMed] [Google Scholar]
- 5.Nelson PT, Abner EL, Schmitt FA, et al. Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol. 2010;20:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braak H, Thal DR, Ghebremedhin E, Del Tredici K.. Stages of pathologic process in Alzheimer’s disease: Age categories from 1 to 100. J Neuropathol Exp Neurol. 2011;70(11):960–969. [DOI] [PubMed] [Google Scholar]
- 7.Spires-Jones TL, Attems J, Thal DR.. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 2017;134(2):187–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esiri MM, Nagy Z, Smith MZ, Barnetson L, Smith Alzheimer’s disease.. Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer’s disease. Lancet. 1999;354(9182):919–920. [DOI] [PubMed] [Google Scholar]
- 9.Hecht M, Krämer LM, von Armin CAF, Otto M, Thal DR.. Capillary cerebral amyloid angiopathy in Alzheimer’s disease: Association with allocortical/hippocampal microinfarcts and cognitive decline. Acta Neuropathol. 2018;135(5):681–694. [DOI] [PubMed] [Google Scholar]
- 10.Launer LJ, Hughes TM, White LR.. Microinfarcts, brain atrophy, and cognitive function: The Honolulu Asia Aging Study Autopsy Study. Ann Neurol. 2011;70(5):774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA.. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol. 2018; 83(1):74-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyle PA, Wilson RS, Yu L, et al. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann Neurol. 2013;74:478–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic-predominant TDP-43 age-related encephalopathy (LATE): Consensus working group report. Brain. 2019;142(6):1503–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennett DA, Buchman AS, Boyle PA, et al. Religious orders study and rush memory and aging project. J Alzheimers Dis. 2018;64(s1):S161–S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett DA, Schneider JA, Aggarwal NT, et al. Decision rules guiding the clinical diagnosis of Alzheimer’s disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study. Neuroepidemiol. 2006;27:169–176. [DOI] [PubMed] [Google Scholar]
- 16.Wilson RS, Beckett LA, Barnes LL, et al. Individual differences in rates of change in cognitive abilities of older persons. Psychol Aging. 2002;17:179–193. [PubMed] [Google Scholar]
- 17.Dawe RJ, Yu L, Leurgans SE, et al. Postmortem MRI: A novel window into the neurobiology of late life cognitive decline. Neurobiol Aging. 2016;45:169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nag S, Yu L, Capuano AW, et al. Hippocampal sclerosis and LATE-NC pathology in aging and Alzheimer disease. Ann Neurol. 2015;77:942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skrobot OA, Attems J, Esiri M, et al. Vascular cognitive impairment neuropathology guidelines (VCING): the contribution of cerebrovascular pathology to cognitive impairment. Brain. 2016;139(11):2957–2969. [DOI] [PubMed] [Google Scholar]
- 20.Han JW, Maillard P, Harvey D, et al. Association of vascular brain injury, neurodegeneration, amyloid and cognitive trajectory. Am Acad Neurol. 2020;95(19):e2622–e2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Josephs KA, Whitwell JL, Parisi JE, et al. Argyrophilic grains: A distinct disease or an additive pathology? Neurobiol Aging. 2008;29(4):566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomé SO, Vandenberghe R, Ospitalieri S, et al. Distinct molecular patterns of TDP-43 pathology in Alzheimer’s disease: Relationship with clinical phenotypes. Acta Neuropathol Commun. 2020;8(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jospehs KA, Whitwell JL, Weigand SD, et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 2014;127(6):911–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bennett DA.Mixed pathologies and neural reserve: Implications of complexity for Alzheimer disease drug discovery. PLoS Med. 2017;14:e1002256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu L, Lutz MW, Farfel JM, Wilson RS, Burns DK, et al. Neuropathologic features of TOMM40 '523 variant on late-life cognitive decline. Alzheimers Dement. 2017; 13(12):1380–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson RS, Nag S, Boyle PA, et al. Neural reserve, neuronal density in the locus ceruleus, and cognitive decline. Neurology. 2013;80:1202–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu L, Petyuk VA, Gaiteri C, et al. Targeted brain proteomics uncover multiple pathways to Alzheimer's dementia. Ann Neurol. 2018;84(1):78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennett DA, Arnold SE, Valenzuela MJ, Brayne C, Schneider JA.. Cognitive and social lifestyle: Links with neuropathology and cognition in late life. Acta Neuropathol. 2014;127(1):137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyle PA, Buchman AS, Wilson RS, Yu L, Schneider JA, Bennett DA.. Effect of purpose in life on the relation between Alzheimer disease pathologic changes on cognitive function in advanced age. Arch Gen Psychiatry. 2012;69(5):499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson RS, Yu L, Leurgans SE, et al. Proportion of cognitive loss attributable to terminal decline. Neurology. 2020;94(1):e42–e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wallace LMK, Theou O, Godin J, et al. Investigation of frailty as a moderator of the relationship between neuropathology and dementia in Alzheimer's disease: A cross-sectional analysis of data from the Rush Memory and Aging Project. Lancet Neurol. 2019;18:177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jellinger KA, Attems J.. Prevalence and of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol. 2008;115(4):427–436. [DOI] [PubMed] [Google Scholar]
- 33.Attems J, Neltner JH, Nelson PT.. Quantitative neuropathological assessment to investigative cerebral multi-morbodity. Alzheimers Res Ther. 2014;6(9):85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson JL, Corrada MM, Kovacs GG, et al. Non-Alzheimer’s contributions to dementia and cognitive resilience in the 90+ Study. Acta Neuropathol. 2018;136(3):377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toledo JB, Gopal P, Raible K, et al. Pathological α-synuclein distribution in subjects with coincident Alzheimer’s and Lewy body pathology. Acta Neuropathol. 2016;131(3):393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yarchoan M, Xie SX, Kling MA, et al. Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain. 2012;135(Pt 12):3749–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thal DR, Schultz C, Botez G, et al. The impact of argyrophilic grain disease on the development of dementia and its relationship to concurrent Alzheimer’s disease-related pathology. Neuropathol Appl Neurobiol. 2005;31(3):270–279. [DOI] [PubMed] [Google Scholar]
- 38.Sahoo A, Bejanin A, Murray ME, Tosakulwong N, et al. TDP-43 and Alzheimer’s disease pathological subtype in non-amnestic Alzheimer’s disease dementia. J Alzheimers Dis. 2018;64(4):1227–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ortiz CA, Lin WL, Ahmed Z, et al. TDP43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol. 2007;61(5):435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available upon request via the Rush Alzheimer’s Disease Center Resource Sharing Hub (www.radc.rush.edu).