

Abstract
Natural killer (NK) cells can recognize virus-infected and stressed cells1 using activating and inhibitory receptors, many of which interact with HLA class I. Although early studies also suggested a functional impact of HLA class II on NK cell activity2,3, the NK cell receptors that specifically recognize HLA class II molecules have never been identified. We investigated whether two major families of NK cell receptors, killer-cell immunoglobulin-like receptors (KIRs) and natural cytotoxicity receptors (NCRs), contained receptors that bound to HLA class II, and identified a direct interaction between the NK cell receptor NKp44 and a subset of HLA-DP molecules, including HLA-DP401, one of the most frequent class II allotypes in white populations4. Using NKp44ζ+ reporter cells and primary human NKp44+ NK cells, we demonstrated that interactions between NKp44 and HLA-DP401 trigger functional NK cell responses. This interaction between a subset of HLA-DP molecules and NKp44 implicates HLA class II as a component of the innate immune response, much like HLA class I. It also provides a potential mechanism for the described associations between HLA-DP subtypes and several disease outcomes, including hepatitis B virus infection5–7, graft-versus-host disease8 and inflammatory bowel disease9,10.
With the description of the class Ib family protein HLA-F as a ligand for KIR3DS1 (ref. 11), activating and inhibitory NK cell receptors for all HLA class I molecules have been identified. However, HLA class II molecules, which have been associated with the outcome of many inflammatory diseases, have never been shown to serve as ligands for NK cell receptors; thus, their role in the innate immune response has not been investigated. To assess binding of NK cell receptors to HLA class II molecules, we used recombinant human Fc constructs, consisting of the extracellular domain of NK cell receptors fused to the human IgG1 Fc domain, in an HLA class II bead-based screening assay. Binding of ten NK cell receptor (NKp30, NKp44, NKp46, KIR2DL3, KIR2DL1, KIR3DL2, KIR3DL1, KIR2DL4, KIR3DS1 and KIR2DS1) Fc constructs to 95 different HLA class II molecules was investigated. Lymphocyte-activation gene 3 (LAG-3) protein, which has been previously described to be closely related to CD4 and to bind with high affinity to HLA class II molecules12, was used as a positive control. The LAG-3 Fc construct did indeed interact with all three HLA class II subtypes, HLA-DR, HLA-DQ and HLA-DP, while NKp30 and NKp46 Fc constructs did not bind to any of the tested HLA class II molecules. Furthermore, the different KIR Fc constructs tested did not exhibit significant binding to any of the HLA class II-coated beads above negative control levels (Fig. 1a and Supplementary Fig. 1). In contrast, the NKp44 Fc construct displayed significant binding to a subset of HLA-DP molecules, including the HLA-DP401 molecule (molecule 8 in Fig. 1b), which is highly prevalent in white populations4, but not to any of the HLA-DR or HLA-DQ molecules tested (Fig. 1a). Using surface plasmon resonance (SPR), we confirmed binding of NKp44, but not NKp46, to HLA-DP401 with an affinity of 42.6 ± 16.2 µM (Fig. 1c,d), which is within the range typically observed for immune receptor–ligand interactions13–16. In contrast, NKp44 exhibited weak or no binding to HLA-DP301 and HLA-DQ2, respectively (Fig. 1c,d). Taken together, these binding experiments identified an interaction between the NK cell receptor NKp44 and a subset of HLA-DP molecules, including direct and specific binding to the ectodomain of HLA-DP401 by SPR.
Fig. 1 |. Binding of LAG-3, KIR and NCR Fc constructs to HLA class II-coated beads.
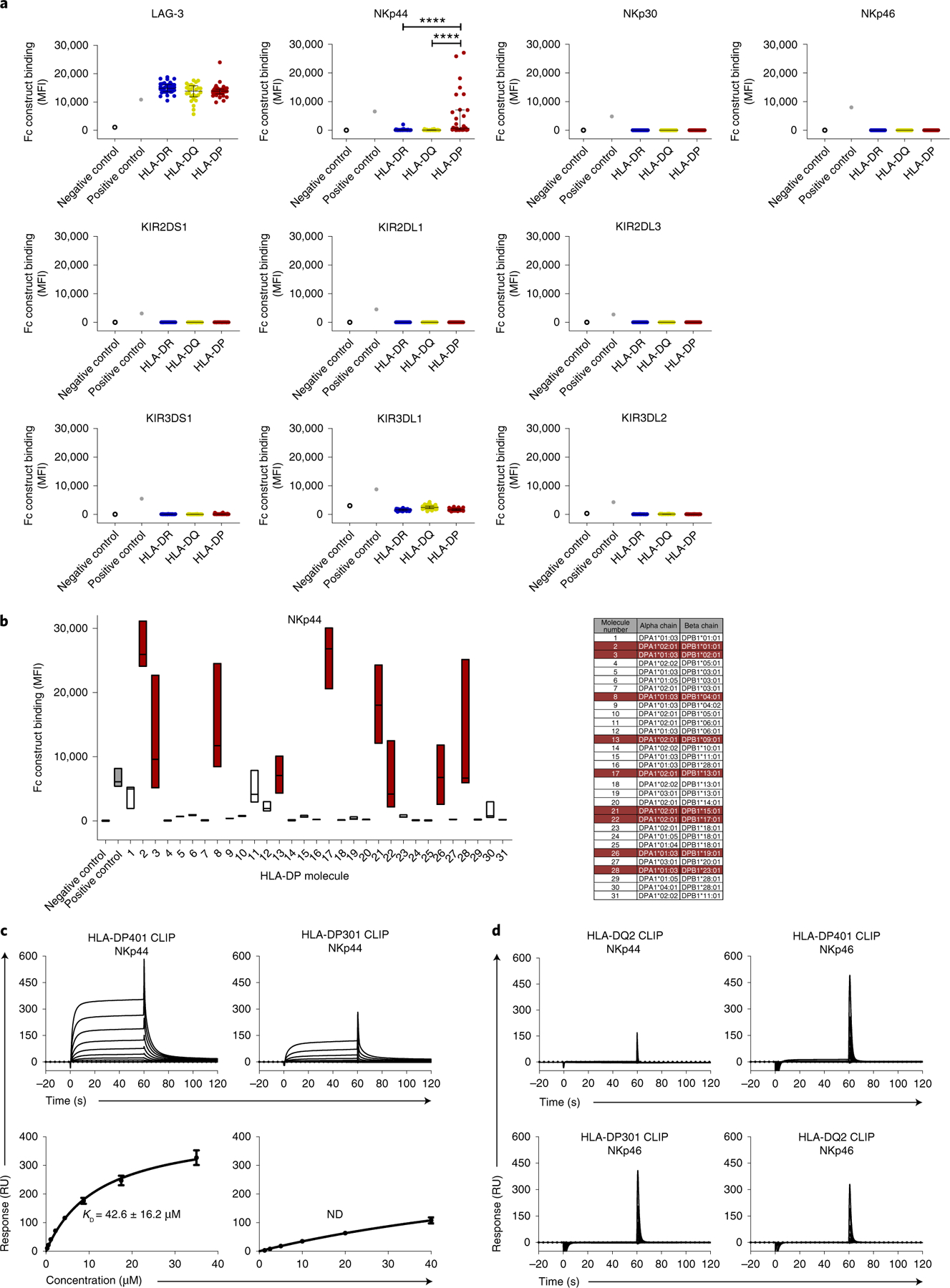
a, Fc construct binding to HLA class II-coated beads is plotted as median fluorescence intensity (MFI). All Fc constructs, except KIR3DL1 Fc construct (10 µg ml−1), were used at a final concentration of 30 µg ml−1. Each dot indicates an individual HLA class II allele. Binding of LAG-3, NKp30, NKp46 and KIR Fc constructs to HLA class II-coated beads was determined in one single experiment (n = 1). Binding of NKp44 Fc construct to HLA class II beads was determined in three independent biological replicates and MFI values of all experiments (n = 3) are depicted as mean values for each allele. The 95 HLA class II alleles were grouped according to HLA-DR, HLA-DQ and HLA-DP. The horizontal line shows the median of each HLA class II group. Error bars depict the interquartile range of each group. Statistical comparisons between HLA molecule types were done using data from each replicate in a mixed effects linear regression model. ****P < 0.0001. b, NKp44 Fc construct binding to different HLA-DP-coated beads was determined in three independent biological replicates and data of all independent experiments (n = 3) are depicted as MFI. Floating bars indicate the median, minimum and maximum MFI of each HLA-DP allele tested. HLA-DP molecules that exhibited higher binding to the NKp44 Fc construct than to the positive control in at least one assay are marked in red. c, Representative SPR sensorgrams (top row) and corresponding equilibrium binding curves (bottom row) of NKp44 binding to HLA-DP401 CLIP (left) and HLA-DP301 CLIP (right) molecules. Equilibrium dissociation constants (Kd) and s.e.m. were calculated from three independent analyte preparations (n = 3). Binding curve error bars represent the error of the fits by a single-site binding model. ND, not determined. RU, response unit. d, Representative SPR sensorgrams demonstrating no detectable binding between NKp44 and HLA-DQ2 CLIP, NKp46 and HLA-DP401 CLIP (top row); NKp46 and HLA-DQ2 CLIP; NKp46 and HLA-DP301 CLIP (bottom row). Data are representative of two (n = 2 for HLA-DQ2) or three (n = 3 for NKp46) independent experiments.
To further investigate functional consequences of the identified interactions between NKp44 and HLA-DP molecules, we generated a Jurkat reporter cell line stably expressing an NKp44ζ construct, composed of the extracellular domain of NKp44, the transmembrane domain of KIR3DL1 (to ensure DAP12-independent expression) and the cytoplasmic domain of the CD3ζ chain to mediate cellular activation upon receptor engagement and cross-linking. Upregulation of CD69 on the surface of NKp44ζ+ Jurkat reporter cells was used as an activation readout for ligand engagement11. In addition, NKp46ζ+ and KIR2DL3ζ+ Jurkat reporter cell lines were used as controls. To assess binding of NKp44 to HLA-DP molecules, biotinylated HLA-DP401 molecules (heterodimer of HLA-DPA1*01:03–HLA-DPB1*04:01) were initially used, and HLA-DR7 molecules (heterodimer of HLA-DRA1*01:01–HLA-DRB1*07:01) were used as controls. Both HLA class II molecules were loaded with the same human class II-associated invariant chain peptide (CLIP) (amino acids 87–101: PVSKMRMATPLLMQA). NKp44ζ+ Jurkat reporter cells displayed significant CD69 upregulation after co-incubation with HLA-DP401 CLIP molecules as compared to HLA-DR7 CLIP molecules (Fig. 2a,b). The activation observed on exposure to HLA-DP401 CLIP molecules was specific to NKp44ζ+ Jurkat reporter cells, and not observed with Jurkat cells transduced with other NK cell receptors (NKp46 or KIR2DL3) or untransduced Jurkat cells (Fig. 2 and Supplementary Fig. 2). All Jurkat reporter cell lines showed robust activity to antibodies specific to the respective NK cell receptor that they were transduced with, demonstrating their functionality (Fig. 2a,b). Furthermore, the proportion of NKp44ζ+ Jurkat reporter cells expressing CD69 in response to HLA-DP401 CLIP was reduced when NKp44ζ+ Jurkat reporter cells were pre-incubated with an anti-NKp44 blocking antibody (Fig. 2c). As the HLA-DP allotypes exhibited differential levels of binding to NKp44 Fc constructs in the bead-based screening assay, we furthermore used two distinct HLA-DP molecules that strongly bound to NKp44 Fc constructs (HLA-DP401 and HLA-DP201 (HLA-DPA1*01:03–HLA-DPB1*02:01)), and two HLA-DP molecules that did not strongly bind to NKp44 Fc constructs (HLA-DP601 (HLA-DPA1*01:03–HLA-DPB1*06:01) and HLA-DP301 (HLA-DPA1*01:03–HLA-DPB1*03:01)). We observed HLA-DP molecule-specific activation of NKp44ζ+ Jurkat cells by the two binders (HLA-DP401 more than HLA-DP201), but not by HLA-DP601 and HLA-DP301 (Fig. 2d). Again, none of the HLA-DP allotypes induced activation of NKp46ζ+ Jurkat reporter cells. These data show that NKp44 specifically interacts with certain HLA-DP allotypes, which can trigger functional cellular responses.
Fig. 2 |. NKp44-expressing reporter cells interact with a subset of HLA-DP molecules.
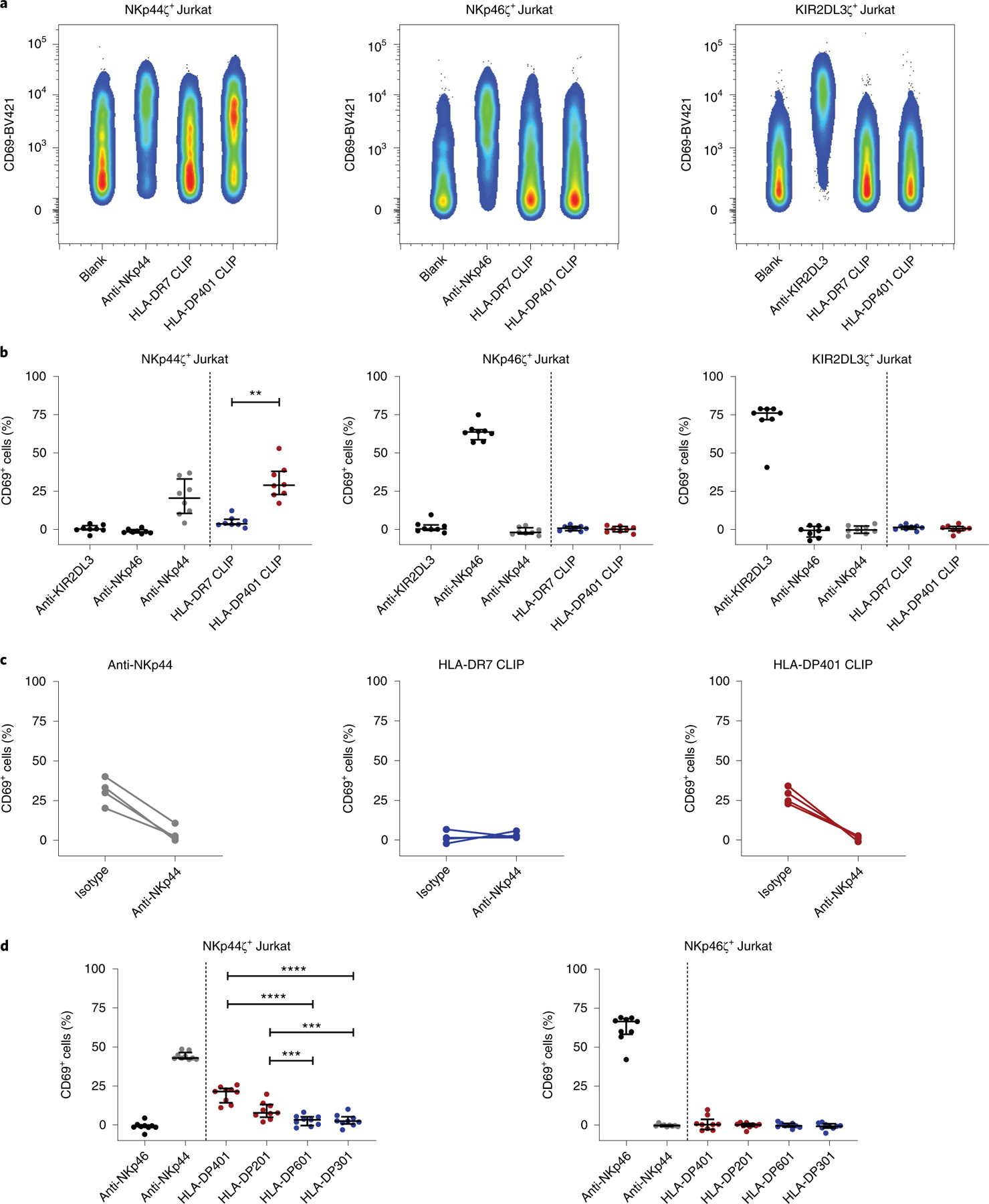
a, Reporter cell activity of NKp44ζ+, NKp46ζ+ and KIR2DL3ζ+ Jurkat reporter cells was determined by percentage of CD69+ cells after co-incubation with plate-coated anti-KIR2DL3, anti-NKp46, anti-NKp44, HLA-DR7 monomers loaded with CLIP peptide, HLA-DP401 monomers loaded with CLIP peptide and non-coated wells (blank). Plots represent one of eight independent experiments. b, Percentage of CD69+ Jurkat reporter cells is depicted after co-incubation with the indicated antibodies and monomers. The percentage of CD69+ cells following incubation on non-coated wells (blank) was subtracted from all samples. Corrected values are illustrated as median with interquartile range as determined in eight independent experiments (n = 8). Two-tailed Wilcoxon matched pairs signed-rank test was used to calculate the difference of CD69+ cells after co-incubation with HLA-DR7 CLIP and HLA-DP401 CLIP molecules. **P = 0.008. c, Reporter cell activity of NKp44ζ+ Jurkat reporter cells in response to anti-NKp44, HLA-DR7 CLIP and HLA-DP401 CLIP was determined in the presence of a purified mouse IgG1 isotype or a purified anti-NKp44 (both at a final concentration of 10 µg ml−1). The percentage of CD69+ cells following incubation on non-coated wells (blank) was subtracted from all samples. Reporter cell activity was determined in four independent experiments (n = 4). Each dot represents one individual experiment and lines connect matched responses determined in IgG1 isotype and anti-NKp44 conditions. d, Activity of NKp44ζ+ and NKp46ζ+ Jurkat reporter cells in response to anti-NKp46, anti-NKp44, HLA-DP401 (HLA-DPA1*01:03–HLA-DPB1*04:01), HLA-DP201 (HLA-DPA1*01:03–HLA-DPB1*02:01), HLA-DP601 (HLA-DPA1*01:03–HLA-DPB1*06:01), HLA-DP301 (HLA-DPA1*01:03–HLA-DPB1*03:01) is displayed as percentage of CD69+ cells. HLA-DP molecules marked in red exhibited binding to the NKp44 Fc construct in the HLA class II-coated bead assay, while HLA-DP molecules marked in blue did not display binding to NKp44 Fc constructs. The percentage of CD69+ cells following incubation on non-coated wells (blank) was subtracted from all samples. Corrected values are illustrated as median with interquartile range. Data were collected in three independent experiments, two of them conducted in quadruplicate (n = 9). Each dot represents one technical replicate. Mixed effects linear regression model was used to calculate differences of CD69+ reporter cells after co-incubation with the different HLA-DP molecules. ***P = 0.0004, ****P < 0.0001.
To further determine the functional response of NKp44-receptor binding to HLA-DP401 molecules in primary human innate immune cells, the response of NKp44+ NK cells to plate-coated HLA class II molecules was assessed. NK cells freshly isolated from the peripheral blood of healthy donors did not express NKp44 on the cell surface, as has been previously reported17, and did not degranulate upon co-incubation with anti-NKp44, HLA-DR7 CLIP or HLA-DP401 CLIP (Supplementary Fig. 3). We therefore induced NKp44 expression on the surface of isolated NK cells by incubating NK cells with IL-2 and IL-15 for 7 d (Supplementary Fig. 3a). The functional response of cytokine-treated NK cells to plate-coated anti-NKp44, HLA-DR7 CLIP and HLA-DP401 CLIP was quantified by CD107a expression as a marker for degranulation18. Cytokine-treated NKp44+ NK cells showed an upregulation of CD107a upon co-incubation with HLA-DP401 CLIP molecules, which was notably higher than after co-incubation with HLA-DR7 CLIP molecules (Fig. 3a,b). Furthermore, degranulation detected after incubation with HLA-DP401 CLIP was inhibited when cytokine-treated NK cells were pre-incubated with an anti-NKp44 blocking antibody (Fig. 3b, right panel), indicating that the observed functional response was mediated specifically by NKp44. Similar to the previous results using NKp44ζ+ Jurkat reporter cells, degranulation of primary human NKp44+ NK cells co-incubated with distinct HLA-DP molecules was dependent on the HLA-DP allotype (Fig. 3c). NKp44+ cells displayed the highest degranulation after incubation with HLA-DP401 and HLA-DP201, while minimal degranulation was observed in response to HLA-DP601 and HLA-DP301 molecules. Furthermore, CD107a upregulation in primary NKp44+ NK cells upon engagement with HLA-DP401 or HLA-DP201 was decreased by pre-incubation with an anti-NKp44 blocking antibody. By contrast, the blocking antibody had no effect on CD107a expression in primary NKp44+ cells co-incubated with HLA-DP601 or HLA-DP301 molecules (Fig. 3c). NK cells from each tested donor degranulated robustly after co-incubation with the NK cell-sensitive tumor cell line K562, and responses to K562 cells were not influenced by pre-incubation of NK cells with an anti-NKp44 blocking antibody (data not shown). In summary, these data indicate a functional modulation of NK cell activity by a subset of HLA-DP molecules mediated by the NK cell receptor NKp44.
Fig. 3 |. Stimulated primary NK cells degranulate on incubation with HLA-DP401 monomers.
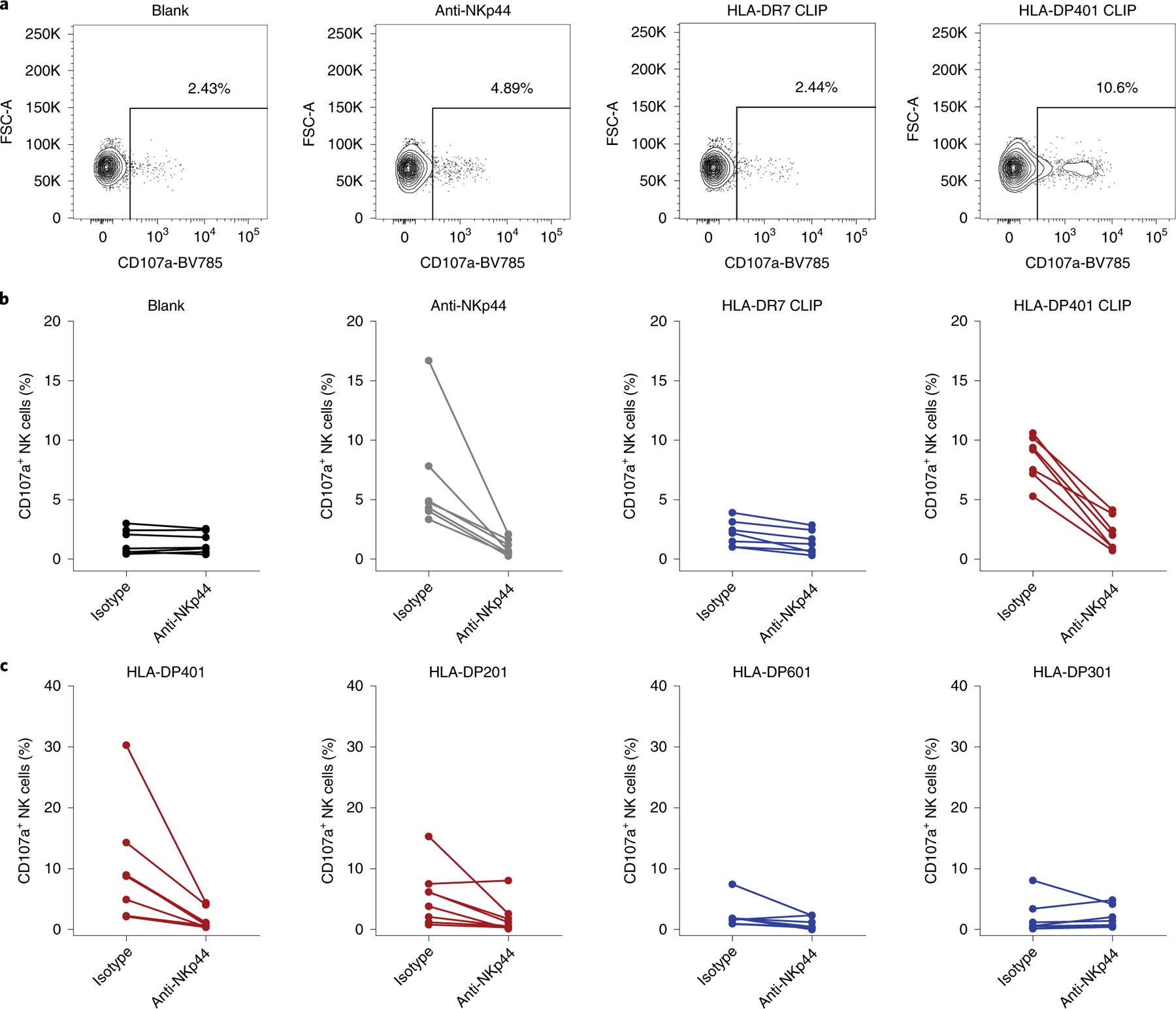
a, Percentage of CD107a+ NK cells was determined after co-incubation with the indicated plate-coated ligands or non-coated wells (blank). Plots represent one of seven independent experiments. b, Degranulation after co-incubation with anti-NKp44, HLA-DR7 and HLA-DP401 monomers loaded with CLIP peptide and non-coated wells (blank) is depicted as percentage of CD107a+ NK cells and was determined in cytokine-treated primary NK cells isolated from seven individual donors (n = 7). Frequency of CD107a+ NK cells was determined in the presence of purified mouse IgG1 isotype or purified anti-NKp44 (both at a final concentration of 10 µg ml−1). Each dot represents one individual donor and lines connect responses from one individual donor. c, Cytokine-treated primary human NK cells derived from eight different donors were co-incubated with distinct plate-coated HLA-DP single-antigen molecules in the presence of purified mouse IgG1 isotype or purified anti-NKp44. Degranulation of primary NK cells is depicted as percentage of CD107a+ NK cells following co-incubation. Each dot represent one individual donor and lines connect responses from one individual donor (n = 8).
HLA-DP molecules can present a large array of different peptides on the cell surface after replacing the CLIP peptide19, and recognition of these peptides has been investigated in the context of T cell receptors expressed on CD4+ T cells20–22. Given the identification of binding of NKp44 to HLA-DP, we tested whether differences in peptides presented by HLA-DP401 have an impact on recognition by NKp44. Using the NKp44ζ+ Jurkat reporter cell assay described above, we compared CD69 expression in response to HLA-DP401 molecules refolded either with CLIP or with a number of pathogen- or self-derived peptides (peptide sequences are shown in Supplementary Table 1). HLA-DR7 molecules refolded with either CLIP or a human immunodeficiency virus 1 (HIV-1)-derived Gag peptide were used as controls. We observed a clear impact of the different peptides bound to HLA-DP401 on activation of NKp44ζ+ Jurkat reporter cells (Fig. 4a), with the HLA-DP401 CLIP complex mediating the strongest activation and the HLA-DP401–Clostridium tetani tetanus toxin (C. tetani TT) peptide complex inducing a significantly lower response. In contrast, HLA-DR7 monomers did not trigger any activation of NKp44ζ+ Jurkat reporter cells, either in complex with CLIP or in complex with the HIV-1 Gag peptide (Fig. 4a). Similar to the observations using NKp44ζ+ Jurkat reporter cells, the activation of primary NKp44+ NK cells incubated with HLA-DP401 was also modulated by the specific HLA-DP-bound peptide. While plate-coated HLA-DP401 CTAG1 and HLA-DP401 HIV-1 Env complexes were able to trigger degranulation of cytokine-treated primary NK cells, HLA-DP401 molecules loaded with C. tetani TT- or human oxytocinase-derived peptides were not (Fig. 4b). In conclusion, engagement of NKp44 by HLA-DP401 induced degranulation of NK cells, and this response was further modulated in a peptide-dependent manner. These results suggest that changes in peptide repertoires that occur during infections or inflammatory processes might impact not only recognition of HLA-DP-expressing cells by CD4+ T cells23,24, but also recognition by NKp44+ innate effector cells. These observations are in line with previous studies that have demonstrated that the sequence of HLA class I-presented peptides has substantial consequences for binding of inhibitory and activating KIRs to their HLA class I ligands, and the activation of KIR+ NK cells25–30.
Fig. 4 |. The interaction between NKp44 and HLA-DP is modulated by the presented peptide.
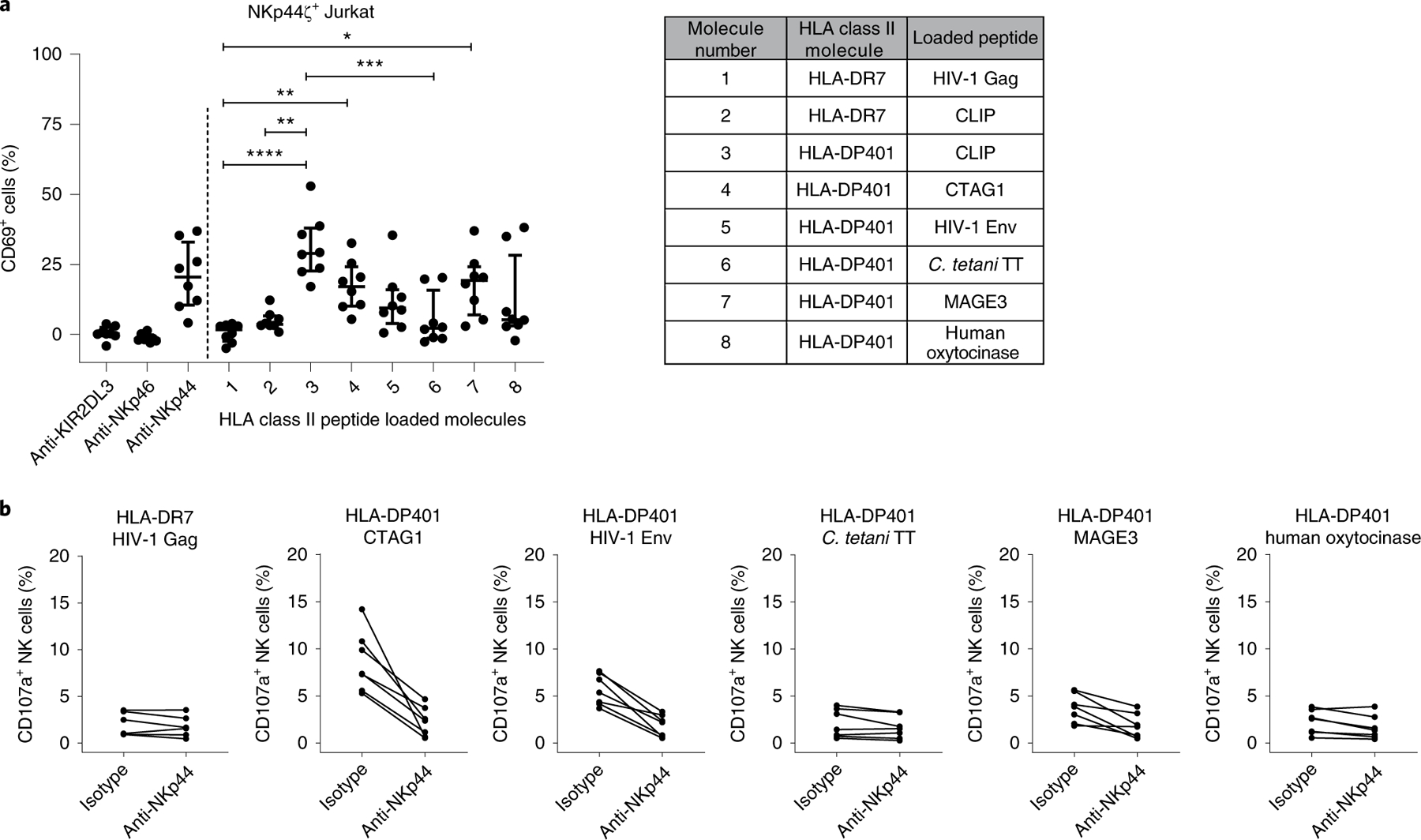
a, Reporter cell activity was determined as percentage of CD69+ cells after co-incubation with plate-coated anti-KIR2DL3, anti-NKp46, anti-NKp44 and HLA class II molecules loaded with the indicated peptides. The percentage of CD69+ cells following incubation on non-coated wells (blank) was subtracted from all samples. Corrected values are illustrated as median with interquartile range as determined in eight independent experiments (n = 8). Two-tailed Friedman test with post-hoc Dunn’s multiple comparison was used to determine statistical differences between percentages of CD69+ cells after co-incubation with the different HLA class II peptide-loaded molecules. Control conditions (anti-KIR2DL3, anti-NKp46, anti-NKp44) were not included in the statistical analysis. *P = 0.03; **P = 0.001 and 0.005; ***P = 0.0005; ****P < 0.0001. b, Percentage of CD107a+ NK cells was determined after co-incubation with different HLA class II molecules loaded with the indicated peptides using cytokine-treated primary NK cells isolated from seven different donors (n = 7). Frequency of CD107a+ NK cells was determined in the presence of purified mouse IgG1 isotype or purified anti-NKp44 (both at a final concentration of 10 µg ml−1). Each dot represents one individual donor and lines connect responses from one individual donor.
To further investigate the physiological interaction of membrane-bound NKp44 with membrane-bound HLA-DP molecules, we generated four HLA-DP-expressing Jurkat (clone E6.1) cell lines, JE6.1-DP401, JE6.1-DP201, JE6.1-DP601 and JE6.1-DP301 (Supplementary Fig. 4a). Jurkat cells are class II major histocompatibility complex transactivator (CIITA)-deficient and therefore lack the expression of all HLA class II molecules31, enabling the controlled and specific investigation of interactions between NKp44 and membrane-bound HLA-DP. In line with our binding data, HLA-DP401-expressing JE6.1 cells induced significantly higher NKp44ζ+ reporter cell activation than HLA-DP601-and HLA-DP301-expressing JE6.1 cells (Fig. 5a). NKp44ζ+ reporter cell activity in response to JE6.1-DP201 cells was less pronounced, yet the percentage of CD69+ reporter cells was higher upon co-incubation with JE6.1-DP201 cells compared to JE6.1-DP601 and JE6.1-DP301 cells. In contrast, NKp46ζ+ Jurkat reporter cells did not display significant upregulation of CD69 after co-incubation with any of the JE6.1-DP cell lines. Since we had observed a peptide-dependent activation of NKp44ζ+ Jurkat reporter cells upon HLA-DP engagement, with HLA-DP401 CLIP molecules inducing the highest activation (Fig. 4a), we subsequently examined the effect of CLIP surface presentation on NKp44ζ+ Jurkat reporter cell activity. Jurkat cells are negative for the HLA class II-associated invariant chain32 and consequently do not display CLIP on their surface, even after transduction with HLA-DP molecules, allowing for external pulsing with CLIP. Addition of CLIP to JE6.1 cells transfected with HLA-DP401, HLA-DP201, HLA-DP301 and HLA-DP601 resulted in enhanced HLA-DP-expression levels (Supplementary Fig. 4b), indicating stabilization of HLA-DP expression by exogenous CLIP. While CLIP-pulsed JE6.1-DP201 cells induced slightly elevated activation of NKp44ζ+ Jurkat reporter cells compared with unpulsed JE6.1-DP201 cells, CLIP pulsing did not increase CD69 expression of NKp44ζ+ Jurkat reporter cells after co-incubation with JE6.1-DP401, JE6.1-DP601 and JE6.1-DP301, compared with the respective unpulsed JE6.1-DP cells. Most importantly, the overall hierarchy of NKp44+ cell activation remained consistent with CLIP-pulsed JE6.1-DP401 and JE6.1-DP201 cells triggering significantly higher CD69 expression on NKp44ζ+ Jurkat reporter cells than CLIP-pulsed JE6.1-DP601 and JE6.1-DP301 cells (Fig. 5b). These data demonstrate that functional responses resulting from membrane-bound NKp44–HLA-DP interactions were more strongly influenced by the respective HLA-DP allotype than by the presentation of CLIP on HLA-DP molecules.
Fig. 5 |. Membrane-bound HLA-DP molecules trigger a functional response of NKp44+ cells in an allotype-dependent manner.
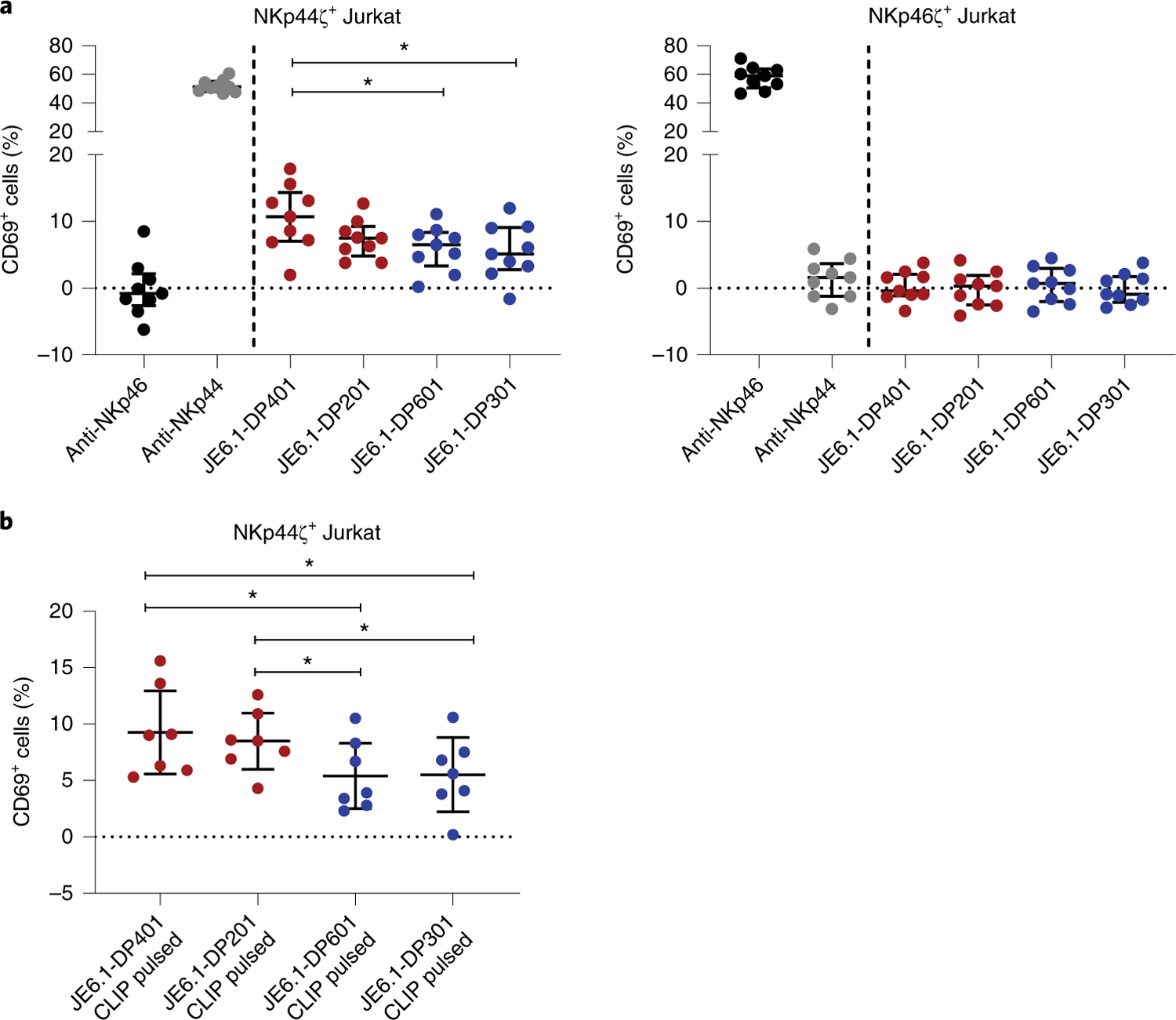
a, Reporter cell activity of NKp44ζ+ and NKp46ζ+ Jurkat cells was determined as percentage of CD69+ cells after co-incubation with plate-coated anti-NKp44 and anti-NKp46 molecules (1 µg ml−1) as well as four distinct HLA-DP-expressing JE6.1 cell lines (JE6.1-DP). The percentage of CD69+ cells following incubation on non-coated wells (blank) was subtracted from all samples. The percentage of CD69+ cells after co-incubation with non-HLA-DP-transduced JE6.1 cells was subtracted from JE6.1-DP-positive samples. Corrected values are illustrated as median with interquartile range as determined in nine independent experiments. Each dot represents one individual experiment (n = 9). Two-tailed Friedman test with post-hoc Dunn’s multiple comparison was used to determine statistical differences between JE6.1-DP-expressing cell lines. Control conditions (anti-NKp46, anti-NKp44) were not included in the statistical analysis. *P = 0.02 and 0.01. b, Activation of NKp44ζ+ Jurkat reporter cells following co-incubation with CLIP peptide-pulsed JE6.1-DP cell lines is depicted as percentage of CD69+ cells. The percentage of CD69+ cells following incubation on non-coated wells (blank) was subtracted from all samples. The percentage of CD69+ cells after co-incubation with CLIP-pulsed non-HLA-DP-transduced JE6.1 cells was subtracted from JE6.1-DP-positive samples. Corrected values are illustrated as median with interquartile range as determined in seven independent experiments (n = 7). Two-tailed Friedman test with post-hoc Dunn’s multiple comparison was used to determine statistical differences between percentages of CD69+ cells following co-incubation with the four different CLIP peptide-pulsed JE6.1-DP cell lines. *P = 0.04.
NKp44 (NCR2) is one of the three NCRs expressed by NK cells17. NKp44+ innate immune cells have been implicated in autoimmune diseases, such as inflammatory bowel disease33,34, and several studies have investigated the functional characteristics of NKp44+ NK cells17,35,36. NKp44 was initially described to be an activating NK cell receptor, although it was subsequently demonstrated that some splice isoforms of NKp44 can also negatively regulate NK cell function37. Although the function of NKp44+ NK cells has been extensively studied, a widely expressed cellular ligand for NKp44 has remained elusive and controversial. Several cellular and viral NKp44 ligands have been proposed, such as viral hemagglutinin38,39, proliferating cell nuclear antigen40 and mixed-lineage leukemia protein 5 (ref. 41), but other studies have failed to reproduce these findings42. Platelet-derived growth factor (PDGF)-DD has been described as a soluble ligand binding for NKp44 (ref. 42). PDGF-DD engagement of NKp44 triggered cytokine secretion by NK cells, and this induced tumor cell-growth arrest42. Here we identified a subset of HLA-DP molecules as cellular ligands for NKp44 and demonstrated that binding of these HLA-DP molecules triggered the activation of NKp44+ reporter cells and primary NK cells expressing NKp44. Neither NKp44 nor HLA-DP exist in mice43,44 and the absence of small-animal models to study NKp44+ NK cells has certainly complicated the identification of its ligand. It has been suggested that expression of NKp44 evolved over the last 23–25 million years, and inefficient transcription of NKp44 in macaques has been described, whereas NKp44 surface expression has been detected on chimpanzee-derived NK cells43. The absence of an NKp44 gene within the murine triggering receptor expressed on myeloid cells (TREM) gene cluster, within which the NKp44 gene is encoded in humans45, and the insufficient transcription within macaques indicate a functional role of NKp44 at later stages in evolution in Homo sapiens sapiens and closely related species. Similarly, the diversification of HLA-DP between different species46 and the interchangeable function of the distinct HLA class II molecules within one species, as described for rodents47, suggests a functional impact of HLA-DP–NKp44 interactions primarily in humans.
HLA-DP is constitutively expressed on antigen-presenting cells and B cells, and HLA-DP molecules have been shown to be upregulated on non-hematopoietic tissues in response to inflammation24. Specific HLA-DP allotypes, as well as the expression levels of HLA-DP molecules, have been associated with a variety of human diseases, including graft-versus-host disease following hematopoietic cell transplantation8, hepatitis B virus (HBV) infection5–7 and autoimmune diseases9,10. In the setting of graft-versus-host disease and HBV infection, it has been suggested that disease outcome is linked to specific single nucleotide polymorphisms (SNPs) in the 3′ untranslated region (UTR) region of the gene encoding the HLA-DP β-chain, which mark differential HLA-DP expression levels on the cell surface7,8. Low expression of HLA-DP was associated with HBV clearance, whereas high expression of HLA-DP was associated with persistence of HBV and a higher risk of developing graft-versus-host disease. Interestingly, HLA-DP401 and HLA-DP201, the two HLA-DP molecules that induced strong NKp44 binding and activation of NKp44ζ+ reporter cells, are both in linkage disequilibrium with the SNP associated with low HLA-DP expression (496A, rs9277534) and recovery from HBV infection. In contrast, the two HLA-DP molecules tested that exhibited no or only limited binding to NKp44 Fc constructs and low activation of NKp44ζ+ reporter cells, HLA-DP601 and HLA-DP301, are associated with high HLA-DP surface expression (496G) and HBV persistence7. These data indicate that the levels of HLA-DP expression and NKp44 binding might have co-evolved, with high-expressed HLA-DP molecules mediating no or little binding to NKp44, possibly to avoid induction of autoimmunity. However, in response to inflammation, such as during HBV infection, HLA-DP molecules have been shown to be upregulated24, and upregulation of normally low-expressed HLA-DP molecules that serve as ligands for NKp44 might enable recognition of infected cells by NKp44+ NK cells and viral clearance. The interaction identified here between a subset of HLA-DP molecules and NKp44 will now allow for functional assessment of the consequences of these interactions for the outcome of diseases that have been associated with HLA-DP genotypes and expression levels.
The precise regions within NKp44 and HLA-DP that are responsible for the binding of these molecules remain unknown. While NKp44 consistently bound to a subset of HLA-DP molecules, binding to these HLA-DP molecules was not solely dependent on either the α- or β-chain of the respective HLA-DP molecule alone, indicating a binding region determined by both chains. Identification of the specific binding sites within the HLA-DP molecules for NKp44, and how binding is further modulated by the HLA-DP-presented peptide, will be a critical step towards understanding the structural requirements for these interactions. It is well established that specific peptides presented by HLA class I molecules impact binding of KIRs25–30, and we also observed an impact of the HLA-DP-presented peptides on the activity of NKp44-expressing cells (Fig. 4). It is therefore possible that NKp44 might bind to additional HLA class II molecules other than the ones identified in this study in situations in which these molecules present specific peptides. However, using membrane-bound HLA-DP molecules presenting a broad array of different peptides, we observed consistent hierarchies of interactions with NKp44, with HLA-DP401 always exhibiting the strongest binding. These data suggest that HLA class II allotypes represent the principal determinants for NKp44 binding, reminiscent of KIR binding to HLA class I, which is strongly defined by the respective HLA class I allotypes48. In conclusion, this study identified an interaction between a subset of HLA-DP molecules and the NK cell receptor NKp44 that has a functional impact on NKp44+ NK cell activity, implicating HLA class II molecules in the regulation of innate immunity.
Methods
Cell lines used.
All Jurkat reporter cell lines (clone E6.1; American Type Culture Collection (ATCC)) used lacked β2-microglobulin (Jurkat-β2mKO cells) and were engineered to express NK cell receptors of interest, as previously described11. The KIR2DL3ζ+ Jurkat reporter cell line was previously generated11. Briefly, NKp44ζ and NKp46ζ constructs were designed by fusing the extracellular domain of the respective NCR molecule to the transmembrane domain of KIR3DL1 and the cytoplasmic domain of the CD3ζ chain. Constructs were synthesized by GeneArt GeneSynthesis (ThermoFisher) and cloned into a lentiviral transfer plasmid encoding a puromycin resistance. To produce lentiviral-like particles, HEK293T cells (ATCC) were transfected using Lipofectamine 2000 (Life Technologies), a VSV-G envelope vector (pHEF-VSVG; NIH AIDS Reagent Program), an HIV-1 Gag-Pol packaging vector (psPAX2; NIH AIDS Reagent Program) and the transfer vector (pSIP-ZsGreen, kindly provided by T. Pertel) encoding the gene of interest. Jurkat-β2mKO cells were transduced with lentiviral-like particles and selected 3 d post transduction with 1 µg ml−1 of puromycin (Sigma-Aldrich). NKp44ζ+, KIR2DL3ζ+ and NKp46ζ+ Jurkat cells were cultured at 37 °C/5% CO2 in RPMI-1640 (Life Technologies) supplemented with 20% heat-inactivated fetal bovine serum (FBS) (Biochrom) and maintained in 1 µg ml−1 puromycin (Sigma-Aldrich). HLA-DP sequences (HLA-DPA1: accession number: P20036, HLA-DPB1: accession number: P04440) containing the mammalian Kozak sequence were synthesized using GeneArt Strings (Life Technologies). DNA fragments encoding for HLA-DPA1*01:03, HLA-DPB1*02:01, HLA-DPB1*04:01, HLA-DPB1*03:01 and HLA-DPB1*06:01 were cloned into a lentiviral transfer vector (pSIP-ZsGreen, kindly provided by T. Pertel) using NEBuilder HiFi DNA Assembly Cloning Kit (New England Biolabs). Lentiviral particles were produced as described for NKp44ζ and NKp46ζ constructs. Wild-type Jurkat E6.1 cells (ATCC) were transduced using HLA-DPA1*01:03 lentiviral-like particles in combination with an HLA-DPB1 molecule to achieve surface expression of the respective HLA-DP molecule. Transduced cells were selected with 1 µg ml−1 puromycin 3 d post transduction and stained for HLA-DP surface expression using an anti-HLA-DP-PE from Leinco Technologies. Cells were cultured at 37 °C/5% CO2 in RPMI-1640 supplemented with 20% heat-inactivated FBS and maintained in 1 µg ml−1 puromycin. K562 cells (DSMZ) were grown in RPMI-1640 supplemented with 10% heat-inactivated FBS at 37 °C/5% CO2. Testing for mycoplasma infection of cell lines was not performed regularly in all cases.
Recombinant human Fc construct binding to HLA class II-coated beads.
Screening of HLA class II-coated beads was performed using the LABScreen Single Antigen HLA class II—Group 1 kit (OneLambda). Recombinant human Fc constructs (LAG-3, NKp30, NKp44, NKp46, KIR2DL3, KIR2DL1, KIR2DS1, KIR3DS1, KIR3DL1, KIR3DL2 and KIR2DL4) were purchased from R&D Systems, diluted in PBS to concentrations ranging from 1 to 100 µg ml−1 and incubated with a mixture of 95 HLA class II-coated beads for 30 min at room temperature. Samples were washed and incubated with F(ab′)2 goat-anti-human IgG PE secondary antibody (Life Technologies) for 30 min at 4 °C. Fc construct binding to HLA class II-coated beads was quantified using Luminex xMAP technology on a Bio-Plex 200 (Bio-Rad Laboratories).
Protein expression and purification.
The genes of NKp44 (S19–S130) and NKp46 (T25–G212) were synthesized by Integrated DNA Technologies with an amino (N)-terminal 6xHis tag and cloned into the pET30 vector. NKp44 and NKp46 were expressed as inclusion bodies in Ton A− BL21 (DE3) Escherichia coli and subsequently refolded by rapid dilution at 4 °C. NKp44 was refolded in a buffer containing 20 mM HEPES pH 7.0, 0.4 M l-arginine, 5 M urea, 0.5 mM oxidized glutathione, 5 mM reduced glutathione, 2 mM EDTA and 0.2 mM phenylmethyl sulfonyl fluoride (PMSF), and NKp46 refolded in a buffer consisting of 0.1 M Tris–HCl pH 8.5, 0.9 M l-arginine, 1 M urea, 0.5 mM oxidized glutathione, 5 mM reduced glutathione, 2 mM EDTA and 0.2 mM PMSF. Refolded NKp44 and NKp46 were dialyzed into 5 mM HEPES pH 7.0, 0.3 M NaCl and 10 mM Tris–HCl pH 8.5, 0.15 M NaCl, respectively, before purification via nickel-affinity and size-exclusion chromatography systems (Superdex 75, GE Healthcare). HLA-DPA1*01:03 paired with either HLA-DPB1*04:01 (HLA-DP401 CLIP) or HLA-DPB1*03:01 (HLA-DP301 CLIP), and HLA-DQA1*05:01 paired with HLA-DQB1*02:01 (HLA-DQ2 CLIP), were expressed and purified similarly as previously described49,50. Briefly, the appropriate α- and β-chains were cloned into the pFastBac Dual vector (Invitrogen) with fos/jun leucine zippers, a 7xHis tag at the β-chain carboxy (C) terminus and the CLIP peptide (ATPLLMQALPMGA) linked via the β-chain N terminus. Recombinant baculovirus was generated as per the manufacturer’s instructions and, following transfection and expansion of the baculovirus in Sf9 cells, 1.4–3.3% of P3 viral stock was used to infect Hi5 cells. After incubation for 72–96 h at 27 °C (HLA-DP301 CLIP) or at 21 °C (HLA-DP401 CLIP and HLA-DQ2 CLIP), media containing secreted protein was concentrated and buffer exchanged into 10 mM Tris pH 8.0, 0.5 M NaCl (by tangential flow filtration) and purified using nickel-affinity and size-exclusion chromatography systems (Superdex S200, GE Healthcare). The fos/jun zippers were removed via overnight treatment with enterokinase and separated via anion exchange chromatography (Hitrap Q, GE Healthcare).
SPR.
SPR experiments were performed on a BIAcore T200 at 25 °C using CM5 series S sensor chips (GE Healthcare). In a buffer comprising 10 mM HEPES pH 7.0, 300 mM NaCl, NKp44 and NKp46 were immobilized to the chip surface via amine coupling (according to the manufacturer’s instructions) with capture levels of ~870 response units (RU) (chip one) and ~1,400 RU (chip two). In duplicates, at 10 µl min−1, in a buffer comprising of 10 mM Tris pH 8.0, 150 mM NaCl, 0.5% BSA, analyte proteins (typically 0.31–40 µM) were injected for 60 s, followed by a 300–500-s dissociation time. One preparation of HLA-DP401 had a maximum concentration of 35 µM. The responses on the active flow cells were double referenced by subtracting responses from an ‘empty’ flow cell as well as from buffer-only injections. Equilibrium dissociation constants were derived from fits using a single-site binding model. Data were analyzed using Scrubber v.2.0 (BioLogic Software) and Prism v.7.0 (GraphPad Software).
Jurkat reporter cell assay.
Non-tissue culture-treated plates (Corning Life Sciences) were coated with 5 µg ml−1 biotinylated HLA class II monomers (kindly provided by the NIH Tetramer Core Facility), HLA-DP single antigens (kindly provided by M.O. and J.-H.L. (OneLambda Inc.); not commercially available), 1 µg ml−1 biotinylated anti-NKp44, 1 µg ml−1 biotinylated anti-NKp46 or 1 µg ml−1 biotinylated anti-KIR2DL3 (Biolegend or Miltenyi Biotec) diluted in PBS. One negative control well with only PBS was prepared for each cell line. Coated plates were incubated at 4 °C for a minimum of 24 h. Jurkat reporter cells (2.5 × 104 cells per well) were incubated for 5 h at 37 °C/5% CO2 on coated plates. For blocking experiments, NKp44ζ+ Jurkat reporter cells were pre-incubated with 10 µg ml−1 purified anti-NKp44 (Biolegend) or 10 µg ml−1 purified isotype control antibody (Biolegend) for 30 min at 37 °C/5% CO2 before co-incubation with coated wells; blocking antibodies remained present throughout the co-incubation. After co-incubation, cells were stained with the viability dye Zombie NIR or ZombieAqua (Biolegend) anti-CD3-BUV737 (BD Biosciences) and anti-CD69-BV421 (Biolegend), as well as their respective NCR (anti-NKp44-PE (Biolegend), anti-NKp46-PE (BD Biosciences)) or anti-KIR2DL3-PE (Miltenyi Biotec)) surface molecule and subsequently fixed with 4% paraformaldehyde (PFA). CD69 expression was assessed using a BD LSR Fortessa (BD Biosciences). (A detailed list of antibodies used is given in Supplementary Table 2.)
Isolation of human NK cells.
Human peripheral blood mononuclear cells were isolated from healthy adult donors recruited at the University Medical Center, Hamburg-Eppendorf using Biocoll (Biochrom) density centrifugation. All donors provided written informed consent and studies were approved by the ethical committee of the Ärztekammer Hamburg (PV4780). NK cells were enriched from isolated PBMCs using magnetic labeling and negative selection using the EasySep human NK cell enrichment kit (StemCell Technologies). Isolated NK cells were either directly used for degranulation assays or cultured for 7 d in RPMI-1640 supplemented with 10% FBS, 10 ng ml−1 IL-15 and 250 U ml−1 IL-2 (PeproTech) at a concentration of 2 × 106 cells ml–1.
Degranulation assay.
Non-tissue culture-treated plates (Corning Life Sciences) were coated with 10 µg ml−1 biotinylated HLA class II monomers (kindly provided by the NIH Tetramer Core Facility), HLA-DP single antigens (kindly provided by M.O. and J.-H.L. ((OneLamda Inc.,); not commercially available), 1 µg ml−1 biotinylated anti-NKp44 (Biolegend) diluted in PBS or left uncoated. NK cells were resuspended at a final concentration of 2 × 105 cells ml–1 in assay medium containing RPMI-1640 medium supplemented with 10% FBS, 250 U ml−1 IL-2. Isolated NK cells (2 × 104 cells per well) were distributed on coated plates or co-incubated with K562 cells at an effector to target ratio of 1:5. Brefeldin A (Sigma-Aldrich), to a final concentration of 5 µg ml−1, and anti-CD107a-BV785 (Biolegend) were added to each well. Cells were incubated for 5 h at 37 °C/5% CO2. For blocking experiments, NK cells were pre-incubated with 10 µg ml−1 anti-NKp44 or 10 µg ml−1 IgG isotype control antibody (Biolegend) for 30 min at 37 °C/5% CO2. Blocking antibodies remained present during the following co-incubation. After co-incubation cells were stained with anti-CD3-BV510, anti-CD56-BV605, anti-CD16-FITC, anti-NKp44-AF647, anti-CD69-BV421 and Zombie NIR (all Biolegend). NK cells were fixed using BD Cytofix/Cytoperm Kit (BD Biosciences) and analyzed using a BD LSR Fortessa (BD Biosciences). (A detailed list of antibodies used is given in Supplementary Table 2.)
Jurkat reporter cell–Jurkat-HLA-DP co-incubation assay.
HLA-DP-expressing Jurkat E6.1 cells were co-incubated with NKp44ζ+, NKp46ζ+ Jurkat reporter cells or untransduced Jurkats at an effector to target ratio of 1:10 for 5 h at 37 °C/5% CO2. For peptide-pulsing experiments, HLA-DP-expressing Jurkat E6.1 cell lines were washed twice with serum-free medium and pulsed with 100 µM CLIP peptide (sequence: LPKPPKPVSKMRMATPLLMQALPM; GenScript) or an equivalent amount of DMSO for 18 h at 37 °C/5% CO2. After peptide pulsing, JE6.1-DP cells were washed and co-incubated with Jurkat reporter cell lines. Subsequently, cells were stained with anti-CD3-BUV737 (BD Biosciences), anti-NKp44-PE (Biolegend) or anti-NKp46-PE (BD Biosciences), anti-CD69-BV421 (Biolegend), LiveDead NearIR (Life Technologies) and anti-HLA-ABC-APC (Biolegend), to discriminate between HLA-DP-expressing JE6.1 and Jurkat reporter cells, for 30 min at 4 °C. Cells were fixed with 4% PFA and analyzed on a BD LSR Fortessa. HLA-DP and CLIP surface expression of JE6.1-DP cells was assessed by staining CLIP-pulsed and DMSO-pulsed JE6.1-DP cells with anti-CD3-BUV737 (BD Biosciences), anti-HLA-DP-APC (Leinco Technologies), anti-CLIP-FITC (BD Biosciences) and LiveDead NearIR (Life Technologies) for 30 min at 4 °C. Cells were subsequently fixed and analyzed on a BD LSR Fortessa. (A detailed list of antibodies used is displayed in Supplementary Table 2.)
Data analysis and statistics.
Flow cytometry data were analyzed using FlowJo v.10 (TreeStar). SPR data were analyzed using Scrubber v.2.0 (BioLogic Software). Statistical analyses were done using two-tailed Wilcoxon matched pairs signed-rank test for experiments comparing two conditions, and two-tailed Friedman test with post-hoc Dunn’s multiple comparison for experiments comparing more than two conditions of interest (GraphPad Prism v.7 and v.8). When data from replicates were included (Figs. 1a and 2d), mixed effects linear regression models were used for statistical comparisons (SAS Software v.9.3). This type of model allows the correlation between replicates in these comparisons to be taken into account, as well as the systematic technical batch effect. If not otherwise indicated, the distribution of data from control conditions were assessed visually and not included in statistical comparisons.
Supplementary Material
Acknowledgements
This work has been funded in part by the Pathogenesis and the Viral Latency Programs of the Heinrich Pette Institute, Leibniz Institute for Experimental Virology and the German Center for Infection Research (DZIF) through TTU 04.810. This project has been funded in part with federal funds from the Frederick National Laboratory for Cancer Research, under contract no. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. This research was supported in part by the Intramural Research Program of the National Institutes of Health, Frederick National Laboratory, Center for Cancer Research. W.F.G.-B. was supported by National Institute of General Medical Sciences (T32GM007752) and the NIH (P01-AI104715 and F31AI116366). P.J.N. was supported by NIH U19 NS095774. A.H. was supported by the German Center for Infection Research (DZIF) through an MD/PhD Stipend (TI 07.002) and via the Clinician Scientist Program of the Faculty of Medicine, University Medical Center Hamburg-Eppendorf, Hamburg, Germany. J.R was supported by an Australian Research Council Laureate Fellowship (FL160100049) and R.B was supported by a Career Development Fellowship from the National Health and Medical Research Council of Australia (APP1109901). We would like to thank H. Reid and K. Loh for their kind gift of HLA-DQ2 viral stocks. We would like to thank the NIH Tetramer Core Facility for all provided HLA class II monomers.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of code and data availability and associated accession codes are available at https://doi.org/10.1038/s41590-019-0448-4.
Competing interests
A.N., W.F.G.-B. and M.A. filed a patent application (EP18174760.1) regarding the therapeutic use of anti-NKp44 antibodies for the treatment and/or prevention of graft-versus-host disease. M.O. and J.-H.L. are current employees of OneLambda Inc., a part of Thermo Fisher Scientific. All other authors declare no competing interest.
Supplementary information is available for this paper at https://doi.org/10.1038/s41590-019-0448-4.
Peer review information: Zoltan Fehervari was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Reporting Summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
All codes are available upon request from the corresponding author
Data availability
All figures have associated raw data. All primary data files are available upon request from the corresponding author.
References
- 1.Jost S & Altfeld M Control of human viral infections by natural killer cells. Annu. Rev. Immunol 31, 163–194 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Jiang YZ et al. Interaction of natural killer cells with MHC class II: reversal of HLA-DR1-mediated protection of K562 transfectant from natural killer cell-mediated cytolysis by brefeldin-A. Immunology 87, 481–486 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lobo PI, Chang MY & Mellins E Mechanisms by which HLA-class II molecules protect human B lymphoid tumour cells against NK- and LAK-mediated cytolysis. Immunology 88, 625–629 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.al-Daccak R et al. Gene polymorphism of HLA-DPB1 and DPA1 loci in caucasoid population: frequencies and DPB1-DPA1 associations. Hum. Immunol 31, 277–285 (1991). [DOI] [PubMed] [Google Scholar]
- 5.Guo X et al. Strong influence of human leukocyte antigen (HLA)-DP gene variants on development of persistent chronic hepatitis B virus carriers in the Han Chinese population. Hepatology 53, 422–428 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamatani Y et al. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat. Genet 41, 591–595 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Thomas R et al. A novel variant marking HLA-DP expression levels predicts recovery from hepatitis B virus infection. J. Virol 86, 6979–6985 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petersdorf EW et al. High HLA-DP expression and graft-versus-host disease. N. Engl. J. Med 373, 599–609 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hadley D et al. HLA-DPB1*04:01 protects genetically susceptible children from celiac disease autoimmunity in the TEDDY study. Am. J. Gastroenterol 110, 915–920 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goyette P et al. High-density mapping of the MHC identifies a shared role for HLA-DRB1*01:03 in inflammatory bowel diseases and heterozygous advantage in ulcerative colitis. Nat. Genet 47, 172–179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Beltran WF et al. Open conformers of HLA-F are high-affinity ligands of the activating NK-cell receptor KIR3DS1. Nat. Immunol 17, 1067–1074 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Triebel F et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med 171, 1393–1405 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berry R et al. Targeting of a natural killer cell receptor family by a viral immunoevasin. Nat. Immunol 14, 699–705 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Deuss FA, Watson GM, Fu Z, Rossjohn J & Berry R Structural basis for CD96 immune receptor recognition of nectin-like protein-5, CD155. Structure 27, 219–228.e3 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Rossjohn J et al. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol 33, 169–200 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Vivian JP et al. Killer cell immunoglobulin-like receptor 3DL1-mediated recognition of human leukocyte antigen B. Nature 479, 401–405 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vitale M et al. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J. Exp. Med 187, 2065–2072 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alter G, Malenfant JM & Altfeld M CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 294, 15–22 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Hiltbold EM & Roche PA Trafficking of MHC class II molecules in the late secretory pathway. Curr. Opin. Immunol 14, 30–35 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Wen F, Esteban O & Zhao H Rapid identification of CD4+ T-cell epitopes using yeast displaying pathogen-derived peptide library. J. Immunol. Methods 336, 37–44 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Birnbaum ME et al. Deconstructing the peptide-MHC specificity of T cell recognition. Cell 157, 1073–1087 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lo W-L et al. An endogenous peptide positively selects and augments the activation and survival of peripheral CD4+ T cells. Nat. Immunol 10, 1155–1161 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dai S et al. Crystal structure of HLA-DP2 and implications for chronic beryllium disease. Proc. Natl Acad. Sci. USA 107, 7425–7430 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stevanovic S et al. HLA class II upregulation during viral infection leads to HLA-DP-directed graft-versus-host disease after CD4+ donor lymphocyte infusion. Blood 122, 1963–1973 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Holzemer A et al. Selection of an HLA-C*03:04-restricted HIV-1 p24 Gag sequence variant is associated with viral escape from KIR2DL3+ natural killer cells: data from an observational cohort in South Africa. PLoS Med 12, e1001900 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naiyer MM et al. KIR2DS2 recognizes conserved peptides derived from viral helicases in the context of HLA-C. Sci. Immunol 2, eaal5296 (2017). [DOI] [PubMed] [Google Scholar]
- 27.O’Connor GM et al. Peptide-dependent recognition of HLA-B*57:01 by KIR3DS1. J. Virol 89, 5213–5221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Connor GM et al. Mutational and structural analysis of KIR3DL1 reveals a lineage-defining allotypic dimorphism that impacts both HLA and peptide sensitivity. J. Immunol 192, 2875–2884 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chapel A et al. Peptide-specific engagement of the activating NK cell receptor KIR2DS1. Sci. Rep 7, 2414 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rajagopalan S & Long EO The direct binding of a p58 killer cell inhibitory receptor to human histocompatibility leukocyte antigen (HLA)-Cw4 exhibits peptide selectivity. J. Exp. Med 185, 1523–1528 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holling TM, Schooten E, Langerak AW & van den Elsen PJ Regulation of MHC class II expression in human T-cell malignancies. Blood 103, 1438–1444 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Thompson JA et al. Tumor cells transduced with the MHC class II transactivator and CD80 activate tumor-specific CD4+ T cells whether or not they are silenced for invariant chain. Cancer Res 66, 1147–1154 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Takayama T et al. Imbalance of NKp44+NKp46− and NKp44−NKp46+ natural killer cells in the intestinal mucosa of patients with Crohn’s disease. Gastroenterology 139, 882–892.e3 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Glatzer T et al. RORgammat+ innate lymphoid cells acquire a proinflammatory program upon engagement of the activating receptor NKp44. Immunity 38, 1223–1235 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Campbell KS, Yusa S, Kikuchi-Maki A & Catina TL NKp44 triggers NK cell activation through DAP12 association that is not influenced by a putative cytoplasmic inhibitory sequence. J. Immunol 172, 899–906 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Cantoni C et al. NKp44, a triggering receptor involved in tumor cell lysis by activated human natural killer cells, is a novel member of the immunoglobulin superfamily. J. Exp. Med 189, 787–796 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siewiera J et al. Natural cytotoxicity receptor splice variants orchestrate the distinct functions of human natural killer cell subtypes. Nat. Commun 6, 10183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arnon TI et al. Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur. J. Immunol 31, 2680–2689 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Ho JW et al. H5-type influenza virus hemagglutinin is functionally recognized by the natural killer-activating receptor NKp44. J. Virol 82, 2028–2032 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosental B et al. Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44. J. Immunol 187, 5693–5702 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baychelier F et al. Identification of a cellular ligand for the natural cytotoxicity receptor NKp44. Blood 122, 2935–2942 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Barrow AD et al. Natural killer cells control tumor growth by sensing a growth factor. Cell 172, 534–548.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Maria A et al. NKp44 expression, phylogenesis and function in non-human primate NK cells. Int. Immunol 21, 245–255 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ting JP-Y & Trowsdale J Genetic control of MHC class II expression. Cell 109, S21–S33 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Allcock RJN, Barrow AD, Forbes S, Beck S & Trowsdale J The human TREM gene cluster at 6p21.1 encodes both activating and inhibitory single IgV domain receptors and includes NKp44. Eur. J. Immunol 33, 567–577 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Slierendregt BL, Otting N, Kenter M & Bontrop RE Allelic diversity at the Mhc-DP locus in rhesus macaques (Macaca mulatta). Immunogenetics 41, 29–37 (1995). [DOI] [PubMed] [Google Scholar]
- 47.Nizetic D, Figueroa F, Dembic Z, Nevo E & Klein J Major histocompatibility complex gene organization in the mole rat Spalax ehrenbergi: evidence for transfer of function between class II genes. Proc. Natl Acad. Sci. USA 84, 5828–5832 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vilches C & Parham P KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu. Rev. Immunol 20, 217–251 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Petersen J et al. T-cell receptor recognition of HLA-DQ2-gliadin complexes associated with celiac disease. Nat. Struct. Mol. Biol 21, 480–488 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Henderson KN et al. A structural and immunological basis for the role of human leukocyte antigen DQ8 in celiac disease. Immunity 27, 23–34 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All figures have associated raw data. All primary data files are available upon request from the corresponding author.