

Abstract
Autophagy is a lysosomal catabolic process by which cells degrade or recycle their contents to maintain cellular homeostasis, adapt to stress, and respond to disease. Impairment of autophagy in endothelial cells studied under static conditions results in oxidant stress and impaired nitric oxide (NO) bioavailability. We tested the hypothesis that vascular autophagy is also important for induction of NO production caused by exposure of endothelial cells to shear stress (i.e., 3 h × ≈20 dyn/cm2). Atg3 is a requisite autophagy pathway mediator. Control cells treated with non-targeting control siRNA showed increased autophagy, reactive oxygen species (ROS) production, endothelial NO synthase (eNOS) phosphorylation, and NO production upon exposure to shear stress (p < 0.05 for all). In contrast, cells with >85% knockdown of Atg3 protein expression (via Atg3 siRNA) exhibited a profound impairment of eNOS phosphorylation, and were incapable of increasing NO in response to shear stress. Moreover, ROS accumulation and inflammatory cytokine production (MCP-1 and IL-8) were exaggerated (all p < 0.05) in response to shear stress. These findings reveal that autophagy not only plays a critical role in maintaining NO bioavailability, but may also be a key regulator of oxidant–antioxidant balance and inflammatory–anti-inflammatory balance that ultimately regulate endothelial cell responses to shear stress.
Keywords: blood flow, exercise, mitochondrial turnover, mitophagy, nutrient deprivation, oxidant stress, vascular
Résumé :
L’autophagie est un processus catabolique lysosomal par lequel les cellules dégradent ou recyclent leur contenu afin de maintenir l’homéostasie cellulaire, s’adapter au stress et répondre à une maladie. La défaillance de l’autophagie chez les cellules endothéliales étudiées en conditions statiques provoque un stress oxydant et une diminution de la biodisponibilité d’oxyde nitrique (NO). Les auteurs ont testé l’hypothèse que l’autophagie vasculaire est aussi importante à l’induction de la production de NO provoquée par l’exposition des cellules endothéliales à un stress de cisaillement (par example, 3 h × ≈ 20 dyn/cm2). Atg3 est un médiateur nécessaire de la voie de l’autophagie. L’autophagie, la production d’espèces réactives d’oxygène (ERO), la phosphorylation de la NO synthase endothéliale (eNOS) et la production de NO étaient accrues chez des cellules contrôles traitées avec un pARNi contrôle non-ciblant, exposées à un stress de cisaillement (p < 0,05 pour tous les paramètres). En revanche, la phosphorylation de eNOS était diminuée de façon marquée chez les cellules dont Atg3 avait été inactivé de > 85 % par « knockdown » (au moyen du pARNi Atg3), et ces cellules étaient incapables d’accroitre le niveau de NO en réponse à un stress de cisaillement. De plus, l’accumulation d’ERO et la production de cytokines inflammatoires (MCP-1 et IL-8) étaient accrues (p < 0,05) en réponse à un stress de cisaillement. Ces données révèlent que l’autophagie joue non seulement un rôle dans le maintien de la biodisponibilité de NO, mais qu’elle agit aussi comme régulateur clé de la balance oxydante–anti-oxydante et inflammatoire–anti-inflammatoire, qui régule ultimement les réponses des cellules endothéliales au stress de cisaillement. [Traduit par la Rédaction]
Mots-clés: flux sanguin, exercice, « turnover » mitochondrial, mitophagie, privation nutritionnelle, stress oxydant, vasculaire
Introduction
Autophagy (i.e., “self-eating”) is a highly conserved, ubiquitous lysosomal trafficking pathway of eukaryotic cells that preserves cell health by degrading nonessential or damaged intracellular constituents that accumulate during the normal life of the cell. In addition, autophagy plays a critical role in maintaining homeostasis during many forms of cellular stress, and provides substrates for energy production during periods of nutrient deprivation (Pallauf and Rimbach 2013). Autophagy utilizes a unique double-membrane vesicle, the autophagasome, to engulf cytosolic macromolecules and organelles and transport them to lysosomes. When autophagasomes fuse with lysosomes, their cargoes are degraded by lysosomal acid hydrolases (Mizushima et al. 2008). It is now evident that autophagy plays a vital role in many physiological functions and, thus, may contribute importantly to both the pathogenesis and prevention of disease (Levine and Kroemer 2008; Ravikumar et al. 2010).
In the cardiovascular system, rates of autophagy can be altered rapidly and dramatically in response to physiological stimuli or pathophysiological stressors (Levine and Kroemer 2008; Ravikumar et al. 2010). For instance, autophagy is activated in cardiomyocytes by nutrient deprivation, likely as a survival mechanism to provide energy for vital cellular functions (Zhang et al. 2010). Autophagy is induced in cardiomyocytes exposed to lipotoxic and oxidative stress by treatment with saturated fatty acids, likely as a defense mechanism to clear misfolded proteins and damaged organelles (Russo et al. 2012). Insulin is an important negative regulator of autophagy in many tissues, and insulin-resistant states are associated with increased autophagy in many tissues. Consistent with these observations, obese mice with insulin resistance resulting from high fat feeding exhibit increased autophagy in myocardium (Russo et al. 2012).
Physical exertion and digestion increase arterial blood flow from basal levels in an attempt to meet greater nutrient requirements of metabolically active tissues (i.e., active skeletal and smooth muscle, respectively). The greater perfusion increases “endothelial shear stress” from basal levels. Endothelial shear stress is the product of blood viscosity and velocity that is sensed by mechanoreceptors on the surface of endothelial cells (Davies 1995; Boo and Jo 2003; Sessa 2004; Wentzel et al. 2012). Endothelial shear stress induces both reactive oxygen species (ROS) production (Chin et al. 2011) and, simultaneously, anti-oxidative and anti-inflammatory gene expression (Zhang and Friedman 2013). These responses enhance phosphorylation state and activity of endothelial nitric oxide (NO) synthase (eNOS) (Ives et al. 2012), increasing NO bioavailability to enhance vessel dilation and facilitate functional hyperemia to metabolically active tissues (Zhang et al. 2009). Whether autophagy plays a role in mediating these responses to shear stress in endothelial cells has not been determined.
Several lines of evidence suggest that autophagy might be induced by endothelial shear stress. Compressive mechanical force (i.e., 0.2–1.0 kPa for 10 min) increases autophagy by 20-fold in mammalian cells (King et al. 2011). In addition, ROS such as superoxide anion (O2−) and hydrogen peroxide (H2O2) are known to induce autophagy (Bensaad et al. 2009; Shafique et al. 2013). Here, we test the hypothesis that autophagy is required for shear-stress-induced increases in NO bioavailability in bovine aortic endothelial cells (BAECs). Our findings indicate that in BAECs with impaired autophagy, eNOS phosphorylation and NO bioavailability are attenuated, whereas ROS accumulation and indices of inflammation are exaggerated. These findings suggest that autophagy may be an important regulator of NO bioavailability in endothelial cells, and could potentially mediate vascular responses in vivo to conditions such as exercise, atherosclerosis, and hyperviscosity syndromes (e.g., Waldenström’s macroglobulinemia, multiple myeloma), which are associated with increased blood flow and (or) endothelial shear stress.
Research design and methods
Cell culture
BAECs obtained commercially (Lonza Inc., Allendale, New Jersey, USA) were grown to 70%–80% confluency in high (25 mmol/L) glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Hyclone Laboratories Inc., Logan, Utah, USA) in a humidified atmosphere (5% CO2, 95% O2) at 37 °C (Symons et al. 2009; Zhang et al. 2009, 2012).
Autophagy and nutrient deficiency
Cells were incubated with DMEM containing amino acids (nutrient-replete; NR) or Hanks balanced salt solution (HBSS) without glucose and amino acids (nutrient depleted; ND). Both solutions contained 0.5 mmol/L pyruvate to maintain mitochondrial metabolism and 0.04 mmol/L phenol red (Zhang et al. 2010).
To evaluate the time course of autophagy induction in endothelial cells, BAECs were incubated in NR or ND medium for 0.5, 1, 1.25, 1.5, 2, 3, 4, 5, 6, 12, and 24 h. Autophagy was maximal at 2 h. To test whether the induction of autophagy could be reversed, glucose and amino acids were restored (R) to ND medium for 1 h following 2 h of exposure to ND medium. To determine autophagic flux, bafilomycin A1 (baf A1, 100 nmol/L) was directly added to NR and ND medium for 2 h to block lysosomal degradation of autophagosomes (Klionsky et al. 2008).
Autophagy and shear stress
BAECs were exposed to no shear stress (−shear) or ≈20 dyn/cm2 for 3 h (+shear). Arterial shear rates are reported to range from ≈5 dyn/cm2 in descending aorta to ≈55 dyn/cm2 in arterioles (Resnick et al. 2003; Papaioannou and Stefanadis 2005). In our system, shear stress was estimated as: τmax = α √(p × η(2πf)3), where α is the orbital radius of rotation (2.5 cm), p is the medium density (1.01 g/mL), η is the viscosity of the medium (0.01 poise measured with a rheometer (model AR550; TA Instruments Inc., Newcastle, Delaware, USA)), and f is the frequency of rotation (rotations/s) (Bevan et al. 2011). Relative to results from control cells at rest, results from cells receiving shear stress consistently showed a robust upregulation of p-eNOS S1177 similar to results we have reported previously in cells exposed to flow (Ives et al. 2012) and vessels from mice after 60 min of treadmill running (Zhang et al. 2009).
Indices of autophagy
Microtubule-associated protein light chain (LC3) participates in the extension of the phagophore and is found both in the internal and external membranes of the autophagosome. Basal autophagic flux was assessed as increased levels of the lipidated or phosphatidylethanolamine conjugated form of LC3-I (i.e., LC3-II) vs. the non-lipidated LC3-I. Additionally, degradation of the autophagic cargo protein sequestosome (p62) was used to assess autophagy (Kabeya et al. 2004).
Indices of mitochondrial turnover
Mitochondrial turnover, i.e., mitophagy, was estimated by quantifying the degradation of mitochondrial matrix proteins [aconitase (m-aconitase) and heat shock protein 60 (HSP60)] and mitochondrial outer membrane proteins [translocase of outer mitochondrial membrane 20 homolog (Tom 20) and voltage-dependent anion channel (VDAC)] (Kubli and Gustafsson 2012) in cells and vessels.
Immunoblotting
Western blotting was used to assess (i) p-eNOS at S1177 normalized to total eNOS, (ii) LC3-II:LC3-I, and (iii) p62, HSP60, m-aconitase, Tom20, and VDAC normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using standard procedures. Antibodies for p-eNOS S1177, eNOS (1:500; Cell Signaling Technologies, Boston, Massachusetts, USA), LC3, HSP60, VDAC, m-aconitase, Tom20 (1:1000; Santa Cruz Biotechnology Inc., Dallas, Texas, USA), and p62 (1:1000; Abnova, Taipei, Taiwan) were obtained commercially. Densitometry of the immunoblot bands was quantified using Odyssey 3.1 (Li-cor, Lincoln, Nebraska, USA) (Symons et al. 2009; Zhang et al. 2009, 2010, 2012).
Small interference RNA (siRNA) mediated silencing
siRNA knockdown of the requisite autophagy pathway mediator, Atg3, was performed to impair autophagy in endothelial cells. Transfection of siRNA was performed with Lipofectamine 2000 (Invitrogen Life Technologies, Grand Island, New York, USA). The siRNA sequences are Atg3 siRNA: sense, 5′-CCCAGAAGAGUUUG UGGCAGCUGGA-3′; antisense, 5′-UCCAGCUGCCACAAACUCUUCUG GG-3′. The non-targeting siRNA (control-siRNA) is the property of Dharmacon (Pittsburgh, Pennsylvania, USA) (sense, 5′-UAAGGCUAU GAAGAGAUAC-3′; antisense, 5′-GUAUCUCUUCAUAGCCUUA-3′) (Zhang et al. 2012).
NO detection
DAF-FM diacetate (4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate; Life Technologies, Grand Island, N.Y.) was used to detect NO production. (Kojima et al. 1998) DAF-FM diacetate is non-fluorescent. When it crosses the plasma membrane, it interacts with NO, is oxidized to a triazole product, and becomes fluorescent. In parallel experiments, cells were treated with Hoechst nuclear stain (1:500 dilution; Molecular Probes, Eugene, Oregon, USA) to demonstrate that any treatment-induced alterations of fluorescence did not result from a heterogeneous number of cells. Then, the appropriate treatments cells were imaged using an Olympus IX-70 microscope with a charge-coupled device (CCD) camera. Ten fields × 10 cells per field were counted for each sample. An additional method of analysis was used for NO (i.e., fluorescence was detected using a fluorescence microplate reader). 500 μmol/L diethylammonium(Z)-1-(N,N-diethylamino) diazen-1-ium-1,2-diolate (DEA NONOate; a NO donor) served as a positive control. Cells incubated ± DEA NONOate ± 500 μmol/L NG monomethyl-L-arginine (L-NMMA; a NOS inhibitor) served as a specificity control. Unstained cells were included to provide a background fluorescence control (Zhang et al. 2012).
ROS detection
2′,7′-Dichlorofluorescein diacetate(DCFDA) (Sigma–Aldrich, St. Louis, Missouri, USA) is non-fluorescent. However, when the acetate group is cleaved by intracellular esterases, subsequent oxidation results in the formation of the highly fluorescent 2′,7′-dichlorofluorescein (DCF) which has an excitation and emission wavelength of 488 and 535 nm, respectively. Cells subjected to different treatments were prepared appropriately and read using a fluorescent microplate reader (Symons et al. 2009; Zhang et al. 2012).
Indices of inflammation
ELISA assays for the inflammatory markers MCP-1 (Kingfisher Biotech Inc., Saint Paul, Minnesota, USA) and IL-8 (Genorise, Berwyn, Pennsylvania, USA) were performed on cells treated ± shear ± Atg3 siRNA (Babu et al. 2012a, 2012b).
Indices of cell death
Percent cell death was estimated as [(no. of cells per field before treatment − no. of cells per field after treatment) divided by no. of cells per field before treatment] × 100 (Symons et al. 2009; Zhang et al. 2009, 2012).
Animals
Animal studies were performed in Association for Assessment and Accreditation of Laboratory Animal Care-accredited facilities using protocols approved by the Institutional Animal Care and Use Committee. Eight-week-old C57BL/6 male and female mice were used.
Food restriction
Mice were housed in new cages with fresh water (with or without chow) at 1800 h for 14, 24, or 40 h. At the appropriate time, mice were anesthetized, and the heart and blood vessels (i.e., entire aorta, iliac arteries) were obtained, snap frozen in Eppendorf tubes, and used later for immunoblotting (Symons et al. 2009; Zhang et al. 2009, 2012). To determine whether the process was reversible, chow was offered to a subset of mice for 1 h after 14 h fasting, and the heart and vessels were obtained as described earlier (Zhang et al. 2010).
Statistical analysis
Comparison of one endpoint among treatments was made using a one-way analysis of variance (ANOVA). If necessary, a Tukey post-hoc test was performed to identify the location of differences. An unpaired t test was used to compare one endpoint between 2 groups. Significance was accepted when p < 0.05. Data are the mean ± SE.
Results and discussion
Autophagy is induced in endothelial cells by nutrient depletion and this response is normalized by the restoration of nutrients
We determined the extent and time-course of autophagy induction in endothelial cells using a known stimulus (e.g., nutrient-depletion (ND); Zhang et al. 2010). Compared with cells incubated in nutrient-replete (NR) medium, the LC3-II:LC3-I ratio increased (p < 0.05) after 30 min of ND, the response was maximal at 2 h of ND, and the elevation in LC3-II:LC3-I ratio was maintained for up to 6 h (data not shown). Next, we assessed whether autophagy induction was a reversible process. Compared with cells incubated in NR medium, increases (p < 0.05) in LC3-II:LC3-I (Fig. 1a) and p62 degradation (Fig. 1b) were observed in cells after 2 h exposure to ND medium, and both responses were normalized when nutrients were restored (R) to the cellular medium. To address whether elevated LC3-II:LC3-I ratio observed in cells exposed to ND medium resulted from increased synthesis and (or) decreased degradation, LC3-II/autophagosome degradation was clamped using bafilomycin A1 (Baf A1). Baf A1 inhibits the vacuolar H+ ATPase (V-ATPase), prevents acidification of lysosomes, and inhibits LC3-II degradation. Comparing accumulation of LC3-II under Baf A1-treated and non-treated conditions provides an index of autophagic flux. Elevated (p < 0.05) LC3-II:LC3-I ratio in ND vs. NR cells was exaggerated (p < 0.05) in the presence of Baf A1, indicating that ND increases autophagic flux in endothelial cells (Fig. 1c).
Fig. 1.
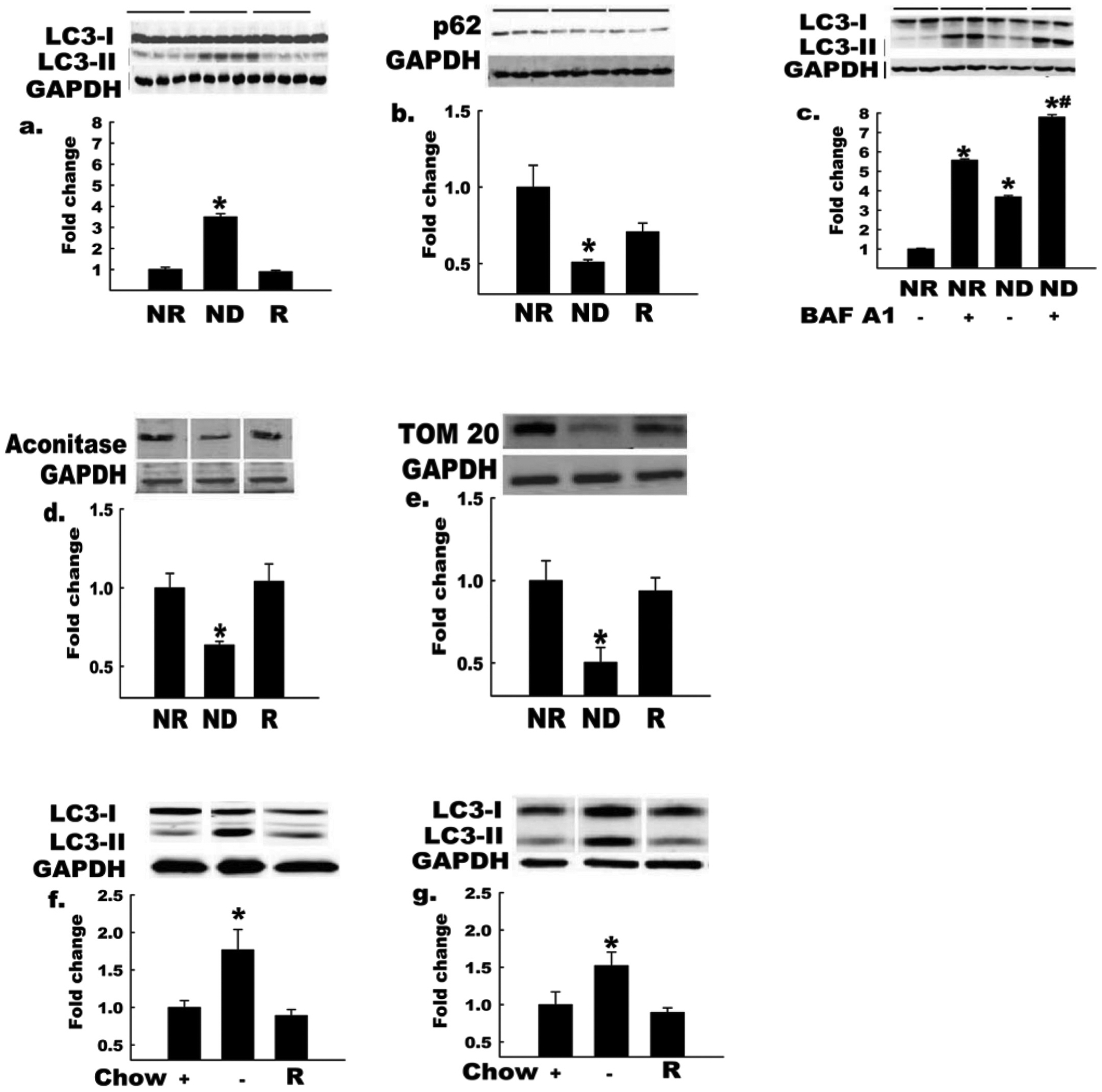
Autophagy is increased in endothelial cells and intact arteries by nutrient deprivation. Bovine aortic endothelial cells (BAECs) incubated in nutrient-deplete (ND) or nutrient-replete (NR) medium for 2 h. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. LC3-II:LC3-I ratio (a) increased and p62 protein level (b) decreased (*, p < 0.05) in BAECs incubated in ND vs. NR medium (n = 4). These responses were restored when nutrients were added back to ND medium (R, third histogram). Horizontal bars above the images indicate the respective groups. LC3-II:LC3-I ratio (c) showed a greater increase in BAECs incubated with ND medium in the presence of the lysosomal proton pump inhibitor BAF A1, owing to the expected effect of this treatment to block lysosomal degradation of LC3-II (n = 3 per condition; *, p < 0.05 for BAF A1 effect; #, p < 0.05 for −AA effect). Protein levels of the mitochondrial markers m-aconitase and TOM 20 (d, e) decreased in cells incubated in ND vs. NR medium (n = 4, p < 0.05), suggesting increased mitochondrial turnover; in both cases (d, e), responses were reversed when nutrients were restored to ND medium (R). LC3-II:LC3-I increased (*, p < 0.05) in hearts (f) and arteries (g) from in vivo fasted (14 h) vs. fed mice, and these responses were reversed by 1 h re-feeding (R, n = 3 per group). Histograms (below) represent the mean ± SE of densitometry (above). *, p < 0.05 for ND medium (d,e) or fasting (Chow −) (f, g) effect. For Figs. 1a–1e, each n refers to one 10 cm Petri dish. For Figs. 1f and 1g, each n refers to one mouse.
A major, but not exclusive, source of ROS under physiological situations is the electron transport chain where ≈1%–3% of the oxygen used by mitochondria is converted to ROS (Balaban et al. 2005). When the production of ROS is not balanced effectively by the cellular antioxidant environment, oxidative stress is said to occur. Fasting increases ROS in a PI3K-dependent manner, and antioxidant treatment negates fasting-induced autophagy (Scherz-Shouval et al. 2007). Autophagy activation might occur in the context of nutrient deprivation as a defensive response to remove ROS generating mitochondria and/or to recycle/degrade mitochondria that have been damaged by elevated oxidant stress (Kubli and Gustafsson 2012). Indeed, autophagy is the sole pathway for degradation of intact mitochondria, or mitophagy (Kubli and Gustafsson 2012). One hypothesis to explain how damaged mitochondria are identified for sequestration and removal via autophagy involves ubiquitination. The serine/threonine kinase PINK1 is normally imported into mitochondria, where it is cleaved, and degraded in the inner membrane space. However, this process is interrupted upon disruption of the mitochondrial membrane potential, which causes recruitment of PINK1 to the mitochondrial outer membrane, where it serves to recruit Parkin to mitochondria. Complexed to PINK1, Parkin ubiquitinates multiple mitochondrial outer membrane proteins (including Mfn1, Mfn2, VDAC, MARF, and Tom20) that can subsequently interact with adaptor proteins such as p62 and (or) neighbor of Breast Cancer 1 (NBR1) that simultaneously bind ubiquitin and LC3-II, targeting the ubiquitinated mitochondria to nascent autophagasome membranes (Geisler et al. 2010). We observed greater (p < 0.05) degradation of mitochondrial markers m-aconitase and Tom 20 in cells after 4 h of exposure to ND vs. NR medium, and both responses were reversed by restoring nutrients (R; Figs. 1d, 1e).
Autophagy is induced in blood vessels by food restriction and normalized by refeeding
Fasting for 24 h is a potent stimulus that activates autophagic flux in intact murine hearts (Zhang et al. 2010). To determine whether similar responses would be observed in blood vessels, mice were fasted for 14, 24, or 40 h starting at 1800 h. At the appropriate time, the heart (positive control) and arteries (entire aorta, both iliac arteries) were obtained, and the LC3-II:LC3-I ratio was assessed. Robust autophagy was observed in the heart at 14 h (Fig. 1f) and 24 h but not at 40 h. Vascular autophagy was maximal at 14 h (Fig. 1g). Consistent with observations from endothelial cells exposed to ND vs. NR medium, we found that fasting-induced (14 h) autophagy was normalized in heart (Fig. 1f) and vessels (Fig. 1g) by re-feeding (R) for 1 h.
Collectively, data from Fig. 1 indicate that vascular autophagy is upregulated in response to ND in vitro (cells) or fasting in vivo (blood vessels), and this response in cells is associated with evidence of mitochondrial degradation (i.e., mitophagy). Autophagy and mitochondrial degradation might represent 2 portions of a comprehensive response to cellular stress that are concerned, at least in part, with (i) clearing damaged and (or) dysfunctional organelles (e.g., mitochondria) that are a source of injurious levels of ROS and (or) (ii) degrading intracellular contents to provide substrates for energy production when nutrients are absent. We next sought to determine the extent of autophagy induction in endothelial cells exposed to shear stress.
Shear stress increases endothelial cell autophagy and NO production
Results from previous experiments indicate that endothelial cells exposed to shear stress demonstrate increased ROS generation (Chin et al. 2011), induction of anti-inflammatory and antioxidative genes (Zhang and Friedman 2013), and increased activation via S1177 phosphorylation of eNOS (Ives et al. 2012), all occurring in a coordinated manner that increases NO bioavailability. As a first step to addressing our hypothesis, it was necessary to characterize our cell culture model. Endothelial cells exposed to physiologically relevant shear forces (Resnick et al. 2003; Papaioannou and Stefanadis 2005) for 3 h exhibited increased (p < 0.05) autophagy, indicated by an increased LC3-II:LC3-I ratio (Fig. 2a), and evidence for mitochondrial degradation, indicated by decreased protein levels of the mitochondrial content markers m-aconitase (Fig. 2b) and Tom20 (Fig. 2c). As expected, shear stress induced ROS generation (Fig. 2d), phosphorylation of eNOS at S1177 (Fig. 2e), and increased levels of NO (Fig. 2f). Importantly, shear-induced increases in NO were prevented by the NOS inhibitor L-NMMA (Figs. 2f, 2g). These data are consistent with prior observations that mechanical stress can induce autophagy (King et al. 2011) and extend these observations to the important physiological context of endothelial shear stress, which appears capable of activating both autophagy and mitochondrial degradation.
Fig. 2.
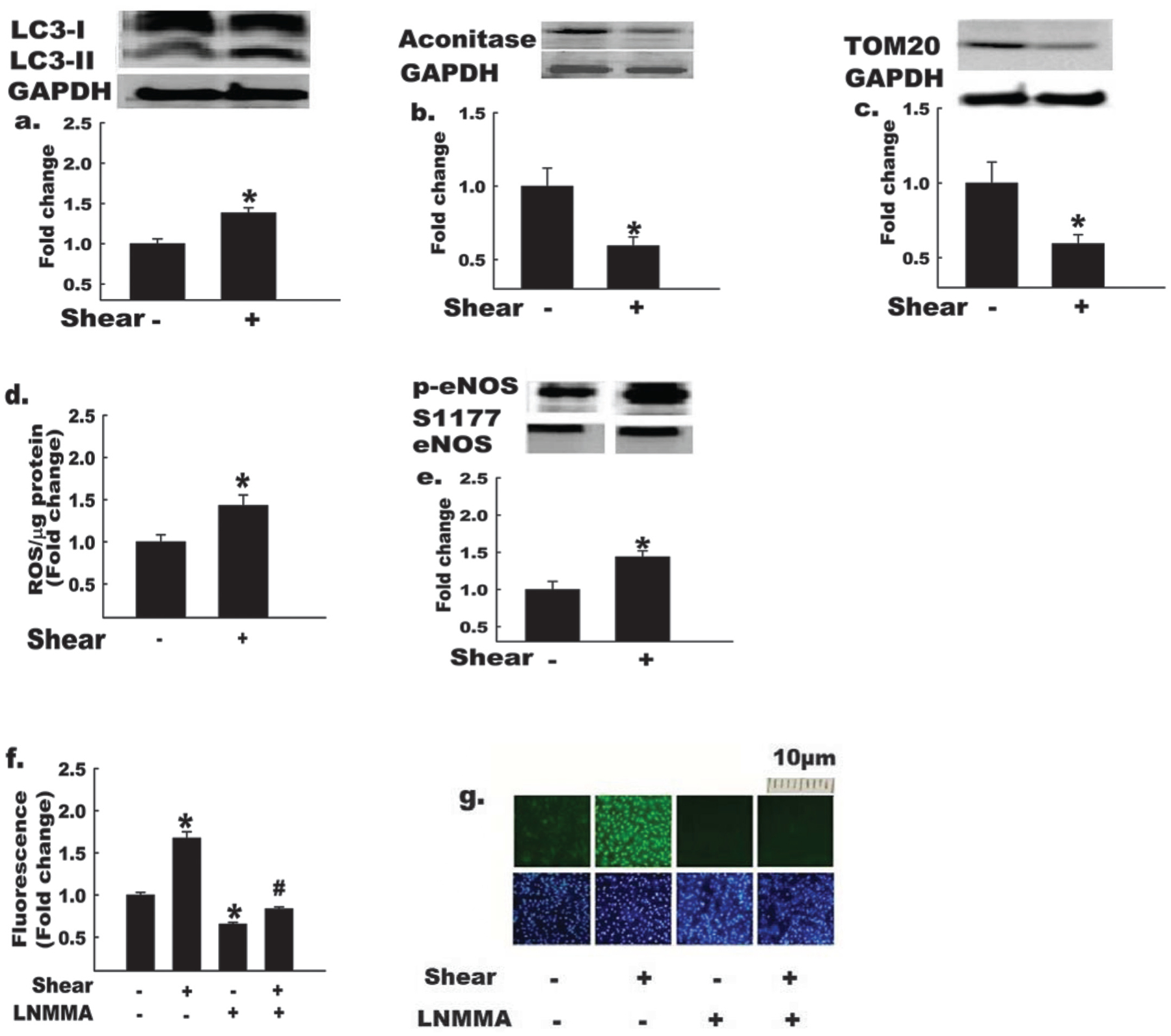
Autophagy and nitric oxide (NO) bioavailability are elevated in endothelial cells exposed to shear stress. Bovine aortic endothelial cells (BAECs) were exposed to shear stress (shear +) or static conditions (shear −) for 3 h, as indicated. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. Shear + BAECs show increased (*, p < 0.05) LC3-II:LC3-I ratio (a), and decreased protein levels of mitochondrial markers, m-aconitase (b), and TOM20 (c); in addition, shear + BAECs exhibit increased reactive oxygen species (ROS) production (d), p-eNOS S1177 : total eNOS (e), and NO production (mean data (panel f), representative images of DAF-FM fluorescence (top row, panel g)) in comparison with shear – BAECs (n = 4–6 per experiment). Treatment of BAECs with the NO synthase inhibitor, monomethyl-L-arginine (L-NMMA), reduced (p < 0.05) NO production ± shear stress (mean data (panel f), representative images of DAF-FM (panel g, top row) and DAPI nuclear fluorescence (panel g, bottom row)). The bottom row of panel g indicates that cell density was similar among treatments. For Figs. 2a, 2b, 2c, and 2e, the histograms (below) represent the mean ± SE of densitometry (above). *, p < 0.05 for shear effect; #, p < 0.05 for L-NMMA effect. For Figs. 2a, 2b, 2c, and 2e, each n refers to one 10 cm petri dish. For Figs. 2d, 2f, and 2g, each n refers to one well of a 6-well plate.
Among ROS species, H2O2 can disrupt the electron transfer process by modifying mitochondrial proteins to an extent that O2− generation is exaggerated. O2− can degrade NO and (or) combine with NO to form reactive nitrogen species such as peroxynitrite (ONOO−) that ultimately disrupt eNOS dimer formation, eNOS enzyme function, and NO bioavailability. Therefore, a potential function of the autophagy and mitochondrial degradation induced by shear stress is to enhance the ability of endothelial cells to clear misfolded proteins and (or) damaged organelles that result from and (or) further contribute to oxidative stress, maintaining the critical balance between NO synthesis and destruction during exposure to shear stress.
Shear-stress-induced NO production in endothelial cells is prevented when autophagy is disrupted
Data are emerging indicating that dysregulated autophagy contributes to and (or) precipitates disease progression in many different organs and cell types (Levine and Kroemer 2008). For example, endothelial cells obtained from aged humans and mice demonstrate impaired autophagy, exaggerated oxidant stress, and lower NO bioavailability vs. age-matched controls (Larocca et al. 2012). We have obtained preliminary data from homogenates of aorta and iliac arteries that are congruent with these observations. Specifically, aortic and (or) iliac homogenates from old (27-month) mice show decreased LC3-II:LC3-I, greater levels of p62 autophagic cargo protein, and reduced p-eNOS S1177 (all p < 0.05, data not shown), in comparison with arteries from young (6-months) mice (n = 4 per group). When aging-induced reductions in endothelial cell autophagy were reversed pharmacologically (Larocca et al. 2012), oxidative stress was attenuated and NO bioavailability was restored. This study also showed that impairment of autophagy was sufficient to reduce NO production in vitro; specifically, O2− was greater and NO bioavailability was lower after knockdown of Atg12 protein expression in human umbilical vein endothelial cells (HUVECs) vs. control cells under static conditions. We explored whether autophagy induction in endothelial cells is important during dynamic conditions to maintain NO bioavailability. To do so, we silenced (>85% knockdown; Fig. 3a) the expression of Atg3, a requisite autophagy protein that mediates lipidation of LC3-I to form LC3-II, which is required for autophagasome formation. Next, cells treated with scrambled, nonspecific siRNA (i.e., −Atg3 siRNA) or Atg3-specific siRNA (i.e., +Atg3 siRNA) were exposed to static or dynamic conditions. Control cells responded as expected; i.e., shear stress caused increased autophagy (Fig. 3b), ROS production (Fig. 3c), p-eNOS S1177 (Fig. 3d), and NO production (Figs. 3e, 3f) (all p < 0.05 vs. static condition). In contrast, cells with impaired autophagy (+Atg3 siRNA) exhibited a profound impairment of p-eNOS S1177 and were incapable of increasing NO in response to shear stress. Moreover, ROS accumulation was exaggerated (Fig. 3c; p < 0.05) and inflammatory cytokine production, e.g., MCP-1 and IL-8 (Figs. 3g, 3h), was unmasked (p < 0.05) in response to shear stress. These findings reveal that autophagy not only plays a critical role in maintaining NO bioavailability, but may also be a key regulator of oxidant–antioxidant balance and inflammatory–anti-inflammatory balance that ultimately regulate endothelial cell responses to shear stress.
Fig. 3.
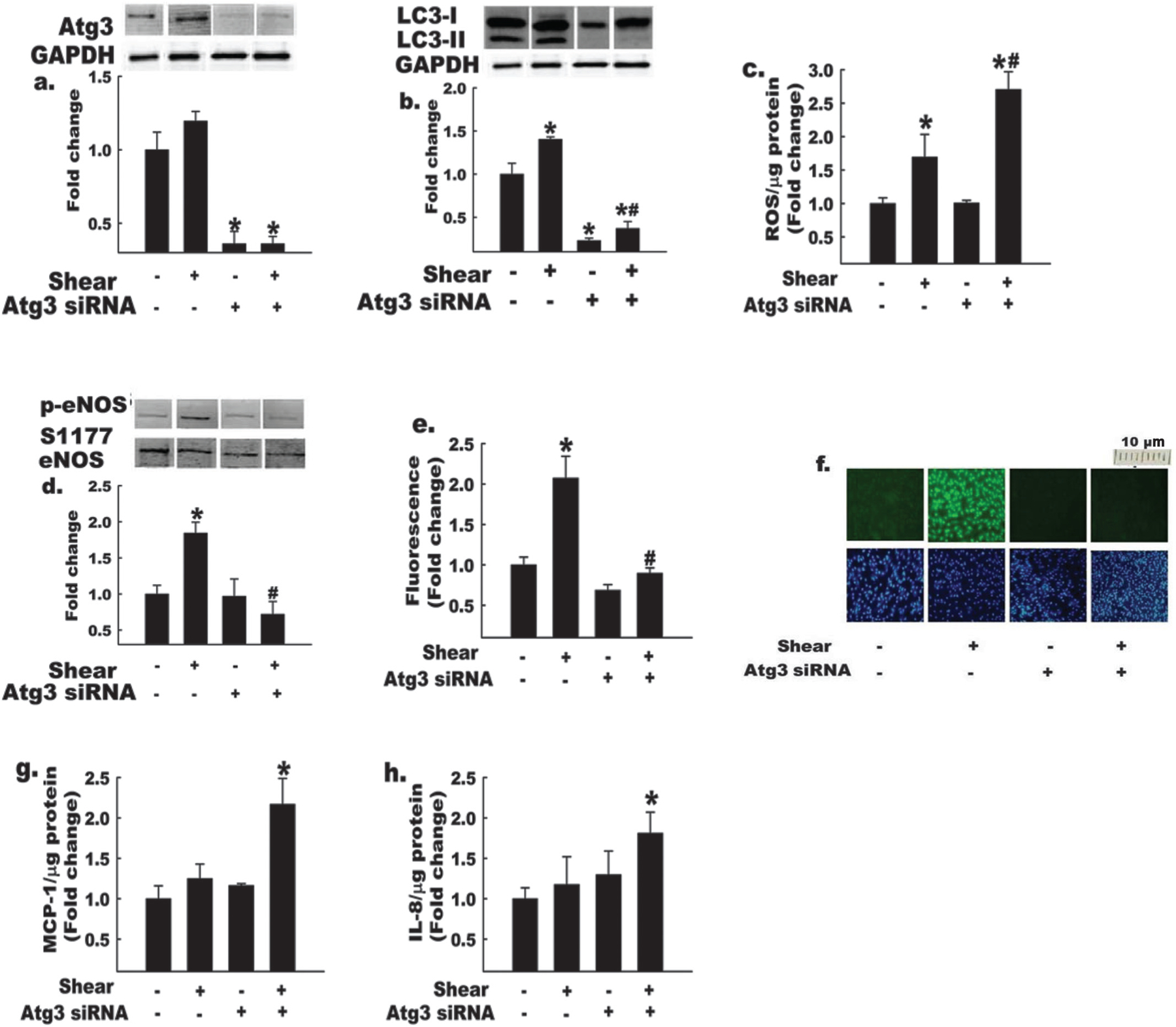
Impairment of autophagy impairs shear-stress-induced increases in eNOS S1177 phosphorylation and nitric oxide (NO) bioavailability. Bovine aortic endothelial cells (BAECs) treated with Atg3 siRNA (+) or control scrambled siRNA (−) were exposed to shear stress (shear +) or static conditions (shear −), as indicated. Atg3 protein knockdown efficiency (a) was identical for each condition (*, p < 0.05 for Atg3 siRNA effect; p = NS for static vs. shear-stress comparisons). Basal and shear-stress-induced autophagy (assessed by LC3-II : LC3-I ratio) were suppressed + Atg3 siRNA (b) under both static and shear-stress conditions. In addition, Atg3 siRNA-treated BAECs exhibited increased (c) reactive oxygen species (ROS) production and decreased (d) p-eNOS S1177 : total eNOS in response to shear stress. NO levels (e) were decreased in Atg3 siRNA-treated BAECs ± shear stress. Mean data (panel e) and representative images of DAF-FM (panel f, top row) and DAPI nuclear fluorescence (panel f, bottom row) are shown. The bottom row of panel f indicates that cell density was similar among treatments. Atg3 siRNA-treated BAECs exhibit increased expression of proinflammatory cytokines (g) MCP-1 and (h) IL8, after exposure to shear stress. For Figs. 3a, 3b, and 3d, histograms (below) represent the mean ± SE of densitometry of 4–6 experiments (above). *, p < 0.05 for shear effect; #, p < 0.05 for Atg3 effect. For Figs. 3a, 3b, and 3d, each n refers to one 10 cm petri dish. For Figs. 3c, 3e, 3f, 3g, and 3h, each n refers to one well of a 6-well plate.
Significance
Our data, in conjunction with evidence that decreased endothelial autophagy is a feature of aging vasculature (Larocca et al. 2012), support the possibility that autophagy may play a critical role in maintaining normal endothelial responses to shear stress during aging. Moreover, because autophagy is a fundamental cellular function influenced by nutrient availability, lipotoxicity, and insulin, our findings raise the intriguing possibility that endothelial dysfunction associated with obesity, insulin resistance, and (or) aging, could relate to altered regulation of endothelial cell autophagy. Very recent data provide important proof of concept for this possibility. Shafique et al. generated a conditional transgenic mouse that induces endothelial cell specific over-expression of Nox2/gp91 when tetracyclin is withdrawn (Tet-OFF mice) (Shafique et al. 2013). Compared with littermate controls (i.e., Tet-ON mice) endothelial ROS production is ≈2-fold greater in Tet-OFF animals. Using endothelial cells isolated from hearts of these mice (MHECs), the authors demonstrated that signaling to eNOS via 5′-adenosine monophosphate-activated protein kinase (AMPK) increased, mechanistic target of rapamycin (mTOR) signaling decreased, and autophagic flux was induced in MHECs isolated from Tet-OFF vs. Tet-ON mice (Shafique et al. 2013). The ability of ROS to induce autophagy in MHECs from Tet-OFF animals was confirmed using molecular (i.e., Nox2 siRNA) and pharmacological (i.e., N-acetylcysteine) approaches. Of clinical importance, when autophagosome–lysosome fusion was prevented in MHECs of Tet-OFF mice (i.e., those with elevated ROS), apoptotic cell death was greater (p < 0.05) than observed in Tet-OFF mice with intact autophagy. Collectively, autophagy appears to play a critical role in maintaining cellular viability in response to endogenous ROS generation by endothelial cells in response to shear stress (present study), aging (Larocca et al. 2012), and manipulation of NADPH oxidase expression (Shafique et al. 2013). Future investigation is warranted to determine the precise intravascular signaling mechanism(s) involved in these processes, and whether dysregulated vascular autophagy might contribute to arterial dysfunction that is observed in the context of diet-induced obesity, insulin resistance, and type 2 diabetes — conditions associated with elevated ROS (Symons et al. 2009; Zhang et al. 2012; Symons and Abel 2013).
Acknowledgements
L.P.B. was invited to submit this manuscript as the Grant Pierce Biomedical Award winner at the Cardiovascular Forum for Promoting Centers of Excellence and Young Investigators Conference in Louisville, KY (August 2013). J.D.S. is supported by the American Diabetes Association (ADA: 1-12-BS-208, ADA: 7-08-RA-164), the University of Utah College of Health and School of Medicine, and the National Institutes of Health (NIH: 2R15HL091493). T.E.G. is supported by American Diabetes Association (ADA: 7-13-BS-05), National Institutes of Health (NIDDK R01DK100826); in addition, this material is the result of work supported by the Veterans Health Administration Office of Research and Development via BLR&D Merit Review Award No. 5I01BX000937. T.R., D.K., R.G., and L.D. are, or were, supported by the University of Utah Undergraduate Research Opportunities Program. T.R. was supported by an American Heart Association, Western States Affiliate, Undergraduate Summer Research Fellowship (2011, 2012, 2013) and by an American Physiological Society, Undergraduate Research Excellence Fellowship (2014).
Footnotes
This article is one of a selection of papers from research presented at “The Cardiovascular Forum for Promoting Centers of Excellence and Young Investigators” held in Louisville, Kentucky, USA, on 15–17 August 2013.
Contributor Information
Leena P. Bharath, College of Health, University of Utah, Salt Lake City, UT 84112, USA; Division of Endocrinology, Metabolism, and Diabetes, University of Utah School of Medicine, Salt Lake City, UT 84112-9003, USA.
Ashot Sargsyan, Division of Endocrinology, Metabolism, and Diabetes, University of Utah School of Medicine, Salt Lake City, UT 84112-9003, USA; Program in Molecular Medicine, University of Utah, Salt Lake City, UT 84112, USA..
Pon Velayutham Anandh Babu, College of Health, University of Utah, Salt Lake City, UT 84112, USA..
Timothy E. Graham, Division of Endocrinology, Metabolism, and Diabetes, University of Utah School of Medicine, Salt Lake City, UT 84112-9003, USA; Program in Molecular Medicine, University of Utah, Salt Lake City, UT 84112, USA.
J. David Symons, College of Health, University of Utah, Salt Lake City, UT 84112, USA; Division of Endocrinology, Metabolism, and Diabetes, University of Utah School of Medicine, Salt Lake City, UT 84112-9003, USA..
References
- Babu PV, Si H, Fu Z, Zhen W, and Liu D 2012a. Genistein prevents hyperglycemia-induced monocyte adhesion to human aortic endothelial cells through preservation of the cAMP signaling pathway and ameliorates vascular inflammation in obese diabetic mice. J. Nutr 142(4): 724–730. doi: 10.3945/jn.111.152322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu PV, Si H, and Liu D 2012b. Epigallocatechin gallate reduces vascular inflammation in db/db mice possibly through an NF-kappaB-mediated mechanism. Mol. Nutr. Food Res 56(9): 1424–1432. doi: 10.1002/mnfr.201200040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, and Finkel T 2005. Mitochondria, oxidants, and aging. Cell, 120(4): 483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Bensaad K, Cheung EC, and Vousden KH 2009. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 28(19): 3015–3026. doi: 10.1038/emboj.2009.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan HS, Slater SC, Clarke H, Cahill PA, Mathieson PW, Welsh GI, et al. 2011. Acute laminar shear stress reversibly increases human glomerular endothelial cell permeability via activation of endothelial nitric oxide synthase. Am. J. Physiol. Renal Physiol 301(4): F733–F742. doi: 10.1152/ajprenal.00458.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boo YC, and Jo H 2003. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am. J. Physiol 285: C499–C508. doi: 10.1152/ajpcell.00122.2003. [DOI] [PubMed] [Google Scholar]
- Chin LK, Yu JQ, Fu Y, Yu T, Liu AQ, and Luo KQ 2011. Production of reactive oxygen species in endothelial cells under different pulsatile shear stresses and glucose concentrations 1. Lab Chip, 11(11): 1856–1863. doi: 10.1039/c0lc00651c. [DOI] [PubMed] [Google Scholar]
- Davies PF 1995. Flow-mediated endothelial mechanotransduction. Physiol. Rev 75: 519–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. 2010. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol 12(2): 119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Ives SJ, Andtbacka RH, Kwon SH, Shiu YT, Ruan T, Noyes RD, et al. 2012. Heat and alpha1-adrenergic responsiveness in human skeletal muscle feed arteries: the role of nitric oxide. J. Appl. Physiol 113(11): 1690–1698. doi: 10.1152/japplphysiol.00955.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, and Yoshimori T 2004. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci 117(13): 2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- King JS, Veltman DM, and Insall RH 2011. The induction of autophagy by mechanical stress. Autophagy, 7(12): 1490–1499. doi: 10.4161/auto.7.12.17924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Elazar Z, Seglen PO, and Rubinsztein DC 2008. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy, 4(7): 849–850. [DOI] [PubMed] [Google Scholar]
- Kojima H, Nakatsubo N, Kikuchi K, Kawahara S, Kirino Y, Nagoshi H, et al. 1998. Detection and imaging of nitric oxide with novel fluorescent indicators: diaminofluoresceins. Anal. Chem 70(13): 2446–2453. doi: 10.1021/ac9801723. [DOI] [PubMed] [Google Scholar]
- Kubli DA, and Gustafsson AB 2012. Mitochondria and mitophagy: the yin and yang of cell death control. Circ. Res 111(9): 1208–1221. doi: 10.1161/CIRCRESAHA.112.265819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larocca TJ, Henson GD, Thorburn A, Sindler AL, Pierce GL, and Seals DR 2012. Translational evidence that impaired autophagy contributes to arterial ageing. J. Physiol 590(14): 3305–3316. doi: 10.1113/jphysiol.2012.229690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, and Kroemer G 2008. Autophagy in the pathogenesis of disease. Cell, 132(1): 27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, and Klionsky DJ 2008. Autophagy fights disease through cellular self-digestion. Nature, 451(7182): 1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallauf K, and Rimbach G 2013. Autophagy, polyphenols and healthy ageing. Ageing Res. Rev 12(1): 237–252. doi: 10.1016/j.arr.2012.03.008. [DOI] [PubMed] [Google Scholar]
- Papaioannou TG, and Stefanadis C 2005. Vascular wall shear stress: basic principles and methods. Hellenic J. Cardiol 46(1): 9–15. [PubMed] [Google Scholar]
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. 2010. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev 90(4): 1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- Resnick N, Yahav H, Shay-Salit A, Shushy M, Schubert S, Zilberman LC, et al. 2003. Fluid shear stress and the vascular endothelium: for better and for worse. Prog. Biophys. Mol. Biol 81(3): 177–199. doi: 10.1016/S0079-6107(02)00052-4. [DOI] [PubMed] [Google Scholar]
- Russo SB, Baicu CF, Van LA, Geng T, Kasiganesan H, Zile MR, et al. 2012. Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J. Clin. Invest 122(11): 3919–3930. doi: 10.1172/JCI63888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, and Elazar Z 2007. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 26(7): 1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa WC 2004. eNOS at a glance. J. Cell Sci 117: 2427–2429. doi: 10.1242/jcs.01165. [DOI] [PubMed] [Google Scholar]
- Shafique E, Choy WC, Liu Y, Feng J, Cordeiro B, Lyra A, et al. 2013. Oxidative stress improves coronary endothelial function through activation of the pro-survival kinase AMPK. Aging (Albany NY), 5(7): 515–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons JD, and Abel ED 2013. Lipotoxicity contributes to endothelial dysfunction: A focus on the contribution from ceramide. Rev. Endocr. Metab. Disord doi: 10.1007/s11154-012-9235-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons JD, McMillin SL, Riehle C, Tanner J, Palionyte M, Hillas E, et al. 2009. Contribution of insulin and Akt1 signaling to endothelial nitric oxide synthase in the regulation of endothelial function and blood pressure. Circ. Res 104(9): 1085–1094. doi: 10.1161/CIRCRESAHA.108.189316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzel JJ, Chatzizisis YS, Gijsen FJ, Giannoglou GD, Feldman CL, and Stone PH 2012. Endothelial shear stress in the evolution of coronary atherosclerotic plaque and vascular remodelling: current understanding and remaining questions. Cardiovasc. Res 96(2): 234–243. doi: 10.1093/cvr/cvs217. [DOI] [PubMed] [Google Scholar]
- Zhang J, and Friedman MH 2013. Adaptive response of vascular endothelial cells to an acute increase in shear stress frequency. Am. J. Physiol. Heart Circ. Physiol 305(6): H894–H902. doi: 10.1152/ajpheart.00174.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QJ, McMillin SL, Tanner JM, Palionyte M, Abel ED, and Symons JD 2009. Endothelial nitric oxide synthase phosphorylation in treadmill-running mice: role of vascular signalling kinases. J. Physiol 587(15): 3911–3920. doi: 10.1113/jphysiol.2009.172916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QJ, Wende AR, Guo A, Sena S, Symons JD, and Abel ED 2010. Inhibition of starvation-induced autophagy in cardiomyocytes by insulin signaling is independent of nutrient availability. Diabetes, 59: 43–OR.19846801 [Google Scholar]
- Zhang QJ, Holland W, Wilson L, Tanner J, Kearns D, Cahoon JM, et al. 2012. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes, 61: 1848–1859. doi: 10.2337/db11-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]