

Abstract
In cancer, linking epigenetic alterations to drivers of transformation has been difficult, in part because DNA-methylation analyses must capture epigenetic variability, which is central to tumour heterogeneity and tumour plasticity. Here, by conducting a comprehensive analysis, based on information theory, of differences in methylation stochasticity in samples from patients with paediatric acute lymphoblastic leukaemia (ALL), we show that ALL epigenomes are stochastic, and marked by increased methylation entropy at specific regulatory regions and genes. By integrating data methylation and single-cell gene-expression data, we arrived at a relationship between methylation entropy and gene-expression variability, and found that epigenetic changes in ALL converge on a shared set of genes that overlap with genetic drivers involved in chromosomal translocations across the disease spectrum. Our findings suggest that an epigenetically driven gene- regulation network, with UHRF1 (ubiquitin-like with PHD and RING finger domains 1) as a central node, links genetic drivers and epigenetic mediators in ALL.
Pre-B cell acute lymphoblastic leukemia (ALL) is the most common form of cancer in children1,2. Epigenetic alterations are especially relevant to ALL, as well as to pediatric tumors in general, which are often characterized by a low mutational burden3,4. Genome-wide methylation profiling at various stages of normal B lymphocyte maturation suggests that DNA methylation changes in B-cell neoplasms occur in regions undergoing dynamic methylation adjustments during normal differentiation5. Moreover, array-based methylation analysis techniques have revealed that alterations in DNA methylation discriminate between cytogenetic subtypes of pediatric pre-B ALL and may hold prognostic value6,7. Mutational analyses of paired diagnostic and relapsed pre-B ALL samples have also identified significant enrichment of mutations in epigenetic regulators, such as SETD2, CREBBP, and KDM6A, at relapse8,9. These results suggest a critical role for epigenetic dysregulation in both the etiology and progression of pediatric pre-B ALL that requires thorough investigation.
Previous studies aiming to define epigenetic drivers of pediatric ALL have been limited by an emphasis on array-based techniques assessing comparatively few target regions6,7,10–12, the paucity of available whole-genome bisulfite sequencing (WGBS) data12,13, as well as by statistical limitations of methylation inferences based on marginal or empirical approaches, which are not capable of validly summarizing stochastic cell-to-cell variation in DNA methylation14,15. Only one study has considered methylation stochasticity in ALL13, which was focused on CpG islands and a method of analysis based on empirically computing a marginal measure of methylation variance. However, we have previously shown that marginal or empirical methods for the analysis of methylation stochasticity can produce unreliable statistical evidence, especially under routine sequencing coverage conditions or within genomic regions that exhibit correlated methylation, since these approaches can accrue large statistical uncertainty and exhibit low sensitivity (true positive rate) and specificity (true negative rate)14,15.
Here we introduce a novel information-theoretic approach for integrating methylation and gene expression data in pediatric ALL which comprehensively maps and localizes differences in methylation stochasticity (which we simply refer to as ‘methylation discordance’) at high resolution throughout the entire genome. By employing recent developments14–16, as well as new methods introduced in this paper, we performed a whole-genome analysis of methylation stochasticity with a systematic application to four cytogenetically-defined and clinically relevant subsets of ALL that included 10 primary diagnostic ETV6-RUNX1 in-frame fusion patient samples, 11 hyperdiploid samples, 6 TCF3-PBX1 in-frame fusion samples, and 4 dicentric chromosome (9;20) samples involving the PAX5 gene (Supplementary Table 1). Our analysis integrated potential energy landscapes, defined through the Ising model of statistical physics and computed genome-wide from WGBS samples at high resolution covering the majority of CpG sites in the human genome (Supplementary Table 1, Supplementary Fig. 1), with bulk and single-cell RNA sequencing data. This is in contrasts to previous studies which performed methylation analysis on a small fraction of CpG sites (less than 2%) using methylation arrays7,12 or a small number (four or fewer) of WGBS samples12,13.
Results
Potential energy landscapes explain DNA methylation stochasticity in ALL.
Characterizing DNA methylation stochasticity requires assessment of the probability that a specific methylation pattern is observed within a genomic region of interest14,17,18. In our previous work14, we computed the probability of the stochastic methylation pattern within a genomic region that contains CpG sites to take value by
Where equals 1 or 0 depending on whether the n-th CpG site is methylated or unmethylated, respectively, is the potential energy of the methylation pattern , and
is a normalizing constant known as the partition function. By imposing consistency with methylation means and nearest-neighbor correlations, and by taking into account non-cooperative and cooperative factors in methylation, we set14
Where , , and are parameters characteristic to the genomic region, computed from WGBS data within non-overlapping estimation regions of the genome via maximum-likelihood, is the CpG density, given by , and is the distance between CpG sites and , defined by . The resulting probability distribution is associated with the one-dimensional Ising model of statistical physics that can effectively capture methylation stochasticity present in WGBS data and leads to informME, a powerful approach for genome-wide modeling and analysis of WGBS data which outperforms pre-existing methods of methylation analysis by producing reliable statistical evidence with high sensitivity and specificity14,15.
Coordinated patterning information present in WGBS data within a genomic region is used by informME to compute a potential energy landscape for DNA methylation, given by , where is the most probable methylation pattern. In this landscape, each methylation pattern is assigned a potential value , with smaller values indicating that the methylation pattern can be observed with higher probability. Notably, the presence of a deep and narrow ‘potential well’ in the energy landscape located at implies low methylation stochasticity, since only a small number of methylation patterns, which must necessarily exhibit low variation from due to the well’s narrowness, will be associated with low potential values and therefore will be observed with high probability (Fig. 1a, left). On the other hand, a shallow and wide ‘potential well’ points to high methylation stochasticity, since a large number of methylation patterns will now be associated with low potential values, implying that any of these patterns will be observed with almost equal probability (Fig. 1b, right).
Fig. 1 |. Potential energy landscapes explain DNA methylation stochasticity in normal and cancer cells.
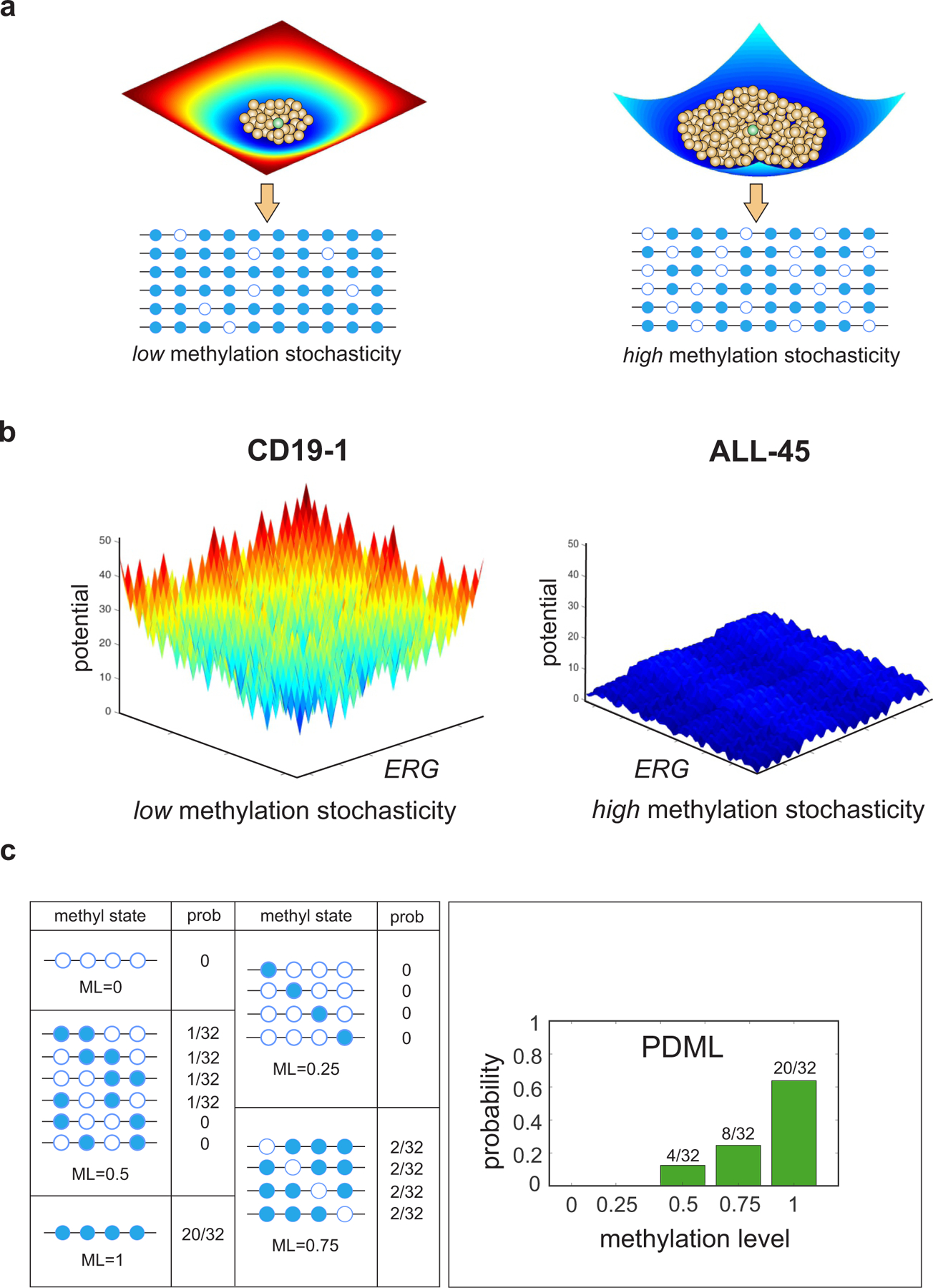
a, Illustration of potential energy landscapes. Each methylation pattern (brown balls) is assigned a potential value that provides a measure of its improbability to be observed relative to the most probable pattern (green ball), which is assigned zero potential. A deep and narrow ‘potential well’ indicates low methylation stochasticity in a cell population (left), whereas a shallow and wide ‘potential well’ points to high methylation stochasticity (right). b, Potential energy landscapes associated with twelve contiguous CpG sites within ERG [chr21: 39,830,065 – 39,830,570] demonstrate increased methylation stochasticity for ERG in ALL, in agreement with observed WGBS data (Supplementary Fig. 2). Here the methylation patterns are assigned to points in a two-dimensional state-space using Gray’s code (Methods). c, Within an analysis region, methylation patterns are grouped in terms of their methylation level (ML). The probability distribution of the methylation level (PDML) is then evaluated by summing the probabilities associated with the methylation patterns within each group. The depicted probability distribution points to an analysis region that is most likely methylated.
We initially applied informME on ten WGBS primary diagnostic patient samples from pre-B cell ALL that carry the most common chromosome translocation seen in this disease, t(12;21)(p13;q22), resulting in ETV6-RUNX1 in-frame fusion (Supplementary Table 1). We included two cell types as controls: (1) normal B cell precursors (two replicates of normal flow sorted pre-B2 cells from fetal bone marrow, defined as CD34-, CD19+, sIgM-)5, and (2) three replicates of immunomagnetically separated umbilical cord blood (UCB) CD19+ B cells (Supplementary Table 1). We used pre-B2 cells as controls since they are a close corresponding normal cell type to pre-B ALL5,12. We also chose UCB CD19+ cells as additional controls since they are readily available and include a range of CD19+ cells that are appropriate normal controls for pre-B ALL, although more mature than pre-B2 cells.
We performed methylation analysis genome-wide and show in Fig. 1b an example of computed potential energy landscapes at a locus inside ERG, a target of genetic alterations in ALL19,20 that encodes an ETS-family transcription factor with critical roles in hematopoiesis and leukemogenesis21, which was identified by our analysis to exhibit significant and consistent methylation discordance in the data (see discussion later in this paper). In normal CD19+ cells, only a small number of methylation patterns are associated with low potential values and these patterns vary only moderately from the most probable pattern of zero potential (which is fully methylated in this case) resulting in low methylation stochasticity (Fig. 1b, left and Supplementary Fig. 2). By contrast, the potential energy landscape flattens in ALL with the most probable pattern becoming fully unmethylated while most other methylation patterns exhibit low potential values, resulting in high methylation stochasticity (Fig. 1b, right and Supplementary Fig. 2).
To facilitate genome-wide analysis, given the combinatorial nature of the space of methylation patterns ( CpG sites are associated with distinct patterns), informME partitions the genome into small (150 bp) non-overlapping analysis regions and performs methylation analysis by quantifying methylation within each analysis region that contains CpG sites using the methylation level (average methylation) . Notably, the probability distribution , , of the methylation level can be evaluated by grouping the methylation patterns within the analysis region using their methylation level and by summing the probabilities associated with the methylation patterns within each group (Fig. 1c)14,15.
By employing this approach, we characterized methylation stochasticity within each analysis region using the probability distribution of the methylation level and employed the mean methylation level to measure average methylation, the normalized methylation entropy of the methylation level to quantify the amount of methylation stochasticity, and the methylation sensitivity index to assess the robustness of the probability distribution of the methylation level to changes in the values of parameters , and of the potential energy landscape, with higher values indicating less robust (more sensitive) behavior pointing to a more ‘responsive’ analysis region in which small changes in parameter values can produce larger changes in methylation stochasticity (Methods). Such changes could be the result of local modifications in the biochemical environment provided by the methylation machinery that can alter the methylation landscape. Using methylation levels instead of methylation patterns is a form of ‘coarse graining’ resulting in computational advantages that allow informME to be applied genome wide. However, both approaches lead to a similar behavior producing identical values for the mean methylation level and comparable values for the normalized methylation entropy (Supplementary Fig. 3).
Genome-wide distributions of mean methylation levels and normalized methylation entropies for the ETV6-RUNX1 ALL and normal control samples showed hypomethylation in ALL and a global gain in entropy (Supplementary Figs. 4a and 5a,b), whereas genome-wide distributions of the values of the methylation sensitivity index globally demonstrated no considerable changes in sensitivity (Supplementary Fig. 4a). By also examining distributions over selected genomic features (Methods), we observed hypermethylation over GpG islands (CGIs) in the ETV6-RUNX1 samples versus the normal controls but hypomethylation over CGI shores, shelves, open sea, gene bodies, exons, introns, and intergenic regions (Supplementary Figs. 4b and 5a,b), in agreement with previous observations7. Notably, the normalized methylation entropy was increased over these features in ALL, while the methylation sensitivity index was higher over CGIs than over other genomic features in both the ETV6-RUNX1 and the control samples (Supplementary Figs. 4b and 5a,b). Interestingly, among classes of enhancers that showed hypomethylation in ALL, Transcription 5’ Enhancers (TxEnh5’) demonstrated a marked gain in normalized methylation entropy (Supplementary Figs. 4b and 5b). Moreover, bivalent promoters (PromBiv) defined by a co-enrichment of the repressive H3K27me3 and activating H3K4me3 histone tail modifications, showed considerable hypomethylation and gain in methylation sensitivity in the control samples, when compared to other genomic features (Supplementary Figs. 4b and 5a,b), consistent with previous observations that bivalent domains in normal cells typically have low levels of DNA methylation22,23 and may confer responsiveness to certain environmental changes24. However, these regions exhibited substantial hypermethylation in the ETV6-RUNX1 samples, as well as increased entropy and loss in sensitivity (Supplementary Figs. 4b and 5a,b).
Comparative analysis identifies methylation discordance in ALL localizing to distinct features and regions of the genome informative of the phenotype.
We next investigated the relationship between the methylation level and the phenotype when comparing ETV6-RUNX1 ALL to CD19/pre-B2 samples. By using the estimated probability distributions of the methylation levels within analysis regions, we first computed their Jensen-Shannon distance between ALL and normal control samples, an information-theoretic measure of dissimilarity between two probability distributions (Methods) that captures methylation discordance due to differences in mean methylation level, normalized methylation entropy, or other statistical factors (Fig. 2a). We then evaluated the degree of mutual dependence between the mean methylation level and the phenotype within genomic regions using the average mutual information between the methylation level and the phenotype, computed as the square of the Jensen-Shannon distance magnitude inside these regions (Methods). We also examined the detailed structure of methylation discordance by computing differences in mean methylation level, normalized methylation entropy, and methylation sensitivity. In addition, we extended our local analysis to larger scales by detecting long contiguous discordantly methylated regions (DMRs) between ALL and normal control samples using the Jensen-Shannon distance (Methods), an approach that was previously shown to outperform pre-existing DMR finders in terms of statistical uncertainty, sensitivity, and specificity15. Due to the previously mentioned relationship between the average mutual information and the Jensen-Shannon distance, DMRs identified by our method point to ‘informative’ regions of the genome in ALL, characterized by statistically significant values of the average mutual information between the methylation level and the phenotype, in which there is a significant degree of mutual dependence between the methylation level and the phenotype.
Fig. 2 |. Differential analysis localizes methylation discordance in ALL.
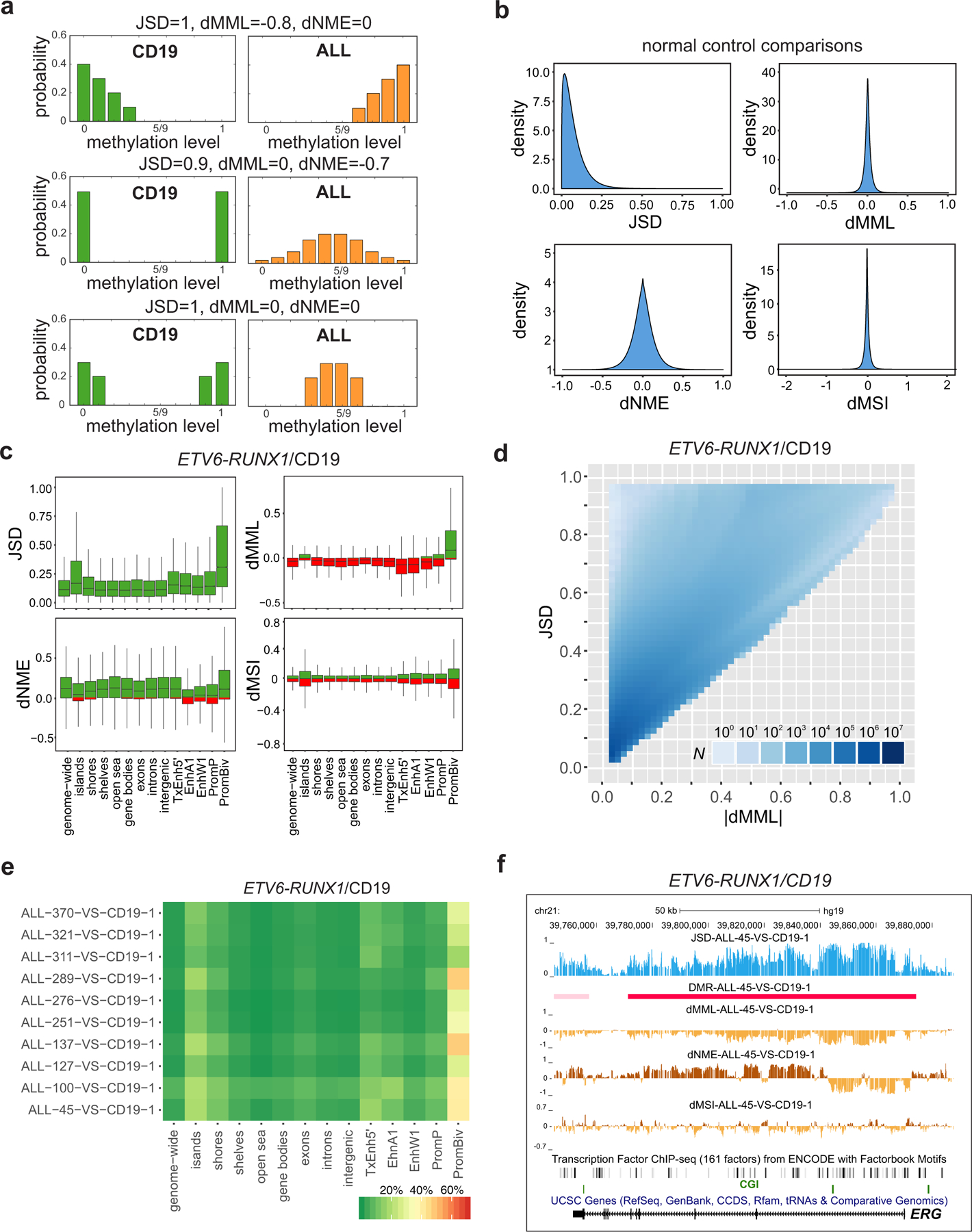
a, The Jensen-Shannon distance (JSD) captures methylation discordance within an analysis region by evaluating differences in the location and shape of the probability distributions of the methylation levels associated with a test (ALL) and a reference (CD19/pre-B2) sample. Methylation discordance can be due to a difference in mean methylation level (dMML – top row), in normalized methylation entropy (dNME – middle row), or due to other statistical factors (bottom row). b, Densities of JSD, dMML, and dNME, as well as of differences in the values of the methylation sensitivity index (dMSI), when comparing all normal CD19 and pre-B2 samples, show relatively small discordances associated with biological, statistical, and technical variability in these samples. c, Distributions of JSD, dMML, dNME, and dMSI values genome-wide and within selected genomic features in an ETV6-RUNX1/CD19 comparison (ALL-45 vs. CD19–1). Green, positive values; red, negative values; center lines, median; boxes, interquartile range (IQR); whiskers, 1.5 × IRQ. d, Distribution of the number of analysis regions with respect to their JSD and absolute dMML values, computed from all ETV6-RUNX1/CD19–1 comparisons genome-wide. Many analysis regions that exhibit similar absolute differences in mean methylation level are associated with a wide range of Jensen-Shannon distance values demonstrating that the mean methylation level is not the only statistical factor influencing methylation discordance in ALL. e, Percentage of analysis regions with significant methylation discordance within selected genomic features when comparing ETV6-RUNX1 ALL with CD19. f, UCSC genome browser images of a chromosomal region associated with ERG exhibiting significant Jensen-Shannon distance values in an ETV6-RUNX1/CD19 comparison (ALL-45 vs. CD19–1), and thus being significantly informative of the phenotype. This region exhibits consistent reduction in mean methylation level and loss in methylation sensitivity, but localized gain or loss in normalized methylation entropy.
When comparing the normal CD19/pre-B2 samples, computed distributions of our differential statistics showed relatively small methylation discordances (Fig. 2b), confirming the appropriateness of these samples as normal controls and providing a quantitative assessment of normal biological, statistical, and technical variability in our analyses. Notably, the normal controls used in this study included highly pure pre-B2 cell populations and umbilical cord blood CD19+ samples that included mature B cells as well as progenitors. This allowed us to assess the suitability of these cell types as normal controls due to differences in purity. As shown in Fig. 2b and Supplementary Fig. 4, distributions of methylation statistics computed from the control samples were in close agreement to each other. Moreover, the control samples exhibited much lower normalized methylation entropies than those associated with the highly pure ALL blasts, providing no evidence of significant influence of purity differences in the control samples on methylation analysis.
Distributions of differential methylation statistics within selected genomic features confirmed gains in Jensen-Shannon distance values, especially within bivalent promoters and CGIs as compared to other features (Fig. 2c), showing that these features exhibit statistically significant methylation discordance. For most genomic features, these gains were consistently associated with hypomethylation and increased normalized methylation entropy in ALL although gains in Jensen-Shannon distance values for most bivalent promoters were associated with hypermethylation and increased normalized methylation entropy in ALL (Fig. 2c). Some CGIs, weak enhancers, and poised promoters exhibited hypermethylation in ALL, whereas some CGIs, shores, gene bodies, exons, introns, enhancers, and poised promoters exhibited reduction in normalized methylation entropy (Fig. 2c). Moreover, distributions of computed values of the methylation sensitivity index demonstrated both losses and gains in methylation sensitivity over all genomic features (Fig. 2c), in agreement with our genome-wide observations (Supplementary Fig. 4).
Focusing on the relationship between the average mutual information and the Jensen-Shannon distance (Methods), we sought to identify informative genes in ALL (i.e., genes exhibiting a significant degree of mutual dependence between the methylation level and the phenotype). We therefore computed average mutual information values within gene promoters from a single ETV6-RUNX1/CD19 comparison, evaluated their statistical significance while controlling for the false-discovery rate (FDR), and ranked genes using the computed Q-values (Supplementary Table 2a). We found a set of 1960 genes to be significantly informative of the phenotype (Q-value ≤ 0.05) with important developmental genes, such as EBF2, HOXD1, MAFB, OTX2, SALL1, and ZIC1, as well as members of the PAX family of transcription factors, being at the top of the list. By computing overlaps with the ‘GO gene sets’ (C5) in the Molecular Signatures Database (MSigDB) using gene set enrichment analysis (GSEA), we found enrichments in genes related to phenotypic determination and regulation, such as gene expression, development, differentiation, morphogenesis, and cell fate (Supplementary Table 2b), thus further confirming the informative nature of the previous set of genes. When we computed overlaps with the ‘curated gene sets’ (C2) in MSigDB, we also found striking enrichments of genes possessing the chromatin repressive mark H3K27me3 and genes possessing the bivalent marks H3K4me3 and H3K27me3 in their promoters (Supplementary Table 2c), as well as a striking enrichment in PRC2 (EED, SUZ12) targets, in agreement with previous results7,12.
We also examined Jensen-Shannon distances and differential mean methylation levels from all ETV6-RUNX1/CD19–1 comparisons genome-wide and within selected genomic features and found that Jensen-Shannon distances associated with absolute differential mean methylation level values vary widely, especially at low such values (Fig. 2d and Supplementary Fig. 6). To investigate the importance of this finding, we performed an ETV6-RUNX1/CD19 comparison and identified a set of 411 genes exhibiting significant magnitudes in Jensen-Shannon distance (Q-values ≤ 0.05) but no significant differences in mean methylation level within their promoters (Supplementary Table 3a). Interestingly, by computing overlaps with the ‘GO gene sets’ (C5) in MSigDB using GSEA, we found enrichment in genes related to development, differentiation, morphogenesis and signaling, such as HOXA cluster and WNT/FZD pathway genes (Supplementary Table 3b), whereas computed overlaps with the ‘curated gene set’ (C2) in MSigDB revealed again striking enrichments of genes possessing the chromatin repressive mark H3K27me3 and genes possessing the bivalent marks H3K4me3 and H3K27me3 in their promoters (Supplementary Table 2c), as well as enrichment in PRC2 (EED, SUZ12) targets and WNT pathway genes (Supplementary Table 3c), confirming the importance of genes that exhibit significant methylation discordance within their promoters for factors other than differences in mean methylation, whose detection is expected to be missed by a mean-based hypothesis testing approach.
Notably, our ETV6-RUNX1 samples consistently exhibited strong methylation discordance within specific genomic regions, as illustrated by the large values of the computed Jensen-Shannon distances and the significant DMRs observed over ERG across samples (Supplementary Fig. 7). Due to the relationship between the average mutual information and Jensen-Shannon distances (Methods), these genomic regions are informative, exhibiting a significant degree of mutual dependence between their methylation level and the phenotype. Large scale analysis of the ETV6-RUNX1 ALL data produced a higher percentage of DMRs overlapping bivalent promoters and CGIs, and to a lesser extent CGI shores, TxEnh5’ enhancers, and EnhA1 enhancers (Fig. 2e). We also assessed methylation discordance within regulatory elements by evaluating significant DMRs at the scale of genes. A notable example of a statistically significant DMR is the one localized at ERG (Fig. 2f), which exhibited consistent hypomethylation. A DMR region upstream the 5’ end of this gene was associated with a consistent loss in normalized methylation entropy in ALL, indicating decreased methylation stochasticity, whereas a DMR region downstream the 3’ end was associated with a consistent gain in entropy, implying increased methylation stochasticity.
Other examples of informative regions of the genome identified by significant DMRs localizing at finer genomic features include those mapping to promoters, such as over the promoter of EBF2 (Early B-cell Factor 2), which exhibited consistent hypermethylation, increased normalized methylation entropy, and loss in methylation sensitivity in ALL (Supplementary Fig. 8a), as well as those that overlap known regulatory elements at the EBF2 and NR2F2 (nuclear receptor subfamily 2 group F member 2) genes, such as binding sites for EZH2 and SUZ12, two transcription factors that are functional enzymatic components of the polycomb repressive complex 2 (PRC2) regulating heterochromatin formation (Supplementary Figs. 8a,b).
To further investigate regulatory influences of regions of significant methylation discordance between ETV6-RUNX1 ALL and normal CD19+ cells, we carried out odds ratio enrichment analysis among detected DMRs (Supplementary Table 4a) and found a consistently significant enrichment of SUZ12 targets (16.10 ≤ OR ≤ 91.78, hypergeometric P-value ), in agreement with previous observations12, and EZH2 targets (9.71 ≤ OR ≤ 64.38, hypergeometric P-value ), as well as H3K4me3 (1.70 ≤ OR ≤ 6.29, hypergeometric P-value ) and H3K27me3 domains (1.85 ≤ OR ≤ 12.95, hypergeometric P-value ), in agreement with previous results7. Finally, by analyzing DMRs in ETV6-RUNX1 ALL using the genomic regions enrichment of annotations tool (GREAT)25, we showed enrichments for lymphoblastic leukemia disease ontology genes, for known B lymphocyte progenitor genes, for genes annotated to be related to B cell/hematopoietic development phenotypes in mouse knockout studies, and for MSigDB perturbation gene sets related to PRC2/H3K27me3 and B cell progenitor phenotypes (Supplementary Table 4b).
Taken together, the previous results show that significant methylation discordance in ETV6-RUNX1 ALL localizes to informative features and regions of the genome that exhibit a significant degree of dependence between the methylation level and the phenotype, raising the possibility that their epigenetic control plays an important role in ALL. In addition, our results show that performing methylation analysis using only mean methylation levels may not be sufficient in comparative studies, since methylation discordance may be influenced by other statistical factors, whereas identification of informative regions of the genome is not possible by a mean-based approach.
DNA methylation stochasticity in ALL relates to gene expression.
We also assessed the influence of DNA methylation stochasticity on gene expression in ETV6-RUNX1 ALL by performing bulk and single-cell RNA sequencing and its associated statistical analysis (Methods, Supplementary Tables 5a,b and 6a–n). Notably, the four samples used for single-cell analysis (Supplementary Table 6a) were found to be relatively homogenous in terms of gene expression (Supplementary Fig. 9), thus confirming that cell purity does not affect our results.
Globally, we observed an increase in gene expression mean and a larger increase in expression variance in ETV6-RUNX1 ALL cells when comparing to normal CD19+ cells (Supplementary Fig. 10). By dividing genes into quartiles of gene expression mean and variance and by using two ETV6-RUNX1 samples, we demonstrated a relationship between DNA methylation stochasticity and stochastic gene expression in ETV6-RUNX1 ALL (Fig. 3). To further investigate this relationship, we identified differentially methylated genes (DMGs), where a DMG is defined here as a gene whose promoter or body overlaps a region of significant methylation discordance as determined by our DMR analysis using the Jensen-Shannon distance. We found significant enrichment of DMGs in differentially expressed genes (OR 1.84, one-sided Fisher’s exact test P-value ) when comparing ALL-45 to CD19–1, and similarly when comparing ALL-289 to CD19–1 (Supplementary Tables 7a,b). By computing overlaps with the ‘GO gene sets’ (C5) in MSigDB using GSEA, we found that differentially expressed DMGs in ALL exhibited enrichments in genes related to phenotypic determination and regulation, such as development, differentiation, morphogenesis, and signaling (Supplementary Tables 7c,d). When computing overlaps with the ‘curated gene sets’ (C2) in MSigDB, we found relationships to stem cell signatures, including hematopoietic and leukemia stem cells, in genes related to B lymphocyte progenitors, in genes normally downregulated in response to glucocorticoids in ALL, in genes associated with AML, and in genes associated with cell cycle control (Supplementary Tables 7e,f).
Fig. 3 |. DNA methylation stochasticity relates to gene expression in ETV6-RUNX1 ALL.
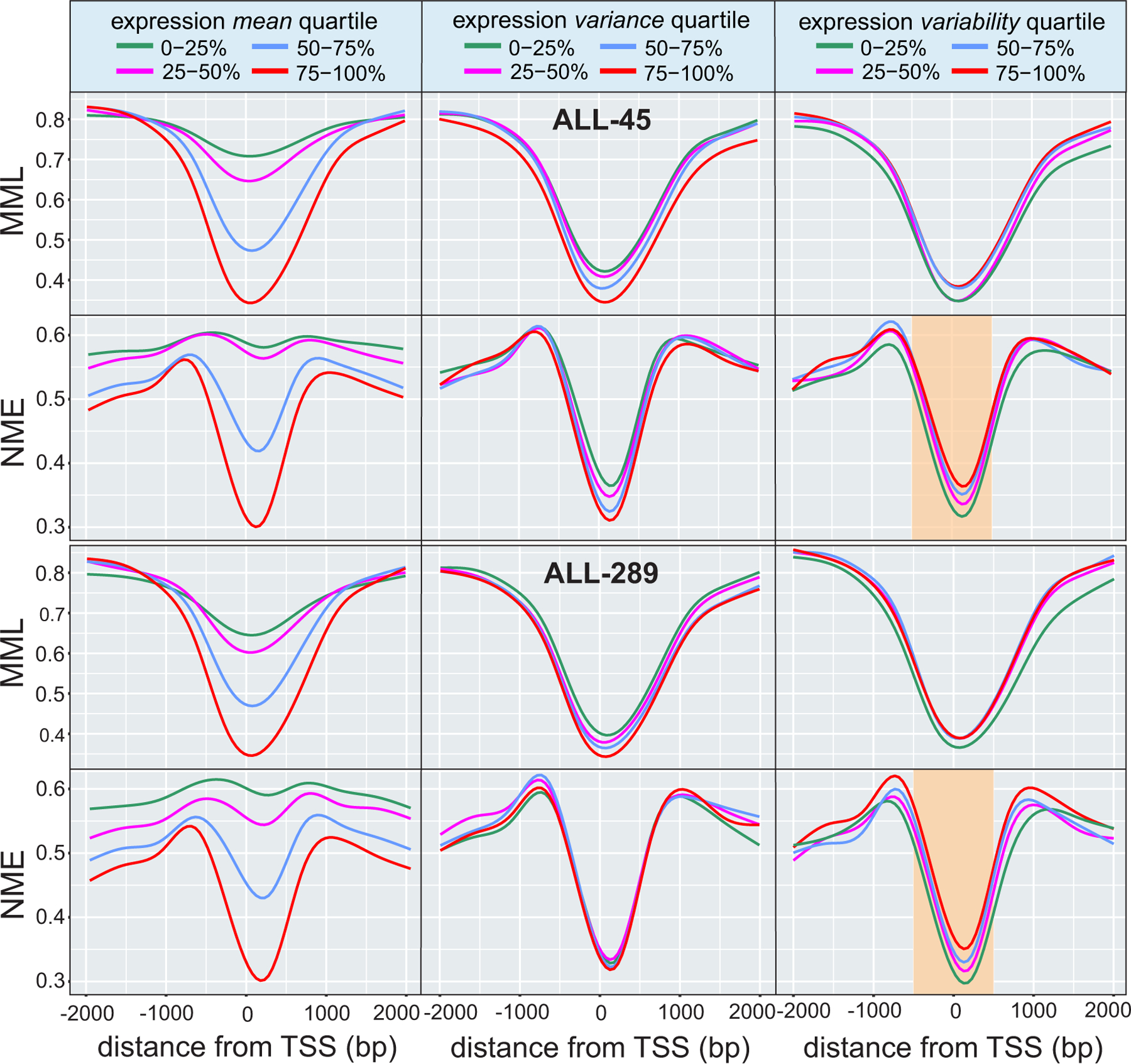
Average relationships in ETV6-RUNX1 ALL between mean methylation level (MML) and normalized methylation entropy (NME) within gene promoters as a function of distance from the transcription start site (TSS). The results correspond to quartiles of gene expression mean (left column), variance (middle column), and variability level (right column). Lower mean expression associates with higher levels of mean methylation (left column, first and third rows), confirming a known relationship between promoter methylation and gene expression. However, lower levels of mean expression are associated with higher levels of normalized methylation entropy (left column, second and fourth rows) implying that promoters of genes with lower gene expression are associated with higher levels of methylation stochasticity. Higher expression variance also relates to reduced levels of mean methylation level and normalized methylation entropy, but these associations can be weak (center column). Although a measure of expression variability (Methods) does not clearly associate with mean methylation level (right column, first and third rows), it relates to normalized methylation entropy, with higher entropy near the TSS being identified with statistically significant gains in expression variability (right column, second and fourth rows - highlighted: two-sided Wilcoxon rank sum test on medians within from the TSS, P-values for second vs. first quartile, third vs. second quartile, and fourth vs. third quartile). This implies that promoter regions of genes with higher expression variability exhibit higher levels of methylation stochasticity near the TSS.
Motivated by the observed association between bivalent promoters and genomic regions exhibiting methylation discordance, we also sought to investigate the relationship between bivalency, DNA methylation stochasticity, and gene expression. We identified bivalent genes in CD19 as those genes whose bodies or promoters overlapped with bivalent domains and found that there is no significant enrichment of bivalent genes in differentially expressed genes (Supplementary Table 8a) when comparing ETV6-RUNX1 ALL to CD19 (OR 1.05, one-sided Fisher’s exact test P-value ). From the 287 bivalent genes in CD19 identified as being overexpressed in ALL, we found 155 (54%) genes exhibiting significant methylation discordance in ALL, whereas the remaining 132 (46%) genes showed no difference in methylation stochasticity. Moreover, none of the 292 bivalent genes in CD19 that were underexpressed in ALL were differentially methylated. GSEA analysis of the 155 overexpressed and differentially methylated bivalent genes using the ‘GO gene sets’ (C5) and the ‘curated gene sets’ (C2) in MSigDB showed overlaps with gene sets associated with development, morphogenesis, cell signaling, differentiation, motility, growth, and apoptosis (Supplementary Table 8b), as well as with gene sets associated with stemness, B lymphocyte progenitors, responses to glucocorticoids in ALL, and AML (Supplementary Table 8c). Moreover, and in addition to these results, the 132 overexpressed but non-differentially methylated bivalent genes showed overlaps with gene sets associated with cell proliferation but not cell growth and apoptosis when using the ‘GO gene sets’ (Supplementary Table 8d), and did not overlap with the gene set associated with responses to glucocorticoids in ALL when using the ‘curated gene sets’ (Supplementary Table 8e). Importantly, by identifying genes exhibiting differential gene expression variability in ALL (Methods and Supplementary Tables 6k–n, 8a), we found a significant enrichment of bivalent genes in CD19 (OR 1.74, one-sided Fisher’s exact test P-value ), even though bivalent genes in CD19 were not enriched for significant differences in mean expression (Supplementary Table 8a).
The previous results show that DNA methylation stochasticity influences gene expression in ALL demonstrating that, on the average, lower mean expression relates to higher mean methylation level and higher normalized methylation entropy, whereas gene expression variability relates only weakly to mean methylation level but associates more tightly to normalized methylation entropy. Importantly, our data indicate that critical regulators of the leukemic phenotype identify with informative genes, thus providing evidence that genes exhibiting statistically significant values in mutual information between the methylation level and the phenotype within their promoters may play a key role in leukemogenesis.
Methylation discordance associates with bona fide driver genes of ALL across cytogenetic subtypes.
Pediatric B-cell precursor ALL includes multiple subtypes characterized by distinct somatic structural chromosomal alterations, including chromosome translocations resulting in in-frame gene fusions, such as ETV6-RUNX1 and TCF3-PBX1, dicentric chromosomes, or chromosome number alterations, such as hyperdiploidy19,26–30. While these are known to serve as initiating lesions, further cooperating alterations are essential for leukemogenesis31.
To determine whether methylation discordance is an important epigenetic driver across distinct molecular subtypes of ALL, we performed methylation analysis on 21 additional primary diagnostic patient samples of pre-B cell ALL (Supplementary Table 1) with TCF3-PBX1 in-frame fusion (6 samples), hyperdiploidy (11 samples), and dicentric chromosome (9;20) involving the PAX5 gene (4 samples). Notably, the DNA methylation landscape of these additional subtypes of ALL behaved similarly to that of ETV6-RUNX1, with hypomethylation and increased normalized methylation entropy relative to control samples, as well as gains in normalized methylation entropy and losses in methylation sensitivity over Transcription 5’ Enhancers and bivalent promoters (Supplementary Figs. 2, 11–13). Odds ratio enrichment analysis among detected DMRs consistently revealed significant enrichments of SUZ12 and EZH2 targets in all leukemic subtypes and samples (Supplementary Table 9), although some subtype-specific differences were also observed, as for example enrichments of H3K4me3 domains among DMRs in ETV6-RUNX1 ALL.
To investigate co-occurrence of methylation discordance across distinct cytogenetic subtypes of pre-B ALL, we identified common DMGs using four representative samples from each subtype group (Fig. 4a and Supplementary Table 10a). Interestingly, we found a set of 1290 DMGs exhibiting statistically significant methylation discordance in all four samples (Methods, Monte Carlo P-value ), pointing to a possibly common epigenetic basis for dysregulation of gene expression across cytogenetic subtypes of ALL. Gene set enrichment analysis (GSEA) of this set of genes using the ‘GO gene sets’ (C5) in MSigDB revealed enrichments in genes related to development, differentiation, morphogenesis and gene expression (Supplementary Table 10b). Moreover, GSEA using the ‘curated gene sets’ (C2) in MSigDB showed a striking enrichment of PRC2 targets (Supplementary Table 10c), reinforcing similar results obtained by our previous odds ratio analyses using all samples (Supplementary Table 9) and by the GREAT enrichment analysis using an ETV6-RUNX1 sample (Supplementary Table 4b), raising the possibility that alterations in methylation stochasticity over targets of PRC2 is common across the four cytogenetic subtypes of ALL. However, it was surprising to discover that the set of 1290 common DMGs included 16 genes (Supplementary Table 10a) that have been previously reported in in-frame chromosomal translocations across the disease spectrum of ALL19.
Fig. 4 |. Methylation discordance and four cytogenetic subtypes of ALL.
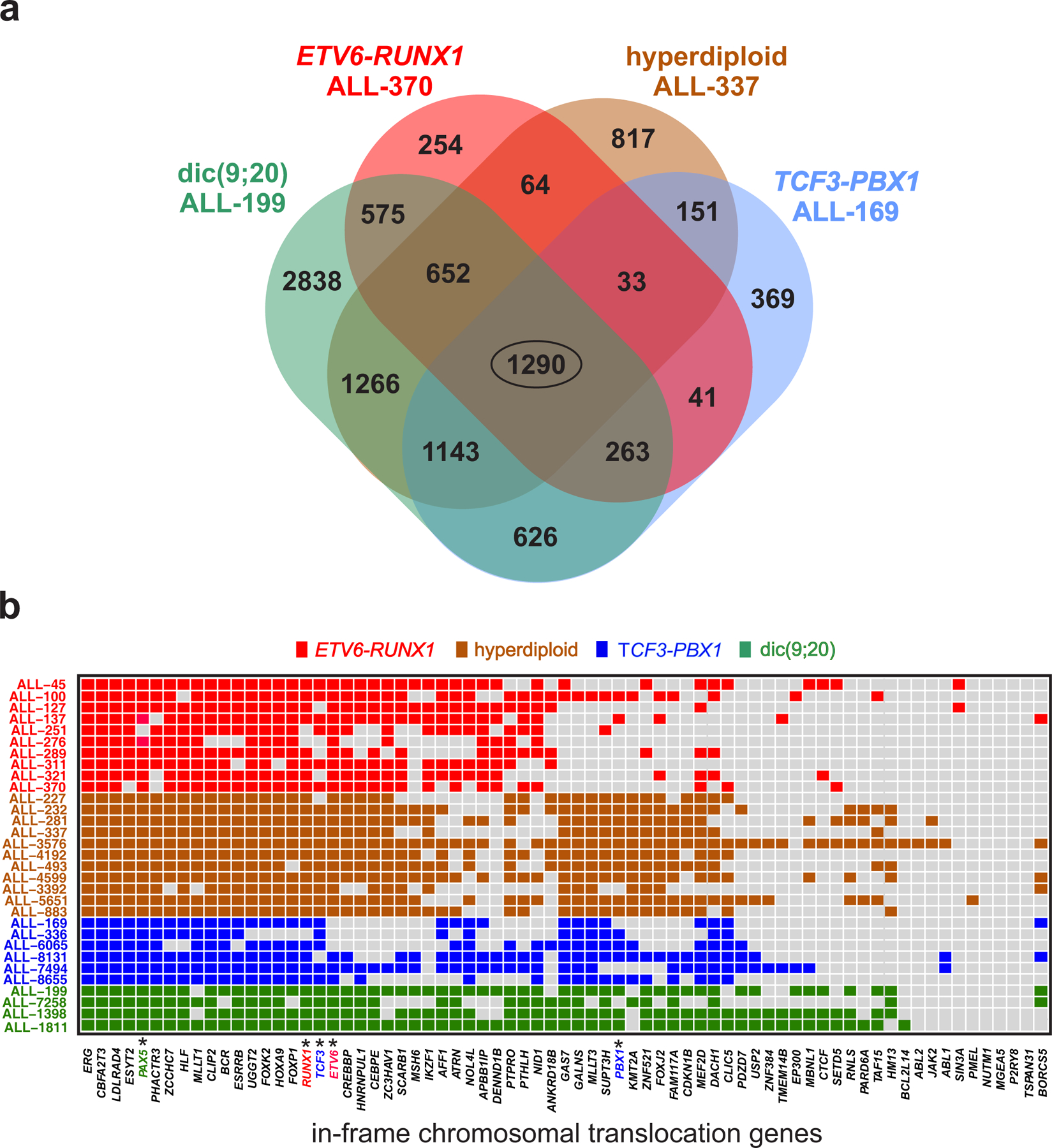
a, Numbers of differentially methylated genes (DMGs) common to four groups of cytogenetic subtypes of ALL as indicated. The corresponding genes are listed in Supplementary Table 10a. b, Differentially methylated regions detected in individual samples (rows) identify in-frame chromosomal translocation genes in ALL exhibiting significant methylation discordance (columns). Colors indicate the underlying cytogenetic abnormality for each sample as well as the genes involved in a sample-specific translocation. Marked columns (*) pinpoint to genes undergoing chromosomal translocation in a specific cytogenetic abnormality.
Motivated by this observation, we sought to examine the possibility that there is a greater than expected chance for translocation genes to exhibit significant methylation discordance (i.e., to be DMGs) in pre-B ALL. Towards this goal, we used previously identified in-frame fusion genes, known to be potent driver lesions in cancer, and constructed a target list of 71 known in-frame chromosomal translocation genes (henceforth referred to as translocation genes) across the disease spectrum of ALL (Fig. 4b and Methods). We then established bona fide drivers of ALL by identifying translocation genes that were classified as DMGs by our DMR analysis. Surprisingly, we found that most translocation genes (41/71) in the ETV6-RUNX1 ALL-45 sample were DMGs, which – except for ETV6 and RUNX1 – were not subject to chromosomal rearrangement in that patient, and obtained similar results for the remaining ALL samples (Fig. 4b). To evaluate the statistical significance of this outcome, we tested for each ALL sample against the null hypothesis that associations between translocation genes and DMGs were generated by chance and found a significant enrichment of DMGs in the list of translocation genes (Supplementary Table 10d; hypergeometric tests combined with Fisher’s meta-analysis; P-values ), including those driven by ETV6-RUNX1 and TCF3-PBX1 rearrangements, dicentric chromosome (9;20) involving the PAX5 gene, and cases driven by changes in chromosome number characterized by hyperdiploidy.
The previous analysis shows that, more often by chance, cytogenetic alterations in ALL concur with significant methylation discordance over a set of genes known to be in-frame bona fide genetic drivers of ALL, suggesting that changes in DNA methylation stochasticity is an important epigenetic driver across different cytogenetic subtypes of ALL. It also raises the possibility that these genes are co-regulated by an underlying gene regulatory network that exhibits significant dysregulation in gene expression due to a common epigenetic disruption in ALL.
Methylomic landscapes identify UHRF1 as a potential driver of ETV6-RUNX1 ALL.
Considering the enrichment of bona fide drivers of ALL observed in our common list of DMGs, we were curious to identify other genes in this list known to be involved in the pathogenesis of cancer that could be candidate drivers of ALL. By using the Jensen-Shannon distance, we computed the average mutual information within promoters and gene bodies and ranked genes in terms of the statistical significance of the degree of mutual dependence between the mean methylation level and the phenotype within these genomic features (Methods and Supplementary Tables 11a,b). We found UHRF1 (ubiquitin-like containing PHD and RING finger domains 1), one of the DMGs in our common list (Supplementary Table 10a), to be the most informative gene (associated with the smallest Q-value), within its gene body in 9/10 ETV6-RUNX1/CD19–1 comparisons (Supplementary Table 11a, Q-values ) but not within its promoter (Supplementary Table 11b). We also obtained similar results when comparing other cytogenetic subtypes to CD19–1 (Supplementary Tables 11c–h), implying that the methylation level within UHRF1’s body is highly informative of the phenotype in most of our samples. We also identified additional informative genes located near the top of the ‘body’ ranked lists known to be involved in ALL chromosome translocations, including CBFA2T3, ERG, LDLRAD4, BCR, and RCSD119,32, as well as other genes associated with leukemia, such as UBASH3B33, MLXIP34, and MME (an important cell surface marker of ALL).
UHRF1 is an important regulator of the epigenome, which maintains DNA methylation and histone modifications in cells by encoding for a multi-domain E3 ubiquitin ligase that coordinates the recognition of repressive histone H3K9 modifications and recruitment of the DNA methyltransferase DNMT1 to regulate chromatin structure and gene expression. Although dysregulation of UHRF1 expression has been implicated in some solid tumors35–38, it has not been sufficiently investigated in ALL. Motivated by the role of UHRF1 in DNA methylation maintenance and chromatin regulation, the consistently exceptional informational content within its body across our pre-B samples, and the fact that UHRF1 consistently exhibited significant upregulation of expression mean and variance in our bulk and single-cell sequencing data (Supplementary Tables 5b, 6b–e), we sought to evaluate the impact of UHRF1 loss on DNA methylation stochasticity.
To this end, we employed CRISPR/Cas9 mediated genome editing to silence UHRF1 in the Reh cell line, a pediatric ALL cell line carrying the ETV6-RUNX1 translocation. Western blot experiments confirmed the targeted loss of UHRF1 protein in a clonal Reh UHRF1 knockout (Reh-UHRF1-KO) cell line (Supplementary Fig. 14a), which exhibited a complete abrogation of clonogenicity (a fundamental property of cancer) as compared to non-targeted (Reh-NT) controls (Supplementary Figs. 14b,c), thus confirming the importance of UHRF1 expression for maintaining the self-renewal capacity of leukemia cells.
By carrying out WGBS of the Reh-NT and Reh-UHRF1-KO cells and by performing analysis of methylation stochasticity using informME, we found that UHRF1 silencing resulted in a profound disruption of the methylation landscape that led to a global loss in mean methylation level and a global increase in normalized methylation entropy (Fig. 5a), indicating that UHRF1 expression plays a major role in globally regulating methylation stochasticity in ALL. By also investigating the effect of UHRF1 disruption on computationally predicted A/B chromatin domains39 in Reh-NT cells (Methods), we observed an altered chromatin structure in which gene rich and transcriptionally active euchromatic A domains exhibited profound hypomethylation and gain in normalized methylation entropy in Reh-UHRF1-KO cells (Figs. 5b,c), in agreement with our global results. However, we were surprised to find a preferential and almost complete loss in mean methylation level and normalized methylation entropy localized at gene poor, transcriptionally inactive, and normally highly entropic heterochromatic B domains (Figs. 5b,c), suggesting that UHRF1 expression exerts substantial control on the methylation state within these domains in ALL (Fig. 5b).
Fig. 5 |. UHRF1 is a target of epigenetic disruption in ALL.
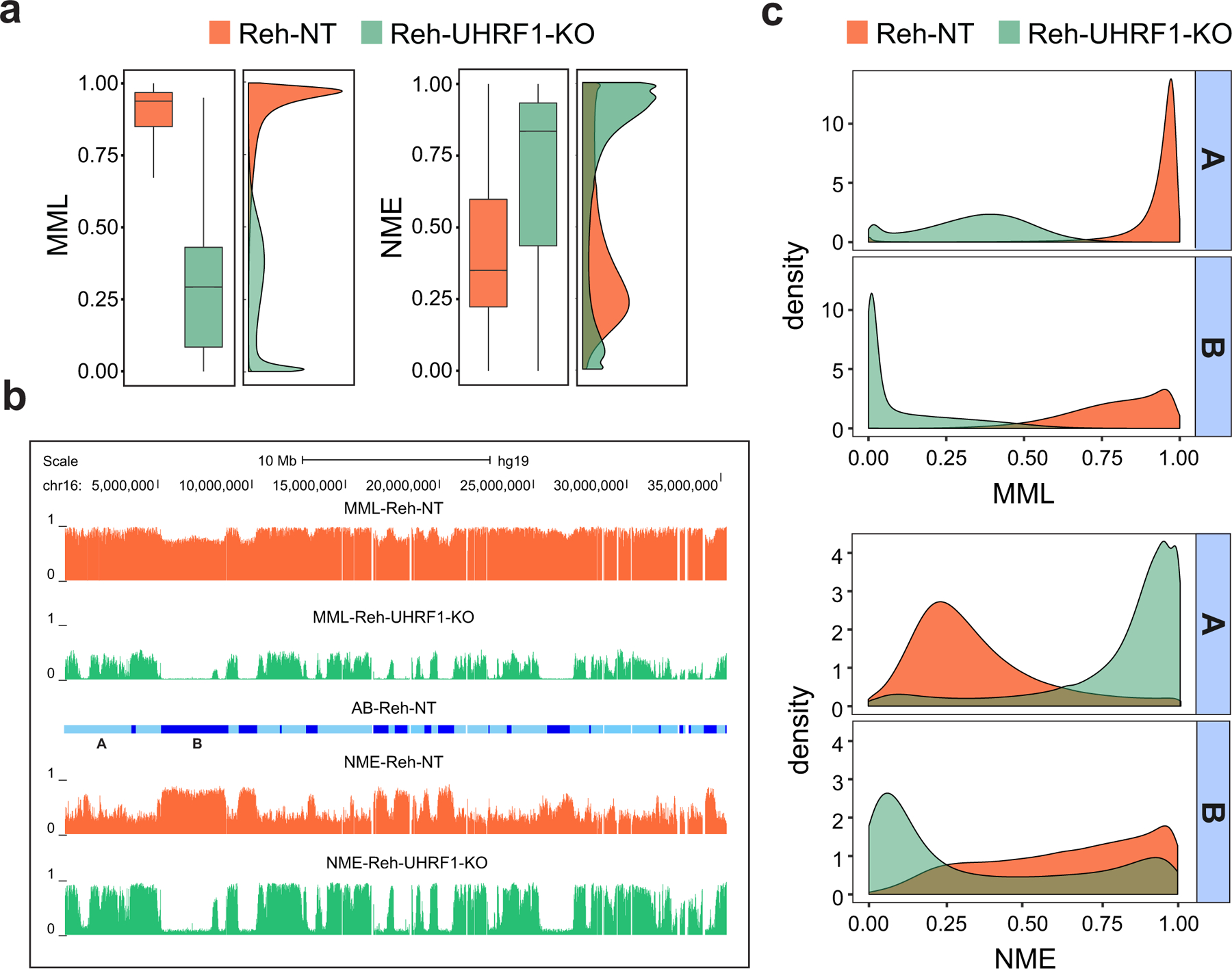
a, Boxplots and densities of genome-wide distributions of mean methylation level (MML) and normalized methylation entropy (NME) values in NT and UHRF1-KO WGBS samples show that UHRF1 silencing results in profound global hypomethylation and marked gain in normalized methylation entropy. Center lines, median; boxes, interquartile range (IQR); whiskers, 1.5 × IRQ. b, UCSC genome browser example showing that chromosome 16 exhibits profound hypomethylation and marked gain in normalized methylation entropy in UHRF1-KO Reh cells over NT associated euchromatic A domains (light blue), as well as almost zero mean methylation level and profound loss in normalized methylation entropy over heterochromatic B domains and over several genomic regions within A domains (dark blue). c, Densities of mean methylation level and normalized methylation entropy within NT associated A/B domains confirm the previous findings genome-wide.
To further explore the previous findings, we ranked genes based on the average mutual information between the methylation level and the phenotype within their promoters in NT and UHRF1-KO Reh cells, as quantified by the square Jensen-Shannon distance magnitude (Supplementary Table 12a), and found several genes with an important role in leukemogenesis, such as PRDM16, SOX8, MEG3, GATA6, and GBX2, to be highly informative of the phenotype (average mutual information , suggesting that UHRF1 exerts substantial control on the epigenetic state of these genes. Notably, although the promoters of these genes were nearly methylated in Reh-NT and exhibited low normalized methylation entropy, they switched to being nearly unmethylated upon UHRF1 silencing. By computing overlaps of the top 1000 genes with the ‘GO gene sets’ (C5) in MSigDB, we found enrichments in genes related to transcription regulation, signaling, development, differentiation, morphogenesis, cell proliferation, and cell fate (Supplementary Table 12b), whereas by using the ‘curated gene sets’ (C2) in MSigDB, we found enrichment of PRC2 (EED, SUZ12) targets, as well as of genes possessing bivalent (H3K4me3 and H3K27me3) marks in their promoters (Supplementary Table 12c). Interestingly, UHRF1 was recently demonstrated to be essential for maintenance of bivalent chromatin domains and regulation of lineage specification in embryonic stem cells40, whereas, UHRF1 silencing was shown to lead to reduced proliferation and increased apoptosis in some types of cancer41–43.
The previous enrichments were strikingly similar to the ones obtained by comparing ETV6-RUNX1 ALL and CD19+ cells (Supplementary Tables 2b,c) providing evidence that some genes whose promoter methylation is regulated by UHRF1 may also exhibit significant methylation discordance within their promoters in ETV6-RUNX1 ALL. To further examine this evidence, we compared the top 1000 genes with the highest methylation discordance within their promoters in the ALL-45/CD19–1 comparison (Supplementary Table 2a) to the top 1000 genes obtained in the Reh-UHRF1-KO/Reh-NT comparison (Supplementary Table 12a). We found 264 common genes that included EBF2, GATA5, GATA6, MAFB, SOX9, SOX14, several FOX family genes, and members of the PAX family of transcription factors implicated in leukemia, which led again to GSEA enrichments in genes related to phenotypic determination and regulation, such as gene expression, differentiation, morphogenesis, and cell fate (Supplementary Table 12d), as well as to PRC2 (EED, SUZ12) targets and genes possessing bivalent marks at their promoters (Supplementary Table 12e).
To gain further insight into the role of UHRF1 in pre-B ALL, we performed an integrative analysis to investigate whether our DNA methylation and gene expression data confirm the possibility that in-frame chromosomal translocation genes in ALL are co-regulated through a gene regulatory network that is driven by UHRF1. By employing our single-cell RNA sequencing data and by exploring statistical dependencies among UHRF1 and the translocation genes in these data using a recent multivariate information-theoretic method (Methods), we surprisingly identified an elaborate regulatory network relationship between UHRF1 and 34 translocation genes (Fig. 6, Supplementary Table 13). In this network, UHRF1 exhibited significant upregulation in expression mean and variance and was predicted to be the gene with the largest ‘betweenness centrality’ (Methods), indicating that it may exert the most influence over the network as compared to other genes. We found UHRF1 to interact with seven translocation genes (AFF1, APBB1IP, BCR, ERG, LDLRAD4, MLLT3, RUNX1), and this was accompanied with significant upregulation of mean expression in 9 translocation genes (BCR, CBFA2T3, CEBPE, CLIP2, ERG, ESYT2, PTHLH, TCF3, ZCCHC7), while the remaining genes exhibited no significant change in mean expression (Fig. 6 and Supplementary Table 13). In addition, 4 translocation genes (ERG, PAX5, CLIP2, CBFA2T3) demonstrated significant upregulation of expression variance in an ETV6-RUNX1/CD19 comparison (AL-45/CD19–4), whereas 18 genes (AFF1, APBB1IP, BCR, CREBBP, ESYT2, ETV6, FOXK2, GALNS, GAS7, IKZF1, LDLRAD4, MLLT3, RUNX1, SCARB1, SUPT3H, TCF3, ZC3HAV1, ZCCHC7) exhibited no significant change in expression variance (Fig. 6, Supplementary Table 13), with similar results obtained from other comparisons.
Fig. 6 |. A plausible regulatory relationship between UHRF1 and 34 in-frame translocation genes identified in ETV6-RUNX1 ALL.
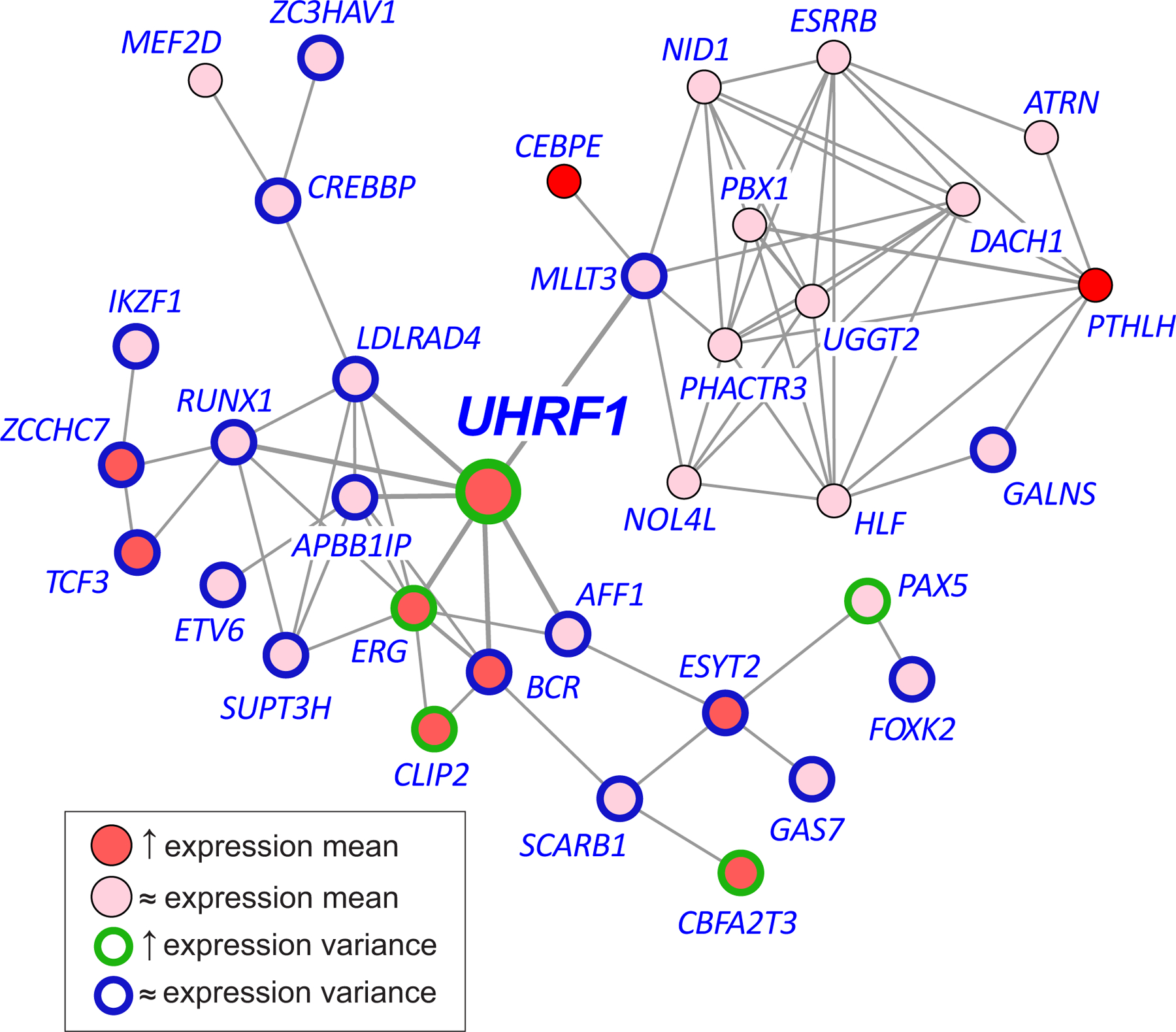
An information-theoretic analysis of single-cell RNA sequencing data identifies a network relationship between UHRF1 and 34 translocation genes driving significant and targeted expression discordance of genetic drivers in ALL. In this network, UHRF1 may play the most influential role as compared to other genes in the network. Red, overexpression; brown, upregulated expression variance; blue, no change in expression variance.
Taken together, these results suggest that dysregulation of DNA methylation stochasticity associated with the highly informative gene UHRF1 and its resultant overexpression may play a crucial role in pediatric ALL, possibly by preferentially altering stochastic epigenetic properties of the chromatin state via regulation of histone modifications and through influencing the expression of key translocation and other genes that serve as genetic drivers of this disease.
Discussion
Structural chromosome alterations act as initiating driver lesions in the majority of ALL cases, with in-frame fusion genes produced by chromosome translocations constituting the largest set44. Identifying the full gamut of driver genes involved in (in-frame) gene fusions across the disease spectrum of pediatric B-precursor ALL has required DNA or RNA sequencing of hundreds of patient samples19,45. Here, by carrying out an integrative analysis of methylation and gene expression data, we identified a surprising convergence of stochastic epigenetic behavior in this genetically diverse disease. Potential energy landscape analysis of methylation discordance using the Jensen-Shannon distance between the probability distributions of the methylation level in even a single ALL/CD19 comparison yielded enrichments of translocation genes that exhibit significant methylation discordance in ALL. Importantly, these translocation genes are informative of the biological state (cancer/normal) in the sense that they exhibit a statistically significant degree of mutual dependence between their methylation level and the phenotype. This novel result identifies non-mutated ALL driver genes as important targets of epigenetic instability and suggests the potential existence of a core gene regulatory network underlying ALL, in which nodes are perturbed either genetically or epigenetically during leukemic transformation.
Investigations across diverse types of cancer have revealed many epigenetic changes in tumor cells due to mutations in genes encoding epigenetic regulators, alterations in DNA methylation, histone modifications, and reorganization of chromatin structure46–48. Recently, array-based DNA methylation analysis across major subtypes of B cell malignancies, including ALL, showed that tumor-specific DNA methylation signatures provide insight into cellular developmental origin and proliferative history and thereby can be predictive of clinical outcome49. It is increasingly recognized that stochastic epigenetic variation is also an important force in cancer by driving phenotypic plasticity that allows for selection of cellular traits promoting growth and survival in a changing environment50–52. For example, using reduced-representation bisulfite sequencing (RRBS) data and two whole-genome bisulfite sequencing (WGBS) samples, it was recently shown that high DNA methylation pattern heterogeneity in chronic lymphocytic leukemia (CLL) could be associated with adverse clinical outcome53. Moreover, DNA methylation analysis of enhanced reduced-representation bisulfite sequencing (ERRBS) data in acute myeloid leukemia (AML) has revealed a link between epigenetic variation and inferior clinical outcome54. Finally, DNA methylation analysis of paired ERRBS samples in diffuse large B-cell lymphoma has also demonstrated that increased methylation heterogeneity at diagnosis is predictive of relapse55. These results affirm the importance of epigenetic stochasticity in cancer and show that understanding its regulatory role is crucial when targeting tumor evolution and resistance to therapy.
Here, we carried out an information-theoretic analysis of a set of WGBS primary ALL and normal CD19+ B/pre-B samples in conjunction with bulk and single-cell gene expression data, and generated a comprehensive map of statistical properties of the methylomic landscape in ALL. We locally estimated probability distributions of the stochastic methylation state which, in addition to computing mean methylation levels, allowed us to quantify the amount of methylation stochasticity within genomic regions using the information-theoretic concept of Shannon entropy, evaluate the robustness of these distributions to changes in the potential energy landscape, and use these statistics to perform differential analysis with respect to normal samples. By employing the information-theoretic concept of mutual information, we also identified ‘informative’ regions of the genome characterized by statistically significant associations between their methylation level and the phenotype in our cancer/normal comparisons. Informative regions are of compelling interest when investigating epigenetic dysregulation in cancer since the probability distribution of the methylation state within these regions is significantly determined by the phenotype, pointing to the possibility that they may play an important role in tumor initiation and progression.
Our analysis revealed that, on the average, DNA methylation stochasticity within gene promoters relates to mean gene expression and gene variability in ALL, with more stochastic methylation associated with genes exhibiting lower mean expression levels and higher expression variability. We also found that significant methylation discordance in ALL localizes within distinct and highly informative regions of the genome, including genes that are critical regulators of the leukemic phenotype. Further investigation revealed a surprising convergence of statistically significant methylation discordance on a shared set of genes, exhibiting overlap with known genetic drivers of ALL (genes involved in in-frame fusions) across different cytogenetic subtypes. This led to the novel hypothesis that these genes may interact through an epigenetically driven regulatory network that exhibits significant dysregulation in pediatric ALL. Finally, we detected enrichment of PRC2 binding sites, as well as H3K4me3 and H3K27me3 domains, in highly informative regions of the genome characterized by a statistically significant degree of mutual dependence between the methylation level and the phenotype.
We also observed profound gains in mean methylation level within regions harboring bivalent chromatin marks in normal B cells, in agreement with a prior WGBS analysis of two ALL samples demonstrating that bivalent domains of embryonic stem cells are hypermethylated in ALL12. Moreover, we consistently found considerable gains in normalized methylation entropy and marked losses in methylation sensitivity within bivalent domains of normal B cells across four cytogenetic subtypes of pre-B ALL. Given the important role of bivalent domains in cell differentiation, gains in methylation stochasticity within these domains raise the possibility of a biological relationship between normal tissue differentiation and stochastic epigenetic variation that may lead to increased phenotypic tumor heterogeneity in pre-B ALL, whereas, losses in methylation sensitivity advocate a reduced ability of the epigenome to regulate methylation stochasticity. Notably, most genes marked by bivalent domains of CD19 that were identified as being overexpressed in ALL exhibited significant methylation discordance. This however was not true for underexpressed bivalent genes, raising the possibility that epigenetic disruption of bivalency in ALL is primarily associated with upregulation of gene expression.
From methylation data alone, our analysis identified UHRF1 as the most informative gene of the leukemic phenotype. Moreover, by using single-cell RNA sequencing data and an information-theoretic network inference method56 that is appropriate for the methylation analysis approach considered in this paper, we found an elaborate regulatory network relationship between UHRF1 and translocation genes in which UHRF1 is the most influential node (i.e., a hub). This suggests that UHRF1 may play an important regulatory role at the heart of ALL. We provided support for this hypothesis by CRISPR/Cas9 silencing of UHRF1 in the Reh cell line, which showed complete abrogation of leukemic clonogenicity. Interestingly, UHRF1 has been reported as a potential translocation target in a case of B-precursor ALL that is due to a rare translocation between chromosomes 19 and 21 resulting in an NRIP1-UHRF1 gene fusion35, and has also been identified in t(1;19) pre-B ALL cells by siRNA screening as being essential for leukemic cell viability38. This is in agreement with previous array-based analysis of DNA methylation and gene expression in B-ALL which reported overexpression of UHRF1 across ALL samples independent of genetic subtype, and demonstrated a strong inverse correlation between methylation β-value and UHRF1 expression7. Our knockout experiments showed a profound reduction of methylation stochasticity in heterochromatic B domains, which are normally characterized by highly disordered methylation. This suggests that UHRF1 expression is essential for faithfully maintaining DNA methylation and for preserving the chromatin state within heterochromatic regions of the genome, which is consistent with its function as a link between repressive histone modifications and the DNA methylation machinery. Ultimate confirmation of the role of UHRF1 in leukemic patients will require mouse knockdown/transplantation and/or design of UHRF1-specific inhibitors for preclinical or clinical investigation, in addition to nonspecific agents which act on this target and are under investigation in prostate and colon cancer57. This however is beyond the scope of the present study.
Notably, the information-theoretic differential analysis of methylation stochasticity performed here identified additional genes that consistently exhibited strong methylation discordance across our cytogenetic subtypes and samples and demonstrated that these genes exhibit high levels of mutual dependence between their methylation level and the phenotype (Supplementary Table 11). For example, we identified genes in the protocadherin families PCDHA and PCDHG, which we found intriguing, given their known stochastic combinatorial expression in defining cell surface diversity and survival during neural system development and in other contexts58, as well as their previously reported role in the epigenetic dysregulation of cancer59. These families are made however by many constituents, which makes targeted gene disruption a much more difficult endeavor in this case when comparing to UHRF1 silencing.
It is also possible to identify additional gene networks that may be significantly dysregulated in ETV6-RUNX1 ALL. To demonstrate this possibility, we integrated existing knowledge of protein-protein interactions in the human interactome with methylation and gene expression data, as well with detected methylation discordance within gene promoters and bodies, and identified a number of functional epigenetic modules (FEMs) in ALL (i.e., gene regulatory networks which are subject to significant epigenetic control in ALL), which included modules associated with TCF3, PLXNB1, LYN, MME, and SLC9A3R2 (Methods, Supplementary Table 14, and Supplementary Fig. 15). Notably, TCF3 is a transcription factor that is critical for regulating lymphoid specification from hematopoietic stem cells60,61 and the target of multiple recurrent structural alterations in ALL, such as TCF3-PBX1 fusions encoded by t(1;19)(q23;p13.3)62 and TCF3-HLF fusions encoded by t(17;19)(q22;p13.3)63.
In conclusion, by integrating WGBS with bulk and single-cell RNA sequencing data, we here conducted a comprehensive and cytogenetically relevant analysis of DNA methylation stochasticity in pediatric ALL using an information-theoretic potential energy landscape approach. This allowed us to quantify informational properties of the methylomic landscape in ALL, identify regulatory underpinnings of malignant transformation, and assess the importance of epigenetic stochasticity as a driver of this disease. Our results demonstrate that cytogenetic alterations in ALL concur with significant dysregulation of methylation stochasticity over a set of in-frame translocation genes whose methylation levels carry significant information about the phenotype. In addition, our analysis predicts that UHRF1 plays a crucial role in pediatric ALL by possibly driving a regulatory network of translocation genes that exhibits significant and targeted dysregulation in ALL. Taken together, our comprehensive genome-wide evaluation of information-theoretic properties of the methylomic landscape in pediatric ALL using our integrated computational method for analyzing DNA methylation stochasticity, which employs potential energy landscapes and uses the Jensen-Shannon distance, mutual information, mean methylation level, normalized methylation entropy, and the methylation sensitivity index to summarize multiple statistical factors influencing methylation stochasticity, shows a great utility for formulating new biological hypotheses that could likely explain diversity in the genetic origins of pediatric ALL and, more generally, for understanding the role of stochastic epigenetic regulation in cancer.
Methods
Sample collection and preparation.
Primary patient ALL samples were collected under the approval of The Johns Hopkins University and Baylor College of Medicine Institutional Review Boards (IRBs), and informed consents were obtained for all research samples. Samples were selected for analysis based on high blast proportion (> 90%) in their clinical characterization. Leukemic blasts were enriched from bone marrow or peripheral blood samples of newly diagnosed patients (collected prior to any chemotherapy) by Ficoll-Hypaque centrifugation. Samples with blast composition < 90% were purified by flow sorting, as follows. Leukemia cell populations were sorted on a FACSMelody (BD) using FACSChorus software (BD). Cells were stained with Live/Dead Fixable Viability Stain 780, PE mouse anti-human CD19 clone HIB19, and APC Mouse Anti-Human CD10 clone HI10a (BD) with the CD19+CD10+ gate defining leukemic cells (Supplementary Fig. 16). The maximum number of cells was acquired, isolated by centrifugation, and cell pellets were immediately flash frozen. Diagnosis and characterization of ALL was based on standard clinical assays, including morphology, flow cytometric immunophenotype, and cytogenetics. Clinical characteristics of primary patient samples are provided in Supplementary Table 1.
Human umbilical cord blood CD19+ primary cells were purchased from Stemcell Technologies, Vancouver, BC, Canada, in three independent lots from distinct donors. The ETV6-RUNX1 pre-B ALL cell line Reh (ATCC CRL-8286) was grown in RPMI 1640 medium with L-glutamine (ThermoFisher), supplemented with 1x penicillin/streptomycin (ThermoFisher) and 10% fetal bovine serum (Gemini Bio-Products, West Sacramento, CA). CRISPR/Cas9 edited Reh cell lines (Reh-NT and Reh-UHRF1-KO) were generated by lentiviral transduction, puromycin selection, and clonal expansion, as described below, and maintained under the same culture conditions.
CRISPR/Cas9 genome editing.
Lentiviral particles were purchased from Sigma-Aldrich using the LentiCRISPR platform. UHRF1 targeting used the gRNA sequence TCCCGTCCATGGTCCGAACC in the pLV-U6g-EPCG vector, with viral titer by p24 antigen ELISA of 8.8 x 106 TU/ml. CRISPR lentivirus non-targeting control particles utilizing the identical pLV-U6g-EPCG vector were also purchased from Sigma-Aldrich (Lot 10201511MN), with p24 antigen ELISA titer 2.9x107 TU/ml. Reh cells (1x105) were combined with viral particles at multiplicity of infection (MOI) 2, in 0.5 ml RPMI media supplemented with 10% FBS and Polybrene 4 μg/ml (Millipore), and centrifuged for 60 minutes at 800x g at 32°C. Cells were grown in RPMI with 10% FBS for 72 hours and subsequently selected with 1 μg/ml Puromycin followed by clonal dilution and evaluation by PCR and Western blot.
Western blot.
Cells were lysed in lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% Triton X-100, 10% glycerol) supplemented with Complete protease inhibitor tablet (Roche) on ice. Lysates were boiled in 1x NuPAGE lithium dodecyl sulfate sample buffer and subjected to SDS-PAGE using the Novex system (Invitrogen) following the manufacturer’s instructions, transferred to nitrocellulose membranes using the iBlot transfer system (Invitrogen), blocked in 5% nonfat milk in Tris-buffered saline/0.1% Tween-20 (TBST) for 1 hour at room temperature, and incubated with primary antibodies in TBST overnight at 4°C. The antibody for UHRF1 (Purified Mouse Anti-ICBP90) was from BD Biosciences (Cat. 612264). The beta-actin antibody was from Thermo (MA1–140 Invitrogen beta-actin antibody, 15G5A11/E2).
Methylcellulose assay.
Methylcellulose 3% stock solution was purchased from R&D Systems (cat. HSC001). A 10x cell suspension in RPMI media was mixed with RPMI supplemented to yield a final concentration of 1.27% methylcellulose and 30% FBS. Methylcellulose colony counts were carried out after 14 days in culture.
Library preparation and sequence data generation.
For primary ALL and umbilical cord blood CD19+ cell samples, genomic DNA and total RNA were extracted using the ZR-Duet DNA/RNA MiniPrep kit (Zymo Research, cat. D7001). RNA preparation included an on-column DNase I digestion (Invitrogen). For cell lines, genomic DNA isolation was carried out using the MasterPure DNA Purification kit (Epicentre). Integrity of genomic DNA was confirmed by gel electrophoresis. RNA was assessed on an Agilent 2100 Bioanalyzer using the Agilent RNA 6000 Nano kit.
WGBS single indexed libraries were generated using NEBNext Ultra DNA library Prep kit for Illumina (New England BioLabs) according to the manufacturer’s instructions with the following modifications. 500 ng input gDNA was quantified by Qubit dsDNA BR assay (Invitrogen) and spiked with 1% unmethylated Lambda DNA (Promega, cat # D1521) to monitor bisulfite conversion efficiency. Input gDNA was fragmented by Covaris S220 Focused-ultrasonicator to an average insert size of 350 bp. Samples were sheared for 60 sec using Covaris microTUBEs, with instrument settings of duty cycle 10%, intensity 5 and cycles per burst 200. Size selection was performed using AMPure XP beads and insert sizes of 300–400bp were isolated. Samples were bisulfite converted after size selection using EZ DNA Methylation-Gold Kit or EZ DNA Methylation-Lightning Kit (Zymo cat#D5005, cat#D5030) following the manufacturer’s instructions. Amplification was performed after the bisulfite conversion using Kapa Hifi Uracil+ (Kapa Biosystems, cat# KK282) polymerase based on the following cycling conditions: 98°C 45s / 8cycles: 98°C 15s, 65°C 30s, 72°C 30s / 72°C 1 min. AMPure cleaned-up libraries were run on the 2100 Bioanalyzer (Agilent) High-Sensitivity DNA assay, and samples were also run on the Bioanalyzer after shearing and size selection for quality control purpose. Libraries were quantified by qPCR using the Library Quantification Kit for Illumina sequencing platforms (Kapa Biosystems, cat#KK4824) and the 7900HT Real Time PCR System (Applied Biosystems). WGBS libraries were sequenced on an Illumina HiSeq4000 instrument using 150 bp paired end indexed reads and 25% of non-indexed PhiX library control (Illumina). Coverage is indicated in Supplementary Table 1. The bisulfite conversion rate of unmethylated Lambda DNA was 99.6% on average.
FASTQ files were processed using Trim Galore! v0.3.6 (Babraham Institute) to perform single-pass adapter- and quality-trimming of reads. FastQC v0.11.2 was employed for quality control of reads. Reads were aligned to the hg19/GRCh37 genome using Bismark v0.14.5 and Bowtie2 v2.2.6. Separate M-bias plots for read 1 and read 2 were generated by running the Bismark methylation extractor using the ‘mbias_only’ flag (Supplementary Fig. 17), and these plots were used to determine how many bases to remove from the 5’ end of reads. The number was generally higher for read 2, known to exhibit a lower quality. The amount of 5’ trimming ranged from 5 bp to 18 bp, consistent with ref. 14. BAM files were subsequently processed with Samtools v0.1.19 for sorting, merging, duplicate removal, and indexing.
Bulk RNA sequencing.
Strand specific mRNA libraries were generated using the TruSeq Stranded mRNA protocol (Illumina, cat# RS-122–2101). Preparation of libraries followed the manufacturer’s protocol (Illumina, Part#15031050) with minor modifications. Input was 500 ng and samples were fragmented for 6 min. The following PCR cycling conditions were used: 98°C 30s / 14 cycles: 98°C 10s, 60°C 30s, 72°C 30s / 72°C 5 min. Stranded mRNA libraries were sequenced on an Illumina HiSeq4000 instrument using 75bp or 125bp paired-end indexed reads and 1% of PhiX control. mRNA sequencing depth ranged from 90–125M reads.
Single-cell RNA sequencing.
Single cells were captured on medium sized Fluidigm C1 Single Cell Integrated Fluidic Circuit (IFC). The SMARTer Ultra Low RNA Kit for Illumina (Clontech) was used for single-cell capture (verified via microscope), on-chip lysis, reverse transcription, and complementary DNA (cDNA) generation. An ERCC Spike-in Mix (Ambion) was used as the technical control per Fluidigm recommendation. Single-cell cDNA quantification was performed using Agilent High Sensitivity DNA Kits and Quant-it Picogreen dsDNA (Invitrogen). cDNA was normalized to 0.20ng/μL with Biomek NXP(Beckman Coulter). The Nextera XT DNA Library Prep Kit (Illumina) was used for dual indexing and amplification following the Fluidigm C1 protocol. Libraries were purified and size selected twice using volume of Agencourt AMPure XP beads (Beckman Coulter). Cleaned libraries were quantified with Quant-it Picogreen dsDNA (Invitrogen) and normalized to 0.3ng/µl with Biomek NXP (Beckman Coulter). Single-cell RNA sequencing libraries were subsequently pooled for 96-plex sequencing. The resulting cDNA libraries were quantified using High Sensitivity DNA Kit (Agilent). The pooled 96x single-cell libraries were sequenced using HiSeq 2500 (Illumina) with paired end 126bp-8bp-8bp-126bp and 14pM-loading concentration with 5% PhiX spike-in.
Genomic features and annotations.
Files and tracks bear genomic coordinates for hg19. CGIs were obtained from ref. 64. CGI shores were defined as sequences flanking 2-kb on either side of islands, shelves as sequences flanking 2-kb beyond the shores, and open seas as everything else. The R Bioconductor package ‘TxDb.Hsapiens.UCSC.hg19.knownGene’ was used for defining genes, exons, introns, and gene bodies. The promoter region of a gene was defined as the 4-kb window centered at the TSS and the gene body region was defined as the remainder of the gene. The enhancer and promoter annotations, as well as other relevant genomic annotations not mentioned above, were obtained from Ernst and Kellis65 using the ChromHMM 25-state reference model. ChromHMM 25-state enhancer and promoter annotations with definitions and emission parameters as previously described were employed66. Specific states such as TxEnh5’ (transcription 5’ enhancer), EnhA1 (active enhancers 1 – enriched in H3K27ac and H3K4me1), EnhW1 (weak enhancers 1 – enriched in H3K4me1 but not in H3K27ac), PromP (poised promoters), and PromBiv (bivalent promoters – enriched in H3K27me3 and H3K4me3), were included in some parts of the data analysis due to their marked methylation discordance when analyzing ETV6-RUNX1 ALL and normal control samples, as compared to the remaining ChromHMM annotations (Supplementary Table 15). Histone marks were obtained from the Roadmap Epigenomics Project23. SUZ12 and EZH2 binding sites were obtained from ENCODE67. A/B chromatin domains for Reh cells were computed using a previously developed random forest approach14 that learns these domains from available ground-truth data.
PEL computation and visualization.
Potential energy landscape (PEL) computations from WGBS data were performed using informME (v0.3.3), a freely available information-theoretic pipeline for analysis of methylation stochasticity based on the one-dimensional Ising model of statistical physics15. To balance computational and estimation performance, it was previously determined15 that a good choice for the length of each genomic region used for parameter estimation is 3-kb. Because of concerns regarding statistical overfitting (i.e., not having enough data for reliable parameter estimation), we did not model genomic regions that had less than 10 CpG sites, for which less than 2/3 of the CpG sites were observed, or for which the average depth of coverage was less than 2.5 observations per CpG site. While CpG sites in very low-density estimation regions were not considered, the vast majority (> 82%) of the CpG sites with data in the WGBS samples were properly modeled (Supplementary Fig. 1). PELs were visualized by employing the two-dimensional version of Gray’s code68 and by assigning to each methylation pattern its potential value. Grays’s code places all possible binary-valued methylation patterns within a genomic region on a two-dimensional pattern space in a manner so that patterns located adjacent to each other in the east/west and north/south directions differ in only one bit.
Methylation level and entropy.
Probability distributions of methylation levels, as well as mean methylation levels and normalized methylation entropies, were computed within each analysis region using informME. The mean methylation level is the expected value of the methylation level within an analysis region, and is given by , where , , is the associated probability distribution of . The normalized methylation entropy is a normalized version of Shannon’s entropy, given by , and was used to quantify the amount of methylation stochasticity observed within an analysis region. It ranges between 0 and 1, taking its maximum value when all methylation levels within an analysis region are equally likely (fully stochastic methylation), and achieving its minimum value only when a single methylation level is observed (perfectly ordered methylation).
Jensen-Shannon distance and mutual information.
Within an analysis region, the Jensen-Shannon distance between the two probability distributions and of the methylation level in a test (ALL) and a reference (CD19/pre-B2) sample was calculated by , where and is the Kullback-Leibler divergence between two probability distributions and (also known as the relative entropy), and was used to quantify dissimilarities between the two probability distributions and . It ranges between 0 and 1, taking its minimum value only when the two probability distributions are identical, in which case no statistical discordance in methylation level is present, and its maximum value of 1 only when the supports of the two probability distributions do not intersect each other, in which case a maximum statistical discordance in methylation level is observed.
Within an analysis region, the mutual information between the methylation level and the phenotype is given by , where for the reference phenotype and for the test phenotype, respectively. We used this quantity to measure, within an analysis region, the degree of mutual dependence between the methylation level and the phenotype, with higher values indicating a stronger mutual dependence. By making the (reasonable) assumption that the test and reference phenotypes are equally probable (i.e., ), it can be shown16 that the average mutual information between the methylation level and the phenotype within a region of the genome that includes analysis regions, given by , where is the methylation level of the -th analysis region, equals the square of the magnitude of the Jensen-Shannon distance within the genomic region, where is the Jensen-Shannon distance within the -th analysis region. Note that the average mutual information ranges between 0 and 1, with higher values indicating a more informative genomic region in which there is a greater degree of mutual dependence between the methylation level and the phenotype. Notably, the previous relationship implies that the mutual information between the methylation level and the phenotype within an analysis region equals the square of the Jensen-Shannon distance between the probability distributions of the methylation level within the region in a test/reference comparison.
Methylation sensitivity.
Local modifications in the biochemical environment produced by the methylation machinery, due for example to environmental variability, can result in a change to the values of parameters of the methylation potential energy landscape. We modeled this change as a perturbation of size applied on , , and , which results in a new probability distribution of the methylation level. We quantified the rate of this change using the methylation sensitivity index, defined as the absolute value of the derivative , evaluated at , where is the Jensen-Shannon distance between the probability distributions and of the methylation level before and after perturbation, respectively. Note that for a small enough , whereas . We therefore set and approximately computed the methylation sensitivity index by . This index assesses robustness of the probability distribution of the methylation level to changes in parameter values of the potential energy landscape, with higher sensitivity indicating less robust behavior pointing to a more ‘responsive’ region in which small variations in parameter values can produce larger changes in the stochastic behavior of the methylation level.
Differential analysis.
Differential analysis between test (ALL) and reference (CD19/pre-B2) WGBS samples was performed using informME by computing, within analysis regions, Jensen-Shannon distances between corresponding probability distributions of the methylation level, as well as by evaluating differences between mean methylation levels, normalized methylation entropies, and methylation sensitivity indices. DMR detection was performed by employing the ‘jsDMR’ utility of informME, which uses the Jensen-Shannon distance to identify regions of the genome with significant methylation discordance. As previously described15, this was done by smoothing the values of the Jensen-Shannon distance using the Nadaraya-Watson kernel regression smother with a Gaussian kernel of a fixed bandwidth (5-kb), by performing multiple hypothesis testing on the smoothed values using, as the null distribution, the density of the Jensen-Shannon distance values obtained from all CD19 control sample comparisons, and by employing the Benjamini-Yekutieli method69 to control the false discovery rate (FDR) at 0.05. Since the mutual information within an analysis region between the methylation level and the phenotype equals the square of the Jensen-Shannon distance, the DMRs identified by the ‘jsDMR’ utility of informME point to informative regions of the genome in which there is a significant degree of mutual dependence between the methylation level and the phenotype.
Hypothesis testing and gene ranking.
In each test/reference comparison, promoter and gene body regions were assigned a score given by the magnitude of the Jensen-Shannon distance or by the magnitude of the differential mean methylation level within each feature, where is the Jensen-Shannon distance and is the differential mean methylation level within the -th analysis region. One-sided hypothesis testing was performed by testing against the null hypothesis that a genomic feature exhibits a Jensen-Shannon distance (or differential mean methylation level) magnitude that can be explained by normal technical, statistical, or biological variability. To do so, a null distribution was constructed for the Jensen-Shannon distance (or differential mean methylation level) magnitude by comparing the normal control samples. To account for variability in the number of analysis regions overlapping each feature, generalized additive models were employed for location scale and shape (GAMLSS)70 with a logit skewed Student’s t-distribution. This was implemented by the R function ‘gamlss’ with the ‘family’ argument set to ‘logitSST’ and the log-number of overlapping analysis regions used as the independent variable. To evaluate the statistical significance of each gene while controlling for the false-discovery rate (FDR), Q-values were computed using the Benjamini-Yekutieli procedure69. In each test/reference comparison, genes were ranked using the calculated Q-values, with genes located higher in the list being associated with lower Q-values. We performed these steps by employing the ‘jsGrank’ utility of informME16.
Gene enrichment analysis.
Gene set enrichment analysis (GSEA) was performed using Broad Institute’s GSEA ‘investigate gene sets’ tool that computes overlaps of a given set of genes in the molecular signatures database (MSigDB) v7.0.
Bulk RNA sequencing analysis.
Bulk RNA sequencing was performed on eight ETV6-RUNX1 ALL samples (ALL-45, −100, −127, −137, −276, −289, −311, −321) and the three normal CD19+ samples used for WGBS (CD19–1, −2, −3). For each bulk RNA sequencing data, paired-end reads were mapped to human genome hg19 using HISAT271. Gene expression values were calculated in TPM (transcripts per million) using StringTie72. The TPM values were log2 transformed (after adding a pseudo-count of 1) and quantile normalized across samples (Supplementary Table 5a). Differentially expressed genes were obtained using the Bioconductor package ‘limma’73 and by performing differential expression analysis between the eight ALL and the three normal CD19+ cell samples based on a moderated t-statistic for differential gene expression (Supplementary Table 5b). To control the false discovery rate (FDR), Q-values were calculated using the Benjamini-Hochberg procedure74.
Single-cell RNA sequencing analysis.
Single-cell RNA sequencing was performed on two ETV6-RUNX1 samples, ALL-45 and ALL-289, and on two normal CD19+ samples, CD19–4 and CD19–5, which were different from the samples used for WGBS and bulk RNA sequencing because single-cell separation required viable cells that were not available for the CD19 samples employed for WGBS. For each cell analyzed, paired-end reads were mapped to human genome hg19 using HISAT2 and gene expression values were calculated in TPM (transcripts per million) using StringTie. Cells with more than 1 million sequenced reads and mapping rate larger than 50% were retained for further analysis. The TPM values were log2 transformed, after adding a pseudo-count of 1 (Supplementary Table 6a). For each sample, genes with average log2 transformed TPM values greater than 1 were retained for subsequent analyses. To adjust for dropout events75, the R package SCDV was used, which applies a two-component mixture model to estimate the true expression value and the dropout probability for each gene.
To compare variation in gene expression between ALL and normal CD19+ cells, the weighted mean and the weighted variance of the expression of each gene within each sample were calculated by and , where is the i-th expression value available for the gene within the sample and is the associated dropout probability. To identify genes with differential gene expression and differential variation while accounting for non-normality, the R package SCDV was used to apply a two-sided permutation test to each gene in each ALL/CD19 comparison, based on a t-statistic for differential weighted mean expression (Supplementary Tables 6b–e) and an F-statistic for differential weighted expression variance (Supplementary Tables 6f–i). Specifically, Welch’s t-test statistic was used, calculated by , where and is the weighted mean and variance in the ALL sample, respectively, and is the weighted mean and variance in the CD19 sample, and , are the corresponding numbers of cells in each sample. Moreover, the F-statistic was computed by the ratio . To control the false discovery rate (FDR), Q-values were calculated using the Benjamini-Hochberg procedure74.
To further characterize variation in gene expression, a generalized version of the dispersion index was employed, given by , where and is the weighted expression mean and variance of a gene, respectively, and is a baseline function so that implies that the expression variance is higher than what is expected for a given expression mean. Taking recovers the traditional dispersion index whose baseline is given by a Poisson process, but here we constructed to account for biases of the single-cell RNA sequencing data sampling process. This leads to , where and , where is the variability level of the gene76. In a given sample, we estimated the function by curve fitting, using local polynomial regression (LOESS), to all pairs of log2-scaled weighted variances and means computed in the sample. All LOESS curves calculated from the ALL and CD19 samples were almost identical to each other (Supplementary Fig. 18), thus permitting comparative analyses. The variability level of a gene was then calculated by computing the vertical distance (i.e., residual) of the log2-weighted variance from the LOESS curve (Supplementary Table 6j).
Finally, to identify a gene that shows differential variability between ALL and normal CD19+ cells, a two-sided permutation test was performed using the R package SCDV based on the computed difference in variability levels (Supplementary Tables 6k–n). To control the false discovery rate (FDR), Q-values were calculated using the Benjamini-Hochberg procedure74.
Statistical analysis of genetic bona fide drivers of ALL.
Evaluation of the statistical significance of the 1290 differential methylated genes (DMGs) in ALL common to four cytogenetic subtypes (Supplementary Table 10a) was performed by testing against the null hypothesis that the DMGs were selected by chance. The number of DMGs common to all samples considered was used as a test statistic and its null distribution was constructed using Monte Carlo iterations. At each iteration and for each cytogenetic sample, a number of genes equal to the number of DMGs in the sample were selected at random and the number of common genes to all samples was computed. The null distribution was then approximated by the empirical distribution of all Monte Carlo values for the number of common genes.
We derived a target list of 71 translocation genes from a published list of in-frame chromosomal translocations19 from which we removed those genes not on autosomes or in the hg19 annotation (e.g., DUX4). Statistical evaluation for enrichment of DMGs in the target list was then carried out using a hypergeometric test in each sample and by combining results across samples using Fisher’s meta-analysis.
Gene network inference.
Inference of the UHRF1 regulatory network was directly performed from the single-cell gene expression data (Supplementary Table 6a) using a previously proposed multivariate information-theoretic method56, which was implemented using the Julia package ‘NetworkInference.jl’ with its default settings. Betweenness centrality of a gene in the network measures the extent to which the gene lies on paths between other genes in the network. We computed this quantity by , where denotes the number of shortest paths from a gene to a gene and denotes the number of shortest paths from to that pass through gene , using the function ‘betweenness centrality’ of the Julia package ‘LightGraphs.jl’.
Inference of functional epigenetic modules (FEMs) was performed using a functional supervised algorithm77 that integrates WGBS and RNA sequencing data in order to identify networks of interacting genes that are significantly dysregulated in a disease, which we modified to fit our information-theoretic framework of methylation analysis. This algorithm employs a known protein-protein interaction (PPI) network and a spin-glass algorithm to identify, for a particular gene of interest (seed), a gene subnetwork (i.e., a FEM) characterized by significant discordance in DNA methylation and gene expression as compared to the rest of the PPI network. For FEM inference, each gene in the PPI network was associated with two P-values, quantifying the significance of its expression and methylation discordance, which were then combined using Fisher’s test statistic.78 The ‘expression’ P-value was computed by analyzing the bulk RNA sequencing data (Supplementary Table 5b). The ‘methylation’ P-value was evaluated by computing, in a single sample comparison, two P-values of methylation discordance (one quantified by the Jensen-Shannon distance within a gene’s promoter and the other within its body), by combining these two P-values using Fisher’s test statistic, and by using again Fisher’s test statistic to combine all P-values obtained from multiple sample comparisons. Implementation of the spin glass algorithm was based on weights assigned to each edge connecting two genes in the PPI network, with each weight computed as the average of the two Fisher test statistics associated with the corresponding genes. This algorithm was applied on each of the top 100 genes ranked by their combined ‘expression/methylation’ P-values obtained using Fisher’s test statistic. Statistical significance of an inferred FEM was assessed via P-values computed by Monte Carlo, which were obtained by testing against the null hypothesis that observed discordance in gene expression and methylation within the FEM was due to chance. FEMs were ranked in terms of their statistical significance evaluated by Q-values, which were computed using the Benjamini-Hochberg procedure74 to control for the FDR that would otherwise be affected by the number of initial hypothesis tests (seeds) used. Only subnetworks containing at least 10 genes and Q-values no more than 0.05 were reported.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
This work was supported by US National Institutes of Health grants R01 CA65438 and DP1 DK119129 to A.P.F, R01 HG006282 to H.J., US National Science Foundation Grant CCF-1656201 to J.G, St. Baldrick’s Foundation fellowship and funding from Unravel Pediatric Cancer to M.A.K. M.A.K. is a Damon Runyon-Sohn Pediatric Cancer Fellow supported by the Damon Runyon Cancer Research Foundation (DRSG-15P-16). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Peer review informationNature Biomedical Engineering thanks the anonymous reviewers for their contribution to the peer review of this work.
Data availability
DNA-methylation and RNA-sequencing data are available at the Gene Expression Omnibus repository, under accession number GSE116229.
References
- 1.Mullighan CG The molecular genetic makeup of acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program 2012, 389–96 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Hunger SP & Mullighan CG Acute lymphoblastic leukemia in children. N Engl J Med 373, 1541–52 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Grobner SN et al. The landscape of genomic alterations across childhood cancers. Nature 555, 321–327 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Ma X et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 555, 371–376 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kulis M et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat Genet 47, 746–56 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Figueroa ME et al. Integrated genetic and epigenetic analysis of childhood acute lymphoblastic leukemia. J Clin Invest 123, 3099–111 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nordlund J et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol 14, r105 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mullighan CG et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471, 235–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mar BG et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat Commun 5, 3469 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milani L et al. DNA methylation for subtype classification and prediction of treatment outcome in patients with childhood acute lymphoblastic leukemia. Blood 115, 1214–25 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Hogan LE et al. Integrated genomic analysis of relapsed childhood acute lymphoblastic leukemia reveals therapeutic strategies. Blood 118, 5218–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee ST et al. Epigenetic remodeling in B-cell acute lymphoblastic leukemia occurs in two tracks and employs embryonic stem cell-like signatures. Nucleic Acids Res 43, 2590–602 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wahlberg P et al. DNA methylome analysis of acute lymphoblastic leukemia cells reveals stochastic de novo DNA methylation in CpG islands. Epigenomics 8, 1367–1387 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Jenkinson G, Pujadas E, Goutsias J & Feinberg AP Potential energy landscapes identify the information-theoretic nature of the epigenome. Nat Genet 49, 719–729 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jenkinson G, Abante J, Feinberg AP & Goutsias J An information-theoretic approach to the modeling and analysis of whole-genome bisulfite sequencing data. BMC Bioinformatics 19, 87 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jenkinson G, Abante J, Koldobskiy MA, Feinberg AP & Goutsias J Ranking genomic features using an information-theoretic measure of epigenetic discordance. BMC Bioinformatics 20, 175 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Landan G et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat Genet 44, 1207–14 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Wang F et al. CellMethy: Identification of a focal concordantly methylated pattern of CpGs revealed wide differences between normal and cancer tissues. Sci Rep 5, 18037 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lilljebjorn H et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun 7, 11790 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet 48, 1481–1489 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsuzuki S, Taguchi O & Seto M Promotion and maintenance of leukemia by ERG. Blood 117, 3858–68 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Meissner A et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–70 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kundaje A et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–30 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharov AA et al. Responsiveness of genes to manipulation of transcription factors in ES cells is associated with histone modifications and tissue specificity. BMC Genomics 12, 102 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McLean CY et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heerema NA et al. Dicentric (9;20)(p11;q11) identified by fluorescence in situ hybridization in four pediatric acute lymphoblastic leukemia patients. Cancer Genet Cytogenet 92, 111–5 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Felice MS et al. Prognostic impact of t(1;19)/ TCF3-PBX1 in childhood acute lymphoblastic leukemia in the context of Berlin-Frankfurt-Munster-based protocols. Leuk Lymphoma 52, 1215–21 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Pui CH, Carroll WL, Meshinchi S & Arceci RJ Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol 29, 551–65 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhojwani D et al. ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: improved outcome with contemporary therapy. Leukemia 26, 265–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paulsson K et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet 47, 672–6 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Greaves M A causal mechanism for childhood acute lymphoblastic leukaemia. Nat Rev Cancer (2018). [DOI] [PMC free article] [PubMed]
- 32.De Braekeleer E et al. Acute lymphoblastic leukemia associated with RCSD1-ABL1 novel fusion gene has a distinct gene expression profile from BCR-ABL1 fusion. Leukemia 27, 1422–4 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Goyama S et al. UBASH3B/Sts-1-CBL axis regulates myeloid proliferation in human preleukemia induced by AML1-ETO. Leukemia 30, 728–39 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wernicke CM et al. MondoA is highly overexpressed in acute lymphoblastic leukemia cells and modulates their metabolism, differentiation and survival. Leuk Res 36, 1185–92 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Zhang R et al. A possible 5’-NRIP1/UHRF1–3’ fusion gene detected by array CGH analysis in a Ph+ ALL patient. Cancer Genet 204, 687–91 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Sidhu H & Capalash N UHRF1: The key regulator of epigenetics and molecular target for cancer therapeutics. Tumour Biol 39, 1–11 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Ashraf W et al. The epigenetic integrator UHRF1: on the road to become a universal biomarker for cancer. Oncotarget 8, 51946–51962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chow M et al. Maintenance and pharmacologic targeting of ROR1 protein levels via UHRF1 in t(1;19) pre-B-ALL. Oncogene 37, 5221–5232 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gibcus JH & Dekker J The hierarchy of the 3D genome. Mol Cell 49, 773–82 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chow ML, Kim D, Kamath S, Peng D & Luu M Use of antiviral medications in drug reaction with eosinophilia and systemic symptoms (DRESS): A case of infantile DRESS. Pediatr Dermatol 35, e114–e116 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Yan F et al. Inhibition effect of siRNA-downregulated UHRF1 on breast cancer growth. Cancer Biother Radiopharm 26, 183–9 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Yan F, Wang X, Shao L, Ge M & Hu X Analysis of UHRF1 expression in human ovarian cancer tissues and its regulation in cancer cell growth. Tumour Biol 36, 8887–93 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Ge TT, Yang M, Chen Z, Lou G & Gu T UHRF1 gene silencing inhibits cell proliferation and promotes cell apoptosis in human cervical squamous cell carcinoma CaSki cells. J Ovarian Res 9, 42 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iacobucci I & Mullighan CG Genetic basis of acute lymphoblastic leukemia. J Clin Oncol 35, 975–983 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lilljebjorn H & Fioretos T New oncogenic subtypes in pediatric B-cell precursor acute lymphoblastic leukemia. Blood 130, 1395–1401 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Reddy KL & Feinberg AP Higher order chromatin organization in cancer. Semin Cancer Biol 23, 109–15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen H & Laird PW Interplay between the cancer genome and epigenome. Cell 153, 38–55 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Timp W et al. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med 6, 61 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duran-Ferrer M et al. The proliferative history shapes the DNA methylome of B-cell tumors and predicts clinical outcome. Nature Cancer 1, 1066–1081 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pujadas E & Feinberg AP Regulated noise in the epigenetic landscape of development and disease. Cell 148, 1123–31 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feinberg AP, Koldobskiy MA & Gondor A Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 17, 284–99 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng SC, Widschwendter M & Teschendorff AE Epigenetic drift, epigenetic clocks and cancer risk. Epigenomics 8, 705–19 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Landau DA et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell 26, 813–25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li S et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med 22, 792–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pan H et al. Epigenomic evolution in diffuse large B-cell lymphomas. Nat Commun 6, 6921 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chan TE, Stumpf MPH & Babtie AC Gene Regulatory Network Inference from Single-Cell Data Using Multivariate Information Measures. Cell Syst 5, 251–267 e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giovinazzo H et al. A high-throughput screen of pharmacologically active compounds for inhibitors of UHRF1 reveals epigenetic activity of anthracycline derivative chemotherapeutic drugs. Oncotarget 10, 3040 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lefebvre JL, Kostadinov D, Chen WV, Maniatis T & Sanes JR Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature 488, 517–21 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.El Hajj N, Dittrich M & Haaf T Epigenetic dysregulation of protocadherins in human disease. Semin Cell Dev Biol 69, 172–182 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Dias S, Mansson R, Gurbuxani S, Sigvardsson M & Kee BL E2A proteins promote development of lymphoid-primed multipotent progenitors. Immunity 29, 217–27 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Semerad CL, Mercer EM, Inlay MA, Weissman IL & Murre C E2A proteins maintain the hematopoietic stem cell pool and promote the maturation of myelolymphoid and myeloerythroid progenitors. Proc Natl Acad Sci U S A 106, 1930–5 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hunger SP et al. The t(1;19)(q23;p13) results in consistent fusion of E2A and PBX1 coding sequences in acute lymphoblastic leukemias. Blood 77, 687–93 (1991). [PubMed] [Google Scholar]
- 63.Inaba T et al. Fusion of the leucine zipper gene HLF to the E2A gene in human acute B-lineage leukemia. Science 257, 531–4 (1992). [DOI] [PubMed] [Google Scholar]
- 64.Wu H, Caffo B, Jaffee HA, Irizarry RA & Feinberg AP Redefining CpG islands using hidden Markov models. Biostatistics 11, 499–514 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ernst J & Kellis M Chromatin-state discovery and genome annotation with ChromHMM. Nat Protoc 12, 2478–2492 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ernst J & Kellis M Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat Biotechnol 33, 364–76 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Press WH, Teukolsky SA, Vetterling WT & Flannery BP Numerical Recipes. The Art of Scientific Computing, (Cambridge University Press, New York, 2007). [Google Scholar]
- 69.Benjamini Y & Yekutieli D The control of the false discovery rate in multiple testing under dependency. Ann Statist 29, 1165–1188 (2001). [Google Scholar]
- 70.Stasinopoulos DM & Rigby RA Generalized additive models for location scale and shape (GAMLSS) in R. Journal of Statistical Software 23, 1–46 (2007). [Google Scholar]
- 71.Kim D, Langmead B & Salzberg SL HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–60 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pertea M et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ritchie ME et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Benjamini Y & Hochberg Y Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57, 289–300 (1995). [Google Scholar]
- 75.Kharchenko PV, Silberstein L & Scadden DT Bayesian approach to single-cell differential expression analysis. Nat Methods 11, 740–2 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guo G et al. Serum-based culture conditions provoke gene expression variability in mouse embryonic stem cells as revealed by single-cell analysis. Cell Rep 14, 956–965 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jiao Y, Widschwendter M & Teschendorff AE A systems-level integrative framework for genome-wide DNA methylation and gene expression data identifies differential gene expression modules under epigenetic control. Bioinformatics 30, 2360–6 (2014). [DOI] [PubMed] [Google Scholar]
- 78.Fisher RA Statistical Methods, Experimental Design, and Statistical Inference, (Oxford University Press, New York, 1990). [Google Scholar]
- 79.Becht E et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol 37, 38–44 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA-methylation and RNA-sequencing data are available at the Gene Expression Omnibus repository, under accession number GSE116229.