

Abstract
Background
The dual-loaded nano-delivery system can realize chemotherapeutic drug and small interfering RNA (siRNA) co-loading as well as enhance the therapeutic effect of drugs on tumors through a synergistic effect, while reducing their toxic and side effects on normal tissues.
Methods
Previously, we developed layered smart nanoparticles (NPs) to co-deliver survivin siRNA as well as small molecule drugs for lung cancer. In this study, we used such smart NPs to co-deliver paclitaxel (PTX) and siRNA against vascular endothelial growth factor (VEGF) gene for breast cancer therapy in mice models. For the prepared NPs, characterizations such as particle size, zeta potential, gel electrophoresis imaging and in vitro stability were investigated. Then, 4T1 cells were used to evaluate the in vitro VEGF silencing capacity, tumor cell inhibitory and anti-apoptotic abilities. Finally, an orthotopic model of mouse breast cancer was established to evaluate the in vivo antitumor effects and safety properties of PTX-siRNAVEGF-NPs.
Results
We prepared PTX-siRNAVEGF-NPs with particle size of 85.25 nm, PDI of 0.261, and zeta potential of 5.25 mV. The NPs with VEGF siRNA effectively knocked down the expression of VEGF mRNA. Cell counting kit-8 (CCK-8) and apoptosis assays revealed that the PTX-siRNAVEGF-NPs exhibited antiproliferation effect of PTX on 4T1 cells. The in vivo anti-tumor study indicated that PTX-siRNAVEGF-NPs could exert an antitumor effect by inhibiting the formation and development of new blood vessels in tumor tissues, thereby cutting off nutrient and blood supplies required for tumor tissue growth. Both the anti-tumor efficacy and in vivo safety of the PTX-siRNAVEGF-NPs group were better than that of the PTX-NPs and siRNAVEGF-NPs groups.
Conclusion
The combination of PTX and VEGF siRNA exerts good antitumor effect on 4T1 tumor cells. This study provides a theoretical and practical basis for breast cancer therapy.
Keywords: paclitaxel, VEGF siRNA, breast cancer, co-delivery
Introduction
Globally, breast cancer is one of the most common cancer type, and the main cause of cancer-associated mortalities in women. As one of the main malignant tumors in women, breast cancer is a serious threat to women’s health.1–4 Chemotherapeutic options for breast cancer include cyclophosphamide, methotrexate, fluorouracil, and other non-anthracycline drugs, anthracyclines, as well as taxanes.5 Paclitaxel (PTX) is a representative taxanes drug. In 1994, the American Food and Drug Administration approved PTX for the treatment of recurrent and metastatic breast cancer while in 2000, it was approved as an adjuvant therapy for early breast cancer surgery.
Nano drug delivery systems have added new vitality to the development of chemotherapeutic drugs in terms of effective drug delivery, improved drug targeting, reduced drug toxicity to normal tissues, and overcoming chemotherapeutic drug resistance. Nanoparticles (NPs), polymer micelles, liposomes, inorganic particles and microemulsion have been used in drug delivery studies.6–13 Danafar et al14,15 prepared anti-tumor nanoparticles using two different pH-responsive materials. They found that the nanoparticles promoted drug release under low pH conditions (pH 5.5), and exhibited an excellent anti-tumor effect on MCF-7 breast cancer cells in vitro. Zheng et al16 confirmed that delivery of PTX by pH-sensitive micelles effectively inhibited primary 4T1 tumor growth in mice and inhibited metastasis to the lungs. The preparation of PTX into smart responsive micelles enhances its cytotoxicity to breast cancer cells, thereby increasing its antitumor effect.17,18
Although chemotherapy is a prominent strategy for effective clinical treatment, its therapeutic efficiency is not satisfactory. This is attributed to non-specific biodistribution-induced therapeutic side effects and limitations of single modal treatment.19 Synergistic effects of small-molecule drugs and genes in cancer treatment have attracted attention because this dual-carrier system can improve chemotherapeutic effects by affecting drug resistance pathways. Small interfering RNA (siRNA) in the dual-carrier system can silence related oncogenes, while chemotherapeutic drugs directly act on tumor cells, thereby exerting a dual inhibitory effect on tumor cell proliferation.20–22 To achieve the best synergistic effects, chemotherapeutic drugs and siRNA must act on the same tumor cells. However, most small molecule drugs are lipophilic, non-specific, and exert toxic side effects on normal tissues.23 Moreover, siRNAs are characterized by a short half-life, poor stability in plasma, and are easy to be degraded by nucleases. In addition, siRNAs are hydrophilic and negatively charged, therefore, they cannot easily penetrate the fat-soluble, negatively charged cell membranes to exert antitumor effects.24 To overcome these limitations, the development of an efficient drug delivery system that can effectively co-load as well as release anticancer drugs and siRNA has become an area of focus in oncology research.
Vascular endothelial growth factor (VEGF) is highly expressed in various solid tumors and is consistent with micro vessel density.25–27 VEGF acts on endothelial cell surfaces in a paracrine or autocrine manner, thereby elevating mitogen levels of endothelial cells, promoting the proliferation, division, and budding growth of vascular endothelial cells and forming new blood vessels.28–30 Tumor formation and growth depend on the blood and oxygen supplied by tumor blood vessels, thereby silencing VEGF can lead to the apoptosis of vascular endothelial cells, while blocking VEGF can directly inhibit tumor growth.25–27 Blood tract metastasis and lymphatic dissemination are the two most important metastatic pathways of breast cancer.31 Elevated expression levels of VEGF in breast cancer indicate a poor prognosis,28 thereby, inhibition of the expression of VEGF in breast cancer tissues is of great significance for effective breast cancer suppression.32
In our previous study, we have developed layered smart NPs self-assembled from amphiphilic copolymer polyethyleneimine-block-polylactic acid (PEI-PLA)/poly (ethylene glycol)-block-poly (L-aspartic acid sodium salt) (PEG-PAsp) and used the prepared NPs to co-deliver survivin siRNA and small molecule drug for lung cancer treatment.33 To investigate the application potential of such nanoparticles in antitumor therapy, this study aimed at using smart NPs to co-load PTX and VEGF-targeted siRNA and to investigate its applicative value in breast cancer therapy. First, the two-block cationic copolymer PEI-PLA was used to achieve the co-delivery of hydrophobic chemotherapeutics and hydrophilic gene drugs. Due to its “proton sponge effect”, PEI promotes endosomal/lysosomal escape, which allows siRNA to migrate into the cytoplasm for gene silencing. A layer of PEG-PAsp anionic polymer was wrapped on the surface of the cationic dual-loaded NPs through electrostatic action to prepare a neutral dual-loaded NPs (Figure 1A). Such neutrally charged particles exhibit the following advantages: i. Circulation in the body is relatively stable, while the outer PEG can increase circulation time in the body and protect the siRNA from degradation by nucleases; ii. Due to enhanced permeability and retention (EPR) effects, nanoscale particles can enter the irregular and highly permeable tumor tissues through the vessel, but not the regularly arranged normal tissue; iii. Since siRNA can specifically silence or inhibit target proteins that are crucial for modulating cellular pathways in cancer, co-loaded polymeric NPs can enhance chemotherapeutic drug effects through the addition of siRNA and inducing a dual inhibitory effect on tumor cell proliferation (Figure 1B).
Figure 1.

(A) PTX/siRNA-loaded layer-by-layer nanoparticle delivery system; (B) schematic of the intracellular therapeutic mechanism of the PTX/siRNA-loaded layer-by-layer nanoparticles.
Experimental
Materials
Poly (lactic acid) with carboxyl groups on one end (PLA-COOH, MW = 1.3 kDa) was supplied by Jinan Daigang Technology Co. (Shandong, China). Branched polyethyleneimine (bPEI1.8k, MW=1.8 kDa) was purchased from Alfa Aesar (Ward Hill, MA, USA). Methoxyl-PEG-PAsp (MW=6.4 kDa) was purchased from Alamanda Polymers (Huntsville, AL, USA). 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS) and SYBR Green qPCR kit were obtained from Sigma-Aldrich (Shanghai, China). Paclitaxel (PTX) was purchased from Meilun Bio Company (Dalian, China). Oregon Green® 488 conjugated paclitaxel (OG-PTX) was purchased from Life Technology (USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was supplied by ZhongmeiTaihe Biotechnology Co. (Beijing, China). VEGF siRNA (5ʹ-CGAUGAAGCCCUGGAGUGCdTdT-3ʹ) and diethyl pyrocarbonate (DEPC) were purchased from GenePharma Co. (Shandong, China). Beta-actin (β-actin) was obtained from Santa (Shanghai, China). Trypsin, RPMI 1640 media and phosphate buffered saline (PBS) were obtained from Thermo Fisher Scientific Co. (Beijing, China). Cell counting kit-8 (CCK-8) was purchased from Dojindo Laboratories (Kumamoto, Japan). The fluorescein isothiocyanate (FITC)-annexin V/propidium iodide (PI) apoptosis detection kit was purchased from KeyGEN Biosciences Company (Nanjing, China). The Western Blot kits were obtained from CoWin Biosciences Co (Beijing, China). The TRIzol reagent was supplied by Invitrogen (Carlsbad, USA). BCA protein concentration determination kit (BCA) was purchased from Tiangen Biological Technology Co. (Beijing, China). All other reagents were of analytical grade and were used without any further purification.
Cell Culture
The 4T1 cells were acquired from the Department of Pathology in the Institute of Medicinal Biotechnology at Peking Union Medical College. A stable luciferase transfected cell line (4T1Luc) was constructed by our laboratory. They were grown in RPMI 1640 media supplemented with 10% fetal bovine serum (FBS) at 37°C in a 5% CO2 atmosphere.
Animals
Female BALB/c nude mice (4–6 weeks old, 18–22 g) were obtained from Vital River Laboratory Animal Technology Co. (Beijing, China). Ethical approval for the use of animals in this study was obtained from the Laboratory Animal Ethics Committee of the Institute of Materia Medica at the Chinese Academy of Medical Sciences (CAMS) and Peking Union Medical College (PUMC). All experimental procedures were performed in conformity with institutional guidelines and protocols for the care and use of laboratory animals.
Synthesis and Characterization of PTX-siRNAVEGF-NPs
Preparation and Evaluation of PTX-siRNAVEGF-NPs
PEI-PLA, a conjugate of PLA-COOH and bPEI1.8k, was synthesized as previously described.33 PEI-PLA (50 mg) and PTX (5 mg) were dissolved in 2 mL of DMSO after which 20 mL of water was added dropwise while stirring. After thorough stirring, the mixture was placed in a dialysis bag with a molecular weight of 7000 Da and dialyzed for 24 h. Then, the mixture was filtered through a 0.45 µM filter membrane and freeze-dried to obtain PTX NPs. Diethyl pyrocarbonate (DEPC) water was used to dissolve the VEGF siRNA dry powder (20 pmol/µL). With an N/P ratio (molar ratio of PEI-PLA nitrogen to siRNA phosphorus) of 30, the prepared PTX-NPs aqueous solution and 20 pmol/µL VEGF siRNA solution were mixed in equal volumes, vortexed for 10 s, and incubated at room temperature for 20 min to obtain composite NPs. Finally, based on the C/N ratio (molar ratio of carboxyl groups of PEG-PAsp to amino groups of PEI-PLA) of 1/5, PEG-PAsp was dissolved in PBS buffer, added to the NPs, mixed well, and incubated at room temperature for 20 min to obtain the final product of co-loaded PTX and siRNA (PTX-siRNAVEGF-NPs, N/P = 30, C/N = 1/5). The PTX-siRNAVEGF-NPs were dissolved in PBS buffer, diluted 10 times, after which particle size distributions and zeta potentials of the composite NPs were determined using a laser particle size analyzer. Using the same method, single-loaded VEGF siRNA NPs (siRNAVEGF-NPs) and single-loaded PTX NPs (PTX-NPs) were prepared.
To determine drug loading (DL) and encapsulation efficiencies (EE), the concentration of PTX was evaluated using an Agilent 1200 LC (Agilent Tech, USA) HPLC system with a Diamonsil C18 column (5 µM, 4.6×250 mm). The mobile phase consisted of a mixture of methyl alcohol, water, and acetonitrile (23:41:36, v/v) delivered at a flow rate of 1.0 mL/min. The injection volume was 20 µL, while the wavelength was set at 227 nm. Each experiment was performed in triplicate, and the mean values±SD calculated using the following formulas:
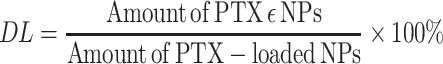 |
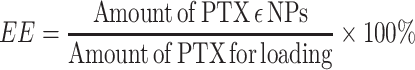 |
Gel Retardation Assay
25 µL of the final product of co-loaded PTX and siRNA (PTX-siRNAVEGF-NPs, N/P = 30, C/N = 1/5) was mixed with an appropriate amount of loading buffer, 10 µL per well was loaded on a 4% agarose gel. Then, 1 × Tris Acetate-EDTA buffer solution (TAE buffer solution) was used as the medium, and electrophoresis was run at 120 mV for 20 min. The sample was stained with 0.5 µg/mL EtBr solution for 30 min and the results were observed using a gel imager.
Plasma Stability of PTX-siRNAVEGF-NPs
100 µL of the final product of PTX-siRNAVEGF-NPs (N/P = 30, C/N = 1/5; PTX content: 6.04%) was diluted in PBS buffer or PBS buffer supplemented with 10% FBS and incubated at 37°C. After 24 h of incubation, particle size changes of composite NPs were determined at different time periods.
In vitro Evaluation of PTX-siRNAVEGF-NPs
Gene Silencing Efficiency Assay
To investigate the siRNA silencing efficacy on VEGF expression, 4T1 cells were incubated in a six-well plate at a density of 1×104 cells/well. Before the experiment, different formulations of the complex NPs were prepared. After incubation at 37°C and 5% CO2 for 24 h, 2 mL of serum-free 1640 media containing blank NPs (PEI-PLA/PEG-PAsp), siRNAVEGF-NPs, PTX-NPs, and PTX-siRNAVEGF-NPs (N/P = 30, C/N =1/5; PTX content: 6.04%; 100 nM siRNA/well) were added to each well. The cells treated with PBS were used as controls and were incubated for 4 h. To inhibit the cytotoxicity associated with PTX, concentrations of PTX in the NPs were as low as 2 nM. After 4 h of incubation, the medium in each well was replaced with fresh medium supplemented with 10% FBS and incubated for another 20 h. Finally, RNA was extracted using the TRIzol reagent and reverse-transcribed into cDNA. The target gene of VEGF and the internal reference of GAPDH upstream and downstream primers were 0.03 µL each. Fluorescence quantitative analysis was performed using the Real-Time PCR Kit according to the manufacturers' instructions. Pre-denaturation was performed at 95°C for 5 min, 95°C for 15 s, and 65°C for 30 s (fluorescence detection) for a total of 40 cycles.
Inhibitory Effects of PTX-siRNAVEGF-NPs on Cell Proliferation
Before the experiment, different formulations of the complex NPs were prepared. 4T1 cells were cultured in a 96-well plate at a density of 3 × 103/well and incubated at 37°C in a 5% CO2 atmosphere. After 24 h, the old media was aspirated, 200 µL of 1640 media containing blank NPs, siRNA negative control NPs (siRNAN.C-NPs), siRNAVEGF-NPs, PTX, PTX-NPs, PTX-siRNAN.C-NPs, and PTX-siRNAVEGF-NPs (N/P = 30, C/N = 1/5; PTX content: 6.04%; 20 nM siRNA/well) were added to each well, and the cells treated with PBS were used as controls. After 48 h of incubation, 10 μL of the CCK-8 reagent was added to each well and further incubated for 3 h. The absorbance value for each well was measured at 450 nm. Untreated cells with 100% viability were used as controls.
To study the cytotoxicity of PTX, PTX-NPs, PTX-siRNAVEGF and A549 cells were seeded at 5×103 cells/well plate. After overnight incubation, the medium was replaced with fresh medium containing different concentrations of with different concentration of PTX (0.05~5 µg/mL) or siRNA (20 nm per well) for 48 h. After the incubation, 10 μL of CCK-8 reagent was added to each well, cultured for 3 h, and the absorbance value of each well was measured at 450 nm. Untreated cells served as controls with 100% viability.
Apoptotic Effects of PTX-siRNAVEGF-NPs
Before the experiment, different formulations of the complex NPs were prepared. The 4T1 cells were incubated in a 12-well plate at a density of 5×104 cells/well, treated with PBS, blank NPs, siRNAVEGF, PTX, siRNAVEGF-NPs, PTX-NPs, and PTX-siRNAVEGF-NPs (N/P = 30, C/N = 1/5; PTX content: 6.04%; 50 nM siRNA/well). The cell apoptosis assay was performed 48 h after drug administration. Cells were harvested using 0.25% trypsin without EDTA, washed using PBS, and resuspended in 0.5 mL of binding buffer after staining with annexin V-FITC/PI for 15 min. Apoptosis was evaluated using a FACSCalibur flow cytometer.
In vivo Anticancer Efficacy
Establishment of the Breast Cancer Orthotopic Model
The 4T1Luc cells in the logarithmic growth phase were digested using trypsin and centrifuged. The supernatant was discarded, and the cells resuspended in PBS. The fourth mammary fat pad of 4–6-week-old female BALB/c mice was inoculated at a density of 1×105 cells/mouse to establish a 4T1 orthotopic tumor model. Tumor masses were measured using a Vernier caliper. Tumor volumes were calculated using the formula:
V = 0.5 × L × D2
whereby V refers to tumor volume, L is the longest tumor diameter while D is the shortest tumor diameter.
Inhibitory Studies on Breast Cancer
Twenty female BALB/c mice (4–6 weeks old) were used in this study. The orthotopic breast cancer models were established as previously described.34 One week after inoculation, the longest and shortest diameters of mouse tumors were measured using Vernier calipers, after which tumor sizes were calculated. Measurements were done after every 2 days. When tumors grew to 130 mm3, tumor-bearing mice were randomly assigned into five groups (n=4/group). Then, normal saline, blank NPs, siRNAVEGF-NPs, PTX-NPs, and PTX-siRNAVEGF-NPs (N/P = 30, C/N =1/5; PTX content: 6.04%) were administered into each group through the tail vein. Administration was done after every three days for a total of four administrations. PTX was administered at a dose of 7.5 mg/kg while siRNA was administered at a dose of 3 mg/kg. During the experiment, body weights and tumor sizes of the mice were determined. After the final administration, 0.1 mL of Luciferin (10 mg/mL) was intraperitoneally injected, after which the mice were anesthetized with 1–2% isoflurane for 10–15 min to detect tumor bioluminescence. Two weeks after the last administration, blood samples were collected from the orbit and alanine aminotransferase (ALT), aspartate aminotransferase (AST), interferon-α (IFN-α), and Interleukin-6 (IL-6) levels measured to evaluate drug toxicity in mice. Mice were euthanized by cervical dislocation after which tumor tissues were resected and fixed in 4% neutral formaldehyde solution for 72 h and subjected to VEGF mRNA expression, VEGF protein expression, and VEGF immunohistochemical assays.
Statistical Analysis
Data are presented as mean ± standard deviation (SD). Significant differences between two groups were evaluated using the Student’s t-test. Comparisons among multiple groups were performed by one-way analysis of variance (ANOVA) with Bonferroni’s post hoc test.
Results and Discussion
Characterization of PTX-siRNAVEGF-NPs
The PEI-PLA copolymer was synthesized through an amino reaction between the carboxyl group of PLA1.3k-COOH and the amino groups of PEI. The specific synthesis method and structure analysis were as previously described.33 In the 1H-NMR spectrum (Figure 2), the PEI peak appeared at approximately 2.6 ppm.34 In PEI-PLA, a new broad peak appeared at 2.3–3.4. ppm, which was attributed to the presence of methylene protons (-CH2CH2-) in PEI. Signals at δ = 1.20 ppm and δ = 4.08 ppm corresponded to -CH3 and (-CH) protons in the PLA block of PEI-PLA, respectively. Strong absorption appears at 1755 cm−1 in the FTIR of PLA-COOH (Figure S1), which is attributed to the stretching vibration absorption peak (VC =O) of the carboxyl group in PLA-COOH. In PEI-PLA (Figure S2), strong absorption appeared at 3300 cm−1 and 1540–1640 cm−1, of which 3300 cm−1 was attributed to the absorption peak of amino on PEI, and 1540–1640 cm−1 was attributed to the characteristic absorption peak of C=O stretching vibration in the amide bond. These findings indicate that the target molecule had been successfully synthesized.
Figure 2.

1H-NMR spectra of PEI-PLA in D2O.
As shown in Figure 3A and B, PTX-siRNAVEGF-NPs (N/P = 30, C/N = 1/5; PTX content: 6.04%) were found to have a particle size of 85.25 nm, a PDI of 0.261, and a zeta potential of 5.25 mV, which was weakly positively charged. The DL % and EE % of PTX in PTX-siRNAVEGF-NPs were 6.04% and 93.03%, respectively. The gel retardation assay revealed that siRNAVEGF-NPs and PTX-siRNAVEGF-NPs (N/P = 30, C/N = 1/5; PTX content: 6.04%) completely condensed the siRNA (Figure 3C). PTX-siRNAVEGF-NPs (N/P = 30, C/N = 1/5; PTX content: 6.04%) were dissolved in PBS buffer and PBS buffer supplemented with 10% FBS, respectively. Particle sizes of the NPs were measured at 1, 3, 6, 12, and 24 h to investigate their stability. Particle sizes of PTX-siRNAVEGF-NPs showed no significant change within the 6 h period. Moreover, changes in particle sizes did not exceed 8 nm within 24 h (Figure 3D). The stability of NPs in vitro and in vivo is a key factor, which plays a significant role in the efficacy of NPs. NPs with positive surface charge have poor stability, which can easily lead to drug inactivation or degradation. In the outer PEG-PAsp, negatively charged PAsp plays a role of neutralizing surface charge, and PEG plays a role of improving drug stability and protecting nanoparticles. The results of stability experiments also confirm this point. Even so, we will need to conduct long-term stability experiments to investigate the factors that change during the storage and transportation of NPs.
Figure 3.

(A) Particle size; (B) zeta potential of PTX-siRNAVEGF-NPs (N/P = 30, C/N = 1/5; PTX content of 6.04%); (C) gel electrophoresis assay for (a) siRNAVEGF-NPs and (b) PTX-siRNAVEGF-NPs (N/P = 30, C/N = 1/5; PTX content of 6.04%); (D) time-dependent colloidal stability of PTX-siRNAVEGF-NPs in PBS or PBS containing 10% FBS at 37°C.
We prepared the siRNA composite nanoparticles through a complex method and synthesized the PEI-PLA polymer. A series of PEI-PLA/siRNA nanoparticles with different N/P ratios were prepared based on the N/P ratio between the amino group of the PEI-PLA polymer and the phosphorus atom of siRNA. There were positive and negative charge interactions between positively charged cationic polymers and negatively charged cell membranes. The more cations the nanoparticles carry, the easier they are to combine with negatively charged cell membranes and enter cells. However, very strong cations exert significant toxic and side effects on cells; therefore, it is necessary to select the appropriate N/P ratio through screening. On the one hand, it is less toxic to cells, on the other hand, it does not affect cell absorption. Through the dialysis method, we prepared PTX loaded nanoparticles from a series of PEI-PLA polymers with different degrees of substitution. We selected a PEI-PLA polymer with the best degree of substitution, which can not only carry high PTX contents, but can also ensure enough surface positive charge. Given that siRNAs will be loaded on PEI-PLA/PTX nanoparticles in future, the positive charge of the system must be large enough. Moreover, due to the intravenous route of administration, particle sizes should not be too large. To exert chemotherapeutic effects, PTX drug loading should also be taken into account. Based on the above factors, we determined that the best carrier material has an N/P ratio of 30 and a C/N ratio of 1/5.
An ideal drug delivery platform for tumors should enhance drug bioavailability, protect the payload from degradation, and directly target the tumor site.35–38 These properties are particularly relevant for the delivery of payloads (chemotherapeutics or siRNA) that are characterized by high toxicities or poor stabilities in harsh biological environments.39,40 The prepared PTX-siRNAVEGF-NPs exhibited the most suitable size (85.25 nm) and zeta potential (5.25 mV) for passive targeting of the EPR effect. It has been postulated that NPs with a weak positive charge and a size range of less than 100 nm can exert an optimal EPR effect and avoid interactions with negatively charged proteins during circulation in vivo.41–43
In vitro Pharmacodynamic Properties of PTX-siRNAVEGF-NPs
Gene Silencing Efficiency
Using human breast cancer 4T1 cells as the subject, the ability of PTX-siRNAVEGF-NPs to silence mRNA expression levels of VEGF in 4T1 cells were measured. It was found that compared to the untreated control group, there were no significant differences in mRNA expression levels of VEGF between the free siRNA group and the blank NPs group. However, compared to the blank NPs group, mRNA expressions in siRNAVEGF-NPs and PTX-siRNAVEGF-NPs treated cells were significantly suppressed (**p < 0.05). Cells treated with PTX-siRNAVEGF-NPs exhibited the lowest VEGF mRNA expression levels (Figure 4).
Figure 4.

Expression of VEGF mRNA determined by quantitative real-time PCR after 48 h of incubation at the final concentrations of different NPs (N/P = 30, C/N = 1/5, PTX content: 6.04%, 100 nM of siRNA per well). **p < 0.01 compared with the controls (n = 3).
Inhibitory Effects of PTX-siRNAVEGF-NPs on Cell Proliferation
To investigate inhibitory effects of PTX and VEGF siRNA on 4T1 breast cancer cells, final concentrations of siRNA were fixed at 20 nM. Cells treated with PBS, blank NPs, and siRNAN.C-NPs were used as control groups. The CCK-8 assay was performed to determine the inhibitory effects of different NPs. It was found that within a certain concentration range, when NPs interacted with cells for 48 h, compared to the control group, cell proliferation inhibitory rates in PTX, PTX-NPs, and PTX-siRNAVEGF-NPs treated groups were significantly elevated (**p < 0.01; Figure 5A). Among them, PTX-siRNAVEGF-NPs exhibited the highest inhibitory rate. Survival rates for cells treated with siRNAVEGF-NPs and PTX-NPs were 77.26% ± 1.23% and 42.66% ± 0.62%, respectively, while that of cells treated with PTX-siRNAVEGF-NPs was 31.55% ± 0.69%. These findings imply that co-delivery of PTX and VEGF siRNA exerted a high antiproliferative effect on 4T1 breast cancer cells. PTX has a direct killing effect on tumor cells, while VEGF siRNA inhibits the expression by silencing related genes, the direct inhibition effect on tumor cell growth mainly depends on PTX. In the cytotoxicity study, the PTX-containing NPs groups showed significant proliferation inhibition compared to the PTX-free NPs groups. In addition, because VEGF siRNA does not have a strong killing effect on cells, the cell inhibition rate in the PTX-siRNAVEGF-NPs did not significantly improve compared with the PTX-NPS group. However, compared with the blank siRNA-loaded NPS group (PTX-siRNAN.C-NPs), the inhibition rate of VEGF siRNA-loaded NPs group (PTX-siRNAVEGF-NPs) decreased from 39.25% to 31.55%, indicating that PTX and VEGF siRNA have a synergistic effect on the proliferation inhibition of 4T1 breast cancer cells in vitro.
Figure 5.

(A) In vitro cytotoxicity of 4T1 cells treated with different formulations. 4T1 cell viability after treatment with different NPs for 48 h (N/P = 30, C/N = 1/5, PTX content: 6.04%, 20 nM of siRNA per well). **p < 0.01 compared with PBS Control, Blank NPs, and siRNAN.C-NPs. (B) 4T1 cell viability after treatment with siRNA and PTX simultaneously by complex NPs. The concentration of PTX varied from 0.05 to 5 µg/mL, and the concentration of VEGF siRNA was 20 nM. **p < 0.01 compared with PTX. Data are presented as the mean ± SEM of quintuplicate experiments.
To investigate the inhibitory effect of PTX on 4T1 cell survival, 4T1 cells were treated with PTX, PTX-NPs, or PTX-siRNAVEGF-NPs (N/P = 30, C/N = 1/5) containing different concentrations of PTX (0.05–5 µg/mL). Cell survival rates decreased with increasing PTX concentrations in a concentration-dependent manner (Figure 5B). Compared to PTX, survival rates of cells treated with PTX-NPs in the range of 0.25–5 µg/mL were significantly suppressed (**p < 0.01). Cell viabilities in the PTX-siRNAVEGF-NPs treatment group (0.05–5 µg/mL) were significantly suppressed (**p < 0.01) while cell survival rates in the PTX-siRNAVEGF-NPs treatment group at each concentration were significantly low (**p < 0.01) compared to the PTX-NPs treatment group.
Cell Apoptosis Effects of the PTX-siRNAVEGF-NPs
Forty-eight h after transfection, apoptosis rates in the PBS control, blank NPs, and naked siRNAVEGF groups were 33.24%, 55.61%, and 32.57%, respectively. The apoptotic rates for cells treated with PTX, siRNAVEGF-NPs, PTX-NPs, and PTX-siRNAVEGF-NPs were 62.56%, 69.55%, 78.93%, and 85.02%, respectively (Figure 6). Compared to the control group, blank NPs and naked siRNAVEGF treated groups, cells in the siRNAVEGF-NPs, PTX, PTX-NPs, and PTX-siRNAVEGF-NPs treatment groups exhibited an apoptotic phenomenon, with cells in the PTX-siRNAVEGF-NPs treatment group exhibiting the highest apoptotic rate. PTX induces apoptosis in various of tumors, including ovarian cancer, lymphoma, leukemia, lung cancer, bowel cancer, and breast cancer. Mechanistically, PTX binds the 31st and 217–231 amino acid positions of the N-terminal of β-tubulin to microtubule polymerization and to stabilize their structures, thereby preventing their depolymerization into subunits. This affects the polymerization and dynamic balance of depolymerization, which inhibits the formation of spindle filaments, leading to cell cycle blockade in the G2/M phase, and suppressing the growth of rapidly dividing cancer cells.44 Therefore, the PTX-siRNAVEGF-NPs promoted cell apoptosis because PTX could interfere with microtubule polymerization required for cell division. In addition, VEGF siRNA inhibited cell migration and invasion. Therefore, the co-loaded PTX and siRNAVEGF promoted 4T1 breast cancer cell apoptosis.
Figure 6.

Cell apoptosis in 4T1 cells 48 h after treating with various NPs (N/P = 30, C/N = 1/5, PTX content: 6.04%, 50 nM of siRNA per well). The cells were stained with Annexin V-FITC/PI for 15 min.
In vivo Anticancer Efficacy
Effects on Tumor Size and Weights
In Figure 7, the color in the image directly represents the number of photons emitted per unit area and indirectly reflects the number of tumor cells. In the color map, the area with a strong signal intensity is shown in red while the area with a weak signal intensity is shown in blue. In Figure 7A and B, control and blank NPs groups exhibited the strongest luminous intensity, both above 5.0×106 p/s/cm2/sr, followed by siRNAVEGF-NPs with a luminous intensity of 1.0×106 p/s/cm2/sr or above. Luminous intensities for mice in the PTX-NPs treatment group were above 0.8×106 p/s/cm2/sr, while luminous intensities for mice in the PTX-siRNAVEGF-NPs treatment group were approximately 0.4×106 p/s/cm2/sr, significantly lower than that of the other groups. These findings show that siRNAVEGF-NPs, PTX-NPs, and PTX-siRNAVEGF-NPs groups exerted inhibitory effects on tumor growth with tumor inhibitory effects of PTX-NPs and PTX-siRNAVEGF-NPs bing significantly better than those of siRNAVEGF-NPs. However, compared to the control group, the intensity of the PTX-siRNAVEGF-NPs treatment group was significantly enhanced (**p < 0.01).
Figure 7.

(A) In vivo bioluminescence imaging analysis in 4T1Luc tumor-bearing mice after the administration of PTX and VEGF siRNA co-delivering NPs (N/P = 30, C/N = 1/5; PTX content: 6.04%) for bioluminescent images of mice after administration with various formulations (n = 4); (B) quantitative estimation by bioluminescent analysis. *p < 0.05, **p < 0.01 vs saline group (n = 4); (C) the changes in tumor volume due to in vivo antitumor effects of PTX and VEGF siRNA co-delivering NPs (N/P = 30, C/N = 1/5; PTX content: 6.04%) in 4T1Luc tumor-bearing mice (n = 4); (D) the weight of tumors due to in vivo antitumor effects of PTX and VEGF siRNA co-delivering NPs (N/P = 30, C/N = 1/5; PTX content: 6.04%) in 4T1Luc tumor-bearing mice. *p < 0.05, **p < 0.01 vs saline group (n = 4).
Figure 7C shows that tumor sizes of the control, blank NPs, and siRNAVEGF-NPs treatment groups increase from 148.88 ± 63.25, 105.32 ± 50.47, and 123.45 ± 39.32 mm3 to 390.02 ± 89.18, 366.80 ± 103.04, and 239.37 ± 55.75 mm3, respectively. Tumor sizes for mice in the PTX-NPs treatment group decreased from 143.73 ± 32.76 to 131.07 ± 43.55 mm3. PTX-siRNAVEGF-NPs exhibited the best tumor suppressive effects, growing from 134.40 ± 33.36 to 99.65 ± 23.45 mm3. Compared to the control group, average tumor weights in the PTX-NPs treatment group decreased significantly (*p < 0.05) as did tumor weights in the PTX-siRNAVEGF-NPs treatment group (**p < 0.01) (Figure 7D). These findings were comparable to those reported by Mu et al45, who evaluated combined in vivo anti-tumor effects of DOX and PD-L1 siRNA co-loaded with multifunctional NPs. Compared to PBS treated groups, free DOX and DOX-loaded NPs moderately inhibited tumor growth. However, the anti-tumor efficacy was significantly enhanced by simultaneous delivery of DOX and PD-L1. This was attributed to the combined effects of tumor chemotherapy and gene therapy. In this study, we found that PTX in NPs is toxic to cancer cells, which inhibits their proliferation, while the function of VEGF siRNA in NPs is to silence gene expression in tumor tissues. Therefore, anti-tumor effects of co-loaded NPs are more obvious than those of single-loaded NPs. This is attributed to the synergistic antitumor effects.
Expression Levels of the VEGF Gene in Tumor Tissues
Figure 8 shows that compared to the control group (2.37 ± 0.20) and the blank NPs group (1.84 ± 0.21), expression levels of VEGF in the PTX-NPs group (1.62 ± 0.22) were significantly suppressed (*p < 0.05). Relative mRNA expression levels of VEGF n the siRNAVEGF-NPs (0.96 ± 0.10) and PTX-siRNAVEGF-NPs groups (0.87 ± 0.05) were significantly suppressed (**p < 0.01). Among them, the PTX-siRNAVEGF-NPs treatment group exhibited the lowest VEGF mRNA expression levels.
Figure 8.

Tumor levels of VEGF mRNA after systemic administration of PTX and VEGF siRNA co-delivery NPs (N/P = 30, C/N = 1/5; PTX content: 6.04%) in 4T1Luc tumor-bearing mice. *p < 0.05, **p < 0.01 vs. Control (n = 4).
Tumor tissues for mice in each group were subjected to immunohistochemical staining to quantitatively determine the expression levels of VEGF. It was revealed that expression levels of VEGF in the siRNAVEGF-NPs, PTX-NPs, and PTX-siRNAVEGF-NPs treatment groups were significantly suppressed than those of the control group (Figure 9 and Table 1; **p < 0.01). Among them, the PTX-siRNAVEGF-NPs treatment group exhibited the lowest VEGF expression levels, consistent with the obtained mRNA expression levels of VEGF in various treatment groups. Therefore, PTX-siRNAVEGF-NPs double-loaded with PTX and VEGF siRNA inhibited tumor cell growth as well as proliferation and mediated specific silencing of VEGF, thereby improving the antitumor efficacy.
Figure 9.

Immunohistochemistry images of representative tumor tissues after staining with VEGF antibody, × 200 (n = 4).
Table 1.
Quantitative Analysis of VEGF Expression in the Tumor Tissues of Different Groups (X ± SD, n = 4)
| Group | Integrated Optical Density (IOD) | Mean Opital Density (MOD) |
|---|---|---|
| Control | 63901.54 ± 5949.08 | 0.0522 ± 0.0097 |
| Blank NPs | 60488.63 ± 4558.26 | 0.0472 ± 0.0087 |
| siRNAVEGF-NPs | 36550.28 ± 6736.87** | 0.0354 ± 0.0079** |
| PTX-NPs | 46294.26 ± 4605.82** | 0.0477 ± 0.0027** |
| PTX-siRNAVEGF-NPs | 34112.55 ± 4569.25** | 0.0284 ± 0.0050** |
Note: **p<0.01 vs control.
As a broad-spectrum antitumor drug, PTX in PTX-siRNAVEGF-NPs has a direct inhibitory effect on the proliferation of 4T1 breast cancer cells in mice. VEGF siRNA inhibits angiogenesis in tumor tissues, thereby cutting off nutrient and blood supplies required for tumor tissue growth. Tumor endothelial cells are more sensitive to chemotherapy when they divide; therefore, continuous low-dose chemotherapy and targeted VEGF therapy exerts a double inhibitory effect on tumor vascular growth.46,47
Safety Evaluation
During the treatment period, body weights of mice were slightly affected (Figure 10). Compared to the control group, body weights of the mice in the PTX-NPs and PTX-siRNAVEGF-NPs treatment groups decreased, however, this decrease was small and not significant. As a cytotoxic antitumor drug, PTX is more toxic than other drugs; however, its toxicity is significantly reduced by NPs. Elevated expression levels of ALT and AST indicated that liver function was impaired. When liver cells are affected by toxic substances, they undergo degeneration, necrosis, or poisoning, thereby releasing transaminases into blood and increasing serum transaminase levels.48 We found that serum ALT and AST levels in the PTX-siRNAVEGF-NPs treatment group did not increase but were decreased (*p < 0.05; Figure 11A and B). This was attributed to liver damage after tumor cell inoculation into mice, causing an increase in transaminase levels. However, after treatment with PTX-siRNAVEGF-NPs, they recovered and liver injury was relieved, implying that the NPs did not induce cytotoxicity in mice. IFN-α has antiviral, antitumor, and immunomodulatory effects.49,50 IL-6 can stimulate the proliferation, differentiation, and function of cells involved in immune responses.51 When the body is traumatized or cells are damaged, circulation IFN-α and IL-6 levels decrease and so does immune function. However, levels of IFN-α and IL-6 did not change among the groups in this experiment (Figure 11C and D). Based on the above changes in body weights of mice during the administration period and alterations in serum ALT, AST, IFN-α, and IL-6 levels after administration, neither the drug delivery vehicle nor the drug-loaded NPs exert toxic effects.
Figure 10.

The changes of body weight of in vivo anti-tumor effect of systemic administration of PTX and VEGF siRNA co-delivery NPs (N/P = 30, C/N = 1/5; PTX content: 6.04%) in 4T1Luc tumor-bearing mice. (n = 4).
Figure 11.

(A) Mouse ALT levels in the serum of the tumor-bearing mice at the end time point of the animal experiment were determined by ELISA (n = 4). Data are provided as mean ± SD. *p < 0.05 vs saline; (B) mouse AST levels in the serum of the tumor-bearing mice at the end time point of the animal experiment were determined by ELISA (n = 4). Data are provided as mean ± SD. *p < 0.05 vs saline; (C) mouse IFN-α levels in the serum of the tumor-bearing mice at the end time point of the animal experiment were determined by ELISA (n = 4). Data are provided as mean ± SD. *p < 0.05 vs saline; (D) mouse IL-6 levels in the serum of the tumor-bearing mice at the end time point of the animal experiment were determined by ELISA (n = 4). Data are provided as mean ± SD. *p < 0.05 vs saline.
Even though we partially evaluated the safety of PTX-siRNAVEGF-NPs, a systematic toxicological evaluation of its safety and pharmacokinetic experiments are still needed. We evaluated the synergistic therapeutic effects of siRNA/PTX-NPs on breast cancer in vivo and in vitro. Their antitumor effects should also be determined on other tumor types such as lung cancer and gastrointestinal tumors.
Conclusions
We successfully prepared PTX-siRNAVEGF-NPs and evaluated their physicochemical properties. The composite NPs had a moderate particle size of 85.25 nm, zeta potential of 5.25 mV, and good plasma stability. In vitro pharmacodynamic evaluation showed that PTX-siRNAVEGF-NPs effectively silenced the expression levels of VEGF mRNA in 4T1 cells, inhibited cell growth, and promoted apoptosis. The 4T1 breast cancer mouse orthotopic model was used to evaluate the anti-tumor efficacy of PTX-siRNAVEGF-NPs in vivo. The results showed that the anti-tumor efficacy of PTX-siRNAVEGF-NPs group was better than that of PTX-NPs and siRNAVEGF-NPs group. The possible reason was that PTX in PTX-siRNAVEGF-NPs directly inhibited tumors on 4T1 breast cancer mice, while VEGF siRNA cut off the nutrition and blood supply required for tumor tissue growth by reducing the expression of VEGF in tumor tissues, thus exhibiting a common anti-tumor effect. Therefore, when co-loaded with small molecule drugs and siRNA, the PEI-PLA/PEG-PAsp drug delivery system is a safe and efficient nanodrug delivery system.
Acknowledgments
This work was supported by Beijing Key Laboratory of Drug Delivery Technology and Novel Formulations, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College.
Funding Statement
This work was financially supported by National Natural Science Foundation of China (No. 81373342), the Drug Innovation Major Project (2018ZX09711001-002-005, China) and CAMS Innovation Fund for Medical Sciences (2019-I2M-1-005, China).
Disclosure
The authors declare no conflicts of interest.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 2.Tsang JYS, Tse GM. Molecular classification of breast cancer. Adv Anat Pathol. 2020;27(1):27–35. doi: 10.1097/PAP.0000000000000232 [DOI] [PubMed] [Google Scholar]
- 3.Viale G. The current state of breast cancer classification. Ann Oncol. 2012;23(Suppl 10):x207–210. doi: 10.1093/annonc/mds326 [DOI] [PubMed] [Google Scholar]
- 4.Januskeviciene I, Petrikaite V. Heterogeneity of breast cancer: the importance of interaction between different tumor cell populations. Life Sci. 2019;239:117009. doi: 10.1016/j.lfs.2019.117009 [DOI] [PubMed] [Google Scholar]
- 5.Seidman AD, Tiersten A, Hudis C, et al. Phase II trial of paclitaxel by 3-hour infusion as initial and salvage chemotherapy for metastatic breast cancer. J Clin Oncol. 1995;13(10):2575–2581. doi: 10.1200/JCO.1995.13.10.2575 [DOI] [PubMed] [Google Scholar]
- 6.Unsoy G, Gunduz U. Smart drug delivery systems in cancer therapy. Curr Drug Targets. 2018;19(3):202–212. doi: 10.2174/1389450117666160401124624 [DOI] [PubMed] [Google Scholar]
- 7.Gao W, Zhang Y, Zhang Q, Zhang L. Nanoparticle-hydrogel: a hybrid biomaterial system for localized drug delivery. Ann Biomed Eng. 2016;44(6):2049–2061. doi: 10.1007/s10439-016-1583-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.George A, Shah PA, Shrivastav PS. Natural biodegradable polymers based nano-formulations for drug delivery: a review. Int J Pharm. 2019;561:244–264. [DOI] [PubMed] [Google Scholar]
- 9.Roviello G, Conter FU, Mini E, et al. Nanoparticle albumin-bound paclitaxel: a big nano for the treatment of gastric cancer. Cancer Chemother Pharmacol. 2019;84(4):669–677. doi: 10.1007/s00280-019-03887-2 [DOI] [PubMed] [Google Scholar]
- 10.Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. [DOI] [PubMed] [Google Scholar]
- 11.Cavallaro G, Lazzara G, Fakhrullin R. Mesoporous inorganic nanoscale particles for drug adsorption and controlled release. Ther Deliv. 2018;9(4):287–301. doi: 10.4155/tde-2017-0120 [DOI] [PubMed] [Google Scholar]
- 12.De Vita A, Liverani C, Molinaro R, et al. Lysyl oxidase engineered lipid nanovesicles for the treatment of triple negative breast cancer. Sci Rep. 2021;11(1):5107. doi: 10.1038/s41598-021-84492-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gharbavi M, Fath S, Oruji H, Pakzad HKM. Formulation and biocompatibility of microemulsion-based PMBN as an efficient system for paclitaxel delivery. J Appl Biotechnol Rep. 2021;8(1):51–62. [Google Scholar]
- 14.Danafar H, Asadi F, Sharafi A, Manjili HK. Preparation and evaluation of pH sensitive novel anticancer drug carrier based on magnetic chitosan quartets. Drug Res (Stuttg). 2019;69(9):496–504. doi: 10.1055/a-0855-8464 [DOI] [PubMed] [Google Scholar]
- 15.Nosrati H, Adibtabar M, Sharafi A, Danafar H, Hamidreza Kheiri M. PAMAM-modified citric acid-coated magnetic nanoparticles as pH sensitive biocompatible carrier against human breast cancer cells. Drug Dev Ind Pharm. 2018;44(8):1377–1384. doi: 10.1080/03639045.2018.1451881 [DOI] [PubMed] [Google Scholar]
- 16.Zheng Z, Lang T, Huang X, et al. Calcitriol-loaded dual-ph-sensitive micelle counteracts pro-metastasis effect of paclitaxel in triple-negative breast cancer therapy. Adv Healthc Mater. 2020;9(12):e2000392. doi: 10.1002/adhm.202000392 [DOI] [PubMed] [Google Scholar]
- 17.Rehan F, Ahemad N, Islam RA, Gupta M, Gan SH, Chowdhury EH. Optimization and formulation of nanostructured and self-assembled caseinate micelles for enhanced cytotoxic effects of paclitaxel on breast cancer cells. Pharmaceutics. 2020;12(10):984. doi: 10.3390/pharmaceutics12100984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cai Y, Xu Z, Shuai Q, et al. Tumor-targeting peptide functionalized PEG-PLA micelles for efficient drug delivery. Biomater Sci. 2020;8(8):2274–2282. doi: 10.1039/C9BM02036E [DOI] [PubMed] [Google Scholar]
- 19.Liu J, Liu K, Zhang L, et al. Heat/pH-boosted release of 5-fluorouracil and albumin-bound paclitaxel from Cu-doped layered double hydroxide nanomedicine for synergistical chemo-photo-therapy of breast cancer. J Control Release. 2021;335:49–58. doi: 10.1016/j.jconrel.2021.05.011 [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Thambi T, Lee DS. Co-delivery of drugs and genes using polymeric nanoparticles for synergistic cancer therapeutic effects. Adv Healthcare Mater. 2018;7(1):1700886. doi: 10.1002/adhm.201700886 [DOI] [PubMed] [Google Scholar]
- 21.Sun H, Yarovoy I, Capeling M, Cheng C. Polymers in the co-delivery of siRNA and anticancer drugs for the treatment of drug-resistant cancers. Top Curr Chem (Cham). 2017;375(2):24. doi: 10.1007/s41061-017-0113-z [DOI] [PubMed] [Google Scholar]
- 22.Aji Alex MR, Nehate C, Veeranarayanan S, Kumar DS, Kulshreshtha R, Koul V. Self assembled dual responsive micelles stabilized with protein for co-delivery of drug and siRNA in cancer therapy. Biomaterials. 2017;133:94–106. doi: 10.1016/j.biomaterials.2017.04.022 [DOI] [PubMed] [Google Scholar]
- 23.Norouzi M, Nazari B, Miller DW. Injectable hydrogel-based drug delivery systems for local cancer therapy. Drug Discov Today. 2016;21(11):1835–1849. doi: 10.1016/j.drudis.2016.07.006 [DOI] [PubMed] [Google Scholar]
- 24.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8(2):129–138. doi: 10.1038/nrd2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307. doi: 10.1038/nature10144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leite de Oliveira R, Hamm A, Mazzone M. Growing tumor vessels: more than one way to skin a cat - implications for angiogenesis targeted cancer therapies. Mol Aspects Med. 2011;32(2):71–87. doi: 10.1016/j.mam.2011.04.001 [DOI] [PubMed] [Google Scholar]
- 27.Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8(8):579–591. doi: 10.1038/nrc2403 [DOI] [PubMed] [Google Scholar]
- 28.Melincovici CS, Bosca AB, Susman S, et al. Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom J Morphol Embryol. 2018;59(2):455–467. [PubMed] [Google Scholar]
- 29.Caporarello N, Lupo G, Olivieri M, et al. Classical VEGF, Notch and Ang signalling in cancer angiogenesis, alternative approaches and future directions (Review). Mol Med Rep. 2017;16(4):4393–4402. doi: 10.3892/mmr.2017.7179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thurston G, Kitajewski J. VEGF and Delta-Notch: interacting signalling pathways in tumour angiogenesis. Br J Cancer. 2008;99(8):1204–1209. doi: 10.1038/sj.bjc.6604484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Linderholm B, Tavelin B, Grankvist K, Henriksson R. Vascular endothelial growth factor is of high prognostic value in node-negative breast carcinoma. J Clin Oncol. 1998;16(9):3121–3128. doi: 10.1200/JCO.1998.16.9.3121 [DOI] [PubMed] [Google Scholar]
- 32.Zhang YQ, Chen WL, Zhang F, et al. Over-expression of both VEGF-C and Twist predicts poor prognosis in human breast cancer. Clin Transl Oncol. 2019;21(9):1250–1259. doi: 10.1007/s12094-019-02051-9 [DOI] [PubMed] [Google Scholar]
- 33.Jin M, Jin G, Kang L, Chen L, Gao Z, Huang W. Smart polymeric nanoparticles with pH-responsive and PEG-detachable properties for co-delivering paclitaxel and survivin siRNA to enhance antitumor outcomes. Int J Nanomedi. 2018;13:2405–2426. doi: 10.2147/IJN.S161426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ding X, Wang W, Wang Y, et al. Versatile reticular polyethylenimine derivative-mediated targeted drug and gene codelivery for tumor therapy. Mol Pharm. 2014;11(10):3307–3321. doi: 10.1021/mp5001263 [DOI] [PubMed] [Google Scholar]
- 35.Molinaro R, Martinez JO, Zinger A, et al. Leukocyte-mimicking nanovesicles for effective doxorubicin delivery to treat breast cancer and melanoma. Biomater Sci. 2020;8(1):333–341. doi: 10.1039/C9BM01766F [DOI] [PubMed] [Google Scholar]
- 36.Svenson S. Clinical translation of nanomedicines. Curr Opin Solid State Mater Sci. 2012;16(6):287–294. doi: 10.1016/j.cossms.2012.10.001 [DOI] [Google Scholar]
- 37.Zinger A, Koren L, Adir O, et al. Collagenase nanoparticles enhance the penetration of drugs into pancreatic tumors. ACS Nano. 2019;13(10):11008–11021. doi: 10.1021/acsnano.9b02395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. doi: 10.1038/nrd2614 [DOI] [PubMed] [Google Scholar]
- 39.Varkouhi AK, Scholte M, Storm G, Haisma HJ. Endosomal escape pathways for delivery of biologicals. J Control Release. 2011;151(3):220–228. doi: 10.1016/j.jconrel.2010.11.004 [DOI] [PubMed] [Google Scholar]
- 40.Lammers T, Hennink WE, Storm G. Tumour-targeted nanomedicines: principles and practice. Br J Cancer. 2008;99(3):392–397. doi: 10.1038/sj.bjc.6604483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao N, Cheng D, Zou S, Ai H, Gao J, Shuai X. The synergistic effect of hierarchical assemblies of siRNA and chemotherapeutic drugs co-delivered into hepatic cancer cells. Biomaterials. 2011;32(8):2222–2232. doi: 10.1016/j.biomaterials.2010.11.061 [DOI] [PubMed] [Google Scholar]
- 42.Ishida O, Maruyama K, Sasaki K, Iwatsuru M. Size-dependent extravasation and interstitial localization of polyethyleneglycol liposomes in solid tumor-bearing mice. Int J Pharm. 1999;190(1):49–56. doi: 10.1016/S0378-5173(99)00256-2 [DOI] [PubMed] [Google Scholar]
- 43.Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9(8):615–627. doi: 10.1038/nrd2591 [DOI] [PubMed] [Google Scholar]
- 44.Xie J, Wang CH. Self-assembled biodegradable nanoparticles developed by direct dialysis for the delivery of paclitaxel. Pharm Res. 2005;22(12):2079–2090. doi: 10.1007/s11095-005-7782-y [DOI] [PubMed] [Google Scholar]
- 45.Mu X, Zhang M, Wei A, et al. Doxorubicin and PD-L1 siRNA co-delivery with stem cell membrane-coated polydopamine nanoparticles for the targeted chemoimmunotherapy of PCa bone metastases. Nanoscale. 2021;13(19):8998–9008. doi: 10.1039/D0NR08024A [DOI] [PubMed] [Google Scholar]
- 46.Browder T, Butterfield CE, Kraling BM, et al. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60(7):1878–1886. [PubMed] [Google Scholar]
- 47.Kerbel RS, Kamen BA. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer. 2004;4(6):423–436. doi: 10.1038/nrc1369 [DOI] [PubMed] [Google Scholar]
- 48.Cao Z, Fang Y, Lu Y, et al. Melatonin alleviates cadmium-induced liver injury by inhibiting the TXNIP-NLRP3 inflammasome. J Pineal Res. 2017;62(3):e12389. doi: 10.1111/jpi.12389. [DOI] [PubMed] [Google Scholar]
- 49.Gibbert K, Schlaak JF, Yang D, Dittmer U. IFN-alpha subtypes: distinct biological activities in anti-viral therapy. Br J Pharmacol. 2013;168(5):1048–1058. doi: 10.1111/bph.12010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Medrano RFV, Hunger A, Mendonca SA, Barbuto JAM, Strauss BE. Immunomodulatory and antitumor effects of type I interferons and their application in cancer therapy. Oncotarget. 2017;8(41):71249–71284. doi: 10.18632/oncotarget.19531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin Sci (Lond). 2012;122(4):143–159. doi: 10.1042/CS20110340 [DOI] [PubMed] [Google Scholar]