

Abstract
Mutations in a number of genes related to chromosomal segregation reportedly cause developmental disorders, e.g., chromosome alignment-maintaining phosphoprotein 1 (CHAMP1). We report on an 8-year-old Japanese girl who presented with a developmental disorder and microcephaly and carries a novel nonsense mutation in CHAMP1. Therefore, CHAMP1 mutation should be considered as a differential diagnosis of global developmental delay and microcephaly.
Subject terms: Neurodevelopmental disorders, Genetics of the nervous system
Chromosome alignment-maintaining phosphoprotein 1 (CHAMP1) is a zinc finger protein that functions in kinetochore-microtubule attachment and regulates chromosome segregation. Pathogenic variants of CHAMP1 have been reported in patients with intellectual disability and other signs, such as microcephaly, muscular hypotonia, facial dysmorphism, and eye anomalies.1–3 Here, we report a novel nonsense mutation in CHAMP1, NM_001164144.2:c.1465C>T, p.(Gln489*).
The patient was an 8-year-old Japanese girl born to nonconsanguineous parents without neurological abnormalities, such as intellectual disability. She was delivered at full term by emergency Cesarean section because of umbilical cord entanglement without newborn asphyxia. Her birth weight was 2755 g (−0.47 standard deviation [SD]), her length was 46.8 cm (−1.5 SD), and her occipital frontal circumference was 31.8 cm (−0.9 SD). Her physical examination at birth was normal. She could control her head at 4 months of age and sit stably at 10 months. At 10 months of age, she developed acute encephalopathy after infection with respiratory syncytial virus and was treated with steroid pulse therapy in another hospital. She recovered without sequelae. However, since her recovery, developmental delay with acquired microcephaly (−2.4 SD) has become evident. Head magnetic resonance imaging was performed when she was 2 years old, and the scan showed mild atrophy of the cerebrum and cerebellum. She could walk independently at 3.5 years of age.
At 4 years of age, she came to our hospital because of global intellectual disability and acquired microcephaly. At the time of the visit, she had severe intellectual disability and could speak no meaningful words. A physical examination revealed no abnormalities. She was very friendly and displayed no dysmorphic facial features. A neurological examination showed normal muscle tone and tendon reflexes. No pyramidal tract signs, dystonia, or involuntary movements were observed. Her intelligence quotient according to a Tanaka-Binet test was 35. As the acute encephalopathy caused no brain magnetic resonance imaging abnormalities or sequelae, we searched for the cause of her intellectual disability and microcephalus. We initially suspected Angelman syndrome; however, a fluorescence in situ hybridization analysis failed to show a microdeletion at 15q11.2. After receiving informed consent, we performed whole-exome sequencing as previously described.4 We identified a novel de novo nonsense heterozygous variant, c.1465C>T, p.(Gln489*), in CHAMP1 (Fig. 1).
Fig. 1. Diagram of CHAMP1 variants.
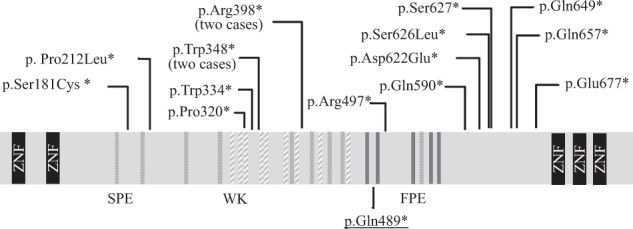
Diagram of previously reported CHAMP1 variants (upper panel) and the mutation identified in this case (lower panel, underlined). CHAMP1 consists of five zinc-finger domains (ZNF) and several motifs; SPE (consensus: PxxSPExxK; dots), WK (SPxxWKxxP; diagonal lines), and FPE (FPExxK; grey bar).
In addition to CHAMP1,1–3 a number of mutations in genes related to chromosome alignment and/or spindle function, including Pogo transposable element-derived with ZNF domain (POGZ), tubulin gamma 1, dynein cytoplasmic 1 heavy chain 1, kinesin family member 5C (KIF5C), KIF2A, KIF4A, and centromere protein E, have been shown to cause various developmental disorders.5–9 CHAMP1 contains several repeat motifs, i.e., SPE, WK, and FPE motifs, and regulates kinetochore-microtubule attachment and chromosome alignment (Fig. 1). These motifs reportedly play an important role in spindle localization and kinetochore-microtubule interactions.10 The C-terminal region of CHAMP1, which contains the zinc-finger domain, has been predicted to be crucial for its proper localization to chromosomes and for mitotic spindle function.1 Functional studies using various deletion mutants of CHAMP1 have shown that lack of the C-terminal region of CHAMP1 prevents proper chromosomal localization.10 Similar to the CHAMP1 mutation in this case, all reported mutations are truncating mutations, i.e., nonsense or frameshift mutations that cause the loss of the C-terminal zinc-finger domain (Table 1). Although the biological mechanism that links CHAMP1 mutations and intellectual disability is unknown, the pathogenic mechanism appears to be a result of the loss of the C-terminal region of CHAMP1. POGZ binds to the C-terminal region of CHAMP1 and is critical for proper chromosome segregation.3,11 Therefore, loss of the C-terminus may impair CHAMP1 chromosomal localization or/and its binding to other proteins, such as POGZ. Mitotic defects in neural progenitor cells may cause a decrease in the number of neural cells and defective neural development, resulting in intellectual disability.3
Table 1.
CHAMP1 variants and clinical presentation.
| Patients | Sex | Mutation | Muscle hypotonia | Microcephaly | Delay in walking (>18m) | Impaired speech development | Eye anomalies | Brain MRI | Seizure | Abnormal behavior | Dysmorphic facial features | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | c.542_543delCT p.Ser181* | − | + | + | + | Strabismus | N/A | Febrile seizures | Skin-picking, rituals, food-foraging | N/A | Tanaka et al.2 |
| 2 | F | c.635delC p. Pro212* | + | + | − | + | Hyperopia, astigmatism | Normal | − | Friendly, hand stereotypy, tacle hypersensitivity, sexual self-stimulation | + | Hempel et al.1 |
| 3 | F | c.958_959delCC, p.Pro320* | N/A | − | + | + | N/A | non specific patchy signal abnormalities in right parieto-occipital subcortical white matter, bulky corpus callosum autistic behavior | − | + | + | Isdor et al.3 |
| 4 | M | c.1002G>A p.Trp334* | N/A | − | + | + | Hyperopia, strabismus | Normal | − | - | − | Isdor et al.3 |
| 5 | F | c.1043G>A, p.Trp348* | N/A | + | + | + | Hyperopia, astigmatism | thickening of the corpus callosum, subtle hypoplasia of the left temporal lobe | − | + | + | Isdor et al.3 |
| 6 | F | c.1044delG p.Trp348* | + | + | − | + | − | Hypoplastic corpus callosum | − | Aggressive, occasionally self injurious | N/A | Tanaka et al.2 |
| 7 | M | c.1192C>T p.Arg398* | + | + | + | + | Strabismus, hyperopia | Normal | − | Friendly | + | Hempel et al.1 |
| 8 | F | c.1192C>T p.Arg398* | + | − | − | + | Hyperopia, astigmatism | Normal | − | Friendly | + | Hempel et al.1 |
| 9 | F | c.1465C>T, p.Gln489* | − | + | + | + | − | mild atrophy of the cerebrum and cerebellum | − | Friendly | − | This case |
| 10 | F | c.1489C>T, p.Arg497* | N/A | − | + | + | Amblyopia | N/A | N/A | − | + | Isdor et al.3 |
| 11 | M | c.1768C>T p.Gln590* | + | + | + | + | Impaired | delayed myelination | Fromtotemporal epilepsy | Hand stereotypy, friendly | + | Hempel et al.1 |
| 12 | M | c.1866_1867delCA p.Asp622* | + | + | + | + | Strabismus, hyperopia | Mild brain atrophy and cerebellar cortical dysplasia | − | Hand stereotypy, friendly | + | Hempel et al.1 |
| 13 | F | c.1876_1877delAG; p.Ser626* | N/A | + | + | + | Hyperopia, astigmatism | arachnoid cyst | + | + | + | Isdor et al.3 |
| 14 | M | c.1880C>G p.Ser627* | N/A | + | + | + | Hyperopia, astigmatism, strabismus | Normal | − | − | + | Isdor et al.3 |
| 15 | F | c.1945C>T p.Gln649* | + | − | + | + | Ocuular albinism | Normal | − | ADD/ADHD | N/A | Tanaka et al.2 |
| 16 | F | c.1969C>T p.Gln657* | + | + | + | + | Strabismus | Mild decreased white matter | Seizure at 3 yo | Inapropriate laughter | N/A | Tanaka et al.2 |
| 17 | F | c.2029G>T p.Glu677* | + | + | + | + | Strabismus | Mild cerebellar atrophy | − | Hyperactivity | N/A | Tanaka et al.2 |
+ present, − absent, N/A not available, ADD/ADHD attention deficit disorder/attention deficit hyperactivity disorder.
The clinical presentation of CHAMP1 mutation includes intellectual disability (17/17), motor development delay (14/17), facial dysmorphism (10/12), eye anomalies (14/16), and microcephaly (12/17). For some individuals, anomalies are evident on magnetic resonance imaging scans of the brain (9/15) and abnormal behavior varies from mild to severe (14/17) (Table 1).1–3 Some patients present with a feeding disorder. Considering these symptoms, Angelman syndrome and Prader-Willi syndrome are the most important differential diagnoses.1
In conclusion, we report a novel nonsense mutation in CHAMP1, c.1465C>T, p.(Gln489*). The patient had intellectual disability, motor development delay, and microcephaly but no facial dysmorphism or eye anomalies. CHAMP1 mutation should be considered when a patient presents with global developmental delay and microcephaly.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at 10.6084/m9.figshare.hgv.3081.
Acknowledgements
This work was supported in part by the Japan Agency for Medical Research and Development (AMED) under Grant numbers JP20ek0109486, JP21ek0109549, JP21cm0106503, and JP21ek0109493 (N.M); the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant number JP20K08164 (T.M.); and the Takeda Science Foundation (T.M.).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hempel M, et al. De novo mutations in CHAMP1 cause intellectual disability with severe speech impairment. Am. J. Hum. Genet. 2015;97:493–500. doi: 10.1016/j.ajhg.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tanaka AJ, et al. De novo pathogenic variants in CHAMP1 are associated with global developmental delay, intellectual disability, and dysmorphic facial features. Cold Spring Harb. Mol. Case Stud. 2016;2:1–8. doi: 10.1101/mcs.a000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Isidor B, et al. De novo truncating mutations in the kinetochore-microtubules attachment gene CHAMP1 cause syndromic intellectual disability. Hum. Mutat. 2016;37:354–358. doi: 10.1002/humu.22952. [DOI] [PubMed] [Google Scholar]
- 4.Aoi H, et al. Whole exome sequencing of fetal structural anomalies detected by ultrasonography. J. Hum. Genet. 2021;66:499–507. doi: 10.1038/s10038-020-00869-8. [DOI] [PubMed] [Google Scholar]
- 5.Fitzgerald TW, et al. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–228. doi: 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poirier K, et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 2013;45:639–647. doi: 10.1038/ng.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Willemsen MH, et al. Involvement of the kinesin family members KIF4A and KIF5C in intellectual disability and synaptic function. J. Med. Genet. 2014;51:487–494. doi: 10.1136/jmedgenet-2013-102182. [DOI] [PubMed] [Google Scholar]
- 8.Mirzaa GM, et al. Mutations in CENPE define a novel kinetochore-centromeric mechanism for microcephalic primordial dwarfism. Hum. Genet. 2014;133:1023–1039. doi: 10.1007/s00439-014-1443-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanks S, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genet. 2004;36:1159–1161. doi: 10.1038/ng1449. [DOI] [PubMed] [Google Scholar]
- 10.Itoh G, et al. CAMP1 (C13orf8, ZNF828) is a novel regulator of kinetochore-microtubule attachment. EMBO J. 2011;30:130–144. doi: 10.1038/emboj.2010.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nozawa R, et al. Human POGZ modulates dissociation of HP1α from mitotic chromosome arms through Aurora B activation. Nat. Cell Biol. 2010;12:719–727. doi: 10.1038/ncb2075. [DOI] [PubMed] [Google Scholar]