

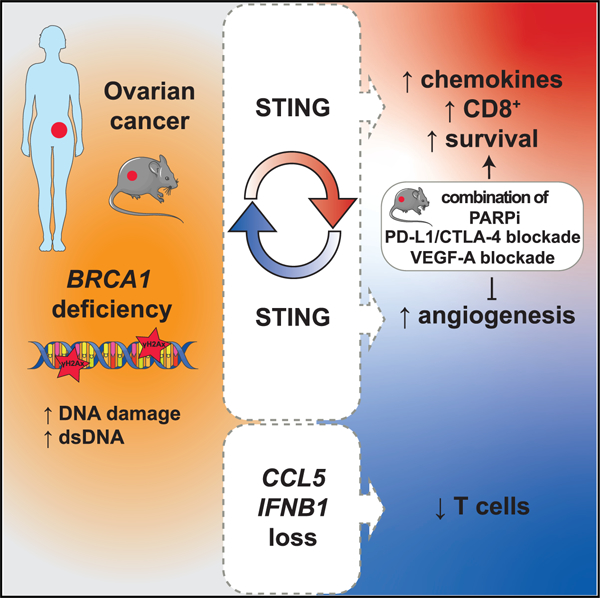
SUMMARY
In this study, we investigate mechanisms leading to inflammation and immunoreactivity in ovarian tumors with homologous recombination deficiency (HRD). BRCA1 loss is found to lead to transcriptional reprogramming in tumor cells and cell-intrinsic inflammation involving type I interferon (IFN) and stimulator of IFN genes (STING). BRCA1-mutated (BRCA1mut) tumors are thus T cell inflamed at baseline. Genetic deletion or methylation of DNA-sensing/IFN genes or CCL5 chemokine is identified as a potential mechanism to attenuate T cell inflammation. Alternatively, in BRCA1mut cancers retaining inflammation, STING upregulates VEGF-A, mediating immune resistance and tumor progression. Tumor-intrinsic STING elimination reduces neoangiogenesis, increases CD8+ T cell infiltration, and reverts therapeutic resistance to dual immune checkpoint blockade (ICB). VEGF-A blockade phenocopies genetic STING loss and synergizes with ICB and/or poly(ADP-ribose) polymerase (PARP) inhibitors to control the outgrowth of Trp53−/−Brca1−/− but not Brca1+/+ ovarian tumors in vivo, offering rational combinatorial therapies for HRD cancers.
Graphical abstract
In brief
Bruand et al. provide insights into the dual role of STING in promoting tumor-intrinsic mechanisms of both immunoreactivity, driven by DNA sensing and type I IFN, and also VEGF-A-driven immune resistance in BRCA1mut ovarian cancers. STING elimination reduces neoangiogenesis, increases CD8+ T cell infiltration, and reverts therapeutic resistance to dual ICB.
INTRODUCTION
A positive correlation between the presence of intraepithelial tumor-infiltrating lymphocytes (ieTILs) and survival has been reported in ovarian cancer (OC) (Goode et al., 2017; Zhang et al., 2003) and other tumors (Gooden et al., 2011). Most high-grade serous OCs (HGSOCs) with homologous recombination deficiency (HRD) carry BRCA1 or BRCA2 mutations (Cancer Genome Atlas Research Network, 2011; Konstantinopoulos et al., 2015). These display chromosomal instability and copy number alterations (CNAs) (Macintyre et al., 2018; Wang et al., 2017). BRCA1 and BRCA2 are implicated in error-free repair of double-strand DNA (dsDNA) breaks by HR repair, as well as non-homologous end joining and base-excision repair mechanisms (Konstantinopoulos et al., 2015). BRCA-mutated (BRCAmut) HGSOCs exhibit immunoreactive gene signatures (George et al., 2013) and increased ieCD8+ TILs (McAlpine et al., 2012; Strickland et al., 2016). Intriguingly, BRCAmut OCs have an increased microvascular density and VEGF-A expression (Ruscito et al., 2018), which is known to mediate tumor immune escape (Buckanovich et al., 2008; Motz and Coukos, 2011).
Chromatin instability, a common hallmark of tumors, causes release of dsDNA from ruptured ectopic micronuclei into the cytoplasm, leading to activation of DNA-sensing cyclic guanosine monophosphate-AMP synthase (cGAS) and stimulator of interferon genes (STING), and, only in some cancers, downstream activation of the interferon (IFN) response (Harding et al., 2017; Härtlova et al., 2015). The generation of micronuclei in senescent cells activates STING and nuclear factor κB (NF-κB), but not IFN, likely through p38 mitogen-activated protein kinase (MAPK) activation (Dou et al., 2017). Metastatic tumor cells may cope with cGAS/STING signaling by preferential activation of NF-κB and not IFNs, which promotes metastasis (Abe and Barber, 2014; Bakhoum et al., 2018). Indeed, STING activation may drive carcinogenesis through inflammation (Ahn et al., 2014).
Opposing evidence shows that chromatin instability from BRCA loss is consistently associated with activation of the IFN response. Indeed, knockdown of BRCA2 in vitro induced micronuclei and activation of the IFN response via cGAS/STING (Heijink et al., 2019; Reisländer et al., 2019). Similarly, HRD breast cancer cells exhibited cytoplasmic DNA (cytDNA) and activation of cGAS/STING and IFN, with downstream activation of chemokines CCL5 and CXCL9–11 (Parkes et al., 2016). This pathway is exploited by poly(ADP-ribose) polymerase inhibitors (PARPis), which exacerbate accumulation of cytDNA, activation of cGAS/STING, and immune reactivity (Chabanon et al., 2019; Pantelidou et al., 2019).
The mechanisms leading to TIL infiltration in tumors are under intense investigation since those with pre-existing TILs are more likely to respond to immune checkpoint blockade (ICB) therapy (Thommen et al., 2018; Tumeh et al., 2014). Given the extraordinary tumor cell plasticity, it appears paradoxical that cancers can afford to carry an immunoreactive phenotype, suggesting the implication of important mechanisms that mediate resistance to immune rejection. In this study, we asked how cytDNA sensing drives IFN activation in BRCA1mut ovarian tumors, unlike in other cancer types with chromatin instability, and investigated the mechanisms of resistance to T cell-mediated rejection. Our study provides insights into the pleiotropic roles of STING in promoting tumor-intrinsic mechanisms of both immunoreactivity and immune resistance.
RESULTS
BRCA1 loss leads to cell-autonomous inflammatory activation through dsDNA sensing and transcriptional reprogramming
To investigate the tumor-intrinsic effects of BRCA1 loss in HGSOCs, we analyzed the UWB1.289 OC cell line, along with its isogenic counterpart with forced expression of BRCA1 wild-type (BRCA1WT) (DelloRusso et al., 2007) (Figure 1A). By RNA sequencing (RNA-seq) and mass spectrometry (MS) shotgun proteomics we detected important differences, revealing activation of key inflammatory pathways in BRCA1mut cells, which were effectively suppressed by forced expression of BRCA1WT (Figures 1B, 1C, S1A, and S1B; Tables S1 and S2). We saw mixed inflammatory activation involving the NF-κB and canonical type I IFN pathways. Numerous IFN-inducible factors were upregulated both at the mRNA and protein levels in BRCA1mut relative to BRCA1WT cells (Figures 1B and 1C; Tables S1 and S2). We confirmed overexpression of IFNB1 and IFN-inducible genes and increased secretion of related inflammatory mediators (e.g., IFNα and CXCL10) (Figures S1C and S1D). Consistent with other tumors (Dou et al., 2017; Bakhoum et al., 2018), we observed upregulation of NFKB1, nuclear translocation of NF-kB, and increased secretion of tumor necrosis factor (TNF)-α, but also interleukin (IL)-1α, IL-6, CXC3L1, and granulocyte-macrophage colony-stimulating factor (GM-CSF) in BRCA1mut cells (Figures 1B, S1D, and S1E).
Figure 1. BRCA1 loss leads to transcriptional reprogramming of ovarian cancer cells and results in overexpression of the DS/IFN pathway.
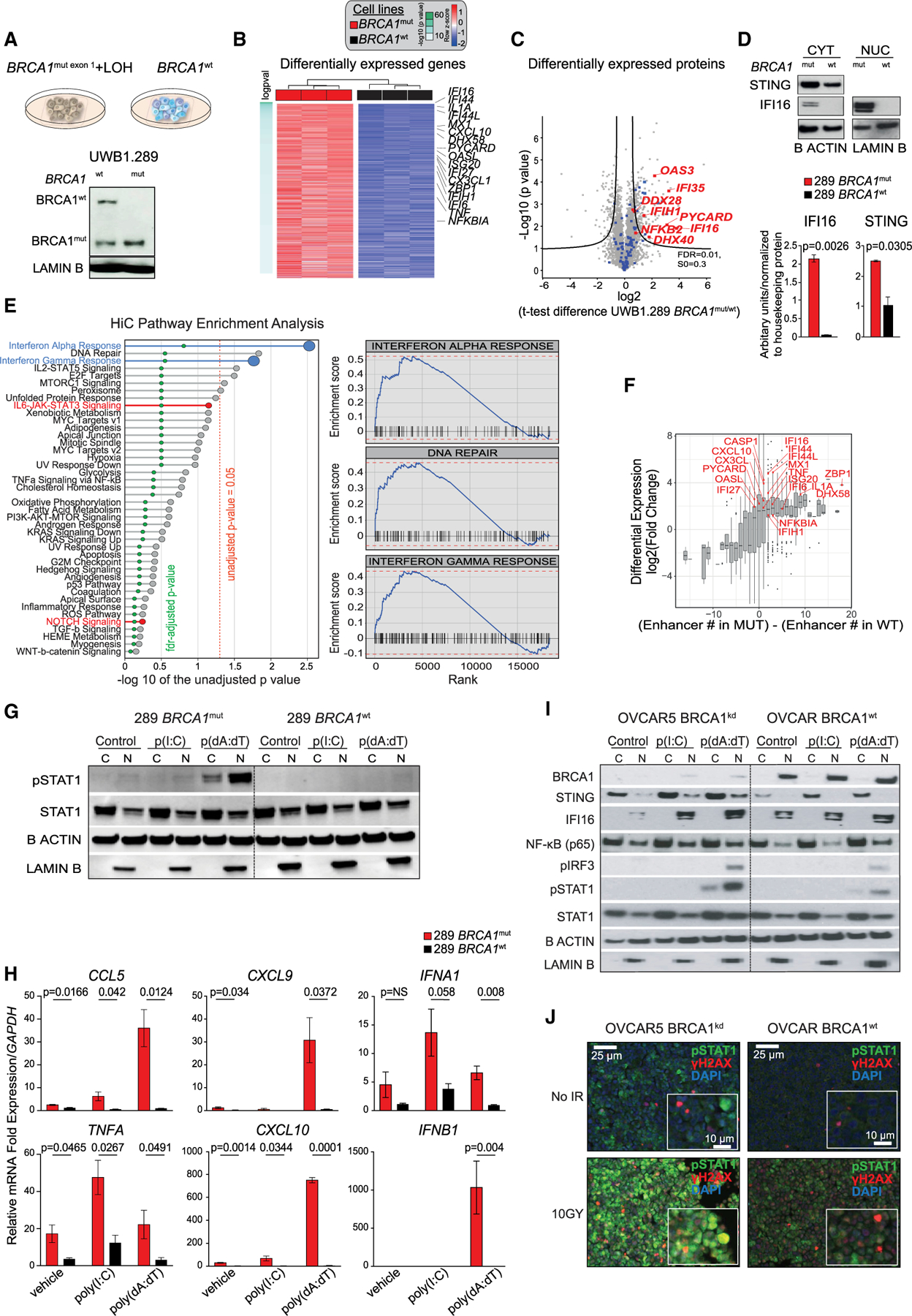
(A) (Upper) UWB1.289 cell lines: UWB1.289 BRCA1mut with concomitant loss of wild-type (WT) allele through LOH and WT BRCA1-reconstituted isogenic cell line. (Lower) Western blot (WB) analysis of BRCA1 in nuclear (N) extracts of BRCA1mut and BRCA1WT cells.
(B) Heatmap of hallmark signatures with significantly different enrichment score between BRCA1mut and BRCA1WT isogenic cell lines (adjusted p value of <0.05 after linear regression) at the proteomics level.
(C) Volcano plot of differentially expressed proteins in the BRCA1mut and BRCA1WT cell lines by MS. Immune-related proteins are highlighted in blue; selected genes of interest are highlighted in red. The position on the right side of the plot indicates higher expression in the BRCA1mut cell line. Black curves represent significance cutoff (t test permutation-based false discovery rate [FDR] < 0.01, S0 = 0.3).
(D) WB analysis for STING and IFI16 in cytoplasmic (CYT) and nuclear (NUC) extracts of BRCA1mut and BRCA1WT cells (n = 3). The signal obtained for each protein was normalized to that of housekeeping β-actin and lamin B in CYT and NUC, respectively. Data are presented as mean ± SEM. p values were calculated by an unpaired t test.
(E) Pre-ranked gene set enrichment analysis using the difference in the number of enhancers between BRCA1mut and BRCA1WT cell lines as a ranking factor. Pathways enriched in BRCA1WT are in red. All of the others were found in the BRCA1mut. Right panels display the three most enriched pathways in BRCA1mut.
(F) Association of differentially expressed genes and the presence of enhancers in the BRCA1mut versus the BRCA1WT cells by RNA-seq, Hi-C, and H3K27ac ChIP-seq. Genes in red are those implicated in the DS/IFN pathway.
(G) WB analysis for pSTAT1 and total STAT1 in the cytoplasmic (C) and nuclear (N) fractions of BRCA1mut and BRCA1WT cells treated with liposomes or poly(I:C)- or poly(dA:dT)-loaded liposomes. β-Actin and lamin B were used as protein loading controls in C and N, respectively.
(H) RT-PCR analysis of CCL5, CXCL9, IFNA1, TNFA, CXCL10, and IFNB1 in BRCA1mut and BRCA1WT cells treated with liposomes or poly(I:C)- or poly(dA:dT)-loaded liposomes (n = 3). Data are presented as mean ± SEM. p values were calculated by an unpaired t test.
(I)WB analysis of BRCA1, STING, IFI16, NF-κB, pIRF3, pSTAT1, and total STAT1 in the C and N fractions of OVCAR5 BRCA1kd and BRCA1WT cells treated with liposomes or poly(I:C)- or poly(dA:dT)-loaded liposomes.
(J)Immunofluorescence (IF) analysis of pSTAT1 (green), γH2AX (red), and DAPI (blue) in BRCA1kd and BRCA1WT cells 48 h after irradiation (10 Gy). Scale bars, 25 mm (10 μm in insets).
Inflammatory activation can be mediated by DNA-sensing molecules. BRCA1mut cells upregulated the machinery for cytoplasmic dsDNA sensing (Figures 1B and S1C; Tables S1 and S2). IFI16 was among the most overexpressed mRNAs and proteins in BRCA1mut cells (Figures 1B and 1C; Tables S1A and S1B): it was undetectable in BRCA1WT cells, but was detected both in the nucleus and the cytoplasm in BRCA1mut cells, indicating cytoplasmic translocation (Figure 1D). STING was also overexpressed in the cytoplasm of BRCA1mut cells (Figure 1D).
To further understand the transcriptional reprogramming in BRCA1mut cells, we mapped chromatin structure through Hi-C and surveyed chromatin-wide transcriptional activity through H3K27acchromatin immunoprecipitation sequencing (ChIP-seq). We ran a pre-ranked gene set enrichment analysis (GSEA) using as a metric the difference between BRCA1mut and BRCA1WT cells in the number of enhancers per gene: the IFNα, IFNγ, and DNA-repair pathways were the three best ranking pathways enriched in BRCA1mut cells (Figure 1E). Furthermore, we found that BRCA1mut cells exhibited more active enhancers in key genes of the DNA sensing pathway and downstream inflammatory effectors (e.g., IFI16, DHX58, or ZBP1) compared to BRCA1WT cells. We detected 6 additional enhancer regions for IFI16 and 18 additional enhancers for ZBP1, both of which were overexpressed in BRCA1mut cells compared to BRCA1WT (Figure 1F). Thus, BRCA1 loss of function leads to chromatin reorganization and transcriptional reprogramming, resulting in overexpression of the DNA sensing and inflammatory (DS/IFN) pathways. This chromatin state was reversed by the re-expression of WT BRCA1.
BRCA1mut cells also exhibited hyperresponsiveness to exogenous dsDNA analog poly(dA:dT), which induced phosphorylation of STAT1 and overexpression of type I IFNs and TNFA, as well as IFN-inducible genes and downstream T cell recruiting chemokines (Figures 1G and 1H). Forced BRCA1 expression suppressed the response to poly(dA:dT). Interestingly, poly(I:C), a dsRNA analog, did not elicit this response.
We reproduced these results with the reverse approach, i.e., by knocking down BRCA1 (BRCA1kd) in the OVCAR5 cell line, which harbors BRCA1WT (Stordal et al., 2013). BRCA1kd cells phenocopied BRCA1mut cells, exhibiting increased expression of STING and cytoplasmic translocation of IFI16 as well as cell-autonomous inflammatory activation (Figures S1F–S1H).
BRCA1kd cells, similar to BRCA1mut cells, exhibited hyperresponsiveness to exogenous poly(dA:dT), which increased STING and IFI16 expression, IRF3 and STAT1 phosphorylation, transcription of IFNB1 and downstream MX1 and CCL5, and nuclear translocation of NF-κB (Figures 1I and S1I). In addition, BRCA1kd exhibited hyperresponsiveness to endogenous dsDNA generated by ionizing radiation, which increased STAT1 phosphorylation colocalized with phosphorylated histone H2AX (γH2AX) (Figure 1J). Ionizing radiation also increased CCL5 and MX1 in BRCA1kd cells (Figure S1J). BRCA1kd cells did not respond to dsRNA analog poly(I:C). These events were minimally detected in OVCAR5 cells with intact BRCA1WT expression (Figures 1I and S1I). Taken together, the data prove that BRCA1-deficient OC cells exhibit a cell-autonomous inflamed state, driven by hypersensitivity to dsDNA, due to orchestrated upregulation of the DNA-sensing pathway.
Chromatin dsDNA fragments engage TBK1 in the cytoplasm of BRCA1-deficient cells
We looked for ectopic dsDNA in the cytoplasm of OC cells by anti-dsDNA-specific antibody and DAPI staining (Figure 2A). We detected significantly more cytoplasmic DNA dots and/or micronuclei in UWB1.289 BRCA1mut or OVCAR5 BRCA1kd cells relative to BRCA1WT cells (Figures 2B, 2C, and S2A–S2D). Consistent with chromatin origin, γH2AX colocalized in the cytoplasm with DNA dots at baseline. Further confirming that cytoplasmic dsDNA dots originate from chromatin instability, the PARPi olaparib significantly increased ectopic dsDNA in BRCA1-deficient but not in BRCA1-proficient cells (Figures 2B, 2C, and S2A–S2D). Olaparib significantly increased gH2AX+ DNA dots selectively in BRCA1-deficient cell lines (Figures 2D and S2E–S2G).
Figure 2. BRCA1 loss in ovarian cancer cells leads to a cell-autonomous inflammatory state through tumor-cell-intrinsic STING/pTBK1 pathway activation, which is exacerbated by PARP inhibition.
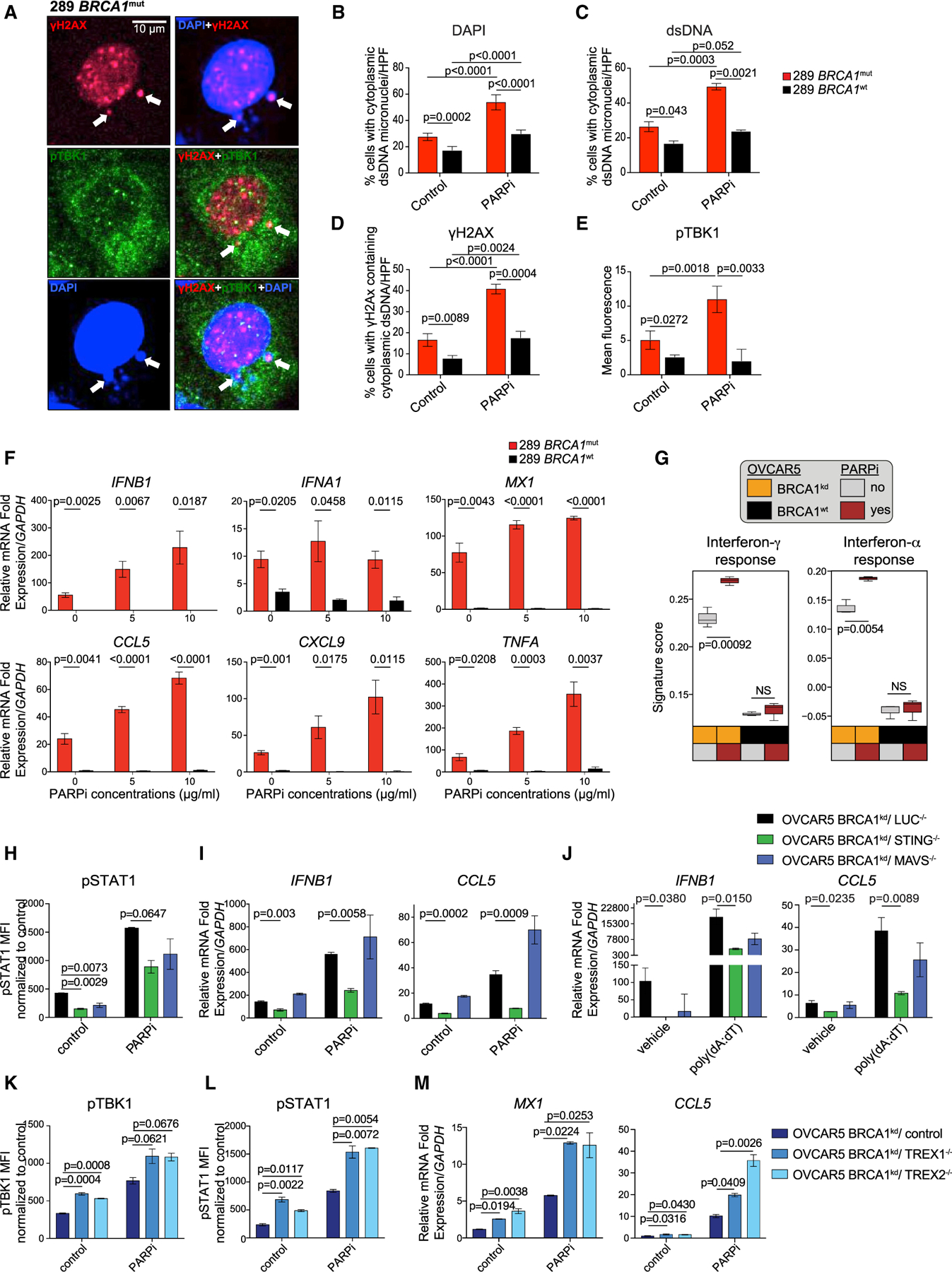
(A) High magnification of a cell with histone-derived cytoplasmic dsDNA micronuclei stained by anti-phospho-histone H2AX (Ser139) (red) and DAPI (blue), and cytosolic expression of pTBK1 (green) in BRCA1mut cells (white arrow). Scale bar, 10 μm.
(B and C) Percentage of cells/high-power field (HPF) carrying cytoplasmic dsDNA micronuclei as detected by DAPI (B) and anti-dsDNA-specific antibody (C) in BRCA1mut and BRCA1WT cell lines treated with DMSO or olaparib (n = 4). Data are presented as mean ± SEM. p values were calculated by an unpaired t test.
(D) Percentage of cells/HPF carrying histone-derived cytoplasmic γH2AX+ dsDNA in BRCA1mut and BRCA1WT cells treated with DMSO or olaparib (n = 3). Data are presented as mean ± SEM. p values were calculated by an unpaired t test.
(E) Mean fluorescence intensity (MFI) of pTBK1 staining in BRCA1mut and BRCA1WT cells treated with DMSO or olaparib (n = 3). Data are presented as mean ± SEM. p values were calculated by an unpaired t test.
(F)RT-PCR of IFNB1, IFNA1, MX1, CCL5, CXCL9, and TNFA in BRCA1mut and BRCA1WT cells 48 h after DMSO or olaparib (n = 3). Data are presented as mean ± SEM. p values were calculated by an unpaired t test.
(G) MS of IFNγ and IFNα responses in OVCAR5 BRCA1 isogenic cell lines (n = 3) 48 h after DMSO or olaparib. Boxplots represent the 25th and 75th percentiles, with the midline indicating the median; whiskers extend to the lowest/highest values. p values were computed by ANOVA followed by a Tukey’s post hoc test for multiple comparison correction.
(H and I) MFI quantified by FACS for pSTAT1 (H) and RT-PCR analysis of IFNB1 and CCL5 expression (I) in OVCAR5 BRCA1kd cells crispered for luciferase, STING, and MAVS 48 h after DMSO or olaparib (n = 3). Data are presented as mean ± SEM. p values were assessed by a one-way ANOVA.
(J) RT-PCR analysis of IFNB1 and CCL5 expression in OVCAR5 BRCA1kd cells crispered for luciferase, STING, and MAVS treated with liposomes or poly(dA:dT)-liposomes (n = 3). Data are presented as mean ± SEM. p values were assessed by a one-way ANOVA.
(K–M) MFI quantified by FACS for pTBK1 (K) and pSTAT1 (L) and RT-PCR analysis of MX1 and CCL5 expression (M) in OVCAR5 BRCA1kd cells transduced with a control, TREX1, or TREX2 CRISPR 48 h after DMSO or PARPi treatment (n = 3). Data are presented as mean ± SEM. p values were assessed by a one-way ANOVA.
See also Figures S2 and S3.
Providing further evidence that chromatin-derived dsDNA activates cytoplasmic DNA sensors in BRCA1-deficient cells, we detected significantly higher levels of perinuclear and cytoplasmic phosphorylated TANK-binding kinase 1 (pTBK1) in BRCA1-deficient relative to BRCA1WT cells at the steady state (Figures 2E and S3A–S3C). Consistent with activation by chromatin DNA damage, olaparib induced a further increase in perinuclear and cytoplasmic pTBK1, specifically in BRCA1-deficient cells (Figures 2E and S3A–S3C). Importantly, we detected pTBK1 in association with ectonuclear DNA (revealed by DAPI staining) mainly in BRCA1-deficient cells exhibiting cytoplasmic γH2AX, but not in cells exhibiting only nuclear γH2AX (Figures 2A, S3D, and S3E). Thus, chromatin-derived dsDNA fragments generated due to BRCA1 loss, and exacerbated by a PARPi, translocate to the cytoplasm and trigger DNA sensing in BRCA1-deficient cells by engaging TBK1. Confirming the effect of the PARPi, we detected a significant increase in IFN response genes and TNFA in BRCA1-deficient cells treated with the PARPi (Figures 2F and S3F), validated by MS analysis (Figure 2G).
STING and TREX1/2 regulate the DS/IFN response in BRCA1-deficient OC cells
STING is a master mediator of IFN activation by cytoplasmic DNA (Liu et al., 2015). We excised STING using CRISPR-Cas9 in OVCAR5 BRCA1 isogenic cells. Demonstrating dependence of dsDNA sensing in BRCA1-deficient cells on STING, its knockdown significantly reduced baseline pSTAT1 and expression of IFNB1 and CCL5 (Figures 2H, 2I, and S3G). STING ablation also attenuated the IFN response to olaparib and poly(dA:dT) (Figure 2J). Consistent with the specific hyperresponsiveness to dsDNA and not to dsRNA, excision of MAVS (Chiu et al., 2009) had no impact on IFN pathway activation at baseline or its further induction by olaparib in BRCA1-deficient cells (Figures 2H and 2I). Thus, chromatin-derived cytoplasmic dsDNA did not require transcription to ectopic dsRNA to activate the IFN response in BRCA1-deficient cells.
We asked whether deletion of the 3′–5′ DNA exonucleases TREX1 and TREX2, which prevent aberrant nucleic acid sensing and autoimmunity (Cheng et al., 2018), exacerbated the IFN response in BRCA1kd cells. Excision of TREX1 or TREX2 in BRCA1kd cells increased TBK1 and STAT1 phosphorylation, and it triggered MX1 and CCL5 upregulation at baseline and upon olaparib ola (Figures 2K–2M and S3H). Thus, BRCA1 loss drives cell-autonomous inflammatory activation mediated by ectopic chromatin dsDNA and activation of the DNA-sensing pathway, which can be negatively regulated by TREX enzymes (Figure S3I).
The DS/IFN pathway is activated in human BRCA1mut HGSOCs in situ
We sought to document the topologic distribution of DS/IFN activation in HGSOCs in situ. We used multispectral imaging to covisualize within the same cells γH2AX (DNA damage), STING (DNA sensing), and pSTAT1 (IFN activation), first validated on OVCAR5 BRCA1kd cells in vitro (Figure 3A). Comparing 25 treatment-naive HGSOCs with documented germline or somatic BRCA1 mutations and 54 homologous recombination-proficient (HRP) HGSOCs (Pennington et al., 2014), we found prevalent cytokeratin-positive (CK+) cancer cells expressing γH2AX, STING, and/or pSTAT1 in BRCA1mut tumors. Triple-positive CK+ cells were almost uniquely seen in BRCA1mut tumors, and they were rare (<0.1% of cells) in HRP tumors (Figures 3B, 3C, and S4A). More rare stroma cells (CK−) positive for STING and/or pSTAT1 were more frequent in BRCA1mut than in BRCA1WT (Figures 3C and S4A). Thus, tumor-cell-intrinsic STING and IFN activation is prevalent in BRCA1-mutated tumors.
Figure 3. BRCA1-mutated HGSOC tumors are inflamed and have DNA damage and IFN activation in situ.
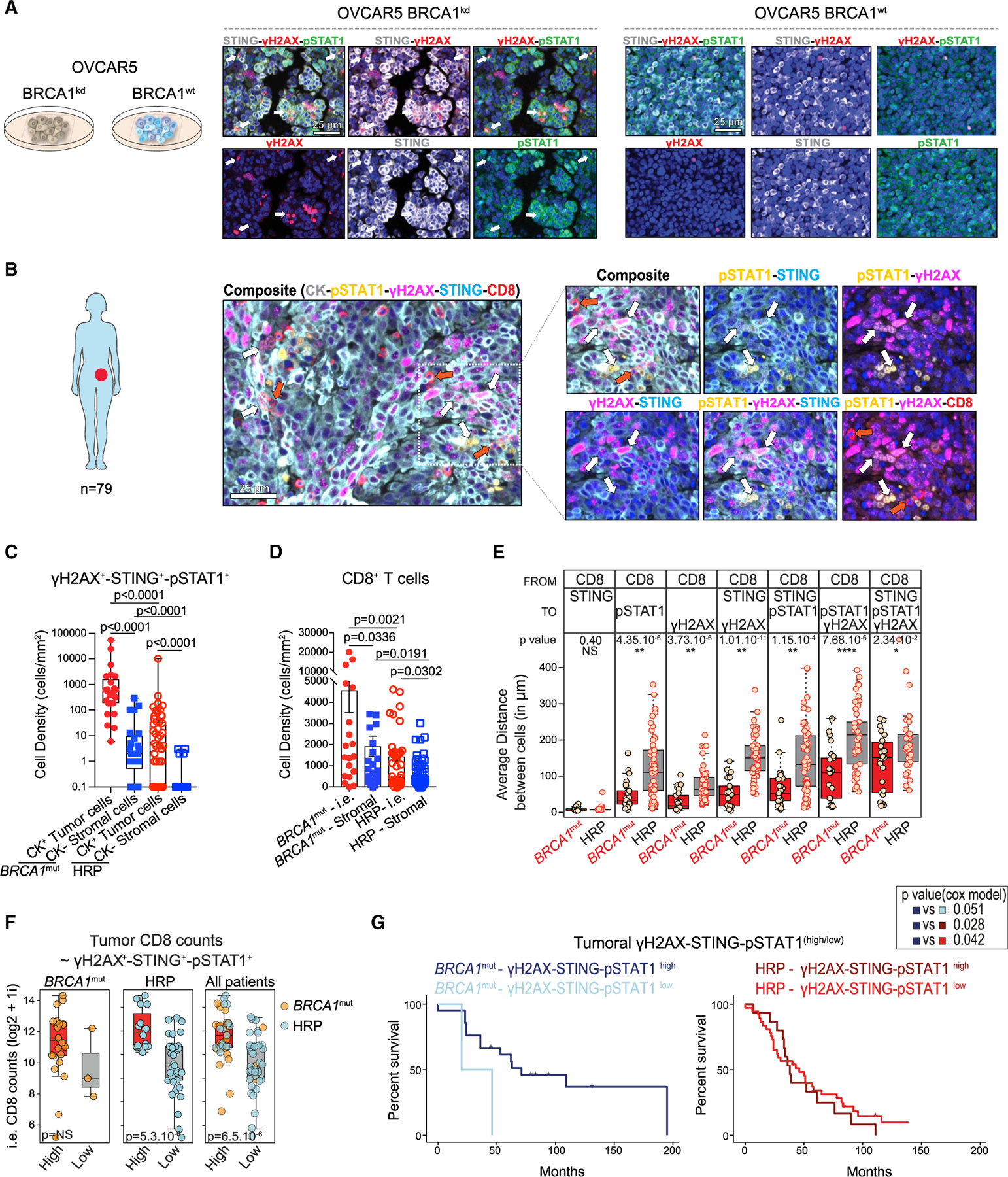
(A) Multiplex IF for STING (gray), pSTAT1 (green), γH2AX (red), and DAPI (blue) in OVCAR5 BRCA1kd and BRCA1WT cells 48 h after irradiation. White arrows show examples of cytoplasmic dsDNA micronuclei labeled by DAPI, γH2AX, and phosphorylated STAT1. Scale bars, 25 μm.
(B) Representative IF image of CK (gray), pSTAT1(yellow), γH2AX (magenta), STING (blue), and CD8 (red) in a case of BRCA1-deficient (BRCA1mut) HGSOC. White arrows indicate CK+γH2AX+STING+pSTAT1+ cells. CD8+ TILs surrounding the γH2AX+STING+pSTAT1+ tumor cells are indicated by red arrows. Scale bar, 25 μm.
(C) Quantification of γH2AX+STING+pSTAT1+ in CK+ and CK− cells in BRCA1mut and HR-proficient (HRP) HGSOCs (n = 25 and 54, respectively). Boxplots represent 25th and 75th percentiles, with the midline indicating the median; whiskers extend to the lowest/highest values, and points indicate values for individual patients. p values were calculated by a Mann-Whitney test.
(D) Quantification of intraepithelial (i.e.) and stromal CD8+ T cells in BRCA1mut and HRP HGSOCs (n = 25, 54, and 5, respectively). Data are presented as mean ± SEM. Each dot represents an individual patient. p values were calculated by a Mann-Whitney test.
(E) Averages of the nearest neighbor distance analysis of CD8+ T cells to STING+, pSTAT1+, and/or γH2AX+ tumor cells in BRCA1mut and HRP HGSOCs. Boxplots represent 25th and 75th percentiles, with the midline indicating the median and each dot representing an individual patient. p values were calculated by a Student’s t tests.
(F) Boxplots showing i.e. CD8+ densities in STING-pSTAT1-γH2AXhigh and STING-pSTAT1-γH2AXlow tumor cells. The triple staining was categorized into high and low groups using the median over the whole cohort. Boxplots represent 25th and 75th percentiles, with the midline indicating the median and each dot representing an individual patient. p values were calculated by Student’s t tests.
(G) Kaplan-Mayer survival analyses of BRCA1mut and HRP HGSOCs expressing high or low levels of tumoral γH2AX, STING, and pSTAT1. p values were extracted from Cox proportional hazards tests.
See also Figure S4.
We sought to understand whether the cell-intrinsic inflammatory state of BRCA1mut tumors in situ provides grounds for increased recruitment of CD8+ T cells. We found a significantly higher frequency of CD8+ T cells in tumor islets than stroma in BRCA1mut tumors (Figures 3D and S4B). Nearest neighbor cell-distance analysis showed that CD8+ T cells were at the highest proximity of tumor cells expressing STING (Figure 3E). Such proximity was noted mostly in BRCA1mut tumors with γH2AX, STING, or pSTAT1 rather than in HRP tumors, which in general also exhibited significantly lower frequencies of ieCD8+ T cells. Among tumors classified as HRP, those with the highest frequency of triple-positive CK+ cells exhibited also the highest frequency of ieCD8+ T cells (Figure 3F).
We observed significant positive correlations between ieCD8+ TILs and pSTAT1+STING+ tumor or stromal cells in patients with BRCA1mut HGSOC, but not in HRP patients (Figure S4C). Importantly, patients with BRCA1mut carcinomas displaying a high frequency of γH2AX+STING+pSTAT1+CK+ cells also exhibited significantly longer survival compared to HRP patients or to BRCA1mut patients with low frequency of these cells (Figure 3G). The frequency of STING+pSTAT1+ stromal cells did not contribute to survival in BRCA1mut patients (Figure S4D). Thus, BRCA1mut HGSOCs display cell-intrinsic DNA sensing and cell-autonomous inflammation in situ, associated with increased immunoreactivity.
HRD HGSOCs exhibit a range of DNA damage, IFN activation, and T cell inflammation
We sought to understand how HRD ovarian carcinomas cope with T cell inflammation. We noticed that the degree of inflammation varied markedly among tumors (Figures 3C, 3D, and S4A). We extended our observations to include 52 treatment-naive OCs with HRD caused by BRCA1 (n = 26) or BRCA2 germline or somatic mutations (n = 17), or BRCA1 (n = 7) or RAD51C (n = 2) methylation, and 49 HRP tumors (i.e., no genetic HRD detected by BROCA; Pennington et al., 2014). We observed high heterogeneity in DNA damage (γH2AX expression) among HRD tumors, with a fraction displaying minimal γH2AX (Figures 4A–4C and S4E), possibly evidence of restored DNA repair (Domchek, 2017; Ray Chaudhuri et al., 2016). Carcinomas displaying increased levels of pSTAT1 exhibited higher γH2AX expression (Figure 4E). However, among γH2AXhi HRD tumors we found marked heterogeneity of pSTAT1 expression (Figure 4F), indicating that HRD tumors with active DNA damage can show variable inflammation. HRD tumors with the highest expression of γH2AX and pSTAT1 harbored more ieCD8+ TILs (Figure 4G). Tumors with higher expression of γH2AX or pSTAT1 exhibited significantly longer survival (Figures 4H and 4I), as did tumors with HRD or higher ieCD8+ TILs (Figures S4F–S4H).
Figure 4. BRCA-deficient HGSOCs exhibit variable degrees of DNA damage, IFN activation, and T cell inflammation.
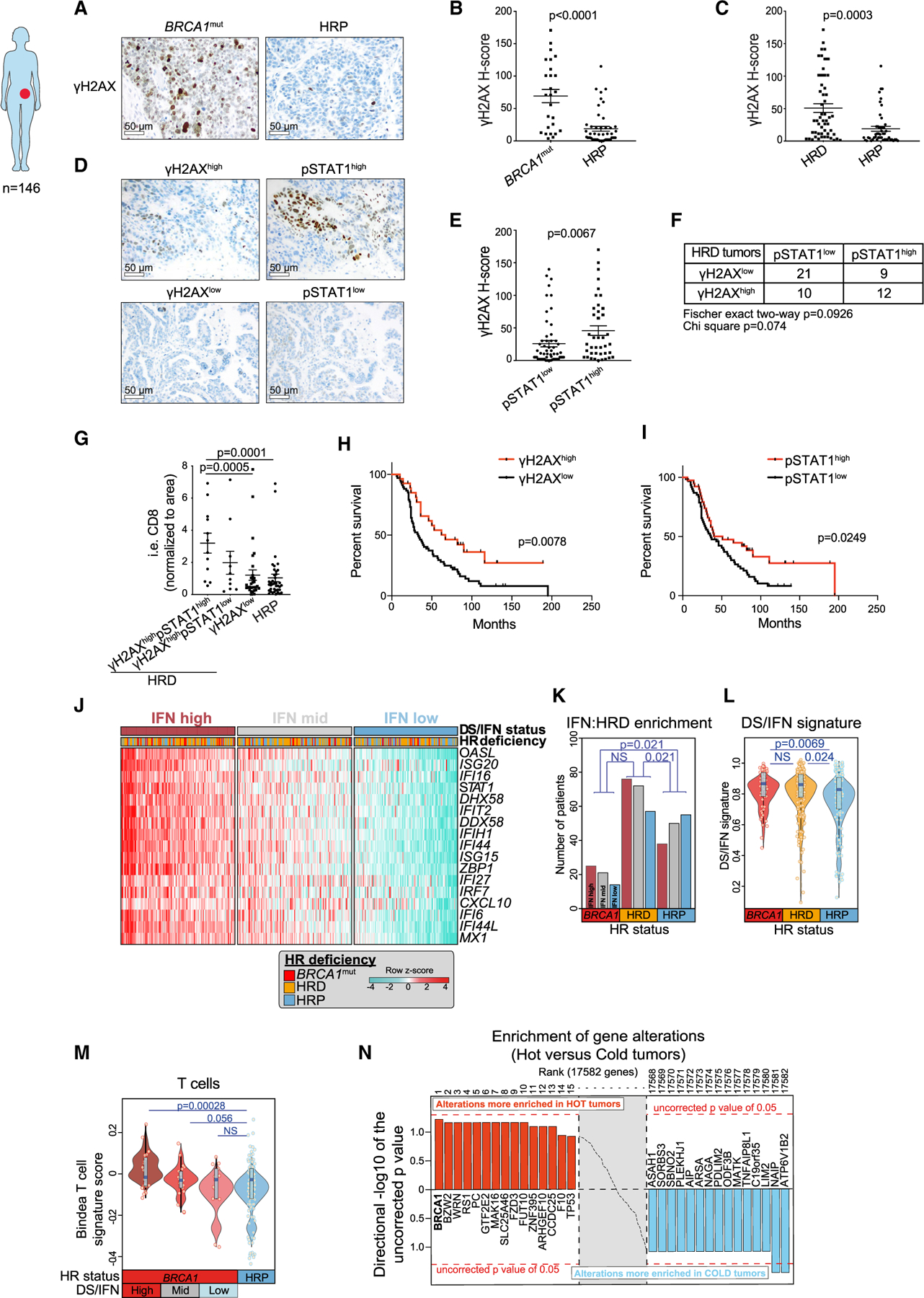
(A) Representative IHC staining of human HGSOC tissue for γH2AX tumor cell expression in BRCA1mut and HRP cancers. Scale bars, 50 mm.
(B and C) Quantification of tumor-expressed γH2AX staining in BRCA1mut (n = 26) and HRP (n = 49) tumors (B) and in HR-deficient (HRD) (n = 53) and HR-proficient (HRP) (n = 49) tumors. Data are presented as mean ± SEM, and points indicate values for individual patients. p values were calculated by a Mann-Whitney test.
(D) Representative IHC staining of pSTAT1 expression in cases of γH2AXhigh and gH2AXlow HGSOCs. Scale bars, 50 μm.
(E) Quantification of γH2AX in pSTAT1high (n = 57) and pSTAT1low (n = 40) HGSOCs. Data are presented as mean ± SEM, and points indicate values for individual patients. p values were calculated by a Mann-Whitney test.
(F) Table summarizing the numbers of γH2AXhigh/low and pSTAT1high/low HRD tumors.
(G) Quantification of i.e. CD8+ T cells in γH2AXhighpSTAT1high (n = 12), γH2AXhighpSTAT1low (n = 30), and γH2AXlow HRD (n = 10) and HRP (n = 39) HGSOCs. Data are presented as mean ± SEM, and points indicate values for individual patients. p values were calculated by a Mann-Whitney test.
(H and I) Kaplan-Mayer survival analyses of OC patients with γH2AXhigh (n = 27) versus γH2AXlow (n = 67) (H) and pSTAT1high (n = 39) and pSTAT1low (n = 61) (I). p values were extracted from Cox proportional hazards tests.
(J) Heatmap showing TCGA ovarian carcinoma cohort (Agilent platform) ranked according to the DS/IFN signature score composed of the genes appearing as rows. The cohort was split into tertiles according to IFN signature score (high, mid, and low). BRCA1 mutant patients include those with somatic and germline mutations in BRCA1, and HRD patients include those with any somatic mutations or copy number loss of any gene implicated in HR (excluding BRCA1 mutations that are taken separately).
(K) Comparison of the numbers of BRCA1mut, HRD, and HRP HGSOCs that express high, medium, and low DS/IFN signature scores, respectively. p values were computed by a Fisher’s exact test comparing high and low IFN groups according to HRD groups.
(L) Comparison of the DS/IFN score between BRCA1mut, HRD, and HRP HGSOCs. Violin plots extend to lowest/highest values, and points indicate values for individual subjects. Median is displayed by middle lines. Each dot represents an individual patient. p values were calculated by a Wilcoxon rank-sum tests.
(M) Comparison of the Bindea T cell signature score in BRCA1mut split by DS/IFN groups and HRP TCGA HGSOCs. Violin plots extend to lowest/highest values, and points indicate values for individual subjects. Median is displayed by middle lines. Each dot represents an individual patient. p values were calculated by a Wilcoxon rank-sum tests.
(N) Unbiased enrichment analysis of gene alterations (CNV loss + mutations) in inflamed (or hot) versus non-inflamed (or cold) tumors of TCGA.
To better understand the variation in DS/IFN activation among HRD HGSOCs, we studied OCs using The Cancer Genome Atlas (TCGA). By comparing BRCA1-deficient and -proficient OC cells we derived a restricted gene signature (Figure S5A) that captured the DS/IFN pathway (Figure 1), partially overlapping with one previously reported (Chiappinelli et al., 2015). We validated it on BRCA1-altered ovarian and breast cancer lines from the Cancer Cell Line Encyclopedia (CCLE) (Figure S5B).
We next interrogated gene expression data from 591 HGSOCs, examining separately Agilent, Affymetrix, and RNA-seq datasets (Table S3). HRD tumors exhibited high HRD scores, telomere allelic imbalance (TAI), large-scale state transition (LST) (González-Martín et al., 2019), loss of heterozygosity (LOH), and mutational and copy number variation (CNV) signatures 3 (Figure S5C) (Alexandrov et al., 2013; Macintyre et al., 2018). Tumors in the highest tertile of DS/IFN activation were enriched for BRCA1 mutations and HRD (Figure 4J). Conversely, BRCA1mut and HRD tumors were enriched for overexpression of the DS/IFN signature (Figures 4K and 4L). Notably, we observed a marked heterogeneity of DS/IFN activation in BRCA1mut and HRD tumors across ovarian datasets (Figures S5D and S5E). In addition, CD8 T cell gene signature scores were significantly higher in HRD or BRCA1mut tumors with high DS/IFN activation (Figures 4M and S5F). In an alternative comparison of any DNA alteration between T cell-inflamed and non-inflamed tumors, BRCA1 was the most frequently associated alteration in inflamed compared to non-inflamed tumors (Figure 4N). These tumors also exhibited higher scores for T cell subsets and activated dendritic cells (DCs) (Figures S5F and S5G). Thus, BRCA1mut and HRD HGSOCs exhibit a broad range of DNA damage/IFN activation, and retention of the tumor-intrinsic IFN response is associated with T cell recruitment.
Deletion or epigenetic silencing of CCL5 and DS/IFN genes is associated with attenuated T cell infiltration in HRD HGSOCs
We searched for genetic or epigenetic alterations that could explain the attenuation of the DS/IFN signature in BRCA1mut and HRD HGSOCs. We found deletions in genes implicated in DS/IFN signaling (Table S4) in 24 out of 245 HRD cases (10%) from TCGA (Figures 5A and S5H). Deletions associated with downregulation of the DS/IFN signature were found mostly in BRCA1mut/HRD and not HRP cancers and involved key genes of the pathway; for example, half involved NFKB1, IFNB1, or CCL5 (Figures 5B and S5I). Importantly, HRD cancers with deletion of IFNB1 or CCL5 exhibited an attenuated CD8 T cell signature (Figure 5C).
Figure 5. Loss of tumor-intrinsic CCL5 or type I IFN is associated with attenuated inflammation.
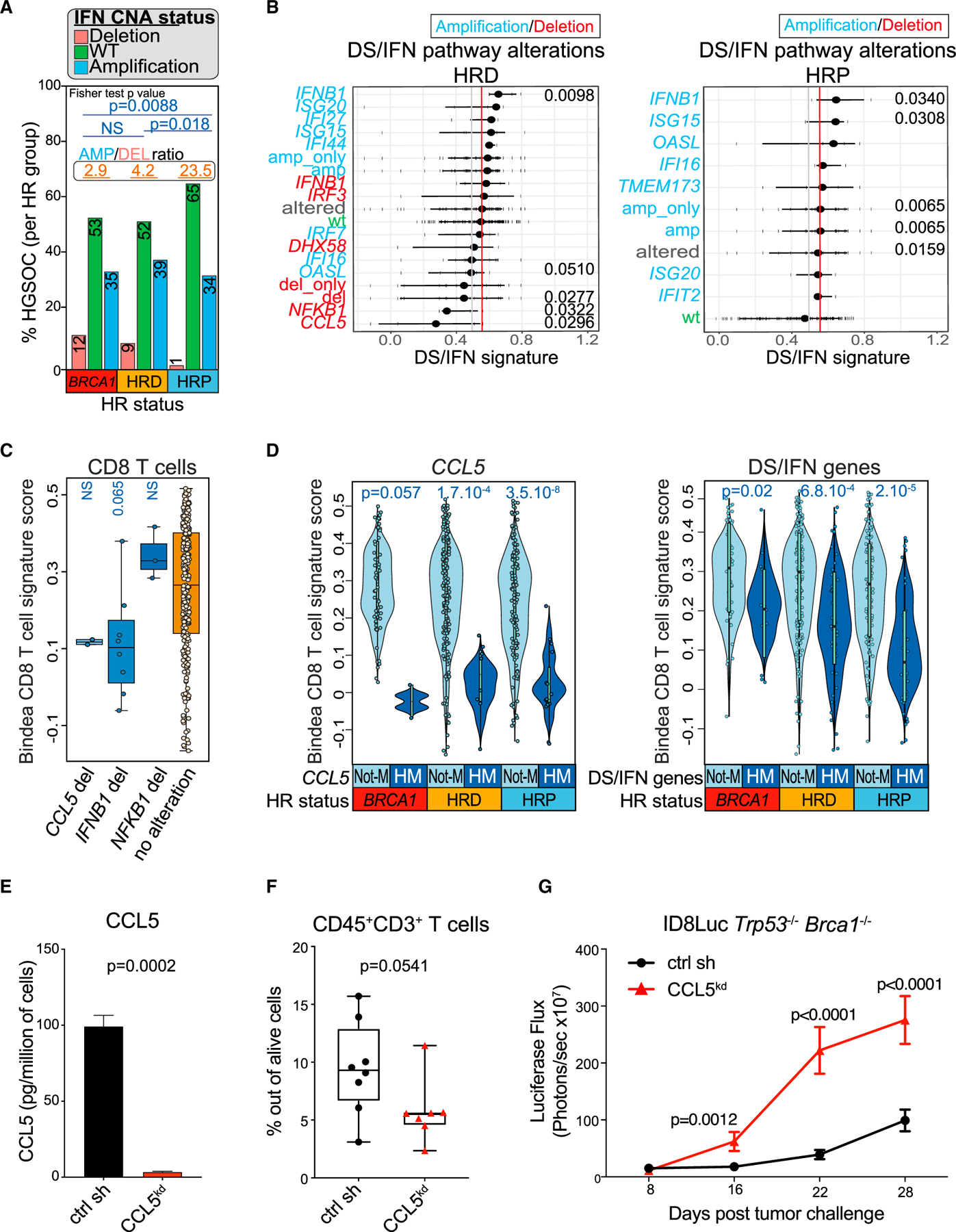
(A) Comparison of the DS/IFN gene CNA status in BRCA1 mutants, HRD and HRP HGSOCs. Ratio between amplified and deleted IFN groups are displayed at the top of each HRD group and number of patients in each bar graph. p values were calculated by a Fisher’s exact test.
(B) Association of DS/IFN gene impairment (deletion/amplification) in y axis and IFN pathway activation (DS/IFN signature score expression) in x axis in HRD and HRP TCGA HGSOCs from the Affymetrix platform. Whiskers represent 25th and 75th percentiles, with middle dots indicating the median. Each dot represents an individual tumor. Significant p values are shown in the graphs.
(C) Bindea CD8 T cell signature score (Bindea et al., 2013) in HRD tumors from patients carrying deletions in the CCL5, IFNB1, or NFKB1 genes versus not. Boxplots represent 25th and 75th percentiles, with the midline indicating the median; whiskers extend to the lowest/highest values, and points indicate values for individual subjects. p values were computed by a Wilcoxon rank-sum tests.
(D) Comparison of the Bindea CD8 T cell signature score in BRCA1mut, HRD, and HRP HGSOCs with HM or without HM (Not-M) of CCL5 or DS/IFN genes. Violin plots extend to lowest/highest values, and points indicate values for individual subjects. Median is displayed by middle lines. p values were computed by Wilcoxon rank-sum tests.
(E) CBA quantification of CCL5 in cell-free supernatants of Trp53−/−Brca1−/− CCL5kd and scr sh. The concentration (pg/mL) was normalized to cell number (n = 3). Data are presented as mean ± SEM. p values were computed by an unpaired t test.
(F) FACS quantification of CD3+ TILs in Trp53−/−Brca1−/− CCL5kd (n = 8) and scr sh (n = 7) intraperitoneal (i.p.) syngeneic tumors. Boxplots represent 25th and 75th percentiles, with the midline indicating the median; whiskers extend to the lowest/highest values, and points indicate values for individual subjects. p values were computed by a Mann-Whitney test.
(G) Luciferase-based tumor growth kinetics of ID8Luc Trp53−/−Brca1−/− CCL5kd (n = 8) and scr sh (n = 8) cancers. Data are presented as mean ± SEM. p values were computed by a two-way ANOVA.
See also Figures S5 and S6 and Table S4.
Hypermethylation (HM) of CCL5 and other genes of the DS/IFN signature also occurred frequently and was similarly associated with marked loss of the signature: DS/IFN gene HM occurred in 46% of tumors with a low DS/IFN score and only 1.4% of tumors with a high DS/IFN score, while CCL5 HM occurred in 18.25% of tumors with a low DS/IFN score and 0.7% of tumors with a high score, respectively (Figure S5J). Similarly, HM of CCL5 or other DS/IFN genes was associated with significant loss of the T cell signature (Figure 5D). Thus, genetic alteration or epigenetic silencing of CCL5 or other DS/IFN genes occurs frequently and is associated with reduced and T cell inflammation in HGSOCs.
Loss of tumor-intrinsic CCL5 attenuates immunoreactivity in murine Brca1-deficient ovarian tumors
To test the significance of the above findings, we adopted a syngeneic orthotopic ID8 mouse model deficient for Trp53 or Trp53 and Brca1 (Figure S6A) (Walton et al., 2016, 2017). Knockout of Brca1 and Trp53 in ID8 cells phenocopied human BRCA1mut HGSOC for sensitivity to PARPis and cell-autonomous inflammatory activation in vitro (Figures S6B and S6C) and baseline tumor immune reactivity in vivo (Figures S6D–S6H). To understand the relevance of losing key DS/IFN mediators in the context of HRD, we knocked down CCL5 in ID8 Trp53−/−Brca1−/− cells (Figure 5E). Phenocopying the attenuated DS/IFN activation in human HRD HGSOCs with CCL5 loss, Trp53−/−Brca1−/− CCL5kd tumors exhibited significantly reduced T cell infiltration and grew more rapidly than their counterparts (Figures 5F and 5G). Thus, loss of key inflammatory mediators such as CCL5 is an important mechanism through which BRCA1mut HGSOCs attenuate inflammation. Since we have previously demonstrated that loss of CCL5 leads to progressive loss of TILs, which then leads to resistance to ICB (Dangaj et al., 2019; Duraiswamy et al., 2014), these results also explain how some BRCA1mut HRD tumors specifically may evolve to eliminate T cell inflammation and become insensitive to ICB.
Immune checkpoint inhibitors and VEGF-A create immune resistance in Brca1-deficient tumors
We next investigated mechanisms driving immune resistance in inflamed HRD tumors. As expected, BRCA1mut and other HRD HGSOCs with DS/IFN activation significantly overexpressed known T cell inhibitory receptors and cognate checkpoint ligands (Figure S7A). HRD parameters such as HRD score, LOH, and LST correlated with an immune checkpoint signature (Figure S7B), suggestive of immune activation but also immune escape. As in human BRCA1mut HGSOCs, we found that ID8 Trp53−/−Brca1−/− tumors expressed high levels of PD-L1 immune inhibitory ligand, which were further increased by PARPi treatment (Figure S7C).
BRCA1 loss was also associated with upregulation of proangiogenesis transcriptional programs in UWB1.289 OC cells (Figures 6A, S1A, S1B, and S7D), validated in BRCA1-deficient cell lines of the CCLE database (Figures 6B and S7E). Loss of WT BRCA1 in human OVCAR5 and mouse ID8 cells markedly upregulated VEGF-A as well as VEGF-B (but not VEGF-C) expression (Figures 6C, 6D, S7F, and S7G). These results could explain the increased microvascular density found in BRCA1/2mut HGSOCs (Ruscito et al., 2018). Importantly, PARPi further enhanced VEGF-A expression in BRCA1-deficient cells (Figures 6C and 6D).
Figure 6. Immune checkpoint inhibitors and VEGF-A create immune resistance in Brca1-deficient tumors.
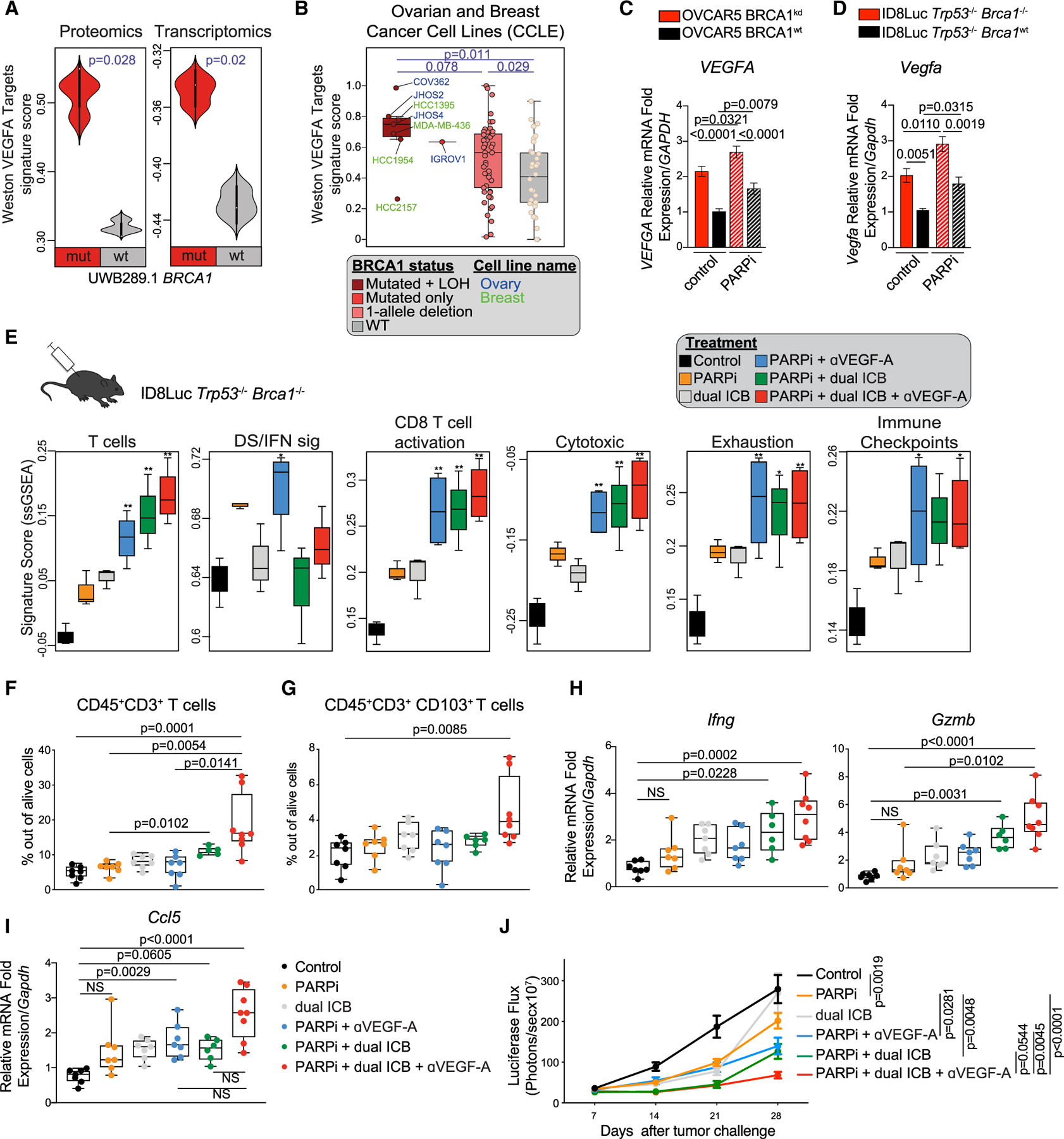
(A) Comparison of the “Weston VEGF targets 12hr” signature score (Weston et al., 2002) at the proteomic and transcriptomic levels between UWB1.289 BRCA1mut and BRCA1WT cell lines (n = 3). Violin plots extend to lowest/highest values, and embedded vertical boxes represent the 25th and 75th percentiles. Median is displayed by middle lines. Statistical significance was assessed by a Wilcoxon rank-sum tests.
(B) Comparison of the Weston VEGF targets 12hr signature score (Weston et al., 2002) in ovarian and breast cancer lines from the CCLE carrying BRCA1 mutations and LOH (complete BRCA1 functional loss), BRCA1 mutation only, CNV only (one allele deletion), or no alteration in BRCA1 (WT). Boxplots represent 25th and 75th percentiles, with the midline indicating the median; whiskers extend to the lowest/highest values, and points indicate values for individual cell lines. Statistical significance was assessed by Wilcoxon rank-sum tests.
(C and D) RT-PCR analysis of VEGFA and Vegfa in isogenic OVCAR5 (n = 8) (C) and ID8 (n = 4) (D) cell lines, respectively with or without PARPi treatment. Data are presented as mean ± SEM. p values were computed by a one-way ANOVA test.
(E) Comparison of the immune signature scores (Azizi et al., 2018; Bindea et al., 2013) between ID8Luc Trp53−/−Brca1−/− tumors treated as indicated. Boxplots represent 25th and 75th percentiles, with the midline indicating the median; whiskers extend to the lowest/highest values. p values were computed by ANOVA followed by a Tukey’s post hoc test for multiple comparison correction. *p < 0.05, **p < 0.01.
(F and G) Ex vivo quantification of CD3+ TIL (F) and CD103+ CD3+ TIL frequencies (G) in ID8Luc Trp53−/−Brca1−/− tumors treated with control, PARPis, dual ICB, anti-VEGF-A (aVEGF-A), or their combinations (n = 6–8 per group). All boxplots represent 25th and 75th percentiles, with the midline indicating the median; whiskers extend to the lowest/highest values. Points indicate values for individual subjects. p values were calculated by a Kruskal-Wallis test.
(H and I) RT-PCR analysis of Ccl5 (H) and Ifng and Gzmb (I) expression in ID8Luc Trp53−/−Brca1−/− i.p. tumors treated with the indicated regimen (n = 6–8 per group). All boxplots represent 25th and 75th percentiles, with the midline indicating the median; whiskers extend to the lowest/highest values. Points indicate values for individual subjects. p values were calculated by a Kruskal-Wallis test.
(J) i.p. tumor growth kinetics of ID8Luc Trp53−/−Brca1−/− cancers during treatment with control, olaparib olapaola, dual ICB, αVEGF-A, or their combinations (n = 7–15 per group). p values were calculated by a two-way ANOVA.
See also Figure S7.
Resembling those patients with BRCA1-deficient tumors who do not benefit from ICB (Konstantinopoulos et al., 2019; Vinayak et al., 2019), we noticed that ID8 Trp53−/−Brca1−/− tumors were intrinsically resistant to dual ICB treatment and there was no significant increase in TIL infiltration upon dual ICB (Figures 6E–6G and S7H). Given that PARPi increased both immune checkpoints and VEGF-A, we next investigated whether the combination of PARPi, VEGF-A blockade, and dual ICB could result in positive therapeutic interactions in vivo. Thus, we treated mice bearing orthotopic ID8 Trp53−/−Brca1−/− tumors with anti-VEGF-A anti-body, PARPi, and/or dual ICB. As expected, PARPi increased the DS/IFN signature (Figure 6E). Addition of dual ICB or VEGF blockade to PARPi further enhanced CD8 signatures, while the combination of PARPi, ICB, and VEGF-A blockade produced the highest activation of tumor T cell signatures (Figure 6E). These results were confirmed by fluorescence-activated cell sorting (FACS) analysis showing that combination therapy elicited the highest infiltration of CD3+, CD4+, and CD8+ T cells, which also expressed CD103, while tumors exhibited the highest expression of Ifng and Gzmb (Figures 6F–6I and S7H). Under-scoring the role of CCL5 in this process, combination therapy also induced maximal Ccl5 expression (Figure 6H). PARPi plus dual ICB or anti-VEGF-A resulted in comparable and significant restriction of tumor growth, while the combination of PARPi with dual ICB and anti-VEGF-A exerted maximal tumor growth suppression of ID8 Trp53−/−Brca1−/− cancers (Figures 6J and S7I). Importantly, this therapeutic interaction was not observed in ID8 Trp53−/−Brca1WT tumors (Figure S7J).
Tumor-intrinsic STING promotes resistance to dual ICB therapy via VEGF-A
We next wondered what drives tumor-intrinsic VEGF-A expression in BRCA1-deficient cancers. RNA-seq analysis of treated ID8 Trp53−/−Brca1−/− tumors revealed a correlation between the STING-mediated immune reactome and angiogenic signatures (Figure 7A). Furthermore, STING HM was associated with downregulation of proangiogenesis transcriptional programs in human tumors (Figures 7B, 7C, and S7K). Thus, we investigated whether STING played a role in generating immune resistance. To assess this, we knocked down STING in ID8 Trp53−/− Brca1−/− tumors (Figures S7L and S7N). Consistent with human data (Figures 2H–2J), Trp53−/−Brca1−/− STINGkd tumor cells expressed lower levels of pTBK1 and pSTAT1 and lost the DS/IFN activation in response to exogenous dsDNA or olaparib (Figures S7L and S7M). STING knockdown in ID8 Brca1-deficient cells reduced Vegfa expression in vitro at the steady state and abrogated its upregulation upon exposure to PARPi (Figures 7D and S7O). Our data suggest that there is a direct link between VEGF-A and STING signaling in tumors with BRCA1 loss.
Figure 7. Tumor-intrinsic STING promotes tumor survival and resistance to dual ICB through increased angiogenesis programs in Brca1-deficient tumors.
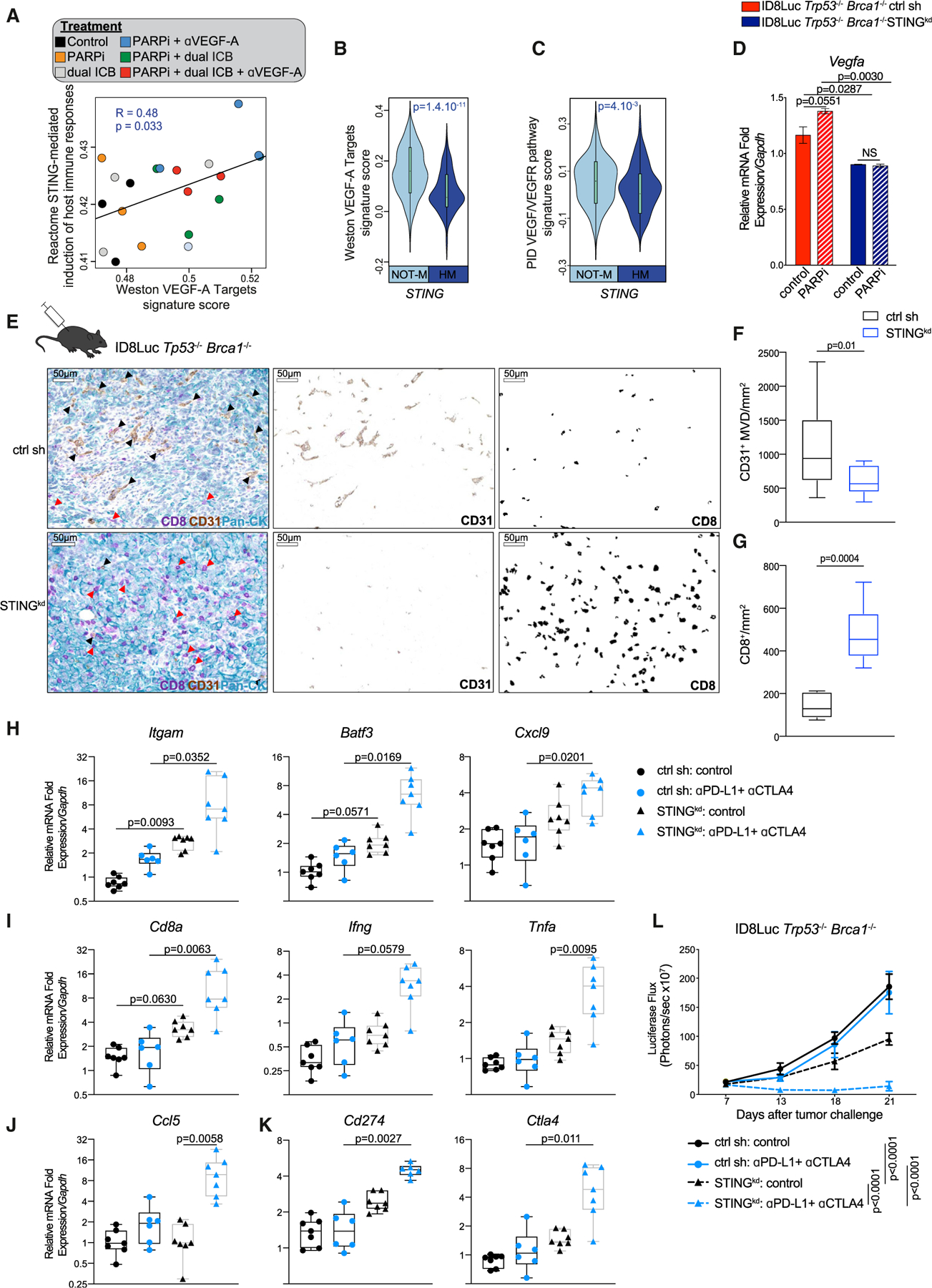
(A) Scatterplot showing the association between Reactome STING signature score and the Weston VEGF targets signature score in mouse ID8Luc Trp53−/− Brca1−/− tumor RNA sequencing treated with the indicated treatments. Statistical significance was assessed by correlation testing (Pearson).
(B and C) Comparison of the Weston VEGF targets signature score (B) and PID VEGF/VEGFR pathway signature score (Schaefer et al., 2009) (C) between TCGA HGSOCs with (HM) or without STING hypermethylation (Not-M). Violin plots extend to lowest/highest values, and embedded vertical boxes represent the 25th and 75th percentiles. Median is shown by a middle line. Statistical significance was calculated by a Wilcoxon rank-sum tests.
(D) RT-PCR analysis of Vegfa expression in ID8Luc Trp53−/−Brca1−/− STINGkd and scr sh isogenic cell lines 48 h after treatment with PARPi (n = 4). Data are presented as mean ± SEM. p values were calculated by a one-way ANOVA.
(E) Multiplex IHC staining and deconvolution staining for CD8, CD31, and pan-cytokeratin of ID8Luc Trp53−/−Brca1−/− STINGkd and scr sh i.p. tumors. Black arrows show examples of CD31+ vessels, and red arrows show CD8+ T cells. Scale bars, 50 μm.
(F and G) Quantification of CD31+ microvasculature density (F) and CD8+ T cells per mm2 (G) in ID8Luc Trp53−/−Brca1−/− STINGkd (n = 7–14) and scr sh (n = 7–14) i.p. tumors. All boxplots represent 25th and 75th percentiles, with the midline indicating the median; whiskers extend to the lowest/highest values. p values were computed by a Mann-Whitney test.
(H–K) RT-PCR analysis of Itgam (CD11b), Batf3, Cxcl9 (H), Cd8, Ifng, Tnfa (I), Ccl5 (J), Cd274 (PD-L1), and Ctla4 (J) expression in ID8Luc Trp53−/−Brca1−/− STINGkd and scr sh cancers treated with vehicle or dual ICB (n = 7, 6, 7, and 7, respectively). All boxplots represent 25th and 75th percentiles, with the midline indicating the median; whiskers extend to the lowest/highest values. Points indicate values for individual subjects. p values were computed by a Kruskal-Wallis test.
(L) Luciferase-based tumor growth kinetics of ID8Luc Trp53−/−Brca1−/− STINGkd and scr sh cancers treated as indicated (n = 7, 6, 7, and 7, respectively). Data are presented as mean ± SEM. p values were calculated by a two-way ANOVA.
To examine the effect of this axis in tumor vasculature, we compared the microvascular density and CD8+ T cell infiltration in WT and STINGkd tumors. STINGkd tumors had significantly lower CD31+ microvasculature density (Figures 7E and 7F), suggesting that forced loss of STING, and thus reduced VEGF-A, diminished neovascularization. Strikingly, STINGkd tumors had significantly more infiltrating CD8+ TILs at the steady state (Figures 7E and 7G).
We then treated these tumors with PD-L1/CTLA-4 blockade. Knockdown of STING was associated with a significant increase in CD8+ T cell and Batf3+ antigen-presenting cell infiltration in these tumors in response to PD-L1/CTLA-4 blockade as revealed by the mRNA levels of the lineage markers Cd8a, Itgam (CD11b), and Batf3, respectively (Figures 7H and 7I). Consistent with the response to dual ICB, we observed a clear upregulation of Ccl5, Ifng, Cxcl9, and Tnfa upon immunotherapeutic treatment (Figures 7H–7J). Cd274 (PD-L1) and Ctla4 were also upregulated but only in STINGkd tumors upon dual ICB (Figure 7K). These results combined are evidence of an increased infiltration of activated TILs (Nesbeth et al., 2010; Swanson et al., 2002). Importantly, tumor-intrinsic loss of STING reversed therapeutic resistance, leading to profound suppression of tumor growth by dual ICB (Figure 7L). Thus, tumor STING mediates tumor protection from immune attack in the context of BRCA1 loss and cell-autonomous inflammation.
DISCUSSION
In this study we demonstrate that BRCA1 loss reprograms OC cells toward an obligatory cell-autonomous inflammatory state, maintained by the simultaneous upregulation of the dsDNA sensing pathway and the oversupply of cytoplasmic dsDNA converging on STING. Hyperresponsiveness to cytoplasmic dsDNA is ensured by spatial chromatin remodeling and transcriptional reprogramming, which result in enhancer enrichment and transcriptional amplification of key genes in the DNA sensing and IFN response. In BRCA1-deficient OC cells, sensing of endogenous cytoplasmic dsDNA was exacerbated by PARPis, similarly to other tumors types (Pantelidou et al., 2019; Parkes et al., 2016; Sen et al., 2019). We confirmed the key roles of STING, TBK1, IRF3, and STAT1 in mediating the response to ectopic cytoplasmic dsDNA (Ding et al., 2018; Pantelidou et al., 2019; Sen et al., 2019; Wang et al., 2019). In addition, we show that the state of hyperresponsiveness to cytoplasmic dsDNA in BRCA1-deficient cells is tonically countered by TREX nucleases, which can be activated by high but not low doses of irradiation (Vanpouille-Box et al., 2017). This inflammatory cell state was retained in vivo and is at the base of T cell recruitment to tumors. While evidence in mouse OC has suggested that STING activation occurs mainly within stromal DCs (Ding et al., 2018), our work establishes that the DS/IFN pathway is activated intrinsically in OC cells, committing tumors to an inflammatory state associated with T cell infiltration. The fact that BRCA1 loss is a founding oncogenic event in these tumors explains how these tumors may be “locked” in a default inflamed state despite their evolutionary plasticity afforded by chromatin instability.
The above findings raised important questions: (1) how may HRD tumors escape immune elimination at the steady state, and (2) why has the combination of PARPis and ICB not produced more dramatic responses in patients with BRCA1mut HGSOC HRD tumors exhibited wide heterogeneity in DNA damage and inflammation in situ. We identified two pathways explaining how BRCA1mut cells can manage to either quench cell-autonomous inflammation or, alternatively, exploit inflammation to escape immune attack. First, elimination of tumor-intrinsic CCL5 markedly reduced T cell inflammation in HRD tumors. Indeed, CNAs and more frequently HM leading to CCL5 or DS/IFN signature downregulation in human HRD HGSOCs, or engineered knockdown of CCL5 in mouse Brca1-deficient OC cells, largely attenuated inflammation and T cell infiltration, and rendered Trp53−/−Brca1−/− tumors resistant to dual ICB. This evidence complements previous evidence that methylation of the CCL5 locus is prevalent in HGSOCs lacking ieCD8+ TILs (Dangaj et al., 2019), and positions CCL5 as a master regulator of T cell inflammation and targets for oncogenic pathways.
Second, we showed that in tumors with active DS/IFN signaling, STING not only drives T cell inflammation, with the expected upregulation in multiple immune checkpoints, but it also promotes tumor angiogenesis through intrinsic overexpression of VEGF-A, known to mediate tumor immune escape (Buckanovich et al., 2008; Motz and Coukos, 2011). Strikingly, STING elimination markedly attenuated tumor growth at the steady state and abrogated therapeutic resistance to dual ICB, which was also phenocopied by VEGF-A blockade. Our study is in agreement with recent studies attributing a protumoral role to STING through chronic NF-κB-driven inflammation (Bakhoum et al., 2018; Dou et al., 2017). Indeed, we also found that BRCA1 loss activates NF-κB in OC cells and that NFKB1 deletion was, along with IFNB1 and CCL5, the most commonly deleted gene in HRD tumors lacking DS/IFN activation. Furthermore, NF-κB inhibition attenuated VEGF-A expression at the steady state and upon exposure to PARPi in BRCA1-deficient OC cells (data not shown). The coexistence of BRCA1 loss with immune resistance mediated by ICB and enhanced angiogenesis creates the basis for therapeutic combinations targeting these pathways, which was indeed effective in the ID8 Trp53−/−Brca1−/− murine tumor model. This evidence explains the recently reported benefit of combining PARPis and bevacizumab specifically seen in patients with BRCA1-mutated and HRD tumors but not those with HRP tumors (Ray-Coquard et al., 2019), and it has important implications for ongoing clinical studies testing the combination of PARPis, ICB, and bevacizumab in HGSOCs.
Study limitations
We underline that despite recent findings about the controversial nature of the OVCAR5 cell line (Blayney et al., 2016), BRCA1 knockdown in this cell line recapitulated all of our findings regarding the activation of the dsDNA/IFN pathway through STING as initially observed in the UWB1.289 isogenic cell lines. Those findings were also validated in HGSOC in situ and also recapitulated in vitro and in vivo in the mouse ID8 isogenic cell lines. Furthermore, our studies could not fully dissect how loss of BRCA1 drives inflammatory transcriptional reprogramming of cells. BRCA1 is required for DNA condensation and satellite repression (Zhu et al., 2011), which could partly explain our findings. Furthermore, direct functions of BRCA1 in transcriptional regulation (Zhang and Li, 2018) could also play a role.
To document activation of the DS/IFN pathway in situ, we analyzed the coexpression of STING, γH2AX, and pSTAT1 in human HGSOCs and correlated it with patients’ survival. A multi-variate analysis to account for clinical parameters such as optimal debulking surgery was not applied in our data. Although pSTAT1 may exhibit pleiotropic roles in different physiologies, pSTAT1 is the most downstream transcription factor and biomarker of this pathway due to cell-intrinsic or -extrinsic type I IFN binding through IFNAR as supported by others (Cardenas, 2019). Nevertheless, exploring also pTBK1/pIRF3 activation in situ could complement these data.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contacts, George Coukos (george.coukos@chuv.ch) and Denarda Dangaj (denarda.dangaj@chuv.ch)
Materials availability
Any new generated material from this study can be shared upon request to the Lead contacts.
Data and code availability
All data are deposited in the GEO repositories and accession numbers are stated in the Key Resources Table. This study did not generate new unique code.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| rat anti-CD31, clone SZ31 | Dianova | Cat#DIA-310; RRID:AB_2631039 |
| rat anti-CD8, clone 4SM15 | Thermo fisher) | Cat#14–0808-82; RRID: AB_2572861 |
| rabbit anti-PanCK, polyclonal | Novus Biologicals | Cat#NBP2–44368 |
| anti-mouse CD45-BV650, clone 30F11,1:400 | Biolegend | Cat#103151; RRID: AB_2565884 |
| anti-mouse CD3e-PECy5.5,clone 145–2C11, 1:100 | Invitrogen | Cat#;35–0031-80; RRID: AB_11218085 |
| anti-mouse CD4-PB, clone GK1.5, 1:100 | Biolegend | Cat#100428; RRID: AB_493647 |
| anti-mouse CD8-BV711, clone 53.6.7, 1:50 | Biolegend | Cat#100748; RRID: AB_2562100 |
| anti-mouse CD11b-PECy7, clone M1/70,1:200 | eBioscience | Cat#25–0112-81; RRID: AB_469587 |
| anti-mouse CD11c-BV60, clone N418 | Biolegend | Cat#117334; RRID: AB_2562415 |
| anti-mouse F4/80-APCCy7, clone BM8,1:200 | Biolegend | Cat#123118; RRID: AB_893477 |
| anti-mouse F4/80-Alexa647, clone F4/80,1:50 | home-made | |
| anti-mouse PD-L1-BV711, clone 10F.9G2,1:200 | Biolegend | Cat#124319; RRID: AB_2563619 |
| anti-mouse Ki67-APC, clone 16A8, 1:200 | Biolegend | Cat#652406; RRID: AB_2561930 |
| anti-mouse CD80-PEDazzle,clone 16–10A1, 1:100 | Biolegend | Cat#104737; RRID: AB_2564174 |
| anti-mouse CD86-APCCy7, clone GL-1,1:100 | Biolegend | Cat#105030; RRID: AB_2244452 |
| anti-mouse CD103-PE, clone 2E7, 1:200 | Biolegend | Cat#121405; RRID: AB_535948 |
| Anti-Mouse CD16/CD32 (Mouse BD Fc Block), Clone 2.4G2, 1:100 | BD | Cat#553142; RRID:AB_394657 |
| LIVE/DEAD Fixable Aqua Dead Cell Stain, 1:500 | Invitrogen | Cat#L34957 |
| anti-human BRCA1, clone MS110, 1:500 | Millipore | Cat#MABC199; RRID: AB_213438 |
| anti-human IFI16, clone 1G7, 1:500 | Santa Cruz Biotechnology | Cat#sc-8023; RRID: AB_627775 |
| anti-human and mouse NF-κB, clone C-20, 1:500 | Santa Cruz Biotechnology | Cat#sc-372; RRID: AB_632037 |
| anti-human and mouse phosphor-IRF3, clone 4D4G, 1:500 | Cell Signaling Technology | Cat#4947S;,RRID: AB_823547 |
| anti-human and mouse STING, clone D2P2F, 1:500 | Cell Signaling Technology | Cat#13647S; RRID: AB_2732796 |
| anti-human and mouse phospho-STAT1, clone D4A7, 1:500 | Cell Signaling Technology | Cat#7649S; RRID: AB_10950970 |
| anti-human and mouse STAT1, clone D4Y6Z, 1:500 | Cell Signaling Technology | Cat#14995S; RRID: AB_2716280 |
| anti-human and mouse Lamin B, clone M-20, 1:1000 | Santa Cruz Biotechnology | Cat#sc-6217; RRID: AB_648158 |
| anti-human and mouse B-ACTIN, clone C4, 1:500 | Santa Cruz Biotechnology | Cat#sc-47778; RRID: AB_2714189 |
| dsDNA Marker, HYB331–01, 1:1000 | Santa Cruz Biotechnology | Cat#sc-58749; RRID: AB_783088 |
| anti-phospho-Histone H2A.X (Ser139), clone JBW301, 1:500 | Millipore | Cat#05–636; RRID: AB_309864 |
| anti-human and mouse phospho-TBK1/NAK(Ser172), clone D52C2, 1:100 | Cell Signaling Technology | Cat#5483; RRID: AB_10693472 |
| anti-human CD8, clone SP16, 1:100 | ThermoFisher Scientific | Cat#MA5–14548; RRID: AB_10984334 |
| anti-human and mouse STAT1, 1:2000 | Abcam | Cat#ab47425; RRID: AB_882708 |
| anti-human and mouse phospho-STAT1, Clone 58D6 | Cell Signaling Technology | Cat#9167S; RRID: AB_561284 |
| anti-human pan Cytokeratin, clone AE1/AE3 | DAKO | Cat#M351501–2; RRID: AB_2631307 |
| anti-human and mouse phospho-Stat1-AF488, clone 58D6, 1:100 | Cell Signaling Technology | Cat#9174S; RRID: AB_2198287 |
| anti-human and mouse phospho-TBK1-AF647,clone D52C2, 1:100 | Cell Signaling Technology | Cat#14590S; RRID: AB_2798527 |
| InVivoMAb anti-mouse CTLA-4, clone 9D9 | BioXCell | Cat#BE0164; RRID: AB_10949609 |
| InVivoMAb anti-mouse PD-L1, clone 10F.9G2 | BioXCell | Cat#BE0101; RRID: AB_10949609 |
| anti-VEGFA mAb, clone B20–4.1.1 | Genentech | a kind gift from Genentech |
| InVivoMAb anti-mouse IFNAR-1, clone MAR1–5A3 | BioXCell | Cat#BE0241; RRID: AB_2687723 |
| InVivoMAb IgG1 Isotype control, clone MOPC-21 | BioXCell | Cat#BE0083; RRID: AB_1107784 |
| H3K27ac pAb | Active Motif | Cat#31933 |
|
| ||
| Biological samples | ||
|
| ||
| HGSOC specimens | (Bernards et al., 2016; Pennington et al., 2014); https://doi.org/10.1016/j.ygyno.2015.12.017 https://doi.org/10.1158/1078–0432.CCR-13–2287 | |
| ovarian carcinomas TCGA Consortium | TCGA | (Cancer Genome Atlas Research Network, 2011); https://doi.org/10.1038/nature10166 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Spectral DAPI, 1:5 | PerkinElmer | Cat#FP1490 |
| IgG Goat Blocker | Ventana Roche | Cat#760–6008 |
| DISCOVERY OmniMap anti-rabbit HRP | Ventana Roche | Cat#760–4311 |
| DISCOVERY OmniMap anti-mouse HRP | Ventana Roche | Cat#760–4310 |
| OPAL 520 | PerkinElmer | Cat#FP1487001KT |
| OPAL 570 | PerkinElmer | Cat#FP1488001KT |
| OPAL 620 | PerkinElmer | Cat#FP1495001KT |
| OPAL 690 | PerkinElmer | Cat#FP1497001KT |
| Olaparib, AZD2281 | Selleckchem | Cat#S1060 |
| poly(dA:dT) | Invivogen | Cat# tlrl-patn, tlrl-patn-1 |
| poly(I:C) | Invivogen | Cat#tlrl-pic |
| Turbofect | ThermoFischer Scientific | Cat#R0532 |
| D-luciferin | Biosynth | Cat#L-8220 |
| Liberase TL | Roche | Cat#540102001 |
| Dnase I | Sigma Aldrich | Cat#D4527 |
| Hygromycin | Millipore | Cat#400052 |
| Puromycin | Invivogen | Cat#ant-pr-1 |
| TRIzol reagent | Invitrogen | Cat#15596026 |
| DMEM | ThermoFischer Scientific | Cat#41966–029 |
| RPMI 1640 | ThermoFischer Scientific | Cat#1870010 |
| Opti-MEM I Reduced Serum Medium, no phenol red | ThermoFischer Scientific | Cat#11058021 |
| TaqMan® Fast Universal PCR reagents | Applied Biosystems | Cat#4366072 |
| Shandon Shandon-Mount | ThermoFischer Scientific | Cat#1900331 |
|
| ||
| Recombinant DNA | ||
|
| ||
| MISSION pLKO.1-puro non-target shRNAcontrol | Sigma | Cat#SHC016 |
| BRCA1 MISSION shRNA | Sigma | Cat#TRCN0000244984 |
| TMEM173 MISSION shRNA Bacterial Glycerol Stock transmembrane protein 173 (STING kd1) | Sigma | Cat#TRCN0000346266 |
| TMEM173 MISSION shRNA Bacterial Glycerol Stock transmembrane protein 173 (STING kd2) | Sigma | Cat#TRCN0000346320 |
| CCL5 MISSION shRNA Bacterial GlycerolStock chemokine (C-C motif) ligand 5 | Sigma | Cat#TRCN0000068102 |
| MSCV Luciferase PGK-hygro constrcuct | Addgene | Cat#18782 |
|
| ||
| Commercial assays | ||
|
| ||
| Human IFNa Flex Set | BD | Cat#560379 |
| Human IL-1a Flex Set | BD | Cat#560153 |
| Human IL-1b Flex Set | BD | Cat#561509 |
| Human TNFa Flex Set | BD | Cat#561516 |
| Human CXCL10 Flex Set | BD | Cat#558280 |
| Human Fractalkine (CX3CL1) Flex Set | BD | Cat#560265 |
| Human IL-6 Flex Set | BD | Cat#558276 |
| Human GM-CSF Flex Set | BD | Cat#558335 |
| Mouse RANTES (CCL5) Flex Set | BD | Cat#558345 |
| MTT Cell Proliferation Assay kit | R&D Systems | Cat#4890–025-K |
| RNA Easy Mini Kit | QIAGEN | Cat#74104 |
| PrimeScript First Strand cDNA Synthesis Kit | Takara | Cat#6110A |
| NE-PER Nuclear and cytoplasmic Extraction Kit | ThermoFischer Scientific | Cat#78833 |
| Halt Protease and Phosphatase Inhibitor Cocktail (100X) | ThermoFischer Scientific | Cat#78440 |
| NucleoSpin Gel and PCR-Clean-up | Macherey-Nagel | Cat#740609.50 |
| Illumina TruSeq Stranded Total RNA reagents | Illumina | Cat#RS-122–2201 |
| Illumina HiSeq PE Cluster Kit v4 cBot reagents | Illumina | Cat#PE-401–400 |
| HiSeq SBS Kit V4 reagents | Illumina | Cat#FC-401–4002 |
| Mouse VEGF-A ELISA Kit | Abcam | Cat#ab209882 |
|
| ||
| Deposited data | ||
|
| ||
| ChIPSeq data | GEO | GSE122155; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122155 |
| HiC data | GEO | GSE122155; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122155 |
| Human RNA sequencing data | GEO | GSE120792; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120792 |
| Mouse RNA sequencing data | GEO | GSE162935; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162935 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| OVCAR5 BRCA1wt and OVCAR5 BRCA1kd | our own laboratory | |
| UWB1.289 (ATCC® CRL-2945), BRCA1mut | ATCC | RRID:CVCL_B079 |
| UWB1.289 BRCA1+ (ATCC® CRL-2946), BRCA1wt | ATCC | RRID:CVCL_B078 |
| ID8 Trp53−/− and ID8 Trp53−/−Brca1−/− | Prof. Iain A. McNeish lab | (Walton et al., 2016; Walton et al., 2017); https://doi.org/10.1158/0008–5472. CAN-16–1272 https://doi.org/10.1038/s41598–017-17119–1 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57/BL6 | Envigo | 057 |
|
| ||
| Oligonucleotides | ||
|
| ||
| CCL5 | Applied Biosystems | Cat#Hs00982282_m1 |
| CXCL9 | Applied Biosystems | Cat#Hs00171065_m1 |
| CXCL10 | Applied Biosystems | Cat#Hs01124251_g1 |
| CD8A | Applied Biosystems | Cat#Hs00233520_m1 |
| IFNB1 | Applied Biosystems | Cat#Hs01077958_s1 |
| IFNA1 | Applied Biosystems | Cat#Hs00256882_s1 |
| TNFA | Applied Biosystems | Cat#Hs01113624_g1 |
| ISG20 | Applied Biosystems | Cat#Hs00158122_m1 |
| ISG15 | Applied Biosystems | Cat#Hs01921425_s1 |
| VEGFA | Applied Biosystems | Cat#Hs03929036_s1 |
| VEGFB | Applied Biosystems | Cat#Hs00173634_m1 |
| VEGFC | Applied Biosystems | Cat#Hs01099203_m1 |
| GAPDH | Applied Biosystems | Cat#Hs03929097_g1 |
| Cd8a | Applied Biosystems | Cat#Mm01188922_m1 |
| Ccl5 | Applied Biosystems | Cat#Mm01302427_m1 |
| Cxcl9 | Applied Biosystems | Cat#Mm00434946_m1 |
| Stat1 | Applied Biosystems | Cat#Mm01257286_m1 |
| Gzmb | Applied Biosystems | Cat#Mm00442837_m1 |
| Ifng | Applied Biosystems | Cat#Mm01168134_m1 |
| Itgax | Applied Biosystems | Cat#Mm00498701_m1 |
| Batf3 | Applied Biosystems | Cat#Mm01318275_m1 |
| Ifnb1 | Applied Biosystems | Cat#Mm00439552_s1 |
| Gapdh | Applied Biosystems | Cat#Mm99999915_g1 |
| Vegfa | Applied Biosystems | Cat#Mm00437306_m1 |
| Vegfb | Applied Biosystems | Cat#Mm00442102_m1 |
| Vegfc | Applied Biosystems | Cat#Mm00437310_m1z |
| Tnfa | Applied Biosystems | Cat#Mm00443258_m1 |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| R | Open source | https://www.r-project.org/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Carcinoma specimens
A cohort of n = 109 of HGSOC specimens were collected with patient consent as approved by the institutional review board and obtained from the University of Washington Gynecologic Oncology Tissue Bank (Seattle, WA). Information about survival of patients was also obtained from the tissue bank. Mutations in the TP53, BRCA1, BRCA2 and RAD51C genes and methylation of BRCA1 and RAD51C were identified as previously described (Pennington et al., 2014) (Bernards et al., 2016).
TCGA ovarian dataset
We considered the molecular data for the set of 437 ovarian carcinomas carrying TP53 mutation curated by The Cancer Genome Atlas (TCGA) Consortium and coming from three different platforms (Agilent: 409 patients; Affymetrix: 393 patients; RNaseq: 237 patients). The publicly available MC3 compendium of somatic point mutation data was retrieved from the Synapse syn7214402 on July 2017 (v. 0.2.8) (https://www.synapse.org/#!Synapse:syn7214402/wiki/405297). Gene expression, copy number (Affymetrix Genome-Wide Human SNP Array 6.0) and methylation data were downloaded from FireHose and gdac repositories on January 2016. Samples TCGA.09.2056.01’, ‘TCGA.24.1544.01’, ‘TCGA.24.1565.01’, ‘TCGA.25.1316.01’, ‘TCGA.61.2095.01’ were excluded from the dataset as they were reclassified as not high grade serous ovarian cancer samples (Zhang et al., 2016). The immune subset analysis of TCGA patients was achieved by computing signature scores for immune subsets using the signatures as published by Bindea and collaborators (Bindea et al., 2013).
Cancer cell line encyclopedia analysis
We used the Cancer Cell Line Encyclopedia (CCLE) in order to interrogate the levels of DS/IFN signature in a panel of breast and ovarian cancer cell lines (https://portals.broadinstitute.org/ccle). Transcriptomics, mutational and copy number variation data were used to identify BRCA1 altered (mutated and/or LOH/CNV) and BRCA1 WT cell lines. The DS/IFN signature was computed using ssGSEA as inferred in the GSVA R package and statistical analyses were done using Wilcoxon rank-sum tests.
Mouse models
C57/BL6 female mice were obtained from Envigo and were maintained in pathogen-free conditions. Age-matched mice between 6 and 8 weeks were used for all experiments. Animal experimentation procedures were performed according to the protocols approved by the Veterinary Authorities of the Canton Vaud (VD2797, VD3480), according to Swiss law.
We injected 5 × 106 ID8 derivative cancer cells expressing luciferase (ID8Luc) i.p. in C57/BL6 female mice. PARPi was administered orally at 40 mg/kg/day. All antibodies were injected i.p. twice a week at the following amounts: 100 μg of aCTLA-4 mAb; 200 μg of αPD-L1 mAb; 20 μg of αVEGFA mAb. The αVEGFA mAb was a kind gift from Genentech. All compounds are list in the Key resource table.
For the evaluation of αVEGFA, Olaparib and dual ICB combination, Olaparib and antibodies were administered in the doses and schedules described above, one week after tumor challenge and continued for 3 weeks when characterizing tumor immune infiltration, or until tumor progression for survival studies.
Mouse health and welfare were monitored regularly. For experiments evaluating survival post-therapy, we used body and health performance score sheets (taking into consideration ascites accumulation) and mice were sacrificed once reaching the equivalent of human endpoints.
Cell cultures
UWB1.289 BRCA1mut and UWB1.289 BRCA1wt were obtained from ATCC and cultured as indicated by manufacturer. OVCAR5 cancer cell lines were obtained from the Ovarian Cancer Research Center cell bank at UPENN. The cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 100 μg/mL penicillin and 100 μg/mL streptomycin, at 37°C in 5% CO2 atmosphere.
ID8 Trp53−/−Brca1wt and Trp53−/− Brca1−/− mouse ovarian cancer cell lines, obtained from the laboratory of Prof. Iain A. McNeish (Institute of Cancer Sciences, University of Glasgow, Scotland) (Walton et al., 2016; Walton et al., 2017), were cultured in DMEM supplemented with 4% FBS, 100 μg/mL penicillin, 100 μg/mL streptomycin, and ITS (5 μg/mL insulin, 5 μg/mL transferrin, and 5ng/mL sodium selenite). All cell lines were negative for Mycoplasma contamination.
METHOD DETAILS
RNA sequencing
RNA from UWB1.289 (BRCA1 2594delCmut, BRCA1mut, n = 3) and UWB1.289 BRCA1+ (BRCA1wt; n = 3) cell lines was extracted using the RNA easy kit. RNA quality was assessed using the Fragment Analyzer. RNA sequencing libraries were prepared using the Illumina TruSeq Stranded Total RNA reagents according to the protocol supplied by the manufacturer and using 1 μg of total RNA. Cluster generation was performed with the libraries using the Illumina HiSeq PE Cluster Kit v4 cBot reagents and sequenced on the Illumina HiSeq 2500 using HiSeq SBS Kit V4 reagents.
Similarly for mouse tissues, bulk RNA was extracted from snap frozen tissues using the RNA easy kit. RNA quality was assessed using the Fragment Analyzer. RNA sequencing libraries were prepared using the Illumina TruSeq Stranded Total RNA reagents according to the protocol supplied by the manufacturer and sequenced using HiSeq 4000 SR.
RNA sequencing data analysis
For human cell line analysis, sequencing data were processed using the Illumina Pipeline Software version 1.84. Initial number of reads averaged 78 ± 35 (standard deviation (s.d.)) million per sample. Reads were first trimmed to remove polyA and Illumina TruSeq adaptor sequences using cutadapt, and aligned to the human reference hGRC37 genome using the STAR aligner (Dobin et al., 2013). The number of counts was summarized at the gene level using featureCounts (Liao et al., 2014). Reads that uniquely mapped to the reference genome averaged 93.6% ± 0.2% (s.d.). The rate of these reads mapping to ribosomal RNA averaged 0.03% ± 0.002% (s.d.) while 97.8% ± 0.2% (s.d.) mapped to exonic protein-coding sequences. Read counts were normalized into reads per kilobase per million (RPKM) and log2 transformed after addition of a pseudocount value of 1. Gene expression data have been deposited in GEO (GSE120792). Differential expression analyses were performed using the edgeR package.
For mouse tumor analysis, Illumina single-end sequencing reads were aligned to the mouse reference GRCm38 genome using STAR aligner (Dobin et al., 2013) and the 2-pass method as briefly followed: the reads were aligned in a first round using the –run-Mode alignReads parameter, then a sample-specific splice-junction index was created using the –runMode genomeGenerate parameter. Finally, the reads were aligned using this newly created index as a reference. The number of counts was summarized at the gene level using htseq-count (Anders, 2015). The number of uniquely-mapped, non-mitochondrial and non-ribosomal reads averaged 28’343’252 ± 2′135’822 (s.d.). Read counts were normalized into reads per kilobase per million (RPKM) and log2 transformed after addition of a pseudocount value of 1. Gene expression data have been deposited in GEO (GSE162935).
Gene expression signatures analyses
Several gene signatures were used in this study. Immune subset gene signatures were directly taken from the Bindea et al. study (Bindea et al.). Hallmarks gene signatures of general biological processes were taken from the MSigDB database (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp). Signatures related to angiogenesis were found as follows: we searched for the terms “angiogenesis” and “VEGF” in the C2 collection from the MSigDB database. We further filtered out non-relevant signatures and then selected two relevant signatures based on curation of the genes and on the experimental method used to obtained such signatures and we retained only the “PID VEGF/VEGFR PATHWAY” (Schaefer et al., 2009) and “WESTON VEGFA TARGETS 12HR” (Weston et al., 2002) signatures. Gene signature score were computed using the ssGSEA as implemented in the GSVA R package (default parameters). Heatmaps were done using the pheatmap R package.
Pathway analyses for mouse tumor were carried out as described for human data except that genesets of human origin were transformed into ortholog mouse genesets. T cell signatures were taken from Bindea et al. (2013); Jerby-Arnon et al. (2018); Azizi et al. (2018). STING reactivity signatures were extracted from Reactome collection of MSigDB.
HR pathway alteration status in TCGA OV cohort
HR pathway alteration status was evaluated by integrating mutation, copy number changes and epigenetic silencing data. First, the list of genes involved in the HR pathway was compiled by manually annotating each gene with its role in the pathway (activating/inhibiting, Table S3). Afterward, molecular data were screened looking for (i) amplification of inhibiting genes (+2 in discrete GISTIC gene level calls), (ii) deep deletions (−2 in discrete GISTIC gene level calls) or truncating mutations (nonsense, frameshift insertions/deletions, splice site events and indels) of activating genes, and (iii) epigenetic silencing (hypermethylation) of BRCA1. BRCA1 hypermethylation status was inferred using RESET, a software to detect functional hyper- and hypo-methylation events (Saghafinia et al., 2018). Briefly, the probes of the Affymetrix Genome-Wide Human SNP Array 6.0 overlapping to any BRCA1 promoter region were considered. Promoter regions for BRCA1 were extracted from the FANTOM5 cohort of robust promoters (Forrest et al., 2014). In total, 5 probes matched a BRCA1 promoter region. For each probe, the hypermethylation status was called by comparing the beta values of the cancer samples versus those of normal samples available in the TCGA ovarian cancer cohort. The functional effect of the hypermethylation was assessed, separately for each probe, by checking whether BRCA1 gene expression was significantly decreased in hypermethylated carcinomas, compared to not hypermethylated ones. Ultimately, we called BRCA1-silenced those samples where 4 out of 5 probes were hypermethylated, as significant downregulation of gene expression was observed only in these cases. In total, 194 out of 360 samples (~54%) had at least one alteration affecting a gene in the HR pathway.
Several metrics were used to assess the HR deficiency and BRCAness: 1) HRD score given by the sum of Large Transition State (LST), LOH and Telomeric Allelic Imbalance (TAI) (taken from (Thorsson et al., 2018)); 2) Mutational Signature 3 as defined by Alexandrov et al. and computed using the YAPSA R package (Alexandrov et al., 2013). 3) Copy Number Signature 3 (taken from (Macintyre et al., 2018).
Sample preparation for MS analysis
Three biological replicates of each cell line, OVCAR5 BRCA1wt and BRCA1kd, UWB.1 289 BRCA1mut and BRCA1wt, were re-suspended in lysis buffer containing 2M Thiourea/6M Urea and 50 mM ammonium bicarbonate at pH8. Cell lysates were sonicated in a Bioruptor instrument for 15 cycles, at maximum mA for 30 s per cycle. The soluble fraction was collected after centrifugation at 20000 g at 4°C for 15 min. Protein concentration of the lysates was measured with a Bradford protein assay. Proteins were then reduced with 10 mM DTT for 30 min at room temperature (RT), followed by alkylation with 55 mM iodacetamide for another 30 min in the dark. Subsequently, digestion was carried out with an endoproteinase Lys-C and Trypsin mix. The first step consists of 1μg endoproteinase Lys-C digestion for 4 h at RT. Four volumes of 50 mM AMBIC were then added and further digested with 1μg Trypsin overnight. On the next day, samples were acidified with 10% trifluoroacetic acid (TFA) and desalted on C18 StageTips. Finally, samples were dried, resuspended in 2% ACN in 0.1% FA and kept at −20°C until MS analysis. Samples were usually injected once at 2.5 μg for MS analysis.
LC-MS/MS analysis of cell lines
Sample acquisition was performed on a nanoflow Ultra-HPLC Easy nLC 1200 (Thermo Fisher Scientific, LC140) coupled online to a Q Exactive HF Orbitrap mass spectrometer with a nanoelectrospray ion source. Peptide separation was achieved using inhouse ReproSil-Pur C18 (1.9 μm particles, 120 Åpore size) packed analytical columns (75 μm i.d. x 50 cm) with a PicoTip 8 mm tip opening. Mounted analytical columns were kept at 50°C using a column oven. The gradient length was 250 min with a buffer B (0.1% FA, 80% ACN) gradient ranging from 2% to 60% at 250 nL /min.
The MS scan range was set to 300 to 1,650 m/z with a resolution of 60000 (200 m/z) at an AGC target value of 3e6. For MS/MS, AGC target value of 1e5 was used with a maximum injection time of 25ms at a set resolution of 15000 (200 m/z). Data were acquired with data-dependent “top15” method, which isolates within a 1.4 m/z window the 15 most abundant precursor ions and fragments them by higher-energy collision dissociation (HCD) at normalized collision energy of 27%. The dynamic exclusion of precursor ions from further selection was set for 20 s.
Proteomics data analysis
We employed the MaxQuant (Cox and Mann, 2008) computational proteomics platform version 1.5.3.2 to search the peak lists against the UniProt databases (Human 2014) and a file containing 247 frequently observed contaminants. N-terminal acetylation and methionine oxidation were set as variable and cysteine carbamidomethylation as fixed modification, respectively. “Trypsin/P” was set for enzyme specificity and a peptide and protein false discovery rate (FDR) of 0.01 was specified. For peptide identification, a minimum number of 7 amino acids was required. ‘Match between runs’ module was enabled which allows the matching of identifications across different replicates of the same biological sample in a time window of 0.5 min and an initial alignment time window of 20 min. Label-free quantification (LFQ) was enabled in the MaxQuant environment (Cox et al., 2014). We used the Perseus computational platform version 1.5.5.3 (Tyanova et al., 2016) for the following statistical analysis. LFQ intensities of proteins were retrieved from the “ProteinGroups” MaxQuant output table. Proteins found as reverse hits, contaminants or only identified by site were filtered out. A filter was set for at least three valid intensity values in at least one group between OVCAR5 BRCA1wt and BRCA1kd, or between the UWB1.289 BRCA1mut and BRCA1wt. Missing intensities were imputed by drawing random numbers from a Gaussian distribution with a standard deviation of 20% in comparison to the standard deviation of measured protein abundances. Proteins were annotated based on “Keywords” in the Perseus gene annotation module. A volcano plot was generated where log2-fold changes of UWB1.289 BRCA1mut versus BRCA1wt group are indicated on the x axis and the corresponding significance levels were calculated by two-sided unpaired t test with an FDR of 0.01 and S0 of 0.3. Protein expression were also subjected to pathway analysis and differential expression in the same way than described for transcriptomics analyses.
ChIP
Harvested cells were washed with PBS, fixed with 1% formaldehyde for 10 min, quenched with 0.125M glycine for 5 min at RT, and washed twice with PBS. Fixed cells were pelleted, flash frozen, and stored at −80°C for further processing. Crosslinked cells were lysed with lysis buffer (1% SDS, 50mM Tris-HCl, pH 8.0, 20 mM EDTA, 1× complete EDTA-free protease inhibitor) on ice for 10 min, diluted with TE buffer, then sheared using a Branson Tip Sonifier 450 on ice (15 cycles, 15 s on, 45 s off/cycle at power 3). For bead preparation, Protein G Dynabeads were incubated with 3 μg of H3K27ac pAb overnight at 4°C. Incubated beads were washed 3 times with PBS with BSA. Detailed protocols for immunoprecipitation and library preparation are available on the ENCODE homepage.
Links to ENCODE homepage
In situ Hi-C
Harvested cells were washed once with PBS, fixed with 2% formaldehyde for 10 min in PBS at RT, quenched with 0.2 M glycine for 5 min. After washing once with PBS, pelleted cells were flash frozen and stored at −80°C for further processing. Nuclei were isolated with lysis buffer (10 mM Tris-HCl (pH 8.0), 10 mM NaCl, 0.2% IGEPAL CA630) with incubation on ice for 5 min, washed once same lysis buffer. Pelleted nuclei were permeabilized with 0.5% SDS for 10 min at 62°C, quenched with Triton X-100 for 15 min at 37°C. Chromatin was digested with a 4-cutter restriction enzyme (MboI, 100U, NEB) overnight at 37°C with mixing. After inactivating digestion at 62°C for 20 min, the 5′ overhangs were filled in with biotinylated-14-dATP (Life Tech) and Klenow (40U, NEB) for 90 min at 37°C with mixing. Biotinylated ends were ligated with T4 DNA ligase (2000U, NEB) for 4 h at RT with mixing. DNA was reverse crosslinked with Proteinase K (400mg, NEB) and 1% SDS for 30 min at 55°C, followed by an overnight incubation at 68°C with NaCl. DNA was purified with ethanol precipitation, and sheared on an ultrasonicator (Covaris S220; duty cycle: 10; intensity: 4; cycles/burst: 200; duration: 55 s; number of cycles: 1) to a fragment range of 300–700bp. Double size selection was performed using SPRI beads (Beckman Coulter). Biotinylated fragments were enriched by pulldown with Dynabeads MyOne T1 Streptavidin beads (Life Technologies), followed by library preparation (Quick Ligation Kit, NEB). Final library was amplified by PCR. Libraries were sequenced on HiSeq4000 Paired-End for 100bp (Illumina).
ChIP-seq analysis
H3K27ac epigenetic marks indicative of active enhancers were located using ChIPSeq. Chromatin precipitation with H3K27ac-specific antibodies on UWB1.289 BRCA1mut and BRCA1wt cells was performed as described above. Two replicates for each sample were processed as follows: ~50 million 50 nucleotide-long sequence reads were aligned to the GRCh38 reference genome using the shortread aligner bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml), with a mapping efficiency of ~90%. Regions enriched “broad peaks” of mapped reads were identified using the MACS2 software (https://mac3-project.github.io/MACS/). The–broad option (for broad peaks) in the callpeak module was used to call significant regions (experimental versus input control) at an FDR (q-value) of 0.05. The PAVIS website (https://manticore.niehs.nih.gov/pavis2) was used to annotate the enriched regions (gene-relative location; chromosome location; transcript ID; gene symbol; strand; distance to TSS). ChIPSeq data have been deposited in GEO (GSE122155).
Hi-C analysis
As a preliminary analysis, the hg38 reference human genome was divided into fragments delimited by GATC sites, which are sites recognized by the MboI restriction enzyme which was used for the HiC protocol. This produced a table for the 24 chromosomes, with start and end position of each fragment. In order to account for potential bias among the fragments, the table also contains the fragment length, the count of GC nucleotide pairs, and a measure of mappability which counts the number of sub-fragments of 36 nucleotides that can be uniquely mapped to each fragment. The table was further annotated by the average nucleotide coverage from two whole genome sequencing runs, one for the mutated BRCA1 condition and one for the rescued wild-type condition. These coverages are useful to determine copy number variations in the genomes of the cell lines.
The number of read pairs received as input for each sample
UWB1.289 BRCA1wt replicate 1: 832,401,844 / UWB1.289 BRCA1wt replicate 2: 730,444,209/ UWB1.289 BRCA1mut replicate 1: 622,942,922/ / SRC195/ UWB1.289 BRCA1mut replicate 2: 735,638,573
The HiC protocol causes two spatially close but genomically distant pieces of DNA to be cut at an enzymatic recognition site and ligated together at the cutting point. Thus, we expect to find GATCGATC motifs in the reads and the genomic origin of the fragments on each side of the motif will be different. At first, we searched for the GATCGATC double motif in each read and discarded the end of the read after the first GATC motif. If the remaining piece became too short (< 32 nucleotides) for unambiguous placement on the reference genome, the whole read pair was discarded. If no double GATCGATC motif was detected, it was assumed that the motif lied within the unsequenced part of the fragment between the two reads of the pair, and so those reads were kept for further processing.
The number of read pairs remaining after this first filtering step was as follows: UWB1.289 BRCA1wt replicate 1: 709,361,979/UWB1.289 BRCA1mut replicate 1: 555,857,566/ UWB1.289 BRCA1wt replicate 2: 639,748,714/ UWB1.289 BRCA1mut replicate 2: 655,781,263
We next mapped the reads on the hg38 human genome reference. We used an in-house derivative of our fetchGWI (Iseli et al., 2007) tool. For each read pair we obtain the position and orientation of the match on the reference genome. We also pre-computed a table containing the positions of all the GATC sites present in the hg38 reference human genome.
The current analysis aimed to determine cis-interactions at a maximum distance of 1,000,000 nucleotides, so we then selected mapped read pairs where both reads were mapped on the same chromosome and at a distance of no longer than 1 million base pairs. Read pairs that formed a usual illumina sequencing fragment (reads in opposing orientation and defining a fragment length of 2 kilobases or less) were discarded as probably not originating from ligation. At this point a table was produced containing the list of remaining read pairs, with their mapping position and orientation, as well as the position of the closest GATC restriction site.
Binning
The genome was divided into consecutive non-overlapping 5kbp bins. Each MboI restriction fragment was assigned to the bin containing the center of the fragment. For each chromosome k, a raw Hi-C count matrix Mk = {Mki,j} was defined, with Mki,j the number of read pairs with one read assigned to bin i and one read assigned to bin j.
Bias correction
Hi-C data are affected by many systematic biases such as mappability, GC content, fragment length (Yaffe and Tanay, 2011) and coverage (Wu and Michor, 2016). To remove these biases, we extended the method proposed by Hu and coauthors (Hu et al., 2012). We used a parametric probabilistic model, in which the raw Hi-C count Mki,j between bins i and j is assumed to follow a negative binomial with mean
and variance . Here is the average GC content, is the average mappability, is the total fragment length, is the number of fragments and is the average coverage for all restriction fragments assigned to bin i of chromosome k. A B-spline function Sk is used to model the expected decrease in the number of contacts between bins i and j when increasing their genomic distance di,j. Parameters , as well as the parameters of the B-spline were obtained by fitting this model to the raw Hi-C count matrix Mk, using pairs of bins on chromosome k separated by a genomic distance < 1Mbp. Model fitting was performed in R 3.4.2 using the MASS package (Venables et al., 2002) function.
Chromatin interactions
To detect chromatin interactions, for each chromosome k and each pair of bins i and j separated by less than 1Mbp on chromosome k, we evaluated the probability (denoted by ) to measure a raw count higher than , assuming that the raw Hi-C count data are explained by the model. A low indicates that the number of read pairs measured between the pair of bins is higher than expected by the model and is interpreted as an interaction between the two bins.
Enhancers
In this work, a genomic region was considered to be an enhancer of a gene if this region was enriched in H3K27ac histone modification, indicative of an active enhancer, and physically interacted (according to Hi-C data) with the transcription start site (Consortium et al.) of the gene.
More precisely, for all genes differentially expressed between wild-type and mutant cell lines (adjusted p value < 0.01, abs(log2(FC))) > 1), we considered all 5kbp bins containing at least one H3K27ac peak in the ChIPSeq data (q-value < 0.01) and located within a 2Mbp window centered around the bin containing the gene TSS.
A bin i with H3K27ac peak and a bin j with a gene TSS on chromosome k were considered to interact in a given cell line if the probability , after correction for multiple testing, was below 0.05 for both replicates of the cell line. Multiple testing correction was done using Benjamini & Hochberg correction, considering only pairs of bins with one bin containing the TSS of a differentially expressed gene and one bin containing a H3K27ac peak and separated by less than 1Mbp. This procedure was applied separately for each cell line to produce two lists of enhancers for all differentially expressed genes. The difference in numbers of enhancers between BRCA1mut and BRCA1wt was further used for preranked geneset enrichment analysis as inferred in the R fgsea package. HiC data have been deposited in GEO (GSE122155).
Cell line transduction
For transduction, the OVCAR5 cell line was seeded in a 6-well plate at a density of 5×105 cells per well and incubated with lentiviruses carrying the BRCA1 short hairpin or a non-specific targeting sequence (Sigma mission shRNA). For the generation of CRISPRED cell lines CRISPR-CAS9 bearing lentiviral constructs with sgRNAs targeting STING, IFI16, MAVS, TREX1 and TREX2. The transduced cells were then selected with puromycin (2 μg/ml final concentration).
Similarly, ID8 Trp53−/−Brca1wt and Trp53−/−Brca1−/− mouse ovarian cancer cell lines were infected with retroviruses carrying the Luciferase gene and selected with hygromycin (400 μg/ml final concentration). MSCV Luciferase PGK-hygro construct was a gift from Scott Lowe.
In order to generate an ID8Luc Trp53−/−Brca1−/− CCL5 and STING knockdown cell line, the lentiviral vector pLKO.1-puro was used. For the propagation of retroviral particles 293T cells were seeded at 6×106 per T75 tissue culture flask in RPMI-10+10% FBS medium 24 h before transfection. Cells were then transfected with 10 mg pLKO.1-puro CCL5 short hairpin plasmid, 1.58 mg pCMV-PAX2 plasmid and 3.125 mg of pMD2G plasmid using 59.175 mL Turbofect. The viral supernatant was harvested at 48 h post-transfection. PLKO.1-puromycin non-specific targeting lentiviral particles were also produced and used as control. The transduction of ID8Luc Trp53−/−Brca1−/− cells with lentiviruses was carried out as described above. The infected cells were then selected with puromycin (2 μg/ml).
Secretion of CCL5 was assessed using the BD cytokine bead array as per manufacturer’s instructions. STING expression was assessed by Western Blot, as described in the methods.
Poly(dA:dT) and poly(I:C) stimulation
Poly(dA:dT) or poly(I:C) and Turbofect were well diluted in serum-free medium (Opti-MEM, ThermoFischer Scientific). The nucleic acid/Opti-MEM mix was then added to the Turbofect/Opti-MEM mix in a drop-by-drop fashion (nucleic acid to Turbofect ratio 3:1) and incubated for 30 min at RT. 70%–80% confluent cells were then washed with PBS and incubated with the transfection reagent/DNA mixture for 6 h at 37°C. The final concentration of nucleic acid was 1 μg/ml. Cell pellets were processed for RNA or protein extraction as described below.
RNA isolation and quantitative real-time PCR
Cells were treated either with DMSO, Olaparib, Turbofect, poly(dA:dT) or poly(I:C) for the indicated times and concentrations. Total RNA was isolated from 100 to 500 mg of frozen tissue or 1×106 cultured cells with TRIzol reagent followed by RNA purification using the RNA Easy Mini Kit. After treatment with RNase-free DNase I, 1 μg of total RNA was reverse-transcribed using PrimeScript First Strand cDNA Synthesis Kit as indicated by manufacturer. Quantitative RT-PCR was performed using TaqMan® Fast Universal PCR reagents according to the manufacturer’s instructions. PCR amplification of the housekeeping gene, GAPDH, was performed for each sample as control for sample loading and to allow normalization among samples. Each sample was run in triplicate and each PCR experiment included three non-template control wells. P values were calculated using Mann–Whitney tests. All probes are listed in the Key resource table.
Western blot
Cells were treated either with DMSO, Olaparib, Turbofect, poly(dA:dT) or poly(I:C) for the indicated times and concentrations. The cells were then lysed with NE-PER Nuclear and cytoplasmic Extraction Kit supplemented with protease and phosphatase inhibitors following the manufacturer’s instructions. Immunoblots were conducted using the Bolt system. A total of 7.5 μg of protein was loaded per sample. All Abs are listed in the Key resource table.
Flow cytometry analysis of human cell lines
Cells were treated either with DMSO or Olaparib (10 μg/ml) for 48 h. Cells were then harvested with Accutase. Cells were permeabilized according to the manufacturer’s protocol (eBiosciences) and Fc receptors were blocked for 15 min at 4°C with anti-human Fc blocking antibody. Cells were fluorescently labeled with antibodies for 1 h at RT with the following antibodies: phospho-Stat1-AF488 and phospho-TBK1-AF647. Cells were then washed and resuspended in permeabilization buffer. Flow cytometric analysis was performed on FACSCanto flow cytometer (BD Biosciences) and analyzed using FlowJo software.
ELISA and cytokine bead array (CBA)
Cytokines concentrations were determined in cell-free supernatants of 48-hour cell cultures using the BD cytokine bead array according to manufacturer’s recommendations. All Flex Sets are listed in the Key resource table.
ID8 cells (1 × 105) were seeded and cultured in 6-well plates and treated with Olaparib (10 μM) for 48 h. Quantitative determination of VEGF-A in cell culture supernatants was assessed using a Mouse VEGF-A ELISA Kit according to the manufacturer’s instructions and after cell number normalization.
MTT assay
PARPi treatment impact on ID8 proliferation/viability was assessed using the Trevigen MTT Cell Proliferation Assay Kit. Cells were seeded at 3000 cells/well in a 96-well plate 24 h before Olaparib treatment. The proliferation assay was then performed after 24 h of treatment according to the manufacturer’s protocol. The optical density value was read at 570 nm in a microplate plate reader.
Surveyor assay
The PCR amplification of Trp53 and Brca1 exons containing the deletions was performed as previously described (Walton et al., 2016; Walton et al., 2017). Briefly, the PCR products were migrated on a 1% agarose gel and purified using the NucleoSpin Gel and PCR-Clean-up. Homoduplexes or heteroduplexes were made from the refined PCR products according to the manufacturer’s instructions of Surveyor Mutation Detection Kit using 100 ng of DNA per sample.
Chromogenic Immunohistochemistry tissue staining
The single chromogenic immunohistochemistry staining was performed on 4 μm formalin-fixed paraffin-embedded (FFPE) sections of ovarian cancer resections using the EnVision-Flex HRP kit on the Dako autostainer Link48. Briefly, slides were heated at 60°C for 1 hour, deparaffinized by immersing the slides in 3 consecutive xylene baths, followed by rehydration by immersing slides in 2 consecutive ethanol decreasing grade baths (100%, 95% and 70%) and in water bath. The antigen retrieval was performed in Citrate buffer (pH6, Dako) for 20 min at RT. Peroxidase blocking was performed for 5 min at RT (Flex Peroxidase Blocking, DAKO), followed by primary Ab incubation (CD8, γH2AX, pSTAT1, total STAT1) for 1 h at RT. After several washes, slides were incubation with the secondary Ab (Flex-HRP, DAKO) for 20 min at RT and Peroxidase were revealed with diaminobenzidine-peroxidase substrate (Flex DAB+Sub chromo, Dako) for 10 min at RT. After several washes, slides were counterstained with Flex-Hematoxylin, dehydrated by immersing slides in successive ethanol baths (70%, 95%, 100%) and xylene baths. The slides were then mounted with Shandon-mount medium and analyzed by microscopic observation and semiquantitative analysis by a qualified pathologist (PGF). All Abs are listed in the Key resource table.
Multiplex chromogenic Immunohistochemistry mouse tissue staining
The triple chromogenic immunohistochemistry assay was performed using the Ventana Discovery ULTRA automate (Roche Diagnostics, Rotkreuz, Switzerland). All steps were performed automatically with Ventana solutions except if specified otherwise. Dewaxed and rehydrated paraffin sections were pretreated with heat using the CC1 solution for 40 minutes at 95°C. Primary antibodies were applied and revealed sequentially either with a rat Immpress HRP (Ready to use, Vector laboratories Laboratories) or a rabbit UltraMap HRP followed by incubation with a chromogen (ChromoMap DAB, Discovery purple and Discovery Teal). A heat denaturation step was performed after every revelation. The primary antibodies sequence was: rat anti-CD31, rat anti-CD8 and rabbit anti-PanCytokeratin. Sections were counterstained with Harris hematoxyline (J.T. Baker) and permanently mounted with Pertex (Sakura). For immunohistochemical quantification of CD8+ cells and CD31+ cells, 10 × 10 tiled bright-field pictures of FPPE sections were taken at 100 mm magnification. Cell counts were obtained using ImageJ software. All Abs are listed in the Key resource table.
Multispectral immunofluorescence tissue staining and image analyses
For the multiplexed staining, FFPE sections of surgical ovarian tumor resections were stained by automated immunostainer (DISCOVERY ULTRA, Ventana Roche). First, the Heat-induced antigen retrieval in EDTA buffer (pH 8.0) was performed for 92 min at 95°C. Multiplex staining was performed in consecutive rounds, each round consisting of protein blocking, primary antibody incubation, secondary HRP-labeled antibody incubation, OPAL detection reagents and then antibodies heat denaturation. The primary antibodies anti-STING, γH2AX, pSTAT1 Ab were incubated at RT for 60 min, and anti-pan Cytokeratin, CD8 at 37°C for 60 min. The signal was revealed with DISCOVERY OmniMap anti-rabbit HRP or anti-mouse incubated for 16 min after 8 min of incubation by the IgG Goat Blocker. Then, the OPAL reagents were incubated for 12 min. The nuclei were visualized with Spectral DAPI after 12 min of incubation.
The Multiplex IF images were acquired on Vectra 3.0 automated quantitative pathology imaging system (Perkin Elmer). Tissue and panel specific spectral library of the specific panel individual fluorophore and tumor tissue autofluorescence were acquired for an optimal IF signal unmixing (individual spectral peaks) and multiplex analysis,
The IF stained slides were pre-scanned at 10× magnification. Using the Phenochart whole-slide viewer, regions of interest containing tumor islets and stroma (at least 100 MSI per sample) were annotated for high-resolution multispectral acquisition of images at 20× magnification. All Abs are listed in the Key resource table.
Confocal microscopy
Cells were plated in chambers slides at a 70%–80% confluency (5×103 cells per chamber), one day prior Olaparib (5 μg/ml) or DMSO treatment. Following 48 h treatment, cells were washed with PBS prior fixation with NBF for 15 min at RT. Cells were then permeabilized with 0.25% Triton X-100 in PBS for 15 min at RT. After blocking with 5% BSA, fixed cells were incubated overnight at 4°C with primary antibodies. Secondary antibodies were incubated for 1 hour at RT. Nuclei were counterstained with DAPI (2 μg/ml in PBS) for 5 min at RT. Slides were then mounted using Fluoromount-G. Confocal images were obtained using a Zeiss LSM 510 META microscope and analyzed using ImageJ (https://imagej.nih.gov/ij/). All Abs are listed in the Key resource table.
Bioluminescence imaging
Tumor growth was also monitored by Bioluminescent imaging (BLI). BLI was performed using Xenogen IVIS® Lumina II imaging system and the photons emitted by the Luciferase-expressing cells within the animal body were quantified using Living Image software. Briefly, mice bearing ID8Luc cancer cells were injected i.p. with D-luciferin (150 mg/kg stock, 100 μL of D-luciferin per 10 g of mouse body weight) resuspended in PBS and imaged under isoflurane anesthesia after 5~10 min. A pseudocolor image representing light intensity (blue, least intense; red, most intense) was generated using Living Image. BLI findings were confirmed at necropsy.
Flow cytometry and analysis of murine samples
At the time of sacrifice, i.p. cancers were dissected. Tumors were digested in 200 μg/ml Liberase TL and 5 units/ml DNase I in DMEM for 1 h at 37°C, with rotation. For ex vivo staining, 1–2×106 cells were stained with LIVE/DEAD Fixable Aqua Dead Cell Stain (1:500). Fc receptors were blocked for 10 min at 4°C with 5 μg/ml Mouse BD FC Block. Cells were fluorescently labeled with antibodies for 30 min at 4°C, washed and resuspended in fixation buffer (1% formaldehyde in PBS) or intracellularly stained according to the manufacturer’s protocol (eBiosciences). Flow cytometric analysis was performed on LSR II flow cytometer and analyzed using FlowJo software. All Abs are listed in the Key resource table.
Statistical analyses
All statistical tests were performed using R (version 3.3.0), Perseus and GraphPad Prism softwares. All of the statistical details of experiments can be found in the figure legends, figures and Results, including the statistical tests used, exact value of n, what n represents (e.g., number of technical and biological replicates, number of animals, etc.), definition of mean or median, and dispersion and precision measures (SD, SEM, confidence intervals).
Supplementary Material
Highlights.
STING and type I IFN pathway activation leads to T cell infiltration in BRCA1mut OC
STING drives VEGF-A upregulation in BRCA1mut tumor cells
STING loss reduces angiogenesis, boosts CD8 T cells, and reverts dual ICB resistance
Anti-VEGF-A, PARPis, and dual ICB combination control Brca1−/− tumor growth in vivo
ACKNOWLEDGMENTS
This study was supported by the Ludwig Institute for Cancer Research, NIH Grant P50 CA083638, SPORE in Ovarian Cancer, the Emma Mouschamp Foundation, the Porphyrogenis Foundation, and by Stand Up To Cancer/Ovarian Cancer Research Fund Alliance/National Ovarian Cancer Coalition Dream Team Translational Cancer Research Grant (SU2C-AACR-DT16–15, to E.S.). We thank Pinelopi Chatziemmanouil, Ekaterina Fortis, and Kariman Chaba for technical assistance; Florence Morgenthaler for advice with confocal microscopy; Genentech for mouse anti-VEGF-A antibody; and the Lausanne Genomic Technologies Facility for RNA-seq analysis. Triple IHC on mouse tumor tissue sections was performed at the EPFL Histology Core Facility, and we thank Dr. Jessica Dessimoz for advice and assistance.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109412.
DECLARATION OF INTERESTS
G. Coukos has received grants from Celgene, Boehringer-Ingelheim, Roche, BMS, Iovance Therapeutics, and Kite Pharma. The institution G. Coukos is affiliated with has received fees for G. Coukos’ participation on an advisory board or for presentation at a company-sponsored symposium from Genentech, Roche, BMS, AstraZeneca, NextCure, Geneos Tx, and Sanofi/Avensis. G. Coukos has patents in the domain of antibodies and vaccines targeting the tumor vasculature as well as technologies related to T cell expansion and engineering for T cell therapy. G. Coukos holds patents around antibodies and receives royalties from the University of Pennsylvania regarding technology licensed to Novartis. The remaining authors declare no competing interests.
REFERENCES
- Abe T, and Barber GN (2014). Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol 88, 5328–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Ruiz P, and Barber GN (2014). Intrinsic self-DNA triggers inflammatory disease dependent on STING. J. Immunol 193, 4634–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. ; Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed]
- Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M., et al. (2018). Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 174, 1293–1308.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernards SS, Norquist BM, Harrell MI, Agnew KJ, Lee MK, Walsh T, and Swisher EM (2016). Genetic characterization of early onset ovarian carcinoma. Gynecol. Oncol 140, 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A., et al. (2013). Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795. [DOI] [PubMed] [Google Scholar]
- Blayney JK, Davison T, McCabe N, Walker S, Keating K, Delaney T, Greenan C, Williams AR, McCluggage WG, Capes-Davis A., et al. (2016). Prior knowledge transfer across transcriptional data sets and technologies using compositional statistics yields new mislabelled ovarian cell line. Nucleic Acids Res. 44, e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, Katsaros D, O’Brien-Jenkins A, Gimotty PA, and Coukos G (2008). Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat. Med 14, 28–36. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas. (2019). Interferon-γ signaling is associated with BRCA1 loss-of-function mutations in high grade serous ovarian cancer. NPJ Precis Oncol. 10.1038/s41698-019-0103-4. [DOI] [PMC free article] [PubMed]
- Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, Lamb A, Hénon C, Dorvault N, Rouanne M., et al. (2019). PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J. Clin. Invest 129, 1211–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HL, Lin CT, Huang KW, Wang S, Lin YT, Toh SI, and Hsiao YY (2018). Structural insights into the duplex DNA processing of TREX2. Nucleic Acids Res. 46, 12166–12176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A., et al. (2015). Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162, 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YH, Macmillan JB, and Chen ZJ (2009). RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, and Mann M (2014). Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, Lanitis E, Duraiswamy J, Tanyi JL, Benencia F., et al. (2019). Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell 35, 885–900.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DelloRusso C, Welcsh PL, Wang W, Garcia RL, King MC, and Swisher EM (2007). Functional characterization of a novel BRCA1-null ovarian cancer cell line in response to ionizing radiation. Mol. Cancer Res 5, 35–45. [DOI] [PubMed] [Google Scholar]
- Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, Li BB, Xie S, Liu JF, Stover EH, et al. (2018). PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep 25, 2972–2980.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domchek SM (2017). Reversion mutations with clinical use of PARP inhibitors: Many genes, many versions. Cancer Discov. 7, 937–939. [DOI] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z., et al. (2017). Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraiswamy J, Freeman GJ, and Coukos G (2014). Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors–Response. Cancer Res 74, 633–634, discussion 635. [DOI] [PubMed] [Google Scholar]
- Forrest AR, Kawaji H, Rehli M, Baillie JK, de Hoon MJ, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, Itoh M., et al. ; FANTOM Consortium and the RIKEN PMI and CLST (DGT) (2014). A promoter-level mammalian expression atlas. Nature 507, 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George J, Alsop K, Etemadmoghadam D, Hondow H, Mikeska T, Dobrovic A, deFazio A, Smyth GK, Levine DA, Mitchell G, and Bowtell DD; Australian Ovarian Cancer Study Group (2013). Nonequivalent gene expression and copy number alterations in high-grade serous ovarian cancers with BRCA1 and BRCA2 mutations. Clin. Cancer Res 19, 3474–3484. [DOI] [PubMed] [Google Scholar]
- González-Martín A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, McCormick C, Lorusso D, Hoskins P, Freyer G., et al. ; PRIMA/ENGOT-OV26/GOG-3012 Investigators (2019). Niraparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med 381, 2391–2402. [DOI] [PubMed] [Google Scholar]
- Goode EL, Block MS, Kalli KR, Vierkant RA, Chen W, Fogarty ZC, Gentry-Maharaj A, Tołoczko A, Hein A, Bouligny AL, et al. ; Ovarian Tumor Tissue Analysis (OTTA) Consortium (2017). Dose-response association of CD8+ tumor-infiltrating lymphocytes and survival time in high-grade serous ovarian cancer. JAMA Oncol. 3, e173290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooden MJ, de Bock GH, Leffers N, Daemen T, and Nijman HW (2011). The prognostic influence of tumour-infiltrating lymphocytes in cancer: A systematic review with meta-analysis. Br. J. Cancer 105, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, and Greenberg RA (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Härtlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kröger A, Nilsson JA, et al. (2015). DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 42, 332–343. [DOI] [PubMed] [Google Scholar]
- Heijink AM, Talens F, Jae LT, van Gijn SE, Fehrmann RSN, Brummelkamp TR, and van Vugt MATM (2019). BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat. Commun 10, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Deng K, Selvaraj S, Qin Z, Ren B, and Liu JS (2012). HiCNorm: Removing biases in Hi-C data via Poisson regression. Bioinformatics 28, 3131–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseli C, Ambrosini G, Bucher P, and Jongeneel CV (2007). Indexing strategies for rapid searches of short words in genome sequences. PLoS ONE 2, e579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997.e24. 10.1016/j.cell.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinopoulos PA, Ceccaldi R, Shapiro GI, and D’Andrea AD (2015). Homologous recombination deficiency: Exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 5, 1137–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, Van Le L, Sachdev JC, Chapman-Davis E, Colon-Otero G., et al. (2019). Single-arm phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol. 5, 1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, and Chen ZJ (2015). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630. [DOI] [PubMed] [Google Scholar]
- Macintyre G, Goranova TE, De Silva D, Ennis D, Piskorz AM, Eldridge M, Sie D, Lewsley LA, Hanif A, Wilson C., et al. (2018). Copy number signatures and mutational processes in ovarian carcinoma. Nat. Genet 50, 1262–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlpine JN, Porter H, Köbel M, Nelson BH, Prentice LM, Kalloger SE, Senz J, Milne K, Ding J, Shah SP, et al. (2012). BRCA1 and BRCA2 mutations correlate with TP53 abnormalities and presence of immune cell infiltrates in ovarian high-grade serous carcinoma. Mod. Pathol 25, 740–750. [DOI] [PubMed] [Google Scholar]
- Motz GT, and Coukos G (2011). The parallel lives of angiogenesis and immunosuppression: Cancer and other tales. Nat. Rev. Immunol 11, 702–711. [DOI] [PubMed] [Google Scholar]
- Nesbeth YC, Martinez DG, Toraya S, Scarlett UK, Cubillos-Ruiz JR, Rutkowski MR, and Conejo-Garcia JR (2010). CD4+ T cells elicit host immune responses to MHC class II-negative ovarian cancer through CCL5 secretion and CD40-mediated licensing of dendritic cells. J. Immunol 184, 5654–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, Visal T, Li MK, Pinto J, Castrillon JA, et al. (2019). PARP inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. 9, 722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, McCloskey KD, Buckley NE, Savage KI, Salto-Tellez M., et al. (2016). Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast cancer. J. Natl. Cancer Inst 109, djw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, Thornton A, Norquist BM, Casadei S, Nord AS, et al. (2014). Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res 20, 764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, Fujiwara K, Vergote I, Colombo N, Mäenpää J., et al. ; PAOLA-1 Investigators (2019). Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N. Engl. J. Med 381, 2416–2428. [DOI] [PubMed] [Google Scholar]
- Reisländer T, Lombardi EP, Groelly FJ, Miar A, Porru M, Di Vito S, Wright B, Lockstone H, Biroccio A, Harris A., et al. (2019). BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun 10, 3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscito I, Cacsire Castillo-Tong D, Vergote I, Ignat I, Stanske M, Vanderstichele A, Glajzer J, Kulbe H, Trillsch F, Mustea A., et al. (2018). Characterisation of tumour microvessel density during progression of high-grade serous ovarian cancer: Clinico-pathological impact (an OCTIPS Consortium study). Br. J. Cancer 119, 330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghafinia S, Mina M, Riggi N, Hanahan D, and Ciriello G (2018). Pan-cancer landscape of aberrant DNA methylation across human tumors. Cell Rep 25, 1066–1080.e8. [DOI] [PubMed] [Google Scholar]
- Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, and Buetow KH (2009). PID: The Pathway Interaction Database. Nucleic Acids Res 37, D674–D679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, Cristea S, Nguyen T, Diao L, Li L., et al. (2019). Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 9, 646–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stordal B, Timms K, Farrelly A, Gallagher D, Busschots S, Renaud M, Thery J, Williams D, Potter J, Tran T., et al. (2013). BRCA1/2 mutation analysis in 41 ovarian cell lines reveals only one functionally deleterious BRCA1 mutation. Mol. Oncol 7, 567–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, Garber JE, Chowdhury D, Wu CJ, D’Andrea AD, et al. (2016). Association and prognostic significance of BRCA1/2-mutation status with neo-antigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 7, 13587–13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson BJ, Murakami M, Mitchell TC, Kappler J, and Marrack P (2002). RANTES production by memory phenotype T cells is controlled by a posttranscriptional, TCR-dependent process. Immunity 17, 605–615. [DOI] [PubMed] [Google Scholar]
- Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, Kiialainen A, Hanhart J, Schill C, Hess C., et al. (2018). A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med 24, 994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. ; Cancer Genome Atlas Research Network (2018). The immune landscape of cancer. Immunity 48, 812–830.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V., et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, and Demaria S (2017). DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun 8, 15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables WN, Ripley BD, and Venables WN (2002). Modern Applied Statistics with S, Fourth Edition (Springer; ). [Google Scholar]
- Vinayak S, Tolaney SM, Schwartzberg L, Mita M, McCann G, Tan AR, Wahner-Hendrickson AE, Forero A, Anders C, Wulf GM, et al. (2019). Open-label clinical trial of niraparib combined with pembrolizumab for treatment of advanced or metastatic triple-negative breast cancer. JAMA Oncol. 5, 1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton J, Blagih J, Ennis D, Leung E, Dowson S, Farquharson M, Tookman LA, Orange C, Athineos D, Mason S., et al. (2016). CRISPR/Cas9-mediated Trp53 and Brca2 knockout to generate improved murine models of ovarian high-grade serous carcinoma. Cancer Res 76, 6118–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton JB, Farquharson M, Mason S, Port J, Kruspig B, Dowson S, Stevenson D, Murphy D, Matzuk M, Kim J., et al. (2017). CRISPR/Cas9-derived models of ovarian high grade serous carcinoma targeting Brca1, Pten and Nf1, and correlation with platinum sensitivity. Sci. Rep 7, 16827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YK, Bashashati A, Anglesio MS, Cochrane DR, Grewal DS, Ha G, McPherson A, Horlings HM, Senz J, Prentice LM, et al. (2017). Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat. Genet 49, 856–865. [DOI] [PubMed] [Google Scholar]
- Wang Z, Sun K, Xiao Y, Feng B, Mikule K, Ma X, Feng N, Vellano CP, Federico L, Marszalek JR, et al. (2019). Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci. Rep 9, 1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston GC, Haviv I, and Rogers PA (2002). Microarray analysis of VEGF-responsive genes in myometrial endothelial cells. Mol. Hum. Reprod 8, 855–863. [DOI] [PubMed] [Google Scholar]
- Wu HJ, and Michor F (2016). A computational strategy to adjust for copy number in tumor Hi-C data. Bioinformatics 32, 3695–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe E, and Tanay A (2011). Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nat. Genet 43, 1059–1065. [DOI] [PubMed] [Google Scholar]
- Zhang X, and Li R (2018). BRCA1-dependent transcriptional regulation: Implication in tissue-specific tumor suppression. Cancers (Basel) 10, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, et al. (2003). Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med 348, 203–213. [DOI] [PubMed] [Google Scholar]
- Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, Zhou JY, Petyuk VA, Chen L, Ray D., et al. ; CPTAC Investigators (2016). Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell 166, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, Gage FH, and Verma IM (2011). BRCA1 tumour suppression occurs via hetero-chromatin-mediated silencing. Nature 477, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are deposited in the GEO repositories and accession numbers are stated in the Key Resources Table. This study did not generate new unique code.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| rat anti-CD31, clone SZ31 | Dianova | Cat#DIA-310; RRID:AB_2631039 |
| rat anti-CD8, clone 4SM15 | Thermo fisher) | Cat#14–0808-82; RRID: AB_2572861 |
| rabbit anti-PanCK, polyclonal | Novus Biologicals | Cat#NBP2–44368 |
| anti-mouse CD45-BV650, clone 30F11,1:400 | Biolegend | Cat#103151; RRID: AB_2565884 |
| anti-mouse CD3e-PECy5.5,clone 145–2C11, 1:100 | Invitrogen | Cat#;35–0031-80; RRID: AB_11218085 |
| anti-mouse CD4-PB, clone GK1.5, 1:100 | Biolegend | Cat#100428; RRID: AB_493647 |
| anti-mouse CD8-BV711, clone 53.6.7, 1:50 | Biolegend | Cat#100748; RRID: AB_2562100 |
| anti-mouse CD11b-PECy7, clone M1/70,1:200 | eBioscience | Cat#25–0112-81; RRID: AB_469587 |
| anti-mouse CD11c-BV60, clone N418 | Biolegend | Cat#117334; RRID: AB_2562415 |
| anti-mouse F4/80-APCCy7, clone BM8,1:200 | Biolegend | Cat#123118; RRID: AB_893477 |
| anti-mouse F4/80-Alexa647, clone F4/80,1:50 | home-made | |
| anti-mouse PD-L1-BV711, clone 10F.9G2,1:200 | Biolegend | Cat#124319; RRID: AB_2563619 |
| anti-mouse Ki67-APC, clone 16A8, 1:200 | Biolegend | Cat#652406; RRID: AB_2561930 |
| anti-mouse CD80-PEDazzle,clone 16–10A1, 1:100 | Biolegend | Cat#104737; RRID: AB_2564174 |
| anti-mouse CD86-APCCy7, clone GL-1,1:100 | Biolegend | Cat#105030; RRID: AB_2244452 |
| anti-mouse CD103-PE, clone 2E7, 1:200 | Biolegend | Cat#121405; RRID: AB_535948 |
| Anti-Mouse CD16/CD32 (Mouse BD Fc Block), Clone 2.4G2, 1:100 | BD | Cat#553142; RRID:AB_394657 |
| LIVE/DEAD Fixable Aqua Dead Cell Stain, 1:500 | Invitrogen | Cat#L34957 |
| anti-human BRCA1, clone MS110, 1:500 | Millipore | Cat#MABC199; RRID: AB_213438 |
| anti-human IFI16, clone 1G7, 1:500 | Santa Cruz Biotechnology | Cat#sc-8023; RRID: AB_627775 |
| anti-human and mouse NF-κB, clone C-20, 1:500 | Santa Cruz Biotechnology | Cat#sc-372; RRID: AB_632037 |
| anti-human and mouse phosphor-IRF3, clone 4D4G, 1:500 | Cell Signaling Technology | Cat#4947S;,RRID: AB_823547 |
| anti-human and mouse STING, clone D2P2F, 1:500 | Cell Signaling Technology | Cat#13647S; RRID: AB_2732796 |
| anti-human and mouse phospho-STAT1, clone D4A7, 1:500 | Cell Signaling Technology | Cat#7649S; RRID: AB_10950970 |
| anti-human and mouse STAT1, clone D4Y6Z, 1:500 | Cell Signaling Technology | Cat#14995S; RRID: AB_2716280 |
| anti-human and mouse Lamin B, clone M-20, 1:1000 | Santa Cruz Biotechnology | Cat#sc-6217; RRID: AB_648158 |
| anti-human and mouse B-ACTIN, clone C4, 1:500 | Santa Cruz Biotechnology | Cat#sc-47778; RRID: AB_2714189 |
| dsDNA Marker, HYB331–01, 1:1000 | Santa Cruz Biotechnology | Cat#sc-58749; RRID: AB_783088 |
| anti-phospho-Histone H2A.X (Ser139), clone JBW301, 1:500 | Millipore | Cat#05–636; RRID: AB_309864 |
| anti-human and mouse phospho-TBK1/NAK(Ser172), clone D52C2, 1:100 | Cell Signaling Technology | Cat#5483; RRID: AB_10693472 |
| anti-human CD8, clone SP16, 1:100 | ThermoFisher Scientific | Cat#MA5–14548; RRID: AB_10984334 |
| anti-human and mouse STAT1, 1:2000 | Abcam | Cat#ab47425; RRID: AB_882708 |
| anti-human and mouse phospho-STAT1, Clone 58D6 | Cell Signaling Technology | Cat#9167S; RRID: AB_561284 |
| anti-human pan Cytokeratin, clone AE1/AE3 | DAKO | Cat#M351501–2; RRID: AB_2631307 |
| anti-human and mouse phospho-Stat1-AF488, clone 58D6, 1:100 | Cell Signaling Technology | Cat#9174S; RRID: AB_2198287 |
| anti-human and mouse phospho-TBK1-AF647,clone D52C2, 1:100 | Cell Signaling Technology | Cat#14590S; RRID: AB_2798527 |
| InVivoMAb anti-mouse CTLA-4, clone 9D9 | BioXCell | Cat#BE0164; RRID: AB_10949609 |
| InVivoMAb anti-mouse PD-L1, clone 10F.9G2 | BioXCell | Cat#BE0101; RRID: AB_10949609 |
| anti-VEGFA mAb, clone B20–4.1.1 | Genentech | a kind gift from Genentech |
| InVivoMAb anti-mouse IFNAR-1, clone MAR1–5A3 | BioXCell | Cat#BE0241; RRID: AB_2687723 |
| InVivoMAb IgG1 Isotype control, clone MOPC-21 | BioXCell | Cat#BE0083; RRID: AB_1107784 |
| H3K27ac pAb | Active Motif | Cat#31933 |
|
| ||
| Biological samples | ||
|
| ||
| HGSOC specimens | (Bernards et al., 2016; Pennington et al., 2014); https://doi.org/10.1016/j.ygyno.2015.12.017 https://doi.org/10.1158/1078–0432.CCR-13–2287 | |
| ovarian carcinomas TCGA Consortium | TCGA | (Cancer Genome Atlas Research Network, 2011); https://doi.org/10.1038/nature10166 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Spectral DAPI, 1:5 | PerkinElmer | Cat#FP1490 |
| IgG Goat Blocker | Ventana Roche | Cat#760–6008 |
| DISCOVERY OmniMap anti-rabbit HRP | Ventana Roche | Cat#760–4311 |
| DISCOVERY OmniMap anti-mouse HRP | Ventana Roche | Cat#760–4310 |
| OPAL 520 | PerkinElmer | Cat#FP1487001KT |
| OPAL 570 | PerkinElmer | Cat#FP1488001KT |
| OPAL 620 | PerkinElmer | Cat#FP1495001KT |
| OPAL 690 | PerkinElmer | Cat#FP1497001KT |
| Olaparib, AZD2281 | Selleckchem | Cat#S1060 |
| poly(dA:dT) | Invivogen | Cat# tlrl-patn, tlrl-patn-1 |
| poly(I:C) | Invivogen | Cat#tlrl-pic |
| Turbofect | ThermoFischer Scientific | Cat#R0532 |
| D-luciferin | Biosynth | Cat#L-8220 |
| Liberase TL | Roche | Cat#540102001 |
| Dnase I | Sigma Aldrich | Cat#D4527 |
| Hygromycin | Millipore | Cat#400052 |
| Puromycin | Invivogen | Cat#ant-pr-1 |
| TRIzol reagent | Invitrogen | Cat#15596026 |
| DMEM | ThermoFischer Scientific | Cat#41966–029 |
| RPMI 1640 | ThermoFischer Scientific | Cat#1870010 |
| Opti-MEM I Reduced Serum Medium, no phenol red | ThermoFischer Scientific | Cat#11058021 |
| TaqMan® Fast Universal PCR reagents | Applied Biosystems | Cat#4366072 |
| Shandon Shandon-Mount | ThermoFischer Scientific | Cat#1900331 |
|
| ||
| Recombinant DNA | ||
|
| ||
| MISSION pLKO.1-puro non-target shRNAcontrol | Sigma | Cat#SHC016 |
| BRCA1 MISSION shRNA | Sigma | Cat#TRCN0000244984 |
| TMEM173 MISSION shRNA Bacterial Glycerol Stock transmembrane protein 173 (STING kd1) | Sigma | Cat#TRCN0000346266 |
| TMEM173 MISSION shRNA Bacterial Glycerol Stock transmembrane protein 173 (STING kd2) | Sigma | Cat#TRCN0000346320 |
| CCL5 MISSION shRNA Bacterial GlycerolStock chemokine (C-C motif) ligand 5 | Sigma | Cat#TRCN0000068102 |
| MSCV Luciferase PGK-hygro constrcuct | Addgene | Cat#18782 |
|
| ||
| Commercial assays | ||
|
| ||
| Human IFNa Flex Set | BD | Cat#560379 |
| Human IL-1a Flex Set | BD | Cat#560153 |
| Human IL-1b Flex Set | BD | Cat#561509 |
| Human TNFa Flex Set | BD | Cat#561516 |
| Human CXCL10 Flex Set | BD | Cat#558280 |
| Human Fractalkine (CX3CL1) Flex Set | BD | Cat#560265 |
| Human IL-6 Flex Set | BD | Cat#558276 |
| Human GM-CSF Flex Set | BD | Cat#558335 |
| Mouse RANTES (CCL5) Flex Set | BD | Cat#558345 |
| MTT Cell Proliferation Assay kit | R&D Systems | Cat#4890–025-K |
| RNA Easy Mini Kit | QIAGEN | Cat#74104 |
| PrimeScript First Strand cDNA Synthesis Kit | Takara | Cat#6110A |
| NE-PER Nuclear and cytoplasmic Extraction Kit | ThermoFischer Scientific | Cat#78833 |
| Halt Protease and Phosphatase Inhibitor Cocktail (100X) | ThermoFischer Scientific | Cat#78440 |
| NucleoSpin Gel and PCR-Clean-up | Macherey-Nagel | Cat#740609.50 |
| Illumina TruSeq Stranded Total RNA reagents | Illumina | Cat#RS-122–2201 |
| Illumina HiSeq PE Cluster Kit v4 cBot reagents | Illumina | Cat#PE-401–400 |
| HiSeq SBS Kit V4 reagents | Illumina | Cat#FC-401–4002 |
| Mouse VEGF-A ELISA Kit | Abcam | Cat#ab209882 |
|
| ||
| Deposited data | ||
|
| ||
| ChIPSeq data | GEO | GSE122155; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122155 |
| HiC data | GEO | GSE122155; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122155 |
| Human RNA sequencing data | GEO | GSE120792; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120792 |
| Mouse RNA sequencing data | GEO | GSE162935; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162935 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| OVCAR5 BRCA1wt and OVCAR5 BRCA1kd | our own laboratory | |
| UWB1.289 (ATCC® CRL-2945), BRCA1mut | ATCC | RRID:CVCL_B079 |
| UWB1.289 BRCA1+ (ATCC® CRL-2946), BRCA1wt | ATCC | RRID:CVCL_B078 |
| ID8 Trp53−/− and ID8 Trp53−/−Brca1−/− | Prof. Iain A. McNeish lab | (Walton et al., 2016; Walton et al., 2017); https://doi.org/10.1158/0008–5472. CAN-16–1272 https://doi.org/10.1038/s41598–017-17119–1 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57/BL6 | Envigo | 057 |
|
| ||
| Oligonucleotides | ||
|
| ||
| CCL5 | Applied Biosystems | Cat#Hs00982282_m1 |
| CXCL9 | Applied Biosystems | Cat#Hs00171065_m1 |
| CXCL10 | Applied Biosystems | Cat#Hs01124251_g1 |
| CD8A | Applied Biosystems | Cat#Hs00233520_m1 |
| IFNB1 | Applied Biosystems | Cat#Hs01077958_s1 |
| IFNA1 | Applied Biosystems | Cat#Hs00256882_s1 |
| TNFA | Applied Biosystems | Cat#Hs01113624_g1 |
| ISG20 | Applied Biosystems | Cat#Hs00158122_m1 |
| ISG15 | Applied Biosystems | Cat#Hs01921425_s1 |
| VEGFA | Applied Biosystems | Cat#Hs03929036_s1 |
| VEGFB | Applied Biosystems | Cat#Hs00173634_m1 |
| VEGFC | Applied Biosystems | Cat#Hs01099203_m1 |
| GAPDH | Applied Biosystems | Cat#Hs03929097_g1 |
| Cd8a | Applied Biosystems | Cat#Mm01188922_m1 |
| Ccl5 | Applied Biosystems | Cat#Mm01302427_m1 |
| Cxcl9 | Applied Biosystems | Cat#Mm00434946_m1 |
| Stat1 | Applied Biosystems | Cat#Mm01257286_m1 |
| Gzmb | Applied Biosystems | Cat#Mm00442837_m1 |
| Ifng | Applied Biosystems | Cat#Mm01168134_m1 |
| Itgax | Applied Biosystems | Cat#Mm00498701_m1 |
| Batf3 | Applied Biosystems | Cat#Mm01318275_m1 |
| Ifnb1 | Applied Biosystems | Cat#Mm00439552_s1 |
| Gapdh | Applied Biosystems | Cat#Mm99999915_g1 |
| Vegfa | Applied Biosystems | Cat#Mm00437306_m1 |
| Vegfb | Applied Biosystems | Cat#Mm00442102_m1 |
| Vegfc | Applied Biosystems | Cat#Mm00437310_m1z |
| Tnfa | Applied Biosystems | Cat#Mm00443258_m1 |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| R | Open source | https://www.r-project.org/ |