

ABSTRACT
Lipid droplets (LDs) contribute to key pathways important for the physiology and pathophysiology of cells. In a homeostatic view, LDs regulate the storage of neutral lipids, protein sequestration, removal of toxic lipids and cellular communication; however, recent advancements in the field show these organelles as essential for various cellular stress response mechanisms, including inflammation and immunity, with LDs acting as hubs that integrate metabolic and inflammatory processes. The accumulation of LDs has become a hallmark of infection, and is often thought to be virally driven; however, recent evidence is pointing to a role for the upregulation of LDs in the production of a successful immune response to viral infection. The fatty acids housed in LDs are also gaining interest due to the role that these lipid species play during viral infection, and their link to the synthesis of bioactive lipid mediators that have been found to have a very complex role in viral infection. This review explores the role of LDs and their subsequent lipid mediators during viral infections and poses a paradigm shift in thinking in the field, whereby LDs may play pivotal roles in protecting the host against viral infection.
Keywords: lipid droplets, immunity, lipid mediators, virus, infection, antiviral
Lipid droplets are key organelles in multiple cellular processes; recent advances in the field have demonstrated that they together with their lipid mediators may play pivotal roles in the outcome of viral infection.
INTRODUCTION
Lipids are a major and diverse class of biomolecules that play an important role in both the physiology and pathophysiology of cells. Generally, lipids are thought to have three main functions within a cell. First, polar lipids make up the matrix of membranes and provide cellular and organelle membranes the ability for budding, tubulation, fusion and fission (reviewed in Casares, Escribá and Rosselló 2019). Specific lipids also enable the formation of highly ordered regions within the plasma membrane (lipid rafts) that serve as key platforms for receptor signaling events (reviewed in Simons and Sampaio 2011). Second, lipids can act as first and second messengers in signal transduction and molecular recognition processes and have signaling roles during infection and inflammation (reviewed in van Meer, Voelker and Feigenson 2008; Fahy et al. 2009; Subramaniam et al. 2011; Teng, Ang and Guan 2017). Lastly, fatty acids can be packaged into lipid droplets (LDs), where they can be used for energy storage. Triglycerides (TGs) and sterol esters (sterols esterified by a fatty acyl chain) are the major components of LDs, where they serve to store energy for a host cell in times of nutrient deprivation (reviewed in Jackson, Walch and Verbavatz 2016). TGs and sterile esters form the neutral lipid core of LDs and are surrounded by a phospholipid membrane monolayer with membrane-bound proteins (Bartz et al. 2007).
The role of lipids in viral infection has been dominated in the literature by reports of viral replication strategies that usurp LDs and alter lipidomic profiles of host cells to enhance their viral life cycles. However, in recent years, there have been many roles described for fatty acids during immune responses, such as their effects on specific immune cell subsets, their effects on intracellular calcium and MAPKs in intracellular signaling pathways, their modulation of lipid rafts, their interactions with nuclear receptors, such as PPARs, and the characterization of anti-inflammatory eicosanoids involved in the resolution of inflammatory responses. Interestingly, there is also evidence for a bidirectional relationship between the immune system and lipids (such as fatty acids and bioactive lipid mediators). There are clear links between antiviral immunity and the induction of cholesterol synthesis, with lowered cholesterol biosynthesis of sterol regulatory element-binding proteins spontaneously engaging type I interferon (IFN) signaling leading to a restriction of viral infection (reviewed in Azzam and Fessler 2012; Josset et al. 2013; O'Neill 2015; York et al. 2015). Notably 25-hydroxycholesterol is in fact an IFN-stimulated gene (ISG) known to restrict the replication of many viruses including Zika virus, rotavirus and norovirus infections (Li et al. 2017; Civra et al. 2018; Shawli et al. 2019). This review will focus on the latest research into the role of LDs and lipid mediators during the innate immune response, and offer insight into how both may govern the outcome of viral infection.
WHAT IS A LIPID DROPLET?
Since their discovery in the 1890s (Altmann 1894), LDs have shown to be highly dynamic, ubiquitous organelles, which are found in virtually all types of cells from prokaryotes to eukaryotes. They consist mainly of TGs and sterol esters, but also harbor other lipid species such as diacylglycerols, retinyl esters and ceramides (Testerink et al. 2012; Preuss et al. 2019), and can also activate the synthesis of bioactive lipid mediators (de Almeida et al. 2018). The size, number, function and composition of LDs can differ considerably not only among different cell types but also in individual cell populations (reviewed in Pol, Gross and Parton 2014). They are involved in many cellular processes, including lipid storage and transport (Liu et al. 2004; Bartz et al. 2007), metabolism (reviewed in Xu, Zhang and Liu 2018) and protein storage and degradation (Klemm, Spooner and Ploegh 2011; Li et al. 2012). LDs have been described to be central in a number of cellular processes with the dysregulation of LDs contributing to a number of diseases, including obesity, fatty liver disease, cardiovascular disease, diabetes and also numerous cancers (reviewed in Onal et al. 2017; Cruz et al. 2020). Typically, it was thought that the function of LDs was to fuel metabolic processes and membrane biosynthesis; however, recently other roles have emerged for LDs, such as their protection against lipotoxicity and mitochondrial damage during autophagy (reviewed in Nguyen and Olzmann 2017), and their role in immune responses.
Within a cell, LDs alternate between periods of growth and depletion. LDs are depleted either through enzymatic hydrolysis mediated by lipases (lipolysis) or through a selective form of autophagy (lipophagy) (reviewed in Olzmann and Carvalho 2019). LD depletion mainly occurs during cell starvation where two lipolytic enzymes mediate LD breakdown: adipose triacylglycerol lipase (ATGL) converts TG to diglyceride (DG) (Haemmerle et al. 2006) and hormone-sensitive lipase (HSL) converts DG to monoglyceride (MG) (Vaughan, Berger and Steinberg 1964). The enzyme phospholipase A (PLA), important for LD homeostasis, also hydrolyzes fatty acids (Kim et al. 2002; Guijas et al. 2014). Although LD depletion is fairly well described, the mechanisms underpinning LD induction appear to be more diverse, and somewhat less understood. There are currently several mechanisms described for the biogenesis of LDs, with the most well-described mechanism regulated by the accumulation of neutral lipids (most commonly TG and sterol esters) between the bilayers of the endoplasmic reticulum (ER) membrane. This accumulation of lipids within the ER membrane leads to neutral lipids producing a sphere of lipids concealed by a phospholipid monolayer of the ER, and then budding off as nascent LDs (Fig. 1) (reviewed in Jackson 2019). Proteins can be embedded into the LD during the biogenesis stage or can be later recruited/trafficked to the LD surface from the cytosol. Proteomic studies have revealed ∼100–150 mammalian LD proteins (Bersuker et al. 2018), and the protein composition of LDs can vary, with cell type and metabolic state of the cell a driver of this heterogeneity. While the function of many of these proteins remains to be fully elucidated, as well as the mechanisms of protein targeting to LDs (reviewed in Dhiman et al. 2020), several have been implicated in lipid synthesis and LD biogenesis (e.g. acyl-CoA synthetase, PLIN2, DGAT2), protein degradation (e.g. AUP1 and ERAD proteins), signaling, membrane trafficking and metabolism (reviewed in Zhang and Liu 2019).
Figure 1.

Homeostatic LD biogenesis from the endoplasmic reticulum (ER), and LD biogenesis from pathogen infection. Homeostatic LD synthesis can occur following three crucial steps. Neutral lipids are synthesized in the ER where these neutral lipids begin to aggregate. When neutral lipid synthesis reaches a threshold, LD budding toward the cytosol occurs forming the beginning of a tethered LD. The tethered LD continues to accumulate neutral lipids resulting in expansion of the LD until a fission-like mechanism results in the detachment of the nascent LD into the cytoplasm. LDs are also induced upon infection with pathogens by the activation of either Toll-like receptors or epidermal growth factor receptor (EGFR).
LDs are highly dynamic organelles, constantly changing in both their size and location. LD size can vary (200–10 000 nm) in a range of different cell types (reviewed in Yang et al. 2012) with the size dependent on the amount of stored neutral lipid, which is the net result of TG synthesis (lipogenesis) and hydrolysis (lipolysis), and the regulation of lipolysis by external factors. Additionally, LDs are usually dispersed throughout the cell and move in small oscillations within a cell; however, they have also been demonstrated to dynamically move around cells during different stress conditions, for example, LDs cluster around microtubule organizing centers during HCV infection (Boulant et al. 2008). In mammalian cells, perilipin1 (PLIN1) expression leads to LD clustering, which is reversed upon perilipin1 phosphorylation (Marcinkiewicz et al. 2006). It is known that LDs can move around the cell environment; however, the reason behind this is not well described in mammalian cells, although recently it was discovered that DipA phosphatase was responsible for the movement and distribution of LDs in the filamentous fungus Aspergillus nidulans model (Salogiannis et al. 2020). A possible reason for LD movement within a cell is the ability of LDs to interact with other organelles, including the ER, endosomes, mitochondria and peroxisomes (Valm et al. 2017). There are several hypotheses as to why LDs may form contacts with other organelles, including the delivery of LDs to lysosomes during autophagy to generate cholesterol (Ouimet et al. 2011), channeling of FAs liberated from lipolysis to sites of oxidation (Blanchette-Mackie and Scow 1983; Binns et al. 2006) and exchange of proteins (Binns et al. 2006; Pu et al. 2011; Wang et al. 2011). The dynamic nature of LDs has been noted in a few disease pathologies, most notably their accumulation following some pathogenic infections.
Even within a cell, the size of LDs may dramatically differ under changing pathophysiological conditions. Increased LD numbers in non-adipocytic cells are commonly observed pathological features of a number of infectious diseases. In recent years there has been a lot of research into the alternate ways in which LDs can be induced, and interestingly it has been discovered that a number of pathogens induced LDs upon infection.
LIPID DROPLETS ARE UPREGULATED DURING VIRAL INFECTIONS
LD formation involves specific and well-regulated mechanisms, and LD biogenesis has been rapidly detected after infection with a number of different viral and non-viral pathogens (Table 1). However, although multiple signaling pathways have been implicated in LD biogenesis, relatively little is known regarding the molecular mechanisms that govern LD biogenesis post-infection with the involvement of both pathogen and host factors usually required.
Table 1.
LD biogenesis by different pathogens.
| Stimulation/infection type | Model | Cell type | Mechanism | Reference |
|---|---|---|---|---|
| Bacteria | ||||
| Salmonella and IFNγ | Murine | Inflammatory monocytes and neutrophils | not determined | (Rosas-Ballina et al. 2015) |
| Leishmaniasp. | Murine | Macrophages | not determined | (Pinheiro et al. 2009; D'Avila et al. 2011; Rabhi et al. 2016) |
| C. pneumonia e | Murine | Macrophages | TLR-2 | (Cao et al. 2007) |
| M. leprae | Human and murine | THP-1, peripheral and peritoneal monocytes and Schwann cells | TLR2, TLR6, PI3K | (Tanigawa et al. 2008; Mattos et al. 2010, 2011) |
| M. bovis bacillus | Murine | Macrophages | TLR-2 | (D'Avila et al. 2006) |
| LPS | Murine | Leukocytes and macrophages | CD14, CD11b/CD18, TLR-4 & MCP-1 | (Pacheco et al. 2002, 2007) |
| Acinetobacter baumannii | Murine | J774 Macrophages | not determined | (Nicolaou, Goodall and Erridge 2012) |
| Chlamydia trachomatis | Human | HepG2, HeLa | IncA (bacterial driven) | (Kumar, Cocchiaro and Valdivia 2006; Cocchiaro et al. 2008) |
| Chlamydia muridarum | Murine | Epithelial cells (cervix) | not determined | (Rank et al. 2011) |
| Klebsiella pneumoniae | Human and murine | Peripheral blood monocytes and J774 macrophages | not determined | (Nicolaou, Goodall and Erridge 2012) |
| E. coli | Murine | J774 macrophages | not determined | (Nicolaou, Goodall and Erridge 2012) |
| Proteus vulgaris | Murine | J774 macrophages | not determined | (Nicolaou, Goodall and Erridge 2012) |
| Pseudomonas aeruginosa | Murine | J774 macrophages | not determined | (Nicolaou, Goodall and Erridge 2012) |
| Pseudomonas diminuta | Murine | J774 macrophages | not determined | (Nicolaou, Goodall and Erridge 2012) |
| Staphylococcus aureus | Murine | J774 macrophages | not determined | (Nicolaou, Goodall and Erridge 2012) |
| Staphylococcus epidermidis | Murine | J774 macrophages | not determined | (Nicolaou, Goodall and Erridge 2012) |
| Staphylococcus salivarius | Human and murine | J774 macrophages and peripheral blood monocytes | not determined | (Nicolaou, Goodall and Erridge 2012) |
| Vibrio cholera | Human | Mucosal mast cells | not determined | (Qadri et al. 2004) |
| Enterobacter cloacae | Mosquito | Aag2 | Toll and IMD pathways | (Barletta et al. 2016) |
| CpG-DNA | Murine | J774 macrophages | not determined | (Nicolaou, Goodall and Erridge 2012) |
| Flagellin | Murine | J774 macrophages | not determined | (Nicolaou, Goodall and Erridge 2012) |
| Parasites | ||||
| Plasmodium berguei | Murine | Hepatocytes, macrophages | not determined | (Rodriguez-Acosta et al. 1998; Borges et al. 2017) |
| Trypanosoma cruzi | Rat | Inflammatory macrophages | not determined | (Melo et al. 2003, 2006; D'Avila et al. 2011) |
| Schistosoma mansoni derivates | Murine | Eosinophils | TLR-2 | (Magalhã es et al. 2010) |
| T. gondii | Human | Fibroblasts | not determined | (Charron and Sibley 2002) |
| Candida albicans derivates | Rat | Macrophages and hepatocytes | not determined | (Paraje et al. 2008) |
| Viruses | ||||
| Hepatitis C (HCV) | Human | CHO & HepG2 | not determined | (Barba et al. 1997) |
| Dengue (DENV) | Hamster | BHK-21 | not determined | (Samsa et al. 2009) |
| Mosquito | Aag2 | Toll and IMD pathways | (Barletta et al. 2016) | |
| Sindbis virus | Mosquito | Aag2 | Toll and IMD pathways | (Barletta et al. 2016) |
| Zika (ZIKV) | Human | Astrocytes | EGFR | (Monson et al. 2020) |
| Herpes simplex-1 (HSV-1) | Human | Astrocytes | EGFR | (Monson et al. 2020) |
| Influenza A (IAV) | Human | Astrocytes | not determined | (Monson et al. 2020) |
| Mouse | Lung tissue | not determined | (Monson et al. 2020) | |
| Human | MDCK cell line | ROS production | (Episcopio et al. 2019) | |
A diverse number of pathogens have been shown to upregulate LDs in numerous animal models and cell lineages following the recognition of either the pathogens themselves, or their pathogen-associated molecular patterns such as proteins, lipopolysaccharides, nucleic acids and other patterns unique from host cells (Table 1). Currently the main documented drivers for LD production following pathogen recognition by a host cell are toll-like receptors (TLRs) in mammals or TLR homologues in non-mammal models. Viral induction of LDs is a more recent phenomenon and has only been shown for a small handful of viruses. Recently, it was described that in an Aedes aegypti model of dengue (DENV) infection, LDs were induced following the activation of TLRs or immune deficiency pathways, with inhibition of these pathways reducing the accumulation of LDs (Barletta et al. 2016). Additionally, influenza, herpes simplex virus and Zika (ZIKV) virus have all been shown to induce LDs in both HeLa and primary immortalized astrocytes in an EGFR-dependent manner, similar to what has previously been demonstrated in a cancer model of LD induction (Penrose et al. 2016; Monson et al. 2020). Interestingly, infection with DENV and ZIKV has also been shown to deplete LDs in some cell types (Heaton and Randall 2010; Chen et al. 2020); however, it is not clear whether this is a cell-type specific response, or whether it might be a timed infection response, with LDs accumulating early as a host response following viral infection, accompanied by viral manipulation of this response, and subsequent depletion of LDs to fuel their viral life cycles.
LDs can also be upregulated following inflammasome stimulation during infection of cells with hepatitis C virus. Upon infection with hepatitis C virus, nod-like receptor protein 3 (NLRP3), a crucial mediator of the innate immune system is activated (Negash et al. 2013). The activation of NLRP3 promotes the activity of the inflammasome, which upregulates the expression of sterol regulatory element-binding proteins (SRBPs) (Im et al. 2011). This cascade enhances the upregulation of lipogenic genes that may result in the accumulation of LDs (Ji, Chan and Kaplowitz 2006). Although LD numbers are now considered an inflammatory hallmark following innate immune activation, as can be seen in Table 1, there are still a lot of unknowns regarding the mechanism of LD induction in an infection setting. This further highlights the importance of understanding these mechanisms and pathways as a potential target of microbial infection. It is also poorly understood if the biogenesis of LDs is host driven, or a mechanism that pathogens have acquired to manipulate natural LD metabolism to promote their survival, or to promote replication and virion maturation to avoid immune clearance from the host innate immune response.
THE ROLE OF LIPID DROPLETS DURING VIRAL INFECTION
Several bacterial, protozoan and viral pathogens are known to exploit host LDs to promote infection, thus emphasizing the importance of LDs at the host–pathogen interface. In particular, associations between LDs and +ssRNA viruses, which exhibit intimate relationships with the host ER, are constantly emerging at varying stages of the virus life cycle. From the Flaviviridae family, WNV, DENV, JEV and ZIKV capsid proteins all co-localize with LDs, and the residues involved in LD binding appear to be conserved amongst flaviviruses (Samsa et al. 2009; Carvalho et al. 2012; Martins et al. 2012, 2019; Shang et al. 2018; Ishida et al. 2019). The association between DENV capsid protein and LDs is particularly strong, and it has been proposed that LDs act as a scaffold for nucleocapsid formation during encapsidation (Samsa et al. 2009). Another study revealed that DENV also upregulates the consumption of LDs, termed ‘lipophagy’, to release lipids for replication and as energy substrates to increase metabolism (Heaton and Randall 2010; Zhang et al. 2018). HCV, another member of the Flaviviridae family, has the most extensively researched associations with LDs, which play a necessary role in morphogenesis and replication (reviewed in Bley, Schöbel and Herker 2020).
A hallmark of +ssRNA viral infection is the induction of the formation of replication platforms within host cells, which involve the rearrangement of host membranes to provide the ideal environment for replication and immune evasion. The ER is usually the preferred membrane and can be the site of viral replication as well as translation, genome packaging and viral assembly. It therefore would seem unlikely that the biogenesis of LDs on the ER coinciding with the integral hijacking of the ER by these viruses would not give rise to intimate associations between LDs and +ssRNA viruses. It has been hypothesized that for HCV infection, proximity between LDs coated in the HCV core protein and viral replication complex was necessary for efficient replication and viral assembly, and recently it has been observed that increased LDs during HCV infection is linked to robust replication complex formation (Bang et al. 2019). These replication platforms contain condensed viral proteins and genetic material, which could also increase the likeliness of viral proteins being trafficked to or interacting with LDs. Notably, different enteroviruses, including poliovirus, have been shown to target viral proteins to LDs, and one protein in particular, 2C, generates membrane contact sites that recruit LDs to the replication complexes. Viral proteins further interact with host machinery to release lipid contents of these LDs to aid in the growth of these complexes (Laufman, Perrino and Andino 2019).
As well as +ssRNA viruses, LD interactions with viral proteins have been uncovered in a diverse spectrum of viruses, illustrating the broad utilization of LDs in viral pathways. Rotaviruses, which also induce membrane rearrangements upon infection, have been shown to recruit LD components to these membrane structures, and to co-localize with major structural and non-structural viral proteins (Cheung et al. 2010; Gaunt et al. 2013). Dispersing LDs and blocking LD formation also led to a significant reduction in the production of infectious virus. The μ1 protein of reoviruses is targeted to LDs and is subsequently involved in the induction of apoptosis (Coffey et al. 2006). Given the burst of findings following focused research on LD–viral interactions, it is likely that emerging roles for LDs in all stages of the viral life cycle will continue to be uncovered with further research.
THE ROLE LIPID DROPLETS PLAY DURING AN ANTIVIRAL HOST RESPONSE
As LDs are induced following a number of viral infections, as well as following infection with other pathogens, it begs the question as to whether this phenomenon is host or pathogen regulated. As mentioned above, LD induction is linked to pathogenic infections, where in most cases, PRRs (mostly TLRs) are activated (Table 1); however, what is less reported is how LDs contribute to an effective host immune response. TLR2, TLR3, TLR4 or TLR7 agonists are known to increase LD numbers and levels of proteins important for LD biogenesis (e.g. PLIN2 or DGAT2), perhaps indicating that LD formation is the result of a host defense mechanism orchestrated by PRR triggering (Feingold et al. 2010; Huang et al. 2014). However, it seems that the contribution of LDs to the immune response is not all that simple, with LDs having been implicated in multiple aspects of immune regulation and function. In the past, the literature has predominantly focused on the role of LDs in pathogen replication and survival; however, recent research has suggested that the early upregulation of LDs may be involved in control of pathogen infection as described below.
Lipid droplet localization with immune proteins
Many of the known LD resident proteins have central roles in lipid metabolism; however, recent work has demonstrated that several LD-resident and transient proteins have known functions outside of lipid homeostasis, having profound impacts on protein abundance and function, as well as roles in innate immune functions. The LD resident proteome is vast, but also contains many documented proteins of a transient nature depending on cellular conditions (Rösch et al. 2016; Menon et al. 2019).
It is well described that LDs are a place of viral assembly for some viruses, mainly members of the Flaviviridae and Reoviridae families, with viral proteins from members of these families being shown to localize with LDs or LD associated proteins during infection (Samsa et al. 2009; Cheung et al. 2010; Herker et al. 2010; Carvalho et al. 2012; Li et al. 2012; Camus et al. 2013). We now know that multiple viral families are able to induce the formation of LDs during infection (Table 1); however, up until recently this was thought to be induced solely by the virus for replication, and did not have a link with host immunity. When cells are infected, they produce and secrete antiviral cytokines, mainly IFNs, and subsequently, a collection of ISGs are expressed (reviewed in Schoggins and Rice 2011). Interestingly, several ISGs are also found to be localized to LDs, a phenomenon that could be a host immune strategy to combat viral propagation.
One of the most well-described ISG proteins to localize to the LD is viperin. Viperin localizes to the LD via its N-terminal amphipathic α-helix and was first found to be antiviral against human cytomegalovirus, but since then has been found to have antiviral properties against multiple viral families (Hinson and Cresswell 2009; Peña Cárcamo et al. 2018; Crosse et al. 2019; Li et al. 2019a). Interestingly, human cytomegalovirus blocks fatty acid β-oxidation by forcing the redistribution of viperin from the LD to the mitochondria resulting in an increase in LD numbers and viral envelope assembly (Seo and Cresswell 2013). This led the field to hypothesize that viperin requires its LD localization to restrict some of these viral families (Seo and Cresswell 2013; Peña Cárcamo et al. 2018; Bai et al. 2019) and also perform important post-translational modifications during antiviral immune pathways (Bai et al. 2019).
Viperin is not the only ISG that has been found on the LD, with members of the immunity related GTPase (IRG) family (IRGM1 and IRGM3) also localizing to this organelle. There is less known about why members of the IRG family of proteins localize to LDs. A few instances have shown they require the LD to bind lipids, modulate host responses and regulate autophagy and lipophagy (Grégoire, Rabourdin-Combe and Faure 2012; Haldar et al. 2013; Lin et al. 2016). Interestingly, the IRGM family of proteins are a common target of RNA viruses, with interactions with 12 viral proteins belonging to five different viruses [Chikungunya virus (ChikV), Mumps virus (MuV), Hepatitis C virus (HCV), Measles virus (MeV) and HIV-1] making it the most targeted autophagy-associated protein by these viruses (Grégoire et al. 2011). A member of this family, IRGM1 is induced by IFNs in a STAT1-dependent manner (MacMicking, Taylor and McKinney 2003; Bafica et al. 2007), and has been shown to mainly have a role in binding selected lipids in mitochondria and phagosomes, which is an essential process for the degradation of pathogen-containing vacuoles via autophagy or fusion with endolysosomes (Tiwari et al. 2009; Singh et al. 2010). Another IRGM family member IRGM3, also localizes to the LD in IFNγ-stimulated mouse DCs. Although a loss of IRGM3 leads to a reduction of LD numbers, there has not been a mechanism of how this modulates the host immune response (Bougnères et al. 2009). Lastly, members of the helicase family are known to localize to LDs during HCV infection (Rösch et al. 2016). Of these helicase family members, DDX3X and DDX1 are involved in innate host responses, particularly the upregulation of IFN-β (Soulat et al. 2008; Li et al. 2015; Zhou et al. 2017; Niu et al. 2019; Xue et al. 2019); however, there are currently no studies demonstrating their reliance on their LD localization to impart their antiviral effects, unlike viperin and IRG family members. Although not in a viral setting, a very recent study has now demonstrated that LDs serve as hubs of innate immune responses to bacterial LPS stimulation. Proteomic analyses of LDs derived from the livers of LPS treated mice demonstrated that multiple proteins involved in a successful antibacterial response, including CAMP and viperin were able to localize to LDs as a function of innate immune activation (Bosch et al. 2020). This recent work paves the way for a better understanding of the role of the LD as a potential functional hub for innate immune proteins.
It is clear that LDs are protein sequestration sites for proteins that have clear roles outside LD function (Cermelli et al. 2006; Welte 2007). LDs have been found to sequester large amounts of histones, particularly in early Drosophila embryos (Cermelli et al. 2006) but also in C. elegans, silkworm and human LD proteomes (Yang et al. 2010; Liu et al. 2012; Wang et al. 2013). Proteomic analysis of LDs isolated from Drosophila embryos confirmed the presence of histone localization via a protein known as jabba (Cermelli et al. 2006; Li et al. 2012). Interestingly, Drosophila embryos lacking jabba and hence histone sequestration were more susceptible to infections than embryos that had no disruption in their ability to sequester histones (Anand et al. 2012). There have not been many reports of histones having antiviral mechanisms; however, viral proteins have been demonstrated to drive the depletion of histones in vitro (Irwin et al. 2018). Interestingly, the M1 protein of influenza A and B virus selectively binds histones H2A, H2B, H3 and H4 (Zhirnov and Klenk 1997), with these histones showing anti-influenza activity against both seasonal H3N2 and H1N1, but not pandemic H1N1 (Hoeksema et al. 2015). Incubation of IAV infected cells with histone H4 results in decreased viral replication via direct effects on viral particles. Although these histones inhibited influenza growth in vitro indicating a possible role for histones in the innate immune response against IAV, in vivo treatment with histones did not yield antiviral effects and instead exacerbated lung pathology. (Ashar et al. 2018) Clearly, more work is needed in this area to elucidate the role of these histones during infection, and uncover the role LDs play in this relationship.
It has also been observed that LDs are vital to protecting the developing zebrafish embryo from pathogens, prior to development of a functional immune system. Zebrafish only develop an adaptive immune system after 4–6 weeks of embryo development, with an intact innate immune system present at fertilization (Lam et al. 2004). LDs have been shown to play a vital role in protecting the zebrafish embryo from bacterial pathogens, with increased LD numbers being highly protective against invading bacterial species (Dutta, Banerjee and Sinha 2018). LDs were found to secrete antimicrobial compounds to afford embryo protection from bacteria, and interestingly, injected exogenous LDs also increased the life expectancy of the embryos strengthening this correlation (Dutta, Banerjee and Sinha 2018). The role of LDs in viral protection of the zebrafish embryo has not been demonstrated to date; however, recent work has shown that LD depletion in the mammalian HeLa and Huh-7 cell lines diminishes the production of type I and III IFN mRNA species following both activation of RNA and DNA PRRs as well as viral infection (Monson et al. 2018).
The ability of LDs themselves, and perhaps the proteins they house to impact viral infection as well as infection of other pathogens, is very interesting; however, the role LDs play in this is not entirely known. One potential hypothesis is that LDs may act as an organelle platform during host defense pathways, hence the requirement for their upregulation during pathogen infection. However, recent evidence suggests that their role may be multi-pronged, and that they may contribute to amplification of an antiviral state in more ways than providing a platform for anti-viral proteins.
Lipid droplets enhance innate immune pathways
Recently, LDs have been described to contribute to an effective immune response. It is now known that LDs play a role in the antiviral response in the mosquito, where viral infection was shown to induce LD formation in the cells of the midgut (Barletta et al. 2016). This LD accumulation was mimicked via synthetic activation of the antiviral innate pathways, Toll and IMD, indicating that accumulation of LDs may be an important antiviral response in the mosquito, independently of viperin expression (Barletta et al. 2016).
The induction of LDs following viral infection has also been shown to drive enhanced interferon output. In an in vitro model, both ZIKV and Herpes simplex virus 1 infected cells promoted an increase in LD accumulation. An enhanced number of LDs resulted in decreased viral replication with heightened interferon production in the early stages of viral replication (Monson et al. 2020). Importantly, this increase in interferon was also abolished when LD induction was inhibited, suggesting that LD induction can drive an effective interferon response following viral infection. Additionally, LD upregulation following viral infection was shown to be regulated by EGFR and subsequent PI3K activation, in an interferon-independent manner, which is an alternative mechanism to what has previously been demonstrated for alternate pathogen induction of LDs. However, the regulation of interferon output and LD communication is a recent finding, with the full mechanisms responsible still unknown, yet it appears there is a clear relationship between LD regulation and orchestrating a successful immune response following infection.
Interestingly, the presence of LDs has been demonstrated to be crucial for dendritic cell cross-presentation, an essential step in the antigen-specific activation of CD8+ T cells during viral infection (Bougnères et al. 2009). These authors were able to demonstrate that dendritic cells with increased LDs are more proficient at antigen cross-presentation, and when LD biogenesis was blocked, this process was inhibited. Genetic removal of LD-localized proteins, IRGM3 or ADRP, from dendritic cells lead to defects in LD formation, which severely impaired cross-presentation (Bougnères et al. 2009). Type I IFN has also been shown to impact cross-presentation by DCs, and the recent finding that LDs may play a role in the production of type I IFN could underpin the role of LDs in dendritic cell antigen cross-presentation (Monson et al. 2020); however, more detailed analysis of the role of LDs is required to fully elucidate the role LDs play in antigen cross-presentation.
The recent literature surrounding the contribution of the LD to an effective antiviral immune response is very understudied and still in its infancy. To further investigate how these organelles may contribute to an effective antiviral response, it is important to look at the make-up of LDs. To date, there are no studies looking at the dynamic exchange of lipids within LDs during an antiviral response; however, it has been demonstrated that this does occur during stress responses (Chitraju et al. 2012; Hartler et al. 2014; Bailey et al. 2015). Interestingly, many lipid classes as well as bioactive lipid signaling mediators are known to play pivotal roles in an immune response and are either packaged within LDs, or directly synthesized via LDs; demonstrating the importance of understanding their role and relationship to LDs to gain a better understanding of the role of LDs during this response (Fig. 2).
Figure 2.
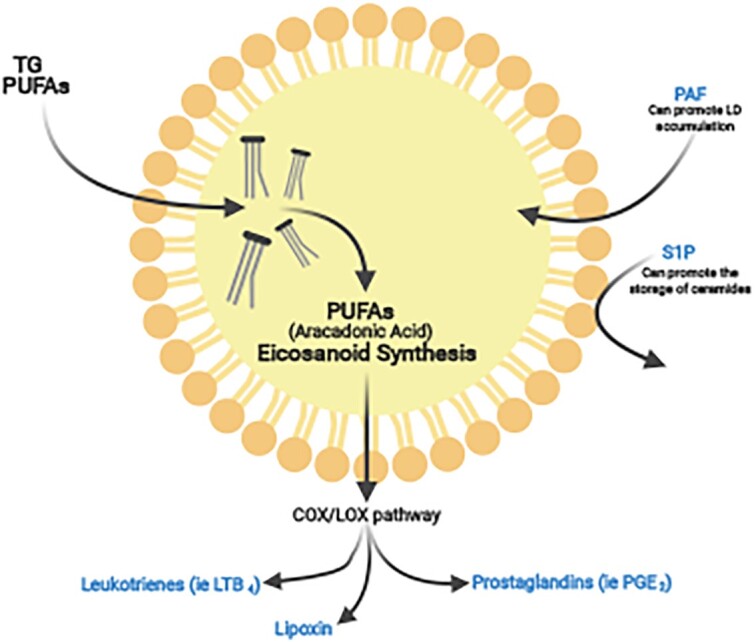
Lipid droplets are sites for lipid mediator synthesis. Polyunsaturated fatty acids (PUFAs; mainly from TGs) such as arachidonic acid can be sequestered into LDs where they can undergo synthesis within the COX/LOX pathway. This synthesis yields bioactive lipid mediators such as prostaglandins, leukotrienes and lipoxins. Sphingosine-1-phosphate (S1P) has been demonstrated to interact with LDs through the promotion of ceramide synthesis and storage as well as platelet-activating factor (PAF), which has been demonstrated to promote LPS- and sepsis-induced LD formation.
THE ROLE OF LIPID SIGNALING MEDIATORS DURING A VIRAL INFECTION
The concept that lipids not only function as structural components in membranes, or act as a source of energy for a cell, but also function directly as intercellular and intracellular signaling mediators has only gained interest in the last 20 years. Lipid mediators have several characteristics that distinguish them from other signaling molecules. First, they are synthesized on demand from precursor membrane lipids. They also have relatively short half-lives (seconds to minutes) and are degraded both enzymatically and non-enzymatically (Corey et al. 1975; Sala et al. 1990; Jones et al. 2005). Lipid mediators can be structurally divided into three categories. Class 1 lipid mediators include well-described arachidonic acid (AA) derived eicosanoids, which include prostaglandins (PGs) and leukotrienes (LTs) and their relatives (Narumiya and FitzGerald 2001; Frank Austen 2008). Class 2 lipid mediators are phospholipids and sphingolipids, and consist of lysophospholipids such as platelet-activating factor (PAF), lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P) and their derivatives (Ishii, Nagase and Shimizu 2002; Takabe et al. 2008; Choi et al. 2010). Class 3 lipid mediators are primarily anti-inflammatory and are derived from ω-3 polyunsaturated fatty acids (PUFAs) such as resolvins and protectins and the AA-derived eicosanoid lipoxin (Ariel and Serhan 2007). Interestingly, members of all three classes of lipid mediators have roles in the effective progression of viruses and/or are involved in an effective host response (Table 2). While it has been known for a long time that viral infections and IFNs interfere with lipid metabolism, including fatty acid (FA) and cholesterol synthesis, it is not well established how lipids can positively contribute to an effective antiviral host response. Interestingly, some of the lipid mediators described below have been associated with LDs, or LD biogenesis pathways; however, a more detailed lipidomic analysis, especially at early stages of infection is required to fully understand the relationship between signaling lipid mediators and LDs, and their subsequent effects on host immunity.
Table 2.
Lipid mediators in viral infection.
| Mediator | Virus | Prohost/provirus | Mechanism | Reference |
|---|---|---|---|---|
| Class 1 | ||||
| PGE2a | HSV-1 | Provirus | Increase viral replication | (Harbour, Blyth and Hill 1978) |
| CMV | Provirus | -PGE2 contributes to immunosuppressive effect | (Nokta et al. 1996; Hooks et al. 2006; Schröer and Shenk 2008) | |
| -PGE2 increases plaque formation and viral DNA copy number | ||||
| -PGE2 plays a role in direct cell-to-cell spreading | ||||
| EBV | Provirus | Lytic reaction via EP signaling pathways | (Gandhi et al. 2015) | |
| Rotavirus | Provirus | Might be required for early infection, i.e. attachment | ||
| Enterovirus 71 | Provirus | PGE2 might be required for replication | (Tung, Hsieh and Yang 2010; Tung et al. 2011; Wang et al. 2015) | |
| Sapovirus | Provirus | PGE2 decreases the production of nitric oxide, leading to an increase in PSAV | (Alfajaro et al. 2017) | |
| VSV | Provirus | -COX inhibitors/antagonist reduced viral production, but the effect is overcome by exogenous PGE2 | (Chen, Warner and Reiss 2000; Chen, Restivo and Reiss 2002) | |
| -COX-2 antagonist decreased viral titers | ||||
| LCMV | Provirus | PGE2i inhibits the survival and effector functions of Tc | (Chen et al. 2015) | |
| HTLV-1 | Provirus | PGE2 causes an increased production of virus | (Kuno et al. 1986) | |
| MDV | Provirus | Promotes virus replication | (Boodhoo et al. 2019) | |
| IAV | Provirus | PGE2 inhibits type-I IFNs and apoptosis pathways | (Coulombe et al. 2014; Park et al. 2016) | |
| RSV | Provirus | PGE2 causes a delayed protective RSV specific immune response | (Bartz et al. 2002) | |
| Coxsackie virus | Prohost | Decrease viral titers | (Xie et al. 2012) | |
| HPIV-3 | Prohost | PGE2 inhibits viral replication | (Luczak et al. 1975) | |
| HBV | Prohost | -PGE2 results in loss of viral replication | (Flowers et al. 1994; Hyman et al. 1999) | |
| -PGE2 decreases viral antigen | ||||
| HIV | Prohost | -PGE2 decreases virion penetration by suppressing expression of CCR5 | (Thivierge et al. 1998; Hayes et al. 2002; Clemente et al. 2014) | |
| -PGE2 inhibits virus replication by protein kinase A-dependent mechanism | ||||
| -PGE2 reduces cell-to-cell spreading | ||||
| HIV | Provirus | -PGE2 enhances HIV-1 long terminal repeat mediated reporter gene activation | (Dumais, Barbeau and Olivier 1998; Fitzgerald et al. 2012; Zambrano-Zaragoza et al. 2014) | |
| -PGE2 has an immunosuppressive effect when co-infected with HPV | ||||
| -PGE2 could play a role in pathogenicity via Th17 cell regulation | ||||
| dmPGA1 | HSV-1 | Prohost | Significantly inhibited viral replication | (Hughes-Fulford et al. 1992) |
| HIV | Significantly inhibited viral replication | (Hughes-Fulford et al. 1992) | ||
| dmPGE2 | HSV | Provirus | Up to a 60% increase in HSV replication compared with that in untreated virus-infected cells | (Hughes-Fulford et al. 1992) |
| dmPGA2 | HSV | Provirus | Up to a 60% increase in HSV replication compared with that in untreated virus-infected cells | (Hughes-Fulford et al. 1992) |
| LTE4 | RSV | Provirus | Urinary LTE4 was 8-fold higher in infants with bronchiolitis than in controls | (Piedimonte et al. 2005) |
| LTB4a | HTLV-1 | Provirus | HTLV-1 infection dysregulates the LT pathway to promote viral replication | (Trindade et al. 2012; Percher et al. 2017) |
| CMV | Prohost | Significant reduction in viral titers | (Gaudreault and Gosselin 2007) | |
| IAV | Prohost | Significant decrease in lung viral loads | (Gaudreault and Gosselin 2008) | |
| HIV | Prohost | LTB4 induced the secretion alpha defensins and MIP-1 beta to activate neutrophils | (Flamand et al. 2004) | |
| EBV | Prohost | LTB4 can induce T-cell activation that inhibits the EBV-induced proliferation of B lymphocytes | (Liu et al. 2008) | |
| Influenza B | Prohost | Significant reduction in viral titers | (Widegren et al. 2011) | |
| Coronavirus | Prohost | Significant reduction in viral titers | (Widegren et al. 2011) | |
| RSV | Prohost | Significant reduction in viral titers | (Widegren et al. 2011) | |
| HSV-1 | Prohost | Increase in antimicrobial proteins to restrict infection | (Flamand, Tremblay and Borgeat 2007) | |
| Class 2 | ||||
| PAFa | HIV | Provirus | Enhances replication | (Lima et al. 2006) |
| RSV | Provirus | Enhances the inflammatory environment | (Villani et al. 1991) | |
| DENV | Provirus | Induces an increase in vascular permeability, hypotension, and production of cytokines | (Souza et al. 2009) | |
| HCV | Provirus | A higher amount of plasma PAF level was observed in infected patients | (Caini et al. 2007) | |
| LPA | HCV | Provirus | Inhibiting LPA signaling reduced HCV replication | (Farquhar et al. 2017) |
| RSV | Provirus | Role in RSV filament formation and virus-induced cell-to-cell fusion | (Gower et al. 2005) | |
| S1Pa | IAV | Provirus | Overexpression of S1P heightened the cells' susceptibility to influenza virus infection | (Seo et al. 2010; Zhao et al. 2019) |
| HSV-1 | Provirus | Facilitates successful HSV-1 replication | (Graber et al. 2020) | |
| DENV | Prohost | Low S1P levels correlated with exaggerated DENV replication | (Gomes et al. 2014; Michels et al. 2015) | |
| Class 3 | ||||
| PD1 | IAV (H5N1) | Prohost | PD1 treatment improved the survival and pathology of severe influenza in mice, decreases in TNF-α and IFN-γ | (Morita et al. 2013) |
| HSV-1 | Prohost | NPD1 significant reduction in the severity and incidence of HSV-induced stromal keratitis | (Rajasagi et al. 2013) | |
| Lipoxina | IAV (H5N1) | Prohost | TNFα and interferon-γ were found to be upregulated | (Cilloniz et al. 2010) |
| RSV | Prohost | Promotes differentiation of alternatively activated macrophages and helps resolve airway hyperreactivity | (Richardson et al. 2005; Shirey et al. 2014) | |
| KSHV | Prohost | Creates an anti-inflammatory environment by decreasing the levels of NF-κB, AKT, ERK1/2, COX-2 and 5-lipoxygenase | (Chandrasekharan et al. 2016) | |
| RvE1 | HSV-1 | Prohost | Reduces proinflammatory mediators and stimulates IL-10 | (Rajasagi et al. 2011) |
| RSV | Prohost | Restored the alternatively activated macrophage phenotype | (Shirey et al. 2014) | |
Found to be produced from LDs or influence LD biogenesis.
Abbreviations: HIV-1: Human immunodeficiency virus 1; CMV: Cytomegalovirus; EBV: Epstein–Barr virus; VSV: Vesicular stomatitis virus; LCMV: Lymphocytic choriomeningitis virus; HTLV-1: Human T-lymphotropic virus 1; MDV: Marek's Disease Virus; IAV: Influenza A virus; RSV: Respiratory syncytial virus; HPIV-3: Human parainfluenza virus-3; HBV: Hepatitis B; DENV: Dengue virus; and KSHV: Kaposi's sarcoma-associated herpesvirus.
Class 1 lipid mediators
Class 1 lipid mediators are the most distinguished family of lipid mediators involved in immunity and inflammation with their main role being the amplification or reduction of inflammation, coordination of immune cell recruitment, cytokine and chemokine production, antibody formation, cell proliferation and migration, and antigen presentation (Funk 2001; Harizi, Corcuff and Gualde 2008) (reviewed in Chiurchiù and Maccarrone 2016). One of the best characterized functions of LDs in cells of the immune response are their capacity to act as platforms for heightened eicosanoid synthesis of leukotrienes and prostaglandins (Bozza, Magalhães and Weller 2009), as well as house enzymes that generate eicosanoids, such as prostaglandin H2 synthase/cyclooxygenase (PGH2 synthase/COX) and lipoxygenases (LO) (Dvorak et al. 1992; Accioly et al. 2008; Weibel et al. 2009) (Fig. 2). Eicosanoids are non-storable, but are promptly synthesized signaling molecules that are synthesized de novo from the enzymatic oxygenation of arachidonic acid, which is released from the LD phospholipid monolayer by PLA2 after cell activation (Nicolaou, Goodall and Erridge 2012; Melo and Weller 2016). It has been well described that LDs release FAs that in turn regulate important signaling pathways such as the PPAR transcriptional network (Zechner et al. 2012) and also store FAs that can be used for conversion into eicosanoids (Weller et al. 1991; Triggiani et al. 1995; Bozza et al. 2011); however, it was only recently discovered that TGs stored in LDs are in fact sources of FA precursors for lipid mediator production and that their availability is controlled by neutral lipases (Dichlberger et al. 2014; Schlager et al. 2015; Gartung et al. 2016; Kuo et al. 2018; Sohn et al. 2018) (Fig. 2).
Class 1 lipid mediators are the most widely studied lipid mediators during a viral infection, and interestingly, have been described to be both beneficial to the virus, and/or beneficial for the host during viral infection (Table 2 and Fig. 3). Of the class 1 mediators, Prostaglandin E2 (PGE2) is the most well described. It has been shown to act in a proviral manner in aiding the progression of multiple viruses such as HSV-1, CMV, EBV, IAV, rotavirus and others (Table 2). The effect of PGE2 on viral infection and replication is cell type dependent and virus family dependent, with it mainly affecting the modulation of immune pathways via the regulation of cytokines (Rowan 2005; Schröer and Shenk 2008; Chen et al. 2015). Interestingly, there are multiple mechanisms in which PGE2 works to modulate these infections with PGE2 working to dampen the host response in different ways and through multiple signaling pathways. For example, during IAV infection, upregulation of PGE2 leads to the inhibition of type I interferon production and apoptosis in macrophages, with antigen presentation and T cell mediated immunity also suppressed, ultimately causing an increase in virus replication (Coulombe et al. 2014). However, during HSV-1 infection, PGE2 increases the spread of HSV-1 between cells, but the exact mechanism is unknown (Harbour, Blyth and Hill 1978). Interestingly, PGE2 has strong suppressive effects on LPS-induced IFN-β production at both the mRNA and protein levels in macrophages; however, the role PGE2 plays on the regulation of IFN-β during viral infection is currently not known (Xu et al. 2008). PGE2 provides a proinflammatory environment in which some viruses such as rotavirus thrive in, thus supporting and promoting their replication (Rossen et al. 2004). Both the innate and adaptive immunity can also be regulated by PGE2, which can either have adverse or beneficial effects on the immune system's ability to fend off pathogens (Kalinski 2012). One of the most well-described effects of PGE2 on the immune system is the effect it has on regulation and activity of T cells, thus affecting the interplay between innate and adaptive immunity. It is well described that PGE2 modulates proliferation, apoptosis and cytokine production of CD4+ T cells; however, it was not until recently that PGE2 was implicated in the enhancement of T helper 2 (Th2) responses via cAMP, promoting infection of Enterovirus 71 (Wang et al. 2015).
Figure 3.

Lipid mediators in viral infection. Lipid mediators can act in both a prohost and proviral sense during infection. Lipid mediators [prostaglandin E2 (PGE2), dimethyl prostaglandin A1 (dmPGA1), leukotriene B4 (LTB4), sphingosine-1-phosphate (S1P), platelet-activating factor (PAF) and lysophosphatidic acid (LPA)] all act to either restrict or foster viral replication. Lipid mediators can also act in a proviral manner to increase the production of inflammatory cytokines, restrict type-1 IFN signaling and increase cell–cell fusion [resolvin E1(RvE1), protectin D1 (PD1) and lipoxins]. On the other hand, lipid mediators can decrease inflammatory cytokines and promote macrophage differentiation.
Very recently, a study has linked a role for TG synthesis within LDs to the effective production of PGE2 (Castoldi et al. 2020). This study has therefore placed LDs, in a position of central importance in inflammatory macrophage activation. The failure of inflammatory macrophages to make PGE2 when TG synthesis is inhibited is critical for this phenotype, as addition of exogenous PGE2 is able to reverse the anti-inflammatory effects of TG synthesis inhibition (Castoldi et al. 2020). These findings suggest that the mechanism by which PGE2 enhances these inflammatory responses to viruses may directly be due to this accumulation of TGs within LDs, therefore, a more detailed analysis of the relationship of lipid classes within LDs during infection will provide insight into this relationship.
Although PGE2 is the most well-described prostaglandin in the context of viral infections, Dimethyl Prostaglandin A1 (dmPGA1) dmPGA, has interestingly been shown to have unusual broad-spectrum antiviral activity against both HSV and HIV-1 (Hughes-Fulford et al. 1992). Interestingly, closely related prostaglandins dmPGE2 and dmPGA2 did not have the same effect on the replication of HSV and their treatment on HSV-1 infected cells resulted in a 60% increase in replication compared with that in untreated virus-infected cells. Another well-studied member of class 1 lipid mediators are the leukotrienes, which are eicosanoid inflammatory mediators secreted to recruit immune cells, mostly leukocytes and eosinophils (Woodward et al. 1991; Medeiros et al. 1999). In particular, LTB4 is a potent chemoattractant that favors the recruitment of leukocytes to inflammatory sites (Canetti et al. 2003; Grespan et al. 2008; Afonso et al. 2012; Lämmermann et al. 2013). LTB4 has been found to inhibit the replication of a number of viruses including Influenza A and B, RSV and Coronaviruses; however, LTB4 secretion was recently demonstrated to facilitate the recruitment of HTLV-1-infected T cells and virus transmission in vitro (Percher et al. 2017) (Table 2).
Class 2 lipid mediators
In comparison to class 1, class 2 lipid mediators have only been found to contribute to a proviral environment (Table 2). Class 2 lipid mediators are mainly membrane phospholipids (particularly phosphatidic acid, phosphatidylcholine, phosphatidylserine and phosphatidylinositol) and sphingolipids (ceramide and sphingosine-1-phosphate) (Hannun and Obeid 2008). They are reservoirs of bioactive metabolites of profound importance to a myriad of cell signaling and cellular processes that are important in immunity, inflammation and inflammatory disorders, including cell growth, survival, immune cell trafficking, and vascular and epithelial integrity (reviewed in Maceyka and Spiegel 2014). They are also known to exert pleiotropic effects such as inflammation, vesicular trafficking, endocytosis, cell cycle and senescence, survival and apoptosis, cell migration and cell-stress responses (El Alwani et al. 2006). Class 2 lipid mediators have been shown to be proviral, with members of this class exacerbating a number of viral infections (Table 2).
The phospholipid platelet-activating factor (PAF) is a proinflammatory class 2 lipid mediator, which has been found to enhance the replication of multiple viral infections such as HIV-1, RSV, DENV and HCV (Table 2). Endogenous release of PAF is related to a number of acute inflammation effects in DENV infection, such as increased vascular permeability, altered leukocyte numbers, thrombocytopenia and degrees of bleeding (Souza et al. 2009).
Class 2 lipid mediators are also made up of sphingolipids, a class of lipids that have several significant roles in regulating cellular functions. The sphingolipid family is made up of diverse members, including sphingomyelin, ceramide, ceramide 1-phosphate, sphingosine and sphingosine 1-phosphate (S1P). Of these, S1P has been implicated in the modulation of cellular processes, including those important for inflammation and immune responses (Singer et al. 2005; Maeda et al. 2007; Hammad et al. 2008; Keul et al. 2011). S1P binding to its receptor (S1PR1) on the endothelial cell surface is a regulator of influenza virus-induced cytokine storm (Marsolais et al. 2009; Teijaro et al. 2011; Walsh et al. 2011) and can limit the migration of effector lymphocytes to immunologic injury areas, down-modulate the number of virus-specific T cells, blunt cytokine/chemokine expression and reduce the supply of innate inflammatory cells (Marsolais et al. 2009; Walsh et al. 2011). Activation of S1PR1 signaling by endogenous S1P inhibits type-I IFN responses. These type-I IFN responses exacerbate numerous pathogenic conditions and by suppressing the auto-amplification loop of these interferons by S1PR1 and the type-I IFN receptor (IFNAR1) in plasmacytoid dendritic cells (pDCs), a specialized DC subset, for robust type I IFN release (Teijaro et al. 2016).
Although S1P is most commonly linked to the progression of Influenza, recently it was also described to facilitate successful HSV-1 replication (Graber et al. 2020). Grabber et al were able to demonstrate that HSV-1 replication in endothelial cells relies on sphingosine kinase activity and S1P-S1PR signaling through activation of phosphatidylinositol-3-kinase (PI3K) and the small GTPase Ras-related C3 botulinum toxin substrate 1 (Rac-1) (Graber et al. 2020). They were able to decrease HSV-1 replication with the utilization of Rac-1 inhibitors or siRNA-meditated reduction of Rac-1-S1P's downstream mediator activity. S1P has also been implicated in the progression of dengue virus infection. S1P levels were significantly lower in patients with more severe forms of dengue infection and the levels remain low throughout the course of the illness (Gomes et al. 2014; Michels et al. 2015). Low S1P levels in acute dengue infection could contribute to the microvascular leak in acute dengue infection exacerbating complications associated with vascular leak such as pleural effusions, ascites, shock and multi organ failure. S1P analogues such as fingolimod could potentially have an effect of reducing vascular leak in acute dengue infection and thus preventing these complications. SK1 converts sphingosine to S1P, and when this is inhibited during DENV infection, although immune responses are largely unchanged, there are adaptive changes in IFNAR1 and IRF1 that compromise DENV-induced type I IFN responses (Aloia et al. 2017).
Class 3 lipid mediators
Class 3 lipid mediators are primarily derived from omega-6 fatty acids (lipoxins), and omega-3 fatty acids (resolvins, protectins and maresins) are produced during an inflammatory response, and act in reducing and/or resolving the inflammatory process in infection by stimulating inflammation resolution pathways (reviewed in Mittal, Ranganath and Nichani 2010; Molfino et al. 2017). As part of the neutrophil-monocyte sequence, the lipoxin signals promote the blocking of the acute inflammatory response. Lipoxins and resolvins stimulate the recruitment of immune cells and regulate the actions of the classic proinflammatory class 1 lipid mediators, prostaglandins and leukotrienes (Colas et al. 2014), reducing the duration of inflammation, and stimulating re-epithelialization, wound healing and tissue regeneration as signs of resolution (reviewed in Serhan 2014). Interestingly, unlike the other two classes of lipid mediators, class 3 lipid mediators have only been demonstrated to have prohost activities against viral infections (Table 2).
Lipoxins are anti-inflammatory metabolites of the arachidonic acid pathway, which are synthesized from arachidonic acid by the action of a series of lipoxygenases (Serhan, Hamberg and Samuelsson 1984). Lipoxins have been shown to alter levels of various transcription factors such as NF-κB, AP-1, PPARγ and Nrf-2, as well as various cytoplasmic signaling molecules such as phosphatidylinositol 3-kinase, AKT, mTOR, Ras, JAK and STAT to create an anti-inflammatory environment (Qiu et al. 2001; Weinberger et al. 2008; Prieto et al. 2010). As lipoxins are eicosanoids like prostaglandins and leukotrienes, they can be synthesized from LDs during host responses; however, a direct link between lipoxins and LDs during a viral infection has not been identified. As lipoxins are anti-inflammatory lipid mediators, they have been found to protect host cells against viral infections such as IAV (H5N1), RSV and Kaposi's sarcoma-associated herpes virus (KSHV) by mainly regulating the production of anti-inflammatory cytokines and decreasing the levels of proinflammatory NF-κB, AKT, ERK1/2, COX-2 and 5-lipoxygenase (Chandrasekharan et al. 2016) (Table 2). Although IAV, RSV and KSHV are the only viruses for which lipoxins have shown an antiviral capacity for, interestingly they have been shown to be upregulated in a range of respiratory viruses (murine parainfluenza virus type 1, rat coronavirus, pneumonia virus of mice and mouse adenovirus) (Kim 1990) as well as following infection of HIV in infected monocytes (Genis et al. 1992). This increase in lipoxins is possibly due to the increase in inflammation, which stimulates their production, and although there are only a handful of studies that demonstrate the upregulation of lipoxins following direct response to viral infection, there is certainly a lot of evidence for their antiviral capacities.
Resolvins and protectins are endogenous lipid mediators that are derived from the omega-3 polyunsaturated fatty acids, which lead to the inhibition of host tissue inflammatory responses, with the release of chemokines and cytokines (reviewed in Bannenberg and Serhan 2010). In regards to viral infection, resolvin E1 (RvE1) therapy has been demonstrated to decrease the influx of effector CD4+ T cells, neutrophils and the production of proinflammatory cytokines during HSV-1 infection, leading to a decrease in the severity of virally induced immunopathological disease (Rajasagi et al. 2011). Likewise, during H1N1 influenza A virus in human lung epithelial cells, protectin D1 (PD1) was found to significantly reduce viral replication in vitro (Morita et al. 2013).
Inflammation is a double-edged sword during viral infection. Controlled proinflammatory responses can enhance host immunity, whereas excessive uncontrolled proinflammatory responses usually facilitate infection. It is clear now that bioactive lipid mediators play a key role in the regulation of these responses in both a prohost and propathogen sense (Fig. 3). More work is needed in this area, as it is now clear that some mediators, most notably the class 1 mediators have been heavily researched in the context of viral infection and have interestingly been demonstrated to play a role on both ends of the immunity spectrum. In saying this, little is known about some of the other lipid mediators and although there is evidence for prohost and proviral mechanisms more work needs to be done in this area to fully elucidate their role. Although there are good techniques to measure lipid mediators effectively (as reviewed in Cottrell, Lin and O'Connor 2015), lipid mediator signatures are distinct across immune cell subsets, meaning studies looking at only one cell type could miss changes in their expression (Giannakis et al. 2019); therefore, a more comprehensive look into the expression of these lipid mediators during infection could provide insights into their role.
POTENTIAL ROLES OF LIPIDS AND LIPID DROPLETS IN THE IMMUNE RESPONSE
The role of lipids and LDs in the viral immunity field is understudied; however, there are many lessons that can be learnt from other recent fields of research. The main constituents of LDs are neutral lipids, specifically triacylglycerols and sterol esters, both lipids that we are only beginning to realize as instrumental in orchestrating pathways in the immune responses. A small and growing body of literature has focused on the role of different lipid species during cellular pathways, such as the immune response. It has also been demonstrated that the combination of neutral lipids packaged into LDs can have differing immune functions. Recently, there has been more attention being paid to LDs, and research has started to find links in the function they have within a cell, however, there are still a lot of unanswered questions around their role in an infection, and whether their role may have a protective mechanism for the host, rather than dominate the driving of inflammation as has been suggested in some other pathologies.
LDs can have both direct and indirect effects on cell signaling and immune pathways. They can affect signaling pathways by actively removing signaling lipids from their bioactive pool to a storage pool, or by regulating their release and production from stored precursors (Coleman and Mashek 2011; Zechner et al. 2012). In addition, by regulating the storage and release of sterols, retinoids and ceramides, LDs also affect various signaling pathways that are directly or indirectly modified by these lipids and their metabolites. Sequestration of these various lipids into the LD core thus reduces their availability for the activation of signaling pathways involved in cellular stress and inflammation, including the activation of PPAR signaling, TLR and NF-κB inflammatory pathways by saturated FAs, the conversion of PUFAs into eicosanoids, the regulation of SREBP signaling by cholesterol and FAs, the activation of protein phosphatase 2A (PP2A) by ceramides, or of protein kinase C by membrane-resident DAG (Shen, Azhar and Kraemer 2016; Yu et al. 2018; Shmarakov et al. 2019; Stith, Velazquez and Obeid 2019). It is also possible that following TLR activation the dramatic increase in de novo lipogenesis and/or lipid uptake increases LDs as a way to protect cells against elevated free fatty acid levels and the toxic effects that this may have on the cells. Another relatively unexplored role of LDs during an infection, is how they contribute to the protection of important fatty acids during an infection. LDs have been demonstrated to be induced by reactive oxygen species (ROS) generated by environmental exposure to hypoxia and other prooxidants in a developing CNS in a Drosophila model (Bailey et al. 2015). LDs formed in the glia of the stem cell (neuroblast) niche during oxidative stress and were shown to limit ROS levels to inhibit the peroxidation of polyunsaturated fatty acids (PUFAs) via the diversion of PUFAs, away from membranes to the core of LDs, where they are less vulnerable to peroxidation. It is well established that activation of PRRs can trigger a ROS response (reviewed in Li et al. 2019b), and it is possible that this protective mechanism is also at play following PRR activation by viral infection, although this has not been explored to date. LDs could thus limit cell damage, prevent cell death and reduce the inflammatory response by sequestering PUFAs and reducing their availability for oxygenation and action as danger signals.
Recently, lipids directly derived from LDs, and separate to the lipid mediators described above, have been involved in the activation of a number of immune cells. Externally acquired fatty acids that are transiently stored in LDs have been demonstrated to be converted into phospholipids to promote the proliferation of innate lymphoid cells (Karagiannis et al. 2020). Innate lymphoid cells are big drivers of inflammation in the lung, with this metabolic program being imprinted by interleukin-33 and regulated by the genes Pparg and Dgat1, which are both controlled by glucose availability and mTOR signaling (Karagiannis et al. 2020). There is also evidence of lipids derived from LDs being packaged into exosomes and resulting in activation of a population of macrophages in adipocytes (Flaherty et al. 2019). In this case, LDs provide specific lipid species to bone marrow progenitor cells promoting their differentiation into macrophages, similar to adipose tissue macrophages (Flaherty et al. 2019). The higher numbers of circulating macrophages increase the likelihood of an enhanced immune response, thus highlighting the immune function of both LDs and lipid species within promoting an efficient immune response following infection. This example also highlights the ability of the lipid content of a LD to act externally to its local environment. Neurons have also been demonstrated to secrete excess FAs, which are taken up by astrocytic LDs during times of enhanced activity (Ioannou et al. 2019). The accumulation of lipids in hyperactive neurons is not only toxic due to the susceptibility of these lipids to peroxidation, but also an overabundance of FAs may enter non-oxidative metabolic pathways triggering excessive ceramide production that is toxic to cells (Guenther et al. 2008). This coordinated mechanism for metabolizing FAs between cell types in the brain could potentially provide a protective mechanism by which cells are able to signal to nearby cells during viral infection, however, needs to be explored in more detail.
While LDs are readily detected intracellularly, their extracellular presence is not widely reported; however, it has been noted (Eyster and Van Camp 2003; Corrêa Soares et al. 2007). We now know that there are several cases of lipids being secreted and taken up by neighboring cells, but if whole LDs are able to be released and potentially taken up by neighboring cells, this could have implications for not only the delivery of FAs but also potentially important proteins during infection. To our knowledge, this is something that has not been examined; however, it could be potentially important for cell-to-cell communication.
CONCLUSIONS/INSIGHTS
The role that lipids play in a viral infection is very complex, and although it is well understood that lipids are essential for cell survival by providing energy, underpinning the integrity and flexibility of cellular structures and contributing to signal transduction, we also know that many viruses are able to alter the cellular lipidome to benefit their own viral life cycles. Excess lipids are stored within LDs in cells, and over the last 15 years the accumulation of LDs has become a hallmark of general infection. The mechanisms driving accumulation of LDs in this sense have traditionally been presumed to be pathogen driven for their own benefit; however, recent work has now identified that LDs can also have a positive role in respect to the antiviral response, as well as influence the synthesis of many bioactive lipid mediators. A better understanding of the fine line between a positive host cell response and the ability of viruses to usurp host lipid synthesis pathways is required for us to fully comprehend the role of lipids in the host response to viral infection.
We are only just beginning to understand the vast diversity and heterogeneity within LDs themselves. LDs contain many different lipid species; however, there has been no assessment of the changing composition of LDs during infection. LDs can vary in number and size between different cell types, but interestingly, there are also very distinct populations of LDs within a cell cytoplasm, with uncertainties remaining over the role of these populations of LDs partly due to heterogeneity in size and function as well as interactions between LDs and other cellular organelles, and how this is associated with discrete functions. It is possible that different populations of LDs play different roles during immune responses; however, we are lacking information regarding these different populations of LDs. More detailed lipidomic analysis of distinct LD populations is required for us to gain a better understanding of the role of LDs during this response; however, the technologies for this are in their infancy.
The heterogeneous nature of LDs may contribute to the differing, and often contradictory roles we see for LDs in infection and immunity, and this may be due to the content of LDs influencing what lipid mediators can arise. Lipid mediators are directly synthesized from the breakdown of lipids such as triglycerides in LDs, and their roles in various aspects of general immune control have been well documented, despite their often opposing roles found in respect to the control of viral infection. However, we are lacking an understanding of what is driving the breakdown of TGs to form lipid mediators (the host or the virus)—whether it is a selective mechanism, or whether it is just a by-product of lipolysis and cell stress. Elucidating the mechanisms by which these lipid mediators can be produced, as well as understanding their role in antiviral responses more definitively through the use of improved model systems, will help us better understand the functional role of these important molecules, which may in turn offer opportunities for directed treatment strategies for viral infections.
It is well known that LDs harbor a myriad of lipid synthesis enzymes and proteins, which regulate their lipid content; however, the changing proteome of LDs during viral infections is very understudied, and may yield critical insight into the role these organelles may play during viral infection. Additionally, there are still questions that need to be addressed to fully understand why there seems to be such a differing role for lipids and their bioactive mediators during the host response to infection. As lipid synthesis machinery is induced very early following infection, it may be that analysis of the role of lipids at earlier time points following viral infection could provide us with a better understanding of the role of lipids for the host response. There also seems to be a balancing act in the fact that an early induction of lipids may help the host initially, but may lead to an exaggerated inflammatory response causing the production of cytokine storms, particularly in the lungs during respiratory viral infections, and a better understanding of what tips this scale may also underpin novel treatment strategies for viral infections moving forward.
It is clear that the field of LDs and their cargo is an emerging field of discovery with many unanswered questions; however, we believe that a better understanding of the roles LDs play in communicating with various immune pathways may assist in informing potential advanced therapeutic approaches to combat pathogenic microorganisms.
ACKNOWLEDGMENTS
We thank BioRender.com for allowing us to create the figures throughout this review. We would also like to thank members of the Helbig and Mackenzie labs for their input on this review.
Contributor Information
Ebony A Monson, School of Life Sciences, La Trobe University, Melbourne, Australia, 3083.
Alice M Trenerry, Department of Microbiology and Immunology, University of Melbourne, at the Peter Doherty Institute for Infection and Immunity, Melbourne, Australia, 3000.
Jay L Laws, School of Life Sciences, La Trobe University, Melbourne, Australia, 3083.
Jason M Mackenzie, Department of Microbiology and Immunology, University of Melbourne, at the Peter Doherty Institute for Infection and Immunity, Melbourne, Australia, 3000.
Karla J Helbig, School of Life Sciences, La Trobe University, Melbourne, Australia, 3083.
FUNDING
This work was funded by the National Health and Medical Research Council of Australia (NHMRC Ideas grant APP1181434) and a La Trobe University Understanding Disease Research Focus Area small grant to KJH. Research performed by JMM was supported by a Melbourne Research Grant Support Scheme grant.
Conflict of Interest
None declared.
REFERENCES
- Accioly MT, Pacheco P, Maya-Monteiro CMet al. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 2008;68:1732–40. [DOI] [PubMed] [Google Scholar]
- Afonso PV, Janka-Junttila M, Lee YJet al. LTB4 is a signal-relay molecule during neutrophil chemotaxis. Dev Cell. 2012;22:1079–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfajaro MM, Choi J-S, Kim D-Set al. Activation of COX-2/PGE2 promotes sapovirus replication via the inhibition of nitric oxide production. J Virol. 2017;91:e01656–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloia AL, Calvert JK, Clarke JNet al. Investigation of sphingosine kinase 1 in interferon responses during dengue virus infection. Clin Transl Immunology. 2017;6:e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmann R. Die Elementarorganismen und ihre Beziehungen zu den Zellen. Leipzig, Germany: Verlag Von Veit & Comp, 1894. [Google Scholar]
- Anand P, Cermelli S, Li Zet al. A novel role for lipid droplets in the organismal antibacterial response. eLife. 2012;1:e00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariel A, Serhan CN. Resolvins and protectins in the termination program of acute inflammation. Trends Immunol. 2007;28:176–83. [DOI] [PubMed] [Google Scholar]
- Ashar HK, Mueller NC, Rudd JMet al. The role of extracellular histones in influenza virus pathogenesis. Am J Pathol. 2018;188:135–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzam KM, Fessler MB. Crosstalk between reverse cholesterol transport and innate immunity. Trends Endocrinol Metab. 2012;23:169–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bafica A, Feng CG, Santiago HCet al. The IFN-inducible GTPase LRG47 (Irgm1) negatively regulates TLR4-triggered proinflammatory cytokine production and prevents endotoxemia. J Immunol. 2007;179:5514–22. [DOI] [PubMed] [Google Scholar]
- Bai L, Dong J, Liu Zet al. Viperin catalyzes methionine oxidation to promote protein expression and function of helicases. Sci Adv. 2019;5:eaax1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey AP, Koster G, Guillermier Cet al. Antioxidant role for lipid droplets in a stem cell niche of Drosophila. Cell. 2015;163:340–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang B-R, Li M, Tsai K-Net al. Regulation of hepatitis C virus infection by cellular retinoic acid binding proteins through the modulation of lipid droplet abundance. J Virol. 2019;93:e02302–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannenberg G, Serhan CN. Specialized pro-resolving lipid mediators in the inflammatory response: an update. Biochim Biophys Acta. 2010;1801:1260–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barba G, Harper F, Harada Tet al. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc Natl Acad Sci USA. 1997;94:1200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barletta ABF, Alves LR, Silva Met al. Emerging role of lipid droplets in Aedes aegypti immune response against bacteria and dengue virus. Sci Rep. 2016;6:19928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartz H, Büning-Pfaue F, Türkel Oet al. Respiratory syncytial virus induces prostaglandin E2, IL-10 and IL-11 generation in antigen presenting cells. Clin Exp Immunol. 2002;129:438–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartz R, Li W-H, Venables Bet al. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J Lipid Res. 2007;48:837–47. [DOI] [PubMed] [Google Scholar]
- Bersuker K, Peterson CWH, To Met al. A proximity labeling strategy provides insights into the composition and dynamics of lipid droplet proteomes. Dev Cell. 2018;44:97–112..e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binns D, Januszewski T, Chen Yet al. An intimate collaboration between peroxisomes and lipid bodies. J Cell Biol. 2006;173:719–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette-Mackie EJ, Scow RO. Movement of lipolytic products to mitochondria in brown adipose tissue of young rats: an electron microscope study. J Lipid Res. 1983;24:229–44. [PubMed] [Google Scholar]
- Bley H, Schöbel A, Herker E. Whole Lotta lipids: from HCV RNA replication to the mature viral particle. Int J Mol Sci. 2020;21:2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boodhoo N, Kamble N, Kaufer BBet al. Replication of Marek's disease virus is dependent on synthesis of de novofatty acid and prostaglandin E2. J Virol. 2019;93:e00352–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges TKS, Alves ÉAR, Vasconcelos HARet al. Differences in the modulation of reactive species, lipid bodies, cyclooxygenase-2, 5-lipoxygenase and PPAR-γ in cerebral malaria-susceptible and resistant mice. Immunobiology. 2017;222:604–19. [DOI] [PubMed] [Google Scholar]
- Bosch M, Sánchez-Álvarez M, Fajardo Aet al. Mammalian lipid droplets are innate immune hubs integrating cell metabolism and host defense. Science. 2020;370:eaay8085. [DOI] [PubMed] [Google Scholar]
- Bougnères L, Helft J, Tiwari Set al. A role for lipid bodies in the cross-presentation of phagocytosed antigens by MHC class I in dendritic cells. Immunity. 2009;31:232–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulant S, Douglas MW, Moody Let al. Hepatitis C virus core protein induces lipid droplet redistribution in a microtubule- and dynein-dependent manner. Traffic. 2008;9:1268–82. [DOI] [PubMed] [Google Scholar]
- Bozza PT, Bakker-Abreu I, Navarro-Xavier RAet al. Lipid body function in eicosanoid synthesis: an update. Prostaglandins Leukot Essent Fatty Acids. 2011;85:205–13. [DOI] [PubMed] [Google Scholar]
- Bozza PT, Magalhães KG, Weller PF. Leukocyte lipid bodies: Biogenesis and functions in inflammation. Biochim Biophys Acta. 2009;1791:540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caini P, Guerra CT, Giannini Cet al. Modifications of plasma platelet-activating factor (PAF)-acetylhydrolase/PAF system activity in patients with chronic hepatitis C virus infection. J Viral Hepat. 2007;14:22–8. [DOI] [PubMed] [Google Scholar]
- Camus G, Herker E, Modi AAet al. Diacylglycerol acyltransferase-1 localizes hepatitis C virus NS5A protein to lipid droplets and enhances NS5A interaction with the viral capsid core. J Biol Chem. 2013;288:9915–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canetti CA, Leung BP, Culshaw Set al. IL-18 enhances collagen-induced arthritis by recruiting neutrophils via TNF-alpha and leukotriene B4. J Immunol. 2003;171:1009–15. [DOI] [PubMed] [Google Scholar]
- Cao F, Castrillo A, Tontonoz Pet al. Chlamydia pneumoniae–induced macrophage foam cell formation is mediated by Toll-like receptor 2. Infect Immun. 2007;75:753–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho FA, Carneiro FA, Martins ICet al. Dengue virus capsid protein binding to hepatic lipid droplets (LD) is potassium ion dependent and is mediated by LD surface proteins. J Virol. 2012;86:2096–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casares D, Escribá PV, Rosselló CA. Membrane Lipid composition: effect on membrane and organelle structure, function and compartmentalization and therapeutic avenues. Int J Mol Sci. 2019;20:2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoldi A, Monteiro LB, van Teijlingen Bakker Net al. Triacylglycerol synthesis enhances macrophage inflammatory function. Nat Commun. 2020;11:4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermelli S, Guo Y, Gross SPet al. The lipid-droplet proteome reveals that droplets are a protein-storage depot. Curr Biol. 2006;16:1783–95. [DOI] [PubMed] [Google Scholar]
- Chandrasekharan JA, Huang XM, Hwang ACet al. Altering the anti-inflammatory lipoxin microenvironment: a new insight into Kaposi's sarcoma-associated herpesvirus pathogenesis. J Virol. 2016;90:11020–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron AJ, Sibley LD. Host cells: mobilizable lipid resources for the intracellular parasite Toxoplasma gondii. J Cell Sci. 2002;115:3049–59. [DOI] [PubMed] [Google Scholar]
- Chen JH, Perry CJ, Tsui Y-Cet al. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat Med. 2015;21:327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Restivo A, Reiss CS. Selective inhibition of COX-2 is beneficial to mice infected intranasally with VSV. Prostaglandins Other Lipid Mediat. 2002;67:143–55. [DOI] [PubMed] [Google Scholar]
- Chen N, Warner JL, Reiss CS. NSAID treatment suppresses VSV propagation in mouse CNS. Virology. 2000;276:44–51. [DOI] [PubMed] [Google Scholar]
- Chen Q, Gouilly J, Ferrat YJet al. Metabolic reprogramming by Zika virus provokes inflammation in human placenta. Nat Commun. 2020;11:2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung W, Gill M, Esposito Aet al. Rotaviruses associate with cellular lipid droplet components to replicate in viroplasms, and compounds disrupting or blocking lipid droplets inhibit viroplasm formation and viral replication. J Virol. 2010;84:6782–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitraju C, Trötzmüller M, Hartler Jet al. Lipidomic analysis of lipid droplets from murine hepatocytes reveals distinct signatures for nutritional stress. J Lipid Res. 2012;53:2141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiurchiù V, Maccarrone M. Bioactive lipids as modulators of immunity, inflammation and emotions. Curr Opin Pharmacol. 2016;29:54–62. [DOI] [PubMed] [Google Scholar]
- Choi JW, Herr DR, Noguchi Ket al. LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol. 2010;50:157–86. [DOI] [PubMed] [Google Scholar]
- Cilloniz C, Pantin-Jackwood MJ, Ni Cet al. Lethal dissemination of H5N1 influenza virus is associated with dysregulation of inflammation and lipoxin signaling in a mouse model of infection. J Virol. 2010;84:7613–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civra A, Francese R, Gamba Pet al. 25-Hydroxycholesterol and 27-hydroxycholesterol inhibit human rotavirus infection by sequestering viral particles into late endosomes. Redox Biol. 2018;19:318–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente MI, Álvarez S, Serramía MJet al. Prostaglandin E2 reduces the release and infectivity of new cell-free virions and cell-to-cell HIV-1 transfer. PLoS One. 2014;9:e85230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchiaro JL, Kumar Y, Fischer ERet al. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc Natl Acad Sci USA. 2008;105:9379–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey CM, Sheh A, Kim ISet al. Reovirus outer capsid protein micro1 induces apoptosis and associates with lipid droplets, endoplasmic reticulum, and mitochondria. J Virol. 2006;80:8422–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colas RA, Shinohara M, Dalli Jet al. Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. Am J Physiol Cell Physiol. 2014;307:C39–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RA, Mashek DG. Mammalian triacylglycerol metabolism: synthesis, lipolysis, and signaling. Chem Rev. 2011;111:6359–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey EJ, Nicolaou KC, Machida Yet al. Synthesis and biological properties of a 9,11-azo-prostanoid: highly active biochemical mimic of prostaglandin endoperoxides. Proc Natl Acad Sci USA. 1975;72:3355–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrêa Soares JBR, Maya-Monteiro CM, Bittencourt-Cunha PRBet al. Extracellular lipid droplets promote hemozoin crystallization in the gut of the blood fluke Schistosoma mansoni. FEBS Lett. 2007;581:1742–50. [DOI] [PubMed] [Google Scholar]
- Cottrell JA, Lin H-N, O'Connor JP. Method for measuring lipid mediators, proteins, and messenger RNAs from a single tissue specimen. Anal Biochem. 2015;469:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe F, Jaworska J, Verway Met al. Targeted prostaglandin E2 inhibition enhances antiviral immunity through induction of type I interferon and apoptosis in macrophages. Immunity. 2014;40:554–68. [DOI] [PubMed] [Google Scholar]
- Crosse KM, Monson EA, Dumbrepatil ABet al. Viperin binds STING and enhances the type-I interferon response following dsDNA detection. Immunol Cell Biol. 2020, 10.1111/imcb.12420. [DOI] [PubMed] [Google Scholar]
- Cruz ALS, Barreto E, de A, Fazolini NPBet al. Lipid droplets: platforms with multiple functions in cancer hallmarks. Cell Death Dis. 2020;11:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Avila H, Freire-de-Lima CG, Roque NRet al. Host cell lipid bodies triggered by Trypanosoma cruzi infection and enhanced by the uptake of apoptotic cells are associated with prostaglandin E2 generation and increased parasite growth. J Infect Dis. 2011;204:951–61. [DOI] [PubMed] [Google Scholar]
- D'Avila H, Melo RCN, Parreira GGet al. Mycobacterium bovis bacillus Calmette-Guérin induces TLR2-mediated formation of lipid bodies: intracellular domains for eicosanoid synthesis in vivo. J Immunol. 2006;176:3087–97. [DOI] [PubMed] [Google Scholar]
- de Almeida PE, Toledo DAM, Rodrigues GSCet al. Lipid bodies as sites of prostaglandin E2 synthesis during Chagas disease: impact in the parasite escape mechanism. Front Microbiol. 2018;9:499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhiman R, Caesar S, Thiam ARet al. Mechanisms of protein targeting to lipid droplets: a unified cell biological and biophysical perspective. Semin Cell Dev Biol. 2020;108:4–13. [DOI] [PubMed] [Google Scholar]
- Dichlberger A, Schlager S, Maaninka Ket al. Adipose triglyceride lipase regulates eicosanoid production in activated human mast cells. J Lipid Res. 2014;55:2471–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumais N, Barbeau B, Olivier M. Prostaglandin E2 up-regulates HIV-1 long terminal repeat-driven gene activity in T cells via NF-κB-dependent and-independent signaling pathways. Journal of Biological. 1998. [DOI] [PubMed] [Google Scholar]
- Dutta A, Banerjee S, Sinha DK. Lipid droplet metabolism dependent microbial defense in pre-immune zebrafish embryos. bioRxiv. 2018:478859. [Google Scholar]
- Dvorak AM, Morgan E, Schleimer RPet al. Ultrastructural immunogold localization of prostaglandin endoperoxide synthase (cyclooxygenase) to non-membrane-bound cytoplasmic lipid bodies in human lung mast cells, alveolar macrophages, type II pneumocytes, and neutrophils. J Histochem Cytochem. 1992;40:759–69. [DOI] [PubMed] [Google Scholar]
- El Alwani M, Wu BX, Obeid LMet al. Bioactive sphingolipids in the modulation of the inflammatory response. Pharmacol Ther. 2006;112:171–83. [DOI] [PubMed] [Google Scholar]
- Episcopio D, Aminov S, Benjamin Set al. Atorvastatin restricts the ability of influenza virus to generate lipid droplets and severely suppresses the replication of the virus. FASEB J. 2019;33:9516–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyster LS, Van Camp LM. Extracellular lipid droplets in Idiosepius notoides, the southern pygmy squid. Biol Bull. 2003;205:47–53. [DOI] [PubMed] [Google Scholar]
- Fahy E, Subramaniam S, Murphy RCet al. Update of the LIPID MAPS comprehensive classification system for lipids. J Lipid Res. 2009;50 Suppl:S9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farquhar MJ, Humphreys IS, Rudge SAet al. Autotaxin-lysophosphatidic acid receptor signalling regulates hepatitis C virus replication. J Hepatol. 2017;66:919–29. [DOI] [PubMed] [Google Scholar]
- Feingold KR, Kazemi MR, Magra ALet al. ADRP/ADFP and Mal1 expression are increased in macrophages treated with TLR agonists. Atherosclerosis. 2010;209:81–8. [DOI] [PubMed] [Google Scholar]
- Fitzgerald DW, Bezak K, Ocheretina Oet al. The effect of HIV and HPV coinfection on cervical COX-2 expression and systemic prostaglandin E2 levels. Cancer Prev Res. 2012;5:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty SE III, Grijalva A, Xu Xet al. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science. 2019;363:989–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamand L, Borgeat P, Lalonde Ret al. Release of anti-HIV mediators after administration of leukotriene B4 to humans. J Infect Dis. 2004;189:2001–9. [DOI] [PubMed] [Google Scholar]
- Flamand L, Tremblay MJ, Borgeat P. Leukotriene B4 triggers the in vitro and in vivo release of potent antimicrobial agents. J Immunol. 2007;178:8036–45. [DOI] [PubMed] [Google Scholar]
- Flowers M, Sherker A, Sinclair SBet al. Prostaglandin E in the treatment of recurrent hepatitis B infection after orthotopic liver transplantation. Transplantation. 1994;58:183–92. [PubMed] [Google Scholar]
- Frank Austen K. The cysteinyl leukotrienes: where do they come from? What are they? Where are they going? Nat Immunol. 2008;9:113–5. [DOI] [PubMed] [Google Scholar]
- Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–5. [DOI] [PubMed] [Google Scholar]
- Gandhi J, Gaur N, Khera Let al. COX-2 induces lytic reactivation of EBV through PGE2 by modulating the EP receptor signaling pathway. Virology. 2015;484:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartung A, Zhao J, Chen Set al. Characterization of eicosanoids produced by adipocyte lipolysis: implication of cyclooxygenase-2 in adipose inflammation. J Biol Chem. 2016;291:16001–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudreault E, Gosselin J. Leukotriene B4-mediated release of antimicrobial peptides against cytomegalovirus is BLT1 dependent. Viral Immunol. 2007;20:407–20. [DOI] [PubMed] [Google Scholar]
- Gaudreault E, Gosselin J. Leukotriene B4 induces release of antimicrobial peptides in lungs of virally infected mice. J Immunol. 2008;180:6211–21. [DOI] [PubMed] [Google Scholar]
- Gaunt ER, Zhang Q, Cheung Wet al. Lipidome analysis of rotavirus-infected cells confirms the close interaction of lipid droplets with viroplasms. J Gen Virol. 2013;94:1576–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genis P, Jett M, Bernton EWet al. Cytokines and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage-astroglia interactions: implications for the neuropathogenesis of HIV disease. J Exp Med. 1992;176:1703–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakis N, Sansbury BE, Patsalos Aet al. Dynamic changes to lipid mediators support transitions among macrophage subtypes during muscle regeneration. Nat Immunol. 2019;20:626–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes L, Fernando S, Fernando RHet al. Sphingosine 1-phosphate in acute dengue infection. PLoS One. 2014;9:e113394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gower TL, Pastey MK, Peeples MEet al. RhoA signaling is required for respiratory syncytial virus-induced syncytium formation and filamentous virion morphology. J Virol. 2005;79:5326–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber K, Khan F, Glück Bet al. The role of sphingosine-1-phosphate signaling in HSV-1-infected human umbilical vein endothelial cells. Virus Res. 2020;276:197835. [DOI] [PubMed] [Google Scholar]
- Grespan R, Fukada SY, Lemos HPet al. CXCR2-specific chemokines mediate leukotriene B4-dependent recruitment of neutrophils to inflamed joints in mice with antigen-induced arthritis. Arthritis Rheum. 2008;58:2030–40. [DOI] [PubMed] [Google Scholar]
- Grégoire IP, Rabourdin-Combe C, Faure M. Autophagy and RNA virus interactomes reveal IRGM as a common target. Autophagy. 2012;8:1136–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grégoire IP, Richetta C, Meyniel-Schicklin Let al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011;7:e1002422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther GG, Peralta ER, Rosales KRet al. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc Natl Acad Sci USA. 2008;105:17402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guijas C, Rodríguez JP, Rubio JMet al. Phospholipase A2 regulation of lipid droplet formation. Biochim Biophys Acta. 2014;1841:1661–71. [DOI] [PubMed] [Google Scholar]
- Haemmerle G, Lass A, Zimmermann Ret al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312:734–7. [DOI] [PubMed] [Google Scholar]
- Haldar AK, Saka HA, Piro ASet al. IRG and GBP host resistance factors target aberrant, “non-self” vacuoles characterized by the missing of “self” IRGM proteins. PLoS Pathog. 2013;9:e1003414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammad SM, Crellin HG, Wu BXet al. Dual and distinct roles for sphingosine kinase 1 and sphingosine 1 phosphate in the response to inflammatory stimuli in RAW macrophages. Prostaglandins Other Lipid Mediat. 2008;85:107–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–50. [DOI] [PubMed] [Google Scholar]
- Harbour DA, Blyth WA, Hill TJ. Prostaglandins enhance spread of herpes simplex virus in cell cultures. J Gen Virol. 1978;41:87–95. [DOI] [PubMed] [Google Scholar]
- Harizi H, Corcuff J-B, Gualde N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol Med. 2008;14:461–9. [DOI] [PubMed] [Google Scholar]
- Hartler J, Köfeler HC, Trötzmüller Met al. Assessment of lipidomic species in hepatocyte lipid droplets from stressed mouse models. Sci Data. 2014;1:140051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MM, Lane BR, King SRet al. Prostaglandin E(2) inhibits replication of HIV-1 in macrophages through activation of protein kinase A. Cell Immunol. 2002;215:61–71. [DOI] [PubMed] [Google Scholar]
- Heaton NS, Randall G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe. 2010;8:422–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herker E, Harris C, Hernandez Cet al. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat Med. 2010;16:1295–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson ER, Cresswell P. The antiviral protein, viperin, localizes to lipid droplets via its N-terminal amphipathic α-helix. Proc Natl Acad Sci USA. 2009;106:20452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeksema M, Tripathi S, White Met al. Arginine-rich histones have strong antiviral activity for influenza A viruses. Innate Immun. 2015;21:736–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooks JJ, Chin MS, Srinivasan Ket al. Human cytomegalovirus induced cyclooxygenase-2 in human retinal pigment epithelial cells augments viral replication through a prostaglandin pathway. Microbes Infect. 2006;8:2236–44. [DOI] [PubMed] [Google Scholar]
- Huang Y-L, Morales-Rosado J, Ray Jet al. Toll-like receptor agonists promote prolonged triglyceride storage in macrophages. J Biol Chem. 2014;289:3001–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes-Fulford M, McGrath MS, Hanks Det al. Effects of dimethyl prostaglandin A1 on herpes simplex virus and human immunodeficiency virus replication. Antimicrob Agents Chemother. 1992;36:2253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman A, Yim C, Krajden Met al. Oral prostaglandin (PGE2) therapy for chronic viral hepatitis B and C. J Viral Hepat. 1999;6:329–36. [DOI] [PubMed] [Google Scholar]
- Im S-S, Yousef L, Blaschitz Cet al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. 2011;13:540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannou MS, Jackson J, Sheu S-Het al. Neuron–astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell. 2019;177:1522–35.e14. [DOI] [PubMed] [Google Scholar]
- Irwin NAT, Martin BJE, Young BPet al. Viral proteins as a potential driver of histone depletion in dinoflagellates. Nat Commun. 2018;9:1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida K, Goto S, Ishimura Met al. Functional Correlation between subcellular localizations of japanese encephalitis virus capsid protein and virus production. J Virol. 2019;93, DOI:10.1128/JVI.00612-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii S, Nagase T, Shimizu T. Platelet-activating factor receptor. Prostaglandins Other Lipid Mediat. 2002;68-69:599–609. [DOI] [PubMed] [Google Scholar]
- Jackson CL, Walch L, Verbavatz J-M. Lipids and their trafficking: an integral part of cellular organization. Dev Cell. 2016;39:139–53. [DOI] [PubMed] [Google Scholar]
- Jackson CL. Lipid droplet biogenesis. Curr Opin Cell Biol. 2019;59:88–96. [DOI] [PubMed] [Google Scholar]
- Ji C, Chan C, Kaplowitz N. Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model. J Hepatol. 2006;45:717–24. [DOI] [PubMed] [Google Scholar]
- Jones PD, Wolf NM, Morisseau Cet al. Fluorescent substrates for soluble epoxide hydrolase and application to inhibition studies. Anal Biochem. 2005;343:66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josset L, Menachery VD, Gralinski LEet al. Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. mBio. 2013;4:e00165–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinski P.Regulation of immune responses by prostaglandin E2. J Immunol. 2012;188:21–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannis F, Masouleh SK, Wunderling Ket al. Lipid-droplet formation drives pathogenic group 2 innate lymphoid cells in airway inflammation. Immunity. 2020;52:620–34.e6. [DOI] [PubMed] [Google Scholar]
- Keul P, Lucke S, von Wnuck Lipinski Ket al. Sphingosine-1-phosphate receptor 3 promotes recruitment of monocyte/macrophages in inflammation and atherosclerosis. Circ Res. 2011;108:314–23. [DOI] [PubMed] [Google Scholar]
- Kim SJ.Elevated formation of lipoxins in viral antibody-positive rat alveolar macrophages. Am J Respir Cell Mol Biol. 1990;3:113–8. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Kim KP, Han SKet al. Group V phospholipase A2 induces leukotriene biosynthesis in human neutrophils through the activation of group IVA phospholipase A2. J Biol Chem. 2002;277:36479–88. [DOI] [PubMed] [Google Scholar]
- Klemm EJ, Spooner E, Ploegh HL. Dual role of ancient ubiquitous protein 1 (AUP1) in lipid droplet accumulation and endoplasmic reticulum (ER) protein quality control. J Biol Chem. 2011;286:37602–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Y, Cocchiaro J, Valdivia RH. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr Biol. 2006;16:1646–51. [DOI] [PubMed] [Google Scholar]
- Kuno S, Ueno R, Hayaishi Oet al. Prostaglandin E2, a seminal constituent, facilitates the replication of acquired immune deficiency syndrome virus in vitro. Proc Natl Acad Sci USA. 1986;83:3487–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo A, Lee MY, Yang Ket al. Caveolin-1 regulates lipid droplet metabolism in endothelial cells via autocrine prostacyclin-stimulated, cAMP-mediated lipolysis. J Biol Chem. 2018;293:973–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam SH, Chua HL, Gong Zet al. Development and maturation of the immune system in zebrafish, Danio rerio: a gene expression profiling, in situ hybridization and immunological study. Dev Comp Immunol. 2004;28:9–28. [DOI] [PubMed] [Google Scholar]
- Laufman O, Perrino J, Andino R. Viral generated inter-organelle contacts redirect lipid flux for genome replication. Cell. 2019;178:275–89..e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Deng Y-Q, Wang Set al. 25-Hydroxycholesterol protects host against Zika virus infection and its associated microcephaly in a mouse model. Immunity. 2017;46:446–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Feng T, Pan Wet al. DEAD-box RNA helicase DDX3X inhibits DENV replication via regulating type one interferon pathway. Biochem Biophys Res Commun. 2015;456:327–32. [DOI] [PubMed] [Google Scholar]
- Li M, Liao Z, Xu Zet al. The interaction mechanism between herpes simplex virus 1 glycoprotein D and host antiviral protein viperin. Front Immunol. 2019a;10:2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima RG, Moreira L, Paes-Leme Jet al. Interaction of macrophages with apoptotic cells enhances HIV Type 1 replication through PGE2, PAF, and vitronectin receptor. AIDS Res Hum Retroviruses. 2006;22:763–9. [DOI] [PubMed] [Google Scholar]
- Lin Y-C, Chang P-F, Lin H-Fet al. Variants in the autophagy-related gene IRGM confer susceptibility to non-alcoholic fatty liver disease by modulating lipophagy. J Hepatol. 2016;65:1209–16. [DOI] [PubMed] [Google Scholar]
- Liu A, Claesson H-E, Mahshid Yet al. Leukotriene B4 activates T cells that inhibit B-cell proliferation in EBV-infected cord blood-derived mononuclear cell cultures. Blood. 2008;111:2693–703. [DOI] [PubMed] [Google Scholar]
- Liu P, Ying Y, Zhao Yet al. Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J Biol Chem. 2004;279:3787–92. [DOI] [PubMed] [Google Scholar]
- Liu X, Lai X, Zhang Set al. Proteomic profile of edible bird's nest proteins. J Agric Food Chem. 2012;60:12477–81. [DOI] [PubMed] [Google Scholar]
- Li Y, Deng S-L, Lian Z-Xet al. Roles of toll-like receptors in nitroxidative stress in mammals. Cells. 2019b;8:576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Thiel K, Thul PJet al. Lipid droplets control the maternal histone supply of Drosophila embryos. Curr Biol. 2012;22:2104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luczak M, Gumulka W, Szmigielski Set al. Inhibition of multiplication of parainfluenza 3 virus in prostaglandin-treated WISH cells. Arch Virol. 1975;49:377–80. [DOI] [PubMed] [Google Scholar]
- Lämmermann T, Afonso PV, Angermann BRet al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. 2013;498:371–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maceyka M, Spiegel S.. Sphingolipid metabolites in inflammatory disease. Nature. 2014;510:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking JD, Taylor GA, McKinney JD. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science. 2003;302:654–9. [DOI] [PubMed] [Google Scholar]
- Maeda Y, Matsuyuki H, Shimano Ket al. Migration of CD4 T cells and dendritic cells toward Sphingosine 1-phosphate (S1P) Is mediated by different receptor subtypes: S1P regulates the functions of murine mature dendritic cells via S1P receptor type 3. J Immunol. 2007;178:3437–46. [DOI] [PubMed] [Google Scholar]
- Magalhã KG, Almeida PE, Atella GCet al. Schistosomal-derived lysophosphatidylcholine are involved in eosinophil activation and recruitment through toll-like receptor-2-dependent mechanisms. J Infect Dis. 2010;202:1369–79. [DOI] [PubMed] [Google Scholar]
- Marcinkiewicz A, Gauthier D, Garcia Aet al. The phosphorylation of serine 492 of perilipin A directs lipid droplet fragmentation and dispersion. J Biol Chem. 2006;281:11901–9. [DOI] [PubMed] [Google Scholar]
- Marsolais D, Hahm B, Walsh KBet al. A critical role for the sphingosine analog AAL-R in dampening the cytokine response during influenza virus infection. Proc Natl Acad Sci USA. 2009;106:1560–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins AS, Carvalho FA, Faustino AFet al. West Nile virus capsid protein interacts with biologically relevant host lipid systems. Front Cell Infect Microbiol. 2019;9:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins IC, Gomes-Neto F, Faustino AFet al. The disordered N-terminal region of dengue virus capsid protein contains a lipid-droplet-binding motif. Biochem J. 2012;444:405–15. [DOI] [PubMed] [Google Scholar]
- Mattos KA, D'Avila H, Rodrigues LSet al. Lipid droplet formation in leprosy: toll-like receptor-regulated organelles involved in eicosanoid formation and Mycobacterium leprae pathogenesis. J Leukoc Biol. 2010;87:371–84. [DOI] [PubMed] [Google Scholar]
- Mattos KA, Lara FA, Oliveira VGCet al. Modulation of lipid droplets by Mycobacterium leprae in Schwann cells: a putative mechanism for host lipid acquisition and bacterial survival in phagosomes. Cell Microbiol. 2011;13:259–73. [DOI] [PubMed] [Google Scholar]
- Medeiros AI, Silva CL, Malheiro Aet al. Leukotrienes are involved in leukocyte recruitment induced by live Histoplasma capsulatum or by the β-glucan present in their cell wall. Br J Pharmacol. 1999;128:1529–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo RCN, D'Avila H, Fabrino DLet al. Macrophage lipid body induction by Chagas disease in vivo: putative intracellular domains for eicosanoid formation during infection. Tissue Cell. 2003;35:59–67. [DOI] [PubMed] [Google Scholar]
- Melo RCN, Fabrino DL, Dias FFet al. Lipid bodies: structural markers of inflammatory macrophages in innate immunity. Inflamm Res. 2006;55:342–8. [DOI] [PubMed] [Google Scholar]
- Melo RCN, Weller PF. Lipid droplets in leukocytes: organelles linked to inflammatory responses. Exp Cell Res. 2016;340:193–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon D, Singh K, Pinto SMet al. Quantitative lipid droplet proteomics reveals Mycobacterium tuberculosisinduced alterations in macrophage response to infection. ACS Infect Dis. 2019;5:559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels M, Japtok L, Alisjahbana Bet al. Decreased plasma levels of the endothelial protective sphingosine-1-phosphate are associated with dengue-induced plasma leakage. J Infect. 2015;71:480–7. [DOI] [PubMed] [Google Scholar]
- Mittal A, Ranganath V, Nichani A. Omega fatty acids and resolution of inflammation: a new twist in an old tale. J Indian Soc Periodontol. 2010;14:3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molfino A, Amabile MI, Monti Met al. Omega-3 polyunsaturated fatty acids in critical illness: anti-inflammatory, proresolving, or both? Oxid Med Cell Longev. 2017;2017:5987082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monson EA, Crosse KM, Das Met al. Lipid droplet density alters the early innate immune response to viral infection. PLoS One. 2018;13:e0190597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monson EA, Crosse KM, Duan Met al. Intracellular lipid droplet accumulation occurs early following viral infection and is required for an efficient interferon response. bioRxiv. 2020:2020.02.12.946749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Kuba K, Ichikawa Aet al. The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell. 2013;153:112–25. [DOI] [PubMed] [Google Scholar]
- Narumiya S, FitzGerald GA. Genetic and pharmacological analysis of prostanoid receptor function. J Clin Invest. 2001;108:25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negash AA, Ramos HJ, Crochet Net al. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013;9:e1003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TB, Olzmann JA. Lipid droplets and lipotoxicity during autophagy. Autophagy. 2017;13:2002–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou G, Goodall AH, Erridge C. Diverse bacteria promote macrophage foam cell formation via Toll-like receptor-dependent lipid body biosynthesis. J Atheroscler Thromb. 2012;19:137–48. [DOI] [PubMed] [Google Scholar]
- Niu Q, Cheng Y, Wang Het al. Chicken DDX3X Activates IFN-β via the chSTING-chIRF7-IFN-β Signaling Axis. Front Immunol. 2019;10:822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nokta MA, Hassan MI, Loesch Ket al. Human cytomegalovirus-induced immunosuppression. Relationship to tumor necrosis factor-dependent release of arachidonic acid and prostaglandin E2 in human monocytes. J Clin Invest. 1996;97:2635–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LAJ. How low cholesterol is good for anti-viral immunity. Cell. 2015;163:1572–4. [DOI] [PubMed] [Google Scholar]
- Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019;20:137–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onal G, Kutlu O, Gozuacik Det al. Lipid droplets in health and disease. Lipids Health Dis. 2017;16:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet M, Franklin V, Mak Eet al. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13:655–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco P, Bozza FA, Gomes RNet al. Lipopolysaccharide-induced leukocyte lipid body formation in vivo: innate immunity elicited intracellular Loci involved in eicosanoid metabolism. J Immunol. 2002;169:6498–506. [DOI] [PubMed] [Google Scholar]
- Pacheco P, Vieira-de-Abreu A, Gomes RNet al. Monocyte chemoattractant protein-1/CC chemokine ligand 2 controls microtubule-driven biogenesis and leukotriene B4-synthesizing function of macrophage lipid bodies elicited by innate immune response. J Immunol. 2007;179:8500–8. [DOI] [PubMed] [Google Scholar]
- Paraje MG, Correa SG, Renna MSet al. Candida albicans-secreted lipase induces injury and steatosis in immune and parenchymal cells. Can J Microbiol. 2008;54:647–59. [DOI] [PubMed] [Google Scholar]
- Park JH, Park EB, Lee JYet al. Identification of novel membrane-associated prostaglandin E synthase-1 (mPGES-1) inhibitors with anti-influenza activities in vitro. Biochem Biophys Res Commun. 2016;469:848–55. [DOI] [PubMed] [Google Scholar]
- Penrose H, Heller S, Cable Cet al. Epidermal growth factor receptor mediated proliferation depends on increased lipid droplet density regulated via a negative regulatory loop with FOXO3/Sirtuin6. Biochem Biophys Res Commun. 2016;469:370–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percher F, Curis C, Pérès Eet al. HTLV-1-induced leukotriene B4 secretion by T cells promotes T cell recruitment and virus propagation. Nat Commun. 2017;8, DOI:10.1038/ncomms15890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña Cárcamo JR, Morell ML, Vázquez CAet al. The interplay between viperin antiviral activity, lipid droplets and Junín mammarenavirus multiplication. Virology. 2018;514:216–29. [DOI] [PubMed] [Google Scholar]
- Piedimonte G, Renzetti G, Auais Aet al. Leukotriene synthesis during respiratory syncytial virus bronchiolitis: influence of age and atopy. Pediatr Pulmonol. 2005;40:285–91. [DOI] [PubMed] [Google Scholar]
- Pinheiro RO, Nunes MP, Pinheiro CSet al. Induction of autophagy correlates with increased parasite load of Leishmania amazonensisin BALB/c but not C57BL/6 macrophages. Microbes Infect. 2009;11:181–90. [DOI] [PubMed] [Google Scholar]
- Pol A, Gross SP, Parton RG. Review: biogenesis of the multifunctional lipid droplet: lipids, proteins, and sites. J Cell Biol. 2014;204:635–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss C, Jelenik T, Bódis Ket al. A new targeted lipidomics approach reveals lipid droplets in liver, muscle and heart as a repository for diacylglycerol and ceramide species in non-alcoholic fatty liver. Cells. 2019;8:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto P, Cuenca J, Través PGet al. Lipoxin A4 impairment of apoptotic signaling in macrophages: implication of the PI3K/Akt and the ERK/Nrf-2 defense pathways. Cell Death Differ. 2010;17:1179–88. [DOI] [PubMed] [Google Scholar]
- Pu J, Ha CW, Zhang Set al. Interactomic study on interaction between lipid droplets and mitochondria. Protein Cell. 2011;2:487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadri F, Bhuiyan TR, Dutta KKet al. Acute dehydrating disease caused by Vibrio cholerae serogroups O1 and O139 induce increases in innate cells and inflammatory mediators at the mucosal surface of the gut. Gut. 2004;53:62–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu FH, Devchand PR, Wada Ket al. Aspirin-triggered lipoxin A4 and lipoxin A4 up-regulate transcriptional corepressor NAB1 in human neutrophils. FASEB J. 2001;15:2736–8. [DOI] [PubMed] [Google Scholar]
- Rabhi S, Rabhi I, Trentin Bet al. Lipid droplet formation, their localization and dynamics during Leishmania major macrophage infection. PLoS One. 2016;11:e0148640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasagi NK, Reddy PBJ, Mulik Set al. Neuroprotectin D1 reduces the severity of herpes simplex virus-induced corneal immunopathology. Invest Ophthalmol Vis Sci. 2013;54:6269–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasagi NK, Reddy PBJ, Suryawanshi Aet al. Controlling herpes simplex virus-induced ocular inflammatory lesions with the lipid-derived mediator resolvin E1. J Immunol. 2011;186:1735–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank RG, Whittimore J, Bowlin AKet al. In vivo ultrastructural analysis of the intimate relationship between polymorphonuclear leukocytes and the chlamydial developmental cycle. Infect Immun. 2011;79:3291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JY, Ottolini MG, Pletneva Let al. Respiratory syncytial virus (RSV) infection induces cyclooxygenase 2: a potential target for RSV therapy. J Immunol. 2005;174:4356–64. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Acosta A, Finol HJ, Pulido-Mendez Met al. Liver ultrastructural pathology in mice infected with Plasmodium berghei. J Submicrosc Cytol Pathol. 1998;30:299–307. [PubMed] [Google Scholar]
- Rosas-Ballina M, Guan XL, Schürmann Net al. Mechanism of lipid droplet accumulation in mononuclear phagocytes during sepsis (IRM9P.461). J Immunol. 2015;194:130.6. [Google Scholar]
- Rossen JWA, Bouma J, Raatgeep RHCet al. Inhibition of cyclooxygenase activity reduces rotavirus infection at a postbinding step. J Virol. 2004;78:9721–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan A. Blocking the route to infection. Nat Rev Drug Discovery. 2005;4:16. [Google Scholar]
- Rösch K, Kwiatkowski M, Hofmann Set al. Quantitative lipid droplet proteome analysis identifies annexin A3 as a cofactor for HCV particle production. Cell Rep. 2016;16:3219–31. [DOI] [PubMed] [Google Scholar]
- Sala A, Voelkel N, Maclouf Jet al. Leukotriene E4 elimination and metabolism in normal human subjects. J Biol Chem. 1990;265:21771–8. [PubMed] [Google Scholar]
- Salogiannis J, Christensen JR, Aguilar-Maldonado Aet al. Regulation of peroxisome and lipid droplet hitchhiking by PxdA and the DipA phosphatase. bioRxiv. 2020:2020.02.03.932616. [Google Scholar]
- Samsa MM, Mondotte JA, Iglesias NGet al. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2009;5:e1000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlager S, Goeritzer M, Jandl Ket al. Adipose triglyceride lipase acts on neutrophil lipid droplets to regulate substrate availability for lipid mediator synthesis. J Leukoc Biol. 2015;98:837–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol. 2011;1:519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröer J, Shenk T. Inhibition of cyclooxygenase activity blocks cell-to-cell spread of human cytomegalovirus. Proc Natl Acad Sci USA. 2008;105:19468–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J-Y, Cresswell P. Viperin regulates cellular lipid metabolism during human cytomegalovirus infection. PLoS Pathog. 2013;9:e1003497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y-J, Blake C, Alexander Set al. Sphingosine 1-phosphate-metabolizing enzymes control influenza virus propagation and viral cytopathogenicity. J Virol. 2010;84:8124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Hamberg M, Samuelsson B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc Natl Acad Sci USA. 1984;81:5335–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Z, Song H, Shi Yet al. Crystal structure of the capsid protein from Zika virus. J Mol Biol. 2018;430:948–62. [DOI] [PubMed] [Google Scholar]
- Shawli GT, Adeyemi OO, Stonehouse NJet al. The oxysterol 25-hydroxycholesterol inhibits replication of murine norovirus. Viruses. 2019;11, DOI:10.3390/v11020097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W-J, Azhar S, Kraemer FB. Lipid droplets and steroidogenic cells. Exp Cell Res. 2016;340:209–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirey KA, Lai W, Pletneva LMet al. Role of the lipoxygenase pathway in RSV-induced alternatively activated macrophages leading to resolution of lung pathology. Mucosal Immunol. 2014;7:549–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmarakov IO, Jiang H, Liu Jet al. Hepatic stellate cell activation: a source for bioactive lipids. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864:629–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Sampaio JL. Membrane organization and lipid rafts. Cold Spring Harb Perspect Biol. 2011;3:a004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer II, Tian M, Wickham LAet al. Sphingosine-1-phosphate agonists increase macrophage homing, lymphocyte contacts, and endothelial junctional complex formation in murine lymph nodes. J Immunol. 2005;175:7151–61. [DOI] [PubMed] [Google Scholar]
- Singh SB, Ornatowski W, Vergne Iet al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol. 2010;12:1154–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn JH, Lee YK, Han JSet al. Perilipin 1 (Plin1) deficiency promotes inflammatory responses in lean adipose tissue through lipid dysregulation. J Biol Chem. 2018;293:13974–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulat D, Bürckstümmer T, Westermayer Set al. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J. 2008;27:2135–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza DG, Fagundes CT, Sousa LPet al. Essential role of platelet-activating factor receptor in the pathogenesis of dengue virus infection. Proc Natl Acad Sci USA. 2009;106:14138–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stith JL, Velazquez FN, Obeid LM. Advances in determining signaling mechanisms of ceramide and role in disease. J Lipid Res. 2019;60:913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S, Fahy E, Gupta Set al. Bioinformatics and systems biology of the lipidome. Chem Rev. 2011;111:6452–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takabe K, Paugh SW, Milstien Set al. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60:181–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanigawa K, Suzuki K, Nakamura Ket al. Expression of adipose differentiation-related protein (ADRP) and perilipin in macrophages infected with Mycobacterium leprae. FEMS Microbiol Lett. 2008;289:72–9. [DOI] [PubMed] [Google Scholar]
- Teijaro JR, Studer S, Leaf Net al. S1PR1-mediated IFNAR1 degradation modulates plasmacytoid dendritic cell interferon-α autoamplification. Proc Natl Acad Sci USA. 2016;113:1351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teijaro JR, Walsh KB, Cahalan Set al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146:980–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng O, Ang CKE, Guan XL. Macrophage–bacteria interactions—a lipid-centric relationship. Front Immunol. 2017;8:1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testerink N, Ajat M, Houweling Met al. Replacement of retinyl esters by polyunsaturated triacylglycerol species in lipid droplets of hepatic stellate cells during activation. PLoS One. 2012;7:e34945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thivierge M, Le Gouill C, Tremblay MJet al. Prostaglandin E2 induces resistance to human immunodeficiency virus-1 infection in monocyte-derived macrophages: downregulation of CCR5 expression by cyclic adenosine monophosphate. Blood. 1998;92:40–5. [PubMed] [Google Scholar]
- Tiwari S, Choi H-P, Matsuzawa Tet al. Targeting of the GTPase Irgm1 to the phagosomal membrane via PtdIns (3, 4) P 2 and PtdIns (3, 4, 5) P 3 promotes immunity to mycobacteria. Nat Immunol. 2009;10:907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triggiani M, Oriente A, Seeds MCet al. Migration of human inflammatory cells into the lung results in the remodeling of arachidonic acid into a triglyceride pool. J Exp Med. 1995;182:1181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trindade BC, Sorgi CA, Nicolete LD, de Fet al. Leukotrienes are upregulated and associated with human T-lymphotropic virus type 1 (HTLV-1)-associated neuroinflammatory disease. PLoS One. 2012;7:e51873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung W-H, Hsieh H-L, Lee I-Tet al. Enterovirus 71 modulates a COX-2/PGE2/cAMP-dependent viral replication in human neuroblastoma cells: role of the c-Src/EGFR/p42/p44 MAPK/CREB signaling pathway. J Cell Biochem. 2011;112:559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung W-H, Hsieh H-L, Yang C-M. Enterovirus 71 induces COX-2 expression via MAPKs, NF-κB, and AP-1 in SK–N–SH cells: role of PGE2 in viral replication. Cell Signal. 2010;22:234–46. [DOI] [PubMed] [Google Scholar]
- Valm AM, Cohen S, Legant WRet al. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature. 2017;546:162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan M, Berger JE, Steinberg D. Hormone-sensitive lipase and monoglyceride lipase activities in adipose tissue. J Biol Chem. 1964;239:401–9. [PubMed] [Google Scholar]
- Villani A, Cirino NM, Baldi Eet al. Respiratory syncytial virus infection of human mononuclear phagocytes stimulates synthesis of platelet-activating factor. J Biol Chem. 1991;266:5472–9. [PubMed] [Google Scholar]
- Walsh KB, Teijaro JR, Wilker PRet al. Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc Natl Acad Sci USA. 2011;108:12018–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Guo Y, Zhou Tet al. In-depth proteomic analysis of the human sperm reveals complex protein compositions. J Proteomics. 2013;79:114–22. [DOI] [PubMed] [Google Scholar]
- Wang H, Sreenivasan U, Hu Het al. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J Lipid Res. 2011;52:2159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhang D, Ge Met al. Formononetin inhibits enterovirus 71 replication by regulating COX- 2/PGE2 expression. Virol J. 2015;12:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel GL, Joshi MR, Wei Cet al. 15(S)-Lipoxygenase-1 associates with neutral lipid droplets in macrophage foam cells: evidence of lipid droplet metabolism. J Lipid Res. 2009;50:2371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger B, Quizon C, Vetrano AMet al. Mechanisms mediating reduced responsiveness of neonatal neutrophils to lipoxin A4. Pediatr Res. 2008;64:393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller PF, Monahan-Earley RA, Dvorak HFet al. Cytoplasmic lipid bodies of human eosinophils. Subcellular isolation and analysis of arachidonate incorporation. Am J Pathol. 1991;138:141–8. [PMC free article] [PubMed] [Google Scholar]
- Welte MA. Proteins under new management: lipid droplets deliver. Trends Cell Biol. 2007;17:363–9. [DOI] [PubMed] [Google Scholar]
- Widegren H, Andersson M, Borgeat Pet al. LTB4 increases nasal neutrophil activity and conditions neutrophils to exert antiviral effects. Respir Med. 2011;105:997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward DF, Krauss AH, Nieves ALet al. Studies on leukotriene D4 as an eosinophil chemoattractant. Drugs Exp Clin Res. 1991;17:543–8. [PubMed] [Google Scholar]
- Xie Y, Chen R, Zhang Xet al. Blockade of interleukin-17A protects against coxsackievirus B3-induced myocarditis by increasing COX-2/PGE2 production in the heart. FEMS Immunol Med Microbiol. 2012;64:343–51. [DOI] [PubMed] [Google Scholar]
- Xue Q, Liu H, Zeng Qet al. The DEAD-box RNA helicase DDX1 Interacts with the viral protein 3D and inhibits foot-and-mouth disease virus replication. Virol Sin. 2019;34:610–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Zhang X, Liu P. Lipid droplet proteins and metabolic diseases. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1968–83. [DOI] [PubMed] [Google Scholar]
- Xu XJ, Reichner JS, Mastrofrancesco Bet al. Prostaglandin E2 suppresses lipopolysaccharide-stimulated IFN-beta production. J Immunol. 2008;180:2125–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Galea A, Sytnyk Vet al. Controlling the size of lipid droplets: lipid and protein factors. Curr Opin Cell Biol. 2012;24:509–16. [DOI] [PubMed] [Google Scholar]
- Yang H, Zhou Z, Zhang Het al. Shotgun proteomic analysis of the fat body during metamorphosis of domesticated silkworm (Bombyx mori). Amino Acids. 2010;38:1333–42. [DOI] [PubMed] [Google Scholar]
- York AG, Williams KJ, Argus JPet al. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell. 2015;163:1716–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Zhang L, Li Yet al. The adrenal lipid droplet is a new site for steroid hormone metabolism. Proteomics. 2018;18:e1800136. [DOI] [PubMed] [Google Scholar]
- Zambrano-Zaragoza JF, Romo-Martínez EJ, Durán-Avelar M, de Jet al. Th17 cells in autoimmune and infectious diseases. Int J Inflam. 2014;2014:651503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner R, Zimmermann R, Eichmann TOet al. Fat signals: lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012;15:279–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Liu P. The new face of the lipid droplet: lipid droplet proteins. Proteomics. 2019;19:e1700223. [DOI] [PubMed] [Google Scholar]
- Zhang J, Lan Y, Li MYet al. Flaviviruses exploit the lipid droplet protein AUP1 to trigger lipophagy and drive virus production. Cell Host Microbe. 2018;23:819–31.e5. [DOI] [PubMed] [Google Scholar]
- Zhao J, Zhu M, Jiang Het al. Combination of sphingosine-1-phosphate receptor 1 (S1PR1) agonist and antiviral drug: a potential therapy against pathogenic influenza virus. Sci Rep. 2019;9:5272. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhirnov OP, Klenk HD. Histones as a target for influenza virus matrix protein M1. Virology. 1997;235:302–10. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Wu W, Xie Let al. Cellular RNA helicase DDX1 is involved in transmissible gastroenteritis virus nsp14-induced interferon-beta production. Front Immunol. 2017;8:940. [DOI] [PMC free article] [PubMed] [Google Scholar]