

Abstract
Activity-dependent insertion of the tropomyosin-related kinase B (TrkB) receptor into the plasma membrane can explain, in part, the preferential effect of brain-derived neurotrophic factor (BDNF) on active neurons and synapses; however, the underlying molecular mechanisms remain obscure. Here, we report a novel function for carboxypeptidase E (CPE) in controlling chemical long-term potentiation stimuli-induced TrkB surface delivery in hippocampal neurons. Total internal reflection fluorescence assays and line plot assays showed that CPE facilitates TrkB transport from dendritic shafts to the plasma membrane. The Box2 domain in the juxtamembrane region of TrkB and the C terminus of CPE are critical for the activity-dependent plasma membrane insertion of TrkB. Moreover, the transactivator of transcription TAT-CPE452-466, which could block the association between CPE and TrkB, significantly inhibited neuronal activity-enhanced BDNF signaling and dendritic spine morphologic plasticity in cultured hippocampal neurons. Microinfusion of TAT-CPE452-466 into the dorsal hippocampus of male C57BL/6 mice inhibited the endogenous interaction between TrkB and CPE and diminished fear-conditioning-induced TrkB phosphorylation, which might lead to an impairment in hippocampal memory acquisition and consolidation but not retrieval. These results suggest that CPE modulates activity-induced TrkB surface insertion and hippocampal-dependent memory and sheds light on our understanding of the role of CPE in TrkB-dependent synaptic plasticity and memory modulation.
SIGNIFICANCE STATEMENT It is well known that BDNF acts preferentially on active neurons; however, the underlying molecular mechanism is not fully understood. In this study, we found that the cytoplasmic tail of CPE could interact with TrkB and facilitate the neuronal activity-dependent movement of TrkB vesicles to the plasma membrane. Blocking the association between CPE and TrkB decreased fear-conditioning-induced TrkB phosphorylation and led to hippocampal memory deficits. These findings provide novel insights into the role of CPE in TrkB intracellular trafficking as well as in mediating BDNF/TrkB function in synaptic plasticity and hippocampal memory.
Keywords: activity, carboxypeptidase E, chemical long-term potentiation, hippocampal memory, surface insertion, TrkB
Introduction
It is now widely accepted that neuronal activity plays a critical role in the modulation of the structure and function of synapses during the development and refinement of neuronal connectivity in adults (Katz and Shatz, 1996). However, the molecular mechanisms that translate patterns of neuronal activity into specific changes in the structures and functions of synapses remain largely unknown. Accumulating evidence suggests that brain-derived neurotrophic factor (BDNF) is involved in synapse development, transmission, and plasticity in an activity-dependent manner (Thoenen, 1995; Bonhoeffer, 1996; Gottschalk et al., 1998; Lu and Chow, 1999; McAllister et al., 1999). When spontaneous electrical activity or synaptic transmission is blocked, the dramatic increase in dendritic arborizations elicited by BDNF is inhibited in the developing cortex (McAllister et al., 1996), which suggests that BDNF acts preferentially on active neurons (Du et al., 2000).
As a diffusible molecule, how does BDNF distinguish active and inactive neurons or synapses and restrict the action of BDNF preferentially on active neurons/synapses? Activity-dependent enhancement of the number of surface tropomyosin-related kinase B (TrkB) receptors defines an important mechanism by which the specificity of BDNF modulation is achieved (Du et al., 2000). TrkB receptors, as high-affinity receptors for BDNF, are abundant in the nervous system and crucial for synaptic plasticity in the CNS (Bramham and Messaoudi, 2005; Nagappan and Lu, 2005; Cohen and Greenberg, 2008). Studies have reported that neuronal activity, such as depolarization by high K+ or tetanic electric stimuli, could enhance neuronal cell surface TrkB insertion (Meyer-Franke et al., 1998; Du et al., 2000). Our previous work showed that the activation of synaptic NMDA receptors by glycine stimulation, a model used to induce chemical long-term potentiation (cLTP) in cultured hippocampal neurons, could significantly increase hippocampal neuronal surface TrkB levels (Musleh et al., 1997; Lu et al., 2001; Zhao et al., 2009). Activity-dependent TrkB membrane insertion can explain, in part, the preferred effect of BDNF on active neurons; however, the TrkB interacting trafficking molecule that mediates its activity-dependent surface insertion has not yet been identified.
Our previous work showed that the TrkB juxtamembrane region (JM) is necessary and sufficient for activity-dependent TrkB surface insertion (Zhao et al., 2009). By using yeast two-hybrid screening with the JM domain of TrkB as bait, we identified carboxypeptidase E (CPE) as a candidate TrkB-associated protein. CPE was first discovered as a prohormone/proneuropeptide processing enzyme that cleaves basic residues from the C terminus of peptide intermediates liberated from the precursors by endoproteases (Fricker and Snyder, 1982; Hook and Loh, 1984; Cawley et al., 2012), and it is also known to be important in processing neuropeptides, sorting neuropeptides to the regulated secretory pathway (RSP), and transporting peptidergic vesicles to the membrane to facilitate peptidergic neurotransmission (Cool et al., 1997; Lou et al., 2005). The absence of CPE in knock-out mice leads to dysfunctional BDNF-TrkB signaling in the hippocampus (Xiao et al., 2017). CPE KO mice exhibit a number of behavioral abnormalities, including deficient learning and memory (Woronowicz et al., 2008) and abnormal mood and emotional responses (Srinivasan et al., 2004; Cawley et al., 2012; Rodriguiz et al., 2013). Hence, CPE has been shown to be a multifunctional protein that plays many essential nonenzymatic roles in the nervous system.
In this study, we investigated whether CPE is involved in activity-dependent TrkB neuronal surface insertion and influences hippocampal-dependent memory.
Materials and Methods
Animals
Male C57BL/6 mice, 8 weeks old and weighing 23–25 g, were used in the experiment. Mice were housed at 22 ± 2°C on a 12 h light/dark cycle condition. Food and water were available ad libitum. All animal procedures were conducted according to the National Institutes of Health's Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Shandong University.
Antibodies and reagents
The following antibodies were used: rabbit anti-Flag from Thermo Fisher Scientific; rabbit anti c-Myc antibodies from Bethyl Laboratories; rabbit anti-TrkB antibody from Millipore; goat anti-TrkB antibodies from R&D Systems; mouse anti-CPE antibodies from BD Transduction Laboratories; rabbit IgG Sepharose, mouse anti-Flag (M2) monoclonal antibody and mouse anti-tubulin antibodies from Sigma-Aldrich; rabbit anti-p44/42 MAP kinase, mouse anti-phospho-p44/42 (Erk1/2; Thr202/Tyr204), mouse anti-phospho-Akt (Ser473) and rabbit anti-pTrkB antibodies from Cell Signaling Technology; mouse anti-Akt1(B-1) antibody from Santa Cruz Biotechnology; chicken anti-TrkB In pAb from Promega; and horseradish peroxidas-conjugated goat anti-mouse or rabbit IgG and horseradish peroxidase-conjugated rabbit anti-goat IgG purchased from Calbiochem. Rabbit anti-NeuN antibody was from Abcam; Alexa Fluor 488- or 594-conjugated goat anti-mouse or rabbit IgG (H + L) was from Invitrogen; and Cy5-conjugated goat anti-mouse or goat anti-rabbit IgG (H + L) was from Jackson ImmunoResearch Laboratories.
Glycine (Gly) was obtained from AMRESCO. Human recombinant BDNF was obtained from PeproTech. Sulfo-NHS-biotin and chemiluminescence were from Pierce Biotechnology. Biotinylated transactivator of transcription (TAT)-CPE452-466 and TAT-Con peptides was synthesized and purified by GL Biochem. The sequence of TAT-CPE452-466 is biotin-GRRRRRRRRR-FSERKEEEKEELMEW, and the sequence of TAT-Con is biotin-GRRRRRRRRR-WEMLEEKEEEKRESF. TAT-like poly arginine membrane permeability sequence (GRRRRRRRRRRR) allowed peptides to penetrate into cells. The restriction enzymes were purchased from MBI Fermentas. The other reagents were from Sigma-Aldrich, and all the cell culture media were purchased from Life Technologies.
Plasmid constructs and small interfering RNAs
The pCDNA3.1-Flag-TrkB (TrkB with Flag antibodies), pCDNA3.1-Flag-T1, pEGFP-Flag-TrkB-GFP, and pEGFP-Flag-T1-GFP constructs were prepared as described previously (Chen et al., 2004; Zhao et al., 2009). All truncated mutants of TrkB-FL including Flag-TrkBΔEX, Flag-ΔExΔJM/ΔTk/ΔCT (JM, from Lys454 to Asn536; tyrosine kinase (TK), from Ile537 to Leu806; C terminus (CT), from Gln807 to Gly821), Flag ΔExΔBox1/ΔBox2/ΔBox3 (Box1, from Lys454 to lLe470; Box2, from Ser471 to Pro508; Box3, from Val509 to Asn536), and Flag-T1 chimeric constructs were subcloned into pCDNA3.1 and pEGFPN1 expression vectors (Invitrogen). The N-terminal Myc epitope tag was added behind the signal peptide of rat CPE. Myc-tagged rat CPE and all its truncated mutants including CPEC25 (C-terminal 25 amino acids, from Phe452 to Phe476), CPEC25Δ4 (deletion of Thr473 to Phe476), CPEC25Δ6 (deletion of Ser471 to Phe476), CPEC25Δ10 (deletion of Trp467 to Phe476), CPEC25Δ15 (deletion of Glu462 to Phe476) were subcloned into pmRFP expression vectors. All the TrkB, CPE deletion constructs, and the T1 chimeric constructs were generated by two-step PCRs. The small interfering RNA targeting the 19 nucleotides (CCTGTCGCAAGAATGATGA) of rat CPE sequence was cloned into pSuper-GFP or pSuper-RFP vector (Oligoengine). The same RNA oligonucleotide [FAM-CPE small interfering RNA (siRNA)] to target CPE and negative controls (FAM-Ctr siRNA) were purchased from GenePharma. All of these constructs were confirmed by DNA sequencing to exclude potential PCR-introduced mutations.
Neuronal cultures and transfection
Hippocampal neurons from timed-pregnant Sprague Dawley rats were cultured as described previously (Chen et al., 2004; Zhao et al., 2009). Briefly, embryos were removed from rats at 18 d of gestation and then were placed into Hank's Balanced Salt Solution without Ca2+ and Mg2+. Hippocampal tissues were dissociated and digested with 0.05% trypsin–EDTA for 5 min at 37°C and isolated into mono cells by mechanical isolation with a sterile, fire-polished glass Pasteur pipette in DMEM/F-12 (Invitrogen) plus 10% fetal bovine serum. Neurons were seeded onto 0.1 mg/ml poly-D-lysine (Sigma-Aldrich) coated coverslips in sixwell plates in Neurobasal medium (Invitrogen) which was supplemented with 2% B27 and 0.5 mm glutamine, and an incubator at 37°C, with 5% CO2, and 95% humidity was used for the culture.
At 7–9 d in vitro (DIV), cultured hippocampal neurons were transfected with Lipofectamine 2000 or Lipofectamine RNAiMAX reagent following the manufacturer's instructions (Invitrogen). Experiments were conducted 48 h after transfection or 18–21 d as indicated after transfection.
Coimmunoprecipitation assay and Western blot analysis
For exogenous coimmunoprecititaton (co-IP), HEK293 cells were electroporated with the indicated constructs (Amaxa). Forty-eight h after transfection, cells were harvested with TNE buffer (10 mm Tris, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1% NP-40, and 10% glycerol with protease inhibitors). Lysates were clarified by centrifugation at 14,000 × g for 15 min at 4°C. For Flag-tagged protein pull-down, the supernatants were incubated with anti-Flag M2 Affinity Gel overnight at 4°C. For c-Myc-tagged protein pull-down, mouse anti-c-Myc antibody was used. Then the immunocomplex was precipitated with protein G-Sepharose (Sigma) overnight at 4°C. After washing with TNE buffer five times, the beads were eluted by boiling in sample buffer and then subjected to separation on sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunobloted with the indicated antibodies.
For endogenous co-IP, mouse hippocampal tissues was homogenized and lysed in TNE buffer containing protease inhibitors (8 ml TNE/g brain lysate) and clarified by centrifugation at 14,000 × g for 15 min at 4°C. The proteins were quantified by bicinchoninic acid protein assay. Ten milligrams of mouse brain lysates incubated with rabbit anti-TrkB antibodies for 2 h, followed by an additional incubation with protein A/G plus agarose beads overnight at 4°C, and the same amount of lysates incubated with rabbit IgG was used as the negative control group. The same procedure was performed as above.
Surface biotinylation
Membrane surface insertion of endogenous TrkB receptors induced by cLTP were performed as previously described (Zhao et al., 2009). Briefly, hippocampal neurons transfected with CPE siRNA or Ctr siRNA were serum starved overnight and then stimulated with or without 200 μm glycine in the Mg2+-free bathing solution containing the following (in mm): 140 NaCl, 1.3 CaCl2, 5.0 KCl, 25 HEPES, 33 glucose, 0.0005 TTX, 0.001 strychine, and 0.02 bicuculline methiodide, pH 7.4, for 10 min to induce cLTP in an incubator at 37°C, 5% CO2, and 95% humidity. At the end of the stimulation, neurons were washed three times with ice-cold PBS buffer (pH 7.4, with 0.1 mm Ca2+ and 1 mm Mg2+), and then incubated in sulfo-NHS-biotin (0.3 mg/ml in cold PBS; Pierce) for 1 h at 4°C to biotinylate surface proteins. Unreacted biotin was quenched and removed by washing twice for 10 min with ice-cold Tris-buffered saline containing 0.1 mm Ca2+, pH 7.4. Cells were then lysed in TNE buffer, which contains 150 mm NaCl, 10 mm Tris, 1 mm EDTA, and 1% NP-40 with protease and phosphatase inhibitors. Extracts were clarified by centrifugation (14,000 × g for 15 min) and an aliquot of each sample was used for the isolation of biotinylated proteins with 30 µl of streptavidin-conjugated Sepharose beads (Pierce) by incubating overnight at 4°C with gentle rotation. The beads were washed three times in TNE Buffer. Biotinylated proteins were eluted from beads by boiling in 4× SDS sample buffer (Invitrogen), and eluted proteins were analyzed by Western blot. The polyvinylidene difluoride membranes were probed with a monoclonal anti-TrkB antibody (1:1000; BD Biosciences) and immunoreactive bands visualized by enhanced chemiluminescence (Pierce) and quantified by ImageJ software (National Institutes of Health).
Immunofluorescence staining
Colocalization studies
To investigate the subcellular colocalization of TrkB and CPE, cultured hippocampal neurons (DIV 7) cotransfected with Flag-TrkB and Myc-CPE, neurons were fixed with 4% paraformaldehyde and permeabilized with 0.4% Triton X-100 in PBS, then stained with mouse anti-Flag M2 antibody (1:1000; Sigma) and rabbit anti-c-Myc antibody combined with the corresponding green or red fluorochrome-conjugated secondary antibody.
Live cell imaging
The hippocampal neurons were seeded in Nunc Glass Base 27 mm dishes (Thermo Fisher Scientific) that had been coated with 0.1 mg/ml poly-D-lysine and were observed with an Andor Dragonfly spinning disk confocal microscope with a motorized Z drive and a 63×, 1.4 numerical aperture (NA) oil-immersion objective lens. The images were acquired every 20 s continuously for 700 s, and the TrkB-GFP and CPE-RFP positive vesicles were tracked and processed with Fusion software. The videos were produced and kymographs generated with ImageJ software.
Surface TrkB receptor fluorescence ratiometric assay
To measure the cell surface TrkB level, a surface TrkB receptor fluorescence ratiometric assay was used as previously described (Zhao et al., 2009). Briefly, Flag-TrkB-GFPs were transfected or cotransfected with other indicated plasmids into hippocampal neurons; after treatments hippocampal neurons were fixed and stained with M2 antibody (1:500; Sigma) without permeabilization and then incubated with Alexa Fluor 594-conjugated or Cy5-conjugated secondary antibody, which represents surface TrkB level, and the GFP fluorescence represented the total TrkB amount.
Surface TrkB internalization measurement
The internalization of TrkB-FL was assessed as described previously (Liu et al., 2015). Cultured hippocampal neurons (7–9 DIV) were cotransfected with Flag-TrkB and the indicated siRNA constructs 48 h before. Neurons were serum starved overnight. Then, the total pool of TrkB initially present at the cell surface was labeled with anti-Flag antibody (M2) conjugated to Alexa Fluor 594 (594-M2) at 4°C for 15 min. After BDNF (50 ng/ml) treatment at 37°C for 15 min, neurons were fixed by 4% PFA followed by labeling with cy5 anti-mouse IgG secondary antibody under nonpermeabilizing conditions. The ratio of cy5/Alexa 594 fluorescence was calculated. The mean cy5/Alexa 594 ratio of the internalized group (I) represented the uninternalized TrkB. For each experiment, there was a parallel control group in which neurons were fixed immediately after the 15 min incubation with 594-M2 and labeled with cy5 anti-mouse IgG antibody, and the mean cy5/Alexa 594 ratio of this group (C) represented the 100% surface TrkB. Thus, the percentage of internalized TrkB receptors was calculated using the following formula: (1 – I/C) × 100.
Localization analysis of TrkB receptors in spines using fluorescence microscopy
To measure the ratio of total TrkB in the spine versus in the corresponding dendritic shaft, CPE siRNA, or Ctr siRNA coupled with GFP was transfected into hippocampal neurons. Neurons were then raised to 18–21 DIV and serum starved overnight, followed by pretreatment with glycine for 10 min, and then were fixed and permeabilized followed by primary antibodies incubation with chicken anti-TrkB in pAb (Promega) and rabbit anti-GFP combined with Alexa 405-conjugated donkey anti-chicken and Alexa 488-conjugated donkey anti-rabbit secondary antibody.
For detection of the spine density or morphology changes, hippocampal neurons pretreated with TAT-Con or TAT-CPE452-466 for 30 min were fixed and performed immunofluorescence staining against GFP as in the above methods.
For NeuN immunohischemstry staining, mice brain slides (40 μm/section) were incubated in 0.4%PBST-diluted donkey serum to 10% to block nonspecific staining for 1 h under room temperature. Then brain slides were stained with Anti-NeuN (1:500; Abcam) as the primary antibody overnight at 4°C, and after that, 0.4%PBST were used to wash them three times, and the slides were incubated in secondary antibody (1:1000; Invitrogen) at room temperature for 1 h. After washing in 0.4% PBST three times, the slides were mounted with cover glasses, n = 4 per group. All the images were captured with a Zeiss LSM 880 confocal microscope at the Translational Medicine Core Facility of Shandong University. Images were analyzed using ImageJ.
Total internal reflection fluorescence microscopy
To observe TrkB and CPE cell membrane-trended vesicles trafficking, hippocampal neurons were seeded onto 0.1 mg/ml poly-D-lysine (Sigma-Aldrich) coated coverslips in six-well plates at a cell density of 3 × 105 cells/ml, and the cells were cotransfected with TrkB-GFP and CPE-RFP with Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen) at DIV 7. Then neurons were serum-free starved overnight and then stimulated with glycine for 10 min in the bathing solution at DIV 10. Neurons were fixed with 4% paraformaldehyde. Neurons were imaged by a high-numerical aperture objective lens [100×, NA = 1.49] that was mounted on an inverted total internal reflection fluorescence (TIRF) microscope (Olympus) equipped with a 488 nm and 561 nm laser, an electron multiplying charged-coupled device (CCD) digital camera, and cellSens Dimension software. All images underwent a transformation to correct for optical cross-talk. Local background, caused by residual out-off focus fluorescence and nonspecific staining, was removed.
Correlation coefficient
We have analyzed the changes in the replication patterns in time by estimating the degree of overlap of the red and green spots. For this we used Pearson's correlation coefficient (r) between the red and the green component of each dual-channel image in the following:
where Ri and Gi are the red and green intensities of voxel i, respectively, and Rav and Gav the average values of Ri and Gi, respectively.
TrkB and downstream signaling Erk1/2 and Akt activation assays
Cultured primary hippocampal neurons were serum-free starved overnight and were then pretreated with TAT-Con or TAT-CPE452-466 for 30 min, followed by glycine stimulation for 10 min and BDNF (50 ng/ml) for 10 min. To detect TrkB phosphorylation, cell lysates were collected and followed by Western blot probed with rabbit anti-pTrkB antibodies (Cell Signaling Technology) and rabbit anti-TrkB antibodies (Millipore), respectively. To detect MAPK/PI3K activation, the membrane was probed with rabbit anti-p44/42 MAP kinase and mouse anti-phospho-p44/42 or mouse anti-phospho-Akt (Ser473) and mouse anti-Akt1(B-1) antibodies. All the above experiments were conducted at least in triplicate. For quantitative analysis of the phosphorylation level of TrkB/Erk1/2/Akt, immunoreactive bands were scanned and performed with densitometric analysis using ImageJ (Scion Image).
Microscopic quantitative analysis
To investigate the surface TrkB expression level, the same methods were adopted as we previously reported (Zhao et al., 2009). In summary, fluorescence images were acquired by a Nikon Eclipse TE 2000-U microscope equipped with a HQ2 cool CCD camera and a motorized Z drive. All the images were acquired through a 63×, 1.4 NA oil-immersion objective lens and processed by MetaMorph software (Universal Imaging). In each experiment, >25 cells were examined at random. The fluorescence intensities of different channels were measured by MetaMorph software by selecting a region of interest. In each experiment, the consistent set of acquisition parameters was used for each set of images. The ratio of the receptor surface versus total fluorescence was measured as previously described (Zhao et al., 2009).
The entire dendritic spine images were acquired with an inverted Carl Zeiss LSM 780 confocal scanning microscope (hardware and software from Carl Zeiss) equipped with a 63×, 1.4 NA oil-immersion objective lens. Dendritic spines on a length of secondary or tertiary proximal dendrites of hippocampal neurons were selected and captured with 1024 × 1024 pixel resolution as a z-stack series in multitrack mode. The snare parameters, such as pinhole, detector gain, and offset, were set for optimum and the same parameter settings were used for all scans.
The line spots method was adopted to measure the total TrkB levels in the dendritic spine and adjacent dendrite shaft (Gerges et al., 2005; Zhao et al., 2009). As illustrated in Figure 3C, fluorescent intensity was line plotted by drawing a line across the dendritic spine head and the adjacent dendritic shaft (arbitrary units). The ratios of total TrkB in the spine versus those in the adjacent dendritic shaft (spine/shaft) are then calculated by the peaks of fluorescence intensity at the spine head divided by that at the dendritic shaft after background subtraction (dotted lines), the fluorescent intensity then was normalized to get the ratio of spine/shaft.
Figure 3.
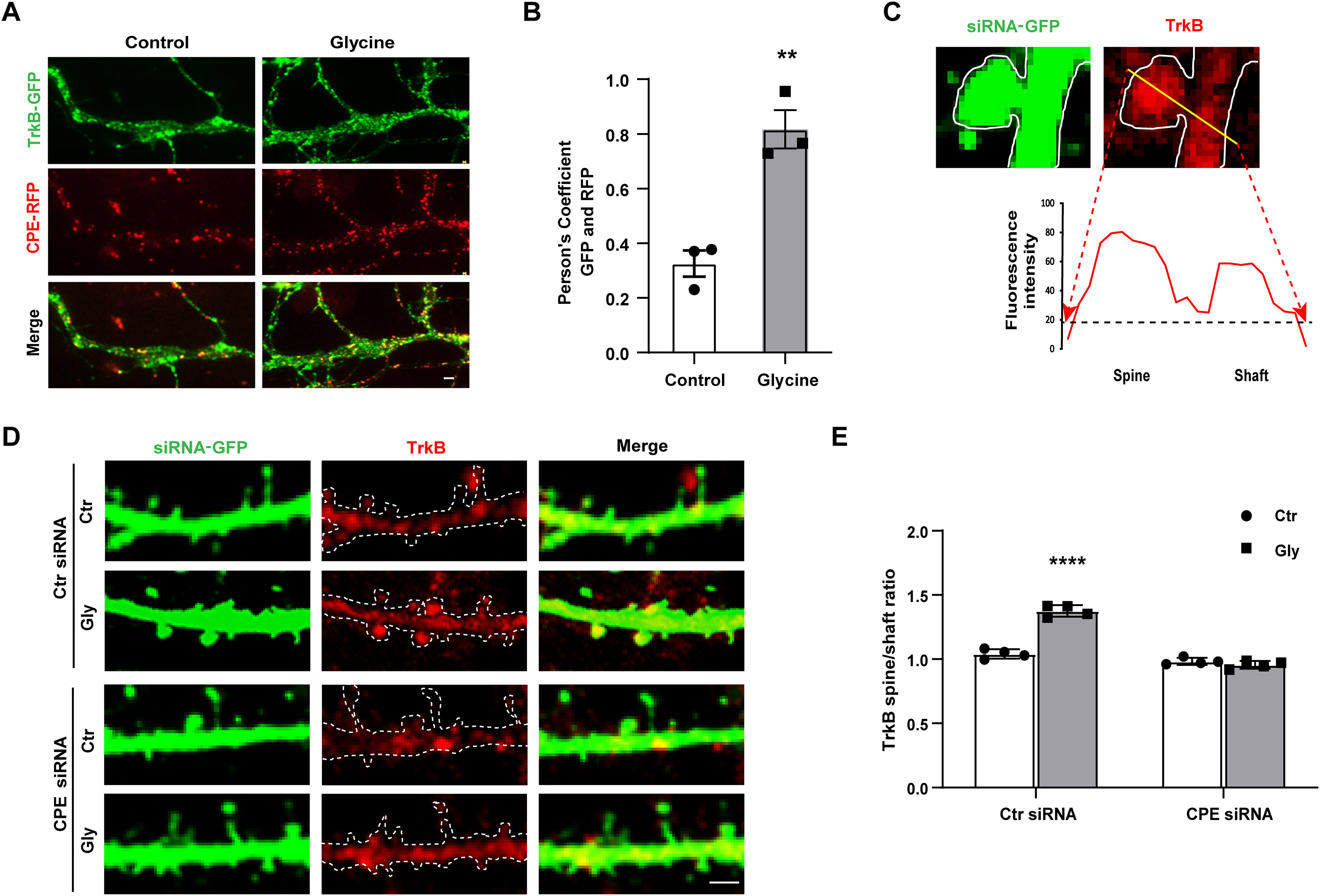
CPE regulates cLTP-enhanced TrkB surface trafficking and translocation into spine. A, Representative TIRF images are shown for hippocampal neurons coexpression of CPE-RFP and TrkB-GFP, in which yellow color points the doubled positive of CPE and TrkB. Scale bar, 2 μm. B, Colocalization of CPE and TrkB was quantified (unpaired t test, t = 5.784, df = 4, **p = 0.0203; n = 4). C, Representative line plot analysis of TrkB receptor (594 signal, red color) across a spine and the adjacent dendritic shaft of siRNA-GFP transfected neurons, as indicated. Values for TrkB receptors were taken from the fluorescence intensity peaks after background subtraction (yellow line). Scale bar, 2 μm. D, Representative immunofluorescence images of total TrkB receptor across a spine and the adjacent dendritic shaft of siRNA-GFP transfected neurons on cLTP stimuli. E, Quantitative analysis of the ratio of TrkB fluorescence in spines and dendrites after normalization (15 dendrites/cell, 15–18 µm2/dendrite). Two-way ANOVA with Bonferroni's multiple comparisons test, F(1,12) = 104.4, ****p < 0.0001; n = 4. All the data are presented as mean ± SEM (n ≥ 25 cells for each condition per experiment).
For analysis of the spine density and head width, only dendritic protrusions from 0.5 to 3 μm lengths were defined as spines. The spine density was calculated by counting the number of spines on >20 neurons and comparing the spine number per 10 μm dendritic segment (∼150 μm total dendritic length per neuron). For analysis of the spine-head width, the largest section of the head was selected, determined through the z stack, and the spine-head diameter was detected by manually drawing a line across the largest section of the head and the spine.
Animal manipulations
All mice surgeries were performed under aseptic conditions. The mice were fixed on a stereotaxic apparatus (RWD Life Science), and the mice were anaesthetized with isoflurane on an anesthetic machine (RWD Life Science). Mice were bilaterally implanted with two 33 gauge guide cannulas to the dorsal hippocampus (DH) following the reference coordinates (anteroposterior: −1.7 mm; ML: ±1.5 mm; DV: −1.8 mm), and the guide cannulas were fixed by curing denture acrylic. To prevent clogging, every guide cannula had one matching cap. Mice were left to recover for 1 week before the animal behavior. The infusion cannula was connected to a 10 μl microsyringe via PE20 tubing controlled by a microinjection pump (KDS 200; KD Scientific). TAT-Con and TAT-CPE452-466 (10 μg/μl, 0.5 μl/side) were microinfused bilaterally 30 min before novel place recognition (NPR) training and fear conditioning training with uniform speed (50 nl/min).
Novel place recognition
The NPR test was used as previously described (Bevins and Besheer, 2006). This experiment included two steps, training and testing. Before training, the mice were put into the experiment environment, an open-field arena (40 cm × 40 cm × 35 cm) for 10 min of habituation. Twenty-four h later, the training process was performed. Two identical objects (A and B) were placed in opposite corners of the open-field arena. Mice were placed in the middle of the two objects for a 10 min exploration, and then they were transferred to their home cages. The testing process started 10 min after training. During testing, object B was moved to a new place, in another corner, and the location of object A remained unchanged. The mice were again put into this chamber for 5 min of free exploration. The exploration time T(A) and T(B) were recorded when the mice sniffed object A or object B. The discrimination index was calculated by T(B)/(T(A)+T(B)).
Contextual fear conditioning and auditory fear conditioning
For contextual fear conditioning (CFC) and auditory fear conditioning (AFC) training, mice were put into a fear box (Context A) with grid floor and black walls controlled by a computer with a fear system (Panlab). First, mice were habituated for 3 min without any stimulation, then mice received an auditory stimulation (3 KHZ, 80 dB) lasting for 30 s and paired with a foot shock (0.75 mA) during the last 1 s. The paired stimulation was repeated three times with interstimulus intervals (ITIs) of 120 s. After the last foot shock, the mice had an additional 120 s, then they were transferred to their home cages. CFC testing was performed 1 h later. The mice were placed in Context A for 5 min without the foot shock, and the freezing was scored. After 1 h, AFC testing was performed. The mice were put into another chamber (Context B) with a floor, color, shape, and smell different from Context A; accordingly, the mice could distinguish Context A and Context B. In addition, after 2 min of habituation in Context B, mice received four tone-alone (20 s) presentations with 30 s ITIs. In this process freezing was only scored while presenting the tone.
Elevated plus maze
The elevated plus maze (EPM) test was used as described previously (Chen et al., 2006; Yu et al., 2012). Briefly, the mouse was placed in the center of the elevated plus maze, which was constructed of black stainless steel and consisted of four arms, two closed arms (30 cm in length × 5 cm in width × 10 cm walls) and two open arms (30 cm in length × 5 cm in width × 0.5 cm in height, no walls). The elevated plus maze was 50 centimeters high. The time mice spent in open arms and the number of entries into the open and closed arms were scored by the video tracking system (Smart) during the 5 min test.
Open field test
The open field test (OFT) took place in an open-field arena (40 cm in length × 40 cm in width × 35 cm in height), mouse was placed into the center of this open field. The Smart video tracking system was used to analyze the time spent in the center zone (20 cm length × 20 cm width) and distance traveled during the 10 min test.
Rapid Golgi impregnation
The tissue impregnation and Golgi impregnation were performed as described previously (Chen et al., 2006; Yu et al., 2012). In brief, Golgi impregnation of all brains was conducted using FD Rapid Golgi Stain Kit (FD NeuroTechnologies). Then brains were embedded in a 3% agarose solution and cut at room temperature (120 μm sections) using a vibratome (VT1200S, Leica). Serial sections were immersed in double distilled water (DDW) three times for 5 min. After 48 h air drying, they were then transferred to a solution of D and E (Golgi kit) for 5–10 min at 4°C, rinsed three times for 5 min each in DDW. Ethanol was then used to dehydrate, and Histo-Clear (three times for 5 min each) was used for clearing. Slices were coverslipped with a DPX mounting medium, n = 3 per group. Z-stack pictures (40×) were captured using a VS120 (Olympus) for morphologies of dendrite analysis, and spines were counted under oil (100×), using light microscopy (Nikon 80i). The entire visible dendritic length was measured by Imaging software (NIS-Elements Basic Research, Nikon).
Experimental design and statistical analysis
For the fluorescence detection experiment in vitro, cells were examined at random and subjected to blind analysis using MetaMorph (Universal Imaging) software; >25 cells were examined in each experiment. Each experiment was repeated at least three times. See below, Results (Figs. 2, 3, 4, 5).
Figure 2.

CPE mediates activity-dependent TrkB membrane surface insertion in hippocampal neurons. A, PC12 cell were transiently transfected with Ctr siRNA or CPE siRNA. After 2 d, cells lysates were immunoblotted with CPE antibody to detect the CPE-knockdown efficiency. Endogenous β-tubulin was used as an expression control. B, Representative immunofluorescence images of TrkB surface levels in neurons (9 DIV) expressing the indicated constructs with or without Gly treatment (200 μm, 10 min). Surface TrkB levels were determined using ratiometric fluorescence assay. The siRNA constructs all expressed fused RFP, and we only captured images of RFP-positive neuron (pseudo blue color). The total TrkB receptors were fused with GFP (green color). The surface TrkB stained with M2 antibody followed by Cy5-conjugated goat anti-mouse antibody (pseudo red color). Anti-FLAG antibody labeled surface TrkB receptors, and the GFP fluorescence represented total receptor levels. Scale bar, 10 μm. C, Surface levels of TrkB receptor were analyzed as in B. Relative surface levels were normalized to that of TrkB-GFP (two-way ANOVA with Bonferroni's multiple comparisons test, F(1,12) = 8.627, Ctr-Ctr siRNA vs Gly-Ctr siRNA **p = 0.0042; n = 4). D, Surface levels of TrkB and T1 receptors were evaluated by surface biotinylation assay in hippocampal neurons transfected with the indicated siRNA. Surface labeled TrKB were detected by streptavidin pull-down followed by anti-TrkB immunoblotting. E, Quantification of the surface levels of TrkB receptors by the ratio of surface to total TrkB intensity normalized to the control group (two-way ANOVA with Bonferroni's multiple comparisons test, F(1,12) = 7.75, Ctr-FAM-Con VS Gly-FAM-CPE siRNA **p = 0.0019; n = 4). F, Quantification of the total TrkB receptors normalized to the control group. Error bars indicate ± SEM. G, Quantification of the surface levels of T1 receptors by the ratio of surface to total T1 intensity normalized to the control group. H, Representative epifluorescence images from the TrkB internalization assay. The Control, Ctr siRNA, and CPE siRNA constructs all express nonfused GFP, and we only captured images of GFP-positive neurons (GFP image not shown). The total pool of Flag-TrkB initially present at the cell surface was labeled with 594-M2 (Internal + surface group, green, pseudo color). After BDNF treatment at 37°C for 15 min, neurons were fixed by 4% PFA under nonpermeabilizing conditions, and the receptors remaining at the cell surface were labeled with Cy5-conjugated secondary antibody (surface group, red, pseudo color). Scale bar, 10 μm. I, Quantitation of internalized TrkB levels in H was performed. All of the data are presented as mean ± SEM determined from analysis of more than three independent experiments (n ≥ 25 cells for each condition per experiment).
Figure 4.

Identification of the key binding domain in TrkB interacting with CPE that mediated activity-dependent surface TrkB recruitment. A, Coimmunoprecipitation was performed in HEK 293 cell expressing Myc-CPEC25-RFP and Flag-TrkB mutants. Cell lysates were immunoprecipitated using anti-Flag antibody, followed by immunoblotting with Myc antibodies, and Myc-RFP is a control protein to confirm RFP couldn't combine with TrkB. B, Co-IP of Myc-CPEC25 and FLAG-TrkB deletion mutants as indicated in HEK 293 cells. C, Summary of TrkB mutants used in B and a summary of the CPEC25 interactions with the mutants of TrkB. FL, full-length domain; EX, Extracellular domain. D, Co-IP of Myc-CPEC25 and Flag-TrkB deletion mutants as indicated in HEK293 cells. E, Summary of TrkB mutants used in D and a summary of the CPEC25 interactions with the mutants of TrkB. A–E, Each co-IP experiment was repeated at least three times, and representative data are shown. F, Representative immunofluorescence images of TrkB mutants or T1 chimeras surface levels in transfected hippocampal neurons before and after glycine treatment. Scale bar, 10 μm. G, Quantification of surface levels of TrkB mutants by the ratio of red to green fluorescence intensity in F (two-way ANOVA with Bonferroni's multiple comparisons test, F(1,12) = 7.75, Ctr-TrkB FL versus Gly-TrkB FL **p = 0.0063; Ctr-TrkBΔBOX1 versus Gly-TrkBΔBOX1 *p = 0.0174; Ctr-TrkBΔBOX3 versus Gly-TrkBΔBOX3 *p = 0.0492; n = 4). H, Quantification of surface levels of T1 chimeras by the ratio of red to green fluorescence intensity in F (two-way ANOVA with Bonferroni's multiple comparisons test, F(3,24) = 3.845, ***p = 0.0007; n = 4). All the data are presented as mean ± SEM (n ≥ 25 cells for each condition per experiment).
Figure 5.
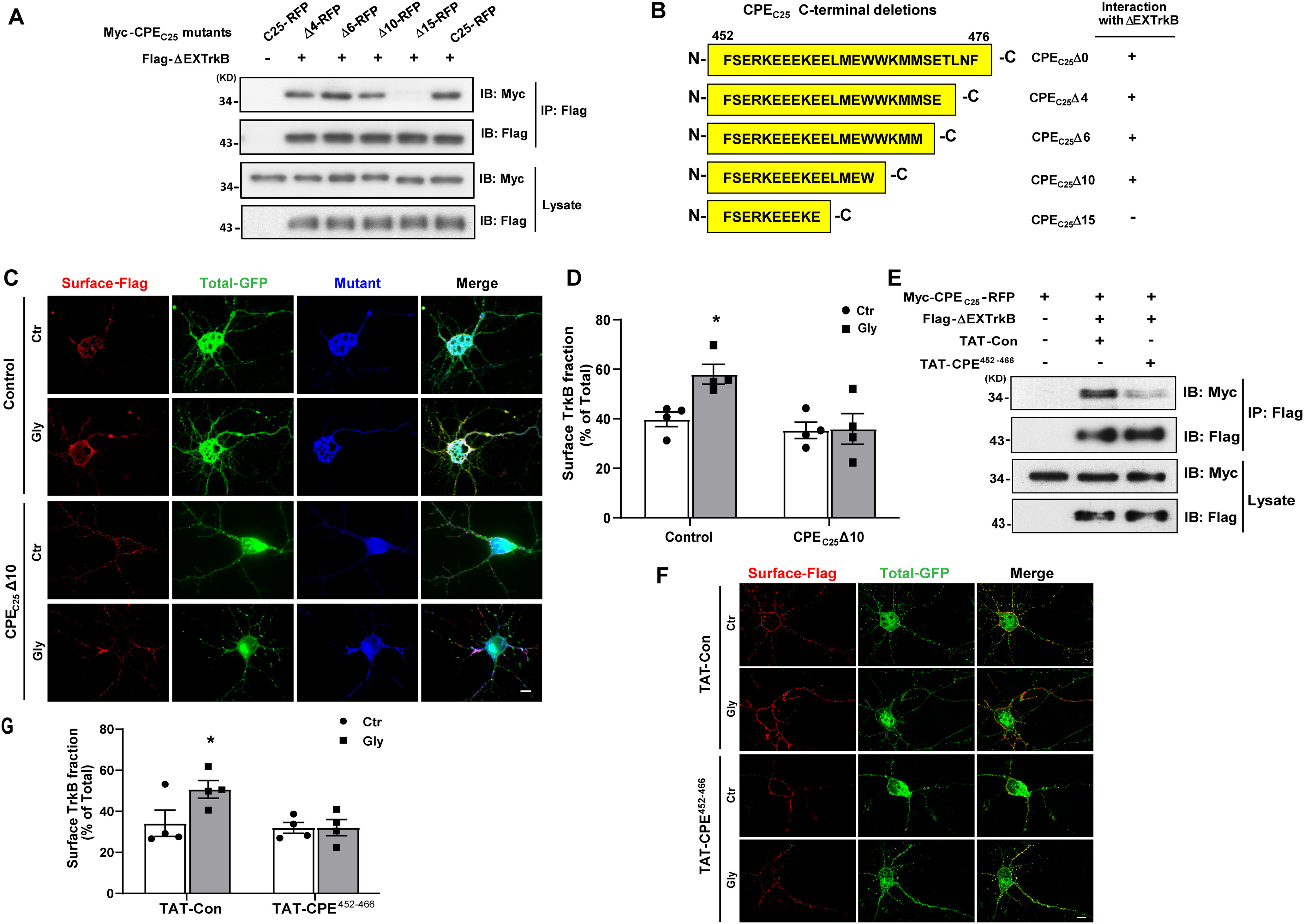
Identification of the key binding domain in CPE interacting with TrkB that mediated activity-dependent surface TrkB recruitment. A, Co-IP was performed in lysates of HEK 293 cells expressing Flag-TrkBΔEX and Myc-CPEC25 truncated mutants. B, Summary of CPE-truncated mutants used in A and a summary of the TrkBΔEX interactions with the mutants of CPE. C, Ratiometric fluorescence assay was performed to examine the influence of CPEC25Δ10-RFP on cLTP-induced surface recruitment of TrkB receptors. Representative epifluorescence images were shown. Scale bar, 10 μm. The Control and CPEC25Δ10-RFP constructs all express fused RFP, and we only captured images of RFP-positive neurons (pseudo blue color). The total TrkB receptors were fused with GFP (green color). The surface TrkB stained with M2 antibody followed by Cy5-conjugated goat anti-mouse antibody (pseudo red color). D, Quantification of TrkB surface levels by the ratio of red to green fluorescence intensity in C (two-way ANOVA with Bonferroni's multiple comparisons test, F(1,12) = 4.193, *p = 0.0225; n = 4). E, TAT-CPE452-466 inhibits the interaction between CPEC25 and TrkBΔEX. HEK293 cells were transfected with Myc-CPEC25 and FLAG-TrkBΔEX. After 2 d cells were treated with TAT-Con or TAT-CPE452-466 (5 μm) for 30 min before lysed. The interaction between CPEC25 and TrkBΔEX was analyzed by IP using anti-Flag antibodies and followed by immunoblotting with Myc antibodies. F, Ratiometric fluorescence assay was performed to examine the influence of TAT-CPE452-466 peptides on glycine-induced surface recruitment of TrkB receptor. Representative images from the ratiometric fluorescence assay are shown. Scale bar, 10 μm. G, Quantification of TrkB surface levels by the ratio of red to green fluorescence intensity in F (two-way ANOVA with Bonferroni's multiple comparisons test, F(1,12) = 3.266, *p = 0.0481; n = 4). All the data are presented as mean ± SEM (n ≥ 25 cells for each condition per experiment).
For co-IP assay and Western blot analysis, the DH regions from mice were separated, and proteins were extracted for the endogenous co-IP and Western blot analysis (n = 4/group). HEK293 cells were prepared for exogenous co-IP assay and Western blot. Each experiment was repeated at least three times.
In all behavioral experiments, the animals were randomly divided into different groups (n = 6–8/group), and observers were blinded to the treatments during the experiments.
In the experiment for analysis of brain slices, NeuN-positive cell was conducted throughout dentate gyrus (DG; from anteroposterior +1.58 mm to anteroposterior +2.46 mm) in five coronal sections for the quantification analysis in each group of mice (n = 4/group). For Golgi impregnated brain sections analysis, 50 neurons from the hippocampal DG subregion for each brain were selected for the spine density and dendrite morphology analysis (n = 3/group).
Data were analyzed with Student's t test, one-way or two-way ANOVA, followed by Tukey's multiple comparisons test or Bonferroni's multiple comparisons test, where appropriate. Student's t test or one-way ANOVA was used for single-factor experiments involving two or more than two groups. For experiments comprising multiple factors, a two-way ANOVA, with test for interaction, was used. Data were displayed as the mean ± SEM, and the significance was set at p < 0.05, analyzed using GraphPad Prism 8.0.
Results
TrkB interacts with carboxypeptidase E
To verify the interaction between CPE and TrkB, co-IP assays were performed in HEK293 cells. As expected, after IP of full-length TrkB (TrkB-FL) with Flag antibodies, an association was observed with CPE, as assessed by Western blot analysis using Myc antibodies (Fig. 1A). The interaction was also confirmed after IP with Myc antibodies and observed by immunoblotting with Flag antibodies (Fig. 1B). To exclude the influence of protein overexpression, endogenous co-IP was performed with rat brain lysate. As shown in Figure 1C, endogenous TrkB receptors coimmunoprecipitated with CPE.
Figure 1.

TrkB receptors interact and colocalize with CPE. A, B, Interaction between TrkB receptors and CPE was assessed in HEK293 overexpressing cDNAs encoding Flag-TrkB, Myc-CPE, and empty vector. Cell lysates were immunoprecipitated (IP) with anti-Flag antibodies (A) or anti-Myc antibodies (B) and immunoblotting (IB) analysis was performed to detect immunoprecipitated proteins. C, Endogenous association of CPE and TrkB receptors. Mice hippocampal tissues lysates were subjected to immunoprecipitation with polyclonal rabbit anti-TrkB antibody or control IgG, followed by immunoblotting with mouse anti-TrkB and mouse anti-CPE antibodies, respectively. A–C, Each co-IP experiment was repeated three times, and representative data are shown. D, Representative immunofluorescence images of the subcellular colocalization of Flag-TrkB and Myc-CPE in hippocampal neurons by confocal microscopy. Lower panels are enlarged images of the framed regions with the white arrows indicating the colocalization of Flag-TrkB and Myc-CPE as shown in yellow. Scale bar, 10 μm. E, Hippocampal neuron cotransfected at DIV 7 with TrkB-GFP and CPE-RFP. Time-lapse images of transport vesicles were acquired at one frame every 20 s for 700 s. F, Corresponding kymographs showing overlapping trajectories of vesicles labeled for TrkB-GFP and CPE-RFP. G, Time-lapse sequence showing a moving vesicle that contains both TrkB-GFP and CPE-RFP, indicated with arrows. Scale bar, 5 µm.
Because subcellular colocalization could provide additional evidence to support the interaction between CPE and TrkB, immunocytochemistry assays and confocal imaging analysis were performed in primary cultured hippocampal neurons (DIV 7) transiently transfected with Flag-TrkB and Myc-CPE. We observed that TrkB was highly expressed as puncta in the neuronal body, axons, and dendrites and distinctly colocalized with CPE in all these regions (Fig. 1D). Moreover, hippocampal neurons cotransfected with TrkB-GFP and CPE-RFP were time-lapse recorded by spinning disk confocal live imaging. Interestingly, we also found that both TrkB-GFP-containing vesicles and CPE-RFP-containing vesicles could be trafficked from the cell body to dendrites via secretory vesicles, and the transport vesicles were partially colabeled for CPE and TrkB. Kymograph and time-lapse sequence analysis of transport events indicated the overlapping trajectories of vesicles labeled for TrkB-GFP and CPE-RFP, as illustrated in Figure 1E–G. Together, these data suggest that CPE might regulate TrkB trafficking from the Golgi to dendrites via secretory vesicles. All these results confirmed that CPE associates with TrkB and might regulate TrkB trafficking in hippocampal neurons.
CPE is involved in the cLTP-induced insertion of TrkB into the plasma membrane
To determine whether CPE is involved in the cLTP-induced insertion of TrkB into the plasma membrane, we first constructed a small interfering RNA (siRNA) targeting CPE to knock down endogenous CPE in PC12 cells using Control (Ctr) siRNA as a control (Fig. 2A). To provide a convenient and visible method for comparing relative receptor cell surface levels, we adopted a surface TrkB ratiometric fluorescence assay described in our previous study (Zhao et al., 2009). In this assay, TrkB receptors were Flag-tagged at the N terminus of the extracellular domain and fused with GFP in the cytoplasmic C terminus. Thus, cell surface receptor levels were quantified by the fluorescence intensity of Flag staining normalized to GFP intensity per cell under nonpermeabilized conditions, which served to control for variable receptor expression levels in each cell because of transient transfection. By performing a surface TrkB ratiometric fluorescence assay, we found that CPE siRNA significantly abolished cLTP-induced TrkB membrane surface insertion in hippocampal neurons expressing Flag-TrkB-GFP (Fig. 2B,C), which suggests that CPE might be involved in activity-dependent TrkB plasma membrane insertion.
Next, the surface insertion of endogenous TrkB in hippocampal neurons was measured using cell surface biotinylation assays. The same results were obtained: FAM-CPE siRNA dramatically inhibited cLTP-induced surface TrkB recruitment but had no influence on total TrkB expression (Fig. 2D–F). However, surface truncated TrkB (TrkB.T1) levels were unchanged (Fig. 2G). T1 is a TrkB isoform and is expressed in neurons because of alternative splicing (Klein et al., 1990; Middlemas et al., 1991); T1 lacks almost all the TrkB-FL intracellular domains but contains a very short isoform-specific cytoplasmic tail. This result is consistent with our previous data that T1 lost its activity-dependent surface increase on cLTP stimuli (Zhao et al., 2009). In addition, membrane surface levels of TrkB receptors depend on receptor internalization. To define whether CPE siRNA affects the internalization of surface TrkB, a quantitative fluorescence assay was employed to measure Flag-TrkB internalization levels in neurons transfected with the corresponding siRNA. The results showed no differences between these two groups (Fig. 2H,I). This result implies that CPE does not affect the internalization of membrane surface TrkB. In conclusion, these results suggest that CPE plays a crucial role in cLTP-induced TrkB surface insertion.
TIRF was applied to limit the visualization of fluorescence to proteins located on the plasma membrane through fluorescently labeled molecules within the 100–200 nm evanescent range underneath the plasma membrane (Schwarz et al., 2011). The colocalization of CPE and TrkB labeled with RFP and GFP, respectively, was calculated using TIRF. Pearson's correlation coefficients established the incidence of colocalization (IOC) between two proteins and had an experimental range of ∼0.1 (segregated) to 0.8 (colocalized), as previously described (Manders et al., 1992; Blum et al., 2014). We observed that the IOC of CPE-RFP with TrkB-GFP increased significantly within the TIRF zone after glycine treatment for 10 min (Fig. 3A). Quantitative analysis demonstrated that cLTP significantly promoted TrkB and CPE movement toward the membrane surface in dendrites (Fig. 3B). This increased amount of the double positive signal within the TIRF zone implies that CPE participates in modulating membrane surface insertion induced by cLTP.
Spines are vital signaling compartments that respond to excitatory inputs for neuronal function. Whether CPE affects the distribution of TrkB in dendritic spines on cLTP was further studied. Here, the relative distribution of TrkB between dendritic spines and adjacent dendritic shafts was investigated with a line plot assay (Gerges et al., 2004, 2005; Fig. 3C). We found that cLTP facilitated more TrkB translocation to the spine region than neighboring dendritic shafts in the control group; however, CPE siRNA significantly blocked this effect (Fig. 3D,E). These findings suggest that CPE is involved in regulating cLTP-induced TrkB translocation to spines from dendritic shafts. Collectively, our data demonstrate that CPE plays a crucial role in cLTP-induced TrkB surface insertion and spine translocation.
Identification of the key binding domain in TrkB interacting with CPE that mediates activity-dependent surface TrkB recruitment
Our previous study showed that the JM region is necessary and sufficient for activity-dependent TrkB surface insertion (Zhao et al., 2009). In this study, we investigated the specific binding domain between CPE and TrkB. First, the interaction between the intracellular domain of TrkB (removal of a portion of the extracellular domain, ΔEXTrkB) and the intracellular domain of CPE (CPEC25) was examined. Since CPEC25 is a very small domain that is difficult to assess by Western blot, RFP was fused to CPEC25, and the inability of RFP to bind to TrkB-FL was confirmed (Fig. 4A). The lysate of HEK293 cells transfected with various TrkB-deleted mutants and Myc-CPEC25-RFP was examined by co-IP assays. We found that CPEC25-RFP associated with TrkB-FL and ΔEXTrkB but not ΔEXT1 (Fig. 4A), which suggests that the intracellular domain of TrkB contains the CPE-interacting motif.
The intracellular domain of the TrkB receptor consists of three regions: the JM, TK, and CT regions. To identify the potential specific region responsible for the interaction, additional co-IP experiments were performed with a series of deletion mutants of the TrkB-FL receptor (ΔEXΔJM, ΔEXΔTK and ΔEXΔCT; Fig. 4B). The co-IP assay in HEK293 cells indicated that CPEC25-RFP was detected in the IP complexes of all TrkB-deleted mutants except for ΔEXΔJM, which carried the JM (amino acids 454–536) deletion, suggesting that CPEC25 may interact with the TrkB JM domain (Fig. 4B,C). This was consistent with our previous studies demonstrating that the TrkB JM region is necessary and sufficient for the activity-dependent recruitment of TrkB to the neuronal surface (Zhao et al., 2009).
To define the minimal region in the TrkB JM domain that interacts with CPEC25, the JM domain was divided into three boxes, Box1, Box2, and Box3 (Fig. 4E). Additional co-IP experiments were performed on a series of TrkB JM deletion mutants (ΔEXΔBox1, ΔEXΔBox2, and ΔEXΔBox3). The co-IP assay in HEK293 cells showed that only the Box2 deletion abolished the association between ΔEXTrkB and CPEC25 (Fig. 4D,E). These data suggest that the JM Box2 region is the crucial and necessary domain for the interaction of TrkB with CPE.
To define whether the Box2 domain plays a relevant role in TrkB activity-dependent plasma membrane insertion, the GFP tag was fused to a series of Flag-TrkBΔBoxn constructs, and surface TrkB recruitment was measured by a ratiometric fluorescence assay. The results showed that only the TrkB mutant lacking the Box2 domain (TrkBΔBox2) abrogated the cLTP-induced TrkB surface increase (Fig. 4F,G). Furthermore, we grafted the different box regions into the C terminus of T1 to generate three chimeric constructs. Immunofluorescence assays showed that only grafting Box2 could confer T1 with a dramatic increase in membrane surface levels in response to neuronal activity (Fig. 4F,H). Together, these results indicate that the Box2 domain is not only necessary but also sufficient for the activity-dependent recruitment of TrkB to the neuronal surface.
Identification of the key binding domain in CPE interacting with TrkB that mediates activity-dependent surface TrkB recruitment
Loh et al. (2004), showed that the 25 C-terminal residues of CPE function as a signal for both raft association and the sorting of CPE to the regulated pathway. To determine the minimum sequence required for the CPE interaction with TrkB, a series of deletion mutants with 4/6/10/15 residues deleted from CPEC25 was constructed and cotransfected with ΔEXTrkB into HEK293 cells. The IP results showed that CPEC25Δ15 lost its interaction with ΔEXTrkB completely, which suggested the essential role of the C-terminal 11th to 15th amino acids of CPEC25 (Glu462-Trp466, named CPEBD) in the CPE/TrkB association (Fig. 5A,B).
To verify the biological function of the key binding domain in CPE, two experiments were performed. First, CPEC25Δ10-RFP was used as a dominant negative construct that could bind to TrkB-FL to compete with endogenous CPE and cause a malfunction in the system. Hippocampal neurons at 7 DIV were cotransfected with CPEC25Δ10-RFP and Flag-TrkB-GFP. Ratiometric fluorescence assays showed that the overexpression of CPEC25Δ10-RFP significantly reduced cLTP-induced TrkB surface insertion (Fig. 5C,D).
Moreover, the 452nd-466th amino acids, including the CPEBD of CPE, were fused with the TAT peptide (TAT-CPE452-466), an efficient cell-penetrating peptide derived from the transactivator of transcription (TAT) of human immunodeficiency virus, and the TAT-CPE452-466 could successfully penetrate into cells (data not shown). We pretreated HEK293 cells transiently overexpressing Flag-TrkBΔEX and Myc-CPEC25-RFP with the TAT-Con/TAT-CPE452-466 peptide (5 μm) for 30 min and performed a co-IP assay. IP analysis showed that TAT-CPE452-466 dramatically decreased the amount of Myc-CPEC25-RFP immunoprecipitated by Flag-TrkBΔEX compared with TAT-Con (Fig. 5E). Furthermore, surface TrkB recruitment was measured by ratiometric fluorescence assays in hippocampal neurons pretreated with TAT-Con/TAT-CPE452-466. The results showed that TAT-CPE452-466 significantly blocked cLTP-enhanced TrkB plasma membrane recruitment (Fig. 5F,G). Therefore, the CPEBD domain was identified as a crucial sequence not only for interacting with TrkB but also for regulating TrkB activity-dependent membrane insertion.
TAT-CPE452-466 attenuates activity-enhanced BDNF signaling in hippocampal neurons
To further investigate the functional consequences of CPE-regulated activity-dependent TrkB surface delivery, we examined BDNF-induced downstream signaling in cultured hippocampal neurons pretreated with TAT-CPE452-466 or TAT-Con. Glycine pretreatment significantly enhanced BDNF-triggered TrkB, ERK1/2, and Akt activation in the TAT-Con group, which underscores the importance of activity-dependent TrkB surface insertion for efficient BDNF-induced signaling (Fig. 6A–D). In contrast, blocking the interaction of CPE and TrkB by pretreatment with TAT-CPE452-466 abolished cLTP stimuli-enhanced BDNF downstream signaling (Fig. 6A–D). Together, these results suggest that TAT-CPE452-466 attenuates activity-enhanced BDNF/TrkB downstream signaling.
Figure 6.
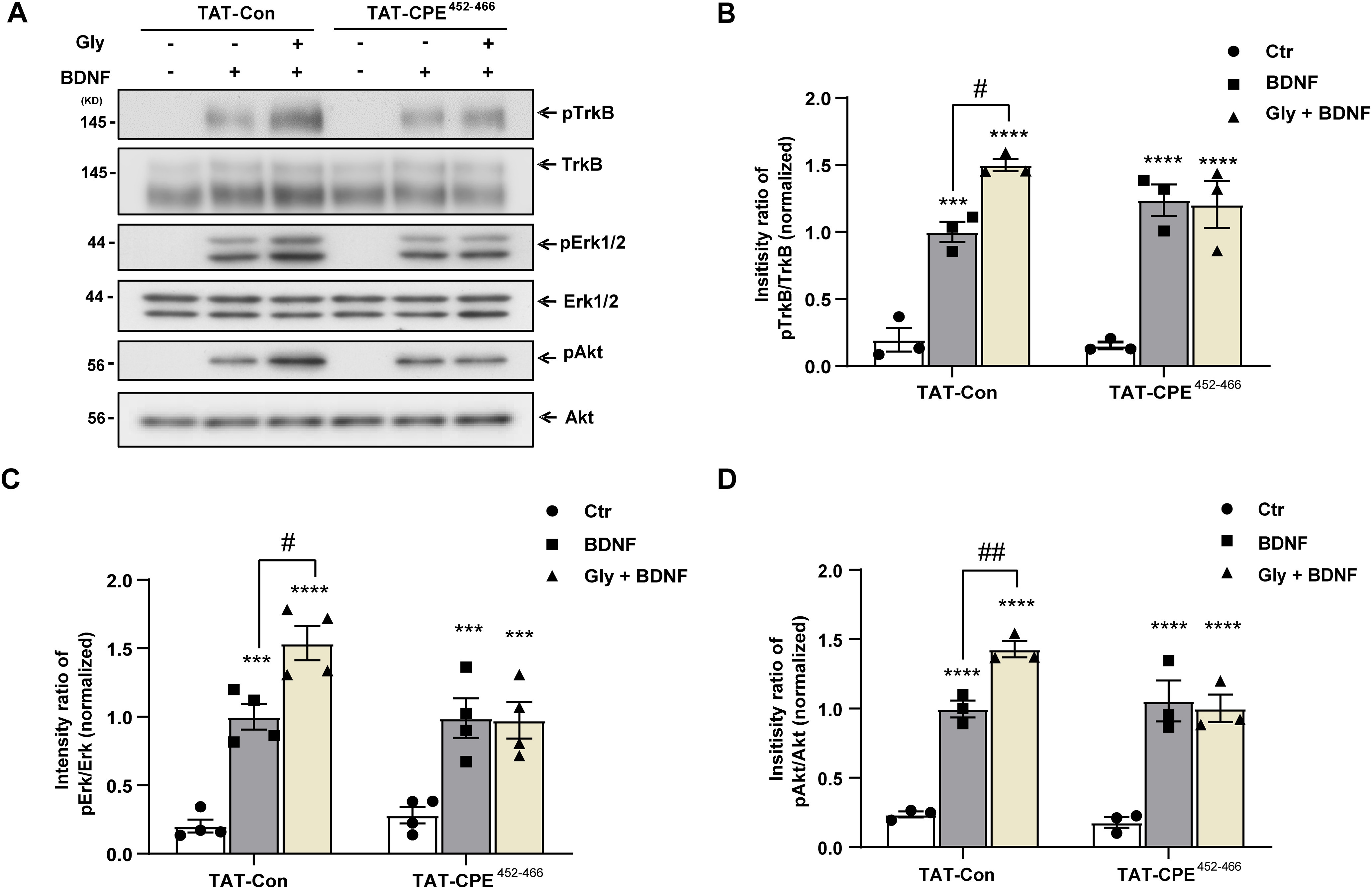
TAT-CPE452-466 attenuates the activity-enhanced BDNF signaling. A, Representative images of immunoblots showing BDNF-induced phosphorylation of TrkB, Akt, and ERK1/2 in cultured hippocampal neurons under the indicated conditions. Neurons at 9 DIV pretreated with TAT-Con or TAT-CPE452-466 for 30 min were stimulated with BDNF and glycine for 10 min. Phosphorylation and total protein levels of TrkB, Erk1/2, and Akt were assessed by immunoblotting. B, Quantitative analysis of pTrkB (two-way ANOVA with Tukey's multiple comparisons test, F(2,12) = 3.489, Ctr-TAT-Con vs BDNF-TAT-Con, ***p = 0.0003; Ctr-TAT-Con vs Gly+BDNF-TAT-Con, ****p < 0.0001; BDNF-TAT-Con vs Gly+BDNF-TAT-Con, #p = 0.0112; Ctr-TAT-CPE452-466 vs BDNF-TAT-CPE452-466, ****p < 0.0001; Ctr-TAT-CPE452-466 vs Gly+BDNF-TAT-CPE452-466, ****p < 0.0001, n = 4). C, Quantitative analysis of pErk (two-way ANOVA with Tukey's multiple comparisons test, F(2,12) = 3.146, Ctr-TAT-Con vs BDNF-TAT-Con, ***p = 0.0004; Ctr-TAT-Con vs Gly+BDNF-TAT-Con, ****p < 0.0001; BDNF-TAT-Con vs Gly+BDNF-TAT-Con, #p = 0.0132; Ctr-TAT-CPE452-466 vs BDNF-TAT-CPE452-466, ***p = 0.0001; Ctr-TAT-CPE452-466 vs Gly+BDNF-TAT-CPE452-466, ***p = 0.0002, n = 4). D, Quantitative analysis of pAkt (two-way ANOVA with Tukey's multiple comparisons test, F(2,12) = 4.723, Ctr-TAT-Con vs BDNF-TAT-Con, ****p < 0.0001; Ctr-TAT-Con vs Gly+BDNF-TAT-Con, ****p < 0.0001; BDNF-TAT-Con vs Gly+BDNF-TAT-Con, ##p = 0.0079; Ctr-TAT-CPE452-466 vs BDNF-TAT-CPE452-466, ****p < 0.0001; Ctr-TAT-CPE452-466 vs Gly+BDNF-TAT-CPE452-466, ****p < 0.0001, n = 4). pTrkB, pErk1/2, and pAkt levels were normalized to the phosphorylation levels in TAT-Con group detected without glycine pretreatment. Graphs represent mean ± SEM.
CPE regulates BDNF-induced spine growth in hippocampal neurons
BDNF has distinct effects on dendritic spine density and morphology in hippocampal pyramidal neurons (Amaral and Pozzo-Miller, 2007). We hence aimed to examine the effect of CPE on BDNF-induced spine growth (density and morphology) in cultured hippocampal neurons treated with TAT-Con or TAT-CPE452-466. Dendritic spines on a length of secondary or tertiary proximal dendrites of hippocampal neurons were selected for analysis (Fig. 7A). As shown in Figure 7, in the TAT-Con group, glycine pretreatment distinctly enhanced the effect of BDNF on the spine density increase (Fig. 7B,C) and spine-head width increase (Fig. 7B,D). However, there were no differences in spine density (Fig. 7B,C) or spine-head width (Fig. 7B,D) in the TAT-CPE452-466 group regardless of glycine treatment. Together, these data suggest that CPE is essential for the cLTP-enhanced dendritic spine density and spine-head width increase on BDNF treatment.
Figure 7.

CPE regulates the BDNF-induced spine growth in hippocampal neurons. A, Confocal image of a representative GFP-transfected hippocampal neuron (green). Bottom, Enlarged image of the framed region indicating the different dendritic spines. B, Hippocampal neurons expressing GFP at DIV 18–21 were pretreated with TAT-Con (5 μm) or TAT-CPE452-466 (5 μm) for 30 min, respectively. Then, neurons were stimulated with glycine (10 min) and/or BDNF (30 min). Higher magnification views of representative segments of dendrites from the neurons. Scale bar, 2 μm. C, Quantification of spine density, expressed per 10 μm of dendrite in B (two-way ANOVA with Bonferroni's multiple comparisons test, F(2,18) = 1.263, Ctr-TAT-Con vs BDNF-TAT-Con, **p = 0.0014; Ctr-TAT-Con versus Gly+BDNF-TAT-Con, ****p < 0.0001; BDNF-TAT-Con vs Gly+BDNF-TAT-Con, #p = 0.0485; Ctr-TAT-CPE452-466 vs BDNF-TAT-CPE452-466, *p = 0.0266; Ctr-TAT-CPE452-466 vs Gly+BDNF-TAT-CPE452-466, ***p = 0.0006, n = 4). D, Quantitation of spine-head width in B. Two-way ANOVA with Bonferroni's multiple comparisons test, F(2,18) = 9.212, Ctr-TAT-Con versus BDNF-TAT-Con, ****p < 0.0001; Ctr-TAT-Con versus Gly+BDNF-TAT-Con, ****p < 0.0001; BDNF-TAT-Con versus Gly+BDNF-TAT-Con, #p = 0.0226; Ctr-TAT-CPE452-466 versus BDNF-TAT-CPE452-466, *p = 0.0394; Ctr-TAT-CPE452-466 versus Gly+BDNF-TAT-CPE452-466, *p = 0.0242, n = 4. All the data are presented as mean ± SEM (n ≥ 25 cells for each condition per experiment).
TAT-CPE452-466 impairs hippocampus-dependent memory
BDNF/TrkB signaling plays a pivotal role in learning and memory in hippocampal- or amygdala-dependent memory processes. On this basis, we further investigated whether CPE-regulated TrkB trafficking is involved in the hippocampus-dependent memory process. We injected the TAT-CPE452-466 peptide into the DH of WT mice, and a series of hippocampus-dependent memory tasks (NPR, CFC), anxiety-related tasks (OFT, EPM), and locomotion tasks (OFT) were performed, as shown in Figure 8A. First, the DH region of TAT-CPE452-466-injected mice was stained with 488-streptavidin to demonstrate that the TAT polypeptide successfully penetrated into the cells in vivo (Fig. 8B). Then, the NPR task was performed 10 min after training. Notably, TAT-CPE452-466-injected mice showed significantly impaired recognition memory compared with TAT-Con-injected mice (Fig. 8C). CFC relies critically on both the DH and the basolateral amygdala (Phillips and LeDoux, 1992; Maren et al., 2013), whereas AFC depends on the auditory cortex and amygdala (Quirk et al., 1995; LeDoux, 2000; Apergis-Schoute et al., 2005). CFC was examined 1 h after training, and TAT-CPE452-466 significantly decreased the freezing time in the CFC test but not in the AFC test (Fig. 8D). These results revealed that TAT-CPE452-466 led to deficits in hippocampus-dependent memory acquisition. We next investigated the role of TAT-CPE452-466 in anxiety-like and locomotion behavior, as well as different memory processes, including consolidation and retrieval, by varying the time point of microinjection. TAT-CPE452-466 was injected into the DH 30 min before the OFT test, and neither the time spent in the center or the distance traveled was different between the TAT-CPE452-466 and TAT-Con groups (Fig. 8E), which suggests that TAT-CPE452-466 microinjection into the DH does not affect anxiety and locomotion. Consistent with the OFT test, TAT-CPE452-466 microinjection had no effect on the time spent in the open arm or entry time into the open arm in the EPM task (Fig. 8G). To reveal the effect of CPE on memory consolidation, we immediately injected TAT-CPE452-466 after fear conditioning and found deficits in CFC but not AFC memory when tested 24 h after training (Fig. 8F). When TAT-CPE452-466 was microinjected into the DH 30 min before memory retrieval, we found that TAT-CPE452-466 had no effect on CFC or AFC memory retrieval (Fig. 8H). Overall, these results suggest that CPE-regulated TrkB trafficking is necessary for hippocampus-dependent memory acquisition and consolidation but not for memory retrieval.
Figure 8.
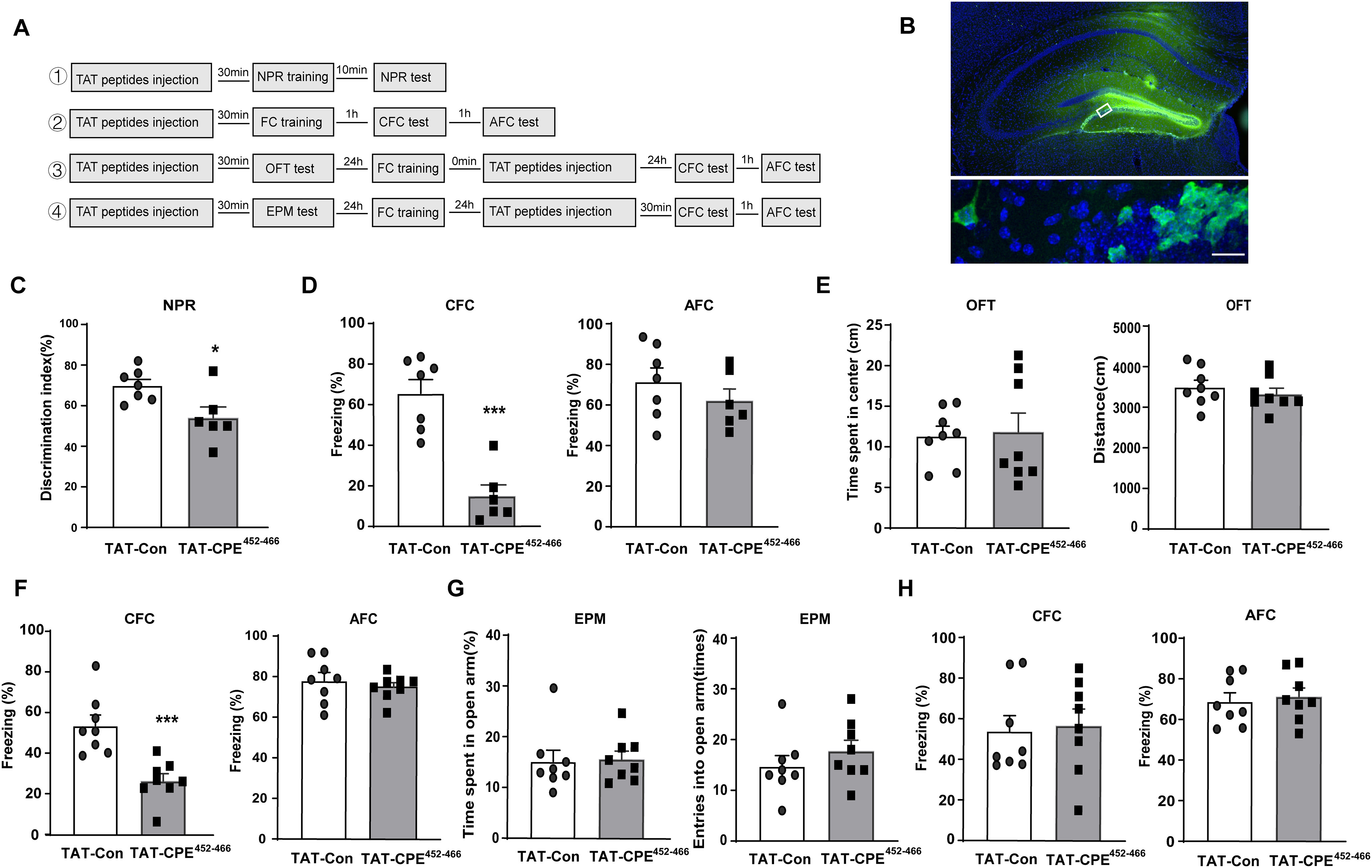
TAT-CPE452-466 treatment impairs hippocampus-dependent memory acquisition and consolidation but not retrieval. A, Schematic representation of the experimental schedules to investigate the effect of TAT-CPE452-466 in the DH on NPR, CFC, AFC, OFT, and EPM behavioral performance. B, Representative image of the DH region 30 min after injection with TAT-CPE452-466. Biotin-TAT-CPE452-466 were stained with 488-Streptavidin and indicated in green representing the infected cells. Nuclei were stained by DAPI and indicated in blue. Scale bar, 20 μm. C, In the NPR tests, compared with TAT-Con-treated mice, TAT-CPE452-466-treated mice show a poorer performance. Unpaired t test, t = 2.709 df = 11, *p = 0.0203; TAT-Con group, n = 7; TAT-CPE452-466 group, n = 6. D, With the microinjection 30 min before FC training, TAT-CPE452-466-treated mice showed lower freezing responses in CFC test but not in the AFC test. Unpaired t test, t = 5.73 df = 11, ***p = 0.0001; TAT-Con group, n = 7, TAT-CPE452-466 group, n = 6. E, In the OFT tests, there is no significant difference between TAT-Con-treated mice and TAT-CPE452-466-treated mice both in time spent in the center and the traveled distance. F, With the microinjection after FC training, TAT-CPE452-466-treated mice showed reduced freezing responses in the CFC test but not in the AFC test. Unpaired t test, t = 4.337 df = 14, ***p = 0.0007; TAT-Con group, n = 8, TAT-CPE452-466 group, n = 8. G, In the EPM tests, TAT-CPE452-466-treated mice showed no significant difference in the time in open arms and entries into open arms. H, With the microinjection 30 min before the CFC test and AFC test, compared with TAT-Con-treated mice, TAT-CPE452-466-treated mice showed similar freezing responses both in the CFC test and AFC test.
TAT-CPE452-466 decreases CFC training-induced TrkB phosphorylation in the dorsal hippocampus
To investigate whether TAT-CPE452-466 could attenuate the interaction of TrkB and CPE in vivo, we injected the TAT-CPE452-466 peptide into the DH 30 min before CFC training. One hour after CFC training, the DH was dissected, and endogenous TrkB and CPE were coimmunoprecipitated. Interestingly, we found that the interaction of TrkB and CPE was increased 1 h after CFC training in the TAT-Con-injected group compared with the no CFC training group, whereas TAT-CPE452-466 injection blocked the interaction of TrkB and CPE compared with TAT-Con injection (Fig. 9A). Previous studies have shown that memory training could induce TrkB phosphorylation (Lin et al., 2015), and we further investigated the phosphorylation levels of TrkB in the DH 1 h after CFC training. Compared with the control group, we found that the phosphorylation levels of TrkB were significantly increased 1 h after CFC training in the TAT-Con-injected group, whereas TAT-CPE452-466 injection decreased the TrkB phosphorylation levels compared with TAT-Con injection (Fig. 9B). These results suggest that TAT-CPE452-466 could block the interaction of TrkB and CPE in vivo, which might lead to impairment in memory-training-induced TrkB plasma insertion and phosphorylation and result in memory deficits. To clarify whether TAT-CPE452-466 could affect the number and morphology of hippocampal neurons, we performed NeuN immunohistochemistry staining and Golgi-Cox staining 1 h after CFC training. We found no difference in the number of NeuN-positive cells between the control group and CFC training group with TAT-Con or TAT-CPE452-466 injection (Fig. 9C). Moreover, the dendritic complexity of the DG and spine density in the basal dendrites of DG pyramidal neurons showed no significant difference in these three groups (Fig. 9D,E). Together, these data suggest that TAT-CPE452-466 microinjection-induced hippocampal memory acquisition deficits might not be because of neuronal number and dendritic morphology alterations.
Figure 9.
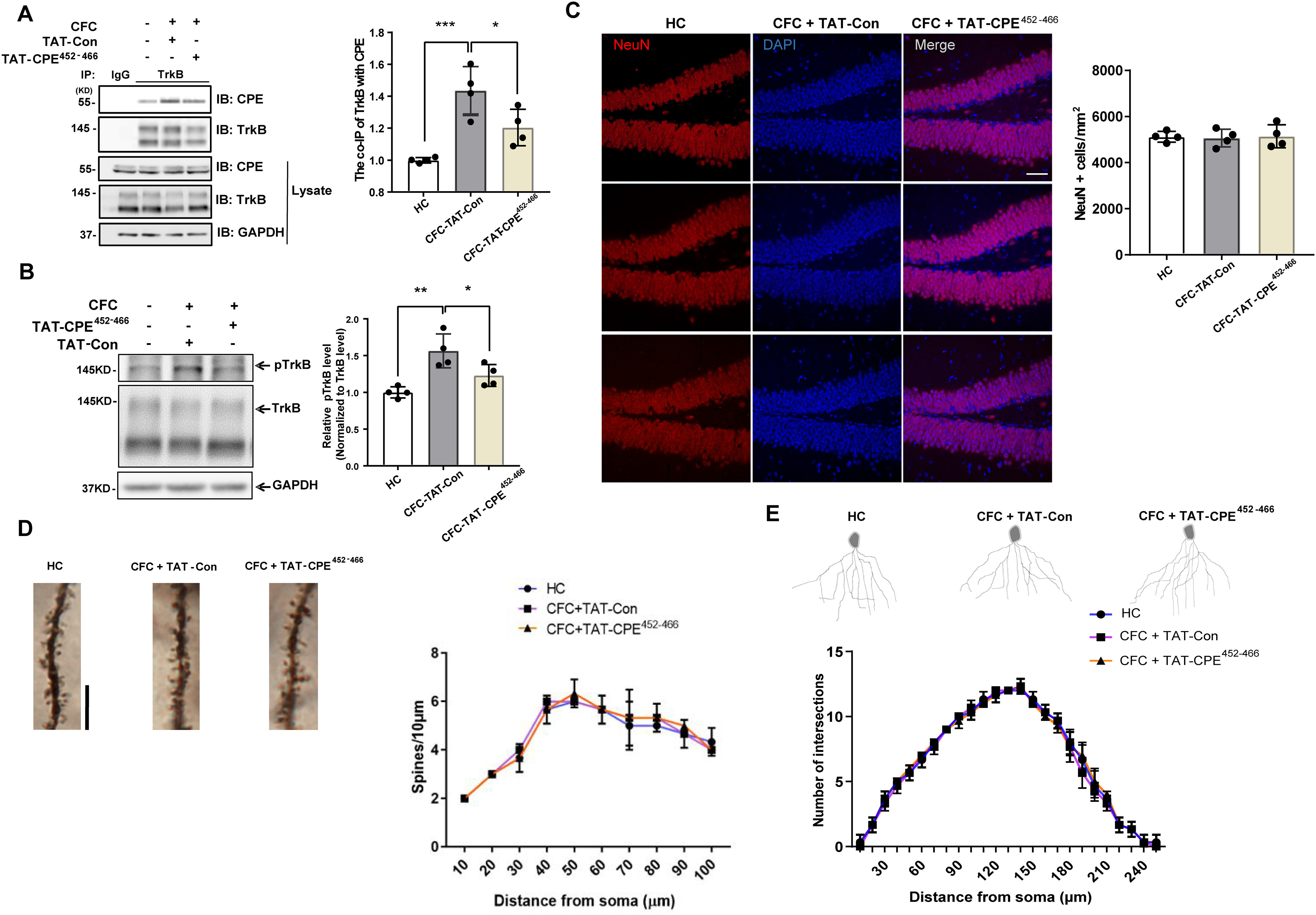
TAT-CPE452-466 blocks the interaction of TrkB and CPE in the DH and inhibits TrkB phosphorylation induced by CFC training. A, TAT-CPE452-466 blocks the interaction between TrkB and CPE. Co-IP was performed 1 h after CFC training with microinjection of TAT-Con or TAT-CPE452-466 into DH 30 min before CFC training with the control of home cage (HC) group. One-way ANOVA with Tukey's multiple comparisons, F(2,9) = 15.77, ***p = 0.0009, *p = 0.0375; n = 4/group. B, TAT-CPE452-466 inhibits the CFC-induced TrkB phosphorylation in DH. Phosphorylation and total protein levels of TrkB were assessed by Western blot 1 h after CFC training with microinjection of TAT-Con or TAT-CPE452-466 into DH 30 min before CFC training. One-way ANOVA with Tukey's multiple comparisons, F(2,9) = 11.86, **p = 0.0024, *p = 0.0445; n 4/group. C, Coronal sections of DG were stained with DAPI and the antibody to a pan-neuronal marker, NeuN, from the group 1 h after CFC training with microinjection of TAT-Con or TAT-CPE452-466 into DH 30 min before CFC training. Scale bar, 20 μm. D, Spine density in basal dendrites of DG pyramidal neurons in the group 1 h after CFC training with microinjection of TAT-Con or TAT-CPE452-466 into DH 30 min before CFC training. Scale bar, 10 μm. E, Examples and Sholl analyses of Golgi-stained dentate gyrus neurons from the group 1 h after CFC training with microinjection of TAT-Con or TAT-CPE452-466 into DH 30 min before CFC training.
Discussion
BDNF has been shown to regulate neuron survival and synaptic plasticity in the CNS in an activity-dependent manner. The preferred biological function of BDNF, in part, relies on TrkB insertion into the plasma membrane on neuronal activity. However, the molecular mechanism underlying neuronal activity-increased TrkB cell surface levels is less understood. Here, we determined that CPE could mediate the cLTP-induced insertion of TrkB into the plasma membrane.
Our results provide several new insights into the mechanisms and significance of neuronal activity-enhanced TrkB surface insertion. First, we provide direct evidence that CPE mediates neuronal activity-regulated TrkB plasma membrane insertion. We verified the reliable interaction between CPE and TrkB-FL but not TrkB.T1 in hippocampal neurons, which was not discovered previously. The cLTP-induced TrkB surface insertion is a regulated secretory process. In general, TrkB trafficking from the Golgi complex to the plasma membrane in response to cLTP may include three steps: sorting from the trans-Golgi network to the reserved secretory pool, transporting from the reserved pool to the plasma membrane region on neuronal activity, and fusing TrkB-containing vesicles with the plasma membrane to insert TrkB into the cell surface. We showed that CPE and TrkB could be trafficked from the Golgi to dendrites in the same vesicles by time-lapse imaging. Depletion of CPE blocked cLTP-induced TrkB translocation into dendritic spines from dendritic shafts, so cLTP could significantly increase the colocalization of TrkB and CPE vesicles beneath the plasma membrane by TIRF. CPE has been demonstrated in many studies to mediate the intracellular trafficking of neuropeptides or transporters. For example, proBDNF is sorted by a sorting signal-CPE-mediated mechanism to vesicles for activity-dependent secretion (Park et al., 2008; Cawley et al., 2012). Enhanced expression of CPE leads to increased trafficking of the dopamine transporter to the presynaptic membrane at the axonal terminus of dopaminergic neurons, thereby enhancing dopamine uptake (Zhang et al., 2009). Moreover, the localization of synaptic vesicles to the preactive zone for the exocytosis of classical neurotransmitters in hypothalamic neurons is also dependent on CPE (Lou et al., 2010). All these studies suggest that CPE might play a multifunctional role in intracellular TrkB transport, which needs further exploration. In addition, our previous study showed that both nocodazole, a microtubule-depolymerizing agent, and cytochalasin D, which inhibits actin polymerization, significantly inhibited cLTP-induced TrkB plasma membrane insertion (Zhao et al., 2009). This suggests that rapid TrkB recruitment to the plasma membrane requires both intact microtubules and microfilaments. The cytoplasmic tail of CPE is known to transport vesicles via interaction with microtubules (Park et al., 2008) or actin via γ-adducin at the active zone of the synapse (Lou et al., 2010). Therefore, it is possible that the cytoplasmic tail of CPE could facilitate the movement of TrkB vesicles to the neuronal surface membrane for fusion and insertion by a similar mechanism.
Second, we identified the minimum binding domain between CPE and TrkB. The JM Box2 (Ser476 to Phe516) of TrkB is necessary and sufficient for cLTP-induced TrkB surface insertion. It has been suggested that the 25 C-terminal amino acids are necessary for delivering CPE to RSP (Dhanvantari et al., 2002). We present evidence of a specific region, Glu462-Trp466, in the C-terminal domain of CPE that is responsible for the interaction with TrkB. We also demonstrated that TAT-CPE452-466, which could block the association between CPE and TrkB, decreased the cLTP-induced insertion of TrkB into the plasma membrane. All these results reveal that the association between CPE and TrkB is essential for TrkB activity-dependent surface insertion.
Third, we hypothesized that CPE-regulated TrkB dendritic spine translocation and cell surface insertion induced by neuronal activity play a role in BDNF synaptic tagging signaling. Previous studies have demonstrated that the activation of the PI3K and ERK pathways plays essential roles in LTP induction and maintenance (Kelleher et al., 2004; Tsokas et al., 2007). Our results proved that specific blockade of CPE-mediated TrkB cell surface insertion with TAT-CPE452-466 abolished neuronal activity-enhanced BDNF downstream signaling. Dendritic spines are small membrane protrusions from the functional compartment of the dendritic membrane. The majority of excitatory synapses build on the spine, and their motile and diverse morphology makes them sensitive structures in response to neuronal activity to burden synaptic plasticity (Alvarez et al., 2007; Matsuzaki, 2007; Bourne and Harris, 2008; De Roo et al., 2008). Spines with a larger head will generally reflect more activity and show greater plasticity (Chidambaram et al., 2019). We found that TAT-CPE452-466 blunted the effect of BDNF-induced increases in spine density and spine-head size on cLTP stimuli. These results demonstrated that CPE-regulated intracellular TrkB trafficking is essential for the role of BDNF in synaptic plasticity.
Finally, we demonstrated that specifically blocking CPE-mediated TrkB cell surface insertion might impair hippocampus-dependent memory. TAT-CPE452-466 microinfusion attenuated the enhanced interaction between TrkB and CPE and increased TrkB phosphorylation levels on CFC training, which suggests that the interaction between the C terminus of CPE and TrkB is involved in hippocampal memory training-induced TrkB surface insertion. Microinjection of TAT-CPE452-466 in the dorsal hippocampus led to defects in novel place recognition and contextual fear memory acquisition and consolidation but not retrieval, which is consistent with the behavioral phenotype in BDNF heterozygous mice (Liu et al., 2004; Heldt et al., 2007; Andero et al., 2014). Microinfusion of BDNF antibodies has also been shown to impair conditioned taste aversion, memory acquisition, and consolidation but not retrieval (Bekinschtein et al., 2008; Ma et al., 2011). When crossed with CaMKII Cre mice, conditional TrkB knock-out mice failed to acquire hippocampus-dependent memory (Minichiello et al., 1999). In addition, CPE knock-out mice were shown to have impairments in hippocampus-dependent memory, including novel object recognition, water maze, and the social transmission of food (Woronowicz et al., 2008). Thus, our data provide direct in vivo evidence that CPE-mediated TrkB cell surface insertion might have physiological relevance to BDNF/TrkB function in the hippocampal memory process. Interestingly, CPE knock-out mice showed decreased locomotion as well as dendritic spine morphology alterations (Woronowicz et al., 2010), and CPEfat/fat mice displayed increased anxiety-like behavior (Cawley et al., 2004; 2012), which was not found in our TAT-CPE452-466-treated mice. These data suggest that CPE-regulated intracellular TrkB trafficking cannot account for the complete function of CPE in behavior regulation as CPE acts as a sorting receptor for other proteins.
In summary, the present study uncovered that CPE has a new and essential role in modulating the cLTP-induced insertion of TrkB into the plasma membrane as well as in mediating the role of BDNF in synaptic plasticity and hippocampal memory. Additionally, these findings shed light on new molecular mechanisms of TrkB cell surface insertion and function underlying BDNF selectively and preferentially acting on active neurons and synapses in the CNS. Future investigations into the mechanism of action of CPE in this respect are necessary and could lead to the identification of targets for enhancing the effect of BDNF on synaptic plasticity.
Footnotes
This study was supported by the National Natural Science Foundation of China (Grant No. 31071254), Natural Science Foundation of Shandong Province (Grant No. ZR2020ZD17), and the Foundation of Advanced Medical Research Institute of Shandong University (Grant No. 22480089398404). We thank Translational Medicine Core Facility of Shandong University for consultation and instrument availability.
The authors declare no competing financial interests.
References
- Alvarez VA, Ridenour DA, Sabatini BL (2007) Distinct structural and ionotropic roles of NMDA receptors in controlling spine and synapse stability. J Neurosci 27:7365–7376. 10.1523/JNEUROSCI.0956-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral MD, Pozzo-Miller L (2007) TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. J Neurosci 27:5179–5189. 10.1523/JNEUROSCI.5499-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andero R, Choi DC, Ressler KJ (2014) BDNF-TrkB receptor regulation of distributed adult neural plasticity, memory formation, and psychiatric disorders. Prog Mol Biol Transl Sci 122:169–192. 10.1016/B978-0-12-420170-5.00006-4 [DOI] [PubMed] [Google Scholar]
- Apergis-Schoute AM, Debiec J, Doyère V, LeDoux JE, Schafe GE (2005) Auditory fear conditioning and long-term potentiation in the lateral amygdala require ERK/MAP kinase signaling in the auditory thalamus: a role for presynaptic plasticity in the fear system. J Neurosci 25:5730–5739. 10.1523/JNEUROSCI.0096-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekinschtein P, Cammarota M, Izquierdo I, Medina JH (2008) BDNF and memory formation and storage. Neuroscientist 14:147–156. 10.1177/1073858407305850 [DOI] [PubMed] [Google Scholar]
- Bevins RA, Besheer J (2006) Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study “recognition memory.” Nature protocols 1:1306–1311. [DOI] [PubMed] [Google Scholar]
- Blum FC, Tepp WH, Johnson EA, Barbieri JT (2014) Multiple domains of tetanus toxin direct entry into primary neurons. Traffic 15:1057–1065. 10.1111/tra.12197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhoeffer T (1996) Neurotrophins and activity-dependent development of the neocortex. Curr Opin Neurobiol 6:119–126. 10.1016/s0959-4388(96)80017-1 [DOI] [PubMed] [Google Scholar]
- Bourne JN, Harris KM (2008) Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci 31:47–67. 10.1146/annurev.neuro.31.060407.125646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E (2005) BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol 76:99–125. 10.1016/j.pneurobio.2005.06.003 [DOI] [PubMed] [Google Scholar]
- Cawley NX, Zhou J, Hill JM, Abebe D, Romboz S, Yanik T, Rodriguiz RM, Wetsel WC, Loh YP (2004) The carboxypeptidase E knockout mouse exhibits endocrinological and behavioral deficits. Endocrinology 145:5807–5819. 10.1210/en.2004-0847 [DOI] [PubMed] [Google Scholar]
- Cawley NX, Wetsel WC, Murthy SR, Park JJ, Pacak K, Loh YP (2012) New roles of carboxypeptidase E in endocrine and neural function and cancer. Endocr Rev 33:216–253. 10.1210/er.2011-1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, Hempstead BL, Lee FS (2006) Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science (New York, NY) 314:140–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Patel PD, Sant G, Meng CX, Teng KK, Hempstead BL, Lee FS (2004) Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J Neurosci 24:4401–4411. 10.1523/JNEUROSCI.0348-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidambaram SB, Rathipriya AG, Bolla SR, Bhat A, Ray B, Mahalakshmi AM, Manivasagam T, Thenmozhi AJ, Essa MM, Guillemin GJ, Chandra R, Sakharkar MK (2019) Dendritic spines: revisiting the physiological role. Prog Neuropsychopharmacol Biol Psychiatry 92:161–193. 10.1016/j.pnpbp.2019.01.005 [DOI] [PubMed] [Google Scholar]
- Cohen S, Greenberg ME (2008) Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu Rev Cell Dev Biol 24:183–209. 10.1146/annurev.cellbio.24.110707.175235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cool DR, Normant E, Shen F, Chen HC, Pannell L, Zhang Y, Loh YP (1997) Carboxypeptidase E is a regulated secretory pathway sorting receptor: genetic obliteration leads to endocrine disorders in Cpe(fat) mice. Cell 88:73–83. 10.1016/s0092-8674(00)81860-7 [DOI] [PubMed] [Google Scholar]
- De Roo M, Klauser P, Garcia PM, Poglia L, Muller D (2008) Spine dynamics and synapse remodeling during LTP and memory processes. Prog Brain Res 169:199–207. 10.1016/S0079-6123(07)00011-8 [DOI] [PubMed] [Google Scholar]
- Dhanvantari S, Arnaoutova I, Snell CR, Steinbach PJ, Hammond K, Caputo GA, London E, Loh YP (2002) Carboxypeptidase E, a prohormone sorting receptor, is anchored to secretory granules via a C-terminal transmembrane insertion. Biochemistry 41:52–60. 10.1021/bi015698n [DOI] [PubMed] [Google Scholar]
- Du J, Feng L, Yang F, Lu B (2000) Activity- and Ca(2+)-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. J Cell Biol 150:1423–1434. 10.1083/jcb.150.6.1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker LD, Snyder SH (1982) Enkephalin convertase: purification and characterization of a specific enkephalin-synthesizing carboxypeptidase localized to adrenal chromaffin granules. Proc Natl Acad Sci U S A 79:3886–3890. 10.1073/pnas.79.12.3886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerges NZ, Backos DS, Esteban JA (2004) Local control of AMPA receptor trafficking at the postsynaptic terminal by a small GTPase of the Rab family. J Biol Chem 279:43870–43878. 10.1074/jbc.M404982200 [DOI] [PubMed] [Google Scholar]
- Gerges NZ, Brown TC, Correia SS, Esteban JA (2005) Analysis of Rab protein function in neurotransmitter receptor trafficking at hippocampal synapses. Methods Enzymol 403:153–166. 10.1016/S0076-6879(05)03013-2 [DOI] [PubMed] [Google Scholar]
- Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B (1998) Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci 18:6830–6839. 10.1523/JNEUROSCI.18-17-06830.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldt SA, Stanek L, Chhatwal JP, Ressler KJ (2007) Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry 12:656–670. 10.1038/sj.mp.4001957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook VY, Loh YP (1984) Carboxypeptidase B-like converting enzyme activity in secretory granules of rat pituitary. Proc Natl Acad Sci U S A 81:2776–2780. 10.1073/pnas.81.9.2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ (1996) Synaptic activity and the construction of cortical circuits. Science 274:1133–1138. 10.1126/science.274.5290.1133 [DOI] [PubMed] [Google Scholar]
- Kelleher RJ 3rd, Govindarajan A, Jung HY, Kang H, Tonegawa S (2004) Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 116:467–479. 10.1016/S0092-8674(04)00115-1 [DOI] [PubMed] [Google Scholar]
- Klein R, Conway D, Parada LF, Barbacid M (1990) The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell 61:647–656. 10.1016/0092-8674(90)90476-u [DOI] [PubMed] [Google Scholar]
- LeDoux JE (2000) Emotion circuits in the brain. Annu Rev Neurosci 23:155–184. 10.1146/annurev.neuro.23.1.155 [DOI] [PubMed] [Google Scholar]
- Lin WJ, Jiang C, Sadahiro M, Bozdagi O, Vulchanova L, Alberini CM, Salton SR (2015) VGF and Its C-terminal peptide TLQP-62 regulate memory formation in hippocampus via a BDNF-TrkB-dependent mechanism. J Neurosci 35:10343–10356. 10.1523/JNEUROSCI.0584-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Geng Z, Yan J, Li T, Chen Q, Zhang Q, Chen Z (2015) Blocking GSK3β-mediated dynamin1 phosphorylation enhances BDNF-dependent TrkB endocytosis and the protective effects of BDNF in neuronal and mouse models of Alzheimer's disease. Neurobiol Dis 74:377–391. 10.1016/j.nbd.2014.11.020 [DOI] [PubMed] [Google Scholar]
- Liu IY, Lyons WE, Mamounas LA, Thompson RF (2004) Brain-derived neurotrophic factor plays a critical role in contextual fear conditioning. J Neurosci 24:7958–7963. 10.1523/JNEUROSCI.1948-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh Y, Kim T, Rodriguez Y, Cawley N (2004) Secretory granule biogenesis and neuropeptide sorting to the regulated secretory pathway in neuroendocrine cells. J Mol Neurosci 22:63–71. 10.1385/JMN:22:1-2:63 [DOI] [PubMed] [Google Scholar]
- Lou H, Kim SK, Zaitsev E, Snell CR, Lu B, Loh YP (2005) Sorting and activity-dependent secretion of BDNF require interaction of a specific motif with the sorting receptor carboxypeptidase e. Neuron 45:245–255. 10.1016/j.neuron.2004.12.037 [DOI] [PubMed] [Google Scholar]
- Lou H, Park JJ, Cawley NX, Sarcon A, Sun L, Adams T, Loh YP (2010) Carboxypeptidase E cytoplasmic tail mediates localization of synaptic vesicles to the pre-active zone in hypothalamic pre-synaptic terminals. J Neurochem 114:886–896. 10.1111/j.1471-4159.2010.06820.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Chow A (1999) Neurotrophins and hippocampal synaptic transmission and plasticity. J Neurosci Res 58:76–87. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT (2001) Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29:243–254. 10.1016/s0896-6273(01)00194-5 [DOI] [PubMed] [Google Scholar]
- Ma L, Wang DD, Zhang TY, Yu H, Wang Y, Huang SH, Lee FS, Chen ZY (2011) Region-specific involvement of BDNF secretion and synthesis in conditioned taste aversion memory formation. J Neurosci 31:2079–2090. 10.1523/JNEUROSCI.5348-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA (1992) Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci 103:857–862. 10.1242/jcs.103.3.857 [DOI] [PubMed] [Google Scholar]
- Maren S, Phan KL, Liberzon I (2013) The contextual brain: implications for fear conditioning, extinction and psychopathology. Nat Rev Neurosci 14:417–428. 10.1038/nrn3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M (2007) Factors critical for the plasticity of dendritic spines and memory storage. Neurosci Res 57:1–9. 10.1016/j.neures.2006.09.017 [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC (1996) Neurotrophin regulation of cortical dendritic growth requires activity. Neuron 17:1057–1064. 10.1016/s0896-6273(00)80239-1 [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC (1999) Neurotrophins and synaptic plasticity. Annu Rev Neurosci 22:295–318. 10.1146/annurev.neuro.22.1.295 [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson MG Jr., Reichardt LF, Barres BA (1998) Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 21:681–693. 10.1016/S0896-6273(00)80586-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middlemas DS, Lindberg RA, Hunter T (1991) trkB, a neural receptor protein-tyrosine kinase: evidence for a full-length and two truncated receptors. Mol Cell Biol 11:143–153. 10.1128/mcb.11.1.143-153.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R (1999) Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24:401–414. 10.1016/s0896-6273(00)80853-3 [DOI] [PubMed] [Google Scholar]
- Musleh W, Bi X, Tocco G, Yaghoubi S, Baudry M (1997) Glycine-induced long-term potentiation is associated with structural and functional modifications of alpha-amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid receptors. Proc Natl Acad Sci U S A 94:9451–9456. 10.1073/pnas.94.17.9451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagappan G, Lu B (2005) Activity-dependent modulation of the BDNF receptor TrkB: mechanisms and implications. Trends Neurosci 28:464–471. 10.1016/j.tins.2005.07.003 [DOI] [PubMed] [Google Scholar]
- Park JJ, Cawley NX, Loh YP (2008) A bi-directional carboxypeptidase E-driven transport mechanism controls BDNF vesicle homeostasis in hippocampal neurons. Mol Cell Neurosci 39:63–73. 10.1016/j.mcn.2008.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE (1992) Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci 106:274–285. 10.1037//0735-7044.106.2.274 [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Repa C, LeDoux JE (1995) Fear conditioning enhances short-latency auditory responses of lateral amygdala neurons: parallel recordings in the freely behaving rat. Neuron 15:1029–1039. 10.1016/0896-6273(95)90092-6 [DOI] [PubMed] [Google Scholar]
- Rodriguiz RM, Wilkins JJ, Creson TK, Biswas R, Berezniuk I, Fricker AD, Fricker LD, Wetsel WC (2013) Emergence of anxiety-like behaviours in depressive-like Cpe(fat/fat) mice. Int J Neuropsychopharmacol 16:1623–1634. 10.1017/S1461145713000059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JP, König I, Anderson KI (2011) Characterizing system performance in total internal reflection fluorescence microscopy. Methods Mol Biol 769:373–386. 10.1007/978-1-61779-207-6_25 [DOI] [PubMed] [Google Scholar]
- Srinivasan S, Bunch DO, Feng Y, Rodriguiz RM, Li M, Ravenell RL, Luo GX, Arimura A, Fricker LD, Eddy EM, Wetsel WC (2004) Deficits in reproduction and pro-gonadotropin-releasing hormone processing in male Cpefat mice. Endocrinology 145:2023–2034. 10.1210/en.2003-1442 [DOI] [PubMed] [Google Scholar]
- Thoenen H (1995) Neurotrophins and neuronal plasticity. Science 270:593–598. 10.1126/science.270.5236.593 [DOI] [PubMed] [Google Scholar]
- Tsokas P, Ma T, Iyengar R, Landau EM, Blitzer RD (2007) Mitogen-activated protein kinase upregulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J Neurosci 27:5885–5894. 10.1523/JNEUROSCI.4548-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woronowicz A, Koshimizu H, Chang SY, Cawley NX, Hill JM, Rodriguiz RM, Abebe D, Dorfman C, Senatorov V, Zhou A, Xiong ZG, Wetsel WC, Loh YP (2008) Absence of carboxypeptidase E leads to adult hippocampal neuronal degeneration and memory deficits. Hippocampus 18:1051–1063. 10.1002/hipo.20462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woronowicz A, Cawley N, Chang S, Koshimizu H, Phillips A, Xiong Z, Loh Y (2010) Carboxypeptidase E knockout mice exhibit abnormal dendritic arborization and spine morphology in central nervous system neurons. J Neurosci Res 88:64–72. 10.1002/jnr.22174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Chang S-Y, Xiong Z-G, Selveraj P, Peng Loh Y (2017) Absence of carboxypeptidase e/neurotrophic factor-Α1 in knock-out mice leads to dysfunction of BDNF-TRKB signaling in hippocampus. J Mol Neurosci 62:79–87. 10.1007/s12031-017-0914-0 [DOI] [PubMed] [Google Scholar]
- Yu H, Wang DD, Wang Y, Liu T, Lee FS, Chen ZY (2012) Variant brain-derived neurotrophic factor Val66Met polymorphism alters vulnerability to stress and response to antidepressants. J Neurosci 32:4092–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li S, Wang M, Vukusic B, Pristupa ZB, Liu F (2009) Regulation of dopamine transporter activity by carboxypeptidase E. Mol Brain 2:10. 10.1186/1756-6606-2-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Sheng AL, Huang SH, Yin YX, Chen B, Li XZ, Zhang Y, Chen ZY (2009) Mechanism underlying activity-dependent insertion of TrkB into the neuronal surface. J Cell Sci 122:3123–3136. 10.1242/jcs.047712 [DOI] [PubMed] [Google Scholar]