

Abstract
Heme-iron oxidoreductases operating through the high-valent FeIVO intermediates perform crucial and complicated transformations, such as oxidations of unreactive saturated hydrocarbons. These enzymes share the same Fe coordination, only differing by the axial ligation, e.g., Cys in P450 oxygenases, Tyr in catalases, and His in peroxidases. By examining ~200 heme-iron proteins, we show that the protein hosts exert highly specific intramolecular electric fields on the active sites, and there is a strong correlation between the direction and magnitude of this field and the protein function. In all heme proteins, the field is preferentially aligned with the Fe–O bond (Fz). The Cys-ligated P450 oxygenases have the highest average Fz of 28.5 MV cm−1, i.e., most enhancing the oxyl-radical character of the oxo group, and consistent with the ability of these proteins to activate strong C–H bonds. In contrast, in Tyr-ligated proteins, the average Fz is only 3.0 MV cm−1, apparently suppressing single-electron off-pathway oxidations, and in His-ligated proteins, Fz is −8.7 MV cm−1. The operational field range is given by the trade-off between the low reactivity of the FeIVO Compound I at the more negative Fz, and the low selectivity at the more positive Fz. Consequently, a heme-iron site placed in the field characteristic of another heme-iron protein class loses its canonical function, and gains an adverse one. Thus, electric fields produced by the protein scaffolds, together with the nature of the axial ligand, control all heme-iron chemistry.
Graphical Abstract
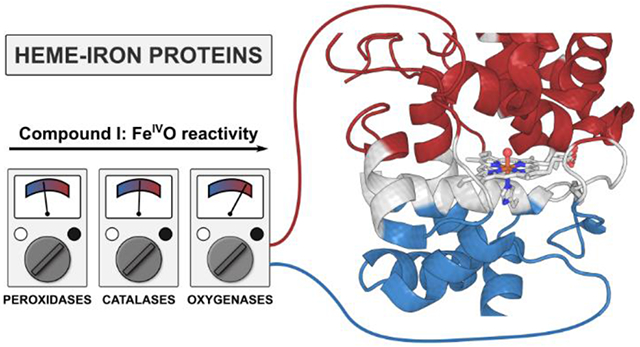
INTRODUCTION
High-valent metal-oxo and metal-hydroxo intermediates are irreplaceable in Nature, as they can catalyze energetically demanding transformations, such as oxidations of (inactivated) saturated hydrocarbons.1–4 Heme-iron proteins may serve as an example using molecular oxygen or hydrogen peroxide to form the reactive species, known as Compound I (CpdI), to oxidize or oxygenate diverse organic substrates (Scheme 1).5–8 Particularly, hydroxylation of the substrates triggered by the cytochrome P450 family of proteins represents a cornerstone reactivity in xenobiotics’ metabolic pathways, making them more soluble and more susceptible for elimination. Although debate remains on which factors dominate (or significantly contribute to) the reactivity of these systems,9–13 the activation barrier is indisputably governed by their ability to activate the strong C–H bonds of the substrates. C–H bond-dissociation free energies (BDFEs) are ca. 80-110 kcal mol−1 and pKAs are above 50,14,15 making the observed rates for transformations all the more impressive, in the range of 107 M−1 s−1.16
Scheme 1.
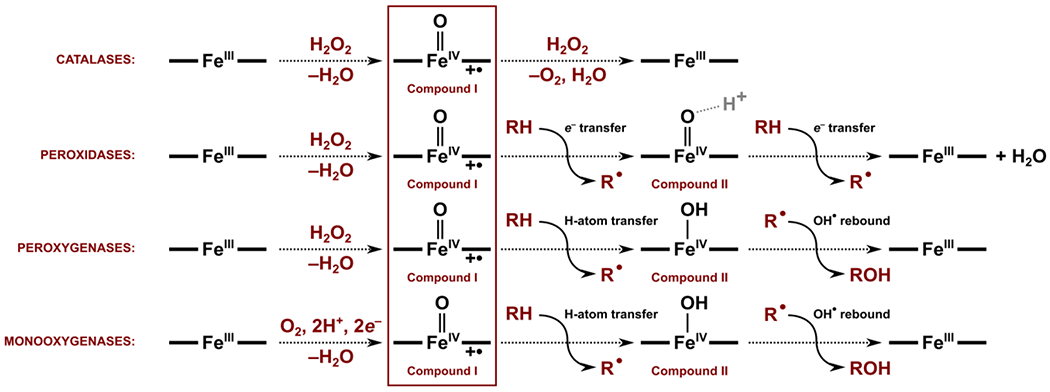
Typical reactions performed by different classes of the heme-iron oxidoreductases. In the absence of a substrate, the resting state of the enzymes is the ferric (FeIII) with a bound water molecule. The resting state is activated either by (i) FeIII binding hydrogen peroxide, leading ultimately to the (Por+•)FeIV=O CpdI intermediate with the release of an H2O molecule, or (ii) FeIII reduction, followed by the FeII binding of molecular oxygen, leading to the same (Por+•)FeIV=O CpdI intermediate through sequential one-electron two-proton transfer and H2O molecule release. Although the nature of the CpdI intermediate is the same, the electronic structure and reactivity may differ as a result of the nonidentical protein environment, such as axial ligands (Cys, His, or Tyr) and the protein fold. In the catalases enzymes, the CpdI reacts with the second molecule of H2O2 to yield O2 and H2O. In the peroxidases and oxygenases subclasses, the second reactive intermediate – the (Por)FeIV=O CpdII or (Por)FeIV–OH CpdII – is proposed to be formed via single-electron transfer (associated with coupled proton transfer from substrate to some nearby amino acid residue) or electron-proton transfer to the CpdI. Lastly, CpdII can also oxidize or oxygenate the organic substrate.
Warshel postulated that the catalysis by enzymes is empowered by electrostatic preorganization: the placement of charged groups throughout the protein in a way that permanently electrostatically favors the transition state compared to the reactants.17–19 In this way, both the enthalpic barrier and the entropic penalty for its crossing are reduced – a phenomenon not achievable for reactions in solution. Electrostatic preorganization has been widely investigated in recent years, focusing in ketosteroid isomerase and several other enzymes.20–29 Given the key role of the FeIV-oxo bond in CpdI and CpdII in the heme-iron catalysis, the well-defined, rigid structure of the heme site presents but a perfect opportunity to apply an intermolecular field to maximize or control the reactivity. The electric field applied to FeIVO may, for example, give the FeIV-oxo bond a stronger oxyl-radical (FeIIIO•−) character, which is often associated with the higher metal-oxo electrophilicity.12,30–34
Indeed, the oriented external electric field (OEEF) parallel to the Fe–O axis was proposed to induce changes in reactivity and selectivity of several heme iron-oxo CpdI models outside of the protein.35–40 Such an OEEF can significantly perturb the electronic structure of CpdI: localization of the unpaired electron in its ground state, and the spin-state (doublet/quartet) ordering and energy splitting, impacting the physicochemical properties.39 In particular, the sign and the magnitude of the OEEF along the Fe–O axis were evidenced to alter the location of the radical site: on the nonbonding a1u/a2u orbital on porphyrin (denoted as Por+•), or on the adjacent Trp residue (denoted as Trp+•), in a pair of His-ligated peroxidases – ascorbate peroxidase (APX) and cytochrome c peroxidase (CcP).39 Note that in Nature, these two peroxidases have different electronic structures, and the ultimate factor differentiating between the Por+• and Trp+• radicals has not been plausibly clarified.6 However, QM/MM calculations of CpdI in CcP and APX correctly reproduced the differences in the electronic structures of their identical QM regions, apparently affected by the protein edifice.41,42 From these findings, the intramolecular local electric fields (LEF) transpire as a strong and thus far not rigorously questioned suspect governing the reactivity and selectivity in the natural heme-iron proteins.
Still using a small model system representing the Cys-ligated CpdI of cytochrome P450, Shaik et al. proposed that the selectivity between the epoxidation and hydroxylation competing pathways could be switched by altering the direction and magnitude of the OEEF.40 A sufficiently large OEEF of ca. F = ± 0.0125 a.u. (~ ±64 MV cm−1) orthogonal to the porphine plane were calculated to realize a 100 % selectivity toward C–H vs. C=C bond activation.40 As of now, it is unclear whether such dramatic fields can be achieved inside the heme-iron proteins. However, it is clear that, since OEEF affects the oxyl-radical character, it should impact the thermodynamic properties of the CpdI and CpdII, such as BDFE(O–H) of CpdII, their electron affinities (reduction potentials), and proton affinities (pKAs) (Scheme 2). Although the overall picture of the high-valent metal-oxo reactivity is extraordinarily convoluted, it can be strongly hypothesized that LEF, on-par with the nature of the axial ligand and its hydrogen-bonding pattern, likely substantially contributes to the oxidative powers and selectivity of CpdI and CpdII, and hence the rates of individual transformations.
Scheme 2.
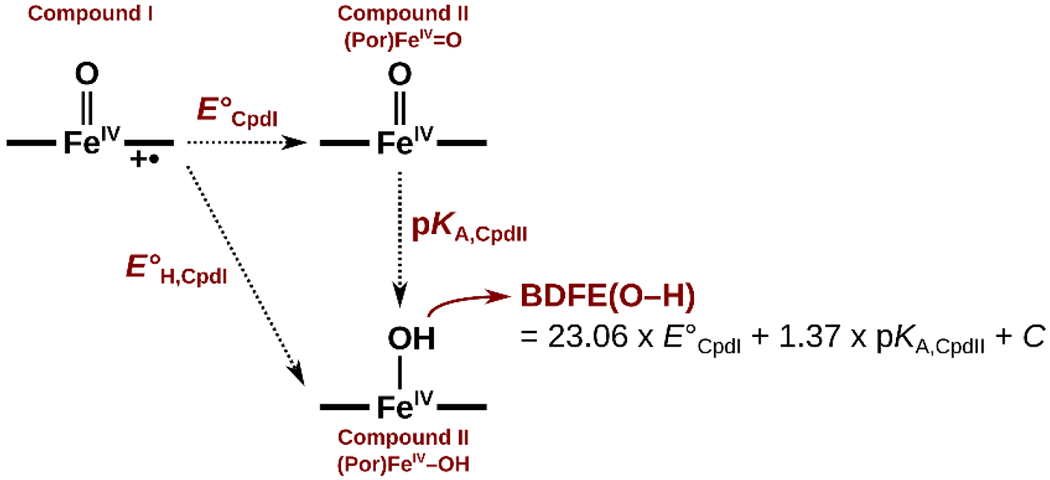
Interconversion of the oxygenated heme-iron CpdI and CpdII intermediates, and the associated thermodynamic quantities.
In this work, we first aim to elucidate what kinds of LEF (directions and magnitudes) the heme-iron sites experience inside the various natural oxidoreductases. We find a wide variety of LEFs, but such that they appear to correlate with the functions of these proteins, and specifically the reactivity of the presumptive CpdI or CpdII intermediates. Next, using the Cys-ligated cytochrome P450 monooxygenases, Tyr-ligated catalases, and His-ligated peroxidases as prototypical examples, we demonstrate that LEF steers the selectivity toward the productive substrate oxidation. Our findings infer that LEFs are indeed evolutionarily altered in heme-iron proteins to stir their reactivity, thus contributing to the understanding of the fundamental aspects of proteins operating through the metal-oxo intermediates.
RESULTS AND DISCUSSION
The local electric fields in the active sites of heme-iron oxidoreductases
We analyzed the crystal structures of ca. 200 individual Fe-containing heme-based oxidoreductases (EC number: 1): catalases, cytochrome P450 monooxygenases and peroxygenases, heme oxygenases, peroxidases, and others, and elucidated the direction and magnitude of the LEF at their Fe centers (see Computational Details section in the SI). Note that the evaluated fields are induced by the entire protein outside of the heme-iron site, i.e., excluding the effects of the site itself and the axial ligand. The axial ligands thus do not contribute to the discussed LEFs. Instead, the LEFs, as presented in the article, precisely correspond to the OEEFs previously applied to the whole ‘axial ligand–porphine–FeO’ models, and thus affect the reactivity on top the effect of the axial ligand. The electric fields in the x,y,z directions (cf. Figure 1) are summarized for each protein in Table S1 in the SI, whereas the proteins, grouped according to their EC numbers and the nature of their axial ligands, are listed in Figures 2–4 and Table 1.
Figure 1.
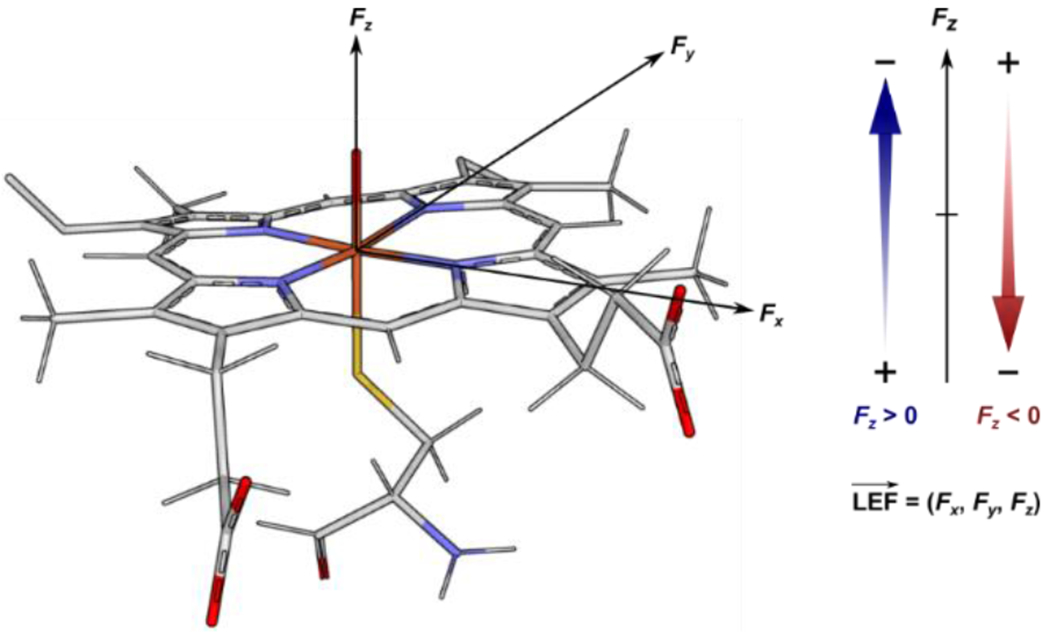
The orientation of the components of the electric field vector in the heme-iron model. The Fx and Fy components are oriented in the porphine plane and the Fe–NA and Fe–NB bonds of the porphyrin. The Fz component is orthogonal to the porphine plane, parallel with the Fe–O bond of the putative CpdI species.
Figure 2.
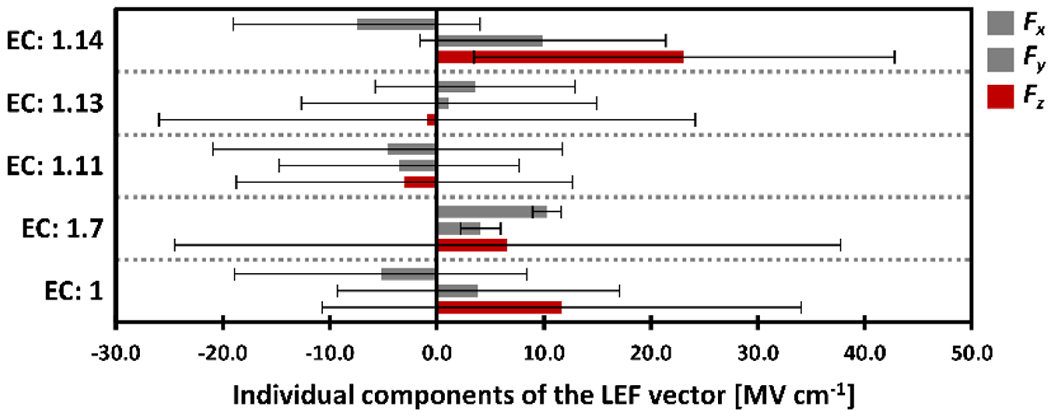
The average values of the Fx, Fy, and Fz components of the LEF vector among the subclasses of the oxidoreductase proteins (EC: 1). For each class, the LEF is displayed from top to bottom as the average Fx, Fy, and Fz components of the LEF vector in MV cm−1 shown as thick bars (Fz is highlighted in red), and the thin black lines represent the standard deviations, σ.
Figure 4.
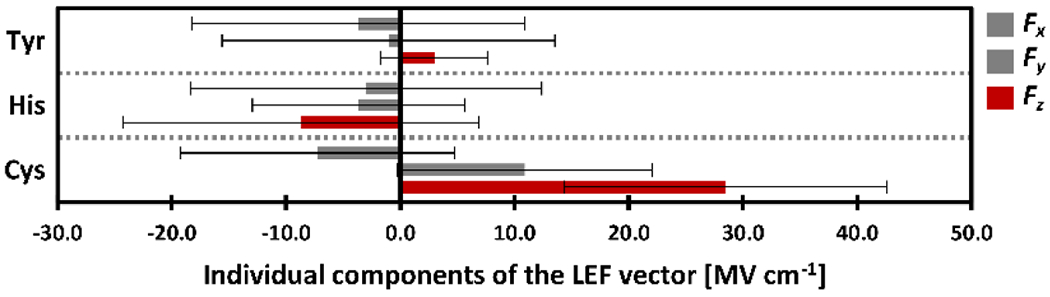
The average values of the Fx, Fy, and Fz components of the LEF vector among the oxidoreductase proteins (EC: 1) with Tyr-, His-, and Cys-axial ligation; average values are shown as thick bars, and thin lines represent standard deviations, σ. Recall that the axial ligand itself is not included in the calculations of LEF; hence, the axial ligand adds to the effect depicted here.
Table 1.
The average components of the LEF vector in the oxidoreductase proteins grouped according to the nature of the heme-iron axial ligand. The individual components of the LEFs are indicated for several examples in each of the groups, as discussed in the text.
| Average Fx [MV cm−1] |
Average Fy [MV cm−1] |
Average Fz [MV cm−1] |
|||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| All proteins | −5.2 ± 13.7 | 3.9 ± 13.2 | 11.7 ± 22.4 | ||||
|
| |||||||
| Cys-ligated | −7.2 ± 12.0 | 10.9 ± 11.1 | 28.5 ± 14.1 | ||||
|
| |||||||
| Examples: | PDB code | EC number | Function |
Fx [MV cm−1] |
Fy [MV cm−1] |
Fz [MV cm−1] |
|
| P. putida P450cam | 1dz9 a | 1.14.15.1 | monooxygenase | −5.1 | −0.3 | 25.2 | |
| S. coelicolor CYP158 | 2d09 | 1.14.19.69 | biflaviolin synthase | −9.4 | 8.3 | 25.7 | |
| H. sapiens CYP7A1 | 3v8d | 1.14.14.23 | monooxygenase | −19.5 | 14.6 | 55.8 | |
|
| |||||||
| Tyr-ligated | −3.7 ± 14.5 | −1.0 ± 14.6 | 3.0 ± 4.7 | ||||
|
| |||||||
| Examples: | PDB code | EC number | Function |
Fx [MV cm−1] |
Fy [MV cm−1] |
Fz [MV cm−1] |
|
| M. luteus KatA | 1gwf a | 1.11.1.6 | catalase | −11.0 | −17.9 | 4.1 | |
| P. mirabilis KatA | 1mqf a | 1.11.1.6 | catalase | −11.1 | −9.1 | 8.6 | |
| H. pvlori KatA | 2a9e a | 1.11.1.6 | catalase | −21.2 | −10.5 | 1.7 | |
| N. crassa KatA | 6nsw a | 1.11.1.6 | catalase | 12.2 | 19.7 | −1.3 | |
|
| |||||||
| His-ligated | −3.0 ± 15.4 | −3.7 ± 9.3 | −8.7 ± 15.6 | ||||
|
| |||||||
| Examples: | PDB code | EC number | Function |
Fx [MV cm−1] |
Fy [MV cm−1] |
Fz [MV cm−1] |
|
| S. cerevisiae CcP | 1ebe a | 1.11.1.5 | peroxidase | −22.1 | −4.8 | −26.7 | |
| A. rusticana HRP | 1hch a | 1.11.1.7 | peroxidase | −5.5 | −9.4 | −36.6 | |
| G. max APX | 5jqr a | 1.11.1.11 | peroxidase | −4.4 | 9.0 | 4.1 | |
| H. sapiens MPO | 5fiw | 1.11.2.2 | peroxygenase | −9.1 | −3.1 | 28.4 | |
| C. hircus LPO | 3rke | 1.11.1.7 | peroxidase | −15.1 | −23.1 | 12.4 | |
| A. ramosus ARP | 2e39 | 1.11.1.7 | peroxidase | −3.9 | −1.1 | −2.1 | |
| L. major LmP | 3riv | 1.11.1.11 | peroxidase | −11.5 | −9.7 | −15.5 | |
| B. pseudomallei KatG | 5sx0 a | 1.11.1.21 | catalase-peroxidase | −17.5 | −9.2 | −15.0 | |
Proteins were crystallized in the CpdI or CpdII form.
A marked preference for the LEF oriented in the z direction across the whole family of proteins is evident from Figure 2. The average values of LEF are Fx = −5.2 MV cm−1, Fy = 3.9 MV cm−1, Fz = 11.7 MV cm−1. A higher preference for the z orientation of the LEF is found for the proteins in the EC subclasses 1.7 (oxidoreductases acting on other nitrogenous compounds as donors), 1.13 (oxidoreductases acting on single donors with incorporation of molecular oxygen (oxygenases)), and 1.14 (oxidoreductases acting on paired donors, with incorporation or reduction of molecular oxygen), indicating a more distinct regulation of the enzymes’ catalytic power by the LEF, as compared to an electrostatically featureless, homogeneous environment. Note that proteins in the EC classes 1.7 and 1.13 have small average Fz (Figure 2), but this is due to the equally large preference for the high positive and high negative Fz (as embodied in the substantial standard deviations of the Fz in Figure 2). The highest average Fz is thus found in the subclass of EC 1.14: Fx = −7.5 MV cm−1, Fy = 9.9 MV cm−1, and Fz = 23.1 MV cm−1. This should come as no surprise, as this class comprises mostly the cytochrome P450 oxygenases, which activate the strong C–H bonds of organic substrates. The mostly positive Fz in this class of proteins (cf. Figure 3) adequately indicates a raised oxygenation reactivity, and thus strongly suggests the careful tuning of the LEF by the evolution. A large standard deviation of the Fz (in the EC: 1.14 class, the σ(Fz) = 19.7 MV cm−1) further suggests a fine-tuning of the proteins’ reactivity or selectivity of the individual proteins through Fz. As a prominent example, the cholesterol 7-alpha-hydroxylase (CYP7A1; PDB code: 3v8d), an important enzyme in the metabolism of cholesterol putatively operating through the CpdI intermediate, attains a strong Fz of 55.8 MV cm−1 (in contrast with the relatively weak Fx = −19.5 MV cm−1, and Fy = 14.6 MV cm−1; cf. Table 1).
Figure 3.
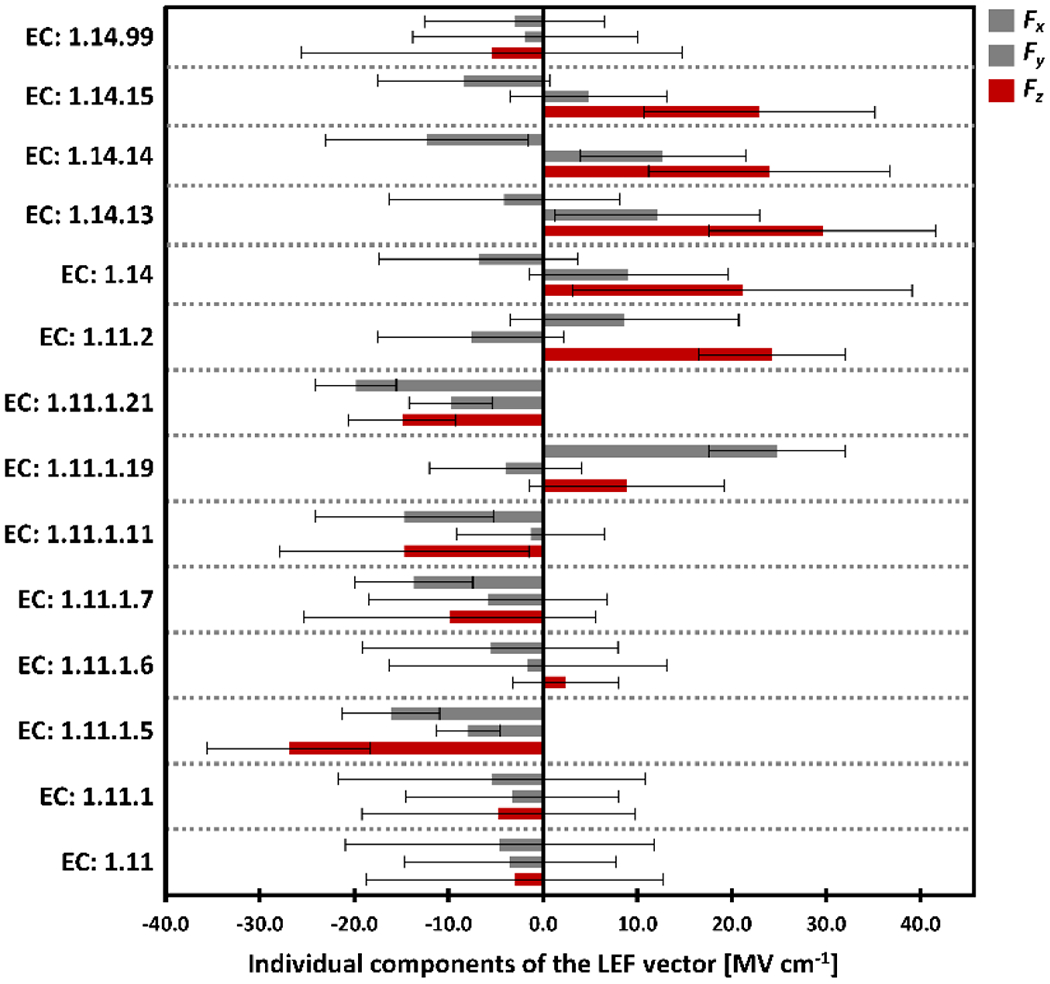
The average values of the Fx, Fy, and Fz components of the LEF vector among the subclasses of the oxidoreductase proteins: (i) acting on a peroxide as acceptor (EC: 1.11) and (ii) acting on paired donors, with incorporation or reduction of molecular oxygen (EC: 1.14); average values are shown as thick bars, and thin lines represent standard deviations, σ.
On the contrary, the oxidoreductases that act on the peroxide (EC 1.11 cf. Figure 2) show a much lower magnitude of the LEF vector with less strictly induced orientation parallel with the Fe–O axis. The average LEF components are: Fx = −4.6 MV cm−1, Fy = −3.5 MV cm−1, and Fz = −3.0 MV cm−1. Besides, Fz is preferably negative in the individual EC: 1.11 subclasses (Figure 3), and that is consistent with a lower reactivity of their CpdI intermediates that are not involved in the H-atom abstractions. In other words, lower Fz is related to a lower CpdI reactivity. This can be supported by the fact that the CpdI intermediates in catalases and peroxidases (both in EC: 1.11.1) were found to be more stable, and as such, more readily isolated in the past than CpdI intermediates in the P450 proteins.43
Importantly, in all of the proteins examined herein, there are only three different axial ligands present – Cys, Tyr, and His, and with certain simplification, we may associate each of the ligands with a specific protein function from Scheme 1. The catalase reactivity (EC: 1.11.1.6) is mostly associated with the Tyr-axial heme-iron ligation, the peroxidase reactivity (EC: 1.11.1., except for 1.11.1.6) - with the His ligation, and the oxygenase/peroxygenase reactivity (EC: 1.11.2. and 1.14, except for 1.14.99) - with the Cys ligation. Figure 4 reveals a pronounced correlation between the proteins LEFs and their function, explicitly evaluating the heme-iron axial ligation (see also Table 1). In particular, the preference for the z component of the LEF is increased in the Tyr < His < Cys series (Figure 4); while the Tyr-ligated proteins have a preference for slightly positive Fz, His-ligated proteins exhibit slightly negative Fz, and Cys-ligated proteins exhibit strictly positive and large Fz (Figure 4).
In detail, the Cys-ligated proteins – consisting mostly of the P450 oxygenases (EC: 1.14) and peroxygenases (EC: 1.11.2) – on average exhibit the Fz = 28.5 MV cm−1. From refs. 40,44, such LEF can be expected to decrease the H-atom-transfer activation barrier by ca. 4 kcal mol−1 owing to an increase in the oxyl-radical character of the FeIVO.
The Tyr-axial ligated proteins (i.e., catalases; EC 1.11.1.6), on the contrary, exhibit a very small field in the positive z orientation: Fz = 3,0 MV cm−1. However, the field in the Tyr-ligated proteins, though minimal in the z direction, is not randomly oriented, either. Instead, the low Fz appears to be genuinely induced by the protein fold, as manifested by the small standard deviation of the σ(Fz) = 4.7 MV cm−1, and compared with the standard deviation of the Fx and Fy components for the whole protein series: σ(Fx = 13.7 MV cm−1 and σ(Fx) = 13.2 MV cm−1, which suggest a greater randomness.
Finally, the His-ligated proteins contain mostly heme oxygenases (EC: 1.14.99) with the average LEF components of Fx = −3.3 MV cm−1, Fy = −2.1 MV cm−1, Fz = −5.9 MV cm−1, dioxygenases (EC: 1.13.11) with Fx = 5.2 MV cm−1, Fy = −2.2 MV cm−1, Fz = −9.5 MV cm−1, and peroxidases (EC: 1.11.1; except for 1.11.1.6) with Fx = −8.0 MV cm−1, Fy = −5.9 MV cm−1, Fz = −9.5 MV cm−1. Noticeably, there is a certain preference for the negative Fz (ca. 71 % of a total of 70 studied His-ligated proteins exhibit negative Fz; see Table S1 in the SI), contradicting the higher oxyl-radical character of the CpdI. Recall that Fz in the Cys-ligated proteins (such as the P450 oxygenases/peroxygenases) is positive in 100 % of 95 of the studied cases. Fz in the His-ligated proteins ranges from Fz = −37 MV cm−1 to Fz = +49 MV cm−1, thus, suggesting a possibility of tuning the CpdI reduction potentials both in a positive and negative direction over a large total range of ca. 500 mV (vide infra), which might be important for activation (oxidation) of a manifold of substrates. This apparent tuning of redox properties is illustrated in Figure 5 (left), correlating the experimental E°cpdI values of various His-ligated peroxidases45 and their LEF Fz established in this work. Specifically, myeloperoxidase (MPO) has E°cpdI = 1.35 V, and Fz = +28.4 MV cm−1; ascorbate peroxidase (APX) has E°cpdI = 1.16 V, and Fz = +4.1 MV cm−1; lactoperoxidase (LPO) has E°cpdI = 1.14 V, and Fz = +12.4 MV cm−1; arthromyces ramosus peroxidase (ARP) has E°cpdI = 0.92 V, and Fz = −2.1 MV cm−1; horseradish peroxidase (HRP) has E°cpdI = 0.90 V, and Fz = −36.6 MV cm−1).
Figure 5.
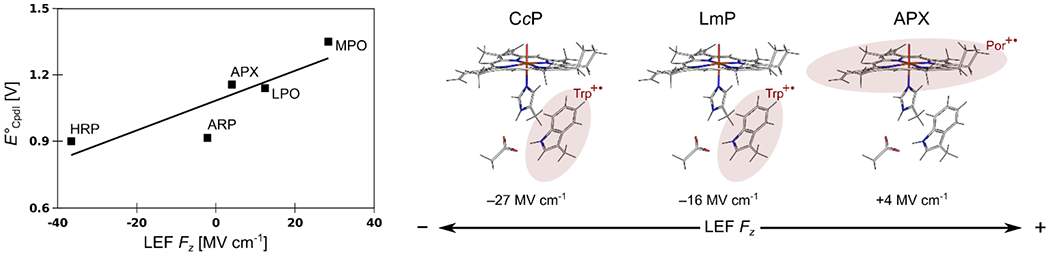
Left: Experimental E°cpdI electrochemical potentials of various peroxidases plotted against the calculated Fz components of their LEFs. Right: Different electronic structure of the (Trp+•)FeIVO CpdI in CcP and LmP vs. (Por+•)FeIVO CpdI in APX as observed in Nature can be associated with the change in the proteins Fz component of the LEF vector. The unpaired radical is shifted from the Trp+• to Por+• as Fz becomes more positive.
At last, the cytochrome c peroxidase (CcP) and leishmania major peroxidase (LmP) exhibit LEF quite different from that in the ascorbate peroxidase (APX), which is otherwise nearly homologous at the active site (Table 1).§ Since the two groups of enzymes have different electronic structures (e.g., the unpaired electron located on Trp+• in CcP and LmP, and on Por+• in APX; cf. Figure 5, right), this finding suggests LEF playing a role in tipping the balance between the two resonant forms. Noticeably, the higher Fz component of the LEF observed in the APX enzyme is indeed consistent with the relocation of the unpaired radical toward the Por+• (vide infra).
The effect of LEF on the physicochemical properties of the CpdI and CpdII in the cytochrome P450 oxygenase, catalase, and peroxidase model systems
The ground spin state of the CpdI can be either quartet (S = 3/2) or doublet (S = 1/2), originating from the ferromagnetic or antiferromagnetic coupling of the ‘Fe–O’ triplet spin state and the unpaired electron located on (i) the porphine ring (Por), (ii) the axial ligand (Ax), or (iii) the adjacent Trp residue in case of the peroxidase model; cf. Figure 5, right. Employing the model systems constructed from the P450 monooxygenase (P450cam), catalase (from P. mirabilis), and peroxidase (APX) protein crystal structures (Figure S1), we next compare the electronic structure properties of their CpdI intermediates. In all of the models, density functional theory, DFT (B3LYP method; see Computational Details in the SI) predicts the quartet and doublet spin states to be nearly degenerate, lying within ca. 1 kcal mol−1 in free energy. In detail, the preferred configuration is calculated to be quartet in Cys- and Tyr-ligated models (by 0.7 kcal mol−1 in both) and doublet in His-ligated model (by 0.3 kcal mol−1). It is evident from the Mulliken spin densities in Table 2 that the radical in CpdI is preferentially located on the axial ligands (or Trp). However, it is also partially displaced to the porphine ring, and that is consistent with an operable mixing of the two resonance forms of the CpdI state, suggested to be a part of tuning of the CpdI properties in the P450 catalysis.46,47 Note that Mulliken spin densities can be only qualitative, due to the DFT delocalization error, which is known to overstabilize the radical-delocalized states.48
Table 2.
The Mulliken spin densities of selected atoms and Fe–O bond distance of the CpdI and CpdII intermediates of the heme-iron P450 monooxygenase, catalase, and peroxidase models calculated at the DFT (B3LYP) level of theory in the zero field (see the Computational Details section in the SI).
| CpdI FeIV=O (S = 1/2) |
CpdI FeIV=O (S = 3/2) |
CpdII (Por)FeIV=O (S = 1) |
CpdII (Por)FeIV-OH (S = 1) |
|
|---|---|---|---|---|
|
Cys-ligated P450 monooxygenase
| ||||
| Fe [e] | 1.24 | 1.22 | 1.32 | 1.82 |
| O [e] | 0.82 | 0.85 | 0.81 | 0.16 |
| Por [e] | −0.33 | 0.24 | −0.11 | −0.14 |
| Cys [e] | −0.68 | 0.67 | −0.02 | 0.15 |
| Fe–O dist. [Å] | 1.65 | 1.66 | 1.69 | 1.84 |
|
| ||||
|
Tyr-ligated catalase
| ||||
| Fe [e] | 1.27 | 1.26 | 1.32 | 1.83 |
| O [e] | 0.78 | 0.82 | 0.77 | 0.13 |
| Por [e] | −0.33 | 0.18 | −0.10 | −0.13 |
| Tyr [e] | −0.72 | 0.74 | 0.01 | 0.17 |
| Fe–O dist. [Å] | 1.63 | 1.65 | 1.67 | 1.82 |
|
| ||||
|
His-ligated peroxidase
| ||||
| Fe [e] | 1.26 | 1.25 | 1.28 | 1.90 |
| O [e] | 0.84 | 0.84 | 0.82 | 0.13 |
| Por [e] | −0.41 | 0.27 | −0.09 | −0.05 |
| Trp [e] | −0.67 | 0.65 | 0.00 | 0.03 |
| Fe–O dist. [Å] | 1.65 | 1.66 | 1.66 | 1.81 |
A one-electron reduction of the (Trp+•/Ax+•/Por+•)FeIV=O CpdI results in the formation of the FeIV=O CpdII in the triplet (S = 1) spin state in all of the cases, where the additional electron is paired with the Trp+•/Ax+•/Por+• radical, not changing the electronic distribution between the Fe and O atoms significantly. In turn, this may be responsible for a small change in the Fe–O bond distance, as also observed in the experiments.49 On the contrary, a transfer of an H atom (i.e., proton and electron pair) to CpdI yields a protonated FeIV-OH CpdII species with a significantly elongated Fe–O bond and electron density that is displaced from the O atom toward the Fe center (cf. Table 2).
The calculated E°H,CpdI potentials of 1.28 V (Cys-ligated heme) and 0.88 V (His-ligated heme) are in excellent agreement with the measured potentials in refs. 45,50 (cf. Cys: E°H,CpdI = 1.28 V vs. 1.22 V for CYP158 in ref. 50; His: E°H,CpdI = 0.88 V vs. ~0.9-1.2 V for various peroxidases with the same His-Asp-Trp/Phe triad in ref. 45). Also, the calculated E°H,CpdI = 1.18 V of the catalase model seems to be realistic, as the electrochemical properties of the negatively charged Cys and Tyr axial ligands in the P450 and catalase enzymes were suspected to be of similar magnitude;51,52 however, since the CpdII is not a native intermediate in the catalase’s enzymatic cycle,53 the E°H,CpdI has no practical (biological) relevance. The calculated values of the E°H,CpdI potentials are also consistent with the highest reactivity of the CpdI in the P450 enzyme and correspond to a decreasing BDFE(O–H) of ca. 97 kcal mol−1 (Cys) > 94 kcal mol−1 (Tyr) > 88 kcal mol−1 (His). We also note that the calculated proton affinities of CpdII are consistent with the trend in the experimentally obtained pKA,CpdII; cf. the proton affinities of −316 kcal mol−1 (Cys), −310 kcal mol−1 (Tyr), and −293 kcal mol−1 (His) vs. the pKA,CpdII of ~12 (CYP158)51, ~13 (H. pylori catalase)52, and ≤4.5 (APX)54. Similarly, the electron affinities of CpdI give the opposite trend of −88 kcal mol−1 (Cys), −91 kcal mol−1 (Tyr), and −101 kcal mol−1 (His).
By applying the oriented external electric field (OEEF) in the Fe–O direction, we next evaluate how the specific values of Fz actually attainable in the proteins impact the electronic structure and physicochemical properties of each proteins’ CpdI. First, the OEEF in the positive Fe–O direction results in the relocation of the radical toward Por+• (Figure 6), as was already described in ref. 40. Besides others, this allows to quantify the effect of the LEF on the peroxidases proteins, CcP, LmP, and APX, and their electronic structure; e.g., the location of the radical in the CpdI intermediates. These peroxidases differ in LEF Fz by ca. 31 MV cm−1 (going from Fz = −26.7 MV cm−1 in CcP to Fz = +4.1 MV cm−1 in APX). Correspondingly, from applying the OEEF of the same magnitudes and directions, we predict the spin densities to be ca. 0.96/0.09 e on Trp/Por in both CcP and LmP, and 0.47/0.59 e on Trp/Por in APX. Therefore, ca. 0.5 e can be expected to transfer from Trp to Por due to the effect of LEF in these proteins.
Figure 6.
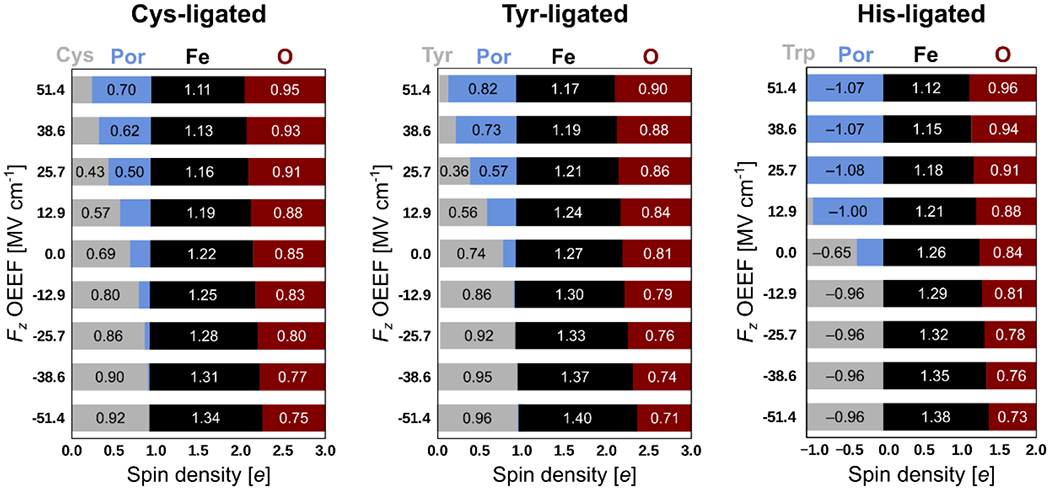
Progression of the Mulliken spin densities in the CpdI intermediates of Cys-, Tyr-, and His-ligated heme-iron models with the change of the Fz component of OEEF.
Additionally, the Fe–O oxyl-radical character is increased as the effect of a higher Fz (cf. the Mulliken spin density progression at the O atom in Figure 6), leading to a higher CpdI electrophilicity. In detail, we observed an increase in the E°H,CpdI of several hundreds of millivolts when the Fz of +0.01 a.u. was applied (= 51.4 MV cm−1; roughly corresponding to the highest LEF observed in the heme-iron oxidoreductases enzymes; vide supra). Since the increase in the E°H,CpdI is equivalent to an increase in the BDFE(O–H), this finding has an important implication for the reactivity of the heme-iron proteins, as it provides a recipe for how the proteins can utilize their sequences and folds to tune BDFE(O–H) over a broad range, while keeping the same heme-CpdI first-shell sphere of ligands. In the presented models, the E°H,CpdI and BDFE(O–H) increase over the range of ca. 10-12 kcal mol−1 as the Fz is altered from −0.01 a.u. to +0.01 a.u. (Figure 7), indicating that change of the E°H,CpdI of 100 mV is induced by the increase in Fz of ca. 20-24 MV cm−1.
Figure 7.

Progression of the BDFE(O–H) in the Cys-, Tyr-, and His-ligated heme-iron models with a change of the Fz component of OEEF. The average values of the LEF Fz observed in the Cys-, Tyr-, and His-ligated proteins in Nature are indicated by dashed vertical lines. Note that the thermal enthalpic, and entropic contributions to the change of the free energy were neglected (see SI for Computational Details).
In more detail, the BDFE(O–H) change with the OEEF is, in fact, a product of the uneven effect of the OEEF on E°CpdI and pKA,CpdII (Figure 8); note that BDFE(O–H) = 23.06 x E°CpdII + 1.37 x pKA,CpdII + 57.6 kcal mol−1. In all three models, E°CpdII and pKA,CpdII exhibit the opposite response to the field: growth in Fz increases E°CpdII and decreases pKA,CpdII, but pKA,CpdII is approximately half as sensitive. Utilizing Cys-ligated model as an example, we note that as Fz increases by 0.01 a.u., E°CpdII increases by 0.38 V (8.7 kcal mol−1), whereas pKA,CpdII decreases by only 4 units (5.5 kcal mol−1). We expect that this differential tuning of E°CpdII and pKA,CpdII by the LEF could also be utilized by the enzymes, to steer the asynchronicity in the H-atom abstraction between the proton and electron transfers, to achieve a higher reactivity/selectivity.13,55
Figure 8.
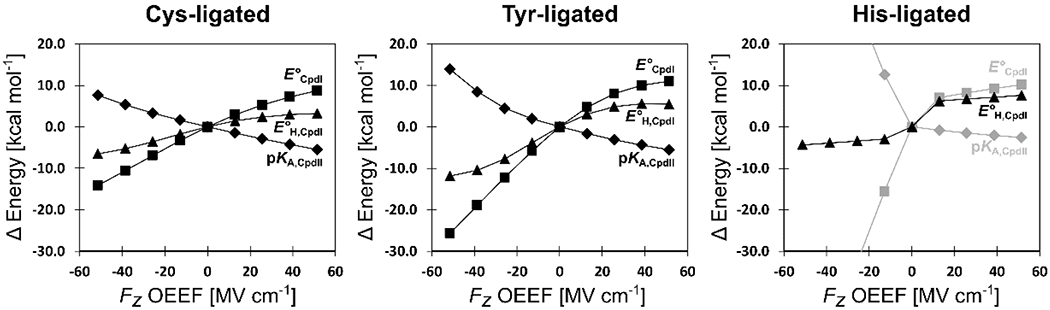
Progression of the physicochemical properties from Scheme 2 of the Cys-, Tyr-, and His-ligated heme-iron models with a change of the Fz component of OEEF. Note that the thermal enthalpic and entropic contributions to the change of the free energy were neglected (see SI for Computational Details).
Finally, we note that the computed properties of all three models (E°CpdII, E°H,CpdI, and pKA,CpdII) respond similarly to positive Fz (Figure 8). However, DFT fails dramatically for E°CpdIdi and pKA,CpdII in the negative Fz, for the His-ligated model. Since the progression of the E°H,CpdI seems to be unaffected by the field orientation, such a failure can be attributed to the incorrect description of the non-protonated FeIVO CpdII state (cf. Scheme 2). Indeed, while the energies of all of the other states from Scheme 2 decrease as the OEEF is applied in either direction (Figure 9), the behavior of His-ligated FeIVO CpdII diverges, showing the largest energy at Fz of ca. −40 MV cm−1. A deviation from (Trp+•/Por+•)FeIV=O CpdI and FeIVOH CpdII thus explains a rational behavior of the E°H,CpdI, but an extraordinary unphysical description of the E°CpdI and pKA,CpdII.
Figure 9.

A: Relative change in the energy of the individual states from Scheme 2 with the variation of the OEEF Fz. The CpdI and FeIVOH CpdII intermediates get stabilized in the positive or negative Fz. In the His-ligated model, the behavior of FeIVO CpdII deviates significantly from the trend. B: Evolution of the Por a1u and πTrp Kohn-Shamm orbital energies in FeIVO CpdII with the change of Fz. We note that the change in E°CpdI (red curve) calculated directly from the energies of the CpdI and FeIVO CpdII intermediates corresponds well with the electron affinity (gray curve) estimated from the simple change in the HOMO energy of the FeIVO CpdII. The change in the HOMO character from πTrp to Por a1u is thus suspected for the unphysical calculated electrochemical behavior in negative Fz.
As analyzed in detail in Figure 9 (see also Supplementary Note S1 in the SI), we assign the failure to an increased multi-reference character and/or improper description of the charge delocalization in the FeIVO CpdII state in the applied field. Although the HOMO (πTrp) and HOMO-1 (a1u) are nearly degenerate in the FeIVO CpdII in the zero field, OEEF breaks the degeneracy, and influences these orbitals very differently. While the Por a1u and a2u Kohn-Sham orbital energies are only slightly altered by the field in both positive and negative direction (the energy is decreased by ~20 kcal mol−1 under OEEF increasing from −0.01 a.u. to +0.01 a.u.), the Trp orbital energy changes dramatically, by as much as 103 kcal mol−1. The HOMO character thus inverts from πTrp in the negative fields to Por a1u in the positive fields, and that crossover introduces complications for single-determinant DFT. The result is insensitive to basis set, type of DFT functional, or change in spin state (see Supplementary Note S1 in the SI). Hence, the asymmetry in the E°cpdI progression at different fields is seen (Figure 8). Finally, to test the hypothesis regarding the multi-reference character, we have evaluated the OEEF effect on the truncated model not including the Trp and Asp residues (Figure S2). This remedied the unphysical behavior in the negative field, but the calculations yield a defective behavior in the positive field, as a result of the incomplete representation of the system. In conclusion, it appears that DFT cannot accurately predict how the thermodynamic properties of the His-ligated peroxidases change with the applied field, but the trends are qualitatively the same for all three model systems. The increase in Fz results in increase of both electron-transfer and proton-coupled electron-transfer potentials, and decrease in the pKA.
Discussion on the heme-iron proteins selectivity related with the local electric fields in their active sites
Comparing the large Fz in the P450 monooxygenases with the intermediate or even negative Fz in some catalases and peroxidases (e.g., ΔFz ~ 95 MV cm−1 between cholesterol 7-alpha-hydroxylase and HRP; vide supra) leads us back to a long-standing question: Why did Nature select a strong electron-donating Cys axial ligand for her most oxidizing heme-iron enzymes?56 We see that LEF in both Cys- vs. His-ligated enzymes can considerably reduce the effect of the axial ligand, bringing the systems closer to oxidative behavior. That is, high Fz in the Cys-ligated cytochrome P450 enzymes (as compared to the lower/negative Fz in peroxidases) increases the electrophilicity of CpdI and decreases the basicity of CpdII (cf. Figure 8), and that eventually “undermines” the effect of the thiolate ligation.8 Specifically, the average fields observed in both protein groups (i.e., Fz = 28.5 MV cm−1 in Cys-ligated vs. Fz = −8.7 MV cm−1 in His-ligated proteins) should be responsible for the shift of ~300–400 mV in E°CpdI and ~3 units in pKA,CpdII. Nevertheless, the experimentally obtained values of reduction potentials and basicities of CpdI and CpdII still demonstrate the significantly higher basicity of the Cys-ligated systems. This was envisaged by Green, who proposed a two-fold effect in the P450 catalysis:51 (i) a negative shift of the reduction potential (as compared to the His-ligated system) is compensated by the increase in the PKA by approximately five units, eventually strengthening the driving force for the H-atom abstraction, via a higher contribution of the basicity component to the BDFE(O–H); and (ii) a lower reduction potential suppresses the unwanted auto-oxidations of the fragile protein framework. Our results from the previous sections, however, suggest that the situation may become much more complicated in the context of the LEF working unequally on the E°CpdI and pKA,CpdII components of the BDFE(O–H).
Specifically, a finding that BDFE(O–H) is additionally increased in the P450 proteins as the effect of high Fz accords with the high reactivity toward the C-H abstractions. However, we also propose that there must exist both upper and lower limits of Fz for the P450 catalysis to occur, due to the trade-off between productive selectivity and sufficient reactivity (see Scheme 3). Following the work of Green, we suggest the upper limit is represented by the increase of the rate of the non-productive oxidation pathway of the protein framework (for the CYP158 protein studied in ref. 51, such non-productive pathway corresponds to the oxidation of the adjacent Tyr residues), which is elevated by decreasing the PKA of the CpdII as Fz increases. A ratio between the non-productive oxidation rate by two different hypothetical CpdI species in otherwise identical systems was derived as: which suggests that an increase in Fz by ca. 17 MV cm−1 should result in an increase in the rate of the non-productive oxidation by one order of magnitude (cf. change in the pKA,CpdII in Figure 8, left). On the contrary, a lower/negative Fz would lead to a higher ratio between the productive and non-productive reactivity owing to the increase in the CpdII basicity. However, given the uneven effect of the field on the E°CpdII and pKA,CpdII, this would simultaneously decrease BDFE(O–H) and, therefore, result in a decreased overall catalytic efficiency. Note that free energy of the reaction is given by: ΔGproductive = BDFE(C–H) − BDFE(O–H); thus, the low reactivity in the productive pathway represents a lower bound of the practicable Fz. All in all, the idea of the confinement of the range of Fz can be supported, e.g., by the positive orientation of the LEF along the Fe–O axis in 100 % of cases in Cys-ligated proteins (ranging from ca. 3 MV cm−1 to 58 MV cm−1), or by a very narrow range of Fz in the catalases proteins, manifested by a standard deviation of σ(Fz) = 4.7 MV cm−1.
Scheme 3.
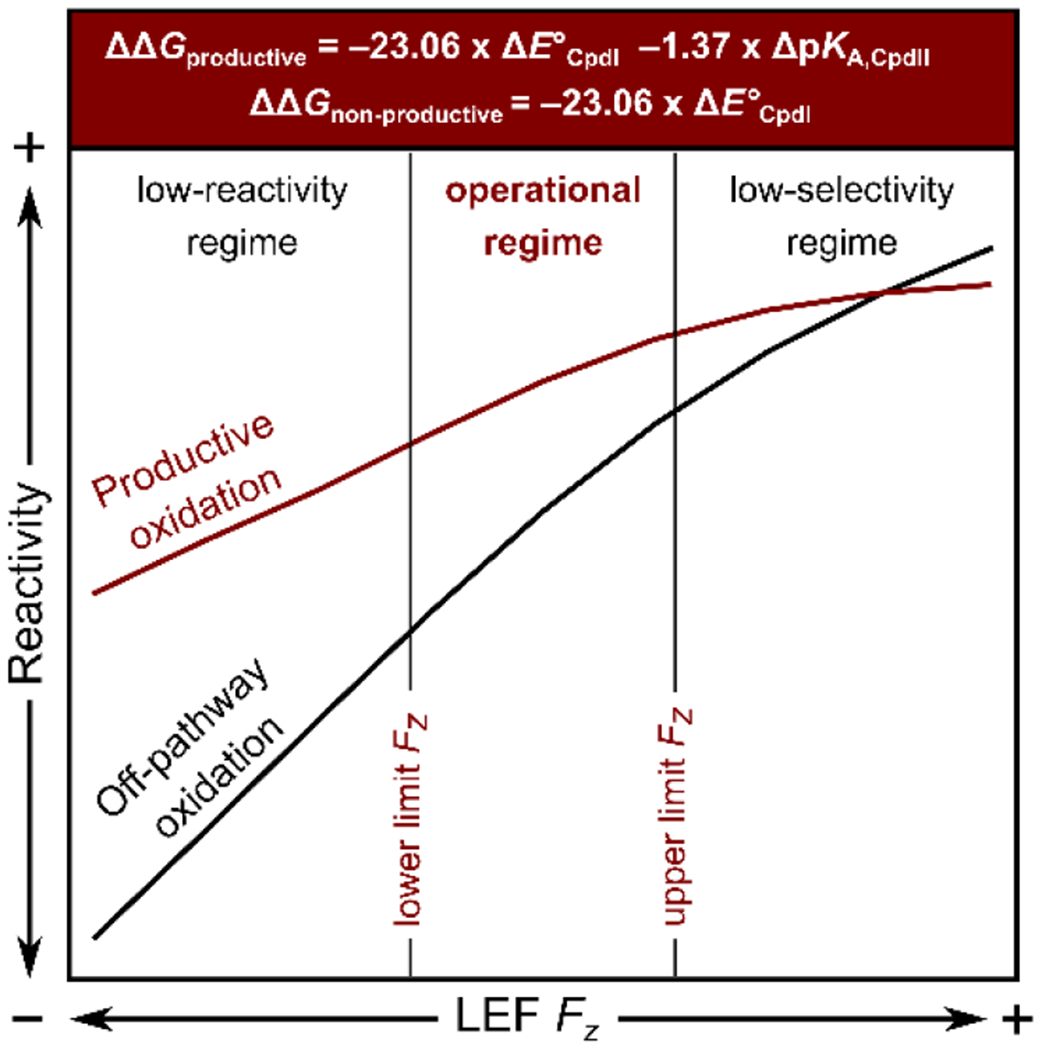
A specific range of the LEF Fz is required to be adopted by a protein to accomplish its biological function. The lower and upper bounds of the Fz are given by the counterbalance between the low reactivity on the one side and the low selectivity on the other. Note that reactivity in both productive and non-productive oxidations in the H-atom abstractions is primarily governed by the E°CpdI term, while the steepness with respect to the Fz is decreased in the productive pathway due to the opposite response of the pKA,CpdII.
We may also speculate that other steps in the catalytic cycles (even those that are not directly connected with the CpdI or CpdII intermediates) are indirectly linked with the Fz component of the LEF, which may be required to adopt a precisely tuned value for the enzyme canonical function. For example, a decrease in the E°H,CpdI and E°CpdI potentials at lower Fz would also likely affect the Fe3+/2+ reduction potential of the enzyme’s resting state, suppressing the electron delivery from the NAD(P)H, which is a prerequisite for the dioxygen binding. Additionally, as suggested by Winkler and Gray, the failure in the substrate oxidation must be under specific time constraints remedied by an electron transfer to the CpdII to regenerate the secure resting state of the enzyme.57–59 Such protection of the protein framework was proposed to be accomplished by the adjacent Tyr or Trp residues, which need to provide reducing equivalents near the CpdII reduction potential. Arguably, the E°CpdII is also tightly coupled with the Fz. Finally, we suggest that proteins’ selectivity can be steered by regulating the disparity between E°CpdI and pKA,CpdII via introducing the asynchronicity in the proton/electron transfer in the H-atom abstraction.13,55
CONCLUSIONS
Utilizing a number of computational approaches on the small model systems, it was previously recognized that relatively large oriented electric fields could significantly contribute to the heme-iron reactivity and selectivity. However, a comprehensive association with the field magnitudes inside the proteins’ active sites was missing, and the true accessibility of such fields in Nature remained unquestioned. Herein, we bridge the pure computational studies to enzymes, and analyze the electric fields actually present in a wide variety of heme-iron proteins. The local electric fields in proteins appear to be of lower magnitude than studied in small models (the average fields are of ~ 20 MV cm−1), suggesting that the possibility of completely reverting the reaction selectivity (as was done in a small computational model) would be rather difficult. However, we demonstrate that the local electric fields can still significantly contribute to several key mechanistic aspects.
Firstly, the field in enzymes is seen not to be randomly oriented, and indeed exhibit a preferred direction along the Fe–O bond of the supposed CpdI FeIVO intermediate. This is consistent with the alteration in the oxyl-radical character of the oxo group, modifying the CpdI reactivity/selectivity. The finding suggests, there is indeed a functional role of the field invoked by Nature.
The field of the highest magnitude and preferred orientation along Fe–O is observed in the Cys-ligated heme-iron proteins, such as P450 oxygenases and peroxygenases. That is consistent with the enhancement of their H-atom-abstraction reactivity as opposed to the strong electron-donation ability of their Cys-axial ligation. We have evaluated that oriented external electric field can increase the bond-dissociation free energy of the FeIVO–H bond of CpdII (i.e., BDFE(O–H) in Scheme 2) by ca. 3 kcal/mol, contributing to the thermodynamic driving force for the H-atom-transfer reactions. Importantly, we noticed that an increase in the BDFE(O–H) is associated with the uneven effect of the field on E°CpdI and pKA,CpdII. Consequently, the rise in the H-atom-transfer reactivity is followed by a decrease in the pKA,CpdII, concurrently aggravating the chemistry toward the off-pathway oxidation. In detail, the increase in the BDFE(O–H) by 3 kcal/mol is associated with the decrease of pKA,CpdII by 4 units, along with the 100x acceleration of the side redox reactions. We thus suggest there must exist an upper bound of the practicable Fz, given by the increase of the side redox reactivity; and the lower bound Fz, provided by the low overall thermodynamic efficiency (Scheme 4; left).
Scheme 4.

The average Fz is indicative of the CpdI reactivity, i.e., the fact that Cys-ligated proteins report the highest Fz is consistent with their ability to activate strong C–H bonds, as compared to a lower CpdI reactivity in Tyr-ligated and His-ligated oxidoreductases; cf. average Fz = 28.5 MV cm−1, 3.0 MV cm−1, and −8.7 MV cm−1 for Cys-, Tyr-, and His-ligated proteins. In each of these protein classes, the operational LEF Fz range is given by the trade-off between the low reactivity of the CpdI intermediate at the more negative Fz and the low selectivity of the reactions at the more positive Fz.
We observed fields of intermediate magnitudes within the His-ligated heme-iron proteins, which are additionally oriented either in the positive, or in the negative direction with respect to the Fe–O bond (Scheme 4; right). While the intermediate magnitudes of the field correspond with the lower reactivity of the CpdI intermediate as compared with the Cys-ligated proteins (i.e., electron transfer vs. H-atom-transfer reactivity), the greater range of Fz suggests the higher tunability of the thermodynamic properties. In view of the peroxidases subclass of proteins, we successfully correlated the observed local electric fields in the active sites with the experimentally determined CpdI reduction potentials. In addition, we believe that the observed fields in various peroxidases, such as cytochrome c peroxidase (CcP), leishmania major peroxidase (LmP), and ascorbate peroxidase (APX), can bring some understanding into the dilemma of the different resonance forms embraced by these proteins. In Nature, the CcP and LmP have unpaired electron located on the adjacent Trp residue and coupled with FeO in Cpd I, whereas APX has a different electronic structure with the unpaired electron located on the porphine ring. We show that the local electric field identified in the peroxidases is consistent with this picture, and is responsible for ~0.5 e spin density translocation from Trp to Por in going from CcP and LmP to APX.
Finally, the lowest magnitude of the field, reporting no preference for the orientation along Fe–O, was observed in the Tyr-ligated heme-iron catalases. The lower magnitude is again consistent with the lower reactivity of the CpdI in catalases, as compared to cytochromes P450. On the other hand, the narrowest range of the LEF Fz in catalases might arise from the strict requirement of the proteins to hold in check the low-enough E°CpdI so that the single-electron pathway and the formation of the CpdII intermediate is suppressed, and the high-enough reactivity of the Fe3+ resting state to alter between the native ferricatalase and CpdI.
To conclude, although the observed LEFs are not so strong as to divert the reactivity and/or selectivity of the heme-iron enzymes (as was previously inferred from the theoretical investigations of the small model systems) they can significantly contribute to many aspects of the heme-iron chemistry. Hence, LEFs inside the proteins’ active sites are an integral part of the metal-oxo reactivity, and as such, they should be included to properly describe the electronic structure, physicochemical properties, and reaction mechanisms of these proteins.
Supplementary Material
ACKNOWLEDGMENTS
Financial support comes from the NSF CHE-1903808 and NIH 1R01GM134047 grants to A.N.A.. We also acknowledge the UCLA-IDRE cluster Hoffman2 and XSEDE for computational resources.
Footnotes
The situation does not change by introducing the K+ cation in the APX enzyme, which is present in the crystal structure, but was omitted in the LEF analysis. We see almost no difference in the LEF component in the z direction (cf. Fz = 4 MV cm−1 without the K+ ion vs. Fz = 2 MV cm−1 with the K+ ion).
REFERENCES
- (1).Guo M; Corona T; Ray K; Nam W Heme and Nonheme High-Valent Iron and Manganese Oxo Cores in Biological and Abiological Oxidation Reactions. ACS Cent. Sci 2019, 5 (1), 13–28. 10.1021/acscentsci.8b00698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ghosh A High-Valent Iron Intermediates in Biology. J. Inorg. Biochem 2006, 100 (4), 419–420. 10.1016/j.jinorgbio.2006.03.001. [DOI] [PubMed] [Google Scholar]
- (3).Sahu S; Goldberg DP Activation of Dioxygen by Iron and Manganese Complexes: A Heme and Nonheme Perspective. J. Am. Chem. Soc 2016, 138 (36), 11410–11428. 10.1021/jacs.6b05251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Krebs C; Galonić Fujimori D; Walsh CT; Bollinger JM Non-Heme Fe(IV)-Oxo Intermediates. Acc. Chem. Res 2007, 40 (7), 484–492. 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Huang X; Groves JT Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev 2018, 118 (5), 2491–2553. 10.1021/acs.chemrev.7b00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Moody PCE; Raven EL The Nature and Reactivity of Ferryl Heme in Compounds i and II. Acc. Chem. Res 2018, 51 (2), 427–435. 10.1021/acs.accounts.7b00463. [DOI] [PubMed] [Google Scholar]
- (7).Poulos TL Heme Enzyme Structure and Function. Chem. Rev 2014, 114 (7), 3919–3962. 10.1021/cr400415k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sono M; Roach MP; Coulter ED; Dawson JH. Heme-Containing Oxygenases. Chem. Rev 1996, 96(7), 2841–2888. 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- (9).Mayer JM Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res 2011, 44 (1), 36–46. 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Dietl N; Schlangen M; Schwarz H Thermal Hydrogen-Atom Transfer from Methane: The Role of Radicals and Spin States in Oxo-Cluster Chemistry. Angew. Chemie Int. Ed 2012, 51 (23), 5544–5555. 10.1002/anie.201108363. [DOI] [PubMed] [Google Scholar]
- (11).Lai W; Li C; Chen H; Shaik S Hydrogen-Abstraction Reactivity Patterns from A to Y: The Valence Bond Way. Angew. Chem. Int. Ed. Engl 2012, 51 (23), 5556–5578. 10.1002/anie.201108398. [DOI] [PubMed] [Google Scholar]
- (12).Saouma CT; Mayer JM Do Spin State and Spin Density Affect Hydrogen Atom Transfer Reactivity? Chem. Sci 2014, 5 (1), 21–31. 10.1039/C3SC52664J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bím D; Maldonado-Domínguez M; Rulísek L; Srnec M Beyond the Classical Thermodynamic Contributions to Hydrogen Atom Abstraction Reactivity. Proc. Natl. Acad. Sci. U S. A 2018, 115 (44), E10287–E10294. 10.1073/pnas.1806399115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lewis JC; Coelho PS; Arnold FH Enzymatic Functionalization of Carbon-Hydrogen Bonds. Chem. Soc. Rev 2011, 40 (4), 2003–2021. 10.1039/c0cs00067a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Warren JJ; Tronic TA; Mayer JM Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev 2010, 110 (12), 6961–7001. 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Rittle J; Green MT Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science (80-. ) 2010, 330 (6006), 933–937. 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- (17).Warshel A Electrostatic Origin of the Catalytic Power of Enzymes and the Role of Preorganized Active Sites. J. Biol. Chem 1998, 273 (42), 27035–27038. 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- (18).Warshel A; Sharma PK; Kato M; Xiang Y; Liu H; Olsson MHM Electrostatic Basis for Enzyme Catalysis. Chem. Rev 2006, 106 (8), 3210–3235. 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- (19).Fried SD; Boxer SG Electric Fields and Enzyme Catalysis. Annu. Rev. Biochem 2017, 86 (1), 387–415. 10.1146/annurev-biochem-061516-044432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Layfield JP; Hammes-Schiffer S. Calculation of Vibrational Shifts of Nitrile Probes in the Active Site of Ketosteroid Isomerase upon Ligand Binding. J. Am. Chem. Soc 2013, 135 (2), 717–725. 10.1021/ja3084384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hennefarth MR; Alexandrova AN Direct Look at the Electric Field in Ketosteroid Isomerase and Its Variants. ACS Catal 2020, 10 (17), 9915–9924. 10.1021/acscatal.0c02795. [DOI] [Google Scholar]
- (22).Kraut DA; Sigala PA; Pybus B; Liu CW; Ringe D; Petsko GA; Herschlag D Testing Electrostatic Complementarity in Enzyme Catalysis: Hydrogen Bonding in the Ketosteroid Isomerase Oxyanion Hole. PLOS Biol. 2006, 4 (4), e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Warshel A; Sharma PK; Chu ZT; Åqvist J Electrostatic Contributions to Binding of Transition State Analogues Can Be Very Different from the Corresponding Contributions to Catalysis: Phenolates Binding to the Oxyanion Hole of Ketosteroid Isomerase. Biochemistry 2007, 46(6), 1466–1476. 10.1021/bi061752u. [DOI] [PubMed] [Google Scholar]
- (24).Fuller J; Wilson TR; Eberhart ME; Alexandrova AN Charge Density in Enzyme Active Site as a Descriptor of Electrostatic Preorganization. J. Chem. Inf. Model 2019, 59 (5), 2367–2373. 10.1021/acs.jcim.8b00958. [DOI] [PubMed] [Google Scholar]
- (25).Kamerlin SCL; Sharma PK; Chu ZT; Warshel A Ketosteroid Isomerase Provides Further Support for the Idea That Enzymes Work by Electrostatic Preorganization. Proc. Natl. Acad. Sci 2010, 107(9), 4075–4080. 10.1073/pnas.0914579107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zoi I; Antoniou D; Schwartz SD Electric Fields and Fast Protein Dynamics in Enzymes. J. Phys. Chem. Lett 2017, 8 (24), 6165–6170. 10.1021/acs.jpclett.7b02989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Adamczyk AJ; Cao J; Kamerlin SCL; Warshel A Catalysis by Dihydrofolate Reductase and Other Enzymes Arises from Electrostatic Preorganization, Not Conformational Motions. Proc. Natl. Acad. Sci 2011, 108 (34), 14115–14120. 10.1073/pnas.1111252108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Welborn VV; Ruiz Pestana L; Head-Gordon T. Computational Optimization of Electric Fields for Better Catalysis Design. Nat. Catal 2018, 1 (9), 649–655. 10.1038/s41929-018-0109-2. [DOI] [Google Scholar]
- (29).Fried SD; Bagchi S; Boxer SG Extreme Electric Fields Power Catalysis in the Active Site of Ketosteroid Isomerase. Science (80-. ). 2014, 346 (6216), 1510–1514. 10.1126/science.1259802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Balcells D; Raynaud C; Crabtree RH; Eisenstein O A Rational Basis for the Axial Ligand Effect in C–H Oxidation by [MnO(Porphyrin)(X)]+ (X = H2O, OH−, O2−) from a DFT Study. Inorg. Chem 2008, 47(21), 10090–10099. 10.1021/ic8013706. [DOI] [PubMed] [Google Scholar]
- (31).Sameera WMC; McGrady JE The Role of Substrate in Unmasking Oxyl Character in Oxomanganese Complexes: The Key to Selectivity? Dalt. Trans 2008, No. 44, 6141–6149. 10.1039/B809868A. [DOI] [PubMed] [Google Scholar]
- (32).Siegbahn PEM; Crabtree RH Manganese Oxyl Radical Intermediates and O–O Bond Formation in Photosynthetic Oxygen Evolution and a Proposed Role for the Calcium Cofactor in Photosystem II. J. Am. Chem. Soc 1999, 121 (1), 117–127. 10.1021/ja982290d. [DOI] [Google Scholar]
- (33).Venturinelli Jannuzzi SA; Phung QM; Domingo A; Formiga ALB; Pierloot K Spin State Energetics and Oxyl Character of Mn-Oxo Porphyrins by Multiconfigurational Ab Initio Calculations: Implications on Reactivity. Inorg. Chem 2016, 55 (11), 5168–5179. 10.1021/acs.inorgchem.5b02920. [DOI] [PubMed] [Google Scholar]
- (34).Shimoyama Y; Kojima T Metal-Oxyl Species and Their Possible Roles in Chemical Oxidations. Inorg. Chem 2019, 58 (15), 9517–9542. 10.1021/acs.inorgchem.8b03459. [DOI] [PubMed] [Google Scholar]
- (35).Shaik S; Danovich D; Joy J; Wang Z; Stuyver T Electric-Field Mediated Chemistry: Uncovering and Exploiting the Potential of (Oriented) Electric Fields to Exert Chemical Catalysis and Reaction Control. J. Am. Chem. Soc 2020, 142 (29), 12551–12562. 10.1021/jacs.0c05128. [DOI] [PubMed] [Google Scholar]
- (36).Shaik S; Mandal D; Ramanan R Oriented Electric Fields as Future Smart Reagents in Chemistry. Nat. Chem 2016, 8(12), 1091–1098. 10.1038/nchem.2651. [DOI] [PubMed] [Google Scholar]
- (37).Shaik S; Ramanan R; Danovich D; Mandal D Structure and Reactivity/Selectivity Control by Oriented-External Electric Fields. Chem. Soc. Rev 2018, 47 (14), 5125–5145. 10.1039/c8cs00354h. [DOI] [PubMed] [Google Scholar]
- (38).De Visser SP What External Perturbations Influence the Electronic Properties of Catalase Compound I? Inorg. Chem 2006, 45 (23), 9551–9557. 10.1021/ic061019r. [DOI] [PubMed] [Google Scholar]
- (39).De Visser SP What Affects the Quartet - Doublet Energy Splitting in Peroxidase Enzymes? J. Phys. Chem. A 2005, 109 (48), 11050–11057. 10.1021/jp053873u. [DOI] [PubMed] [Google Scholar]
- (40).Shaik S; de Visser SP; Kumar D. External Electric Field Will Control the Selectivity of Enzymatic-Like Bond Activations. J. Am. Chem. Soc 2004, 126 (37), 11746–11749. 10.1021/ja047432k. [DOI] [PubMed] [Google Scholar]
- (41).Bathelt CM; Mulholland AJ; Harvey JN QM/MM Studies of the Electronic Structure of the Compound I Intermediate in Cytochrome c Peroxidase and Ascorbate Peroxidase. Dalt. Trans 2005, No. 21, 3470–3476. 10.1039/B505407A. [DOI] [PubMed] [Google Scholar]
- (42).Harvey JN; Bathelt CM; Mulholland AJ QM/MM Modeling of Compound I Active Species in Cytochrome P450, Cytochrome C Peroxidase, and Ascorbate Peroxidase. J. Comput. Chem 2006, 27 (12), 1352–1362. 10.1002/jcc.20446. [DOI] [PubMed] [Google Scholar]
- (43).Huang X; Groves JT Beyond Ferryl-Mediated Hydroxylation: 40 Years of the Rebound Mechanism and C-H Activation. JBIC J. Biol. Inorg. Chem 2017, 22 (2), 185–207. 10.1007/s00775-016-1414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Stuyver T; Huang J; Mallick D; Danovich D; Shaik S TITAN: A Code for Modeling and Generating Electric Fields—Features and Applications to Enzymatic Reactivity. J. Comput. Chem 2020, 41 (1), 74–82. 10.1002/jcc.26072. [DOI] [PubMed] [Google Scholar]
- (45).Efimov I; Papadopoulou ND; McLean KJ; Badyal SK; Macdonald IK; Munro AW; Moody PCE; Raven EL. The Redox Properties of Ascorbate Peroxidase. Biochemistry 2007, 46(27), 8017–8023. 10.1021/bi7006492. [DOI] [PubMed] [Google Scholar]
- (46).Ogliaro F; de Visser SP; Shaik S. The ‘Push’ Effect of the Thiolate Ligand in Cytochrome P450: A Theoretical Gauging. J. Inorg. Biochem 2002, 91 (4), 554–567. 10.1016/S0162-0134(02)00437-3. [DOI] [PubMed] [Google Scholar]
- (47).Schöneboom JC; Lin H; Reuter N; Thiel W; Cohen S; Ogliaro F; Shaik S The Elusive Oxidant Species of Cytochrome P450 Enzymes: Characterization by Combined Quantum Mechanical/Molecular Mechanical (QM/MM) Calculations. J. Am. Chem. Soc 2002, 124 (27), 8142–8151. 10.1021/ja026279w. [DOI] [PubMed] [Google Scholar]
- (48).Lundberg M; Siegbahn PEM Quantifying the Effects of the Self-Interaction Error in DFT: When Do the Delocalized States Appear? J. Chem. Phys 2005, 122 (22), 224103. 10.1063/1.1926277. [DOI] [PubMed] [Google Scholar]
- (49).Green MT Application of Badger’s Rule to Heme and Non-Heme Iron–Oxygen Bonds: An Examination of Ferryl Protonation States. J. Am. Chem. Soc 2006, 128 (6), 1902–1906. 10.1021/ja054074s. [DOI] [PubMed] [Google Scholar]
- (50).Mittra K; Green MT Reduction Potentials of P450 Compounds i and II: Insight into the Thermodynamics of C-H Bond Activation. J. Am. Chem. Soc 2019, 141 (13), 5504–5510. 10.1021/jacs.9b00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Yosca TH; Rittle J; Krest CM; Onderko EL; Silakov A; Calixto JC; Behan RK; Green MT. Iron(IV)Hydroxide PK a and the Role of Thiolate Ligation in C-H Bond Activation by Cytochrome P450. Science (80-. ) 2013, 342 (6160), 825–829. 10.1126/science.1244373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Yosca TH; Langston MC; Krest CM; Onderko EL; Grove TL; Livada J; Green MT. Spectroscopic Investigations of Catalase Compound II: Characterization of an Iron(IV) Hydroxide Intermediate in a Non-Thiolate-Ligated Heme Enzyme. J. Am. Chem. Soc 2016, 138 (49), 16016–16023. 10.1021/jacs.6b09693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Kirkman HN; Rolfo M; Ferraris AM; Gaetani GF Mechanisms of Protection of Catalase by NADPH. J. Biol. Chem 1999, 274 (20), 13908–13914. 10.1074/jbc.274.20.13908. [DOI] [PubMed] [Google Scholar]
- (54).Ledray AP; Krest CM; Yosca TH; Mittra K; Green MT Ascorbate Peroxidase Compound II Is an Iron(IV) Oxo Species. J. Am. Chem. Soc 2020. 10.1021/jacs.0c09108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Bím D; Alonso-Gil S; Srnec M From Synthetic to Biological Fe4S4 Complexes: Redox Properties Correlated to Function of Radical S-Adenosylmethionine Enzymes. Chempluschem 2020, n/a (n/a). 10.1002/cplu.202000663. [DOI] [PubMed] [Google Scholar]
- (56).Groves JT The Bioinorganic Chemistry of Iron in Oxygenases and Supramolecular Assemblies. Proc. Natl. Acad. Sci 2003, 100 (7), 3569–3574. 10.1073/pnas.0830019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Gray HB; Winkler JR Living with Oxygen. Acc. Chem. Res 2018, 51 (8), 1850–1857. 10.1021/acs.accounts.8b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Winkler JR; Gray HB Electron Flow through Metalloproteins. Chem. Rev 2014, 114 (7), 3369–3380. 10.1021/cr4004715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Gray HB; Winkler JR Hole Hopping through Tyrosine/Tryptophan Chains Protects Proteins from Oxidative Damage. Proc. Natl. Acad. Sci. U. S. A 2015, 112 (35), 10920–10925. 10.1073/pnas.1512704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.