

Abstract
Two series of novel N6 derivatives of 8-azapurine I and II were designed as antiplatelet agents. Series I and II were N6 amino derivatives and N6 hydrazone derivatives of 8-azapurine, respectively. The compounds were synthesized in acceptable yields via conventional procedures, including nucleophilic substitution, diazotization, and amination or hydrazonation with amino alcohol and 4,6-dichloropyrimidine as starting materials. To assess the ability of the synthesized compounds as antiplatelet agents, the ADP-induced platelet aggregation assay of Born was performed both in vitro and in vivo using ticagrelor as a reference control substance. The analysis of the structure–activity relationship and molecular docking were also discussed in detail. The results demonstrated that series I and II compounds exhibited antiplatelet activity in vitro and IIh was the most active compound (IC50 = 0.20 μM) among the target compounds, being almost 4-fold better than ticagrelor (IC50 = 0.74 μM). For a preliminary assessment of the safety profile, a bleeding test (mouse tail) and a single-dose toxicity test were conducted. The use of compound IIh resulted in a shorter bleeding time, less blood loss and lower acute toxicity compared to ticagrelor. In addition, a molecular docking study was performed to investigate the binding capacity and binding mode between IIh and P2Y12.
Two series of novel N6 derivatives of 8-azapurine I and II were designed as antiplatelet agents.
1. Introduction
Abnormal platelet adhesion and aggregation is one of the primary causes of serious cardiovascular events such as acute coronary syndrome (ACS) and ischemic stroke.1,2 So far, the main target of antiplatelet agents is the P2Y12 receptor.3–5 Its central role in the formation and stabilization of a thrombus renders P2Y12 an attractive target for antiplatelet drug discovery.6–8 The currently marketed P2Y12 inhibitors include clopidogrel, prasugrel, cangrelor and ticagrelor (Fig. 1).9–13 Clopidogrel and prasugrel are thienopyridine prodrugs and irreversible P2Y12 inhibitors with proven antiplatelet aggregation efficacy.9,10,14 Both drugs require metabolic conversion by the cytochrome P450 (CYP450) system to generate the active metabolite, resulting in a slow onset and offset of their pharmacological action.15–18 Cangrelor was approved as a quick onset/quickly reversed intravenous antiplatelet drug in 2015, but cangrelor must be administered by intravenous injection.19 Ticagrelor, the first orally reversible P2Y12 inhibitor approved by the FDA for the prevention of thrombotic events, exhibits rapid onset/offset effects and has less variability related to genetic polymorphisms for its direct and reversible mode of action.12,20 However, undesirable side effects such as a higher non-lethal bleeding rate and dyspnea limit its widespread application.21 Therefore, the development of novel antiplatelet agents that can offer advantages such as faster onset of action, less inter-individual variability and lower risk of bleeding is highly desirable.
Fig. 1. Structures of popularly used clinical antiplatelet agents.

Purines, as one of the key cellular components, play a significant role in energy transactions, nucleic acid synthesis, and metabolic and other biochemical reactions.22 As purine analogues, 8-azapurine (1,2,3-triazolo-[4,5-d]pyrimidine) derivatives are widely used as antitumor and antiviral agents and in the prevention of thrombotic events.12,23–25 Inspired by the versatility of 8-azapurine derivatives and the excellent antithrombotic activity of ticagrelor, herein a series of N6 amino derivatives of 8-azapurine I were designed and synthesized as antiplatelet agents (Fig. 2). Moreover, hydrazone derivatives also exhibit significant biological activities in the medicinal field due to hydrogen bonds formed with molecular targets.26–30 Especially encouraged by our previous report on a series of 8-azapurine carbocyclic nucleoside hydrazones, which possess excellent anticancer activity,25 another series of N6 hydrazone derivatives of 8-azapurine II have been designed as antiplatelet agents (Fig. 2). The two series of compounds were synthesized by conventional procedures using commercially available amino alcohol 1 and 4,6-dichloropyrimidine 2 as starting materials (Scheme 1). All the synthesized compounds were evaluated for the ability to inhibit human platelet aggregation induced by ADP in vitro using the method of Born as a preliminary screening and ticagrelor as a reference control substance. A detailed discussion of the structure–activity relationship was included. A preliminary assessment of the safety profile, based on a bleeding test (mouse tail) and a single-dose toxicity test, was performed. Also, molecular docking of the title compound with the P2Y12 receptor was performed using the AutoDock software package (ver. 4.2) for better understanding of how it played a role in antiplatelet aggregation activity.
Fig. 2. Design strategies of N6 derivatives of 8-azapurine as antiplatelet agents.

Scheme 1. Synthesis of N6 derivatives of 8-azapurines I and II. Reagents and conditions: (a) 1,4-dioxane, diisopropylethylamine (DIPEA), 100 °C, 16 h; (b) HClaq, MeOH, NaNO2, 0 °C, 1 h; (c) isopropanol, DIPEA, r.t.; (d) NH2NH2·H2O, THF, DIPEA, r.t., 1 h; (e) aromatic aldehyde, r.t., 0.5–1 h.

2. Results and discussion
2.1. Chemistry
The N6 derivatives of 8-azapurine I and II were synthesized via conventional or reported procedures including nucleophilic substitution, diazotization and amination or hydrazonation with amino alcohol 1a–b and 4,6-dichloropyrimidine 2 as starting materials (Scheme 1). Amino pyrimidines 3a–b were prepared by nucleophilic substitution with commercially available 1a–b and 2 in 1,4-dioxane at 100 °C. The triazolopyrimidines 4a–b were synthesized via the diazotization of 3a–b in methanol with hydrochloric acid and sodium nitrite at 0 °C. Compounds I were readily obtained via the reaction of compounds 4a–b and the appropriate amines in isopropanol at room temperature with triethylamine or N,N-diisopropylethylamine as an acid binding agent, and the total yields of series I are summarized in Table 1. Intermediates 5a–b were prepared by filtration from hydrazine hydrate and 4a–b in tetrahydrofuran. The targeted compounds II were obtained by adding the aromatic aldehyde into 5a–b in ethanol and the totals yield of series II are summarized in Table 2. It can be seen from Table 1 and Table 2 that the total yields of the two series of compounds are stable and within a reasonable range from 46–66%. All the products were isolated as solids and characterized by 1H NMR, 13C NMR and HRMS.
Total yields of compounds (series I).

| |||||||
|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | Yield (%) | Compd | R1 | R2 | Yield (%) |
| Ia | CH2CH2OH |

|
66 | Ik | H |

|
61 |
| Ib | CH2CH2OH |

|
64 | Il | H |

|
58 |
| Ic | CH2CH2OH |

|
60 | Im | H |

|
52 |
| Id | CH2CH2OH |

|
66 | In | H |

|
47 |
| Ie | CH2CH2OH |

|
53 | Io | H |

|
52 |
| If | CH2CH2OH |

|
50 | Ip | H |
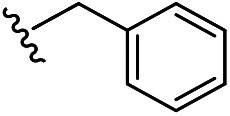
|
55 |
| Ig | CH2CH2OH |

|
57 | Iq | H |

|
56 |
| Ih | CH2CH2OH |

|
60 | Ir | H |

|
54 |
| Ii | CH2CH2OH |

|
55 | Is | H |

|
60 |
| Ij | CH2CH2OH |

|
59 | It | H |

|
58 |
Total yields of compounds (series II).

| |||||||
|---|---|---|---|---|---|---|---|
| Compd | R1 | R3 | Yield (%) | Compd | R1 | R3 | Yield (%) |
| IIa | CH2CH2OH |

|
57 | IIl | H |

|
58 |
| IIb | CH2CH2OH |

|
59 | IIm | H |

|
56 |
| IIc | CH2CH2OH |

|
58 | IIn | H |

|
57 |
| IId | CH2CH2OH |

|
54 | IIo | H |

|
58 |
| IIe | CH2CH2OH |
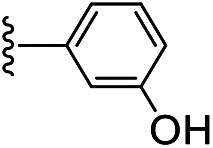
|
46 | IIp | H |

|
63 |
| IIf | H |

|
59 | IIq | H |

|
60 |
| IIg | H |

|
61 | IIr | H |

|
58 |
| IIh | H |

|
62 | IIs | H |

|
61 |
| IIi | H |

|
61 | IIt | H |

|
58 |
| IIj | H |

|
57 | IIu | H |

|
55 |
| IIk | H |

|
59 | ||||
2.2. Inhibition of ADP-induced platelet aggregation in vitro and analysis of the structure–activity relationship
All the synthesized compounds were evaluated for the ability to inhibit human platelet aggregation induced by adenosine diphosphate (ADP) in vitro using Born's method as a preliminary screening and ticagrelor as a reference control.31–33 The assay results are summarized in Tables 3 and 4. Further, the IC50 values were calculated and are presented in Table 5.
Inhibition of compounds Ia–t on ADP-induced platelet aggregationa.
| Compd | Inhibitionb (%) | Compd | Inhibitionb (%) |
|---|---|---|---|
| Ia | 21.4 | Ik | 68.6 |
| Ib | 13.6 | Il | 85.4 |
| Ic | 40.8 | Im | 69.4 |
| Id | 7.8 | In | 70.3 |
| Ie | 15.2 | Io | 86.3 |
| If | 13.9 | Ip | NAc |
| Ig | NAc | Iq | 33.4 |
| Ih | 14.8 | Ir | 68.3 |
| Ii | 8.1 | Is | 76.8 |
| Ij | 5.3 | It | 29.3 |
| Ticagrelor | 92.7 |
ADP-induced platelet aggregation ([ADP] = 10 μM).
Detection of inhibitory activity at a concentration of 10 μM.
NA: Not Active.
Inhibition of compounds IIa–u on ADP-induced platelet aggregationa.
| Compd | Inhibition (%) | Compd | Inhibition (%) | ||
|---|---|---|---|---|---|
| 3 (μM) | 10 (μM) | 3 (μM) | 10 (μM) | ||
| IIa | 46.3 | 85.4 | IIl | 71.4 | 81.5 |
| IIb | 9.1 | 69.1 | IIm | 33.4 | 78.3 |
| IIc | 77.3 | 95.5 | IIn | 62.7 | 96.5 |
| IId | 70.9 | 90.9 | IIo | 51.6 | 83.2 |
| IIe | 85.4 | 100 | IIp | 7.0 | 53.7 |
| IIf | 81.6 | 100 | IIq | NAb | 50.1 |
| IIg | 83.6 | 92.1 | IIr | 76.3 | 88.6 |
| IIh | 93.4 | 100 | IIs | 5.0 | 62.7 |
| IIi | 86.3 | 100 | IIt | 36.4 | 71.7 |
| IIj | 63.4 | 98.3 | IIu | 80.5 | 95.4 |
| IIk | 11.7 | 48.7 | Ticagrelor | 78.6 | 92.7 |
ADP-induced platelet aggregation ([ADP] = 10 μM).
NA: Not Active.
The IC50 for selected compounds which inhibit ADP-induced platelet aggregationa.
| Compd | IC50 (μM) | Compd | IC50 (μM) |
|---|---|---|---|
| Ia | 4.89 | IIg | 0.67 |
| Ic | 3.16 | IIh | 0.20 |
| In | 1.78 | IIi | 0.78 |
| Io | 0.82 | IIl | 1.30 |
| IIe | 0.58 | IIr | 0.66 |
| IIf | 0.45 | IIu | 0.40 |
| Ticagrelor | 0.74 |
ADP-induced platelet aggregation ([ADP] = 10 μM).
The synthesized series I compounds exhibited specific antiplatelet aggregation activity and compound Io was the best in terms of activity (Tables 3 and 5). The inhibition rate of Io was 86.3% which was a little weaker than that of ticagrelor (inhibition 92.7%). Similarly, the IC50 value of Io was 0.82 μM which was slightly less potent than that of ticagrelor (IC50 0.74 μM). Clearly, the structures of R1 and R2 were critical for the activities of series I compounds. When R1 is 2-hydroxyethoxy, compounds Ia–j showed weak to moderate antiplatelet aggregation activity, wherein compound Ic (R1 = CH2CH2OH, R2 = CH2CH2CH2CH3) was proved to be the most potent substance with an inhibition rate and IC50 of 40.8% and 3.16 μM, respectively. When R1 is hydroxyl, compounds Ik–t had higher antiplatelet aggregation activities than Ia–j, and compound Il (R1 = H, R2 = CH2CH2CH2CH3) was the most active, having an inhibition rate of 85.4%. The substituents of R2 also had a remarkable influence on the antiplatelet aggregation activity. Compounds Ik (R1 = H, R2 = CH2CH2CH3) and Il (R1 = H, R2 = CH2CH2CH2CH3) with alkyl substituents exhibited significant potencies with inhibition rates of 68.6% and 85.4%, respectively. Compounds Im (R1 = H, R2 = CH2CH2OH) and In (R1 = H, R2 = CH2CH2CH2OH) with hydroxyalkyl substituents had comparable potencies with inhibition rates of 69.4% and 70.3%, respectively. When R2 was cycloalkyl, the potency decreased accordingly as the size of the ring increased (e.g., potency in the order Ih (R1 = CH2CH2OH, R2 = C3H5, inhibition 14.8%) > Ii (R1 = CH2CH2OH, R2 = C5H9, inhibition 8.1%) > Ij (R1 = CH2CH2OH, R2 = C6H11, inhibition 5.3%)). When R2 was phenylalkyl, the longer side chain conferred a slight decrease of antiplatelet aggregation activity, (e.g., Iq (R1 = H, R2 = CH2CH2Ph, inhibition 33.4%) > It (R1 = H, R2 = CH2CH2CH2Ph, inhibition 29.3%)). The electronic effects on the phenethyl of R2 caused a significant increase in antiplatelet aggregation activity (e.g., Ir (R1 = H, R2 = m-FPhCH2CH2, inhibition 38.3%), Is (R1 = H, R2 = p-OHPhCH2CH2, inhibition 76.8%)).
The synthesized series II showed potent antiplatelet aggregation activity with inhibition rates ranging from 48.7 to 100% at the 10 μM concentration level (Tables 4 and 5). That is to say, the introduction of hydrazone at the N6 position of 8-azapurine resulted in an improvement in the antiplatelet aggregation activities. Herein, compounds IIe, IIf, IIh and IIi all attained a 100% inhibition rate for a concentration of 10 μM; thus the inhibition rate at a concentration of 3 μM was also examined and similar inhibition rates were observed. Different from series I, R1 and the hydrazone moiety R3 played different roles with respect to activity in series II. When R1 was 2-hydroxyethoxy or hydroxyl, compounds IIa–e and compounds IIf–u showed no significant differences in the antiplatelet aggregation activity. However, the modification of the hydrazone moiety R3 had a significant impact on potency. Thus, we next turned our attention to the hydrazone moiety. When the hydrazone moiety R3 was phenyl, compound IIf (R1 = H, R3 = Ph) displayed potent antiplatelet aggregation activity having an inhibition rate of 81.6% at a concentration of 3 μM and an IC50 value of 0.45 μM, which was almost 2-fold better than that of ticagrelor (IC50 = 0.74 μM). This gives us more expectations for the substituted benzene ring. When the hydrazone moiety R3 was hydroxyphenyl, compounds IIg (R1 = H, R3 = o-OHPh), IIh (R1 = H, R3 = m-OHPh) and IIi (R1 = H, R3 = p-OHPh) exhibited potent antiplatelet aggregation activity with inhibition rates of 83.6%, 93.4%, 86.3% and IC50 values of 0.67 μM, 0.20 μM and 0.78 μM, respectively. Compound IIh was the most active, being almost 4-fold better than ticagrelor (IC50 = 0.74 μM). Compared with compounds IIg, IIh and IIi, the hydroxy at the meta-position was much more powerful than that at the ortho- or para-position for the antiplatelet aggregation activity, which revealed that the position of substitution at the phenyl ring has a critical influence on the potency. It was noticed that compound IIj (R1 = H, R3 = 2,4-diOHPh) with 2,4-dihydroxy led to a pronounced loss of potency. When the hydrazone moiety R3 had electron-withdrawing groups F, Cl and NO2, compounds IIn–s exhibited lower antiplatelet aggregation activities than compounds IIg–m with OH and OCH3. When the hydrazone moiety R3 was heteroaromatic, compound IIt (R1 = H, R3 = pyridin-2-yl) exhibited an evident loss of activity, and IIu (R1 = H, R3 = thiophen-2-yl) displayed activities that were close to those of compounds IIf–m when R3 was aromatic.
2.3. Evaluation of antiplatelet aggregation activity in vivo and preliminary assessment of the safety profile of IIh
Based on its excellent in vitro activity in ADP-induced platelet aggregation, compound IIh was selected for further evaluation of antiplatelet aggregation activity in vivo and safety profiling in mice; ticagrelor was selected as a reference control substance. In vivo antiplatelet aggregation activity was determined in mice using the method of Born. Compound IIh was administered by intravenous injection at a dose of 2.5 mg kg−1 and by oral administration at a dose of 5.0 mg kg−1. After 1 hour, the blood samples were collected from the abdominal aorta for the platelet aggregation tests. As illustrated in Table 6, compound IIh exhibited excellent antiplatelet aggregation activity in vivo compared with ticagrelor (for both intravenous injection and oral administration). Following intravenous injection, compound IIh had an inhibition rate of 62.2% compared with 52.2% for ticagrelor. A similar profile was observed for oral administration of compound IIh, the drug candidate exhibiting an inhibition rate of 73.3% which was better than the 69.4% rate for ticagrelor. These results demonstrated that compound IIh and/or its active metabolite were present in mice 1 hour after drug administration and the efficacy was superior to that of ticagrelor.
Inhibition of IIh on platelet aggregation in vivo.
| Compd | Intravenous injection | Oral administration | ||
|---|---|---|---|---|
| Dose (mg kg−1) | Inhibition (%) | Dose (mg kg−1) | Inhibition (%) | |
| IIh | 2.5 | 62.2 | 5.0 | 73.3 |
| Ticagrelor | 2.5 | 52.2 | 5.0 | 69.4 |
The use of antiplatelet agents often causes a concomitant increase in the risk of bleeding events. As one of the primary side effects of antiplatelet agents, it is clearly necessary to evaluate this risk. Therefore, a bleeding test via the tail was performed for compound IIh. As shown in Table 7, IIh and ticagrelor had prolonged bleeding times, but the bleeding time for IIh was 10.4 min, whereas that of ticagrelor was more than 20 min. Moreover, in terms of blood loss, in the absence of any P2Y12 inhibitor treatment (vehicle group), the blood loss was quantified to be 34.92 μL, whereas after administration of ticagrelor, the blood loss was 73.25 μL (20 min). However, for the same dose, the blood loss for IIh was only 44.50 μL. Thus, a shorter bleeding time and a lower blood loss volume point to a superior safety profile and therapeutic window for IIh compared with ticagrelor. Therefore, there may be a wider scope for advocating the use of IIh across a broader spectrum of ACS patients and with fewer contraindications.
Bleeding time and blood loss tests for IIh in mice.
| Compd | Dose (mg kg−1) | Bleeding time (min) | Blood loss (μL) |
|---|---|---|---|
| Vehicle | — | 5.2 | 34.92 |
| Ticagrelor | 5.0 | >20 | 73.25 |
| IIh | 5.0 | 10.4 | 44.50 |
To make a preliminary assessment of the safety profile of IIh, a single-dose toxicity study was undertaken in a laboratory operating to the GLP standard. Forty mice (20 males and 20 females) were randomly divided into two groups (IIh group and vehicle group). The IIh group was treated with IIh at a dose of 1500 mg kg−1 d−1 (the rationale of the dose is 500-fold of the clinically intended dose (reference to ticagrelor)) by oral gavage and the vehicle group was treated with an aqueous solution of sodium carboxymethyl cellulose. The weights of the mice before dosing were recorded. As can be seen from Table 8, no mouse died in the 14 day observation period. Also, there were no significant changes in the weights of the mice, whether male or female, within the 14 days as shown in Fig. 3. The above data indicated that compound IIh had a low acute toxicity profile.
Single-dose toxicity test for compound IIh in mice.
| Compd | Quantity (pcs) | Dose (mg kg−1 d−1) | State (14 days later) |
|---|---|---|---|
| Vehicle | 20 (n = 10 males, 10 females) | — | No deaths |
| IIh | 20 (n = 10 males, 10 females) | 1500 | No deaths |
Fig. 3. Trends for weights of mice in a single-dose toxicity test.

Given the excellent bioactivity of compound IIh, its chemical and metabolic stability in vitro were tested. It can be seen from Table 9, compound IIh exhibited high chemical stability. Recovery after incubation with PBS at pH 7.4 or with fasted-state simulated gastric or intestinal fluid (FaSSGF/FaSSIF) was close to quantitative, indicating excellent chemical stability. Investigation of metabolic stability of IIh in mouse liver microsomes also resulted in a high recovery.
Chemical and metabolic stability of compound IIh.
| Assay | IIh | |
|---|---|---|
| Chemical stability | PBS, 37 °C, 1 week | 98.3% |
| FaSSGF, 1 day | 96.7% | |
| FaSSIF, 1 day | 97.4% | |
| Metabolic stability (MLM, remaining) | 1 h | 81.2% |
| 2 h | 73.7% | |
2.4. Effect on P2Y12 of IIh
The effect of IIh on the human platelet P2Y12 receptor was determined by measuring the P2Y12-mediated decrease in intraplatelet phosphorylated vasodilator stimulated phosphoprotein (VASP).34 VASP phosphorylation was measured by flow cytometry using a kit (BioCytex, Marseilles, France) after P2Y12 activation with 3 mM ADP. The results were reported as summarized in Table 10. Compound IIh displayed good inhibitory activity on the P2Y12 receptor at a low IC50 value of 46 nM, which was in the same order of magnitude as ticagrelor (IC50 = 39 nM). The result indicated that compound IIh might towards the P2Y12 receptor.
Platelet P2Y12 receptor inhibition.
| Compd | P2Y12 inhibition IC50 (nM) |
|---|---|
| IIh | 46 |
| Ticagrelor | 39 |
2.5. Molecular docking study
Based on the excellent platelet aggregation activity in vitro/vivo and the safety profile of compound IIh, molecular docking of IIh to P2Y12 was also performed to better understand how it played the role of an antiplatelet agent. Herein, molecular docking analysis was carried out using the AutoDock software package (ver. 4.2) as implemented through the graphical user interface AutoDock Tool (ADT 1.5.2).35 The protein crystal structure of P2Y12 (PDB: 4PXZ) was chosen as the receptor protein.36 The stacking map showed that the dominant conformation of IIh and the ticagrelor ligand conformation almost completely coincided (Fig. 4A). The results indicated that the design ideas mentioned earlier of replacing cyclopropyl with a similar spatially orientated hydrazone was completely feasible. Moreover, the hydrazone at the N6 position of 8-azapurine was found to effectively engage Asn159 via hydrogen bonding interactions, while the cyclopropyl of ticagrelor only serves as a steric support (Fig. 4B and C). This observation is in agreement with previous predictions that hydrazone was prone to form hydrogen bonds with the molecular target. Simultaneously, the 3-hydroxyphenyl group of IIh also interacted with the conserved Tyr105 through hydrogen bonding, while the 3,4-difluorophenyl of ticagrelor does not form hydrogen bond interactions with amino acids. For the cyclopentyl group, IIh formed five hydrogen bonds with Gln263, Arg256, Ser101, Arg93 and Lys280, whereas ticagrelor only formed four hydrogen bonds with Gln263, Arg256, Lys280 and Glu281. These observed results would likely result in a higher affinity of IIh for P2Y12 compared to that of ticagrelor. Also, the docking results indicated that IIh exhibits a better binding energy (ΔG = −4.14 kcal mol−1) and an inhibition constant (Ki = 0.91 μM) in comparison to ticagrelor (ΔG = −3.03 kcal mol−1; Ki = 6.02 μM). These conclusions are consistent with the test data (IIh: IC50 of 46 nM; ticagrelor: IC50 of 39 nM).
Fig. 4. A: Stacking map of compound IIh (green) and ticagrelor (purple); B: the binding mode of IIh (green) with P2Y12 (PDB: 4PXZ); C: the binding mode of ticagrelor (purple) with P2Y12 (PDB: 4PXZ).

3. Experimental section
3.1. General information
All experiments were performed in accordance with the Helsinki Guidelines, and experiments were approved by the Science and Technology Ethics Committee of Beijing University of Technology. Informed consent was obtained from human participants of this study. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Beijing University of Technology and experiments were approved by the Science and Technology Ethics Committee of Beijing University of Technology (Approval number: HS202103018). All reagents and solvents, including anhydrous solvents, were obtained from commercial sources and used without further purification. When necessary, solvents were dried or purified by standard methods. Room temperature refers to 20–25 °C. Purification of the crude products was performed using flash column chromatography on silica gel (300–400 mesh). All reactions were monitored by thin-layer chromatography (TLC) on 0.25 mm SDS silica gel coated glass plates (60F254) and visualized under UV light (254 nm or 365 nm). Melting points (mp) were determined with an Electrothermal 9100 capillary melting point apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on a Bruker DRX 400 instrument. These were analyzed using the Bruker TOPSPIN 2.1 program. Chemical shifts (δ) are reported in parts per million relative (ppm) to the internal tetramethylsilane standard. And coupling constants (J) are reported in hertz (Hz). The following abbreviations were used to designate the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, quint = quintuplet, m = multiplet, br = broad. High resolution mass spectra (HRMS) were obtained on a Thermo LTQ Orbitrap XL (ThermoFisher, San Jose, CA, USA) spectrometer, equipped with a quadrupole detector. In addition, the synthesis of compounds IIf–u has been reported in our another paper,25 so detailed synthetic procedures and spectral characterization data for compounds IIf–u are included in the ESI.†
3.2. Synthesis
2-(((3aR,4S,6R,6aS)-6-((5-amino-6-chloro-2-(propylthio)pyrimidin-4-yl)amino)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1,3]dioxol-4-yl)oxy)ethan-1-ol (3a)
N,N-Diisopropylethylamine (193.0 g, 1.5 mol) and 4,6-dichloro-2-(propylthio)pyrimidin-5-amine (2) (71.40 g, 0.30 mol) were added to a solution of 2-(((3aR,4S,6R,6aS)-6-amino-2,2-dimethyltetrahydro-4H-cyclopenta[d][1,3]dioxol-4-yl)oxy)ethan-1-ol (1a) (100.61 g, 0.30 mol) in 1,4-dioxane (500 mL), and the mixture was heated to 100 °C for 20 h. After completion of the reaction, the reaction mixture was cooled to r.t and then water (300 mL) was added to the reaction mixture. The aqueous phase was separated and extracted twice with ethyl acetate. The combined organic phases were washed with brine and dried over Na2SO4. After evaporation of the solvent, the sample was recrystallized with ethyl acetate and petroleum ether and dried at 40 °C to afford the white solid 3a (108.11 g, 86.0% yield). 1H NMR (400 MHz, DMSO-d6) δ 0.93 (3H, t, CH3, J = 6.0 Hz), 1.19 (3H, s, CH3), 1.35 (3H, s, CH3), 1.56–1.68 (2H, m, CH2), 1.79–1.89 (1H, m, CH2), 2.14–3.14 (1H, m, CH2), 2.91–2.99 (2H, m, CH2), 3.43–3.55 (4H, m, CH2), 3.83–3.89 (1H, m, CH), 4.29 (1H, s, OH), 4.42–4.48 (1H, m, CH), 4.46–4.54 (1H, m, CH), 4.71 (2H, s, NH2), 4.96–5.01 (1H, m, CH), 6.54 (1H, s, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.2, 22.8, 23.9, 26.3, 32.1, 32.8, 56.5, 60.3, 70.3, 83.1, 83.6, 84.1, 109.9, 119.7, 138.4, 151.9, 155.7. HRMS-ESI (m/z) [M + H]+ calcd for [C17H28ClN4O4S]+ 419.1520, found 419.1518.
(3aR,4S,6R,6aS)-6-((5-amino-6-chloro-2-(propylthio)pyrimidin-4-yl)amino)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1,3]dioxol-4-ol (3b)
To a stirred solution of 4,6-dichloro-2-(propylthio)pyrimidin-5-amine (2) (16.10 g, 67.67 mmol) in 1,4-dioxane (300 mL), (3aR,4S,6R,6aS)-6-amino-2,2-dimethyltetrahydro-4H-cyclopenta[d][1,3]dioxol-4-ol (1b) (20.03 g, 61.47 mmol) and N,N-diisopropylethylamine (43.94 g, 340 mmol) were added in one portion, respectively. The mixture was heated to 100 °C for 20 h, while the colour of the reaction mixture changed to brown. After completion of the reaction, the reaction mixture was cooled to rt and then water (500 mL) was added to the reaction mixture. The aqueous phase was separated and extracted twice with ethyl acetate. The combined organic phases were washed with brine and dried over Na2SO4. The organic solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography (ethyl acetate/hexane = 1 : 5) to afford the brown oil 3b (20.01 g, 87.0% yield). 1H NMR (400 MHz, DMSO-d6) δ 0.97 (3H, t, CH3, J = 7.2 Hz), 1.21 (3H, s, CH3), 1.36 (3H, s, CH3), 1.59–1.71 (2H, m, CH2), 1.67–1.77 (1H, m, CH2), 2.18–2.28 (1H, m, CH2), 2.94–3.02 (2H, m, CH2), 4.08 (1H, s, CH), 4.28(1H, s, CH), 4.42 (1H, d, CH, J = 5.6 Hz), 4.52 (1H, d, CH, J = 5.6 Hz), 4.65 (2H, s, NH2), 5.26 (1H, s, OH), 6.61(1H, s, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 14.5, 23.4, 26.9, 32.6, 36.3, 57.7, 60.2, 75.9, 85.0, 86.2, 139.3, 152.9, 156.6, 170.7. HRMS-ESI (m/z) [M + H]+ calcd for [C15H24ClN4O3S]+ 375.1258, found 375.1251.
(1S,2S,3R,5S)-3-(7-chloro-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-5-(2-hydroxyethoxy)cyclopentane-1,2-diol (4a)
A solution of hydrochloric acid (4.94 mol) in water was added dropwise to a solution of 3a (79.61 g, 0.19 mol) in methanol (600 mL), and the temperature was kept between −5 and 0 °C. After complete addition, a solution of NaNO2 (157.31 g, 2.28 mol) in water was added dropwise at 0 °C. The reaction mixture was stirred at 0 °C for 1 h, then treated with saturated potassium carbonate solution. The aqueous phase was separated and extracted twice with ethyl acetate. The combined organic phases were washed with brine and dried over Na2SO4. After evaporation of the solvent, the sample was recrystallized with ethyl acetate and hexane and dried at 40 °C to afford the white solid 4a (62.22 g, 84% yield). 1H NMR (400 MHz, DMSO-d6) δ 1.01 (3H, t, CH3, J = 7.2 Hz), 1.72–1.77 (2H, m, CH2), 2.11–2.14 (1H, m, CH2), 2.69–2.74 (1H, m, CH2), 3.17–3.34 (2H, m, CH2), 3.47–3.52 (4H, m, CH2), 3.77–3.78 (1H, m, CH), 3.97–4.16 (1H, m, CH), 4.59–4.64 (2H, m, CH, OH), 5.12–5.22 (3H, m, CH, 2OH). 13C NMR (100 MHz, DMSO-d6) δ 13.6, 22.6, 33.3, 33.6, 55.4, 62.4, 71.3, 75.0, 82.2, 119.7, 132.3, 151.2, 152.2, 170.1. HRMS-ESI (m/z) [M + H]+ calcd for [C14H21ClN5O4S]+ 390.1003, found 390.1001.
(1S,2R,3S,4R)-4-(7-chloro-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2,3-triol (4b)
A solution of hydrochloric acid (337 mol) in water was added dropwise to a solution of 3b (18.04 g, 48.01 mol) in methanol (200 mL), and the temperature was kept between −5 and 0 °C. After complete addition, a solution of NaNO2 (9.98 g, 144 mmol) in water was added dropwise at 0 °C. The reaction mixture was stirred at 0 °C for 1 h, then treated with saturated potassium carbonate solution. The aqueous phase was separated and extracted twice with ethyl acetate. The combined organic phases were washed with brine and dried over Na2SO4. After evaporation of the solvent, the sample was recrystallized with ethyl acetate and hexane and dried at 40 °C to afford the white solid 4b (13.98 g, 84% yield). 1H NMR (400 MHz, DMSO-d6) δ 1.00 (3H, t, CH3, J = 7.2 Hz), 1.71–1.77 (2H, m, CH2), 1.88–2.06 (1H, m, CH2), 2.64–2.72 (1H, m, CH2), 3.10–3.20 (2H, m, CH2), 3.81 (1H, s, CH), 3.93–3.98 (1H, m, CH), 4.69–4.75 (1H, m, CH), 4.99 (1H, OH), 4.98–5.04 (1H, m, CH), 5.11(1H, OH), 5.14 (1H, OH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 22.7, 33.3, 36.3, 62.9, 73.6, 75.1, 77.2, 132.2, 151.9, 160.6, 170.0. HRMS-ESI (m/z) [M + H]+ calcd for [C12H17ClN5O3S]+ 346.0741, found 346.0732.
(1S,2S,3R,5S)-3-(7-(ethylamino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-5-(2-hydroxyethoxy)cyclopentane-1,2-diol (Ia)
A solution of ethylamine (54 mg, 1.2 mmol) in isopropanol was added to a mixture solution of 4a (389 mg, 1 mmol) and N,N-diisopropylethylamine (258 mg, 2 mmol) in isopropanol at room temperature, and the mixture was stirred at 25 °C until the starting material had been consumed. Then it was treated with saturated ammonium chloride solution, and the volatiles were removed by rotary evaporation. Water (100 mL) was added to the reaction mixture which was then extracted twice with ethyl acetate (2 × 100 mL). The combined organic phases were washed with brine and dried over Na2SO4. The organic solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography to afford the white solid Ia (362 mg, 91% yield). Mp 154.5–155.2 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.99 (3H, t, CH3, J = 7.2 Hz), 1.20 (3H, t, CH3, J = 7.2 Hz), 1.65–1.77 (2H, m, CH2), 2.01–2.09 (1H, m, CH2), 2.60–2.68 (1H, m, CH2), 3.03–3.12 (2H, m, CH2), 3.47–3.56 (6H, m, CH2), 3.72–3.77 (1H, m, CH), 3.92–3.98 (1H, m, CH), 4.50–4.65 (2H, m, CH, OH), 4.95–5.09 (3H, 2OH, CH), 8.55, 8.94 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 14.7, 23.1, 32.9, 33.6, 35.3, 60.7, 68.7, 71.2, 74.2, 74.7, 82.2, 123.6, 149.7, 153.4, 169.5. HRMS-ESI (m/z) [M + H]+ calcd for [C16H27N6O4S]+ 399.1809, found 399.1808.
(1S,2S,3S,5R)-3-(2-hydroxyethoxy)-5-(7-(propylamino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2-diol (Ib)
The title compound Ib (358 mg) was prepared from 4a (389 mg, 1 mmol) and propylamine (71 mg, 1.2 mmol) using the procedure described for compound Ia in 87% yield as a white solid. Mp 152.7–153.8 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.93 (3H, t, CH3, J = 7.2 Hz), 0.99 (3H, t, CH3, J = 7.2 Hz), 1.60–1.73 (4H, m, CH2), 1.99–2.11 (1H, m, CH2), 2.58–2.66 (1H, m, CH2), 3.04–3.12 (2H, m, CH2), 3.43–3.53 (6H, m, CH2), 3.74–3.79 (1H, m, CH), 3.92–3.98 (1H, m, CH), 4.56–4.61 (2H, m, CH, OH), 4.95–5.10 (3H, m, OH, CH), 8.60, 8.97 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 11.8, 13.7, 22.3, 23.2, 32.9, 33.6, 42.2, 60.7, 60.9, 71.2, 74.2, 74.7, 82.2, 123.5, 149.8, 153.6, 169.5. HRMS-ESI (m/z) [M + H]+ calcd for [C17H29N6O4S]+ 413.1971, found 413.1968.
(1S,2S,3R,5S)-3-(7-(butylamino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-5-(2-hydroxyethoxy)cyclopentane-1,2-diol (Ic)
The title compound Ic (349 mg) was prepared from 4a (389 mg, 1 mmol) and butylamine (88 mg, 1.2 mmol) using the procedure described for compound Ia in 82% yield as a white solid. Mp 153.6–154.2 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.91 (3H, t, CH3, J = 7.2 Hz), 0.99 (3H, t, CH3, J = 7.2 Hz), 1.28–1.41 (2H, m, CH2), 1.55–1.65 (2H, m, CH2), 1.66–1.75 (2H, m, CH2), 2.01–2.08 (1H, m, CH2), 2.59–2.67 (1H, m, CH2), 3.08 (2H, t, CH2, J = 6.0 Hz), 3.49–3.53 (6H, m, CH2), 3.74–3.79 (1H, m, CH), 3.91–3.98 (1H, m, CH), 4.53–4.65 (2H, m, CH, OH), 4.94–4.99 (1H, m, CH), 5.02 (1H, OH), 5.11(1H, OH), 8.57, 8.94 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 14.0, 20.0, 23.2, 32.1, 32.9, 33.6, 49.8, 60.7, 60.9, 71.3, 74.2, 74.8, 82.2, 123.5, 149.8, 153.5, 169.5. HRMS-ESI (m/z) [M + H]+ calcd for [C18H31N6O4S]+ 427.2127, found 427.2130.
(1S,2S,3S,5R)-3-(2-hydroxyethoxy)-5-(7-(isobutylamino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2-diol (Id)
The title compound Id (401 mg) was prepared from 4a (389 mg, 1 mmol) and isobutylamine (88 mg, 1.2 mmol) using the procedure described for compound Ia in 91% yield as a white solid. Mp 137.6–138.1 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.90–1.01 (9H, m, CH3), 1.65–1.77 (2H, m, CH2), 1.96–2.03 (1H, m, CH), 2.04–2.09 (1H, m, CH2), 2.58–2.66 (1H, m, CH2), 3.03–3.09 (2H, m, CH2), 3.48 (2H, d, CH2, J = 4.8 Hz), 3.44–3.58 (4H, m, 2CH2), 3.72–3.77 (1H, m, CH), 3.91–3.97 (1H, m, CH), 4.49–4.63 (2H, m, CH, OH), 4.91–4.96 (1H, m, CH), 5.01(1H, OH), 5.08 (1H, OH), 8.65, 9.02 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 20.5, 23.2, 28.1, 32.9, 33.6, 47.9, 60.8, 60.9, 71.3, 74.2, 74.8, 82.2, 123.5, 149.8, 153.7, 169.4. HRMS-ESI (m/z) [M + H]+ calcd for [C18H31N6O4S]+ 427.2127, found 427.2130.
(1S,2S,3S,5R)-3-(2-hydroxyethoxy)-5-(7-((2-hydroxyethyl)amino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2-diol (Ie)
The title compound Ie (302 mg) was prepared from 4a (389 mg, 1 mmol) and ethanolamine (73 mg, 1.2 mmol) using the procedure described for compound Ia in 73% yield as a white solid. Mp 140.2–141.1 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.99 (3H, t, CH3, J = 7.2 Hz), 1.68–1.73 (2H, m, CH2), 2.01–2.09 (1H, m, CH2), 2.61–2.67 (1H, m, CH2), 3.04–3.12 (2H, m, CH2), 3.51–3.60 (8H, m, 4CH2), 3.74–3.79 (1H, m, CH), 3.94–4.01 (1H, m, CH), 4.57, 4.60 (2H, CH, OH), 4.79 (1H, OH), 4.94–5.02 (1H, m, CH), 5.04 (1H, OH), 5.12 (1H, OH), 8.45, 8.81 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 23.1, 32.9, 33.6, 43.2, 59.6, 60.7, 61.1, 71.2, 74.2, 74.7, 82.2, 123.6, 149.7, 153.8, 169.5. HRMS-ESI (m/z) [M + H]+ calcd for [C16H27N6O5S]+ 415.1763, found 415.1759.
(1S,2S,3S,5R)-3-(2-hydroxyethoxy)-5-(7-((3-hydroxypropyl)amino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2-diol (If)
The title compound If (291 mg) was prepared from 4a (389 mg, 1 mmol) and 3-aminopropano (90 mg, 1.2 mmol) using the procedure described for compound Ia in 68% yield as a white solid. Mp 143.6–144.5 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.99 (3H, t, CH3, J = 7.2 Hz), 1.67–1.79 (4H, m, CH2), 2.01–2.08 (1H, m, CH2), 2.59–2.65 (1H, m, CH2), 3.05–3.11 (2H, m, CH2), 3.47–3.57 (8H, m, 4CH2), 3.73–3.79 (1H, m, CH), 3.92–3.97 (1H, m, CH), 4.50–4.61 (3H, m, CH, 2OH), 4.95–5.01 (1H, m, CH), 5.09 (1H, OH), 5.11 (1H, OH), 8.52, 8.89 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 23.1, 32.2, 32.9, 33.6, 37.9, 59.0, 60.7, 61.0, 71.2, 74.2, 74.7, 82.2, 123.6, 149.7, 153.6, 169.5. HRMS-ESI (m/z) [M + H]+ calcd for [C17H29N6O5S]+ 429.1920, found 429.1914.
(1S,2S,3R,5S)-3-(7-((1-hydroxy-2-methylpropan-2-yl)amino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-5-(2-hydroxyethoxy)cyclopentane-1,2-diol (Ig)
The title compound Ig (345 mg) was prepared from 4a (389 mg, 1 mmol) and 2-amino-2-methyl-1-propanol (107 mg, 1.2 mmol) using the procedure described for compound Ia in 78% yield as a white solid. Mp 86.8–87.4 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.97 (3H, t, CH3, J = 7.2 Hz), 1.45 (6H, s, CH3), 1.64–1.72 (2H, m, CH2), 2.01–2.06 (1H, m, CH2), 2.58–2.64 (1H, m, CH2), 3.02–3.10 (2H, m, CH2), 3.41–4.53 (4H, m, 2CH2), 3.56–3.62 (2H, m, CH2), 3.70–3.75 (1H, m, CH), 3.89–3.95 (1H, m, CH), 4.52–4.58 (2H, OH, CH), 4.90–4.95 (1H, m, CH), 5.02 (1H, OH), 5.08 (1H, OH), 5.13 (1H, OH), 7.77, 8.30 (1H, NH). 1H NMR (600 MHz, D2O, DMSO-d6) δ 0.92 (3H, t, CH3, J = 7.2 Hz), 1.45 (6H, s, CH3), 1.62–1.68 (2H, m, CH2), 1.98–2.03 (1H, m, CH2), 2.56–2.62 (1H, m, CH2), 3.00–3.07 (2H, m, CH2), 3.41–3.50 (4H, m, CH2), 3.57–3.62 (2H, m, CH2), 3.71–3.76 (1H, m, CH), 3.91–3.95 (1H, m, CH), 4.48–4.53 (1H, m, CH), 4.90–4.95 (1H, m, CH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 23.2, 23.8, 32.8, 33.6, 56.7, 60.8, 61.1, 67.9, 71.3, 74.2, 74.7, 82.3, 124.1, 149.6, 153.6, 169.0. HRMS-ESI (m/z) [M + H]+ calcd for [C18H31N6O5S]+ 443.2076, found 443.2071.
(1S,2S,3R,5S)-3-(7-(cyclopropylamino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-5-(2-hydroxyethoxy)cyclopentane-1,2-diol (Ih)
The title compound Ih (337 mg) was prepared from 4a (389 mg, 1 mmol) and cyclopropylamine (69 mg, 1.2 mmol) using the procedure described for compound Ia in 82% yield as a white solid. Mp 135.4–136.9 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.65–0.71 (2H, m, CH2), 0.73–0.78 (2H, m, CH2), 0.73–0.83 (1H, m, CH), 0.99 (3H, t, CH3, J = 7.2 Hz), 1.70–1.75 (2H, m, CH2), 2.02–2.07 (1H, m, CH2), 2.59–2.65 (1H, m, CH2), 3.04–3.10 (2H, m, CH2), 3.47–3.53 (4H, m, 2CH2), 3.74–3.79 (1H, m, CH), 3.93–3.98 (1H, m, CH), 4.51–4.67 (2H, m, CH, OH), 4.95–5.10 (3H, m, 2OH, CH), 8.75, 9.05 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 6.4, 13.7, 23.1, 24.1, 33.0, 33.6, 60.7, 61.0, 71.3, 74.2, 74.7, 82.2, 123.6, 149.7, 154.6, 169.5. HRMS-ESI (m/z) [M + H]+ calcd for [C17H27N6O4S]+ 411.1814, found 411.1806.
(1S,2S,3R,5S)-3-(7-(cyclopentylamino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-5-(2-hydroxyethoxy)cyclopentane-1,2-diol (Ii)
The title compound Ii (333 mg) was prepared from 4a (389 mg, 1 mmol) and cyclopentylamine (102 mg, 1.2 mmol) using the procedure described for compound Ia in 76% yield as a white solid. Mp 127.7–128.9 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.98 (3H, t, CH3, J = 7.2 Hz), 1.56–1.73 (8H, m, 4CH2), 1.93–1.99 (2H, m, CH2), 2.00–2.05 (1H, m, CH2), 2.59–2.66 (1H, m, CH2), 3.03–3.11 (2H, m, CH2), 3.44–3.56 (4H, m, 2CH2), 3.73–3.79 (1H, m, CH), 3.91–3.97 (1H, m, CH), 4.51–3.64 (3H, m, 2CH, OH), 4.92–4.97 (1H, m, CH), 5.01(1H, OH), 5.08 (1H, OH), 8.65, 8.95 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 23.2, 24.0, 32.2, 33.6, 52.3, 60.7, 60.9, 66.6, 71.3, 74.2, 74.7, 82.2, 123.5, 149.8, 153.1, 169.4. HRMS-ESI (m/z) [M + H]+ calcd for [C19H31N6O4S]+ 439.2127, found 439.2121.
(1S,2S,3R,5S)-3-(7-(cyclohexylamino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-5-(2-hydroxyethoxy)cyclopentane-1,2-diol (Ij)
The title compound Ij (367 mg) was prepared from 4a (389 mg, 1 mmol) and cyclohexylamine (119 mg, 1.2 mmol) using the procedure described for compound Ia in 81% yield as a white solid. Mp 120.1–120.5 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.98 (3H, t, CH3, J = 7.2 Hz), 1.12–1.19 (1H, m, CH2), 1.30–1.44 (4H, m, 2CH2), 1.61–1.70 (1H, m, CH2), 1.68–1.78 (4H, m, 2CH2), 1.87–1.91 (2H, m, CH2), 1.99–2.05 (1H, m, CH2), 2.60–2.65 (1H, m, CH2), 3.02–3.11 (2H, m, CH2), 3.45–3.56 (4H, m, 2CH2), 3.72–3.78 (1H, m, CH), 3.91–3.96 (1H, m, CH), 4.05–4.11 (1H, m, CH), 4.51–4.64 (2H, m, CH, OH), 4.92–4.97 (1H, m, CH), 5.01(1H, OH), 5.08 (1H, OH), 8.53, 8.83 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.8, 23.3, 25.4, 25.5, 32.3, 32.9, 33.6, 49.8, 60.7, 60.7, 71.3, 74.2, 74.7, 82.2, 123.4, 149.9, 157.7, 169.4. HRMS-ESI (m/z) [M + H]+ calcd for [C20H33N6O4S]+ 453.2284, found 453.2278.
(1S,2R,3S,4R)-4-(7-(propylamino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2,3-triol (Ik)
The title compound Ik (309 mg) was prepared from 4b (346 mg, 1 mmol) and propylamine (71 mg, 1.2 mmol) using the procedure described for compound Ia in 84% yield as a white solid. Mp 157.8–159.1 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.90 (3H, t, CH3, J = 7.2 Hz), 0.96 (3H, t, CH3, J = 7.2 Hz), 1.59–1.68 (4H, m, 2CH2), 1.91–1.96 (1H, m, CH2), 2.55–2.61 (1H, m, CH2), 3.03–3.11 (2H, m, CH2), 3.41–3.50 (2H, m, CH2), 3.75–3.81 (1H, m, CH), 3.90–3.97 (1H, m, CH), 4.61–4.66 (1H, m, CH), 4.93–5.13 (4H, m, CH, 3OH), 8.65, 9.01 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 11.8, 13.7, 22.3, 23.2, 32.9, 36.3, 42.2, 61.4, 73.6, 74.9, 77.2, 123.5, 149.7, 153.6, 169.4. HRMS-ESI (m/z) [M + H]+ calcd for [C15H25N6O3S]+ 369.1709, found 369.1701.
(1S,2R,3S,4R)-4-(7-(butylamino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2,3-triol (Il)
The title compound Il (302 mg) was prepared from 4b (346 mg, 1 mmol) and butylamine (88 mg, 1.2 mmol) using the procedure described for compound Ia in 79% yield as a white solid. Mp 158.4–159.9 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.84 (3H, t, CH3, J = 7.2 Hz), 0.98 (3H, t, CH3, J = 7.2 Hz), 1.31–1.42 (2H, m, CH2), 1.57–1.71 (4H, m, 2CH2), 1.89–1.94 (1H, m, CH2), 2.57–2.63 (1H, m, CH2), 3.03–3.10 (2H, m, CH2), 3.44–3.52 (2H, m, CH2), 3.76–3.81 (1H, m, CH), 3.87–3.92 (1H, m, CH), 4.63–4.69 (1H, m, CH), 4.91–5.14 (4H, 3OH, CH), 8.61, 8.99 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 14.1, 20.0, 23.2, 31.1, 32.1, 32.9, 36.4, 61.4, 73.6, 74.9, 77.2, 123.5, 149.7, 153.5, 169.4. HRMS-ESI (m/z) [M + H]+ calcd for [C16H27N6O3S]+ 383.1865, found 383.1859.
(1S,2R,3S,4R)-4-(7-((2-hydroxyethyl)amino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2,3-triol (Im)
The title compound Im (263 mg) was prepared from 4b (346 mg, 1 mmol) and ethanolamine (73 mg, 1.2 mmol) using the procedure described for compound Ia in 71% yield as a white solid. Mp 149.7–150.3 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.98 (3H, t, CH3, J = 7.2 Hz), 1.64–1.72 (2H, m, CH2), 1.89–1.94 (1H, m, CH2), 2.57–2.63 (1H, m, CH2), 3.04–3.13 (2H, m, CH2), 3.43–3.55 (4H, m, 2CH2), 3.72–3.77 (1H, m, CH), 3.91–3.97 (1H, m, CH), 4.65–4.84 (2H, m, CH, OH), 4.88–4.97 (2H, m, CH, OH), 5.05 (1H, s, OH), 5.13 (1H, s, OH), 8.50, 8.87 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 23.1, 32.9, 36.4, 43.2, 59.6, 61.5, 73.6, 74.9, 77.2, 123.6, 149.7, 153.8, 169.4. HRMS-ESI (m/z) [M + H]+ calcd for [C14H23N6O4S]+ 371.1501, found 371.1495.
(1S,2R,3S,4R)-4-(7-((3-hydroxypropyl)amino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2,3-triol (In)
The title compound In (246 mg) was prepared from 4b (346 mg, 1 mmol) and 3-aminopropano (90 mg, 1.2 mmol) using the procedure described for compound Ia in 64% yield as a white solid. Mp 144.7–146.2 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.98 (3H, t, CH3, J = 7.2 Hz), 1.75–1.82 (4H, m, 2CH2), 1.92–1.98 (1H, m, CH2), 2.56–2.61 (1H, m, CH2), 3.06–3.12 (2H, m, CH2), 3.44–3.56 (4H, m, 2CH2), 3.72–3.77 (1H, m, CH), 3.91–3.96 (1H, m, CH), 4.53–4.67 (2H, m, CH, OH), 4.90–5.02 (2H, m, CH, OH), 5.05 (1H, OH), 5.13 (1H, OH), 8.55, 8.94 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 23.2, 32.3, 32.9, 36.3, 37.9, 59.0, 61.4, 73.6, 74.9, 77.2, 123.6, 149.7, 153.6, 169.4. HRMS-ESI (m/z) [M + H]+ calcd for [C15H25N6O4S]+ 385.1658, found 385.1651.
(1S,2R,3S,4R)-4-(7-(cyclopropylamino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2,3-triol (Io)
The title compound Io (260 mg) was prepared from 4b (346 mg, 1 mmol) and cyclopropylamine (69 mg, 1.2 mmol) using the procedure described for compound Ia in 71% yield as a white solid. Mp 141.3–143.2 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.71–0.82 (3H, m, CH2, CH2), 0.99 (3H, t, CH3, J = 7.2 Hz), 1.20–1.29 (1H, m, CH2), 1.69–1.81 (2H, m, CH2), 1.92–1.98 (1H, m, CH2), 2.59–2.65 (1H, m, CH2), 3.05–3.20 (2H, m, CH2), 3.38–3.43 (1H, m, CH), 3.76–3.81 (1H, m, CH), 3.90–3.97 (1H, m, CH), 4.66–4.70 (1H, m, CH), 4.94–5.13 (4H, m, 3OH, CH), 8.78, 9.09 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 6.4, 13.6, 23.1, 24.1, 33.1, 36.1, 61.5, 73.6, 74.9, 77.2, 123.6, 149.6, 154.6, 169.5. HRMS-ESI (m/z) [M + H]+ calcd for [C15H23N6O3S]+ 367.1552, found 367.1545.
(1S,2R,3S,4R)-4-(7-(benzylamino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2,3-triol (Ip)
The title compound Ip (316 mg) was prepared from 4b (346 mg, 1 mmol) and benzylamine (128 mg, 1.2 mmol) using the procedure described for compound Ia in 76% yield as a white solid. Mp 127.6–128.5 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.90 (3H, t, CH3, J = 7.2 Hz), 1.54–1.65 (2H, m, CH2), 1.91–1.99 (1H, m, CH2), 2.58–2.64 (1H, m, CH2), 3.00–3.11 (2H, m, CH2), 3.80 (1H, s, CH), 3.92–3.98 (1H, m, CH), 4.64–4.71 (1H, m, CH), 4.65–4.72 (2H, m, CH2), 4.94–5.17 (4H, m, 3OH, CH), 7.21–7.39 (5H, m, Ph–H), 9.12, 9.55 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 23.0, 32.8, 36.4, 43.6, 61.5, 73.6, 74.9, 77.2, 123.5, 127.3, 127.6, 128.7, 139.3, 149.9, 153.5, 169.5. HRMS-ESI (m/z) [M + H]+ calcd for [C19H25N6O3S]+ 417.1709, found 417.1701.
(1S,2R,3S,4R)-4-(7-(phenethylamino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2,3-triol (Iq)
The title compound Iq (331 mg) was prepared from 4b (346 mg, 1 mmol) and 2-phenylethylamine (145 mg, 1.2 mmol) using the procedure described for compound Ia in 77% yield as a white solid. Mp 106.3–108.1 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.97 (3H, t, CH3, J = 7.2 Hz), 1.63–1.75 (2H, m, CH2), 1.87–1.94 (1H, m, CH2), 2.58–2.63 (1H, m, CH2), 2.94 (2H, t, CH2, J = 7.2 Hz), 3.06–2.12 (2H, m, CH2), 3.69–3.81 (3H, m, CH, CH2), 3.90–3.95 (1H, m, CH), 4.62–4.69 (1H, m, CH), 4.93–5.12 (4H, m, 3OH, CH), 7.10–7.40 (5H, m, Ph–H), 8.72, 9.07 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 23.1, 32.9, 35.0, 36.4, 41.9, 61.4, 73.6, 74.9, 77.2, 123.5, 126.6, 128.7, 129.0, 139.6, 149.7, 153.5, 169.5. HRMS-ESI (m/z) [M + H]+ calcd for [C20H27N6O3S]+ 431.1865, found 431.1858.
(1S,2R,3S,4R)-4-(7-((3-fluorophenethyl)amino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2,3-triol (Ir)
The title compound Ir (331 mg) was prepared from 4b (346 mg, 1 mmol) and 3-fluorophenethylamine (167 mg, 1.2 mmol) using the procedure described for compound Ia in 74% yield as a white solid. Mp 113.6–114.9 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.96 (3H, t, CH3, J = 7.2 Hz), 1.65–1.74 (2H, m, CH2), 1.91–1.98 (1H, m, CH2), 2.58–2.63 (1H, m, CH2), 2.96 (2H, t, CH2, J = 7.2 Hz), 3.10 (2H, t, CH2, J = 7.2 Hz), 3.70–3.86 (3H, m, CH, CH2), 3.91–3.97 (1H, m, CH), 4.62–4.67 (1H, m, CH), 4.91–5.12 (4H, m, 3OH, CH), 7.01–7.17 (3H, m, Ph–H), 7.32 (1H, s, Ph–H), 8.70, 9.08 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 23.1, 32.9, 34.6, 36.4, 41.5, 61.4, 73.7, 75.0, 77.2, 113.3(113.4), 115.7(115.8), 123.5, 125.2(125.2), 130.5(130.5), 142.5(142.6), 149.7, 153.5, 161.8(163.4), 169.5. HRMS-ESI (m/z) [M + H]+ calcd for [C20H26FN6O3S]+ 449.1771, found 449.1764.
(1S,2R,3S,4R)-4-(7-((4-hydroxyphenethyl)amino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2,3-triol (Is)
The title compound Is (366 mg) was prepared from 4b (346 mg, 1 mmol) and tyramine (164 mg, 1.2 mmol) using the procedure described for compound Ia in 82% yield as a white solid. Mp 126.3–127.4 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.97 (3H, t, CH3, J = 7.2 Hz), 1.64–1.74 (2H, m, CH2), 1.88–1.95 (1H, m, CH2), 2.56–2.63 (1H, m, CH2), 2.83 (2H, t, CH2, J = 7.2 Hz), 3.09 (2H, t, CH2, J = 7.2 Hz), 3.66 (1H, OH), 3.76–3.81 (1H, m, CH), 3.91–3.97 (1H, m, CH), 4.52–4.59 (1H, m, CH), 4.90–5.11 (6H, m, 3OH, CH, CH2), 6.68 (2H, d, Ph–H, J = 10.4 Hz), 7.37 (2H, d, Ph–H, J = 10.4 Hz), 8.66, 9.04 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 23.1, 32.9, 34.2, 36.4, 42.3, 61.4, 73.7, 75.0, 77.2, 115.6, 123.5, 129.6, 129.9, 149.7, 153.5, 156.1, 169.5. HRMS-ESI (m/z) [M + H]+ calcd for [C20H27N6O4S]+ 447.1814, found 447.1807.
(1S,2R,3S,4R)-4-(7-((3-phenylpropyl)amino)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2,3-triol (It)
The title compound It (351 mg) was prepared from 4b (346 mg, 1 mmol) and 3-phenylpropylamine (162 mg, 1.2 mmol) using the procedure described for compound Ia in 79% yield as a white solid. Mp 103.1–104.6 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.95 (3H, t, CH3, J = 7.2 Hz), 1.61–1.70 (2H, m, CH2), 1.89–2.00 (3H, m, CH2, CH2), 2.58–2.70 (3H, m, CH2, CH2), 3.01–3.09 (2H, m, CH2), 3.49–3.55 (1H, m, CH), 3.81 (1H, OH), 3.92–3.98 (1H, m, CH), 4.62–4.75 (3H, m, CH, CH2), 4.92–5.01 (3H, 2OH, m, CH), 7.15–7.28(5H, m, Ph–H), 8.67, 9.05 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 23.1, 30.7, 32.9, 33.0, 36.4, 43.2, 61.4, 73.7, 75.0, 77.2, 123.6, 126.1, 128.6, 128.7, 141.9, 149.7, 153.6, 169.5. HRMS-ESI (m/z) [M + H]+ calcd for [C21H29N6O3S]+ 445.2022, found 445.2015.
(1S,2S,3R,5S)-3-(7-(2-((E)-3,4-difluorobenzylidene)hydrazinyl)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-5-(2-hydroxyethoxy)cyclopentane-1,2-diol (IIa)
A solution of 4a (390 mg, 1 mmol) in tetrahydrofuran was added dropwise to a mixture solution of hydrazine hydrate (250 mg, 5 mmol) and N,N-diisopropylethylamine (258 mg, 2 mmol) in tetrahydrofuran at 0 °C, and then the mixture was stirred at 25 °C until the starting material had been consumed. After the completion, 5a was obtained by filtration and used for the next step without further purification. 3,4-Difluorobenzaldehyde (284 mg, 2 mmol) and 5a were dissolved in ethanol, and then the reaction mixture was refluxed for 1–2 h. The organic solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography to afford the white solid IIa (397 mg, 78% yield). Mp 147.8–149.2 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.01 (3H, t, CH3, J = 7.2 Hz), 1.68–1.80 (2H, m, CH2), 2.06–2.11 (1H, m, CH2), 2.63–2.69 (1H, m, CH2), 3.10–3.19 (2H, m, CH2), 3.47–3.59 (4H, m, 2CH2), 3.78 (1H, s, CH), 3.98 (1H, s, CH), 4.56–4.68 (2H, m, CH, OH), 5.03–5.18 (3H, m, CH, 2OH), 7.53–7.92 (3H, m, Ph–H), 8.22, 8.48 (1H, =CH), 12.56, 12.70 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 22.7, 32.7, 33.6, 60.7, 61.1, 71.3, 74.2, 74.8, 82.2, 115.4(115.5), 118.4(118.5), 122.8, 124.8, 132.7, 143.8, 149.3(149.4), 150.9(151.1), 151.7, 153.6, 168.7. HRMS-ESI (m/z) [M + H]+ calcd for [C21H26F2N7O4S]+ 510.1735, found 510.1729.
(1S,2S,3R,5S)-3-(7-(2-((E)-4-chlorobenzylidene)hydrazinyl)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-5-(2-hydroxyethoxy)cyclopentane-1,2-diol (IIb)
The title compound IIb (411 mg) was prepared from 4a (390 mg, 1 mmol) and 4-chlorobenzaldehyde (280 mg, 2 mmol) using the procedure described for compound IIa in 81% yield as a white solid. Mp 136.9–138.3 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.00 (3H, t, CH3, J = 7.2 Hz), 1.68–1.80 (2H, m, CH2), 2.06–2.14 (1H, m, CH2), 2.64–2.71 (1H, m, CH2), 3.10–3.18 (2H, m, CH2), 3.47–3.61 (4H, m, 2CH2), 3.79 (1H, s, CH), 3.98 (1H, s, CH), 4.59–3.72 (2H, m, CH, OH), 5.03–5.18 (3H, m, CH, 2OH), 7.51–7.88 (4H, m, Ph–H), 8.25, 8.50 (1H, =CH), 12.50 (1H, br, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 22.8, 33.7, 33.6, 60.8, 61.1, 71.2, 74.3, 74.8, 82.2, 122.8, 129.1, 129.3, 133.7, 134.8, 145.1, 151.7, 153.6, 168.7. HRMS-ESI (m/z) [M + H]+ calcd for [C21H27ClN7O4S]+ 508.1534, found 508.1529.
(1S,2S,3S,5R)-3-(2-hydroxyethoxy)-5-(7-(2-((E)-3-nitrobenzylidene)hydrazinyl)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2-diol (IIc)
The title compound IIc (410 mg) was prepared from 4a (390 mg, 1 mmol) and 3-nitrobenzaldehyde (302 mg, 2 mmol) using the procedure described for compound IIa in 79% yield as a yellow solid. Mp 150.6–152.1 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.01 (3H, t, CH3, J = 7.2 Hz), 1.68–1.79 (2H, m, CH2), 2.10–2.17 (1H, m, CH2), 2.65–2.72 (1H, m, CH2), 3.11–3.20 (2H, m, CH2), 3.47–3.59 (4H, m, 2CH2), 3.79 (1H, s, CH), 4.00 (1H, s, CH), 4.60–3.75 (2H, m, CH, OH), 5.01–5.19 (3H, m, CH, 2OH), 7.70–7.76 (1H, m, Ph–H), 8.21–8.24 (2H, m, Ph–H), 8.35, 8.60 (1H, =CH), 8.68 (1H, s, Ph–H), 12.63, 12.75 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 22.7, 32.7, 33.6, 60.8, 61.1, 71.3, 74.2, 74.8, 82.2, 121.5, 122.8, 124.3, 130.7, 133.4, 136.6, 143.9, 148.6, 151.7, 153.6, 168.8. HRMS-ESI (m/z) [M + H]+ calcd for [C21H27N8O6S]+ 519.1774, found 519.1769.
(1S,2S,3S,5R)-3-(2-hydroxyethoxy)-5-(7-(2-((E)-4-nitrobenzylidene)hydrazinyl)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)cyclopentane-1,2-diol (IId)
The title compound IId (384 mg) was prepared from 4a (390 mg, 1 mmol) and 4-nitrobenzaldehyde (302 mg, 2 mmol) using the procedure described for compound IIa in 74% yield as a yellow solid. Mp 149.3–150.7 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.02 (3H, t, CH3, J = 7.2 Hz), 1.68–1.77 (2H, m, CH2), 2.08–2.14 (1H, m, CH2), 2.66–2.71 (1H, m, CH2), 3.12–3.19 (2H, m, CH2), 3.35–3.53 (4H, m, 2CH2), 3.79 (1H, s, CH), 3.98 (1H, s, CH), 4.59–3.72 (2H, m, CH, OH), 5.04–5.18 (3H, m, CH, 2OH), 8.03–8.11 (2H, m, Ph–H), 8.26–8.33 (2H, m, Ph–H), 8.33 (1H, =CH), 12.75 (1H, br, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 22.7, 32.8, 33.6, 60.7, 61.1, 71.2, 74.2, 74.8, 82.2, 122.7, 124.5, 128.3, 131.0, 141.1, 148.1, 151.7, 153.6, 168.8. HRMS-ESI (m/z) [M + H]+ calcd for [C21H27N8O6S]+ 519.1774, found 519.1769.
(1S,2S,3R,5S)-3-(7-(2-((E)-3-hydroxybenzylidene)hydrazinyl)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-5-(2-hydroxyethoxy)cyclopentane-1,2-diol (IIe)
The title compound IIe (308 mg) was prepared from 4a (390 mg, 1 mmol) and 3-hydroxybenzaldehyde (244 mg, 2 mmol) using the procedure described for compound IIa in 63% yield as a white solid. Mp 208.9–210.5 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.01 (3H, t, CH3, J = 7.2 Hz), 1.68–1.79 (2H, m, CH2), 2.03–2.08 (1H, m, CH2), 2.66–2.72 (1H, m, CH2), 3.10–3.19 (2H, m, CH2), 3.51–3.53 (4H, m, 2CH2), 3.78 (1H, s, CH), 3.97 (1H, s, CH), 4.57–4.68 (2H, m, CH, OH), 4.99–5.18 (3H, m, CH, 2OH), 6.80–6.86 (1H, m, Ph–H), 7.24–7.31 (3H, m, Ph–H), 8.19, 8.43 (1H, =CH), 9.68 (1H, s, Ph–OH), 12.35, 12.53 (1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.7, 22.8, 32.7, 33.5, 60.8, 61.1, 71.2, 74.2, 74.7, 82.2, 113.3, 117.8, 119.2, 122.8, 130.2, 135.9, 146.9, 151.7, 153.5, 158.1, 168.7. HRMS-ESI (m/z) [M + H]+ calcd for [C21H28N7O5S]+ 490.1873, found 490.1867.
3.3. Biological evaluation
3.3.1. Inhibition of ADP-induced platelet aggregation in vitro
All experiments using human subjects were performed in accordance with the Helsinki declaration. Blood was sampled from the cubital vein of healthy volunteers who were not taking aspirin or other nonsteroidal anti-inflammatory drugs for at least 14 days. Participant consent was obtained before blood collection. The blood sample was collected in 50 mL plastic tubes containing 3.8% sodium citrate (1 : 9, v/v) and centrifuged at 300 rpm for 20 min at room temperature to generate platelet-rich plasma (PRP). The supernatant PRP was transferred to a fresh tube, and the residual blood was centrifuged at 900 rpm for 10 min to obtain platelet-poor plasma (PPP), which was transferred into another tube for use in the assay. The platelet aggregation was analyzed using a double-channel aggregometer (model 400VS, Chrono-Log, Havertown, PA) with stirring (900 rpm) at 37 °C; the chart recorder was set for 1 cm min−1. The test compounds and ticagrelor were dissolved in DMSO (final concentration less than 0.5%) and the solutions were added to the PRP and left for 1 min before platelet activation with agonists. The extent of aggregation was quantified by determining the maximum height of the aggregation trace. The aggregometer system was calibrated with PRP (0% aggregation control, baseline) and PPP (100% aggregation control). Fresh PRP (500 μL) was added to a cuvette and incubated at 37 °C for 1 min with stirring before being transferred to the aggregometer. The baseline was set using PPP as the blank. The solution of the test compound was added to the fresh PRP, and then, the mixture was warmed at 37 °C for 2 min. Platelets were stimulated with ADP. Platelet aggregation was monitored for 5 min by recording the variations of optical density according to the method of Born.33 The platelet aggregation inhibitory activity was expressed as %inhibition by comparison with that measured in the presence of an equivalent amount of vehicle (DMSO). The aggregation inhibition rate was calculated as follows: inhibition rate (%) = [(control tube maximal aggregation response − test tube maximum aggregation response)/control tube maximum aggregation response] × 100%.37,38 The IC50 values were calculated by nonlinear regression using XLFit software.
3.3.2. Inhibition of ADP-induced platelet aggregation in vivo
One hour after intravenous injection (2.5 mg kg−1) or oral administration (5 mg kg−1) of the test compound or ticagrelor to male mouse (∼20 g in weight), blood samples were drawn from the abdominal aorta and anticoagulated with 3.8% sodium citrate (1 : 9, v/v). For the centrifuge conditions of 1000 rpm and 7 min, PRP was obtained. The PRP was adjusted with PPP to provide a platelet count of approximately 2 × 106 per mL. The PRP was added to the test cup, and incubated at 37 °C for 10 min. Then the zero setting and 100% settings were adjusted by PRP and PPP, respectively; ADP (final concentration 5 μM) was used as an induction agent, and, in accordance with the turbidimetric method, the platelet aggregation for test samples was measured, with the values being expressed as a % of platelet aggregation.
3.3.3. Mouse bleeding test (tail)
The test compounds, either ticagrelor or vehicle, were administered to the mouse by oral gavage. After 1 h, the mouse was anesthetized by intraperitoneal injection of 2% pentobarbital sodium salt hydrate at a dose of 100 mg kg−1. The animals were then placed on a thermostatically controlled heating table to maintain the body temperature at 36–38 °C and with their tails straightened. The tails were transected 4 mm from the tip with a scalpel blade and immediately immersed into 15 mL centrifuge tubes containing 10 mL of saline and held for 10 minutes at 37 °C. Then the centrifuge tubes were placed in the refrigerator at −80 °C overnight. The next day, the 15 ml centrifuge tubes were removed from the refrigerator and thawed, then 1 ml of solution was taken out and 9 ml red blood cell lysis buffer (10 mM KHCO3, 155 mM NH4Cl, 0.1 mM Na2EDTA, pH 7.30) was added to lyse the red blood cells. After complete dissolution, 200 μL of sample was removed and added to a 96-well plate. The absorbance was determined using a microplate reader at a wavelength of 405 nm; thereafter the blood loss was calculated.
3.3.4. P2Y12-mediated vasodilator-stimulated phosphoprotein (VASP) phosphorylation assay
VASP phosphorylation was measured by flow cytometry using a BioCytex kit, essentially according to the manufacturer's recommendations, except that a small volume of the test compound solution or vehicle (HEPES-saline) was added to each assay tube as previously described.39,40 Analysis was performed in a FACSCalibur (Becton Dickinson) flow cytometer.
3.3.5. Molecular docking study
The crystal structure of P2Y12 in complex with 2-MeSADP was retrieved from the Brookhaven Protein Data Bank (PDB code: 4PXZ).36 The prepared protein structure was used for rigid docking of the synthesized compounds using Autodock 4.2.35 The 3D structures of the compounds were built using Gaussian View, then imported into the Autodock software. The compounds were prepared by adding hydrogen atoms and partial charges followed by merging non-polar hydrogen atoms.41,42 A grid box of 50, 50 and 50 points in the x, y and z directions with a grid spacing of 0.375 Å was used to define the binding site. The binding affinity was reported as the binding free energy (ΔG) and inhibition constant (Ki). The model analysis was performed using the DISCOVERY STUDIO VISUALIZER software (Accelrys, Inc., San Diego, CA, U.S.A.).
4. Conclusion
In summary, based on the significant biological activities in the medicinal field of purine analogues and hydrazone derivatives, forty-one N6 amino derivatives of 8-azapurines I and N6 hydrazone derivatives of 8-azapurines II were designed as antiplatelet agents. The target compounds I and II were synthesized in acceptable yields via a few procedures starting from amino alcohol and 4,6-dichloropyrimidine. All the synthesized compounds were evaluated for the ability to inhibit ADP-induced human platelet aggregation in vitro, with ticagrelor being used as a reference control. The results demonstrated that series I and II compounds showed potent antiplatelet aggregation activity. The structures of R1 and R2 were critical for the activities of series I. Compound Io had an IC50 value of 0.82 μM which was slightly less potent than that of ticagrelor (IC50 = 0.74 μM). The introduction of hydrazone at the N6 position of 8-azapurine thus clearly resulted in an improvement in the activities of series II. When the hydrazone moiety R3 was hydroxyphenyl, compounds IIh, IIg and IIi exhibited potent antiplatelet aggregation activities with IC50 values of 0.20 μM, 0.67 μM and 0.78 μM, respectively. Compound IIh performed best, being almost 4-fold more active than ticagrelor (IC50 = 0.74 μM). Further, the antiplatelet aggregation test in vivo of IIh demonstrated that IIh and/or its active metabolite were present in mice 1 hour after intravenous injection/oral administration and its efficacy was superior to that of ticagrelor. The safety profile of compound IIh was determined by bleeding and single-dose toxicity tests. The results of the bleeding test indicated that IIh had a much shorter bleeding time and less blood loss than ticagrelor. Also, the single-dose toxicity test indicated that IIh had a lower acute toxicity. The molecular docking of IIh with P2Y12 indicated that IIh exhibits superior binding energy (ΔG = −4.14 kcal mol−1) and inhibition constant (Ki = 0.91 mM) relative to ticagrelor (ΔG = −3.03 kcal mol−1; Ki = 6.02 μM). These are consistent with the test data (IIh: IC50 of 46 nM; ticagrelor: IC50 of 39 nM). These results indicate that N6 derivatives of 8-azapurine have the potential to be candidates for the development of novel antiplatelet drugs.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This study was supported by Beijing Natural Science Foundation (grant number 2192004). Thanks to Dr. Xiuqing Song for conducting the NMR tests.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00128k
Notes and references
- Tu W. Fan J. Zhang H. Xu G. Liu Z. Qu J. Yang F. Zhang L. Luan T. Yuan J. Bioorg. Med. Chem. Lett. 2014;24:141–146. doi: 10.1016/j.bmcl.2013.11.055. [DOI] [PubMed] [Google Scholar]
- Horiuchi H. Ann. Med. 2006;38:162–172. doi: 10.1080/07853890600640657. [DOI] [PubMed] [Google Scholar]
- Jacobson K. A. Jarvis M. F. Williams M. J. Med. Chem. 2002;45:4057–4093. doi: 10.1021/jm020046y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gachet C. Thromb. Haemostasis. 2001;86:222–232. [PubMed] [Google Scholar]
- André P. Delaney S. M. LaRocca T. Vincent D. DeGuzman F. Jurek M. Koller B. Phillips D. R. Conley P. B. J. Clin. Invest. 2003;112:398–406. doi: 10.1172/JCI17864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach P. Antonsson T. Bylund R. Björkman J.-A. Österlund K. Giordanetto F. van Giezen J. Andersen S. M. Zachrisson H. Zetterberg F. J. Med. Chem. 2013;56:7015–7024. doi: 10.1021/jm400820m. [DOI] [PubMed] [Google Scholar]
- Parlow J. J. Burney M. W. Case B. L. Girard T. J. Hall K. A. Harris P. K. Hiebsch R. R. Huff R. M. Lachance R. M. Mischke D. A. J. Med. Chem. 2010;53:2010–2037. doi: 10.1021/jm901518t. [DOI] [PubMed] [Google Scholar]
- Storey R. F. Curr. Pharm. Des. 2006;12:1255–1259. doi: 10.2174/138161206776361318. [DOI] [PubMed] [Google Scholar]
- Hollopeter G. Jantzen H.-M. Vincent D. Li G. England L. Ramakrishnan V. Yang R.-B. Nurden P. Nurden A. Julius D. Nature. 2001;409:202–207. doi: 10.1038/35051599. [DOI] [PubMed] [Google Scholar]
- Sugidachi A. Asai F. Ogawa T. Inoue T. Koike H. Br. J. Pharmacol. 2000;129:1439–1446. doi: 10.1038/sj.bjp.0703237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D. E. Ingram K. T. Hosp. Pharm. 2015;50:922. doi: 10.1310/hpj5010-922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springthorpe B. Bailey A. Barton P. Birkinshaw T. N. Bonnert R. V. Brown R. C. Chapman D. Dixon J. Guile S. D. Humphries R. G. Bioorg. Med. Chem. Lett. 2007;17:6013–6018. doi: 10.1016/j.bmcl.2007.07.057. [DOI] [PubMed] [Google Scholar]
- Conroy S. Kindon N. Kellam B. Stocks M. J. J. Med. Chem. 2016;59:9981–10005. doi: 10.1021/acs.jmedchem.5b01972. [DOI] [PubMed] [Google Scholar]
- Jakubowski J. A. Matsushima N. Asai F. Naganuma H. Brandt J. T. Hirota T. Freestone S. Winters K. J. Br. J. Clin. Pharmacol. 2007;63:421–430. doi: 10.1111/j.1365-2125.2006.02792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows T. A. Bhatt D. L. Circ. Res. 2007;100:1261–1275. doi: 10.1161/01.RES.0000264509.36234.51. [DOI] [PubMed] [Google Scholar]
- Hagihara K. Kazui M. Ikenaga H. Nanba T. Fusegawa K. Takahashi M. Kurihara A. Okazaki O. Farid N. Ikeda T. Xenobiotica. 2009;39:218–226. doi: 10.1080/00498250802650077. [DOI] [PubMed] [Google Scholar]
- Mega J. L. Close S. L. Wiviott S. D. Shen L. Hockett R. D. Brandt J. T. Walker J. R. Antman E. M. Macias W. Braunwald E. N. Engl. J. Med. 2009;360:354–362. doi: 10.1056/NEJMoa0809171. [DOI] [PubMed] [Google Scholar]
- Farid N. Payne C. Small D. Winters K. Ernest C. Brandt J. Darstein C. Jakubowski J. Salazar D. Clin. Pharmacol. Ther. 2007;81:735–741. doi: 10.1038/sj.clpt.6100139. [DOI] [PubMed] [Google Scholar]
- Kalra K. Franzese C. J. Gesheff M. G. Lev E. I. Pandya S. Bliden K. P. Tantry U. S. Gurbel P. A. Curr. Atheroscler. Rep. 2013;15:1–13. doi: 10.1007/s11883-013-0371-3. [DOI] [PubMed] [Google Scholar]
- Van Giezen J. Humphries R. G. Semin. Thromb. Hemostasis. 2005;31:195–204. doi: 10.1055/s-2005-869525. [DOI] [PubMed] [Google Scholar]
- James S. K. Roe M. T. Cannon C. P. Cornel J. H. Horrow J. Husted S. Katus H. Morais J. Steg P. G. Storey R. F. BMJ. 2011;342:d3527. doi: 10.1136/bmj.d3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio F. Cancer Res. 2012;72:5441–5447. doi: 10.1158/0008-5472.CAN-12-1600. [DOI] [PubMed] [Google Scholar]
- Vince R. Brownell J. Daluge S. J. Med. Chem. 1984;27:1358–1360. doi: 10.1021/jm00376a025. [DOI] [PubMed] [Google Scholar]
- Vince R. Turakhia R. H. Shannon W. M. Arnett G. J. Med. Chem. 1987;30:2026–2030. doi: 10.1021/jm00394a017. [DOI] [PubMed] [Google Scholar]
- Wang Y. Yan H. Ma C. Lu D. Bioorg. Med. Chem. Lett. 2015;25:4461–4463. doi: 10.1016/j.bmcl.2015.09.002. [DOI] [PubMed] [Google Scholar]
- Küçükgüzel Ş. G. Oruç E. E. Rollas S. Şahin F. Özbek A. Eur. J. Med. Chem. 2002;37:197–206. doi: 10.1016/s0223-5234(01)01326-5. [DOI] [PubMed] [Google Scholar]
- Rollas S. Gulerman N. Erdeniz H. Il Farmaco. 2002;57:171–174. doi: 10.1016/s0014-827x(01)01192-2. [DOI] [PubMed] [Google Scholar]
- Savini L. Chiasserini L. Travagli V. Pellerano C. Novellino E. Cosentino S. Pisano M. B. Eur. J. Med. Chem. 2004;39:113–122. doi: 10.1016/j.ejmech.2003.09.012. [DOI] [PubMed] [Google Scholar]
- He H. Wang X. Shi L. Yin W. Yang Z. He H. Liang Y. Bioorg. Med. Chem. Lett. 2016;26:3263–3270. doi: 10.1016/j.bmcl.2016.05.059. [DOI] [PubMed] [Google Scholar]
- Zhang B. Zhao Y. F. Zhai X. Fan W. J. Ren J. L. Wu C. F. Gong P. Chin. Chem. Lett. 2012;23:915–918. [Google Scholar]
- Gurbel P. A. Becker R. C. Mann K. G. Steinhubl S. R. Michelson A. D. J. Am. Coll. Cardiol. 2007;50:1822–1834. doi: 10.1016/j.jacc.2007.07.051. [DOI] [PubMed] [Google Scholar]
- Nicholson N. S. Panzer-Knodle S. G. Haas N. F. Taite B. B. Szalony J. A. Page J. D. Feigen L. P. Lansky D. M. Salyers A. K. Am. Heart J. 1998;135:S170–S178. doi: 10.1016/s0002-8703(98)70245-5. [DOI] [PubMed] [Google Scholar]
- Born G. V. R. Nature. 1962;194:927–929. doi: 10.1038/194927b0. [DOI] [PubMed] [Google Scholar]
- Aleil B. Ravanat C. Cazenave J. Rochoux G. Heitz A. Gachet C. J. Thromb. Haemostasis. 2005;3:85–92. doi: 10.1111/j.1538-7836.2004.01063.x. [DOI] [PubMed] [Google Scholar]
- Sanner M. F. J. Mol. Graphics Modell. 1999;17:57–61. [PubMed] [Google Scholar]
- Zhang J. Zhang K. Gao Z.-G. Paoletta S. Zhang D. Han G. W. Li T. Ma L. Zhang W. Müller C. E. Nature. 2014;509:119–122. doi: 10.1038/nature13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong D. Xue T. Guo B. Cheng J. Liu S. Wei J. Lu Z. Liu H. Gong G. Lan T. J. Med. Chem. 2019;62:3088–3106. doi: 10.1021/acs.jmedchem.8b01971. [DOI] [PubMed] [Google Scholar]
- Zhang S. Hu L. Du H. Guo Y. Zhang Y. Niu H. Jin J. Zhang J. Liu J. Zhang X. Thromb. Haemostasis. 2010;104:845–857. doi: 10.1160/TH10-05-0285. [DOI] [PubMed] [Google Scholar]
- Chang H. Yanachkov I. Dix E. Li Y. Barnard M. Wright G. Michelson A. Frelinger III A. J. Thromb. Haemostasis. 2012;10:2573–2580. doi: 10.1111/jth.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H. Yanachkov I. B. Michelson A. D. Li Y. Barnard M. R. Wright G. E. Frelinger III A. L. Thromb. Res. 2010;125:159–165. doi: 10.1016/j.thromres.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M. Huey R. Lindstrom W. Sanner M. F. Belew R. K. Goodsell D. S. Olson A. J. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M. Huey R. Olson A. J. Curr. Protoc. Bioinf. 2008;24:8.14.11–18.14.40. doi: 10.1002/0471250953.bi0814s24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.