

Abstract
Recent catalytic methods to construct medium-sized lactams and partially saturated benzazepines and their derivatives are surveyed. The review is divided into the following sections:
1 Introduction
2 Non-Transition Metal Catalyzed Reactions
2.1 Beckmann Rearrangement
2.2 Non-Beckmann Rearrangement Reactions
2.3 Multi-component reactions
3 Transition Metal-Catalyzed Reactions
3.1 Au-catalyzed reactions to access medium-sized N-heterocycles
3.2 Reactions involving a metal η3-complex catalytic intermediate
3.3 Transition metal-catalyzed reactions of strained cycloalkanes
4 Conclusions
Keywords: Beckmann rearrangement, caprolactam, benzazepine, benzazepinone, medium-ring N-heterocycle
Graphical Abstract

Introduction
Medium-ring nitrogen heterocycles are common structural motifs. Benzazepines, in particular, are privileged scaffolds present in a wide range of bioactive compounds.1 Partially saturated benzazepines, or a related variants, are the core structure of many pharmaceutical products: tolvaptan,2 benazepril,3 and zilpaterol,4 a beef improvement agent, are examples of the tetrahydro benzo[b]azepine scaffold (Scheme 1). Despite their proven pharmaceutical worth, partially saturated benzazepines and other larger N-heterocyclic rings are underrepresented in drug discovery libraries.5 Despite N-heterocycles being present in 59% of FDA-approved small molecule drugs, seven- and eight-membered rings are present in only approximately 4% of these compounds.5b Their absence has been attributed to unfavorable enthalpic- and entropic barriers—including developing Pitzer-, Baeyer-, and transannular strain—present in the ring closure transition states that lead to medium-sized rings.6 For these reasons, the development of methods that efficiently construct medium-sized rings remains a long-standing challenge in organic synthesis. Within this review, recent synthetic methods from the past 15 years that address this gap are examined. This review is divided into two parts: the development of non-transition metal-catalyzed reactions and transition metal-catalyzed reactions to construct seven-membered ring N-heterocycles.
Scheme 1.

Bioactive compounds that contain partially saturated benzazepine cores.
Non-Transition Metal-Catalyzed Reactions
Beckmann Rearrangement
Historically, medium-ring lactams were synthesized using the Beckmann rearrangement. This reaction, first reported in 1886,7,8 is an efficient tool that constructs amides from their corresponding oximes by generating an electrophilic nitrogen reactive intermediate that triggers an alkyl- or aryl group migration to create a new C─N bond with concomitant cleavage of the N─O bond (Scheme 2).9 The use of the Beckmann rearrangement for the industrial manufacture of Nylon-6 and Nylon-12 demonstrates its broad impact on society.10 While the Beckmann rearrangement can be catalyzed using oleum, this produces significant amounts of sulfates as by-products.11 Further its relative harsh conditions, such as the use of strong acids and high temperatures, however, has precluded its use to construct complex, functionalized molecules. Together these limitations have spurred the development of organocatalytic- and Lewis acid-catalyzed methods to identify more mild conditions to broaden the substrate scope.
Scheme 2.

Development of cyanuric chloride-catalyzed Beckmann rearrangement.
The first example of an organocatalytic Beckmann rearrangement was reported by Yamamoto and Ishihara in 2005.12 Their work was inspired by the Beckmann rearrangement of O-picrylbenzophenone oxime 3, which afforded N-picrylbenzanilide 5 and benzanilide 6 via picryl N-phenylbenzimidate 4 (Scheme 2).13 The authors recognized that in the absence of water, the benzanilide product might be formed through a nucleophilic attack via a Meisenheimer complex (10) if the aryl moiety was sufficiently electron deficient. After screening several electron-deficient arenes, they found that the Beckmann rearrangement could be catalyzed with as little 2 mol % of cyanuric chloride and 2 mol % of ZnCl2 in refluxing acetonitrile. While their protocol worked well for a variety of ketones, the formation of medium-rings was challenging with ε-caprolactam afforded in only 30%.
Ishii and co-workers built on this strategy with additional catalyst development to improve the yield of medium-sized lactams (Scheme 3).14 The authors performed a solvent screen and identified hexafluoroisopropanol (HFIP) as a more optimal solvent than acetonitrile. Using HFIP as solvent, the authors examined a range of potential electron-deficient arenes as catalysts. They found 1,3,4-triazo-2,4,6-triphosphorine-2,2,4,4,6,6-chloride (TAPC) as the most efficient catalyst for the conversion of ketoximes to lactams. The catalytic performance of several catalysts was examined in both HFIP and acetonitrile: the fluorinated analog of TAPC (TAPF) was found to be inactive, and trichloroisocyanuric acid (TICNA) proved to be highly active in HFIP but much less active in acetonitrile. Following their earlier report,15 the authors attributed the improved yield in HFIP to its weak acidity, which enables reaction without the addition of a strong acid catalyst. The use of the weaker acid was proposed to allow the formation of an active transient intermediate without decomposition.15 While using HFIP as solvent resulted in significantly more efficient catalysis to produce medium-ring lactams, one weakness is that HFIP is significantly more expensive than acetonitrile.
Scheme 3.

Development of TAPC-catalyzed Beckmann rearrangement.
Using TAPC, Ishii and co-workers examined the Beckmann rearrangement of cyclohexanone oxime to ε-caprolactam (Scheme 3).14 As expected from their optimization study, they found that switching from acetonitrile to HFIP improved significantly the reaction conversion and that increasing the reaction time from 2 h to 4 h also had a beneficial effect. They found that the by-product of the reaction, imidate 15, could be hydrolyzed to 11 using MeSO3H in t-BuOH/H2O mixture.
An efficient Beckmann-rearrangement for the synthesis of medium-ring lactams was reported by Lambert and co-workers (Scheme 4).16 Their elegant solution exploited the use of an electrophilic cyclopropene reagent, which they have exploited to develop other dehydrative processes.17 While they found that the dichlorodiphenylcyclopropene 16a mediated the formation of ε-caprolactam 11 in 95%, optimization of the catalyst structure by replacing the phenyl substituents with 2,4,6-trimethylphenyl groups resulted in a more efficient reaction—requiring only 30 min to produce the lactam in 96%. The authors proposed that product formation could occur through either a cyclopropenium-catalyzed mechanism or a self-propagating mechanism. In the former, the cyclopropenium catalyst functions to activate N-hydroxyl group of the oxime to be a good leaving group. Whereas, in a self-propagating mechanism, the reactive intermediate chloroimidate serves to activate the N-hydroxy group. Specifically, the cyclopropenium ion could activate the oxime to produce 17. Elimination of cyclopropenone forms chloroimidate 19, which could be re-captured by the cyclopropenone to afford 20 or the C─O bond could be formed by attack of oxime to produce 21. ε-Caprolactam would be furnished by chloride attack on 20 or 21 to regenerate the cyclopropenium ion catalyst in the activation mechanism or the chloroimidate in the self-propagation mechanism. Irrespective of which mechanism is operating, this variant of the Beckmann rearrangement is significantly milder and results in nearly quantitative yield of ε-caprolactam.
Scheme 4.

Development of a cyclopropenium-mediated Beckmann rearrangement.
Brennan and McLaughlin and co-workers reported a mild Lewis acid-catalyzed Beckmann rearrangement in 2018 (Scheme 5).18 They perceived that existing methods of the Beckmann rearrangement exhibited intolerance to functional groups prevalent in medicinal chemistry studies. To address this limitation, they targeted the use of calcium salts as Lewis acids because of their non-toxic nature and ready availability.19 Their optimization study using benzophenone oxime as the substrate identified Ca(NTf2)2 as a potent catalyst. They found that the combination of 10 mol % of Ca(NTf2)2 and 10 mol % of n-Bu4NPF6 catalyzed the conversion of oxime 22 into lactam 25 in 66% when a 4:1 mixture of DCE and DME was used as the reaction solvent. While the authors did not propose the specific role for the n-Bu4NPF6 co-catalyst, it was critical for the success of the reaction: if it was omitted from the reaction mixture, no reaction occurred. This reaction was temperature dependent with attenuated yields observed below 80 °C. At 80 °C, however, the reaction scope was broad. In particular, the conversion of erythromycin to azaerythromycin 27 showcases its ability to transform complex, functionalized molecules into amides efficiently without being adversely affected by the sensitive saccharide groups. The authors proposed a mechanism where coordination of the oxime oxygen to calcium occurs to give 23. This activation produces a transient calcium hydroxide intermediate which attacks a nitrilium ion to form 24, which dissociates the calcium catalyst and tautomerizes to form the amide product. The authors eliminated C─O bond formation through attack of water as a mechanistic possibility because full conversion to the amide was observed in the presence of 4 Å molecular sieves.
Scheme 5.

Development of Ca-catalyzed Beckmann rearrangement.
Catalysis of the Beckmann rearrangement using a boronic Lewis acids was reported by Hall and co-workers in 2018.20 This report built on earlier reports by the authors using boronic acid catalysis (BAC) to trigger the substitution of allylic alcohols as well as the Friedel–Crafts alkylation of neutral arenes.21 After optimization, the authors found that using 5 mol % of boronic acid 30 in combination with 5 mol % of perfluoropinacol in a 4:1 mixture of HFIP and MeNO2, the author’s method transformed a broad range of oximes into amides. Although their method requires the use of the expensive HFIP, their method was highly effective: medium-ring lactams 11, 12 and 29 were constructed in good yields.
The authors found that the success of the reaction depended on the use of HFIP. The authors attributed its requirement to providing a polar reaction medium as well as activating the boronic acid catalyst. The author’s latter assertion was validated by their observation that adding a substoichiometric amount of perfluoropinacol allowed the amount of the boronic acid catalyst to be reduced to as little as 5 mol %. They interpreted this result to be that the boronic acid was activated through substitution to generate in situ a perfluoronated boronic acid ester (e.g. 31). The increased electrophilicity of this ester would promote the initial transesterification. In line with their hypothesis, the authors independently synthesized 31 and showed that it was a competent catalyst, and that replacing the perfluoronated group with a pinacol resulting in a boronic acid ester that was inert to promote the transesterification yet effectively triggered the Beckmann rearrangement of activated oxime ester 33. Together, this data led the authors to propose that the boronic acid had dual catalyst roles in the reaction. It functioned to not only mediate the transesterification with the oxime, but also to catalyze the Beckmann rearrangement. The author’s kinetic experiments and absence of dimerization by-products provided evidence in line with catalysis as opposed to self-propagation. On the basis of their mechanistic experiments, the authors proposed that the catalytic cycle of their transformation begins with esterification of boronic acid 30 with perfluoropinacol to produce 31, which triggers a rate-limiting transesterification with oxime 7 to form acyl oxime 33. Beckmann rearrangement via TS-34 forms acyl imidate 35, which can undergo solvolysis with HFIP to produce boronic acid 30b or transesterification with oxime to form 33. Either process produces imidate 36 that tautomerizes to ε-caprolactam 11.
Hyodo and co-workers developed a method to directly transform ketones into amides using the Beckmann rearrangement (Scheme 7).22 The authors targeted the development of a transoximation reaction because this strategy would eliminate the preparation and purification of oximes. While the use of O-(mesitylsulfonyl)hydroxylamine (MSH) enables the synthesis of oximes under mild conditions,23 it is explosive.24 On the basis of their earlier work exploiting Brønsted acid-catalyzed transoximation for the synthesis of nitriles from aldehydes using an O-protected oxime,25 the authors envisioned that a Beckmann rearrangement might also be achieved using this tactic if conditions could be found to allow for the use of ketones as substrates. During the course of their optimization, the authors found that the identity of the oxime O-substituent was critical to the success of the reaction with phenylsulfonyl providing the highest yield at room temperature when 1.05 equivalents of water were present. In the absence of water, only a trace amount of lactam was formed. Using these conditions, the authors reported that several medium-ring lactams could be produced in high yield with good migratorial selectivity. In addition to efficiently synthesizing ε-caprolactam 11 in 81% yield from cyclohexanone, the authors also demonstrated that in more functionalized cyclohexanones the 1,2 shift of the more substituted α-carbon was preferred. This preference enabled the synthesis of 37 and 2 from simple α-substituted cyclic ketones.
Scheme 7.

Transoximation to access medium-ring lactams.
The authors proposed that the their transoximation reaction occurred through mechanism outlined in Scheme 7. Protonation of 37 produced iminium 41, which was primed for hydrolysis to produce ethylacetate and PhO2SONH3+ 42. Reaction with cyclohexanone formed iminium 43. Ring expansion through a Beckmann rearrangement formed 44. Hydrolysis of 44 produced imidate 36, which rearranged to ε-caprolactam 11. Critical to the success of this mechanism was the ability of 37 to hydrolyze under mild conditions to PhO2SONH3+ 42 without competing with the ketone substrate for iminium ion formation.
Non-Beckmann Rearrangement reactions to access medium-ring lactams
A cooperative organocatalytic method was reported by Pan, Chi and co-workers to access azepino[1,2-a]indoles from bromoenals 45 and 2-nitrovinylindoles 46 (Scheme 8).26 In this Rauhut–Currier reaction,27 an N-heterocyclic carbene catalyst activates 45 to give 49, which is attacked by nucleophilic 48, generated from the 1,4-addition of benzene sulfinic acid to indole 46. Although a relative high catalyst loading of 20 mol % was required, the authors found that this nucleophilic addition occurred with excellent enantioselectivity across a broad range of substrates. For example, the authors showed a series of enals could be used as substrates as long as the β-substituent was an aryl or electron-rich heteroaryl group to afford 47a – 47e with almost perfect enantioselectivity. The authors also examined the effect on medium-ring N-heterocycle formation when the electronic- and steric nature of the indole component was modified. They found that azepino[1,2-a]indoles 47f – 47h were formed with excellent enantioselectivity. The breadth of the authors cooperative catalysis was demonstrated using pyrroles as substrates to produce 47i, albeit with attenuated enantioselectivity in comparison to the reported azepino[1,2-a]indoles.
Scheme 8.

Cooperative organocatalytic Rauhut–Currier reaction to construct azepino[1,2-a]indoles.
Kundu and co-workers published an efficient and a versatile method for the synthesis of indole-based polycyclic indolo-benzazepines 53 through a water-accelerated cationic π-(7-endo-trig) cyclization.28 To accomplish this, their strategy was to condense the arylamine moieties linked to C-4 in indole or azaindole systems 51 with aryl aldehydes in water containing substoichiometric amount of a Brønsted acid (Scheme 9). The authors reported that using water as the reaction medium was critical to the efficiency of the reaction because using organic solvents resulted in significantly longer reaction times (10 h versus 30 min). While only a representative subset of examples is shown in Scheme 9, their reaction tolerates aryl aldehydes, irrespective of their electronic nature. Alkyl aldehydes could also be used as substrates, however, the N-heterocyclic product (e.g. 53e) is formed in a significantly poorer yield. Despite the requirement of trifluoroacetic acid, pyridyl indoles are competent substrates furnishing medium ring N-heterocycles such as 53f in good yields.
Scheme 9.

Brønsted acid-catalyzed π-(7-endo-trig) cyclization to access medium-ring lactams.
An aza-Piancatelli cyclization-Michael addition tandem reaction of 2-substituted furans 54 and amines was reported by Gandon, Lebœuf and co-workers to construct bicyclic tetrahydrobenzo[b]azepines 56 (Scheme 10). Similar to the Brennan and McLaughlin and co-workers report discussed earlier,18 the authors used the combination of 5 mol % of Ca(NTf2)2 and 5 mol % of n-Bu4NPF6 as a Lewis acid to trigger an aza-Piancatelli cyclization. This reaction generated in situ cyclopentanone 55,29 which was trapped by the proximal aniline to create bicycle 56. The authors noted that alkyl amines were not tolerated, which is a common limitation of the aza-Piancatelli reaction. While only a few representative examples are included in Scheme 10, the authors demonstrated that the scope of their reaction is broad: irrespective of the electronic- or steric nature of the aniline, bicyclic tetrahydrobenzo[b]azepines 56a – 56d were formed in good yields. Secondary aryl amines were also potent nucleophiles to afford 56e although an elevated reaction temperature (60 °C) was required for complete conversion. In contrast, N-alkoxy secondary amines smoothly reacted at 20 °C to give 56f in good yield. The authors demonstrated that their reaction was not limited to secondary furyl alcohols containing an aryl amine substituent, but that substrates bearing an aliphatic amine could effectively trap the cyclopentanone reactive intermediate. These substrates, however, required an increased reaction temperature (40 °C) and afforded the bicyclic product as a mixture of diastereomers (cf. 56g, 69:31 d.r.).
Scheme 10.

Development of an aza-Piancatelli cyclization-Michael addition tandem reaction to access bicyclic medium-ring N-heterocycles.
Chen and co-workers reported a modular asymmetric [4+3] annulation to construct spirocyclic azepines from Morita–Baylis–Hillman carbonates 57 and 1-azadienes 58 (Scheme 11).30 The success of the author’s annulation strategy required the chiral amine catalyst to react with 57 to generate allylic ylide 61. Addition to 58 forms 62, which cyclizes to afford the spirocyclic N-heterocycle 60. The author’s optimization study revealed that tertiary amine 59 triggers the formation of spirocycles 60 with the highest enantioselectivity. Using this catalyst, the scope of the reaction proved to be broad tolerating alkyl-, benzyl- or Boc-substituents on the oxindole nitrogen irrespective of the electronic nature of the oxindole’s aromatic moiety to afford 60a – 60d with excellent enantioselectivities, although a slight diminishment in both the yield and % ee was observed with the electron rich oxindole 57c. Pyridyl oxindoles were also excellent substrates to afford N-heterocycles such as 60e. The reaction also tolerated a range of aryl substituents on the 1-azadiene component including electron-rich arenes, electron-deficient arenes or thiophenes to afford 60f – 60h in good yield and with excellent enantioselectivities.
Scheme 11.

Development of a chiral amine-catalyzed [4+3] cyclization reaction to access spirocyclic azepines.
Multicomponent synthesis of medium-ring lactams
Because of their modularity and inherent efficiency, multicomponent reactions have elicited significant research interest for the synthesis of benzodiazepine derivatives. In 2010, Voskressensky and co-workers reported an azide-Ugi four component reaction of 2-carboxylate arylisocyanides with ketones, ammonium chloride and sodium azide to afford tetrazole 64, which spontaneously lactamized into tetrazolo-1,4-benzodiazepines 65 (Scheme 12).31 While only a few representative examples are shown, the scope of the author’s reaction was relatively broad tolerating a range of ketones including piperidone and halide- or carboxylate-substituents on the arylisocyanide. Missing from their substrate scope, however, were electron-rich arylisocyanides.
Scheme 12.

Development of four component azide-Ugi reactions to access benzodiazepines.
Since this initial report, multicomponent Ugi-cyclization strategies have emerged as a common strategy to construct benzazepinones (Scheme 13). Hulme and co-workers showed that dihydroquinazoline-benzodiazepine tetracycles, such as 67, could be efficiently synthesized in a two-step process where an Ugi reaction stitched together an alkyl isocyanide, alkyl aldehyde, N-Boc anthranilic acid and 2-substituted anilines to form 66, which cyclized to 67 upon exposure to trifluoroacetic acid.32 The groups of Alidadeh and Shaabani reported a four component Ugi reaction using diketone as the electrophilic component to construct peptoids such as 69 via 68 in good yield.33 Yan and co-workers reported fluorous phase technology dould be exploited in a four component Ugi-reaction to construct medium-sized N-heterocycles such as 71 in good yield.34 To showcase the potential power of their method, the authors demonstrated that the perfluorinated sulfonate group of 71 was a competent partner in Suzuki–Miyaura cross-coupling reactions to enable additional functionalization. By incorporating the perfluorinated group, the authors effectively demonstrated that their strategy lends itself well to the combinatorial synthesis of libraries of medium-ring N-heterocycles.
Scheme 13.

Development of four component Ugi reactions.
Organocatalytic methods using chiral amine catalysts were also reported by Romo and co-workers to efficiently access tetrahydrobenzo[c]azocinones (Scheme 14).35 The authors demonstrated that in situ generation of chiral α,β-unsaturated ammonium salt 75 effectively lowered the LUMO to enable 1,4-addition of a malonate. While the authors initially developed this method to construct five- and six-membered N-heterocycles,36 their LUMO-lowering strategy proved amenable for the construction of seven- and eight membered lactams. Although 20 mol % of TMSQN was required, their method constructed chiral, non-racemic eight-membered N-heterocycles in high enantioselectivity from acryloyl chlorides with either methyl- or carboxylate β-substituents. The authors also showed that changing the electronic identity of the aryl malonate did not adversely affect the outcome of the reaction. Remarkably, the aryl moiety on the malonate was not required as a template to achieve synthetically useful yields—using a stronger base (LiHMDS) enabled the formation of chiral non-racemic ε-caprolactams 77a and 77b, and even azocanone (77c) could be formed with excellent enantioselectivity, albeit in a diminished yield.
Scheme 14.

Chiral amine-catalyzed reactions to access medium-ring lactams through LUMO lowering.
Transition metal-catalyzed methods
Interest in developing transition metal-catalyzed reactions to access medium-sized N-heterocycles continues to be motivated by the new reactivity patterns that transition metals can enable. In 2010, Li and co-workers reported an Fe(III)-catalyzed Prins cyclization/halogenation of alkynyl acetals (Scheme 15).37 The authors anticipated that treatment of an acetal tethered to an acetylene (e.g. 78) with a Lewis acid would generate an oxocarbenium ion (79). This electrophile might be trapped by the proximal acetylene through a [2+2] cycloaddition or stepwise cyclization to produce oxonium 80 which would rearrange through an eliminative ring-opening to give 81. The author’s optimization study identified that 20 mol % of ferric chloride hydrate effectively triggered their desired reaction. While this reaction proved efficient to generate a range of 5- and 6-membered cycles, seven- and even eight membered N-heterocycles (81a or 81b) could be constructed, albeit in lower yields.
Scheme 15.

Iron-catalyzed Prins-cyclization/halogenation sequence to access medium ring N-heterocycles.
In 2010, Van der Eycken and co-workers reported an efficient two-step protocol for the synthesis of medium-sized N-heterocycles (Scheme 16).38 Critical to the success of their strategy was identification of the optimal conditions for the three-component coupling between an aldehyde, terminal acetylene and an amine to produce 83. After a screen of reaction conditions, the authors identified the neat combination of 15 mol % of CuI and substrates at 90 °C to provide the best result. The authors reported that the yield of the Cu-catalyzed three-component coupling reaction went down when the use of microwave radiation was replaced with conventional heating. A subsequent Pd-catalyzed intramolecular acetylene hydroarylation reaction constructed the medium-sized N-heterocycle product. The authors found that this two-step protocol worked best with an electron-rich N-substituent on 82 but did not report the effect of changing the identity of the aryl substituents on 82. Outside of these limitations, the scope with regards to the aldehyde and acetylene component was broad: aryl- or alkyl aldehydes could be used, and the aryl substituent on the acetylene could be electron-rich, electron-poor or a heteroarene. In these cases, smooth conversion to benzazepines 84a – 84e was observed. Remarkably, this strategy worked well to construct eight-membered rings (e.g. 84f), albeit with a reduced yield for the Pd-catalyzed hydroarylation.
Scheme 16.

Cu-Catalyzed three component coupling / Pd-catalyzed hydroarylation two-step protocol to construct medium-ring N-heterocycles.
Gold-catalyzed reactions to access medium-sized N-heterocycles.
Zhang and co-workers reported an efficient gold-catalyzed oxidative cyclization to construct tetrahydrobenz[b]azepin-4-ones 86 from tertiary anilines 85 bearing a homopropargylic substituent (Scheme 17).39 As an initial attempt to synthesize the desired benzazepinone, the authors treated N-oxide 87a•HCl with 5 mol % of Ph3PAuNTf2 and observed benzazepinone in 30%. After this initial result, the authors performed an optimization screen where the N-oxide is generated in situ to determine if the reaction yield might be improved, and their screen of a series of Au(I)- and Au(III)-catalysts identified that the Ph3PAuNTf2 to be the best catalyst to produce tetrahydrobenz[b]azepin-4-one 86a in significantly higher yields. Based on their initial result, the authors posited that the aniline nitrogen serves to relay the oxygen from the m-CPBA oxidant to the tethered acetylene to produce 87. Coordination of the π-philic gold catalyst with the terminal acetylene produces 88, which activates it toward attack by the N-oxide to produce 89. Fragmentation of 89 generates gold carbenoid 90, which reacts with the electron-rich arene to yield the N-heterocyclic product. In line with their mechanistic hypothesis, substrates that lack a nitrogen atom were found to be inert. Their reaction is remarkably general tolerating a range of functional groups including iodide-, carboxylate- and heteroaromatic substituents to produce tetrahydrobenz[b]azepin-4-ones such as 86a – 86d. Unfortunately, it is not regioselective. Substrates that contain two nucleophilic sites react to create the medium-ring N-heterocycle(e.g. 86e and 86f) as a mixture of regioisomers. Notably while gold is required for terminal acetylenic substrates, no catalyst is required when the hydrogen is replaced with an electron-withdrawing group.
Scheme 17.

Au-Catalyzed oxidative cyclization of tertiary anilines to synthesize tetrahydrobenz[b]azepin-4-ones.
Zhang and co-workers further advanced this oxygen-atom relay chemistry to construct azepan-4-ones 96 through a two-step N-alkylation-[5+2] annulation reaction sequence (Scheme 18).40 To achieve their desired annulation, the authors screened a variety of Au(I)- and Pt(II) catalysts, and (2-biphenyl)Cy2PAuNTf2 was found to be the best catalyst. A control experiment established that Tf2NH did not catalyze azapanone formation. To account for product formation, the authors proposed that after alkylation of 91, azepanone formation occurred through gold-coordination of the acetylene to afford 93, which activated it towards attack by the pendant N-oxide to produce 94. Ring-opening isomerization then afforded α-gold carbenoid 95. Subsequent functionalization of the aminomethylene C─H bond produced the N-heterocyclic product. Their reaction proved to be remarkably robust tolerating a range of functional group including halogens, silyl ethers and carboxylates. The C─H functionalization step was diastereoselective—irrespective of the substituent position on the cyclic amine—as well as remarkably regioselective. The authors observed preferential functionalization of the secondary methylene over the benzyl C─H bond to provide 96f as the major product, and the gold carbenoid preferred to react with the secondary methylene instead of the α-C─H bond of the carboxylate to provide 96g as the major product.
Scheme 18.

Au-Catalyzed oxidative [5+2] cyclization reaction.
Zhou and Zhang reported that ring-fused tetrahydroazepines 100 could be formed from yne-enones through a gold-catalyzed intramolecular redox reaction (Scheme 19).41 Building on the work of Seidel and co-workers,42 the author’s goal was to directly functionalize an aminomethylene C─H bond by triggering a [1,5] H-shift in a tandem reaction of yne-enone 97. After screening a range of Lewis acids and π-philic metal complexes, the authors found that the reaction outcome depended on the identity of the catalyst. While the use of oxophilic Sc(OTf)3 led to tetrahydroquinolines, the carbophilic IPrAuOTf afforded tetrahydroazepines 100. The authors proposed that this seven-membered-ring N-heterocycle was formed by attack of the carbonyl oxygen on the gold-acetylene complex to generate 98. A 1,5 hydride shift then formed iminium ion 99, which was attacked by the furyl gold to afford azepine 100. The scope of this reaction was broad tolerating a range of aryl- or alkyl acetylenic substituents, although the latter required the reaction temperature to be increased to 40 °C. No ring opening was observed with cyclopropane substituents. The cyclic amine moiety in 97 could also be modified without attenuating the reaction yield. In addition to morpholine, the authors also examined a series of cyclic and acyclic amines and in every case a successful reaction outcome occurred—even for eight-membered rings.
Scheme 19.

Au-Catalyzed intramolecular redox reaction of yne-enones to access tetrahydroazepines.
Reactions involving a metal η3-complex catalytic intermediate
A cooperative approach to the synthesis of benzazepinones was reported by Glorius and co-workers that blended NHC organocatalysis with palladium catalysis to enable the regio- and enantioselective annulation between vinyl benzoxazinanone 101 and enal 102 (Scheme 20).43 Their strategy of cooperative umpolung annulation cleverly eliminated the mutual deactivation of the catalysts and led to the formation of a diverse set of medium-ring N-heterocycles with excellent enantioselectivities. Palladium tetrakistriphenylphosphine (5 mol %) was used to generate the requisite η3-complex from vinyl benzoxazinanone 101, and their optimization experiments identified triazolium 103 (15 mol %) as the best precatalyst to trigger the formation of homoenolate 105 from 102. Using these conditions, the authors demonstrated that their reaction scope was broad. Enals bearing a variety of aryl- and heteroaryl-β-substituents were shown to be smoothly converted to the benzazepinones 106a – 106c with excellent diastereo- and enantioselectivity. Their reaction even tolerated vinyl- and alkyl- β-substituents to afford N-heterocycles 106d and 106e, but the diastereoselectivity and enantioselectivity were slightly diminished in comparison. Changing the electronic- and steric nature of vinyl benzoxazinanone 101 also impacted the reaction—while the enantioselectivity was not affected, lower yields were observed with electron-poor 106f. Increasing the steric nature of the reaction center by adding a methoxy substituent led to reduced yield and reduced diastereoselectivity in 106h. The enantioselectivity, however, remained high.
Scheme 20.

Cooperative catalysis for the regio- and enantioselective annulation of enals and benzoxazinanones to create benzazepinones.
On the basis of their results, the authors proposed that benzazepinones were constructed by two cooperative catalytic cycles (Scheme 21). A palladium-catalyzed decarboxylation of vinyl benzoxazinanone 101 produces CO2 and palladium η3-allyl complex 104 that is intercepted by homoenolate 105, which was generated from the reaction of N-heterocyclic carbene 103 and aldehyde 102. The authors rationalized the stereochemical outcome of the annulation reaction with 107, where the indenyl NHC-substituent blocks approach of the palladium η3-allyl complex from the back face. Alkylation then provides 108, which then undergoes cyclization with the sulfonamide to form benzazepinone 106 and eliminates NHC 103.
Scheme 21.

Cooperative NHC- and Pd catalytic cycles for benzazepinones.
Cooperative catalysis was also reported by Du, Chen and co-workers using the combination of DABCO and a chiral, non-racemic Ir(I) catalyst to produce medium-ring spirocyclic N-heterocycles 112 from Morita–Baylis–Hillman (MBH) adducts oxindoles 109 and allylic carbonates 110 (Scheme 22).44 This work built on an earlier report of the authors that demonstrated that cooperative catalysis could be realized to achieve the chemoselective activations of MBH carbonates from isatins and simple allylic carbonates using phosphines and palladium(0) catalysts.30 The authors were curious if a similar cooperative catalysis strategy could be executed to realize a [4+3] annulation to construct spirocyclic medium-ring N-heterocycles, which would require differentiation of the allylic carbonate motif present in both substrates. After screening a series of palladium(0)- and iridium(I) catalysts as well as both chiral, non-racemic and racemic tertiary amines, the authors identified that 4 mol % of BINOL-Ir(I) BF4 111 and 20 mol % of DABCO effectively catalyzed the desired transformation. The authors noted that switching from DABCO to a chiral, non-racemic amine did not affect the enantioselectivity of the reaction. Remarkably, these two catalysts were chemoselective with DABCO reacting with oxindole 109 to produce ylide 113 and allylic addition of the iridium catalyst produced the η3-complex 114. The authors believed that the [4+3] cyclization occurred from the reaction of ylide 113 with 1,4-metal dipole 114. The authors examined the scope of the reaction and found that a successful reaction could be realized irrespective of the electronic nature of oxindole 109, albeit with reduced yields for electron-poor substrates like 112c. In contrast, the modifying the electronic nature of the aniline component did not impact the yield of the [4+3] annulation to smoothly give 112e and 112f.
Scheme 22.

Cooperative DABCO-Ir(I) catalysis to synthesize
A Pd-catalyzed asymmetric [4+3] cycloaddition of trimethylenemethane 115 and benzofuran-derived azadienes 116 was reported by Yang, Deng and co-workers to efficiently construct benzofuro[3,2-b]azepines 119 (Scheme 23).26 Similar to the report of Du, Chen and co-workers, a chiral, non-racemic metal catalyst was used to achieve an enantioselective reaction. No cooperative catalysis was necessary because the authors envisioned that the requisite 1,4-dipole component might originate from 116 through an aromatization driven charge separated resonance structure 120. Reaction of 120 with a chiral, non-racemic palladium trimethylenemethane (Pd-TMM) 121 in a [4+3] annulation would furnish the fused azepine 119. To accomplish this, the authors screened a series of chiral, non-racemic phosphoramidites using 2.5 mol % of Pd2(dba)3 and identified 10 mol % of L117 or L118 as the most effective ligands to produce 119 in almost perfect enantioselectivity. A phosphoramidite ligand was critical to the success of the reaction. The authors reported that no reaction was observed when BINAP or other chiral, non-racemic diphosphine ligands were used. For 115a, L117 provided the best diastereoselectivity and highest enantioselectivity irrespective of whether an aryl substituent or alkyl substituent was present on 116. When the TMM precursor was switched to 115b, phosphoramidite L118 proved to be the best ligand to access a diverse array of azepines—irrespective of the electronic nature of benzofuran 116—in high yield with nearly perfect enantioselectivity.
Scheme 23.

Pd-Catalyzed asymmetric [4+3] cycloaddition to synthesize benzofuro[3,2-b]azepines.
Transition metal-catalyzed reactions of strained cycloalkanes
A directed carbonylative Rh-catalyzed C─C bond activation of aminocyclopropanes 122 was reported by Bower and Wang to construct benzazepinones 124 (Scheme 24).45 The author’s strategy was to “capture” an in situ generated rhodacyclopentanone 123 with a proximal nucleophile, which would “collapse” to the N-heterocyclic target. This “capture-collapse” approach had been previously employed by the Bower group to create heterobicyclic enones,46 1,3-diazepanes,47 perhydroisoindoles,48 and other complex, functionalized N-heterocycles.49 Their strategy is appealing because it leverages the strain of readily accessible stereodefined aminocyclopropanes to achieve a challenging ring closure. Optimization experiments revealed that N-heterocycle formation could be realized using the combination of 7.5 mol % of [Rh(cod)2]OTf, 15 mol % of p-(4-F3CC6H4)3P, 30 mol % of Na2SO4 under 1 atm of CO and 1 equivalent of ortho-nitrobenzoic acid. Using these conditions enabled the authors to access a range of benzazepinones. For example, a successful reaction outcome was obtained with a N-carboxylate or N-acyl groups as well as electron-rich or electron-deficient N-aryl groups to afford 124a – 124g. Remarkably, the N-aryl group was not a critical structural component: tetrahydroazepine 124i could even be constructed from an enamine cyclopropane.
Scheme 24.

Rh-Catalyzed “capture-collapse” approach to transform aminocyclopropanes into benzazepinones.
On the basis of their mechanistic experiments and reactivity trends, the authors proposed the catalytic cycle in Scheme 24 for benzazepinone formation. A regiospecific and selective rhodium-catalyzed carbonylative C─C bond activation of aminocyclopropane 122h produces only rhodacycle 125h—out of 6 possible regioisomers. This rhodacycle is captured by the N-aryl group to produce 123h through an intramolecular aryl C─H bond functionalization. Rhodacycle 123h then collapses to produce benzazepinone 124h and regenerate the rhodium catalyst.
Exploiting the ring-strain of small carbocycles was also employed by Driver and co-workers to construct benzazepinones 128 from cyclobutanol-substituted aryl azides 126 (Scheme 25).50 The authors were curious if a new reactivity pattern of metal N-aryl nitrene 127 might be realized by leveraging the ring strain in cyclobutanol to construct medium-ring N-heterocycles. After screening a series of established N-atom transfer catalysts, the authors determined that 1 mol % of Rh2(esp)2 efficiently transformed aryl azide 126 into benzazepinone 128. Using this catalyst, the authors found that the scope of the reaction was relatively broad, tolerating both electron-donating- or electron-withdrawing substituents on the aryl azide. The authors also examined the migratorial selectivity of the reaction by synthesizing a series of 2-substituted cyclobutanols. For these intramolecular competition experiments, the authors observed that the more substituted carbon migrated to afford 128f through preferential benzyl migration over methylene and 128g through methine migration instead of methylene. The author’s reaction was also stereospecific: only 128h was produced from the cis-substituted aryl azide 126h.
Scheme 25.

Rh2(II)-Catalyzed reaction of ortho-cyclobutanol substituted aryl azides to construct benzazepinones.
The authors proposed that N-heterocycle formation occurred via rhodium-N-aryl nitrene catalytic intermediate 127 (Scheme 26). Formation of this species from aryl azide 126 triggers a ring-expansion of the cyclobutanol to produce spirocycle 129. Aromaticity is restored upon acylium ion 131 formation, which is trapped by the nitrogen nucleophile to provide 132. Dissociation of the rhodium carboxylate catalyst affords benzazepinone 128. The intermediacy of an N-aryl rhodium nitrene was supported by the reactivity of ortho-cyclobutane-methyl ether 133. Instead of medium-ring N-heterocycle formation, exposure of this azide to reaction conditions produced the C─H bond amination product 135. C─H bond amination was also observed when the analogous cyclopentanol substrate was exposed to reaction conditions.
Scheme 26.
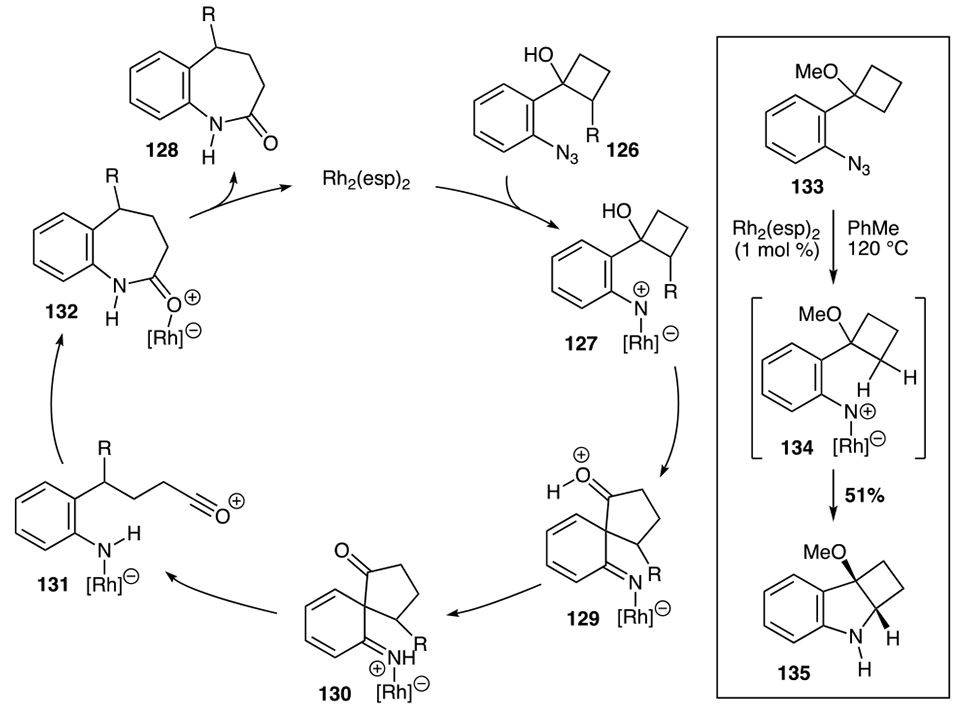
Catalytic cycle involving a putative Rh2(II)- N-aryl nitrene.
Driver and co-workers reported in 2020 that benzazepinones 128 could be constructed from cyclobutanol-substituted anilines (Scheme 27).51 The authors anticipated that the putative N-aryl nitrenoid intermediate might be accessed by a low temperature oxidation of 136 and were curious if it would exhibit similar reactivity as the rhodium N-aryl nitrene generated from the analogous aryl azide. After optimization, the authors determined that exposure of aniline 136 to 1.5 equivalents of PhI(O2CCF3)2 and 10 mol % of Sc(OTf)3 in TFE at 0 °C to room temperature produced benzazepinone 128. Similar to their aryl azide reaction, their reaction tolerates both electron-releasing and electron-withdrawing substituents on the aniline moiety. In contrast to the azide reactivity, benzazepinone 128d was not observed when an additional ortho-substituent was added to the aniline. The migratorial aptitude exhibited by the anilines was similar to that seen by the aryl azides. In every case reported, the more substituted carbon migrated to afford benzazepinones, such as 128f and 128g, as the only product. This oxidative cyclization-migration reaction was also stereospecific affording only 128h from the cis-disubstituted ortho-cyclobutanol aniline 136h.
Scheme 27.

I(III)-Mediated oxidative cyclization-migration to access benzazepinones.
The authors proposed that the mechanism for N-heterocycle formation involved the generation of electrophilic N-aryl iminoiodinane 137 (Scheme 27). The authors speculated that the role of the Lewis acidic Sc(OTf)3 was to promote the substitution of the iodine substituent with the aniline substrate. Ring-expansion to spirocycle 139 could occur from electrophilic N-aryl iminoiodinane 137 or from N-aryl nitrene 138, which would be produced after dissociation of iodobenzene. Proton transfer forms 140 and aromaticity is re-established to afford acylium ion 141, which is trapped by the Lewis basic nitrogen to form benzazepinone 128. In contrast to the reactivity observed with aryl azides, no C─H bond amination products were observed when cyclopentanone-derived aniline 142 was exposed to reaction conditions. The authors interpreted this result as evidence that the reaction proceeded via an iminoiodinane intermediate.
Conclusions
Over the past 15 years, significant progress has been made to develop efficient methods to access medium-ring N-heterocycles in a stereochemically defined manner. This progress was made across a variety of fronts: new variants of the venerable Beckmann rearrangement were discovered; new organocatalytic annulation reactions were reported and efficient, modular multicomponent reactions were reported. New transition metal-catalyzed reactions were also discovered to exploit new reactivity patterns embedded in catalytic intermediates. Together these new methods are addressing the gap in the synthetic repertoire to enable construction of challenging medium-ring lactams, partially saturated benzazepines and their derivatives.
Scheme 6.

Boronic Acid Catalysis (BAC) strategy to assemble medium-ring lactams.
Acknowledgment
The authors are grateful to the National Science Foundation and the National Institutes of Health for supporting their research program.
Funding Information
National Science Foundation, CHE-1564959
National Institutes of Health, NIGMS, R01GM138388.
Biography

Wrickban Mazumdar is currently working as a Postdoctoral Associate at The Scripps Research Institute (CALIBR division) focusing on medicinal chemistry. He finished his Ph.D. in 2020 under the direction of Tom G. Driver at the University of Illinois at Chicago. He received his Bachelor's degree from the Osmania University in 2010 and Master's degree from Vellore Institute of Technology in 2013. Prior to joining UIC in 2014, he also worked at Dr. Reddy Laboratories as a Senior Associate in the Analytical Research & Development division. His research work have been focused on transition metal catalyzed reactions for the synthesis of N-heterocycles & synthesis of bioactive N-heterocyclic chemical probes.

Tom G. Driver is a Professor of Chemistry. He obtained his B.S. in Chemistry from Indiana University, Bloomington in 1999, and his Ph.D. from the University of California, Irvine under the mentorship of K. A. Woerpel in 2004. After an NIH-funded postdoctoral position at Caltech under the supervision of John E. Bercaw and Jay A. Labinger, he began his independent academic career at the University of Illinois at Chicago in 2006. His group’s research program is centered on the development of new reactions that exploit electrophilic N-aryl nitrogen intermediates for the construction of C─N and C─C bonds and the use of these methods in medicinal chemistry applications.
References
- (1).(a) Ryan JH; Green JL; Hyland C; Smith JA; Williams CC In Progress in Heterocyclic Chemistry; Gribble GW, Joule JA, Eds.; Elsevier: 2011; Vol. 23, p 465; [Google Scholar]; (b) Meyer AG; Bissember AC; Hyland CJT; Williams CC; Szabo M; Abel S-AG; Bird MJ; Hyland IK; Pham H In Progress in Heterocyclic Chemistry; Gribble GW, Joule JA, Eds.; Elsevier: 2018; Vol. 30, p 493; [Google Scholar]; (c) Ji Ram V; Sethi A; Nath M; Pratap R In The Chemistry of Heterocycles; Ji Ram V, Sethi A, Nath M, Pratap R, Eds.; Elsevier: 2019, p 393. [Google Scholar]
- (2).Kondo K; Ogawa H; Yamashita H; Miyamoto H; Tanaka M; Nakaya K; Kitano K; Yamamura Y; Nakamura S; Onogawa T; Mori T; Tominaga M Bioorg. Med. Chem 1999, 7, 1743. [DOI] [PubMed] [Google Scholar]
- (3).Hou FF; Zhang X; Zhang GH; Xie D; Chen PY; Zhang WR; Jiang JP; Liang M; Wang GB; Liu ZR; Geng RW N. Engl. J. Med 2006, 354, 131. [DOI] [PubMed] [Google Scholar]
- (4).Lean IJ; Thompson JM; Dunshea FR PLOS ONE 2015, 9, e115904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Roughley SD; Jordan AM J. Med. Chem 2011, 54, 3451; [DOI] [PubMed] [Google Scholar]; (b) Vitaku E; Smith DT; Njardarson JT J. Med. Chem 2014, 57, 10257. [DOI] [PubMed] [Google Scholar]
- (6).(a) Illuminati G; Mandolini L Acc. Chem. Res 1981, 14, 95; [Google Scholar]; (b) Wiberg KB Angew. Chem., Int. Ed. Engl 1986, 25, 312; [Google Scholar]; (c) Weinhold F Nature 2001, 411, 539. [DOI] [PubMed] [Google Scholar]
- (7).Beckmann E Ber. Deutsch. Chem. Ges 1886, 19, 988. [Google Scholar]
- (8).(a) Donaruma LG; Heldt WZ. Org. React 1960, 11, 1. [Google Scholar]; (b) Gawley RE Org. React 1988, 35, 14. [Google Scholar]
- (9).Yamabe S; Tsuchida N; Yamazaki SJ Org. Chem 2005, 70, 10638. [DOI] [PubMed] [Google Scholar]
- (10).(a) Luedeke VD In Encyclopedia of Chemical Processing and Design, Mcketta JJ, Ed.; Marcel Dekker: New York, 1978, 72. [Google Scholar]; (b) Rademacher H In Ullmann’s Encyclopedia of Industrial Chemistry, 5th ed., Gerhartz W, Ed.; Wiley: New York, 1987, Vol. A8, 201. [Google Scholar]; (c) Weber JN In Kirk-Othmer Encyclopedia of Chemical Technology, 4th ed., Kroschwitz JI, Ed.; Wiley: New York, 1990, Vol. 19, 500. [Google Scholar]; (d) Wessermel K; Arpe H-J In Industrial Organic Chemistry, 4th ed., Wiley-VCH: Weinheim, Germany, 2003, 239. [Google Scholar]
- (11).Fisher WB; Crescentini L In Kirk-Othmer Encyclopedia of Chemical Technology, 4th ed., Kroschwitz JI, Ed.; Wiley: New York, 1990, Vol. 4, 827. [Google Scholar]
- (12).Furuya Y; Ishihara K; Yamamoto HJ Am. Chem. Soc 2005, 127, 11240. [DOI] [PubMed] [Google Scholar]
- (13).(a) Lampert BB; Bordwell FG J. Am. Chem. Soc 1951, 73, 2369; [Google Scholar]; (b) Fischer HP Tetrahedron Lett. 1968, 9, 285. [Google Scholar]
- (14).Hashimoto M; Obora Y; Sakaguchi S; Ishii YJ Org. Chem 2008, 73, 2894. [DOI] [PubMed] [Google Scholar]
- (15).Hashimoto M; Sakaguchi S; Ishii Y Chem. Asian J 2006, 1, 712. [DOI] [PubMed] [Google Scholar]
- (16).Vanos CM; Lambert TH Chem. Sci 2010, 1, 705. [Google Scholar]
- (17).Kelly BD; Lambert TH J. Am. Chem. Soc 2009, 131, 13930. [DOI] [PubMed] [Google Scholar]
- (18).Kiely-Collins HJ; Sechi I; Brennan PE; McLaughlin MG Chem. Commun 2018, 54, 654. [DOI] [PubMed] [Google Scholar]
- (19).Harder S Chem. Rev 2010, 110, 3852. [DOI] [PubMed] [Google Scholar]
- (20).Mo X; Morgan TDR; Ang HT; Hall DG J. Am. Chem. Soc 2018, 140, 5264. [DOI] [PubMed] [Google Scholar]
- (21).(a) Zheng H; Lejkowski M; Hall DG Chem. Sci 2011, 2, 1305; [Google Scholar]; (b) Zheng H; Ghanbari S; Nakamura S; Hall DG Angew. Chem. Int. Ed 2012, 51, 6187; [DOI] [PubMed] [Google Scholar]; (c) Mo X; Hall DG J. Am. Chem. Soc 2016, 138, 10762; [DOI] [PubMed] [Google Scholar]; (d) Mo X; Yakiwchuk J; Dansereau J; McCubbin JA; Hall DG J. Am. Chem. Soc 2015, 137, 9694. [DOI] [PubMed] [Google Scholar]
- (22).Hyodo K; Hasegawa G; Oishi N; Kuroda K; Uchida KJ Org. Chem 2018, 83, 13080. [DOI] [PubMed] [Google Scholar]
- (23).Tamura Y; Fujiwara H; Sumoto K; Ikeda M; Kita Y Synthesis 1973, 1973, 215. [Google Scholar]
- (24).Johnson CR; Kirchhoff RA; Corkins HG J. Org. Chem 1974, 39, 2458. [Google Scholar]
- (25).Hyodo K; Togashi K; Oishi N; Hasegawa G; Uchida K Org. Lett 2017, 19, 3005. [DOI] [PubMed] [Google Scholar]
- (26).Liu Y-Z; Wang Z; Huang Z; Zheng X; Yang W-L; Deng W-P Angew. Chem. Int. Ed 2020, 59, 1238. [DOI] [PubMed] [Google Scholar]
- (27).Rauhut MM; Currier H (American Cyanamid Co.) U.S. Patent 307499919630122, 1963; Chem. Abstr 1963, 58, 11224a.
- (28).Saifuddin M; Agarwal PK; Sharma SK; Mandadapu AK; Gupta S; Harit VK; Kundu B Eur. J. Org. Chem 2010, 2010, 5108. [Google Scholar]
- (29).(a) For reviews of the aza-Piancatelli cyclization, see: Piutti C; Quartieri F Molecules 2013, 18, 12290. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Verrier C; Moebs-Sanchez S; Queneau Y; Popowycz F Org. Biomol. Chem 2018, 16, 676.; [DOI] [PubMed] [Google Scholar]; (c) Gomes RFA; Coelha JAS; Afonso CAM Chem. Eur. J 2018, 24, 9170. [DOI] [PubMed] [Google Scholar]
- (30).Chen P; Chen Z-C; Li Y; Ouyang Q; Du W; Chen Y-C Angew. Chem. Int. Ed 2019, 58, 4036. [DOI] [PubMed] [Google Scholar]
- (31).Borisov RS; Polyakov AI; Medvedeva LA; Khrustalev VN; Guranova NI; Voskressensky LG Org. Lett 2010, 12, 3894. [DOI] [PubMed] [Google Scholar]
- (32).(a) Xu Z; Dietrich J; Shaw AY; Hulme C Tetrahedron Lett. 2010, 51, 4566; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dietrich J; Kaiser C; Meurice N; Hulme C Tetrahedron Lett. 2010, 51, 3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).(a) Zohreh N; Alizadeh A; Bijanzadeh HR; Zhu L-G J. Comb. Chem 2010, 12, 497; [DOI] [PubMed] [Google Scholar]; (b) Shaabani A; Maleki A; Hajishaabanha F; Mofakham H; Seyyedhamzeh M; Mahyari M; Ng SW J. Comb. Chem 2010, 12, 186. [DOI] [PubMed] [Google Scholar]
- (34).Zhou H; Zhang W; Yan BJ Comb. Chem 2010, 12, 206. [DOI] [PubMed] [Google Scholar]
- (35).Kang G; Yamagami M; Vellalath S; Romo D Angew. Chem. Int. Ed 2018, 57, 6527. [DOI] [PubMed] [Google Scholar]
- (36).Vellalath S; Van KN; Romo D Angew. Chem. Int. Ed 2013, 52, 13688. [DOI] [PubMed] [Google Scholar]
- (37).Xu T; Yang Q; Li D; Dong J; Yu Z; Li Y Chem. Eur. J 2010, 16, 9264. [DOI] [PubMed] [Google Scholar]
- (38).(a) Mont N; Pravinchandra Mehta V; Appukkuttan P; Beryozkina T; Toppet S; Van Hecke K; Van Meervelt L; Voet A; DeMaeyer M; Van der Eycken EJ Org. Chem 2008, 73, 7509; [DOI] [PubMed] [Google Scholar]; (b) Peshkov VA; Pereshivko OP; Donets PA; Mehta VP; Van der Eycken EV Eur. J. Org. Chem 2010, 2010, 4861. [Google Scholar]
- (39).Cui L; Zhang G; Peng Y; Zhang L Org. Lett 2009, 11, 1225. [DOI] [PubMed] [Google Scholar]
- (40).Cui L; Ye L; Zhang L Chem. Commun 2010, 46, 3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhou G; Zhang J Chem. Commun 2010, 46, 6593. [DOI] [PubMed] [Google Scholar]
- (42).(a) Murarka S; Deb I; Zhang C; Seidel DJ Am. Chem. Soc 2009, 131, 13226; [DOI] [PubMed] [Google Scholar]; (b) Murarka S; Zhang C; Konieczynska MD; Seidel D Org. Lett 2009, 11, 129. [DOI] [PubMed] [Google Scholar]
- (43).Guo C; Fleige M; Janssen-Müller D; Daniliuc CG; Glorius FJ Am. Chem. Soc 2016, 138, 7840. [DOI] [PubMed] [Google Scholar]
- (44).Chen Z-C; Chen Z; Yang Z-H; Guo L; Du W; Chen Y-C Angew. Chem. Int. Ed 2019, 58, 15021. [DOI] [PubMed] [Google Scholar]
- (45).Wang G-W; Bower JF J. Am. Chem. Soc 2018, 140, 2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Shaw MH; Melikhova EY; Kloer DP; Whittingham WG; Bower JF J. Am. Chem. Soc 2013, 135, 4992. [DOI] [PubMed] [Google Scholar]
- (47).McCreanor NG; Stanton S; Bower JF J. Am. Chem. Soc 2016, 138, 11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Wang G-W; McCreanor NG; Shaw MH; Whittingham WG; Bower JF J. Am. Chem. Soc 2016, 138, 13501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).(a) Shaw MH; McCreanor NG; Whittingham WG; Bower JF J. Am. Chem. Soc 2015, 137, 463; [DOI] [PubMed] [Google Scholar]; (b) Shaw MH; Bower JF Chem. Commun 2016, 52, 10817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Mazumdar W; Jana N; Thurman BT; Wink DJ; Driver TG J. Am. Chem. Soc 2017, 139, 5031. [DOI] [PubMed] [Google Scholar]
- (51).Deng T; Mazumdar W; Ford RL; Jana N; Izar R; Wink DJ; Driver TG J. Am. Chem. Soc 2020, 142, 4456. [DOI] [PubMed] [Google Scholar]