

Abstract
Background and objectives
Chronic widespread musculoskeletal pain (CWP) is a symptom of fibromyalgia and a complex trait with poorly understood pathogenesis. CWP is heritable (48%–54%), but its genetic architecture is unknown and candidate gene studies have produced inconsistent results. We conducted a genome-wide association study to get insight into the genetic background of CWP.
Methods
Northern Europeans from UK Biobank comprising 6914 cases reporting pain all over the body lasting >3 months and 242 929 controls were studied. Replication of three independent genome-wide significant single nucleotide polymorphisms was attempted in six independent European cohorts (n=43 080; cases=14 177). Genetic correlations with risk factors, tissue specificity and colocalisation were examined.
Results
Three genome-wide significant loci were identified (rs1491985, rs10490825, rs165599) residing within the genes Ring Finger Protein 123 (RNF123), ATPase secretory pathway Ca 2+ transporting 1 (ATP2C1) and catechol-O-methyltransferase (COMT). The RNF123 locus was replicated (meta-analysis p=0.0002), the ATP2C1 locus showed suggestive association (p=0.0227) and the COMT locus was not replicated. Partial genetic correlation between CWP and depressive symptoms, body mass index, age of first birth and years of schooling were identified. Tissue specificity and colocalisation analysis highlight the relevance of skeletal muscle in CWP.
Conclusions
We report a novel association of RNF123 locus and a suggestive association of ATP2C1 locus with CWP. Both loci are consistent with a role of calcium regulation in CWP. The association with COMT, one of the most studied genes in chronic pain field, was not confirmed in the replication analysis.
Keywords: fibromyalgia, polymorphism, genetic, epidemiology
Key messages.
What is already known about this subject?
Chronic widespread musculoskeletal pain (CWP) is a primary diagnostic feature of fibromyalgia.
CWP is moderately heritable, but precise genes involved in the pathogenesis of CWP are yet to be identified.
What does this study add?
This is the largest genetic study conducted on CWP to date and identified novel genetic risk loci (Ring Finger Protein 123 and ATPase secretory pathway Ca 2+ transporting 1).
The genetic signal points to peripheral pain mechanisms in CWP, and shows genetic correlation with other traits, including body mass index and depression.
How might this impact on clinical practice or future developments?
The findings add to aetiological basis of CWP.
Introduction
Chronic widespread musculoskeletal pain (CWP) is a common complex trait influenced by genetic and environmental factors, most of which have yet to be determined.1 CWP and fibromyalgia syndrome are sometimes used interchangeably, although the latter is generally more severe and includes other features such as sleep disturbance, fatigue and depression.2 It is thought to represent a subgroup at the more severe end of the spectrum of CWP.3 The prevalence of CWP is 10.6% in the world population and 14.2% in the UK population.4 5 It is associated with high societal cost.6 CWP is responsible for excess mortality,7 which is thought to be attributable to cardiovascular disease, respiratory disease and cancer. Females are more affected by CWP than males,4 and the prevalence rises with age.5 In addition to age and sex, a number of exposures have been proposed as risk factors for CWP,8 9 but only increased body mass index (BMI) has been consistently reported across studies, including longitudinal studies.10–12
Broad-sense heritability estimates for CWP range between 48% and 54%, indicating a substantial genetic contribution.13 To date, the candidate gene approach has been extensively applied to identify genetic factors in CWP,14 but few agnostic studies have been published.15 The only genome-wide association study (GWAS) meta-analysis combining 14 studies identified a locus lying on chromosome 5 intergenic to CCT5 and FAM173B.15 CCT5 has previously been implicated in neuropathy16 and there is increasing evidence that small fibre neuropathy underlies a subset of fibromyalgia.17
Genetic factors are known to be shared by chronic pain conditions.18 19 One of the most extensively studied chronic pain-associated genes encodes catechol-O-methyltransferase (COMT), an enzyme which regulates the production of catecholamines that act as neurotransmitters in the central nervous system (CNS) pain tract. A non-synonymous change of A to G encoding a valine (Val) to methionine (Met) substitution at codon 158 (Val158Met; rs4680) reduces the enzymatic activity of COMT. This single nucleotide polymorphism (SNP) has been reported to be associated with CWP in a small study of 122 participants,20 but a subsequent association study of 3017 participants did not confirm earlier findings.21 An inconclusive role of COMT was observed for temporomandibular disorders (TMD) as well.22 23 Further investigation is required to identify genetic variants underlying CWP, which will shed light on the pathophysiological mechanisms underlying the development of chronic pain and may reveal therapeutic targets.
Materials and methods
An overview of study design is presented in figure 1.
Figure 1.

Overview of study design.
Participant selection
For the discovery analysis, we performed a GWAS of CWP using UK Biobank (UKB) comprising 249 843 participants of European descent (6914 CWP cases and 242 929 controls). Independent SNPs passing a threshold p<5.0E-08 were submitted for replication in 43 080 individuals of European ancestry (14 177 CWP cases and 28 903 controls) from six independent cohorts originating in the UK (TwinsUK and The English Longitudinal Study of Ageing (ELSA)), the Netherlands (The Rotterdam Study 1, 2 and 3 (RS-1, RS-2 and RS-3)) and Norway (The Nord-Trøndelag Health Survey (HUNT)). The UKB dataset was used under project #18219. Description of each study cohort is presented in online supplemental text.
annrheumdis-2020-219624supp001.pdf (1MB, pdf)
Phenotype
In UKB, CWP cases were defined by combining self-reported diagnosis of pain all over the body lasting for >3 months; simultaneous pain in the knee, shoulder, hip and back lasting 3+ months and fibromyalgia. Controls comprised those who reported no pain in the last month or reported pain all over the body in the previous month that did not last for 3 months or reported only ≥3 months of non-musculoskeletal pain (headache, facial and abdominal pain). Those reporting a self-reported diagnosis of rheumatoid arthritis, polymyalgia rheumatica, arthritis not otherwise specified, systemic lupus erythematosus, ankylosing spondylitis and myopathy were excluded from the study (online supplemental figure S1). Further phenotype details for UKB and replication cohorts are provided in online supplemental text.
Genotyping and imputation
Genotyping and imputation methods across cohorts are summarised in online supplemental table S1 (online supplemental text).
Statistical analysis and in silico follow-up
The details of statistical analysis, and in silico follow-up are described in online supplemental text. In brief, GWAS in the discovery sample was performed using linear mixed-effects model implemented in BOLT-LMM (V.2.3.2).24 An additive genetic model for SNP effect on CWP was adjusted for age, sex, genotyping platform and the first 10 genetic principal components provided by UKB. A sensitivity GWAS (controls: 223 606 and CWP cases: 6914) was performed excluding participants with chronic non-musculoskeletal pain such as headache, facial and abdominal pain from the controls. Independent SNPs at GWAS significant loci were identified using Conditional and Joint25 analysis and submitted for replication. Independent SNPs across all replication cohorts were meta-analysed using fixed-effects model with both sample size, and inverse-variance weighting implemented in METAL.26 SNP heritability was estimated using BOLT-REML24 and converted to liability scale. Linkage disequilibrium score regression (LDSR)27 was used to estimate inflation in test statistics and genetic correlations. We also estimated partial genetic correlations.28 We used Functional Mapping and Annotation (FUMA) webtool29 for the annotation of functional consequences of CWP-associated SNPs, gene mapping, tissue specificity and gene-set enrichment. Differential expression of replicated independent SNP was assessed using the GTEx V.8 tissues.30 Colocalisation of GWAS-independent SNPs in human skeletal muscle and dorsal root ganglion (DRG) tissues was assessed using publicly available data.30 31 Functional annotation of GWAS-replicated locus was performed using Open Targets Platform.32
Results
Details of the discovery and replication cohorts are presented in table 1. Cases were enriched for females compared with controls in all cohorts (p<0.001) and were on average older in the discovery, and in three replication cohorts (p<0.05). In all cohorts, BMI was significantly higher in cases than controls (p<0.0001) except for RS-3 where a similar but non-significant trend was observed (p=0.0827).
Table 1.
Sample characteristics stratified by case/control status for discovery and replication cohorts
| Cases | Controls | P value | |
| Discovery cohort (UK Biobank) | |||
| Female | 4470 (64.7%) | 128 599 (47.1%) | <0.0001 |
| Male | 2444 (35.3%) | 114 330 (52.9%) | |
| Age (mean±SD) | 57.8±7.45 | 57.0±8.09 | <0.0001 |
| BMI (mean±SD) | 30.02±5.97 | 26.83±4.40 | <0.0001 |
| Replication cohorts | |||
| TwinsUK | |||
| Female | 1041 (93.7%) | 3116 (87.6%) | <0.0001 |
| Male | 70 (6.3%) | 440 (12.4%) | |
| Age (mean±SD) | 54.78±10.48 | 50.12±13.21 | <0.0001 |
| BMI (mean±SD) | 27.39±5.11 | 25.74±4.57 | <0.0001 |
| HUNT | |||
| Female | 6315 | 5836 | <0.0001 |
| Male | 4241 | 7403 | |
| Age (mean±SD) | 55.95±9.48 | 54.82±10.31 | <0.0001 |
| BMI (mean±SD) | 27.37±4.33 | 26.52±3.88 | <0.0001 |
| ELSA | |||
| Female | 1090 (64.9%) | 2660 (50.2%) | <0.001 |
| Male | 589 (35.1%) | 2644 (49.8%) | |
| Age (mean±SD) | 68.10±9.49 | 66.55±9.98 | <0.0001 |
| BMI (mean±SD) | 28.60±4.98 | 27.08±4.22 | <0.0001 |
| RS-1 | |||
| Female | 422 | 1323 | <0.0001 |
| Male | 110 | 1281 | |
| Age (mean±SD) | 64.49±5.30 | 64.60±5.24 | 0.6660 |
| BMI (mean±SD) | 26.98±3.91 | 26.14±3.54 | <0.0001 |
| RS-2 | |||
| Female | 106 | 745 | <0.0001 |
| Male | 38 | 676 | |
| Age (mean±SD) | 61.59±4.59 | 61.93±4.72 | 0.2651 |
| BMI (mean±SD) | 28.54±4.73 | 27.77±3.91 | 0.0363 |
| RS-3 | |||
| Female | 128 | 1516 | <0.0001 |
| Male | 27 | 1263 | |
| Age (mean±SD) | 56.28±5.77 | 56.32±5.46 | 0.0348 |
| BMI (mean±SD) | 28.54±4.86) | 27.71±4.62 | 0.0827 |
BMI, body mass index; ELSA, The English Longitudinal Study of Ageing; HUNT, The Nord-Trøndelag Health Survey; RS-1, RS-2 and RS-3, The Rotterdam Study 1, 2 and 3; SD, Standard deviation.
Discovery genome-wide association study
Three genomic loci tagged by rs1491985, rs10490825 and rs165599 passed genome-wide significance threshold of p<5E-08 (figure 2). Observed inflation in test statistics (λGC=1.146, online supplemental figure S2) was due to polygenicity (LDSR intercept=1.002±0.0085, LDSR ratio=0.0118±0.0497) rather than population stratification. SNP heritability of CWP was 0.05±0.003 on the observed scale, and 0.33±0.0004 on the liability scale meaning that the observed SNPs explain approximately 33% of the variance in CWP risk. Independent SNPs were located in the gene Ring Finger Protein 123 (RNF123) (chromosome 3, rs1491985, intronic variant, p=1.60E-08), ATPase secretory pathway Ca 2+ transporting 1 (ATP2C1) (chromosome 3, rs10490825, intronic variant, p=1.30E-08) and COMT (chromosome 22, rs165599, 3’-untranslated region (3’-UTR) variant, p=2.50E-08), respectively (figure 3A–C; online supplemental table S2). Six additional loci near or within genes HNRNPA1P46, LRRC3B, PDE6A, DPYSL2, ANXA11 and AL138498.1 were identified at suggestive GWAS threshold of p<5E-07. Sensitivity GWAS excluding participants with chronic non-musculoskeletal pain provided similar findings except that COMT locus now became suggestively significant (p=5.3E-08) (online supplemental figure S3).
Figure 2.

Manhattan plot of a genome-wide association analysis of chronic widespread musculoskeletal pain (CWP). Each circle in the plot represents a single nucleotide polymorphism (SNP), which was positioned following genomic build GRCh37. The y-axis shows the corresponding –log10 p values and the x-axis shows chromosome position along with SNPs. The horizontal red dotted line indicates genome-wide significance threshold at p=5.0×10–8. The horizontal blue dotted line indicates suggestive genome-wide significance threshold at p=5.0×10–7. Gene labels represent nearest genes to independent SNPs located at loci associated with p<5.0×10–7.
Figure 3.
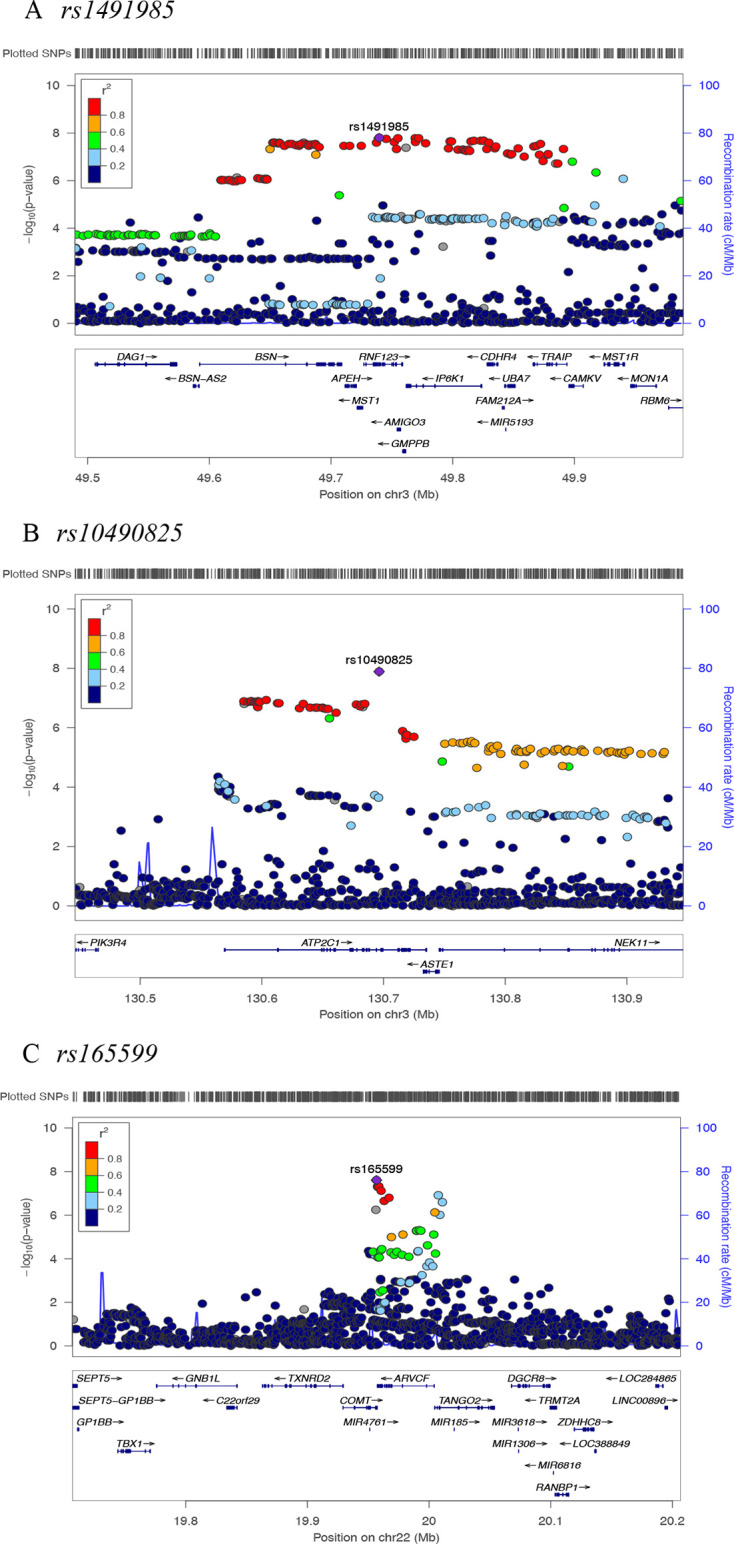
Regional plots for three independent chronic widespread musculoskeletal pain associated single nucleotide polymorphisms (SNPs). Independent SNPs are coloured in purple. Other coloured circles indicate pairwise linkage disequilibrium (LD). The strength of LD (r2) presented in the upper left corner of each plot.
Replication results and meta-analysis
Results are presented in online supplemental table S3, with meta-analysis of the six replication samples as shown in figure 4 (online supplemental tables S4, S5). Given the significance threshold for replication: 0.05/3=0.017, association between CWP and rs1491985 was considered replicated (sample-size based p=0.0002; standard-error based p=0.0003). Rs10490825 showed suggestive association with CWP (sample-size based p=0.0227; standard-error based p=0.0490) and demonstrated a consistent direction of effect in five of the six replication samples. Rs165599 did not replicate (sample-size based p=0.7300; standard-error based p=0.5000) and the direction of effect was not consistent across cohorts: in three cohorts, allele A was protective, while in the other three it was the risk allele. None of the three SNPs displayed statistically significant heterogeneity in the replication cohorts.
Figure 4.

Forest plot for the association of (A) rs1491985, (B) rs10490825, and (C) rs165599 with chronic widespread musculoskeletal pain. X-axis shows effect size measures are presented as beta value. The red square with horizontal black line represents the cohort-specific effect with a corresponding CI for the single nucleotide polymorphism (SNP) of interest. Size of the square indicates the weight of the study and reflects sample size. The vertical black line indicates ‘line of no effect’. Overall effect is presented as a black diamond. Test statistics for each cohort, meta-analysis and heterogeneity are available on the left-hand side. The rs1491985 and rs10490825 were not present in The English Longitudinal Study of Ageing (ELSA); therefore rs9870858 and rs17329848 were used as proxy SNPs, respectively (online supplemental text).
CWP shares genetic components with BMI, depression, age at first birth and years of schooling
Two hundred and nine traits from LD-hub (online supplemental text) were examined for genetic correlation with CWP. We selected traits for which the absolute value of the correlation coefficient (rg) was >0.2, and for which the Bonferroni-corrected p was <0.01/209=4.78E-05. Twenty-three traits fulfilled these criteria (online supplemental figure S4). The highest positive genetic correlation was observed for depressive symptoms (rg=0.65) and the highest negative correlation was observed for college completion (rg=−0.61). Many of the 23 genetically correlated traits were correlated with each other raising concerns about their independency of correlations with CWP. We therefore calculated partial genetic correlations conditionally independent of each other. Using hierarchical clustering of genetic correlations we identified seven clusters (online supplemental figure S5A), with seven traits selected to represent each cluster (BMI, triglycerides, depressive symptoms, coronary artery disease, smoking, age of first birth and years of schooling) to quantify partial genetic correlation with CWP. We found depressive symptoms (rg=0.59), BMI (rg=0.20), age of first birth (rg=−0.26) and years of schooling (rg=−0.17) independently correlated with CWP (online supplemental figure S5B and table S6).
Tissue-specific expression of CWP mapped gene sets
The results of functional consequences of GWAS-independent SNPs and their proxies are presented in online supplemental figure S6 (online supplemental text). Four different gene mapping strategies were implemented in FUMA (genome-wide gene-based association analysis, positional, expression quantitative trait locus (eQTL) and chromatin interaction mapping) linking annotated SNPs to 89 genes of which MST1, GMPPB, APEH, RNF123, ARVCF, AMIGO3, IP6K1, TANGO2 and TRAIP were identified using all four methods (figure 5A–D).33 Mapped genes were investigated for tissue-specific gene expression and gene-set enrichment. In 54 specific GTEx tissues types, differentially expressed gene sets enriched for skeletal muscle, several brain tissues, heart, whole blood, pancreas and transverse colon (figure 6A, online supplemental table S7). In 30 general GTEx tissue types, differentially expressed gene sets enriched for skeletal muscle, pancreas, heart, blood and brain (figure 6B, online supplemental table S8). In both sets of GTEx tissues, overall enrichment for differentially expressed gene sets containing RNF123 and ATP2C1 genes were stronger for skeletal muscle than other tissues. RNF123 was found to be highly expressed in skeletal muscle compared with other tissue types (figure 6C). None of the hallmark gene sets available in the molecular signature database was identified in the analysis.
Figure 5.

(A) Manhattan plot of the genome-wide gene-based association analysis, (B) & (C) The circus plot displaying chromatin interactions (Ci) and expression quantitative trait loci (eQTLs) on chromosomes 3 and chromosomes 22, respectively, (D) Venn diagram showing overlap of genes implicated by genome-wide gene-based analysis implemented in MAGMA, positional mapping (Pos Map), chromatin interaction mapping (Ci Map), and expression quantitative trait locus mapping (eQTL Map). (A) The y-axis shows the ─log10 transformed two-tailed p-value of each gene from a linear model and the chromosomal position on the x-axis. The red dotted line indicates the Bonferroni-corrected threshold for genome-wide significance of the gene-based test. (B, C) The most outer layer of the circus plot displaying Manhattan plot with –log10 p-values for chronic widespread musculoskeletal pain associated independent single nucleotide polymorphisms (SNPs). Each SNP is presented with rsID. Linkage disequilibrium (LD) relationship between independent SNPs at the locus and their proxies are indicated with red (r2 > 0.8) and orange (r2 > 0.6). Grey SNPs indicate minimal LD with r2 ≤0.20. The outer circle represents chromosome with genomic risk loci are highlighted in blue. Either Ci- or eQTL mapped genes are displayed on the inner circle. Ci- and eQTL mapped genes are presented in orange or green color, respectively. Genes mapped with both approaches are colored red.
Figure 6.

(A) Differentially expressed gene (DEG) plots for chronic widespread musculoskeletal pain (CWP) in 54 tissue types from GTEX v8, (B) DEG plots for CWP in 30 general tissue types from GTEX v8 and (C) Differential expression of RNF123 gene across tissue types from GTEX v8. (A, B) In both plots, the y-axis represents the ─log10 transformed two-tailed p value of the hypergeometric test. Significantly enriched DEG sets (Bonferroni-corrected p value <0.05) are highlighted in red. (C) Y-axis represents transcripts per million (TPM) and x-axis represents the GTEx (V.8) tissues. The figure was adapted from GTEx portal (https://www.gtexportal.org/home/gene/ENSG00000164068).
Putative causal genes in RNF123 locus
Colocalisation analysis identified a 93% probability of shared eQTL variant rs6809879, which controls Cadherin Related Family Member 4 (CDHR4) expression in the skeletal muscle and CWP association signal near the RNF123 locus (online supplemental table S9, online supplemental figure S7A). Additionally, significant colocalisation was found for rs13093525, which controls APEH expression in DRG at exon level (72% probability of shared variant with RNF123 locus). Both rs6809879 and rs13093525 were in complete LD with independent SNP rs1491985 (R2=1) (online supplemental table S10, online supplemental figure S7B). No evidence of skeletal muscle or DRG eQTL colocalisation was observed for ATP2C1 and COMT loci. Functional annotation of RNF123 locus identified nine genes (SLC25A20, NDUFAF3, DAG1, HYAL1, GMPPB, TRAIP, RHOA, CACNA2D2 and IMPDH2) specific to musculoskeletal system diseases, of which CACNA2D2, NDUFAF3 and IMPDH2 enriched as druggable targets (online supplemental figure S8).
Discussion
CWP is a prevalent condition with moderate heritability and serves as a cardinal diagnostic feature of fibromyalgia. Therefore, our findings are of importance for better understanding the genetic basis of fibromyalgia. We report here the largest GWAS of CWP to date using 249 843 participants from the UKB, identifying 3 genome-wide significant loci implicating RNF123, ATP2C1 and COMT. The association in RNF123 was replicated, whereas ATP2C1 showed a suggestive association, and the COMT locus did not replicate in 43 080 individuals from independent cohorts.
RNF123 gene encodes E3 ubiquitin-protein ligase, has a role in cell cycle progression, metabolism of proteins and innate immunity.34 35 This gene is highly expressed in skeletal muscle than other tissues. Recent studies involving UKB samples also associated the locus with musculoskeletal pain.19 36 However, it is not clear how RNF123 may contribute to CWP. Using in silico follow-up, we identified CDHR4, APEH, SLC25A20, NDUFAF3, DAG1, HYAL1, GMPPB, TRAIP, RHOA, CACNA2D2 and IMPDH2 genes as putative causal candidates at the locus, of which CACNA2D2, NDUFAF3 and IMPDH2 can be targeted using known drugs.37–39 Notably, CACNA2D2 encodes the alpha-2/delta subunit of the voltage-dependent calcium channel complex, which is a receptor for gabapentinoids,40 used by some in the management of fibromyalgia.41 42 Another prioritised gene CDHR4 belongs to cadherin superfamily has a role in calcium-ion binding to facilitate cadherin-mediated cell-cell interaction.43 44
Additionally, the ATP2C1 locus demonstrated suggestive association in replication (p=0.0227). There was a consistent direction of effect for ATP2C1 locus in six replication cohorts but not ELSA, where we used a proxy SNP, which had close to zero effect size (beta=−0.0004±0.0110). This is the first study to implicate ATP2C1 with musculoskeletal pain using an agnostic approach. The ATP2C1 gene encodes for the ATP-powered magnesium-dependent calcium pump protein hSPCA1, which mediates Golgi uptake of cytosolic Ca(2+) and Mg(2+).45 A loss of function mutation in the ATP2C1 leads to Hailey-Hailey disease (HHD), an autosomal dominant skin condition characterised by blistering and erosion of the epidermis.46 Interestingly, HHD may be treated successfully with low-dose naltrexone, an opioid receptor antagonist, which has also been used in the management of fibromyalgia.47 48 A recent study showed that naltrexone is capable of restoring calcium homeostasis in natural killer cells of patients with chronic fatigue syndrome.49 Additionally, the role of calcium regulation in pain processing is well known.50–52 Taken together, our findings suggest a role in the regulation of calcium influencing CWP/fibromyalgia.
COMT is one of the most studied genes in human pain.53 Almost 30 SNPs and 3 haploblocks of the COMT gene have been studied in acute clinical, experimental and chronic pain. Rs4680 of the COMT gene is extensively studied in many pain phenotypes such as pain sensitivity, TMD and fibromyalgia.54 Across multiple ethnic populations, rs4680 was implicated with fibromyalgia.55 However, a meta-analysis of 8 case-control studies (589 fibromyalgia cases and 527 controls) did not confirm earlier association.56 To date, the largest study that assessed the association between COMT haplotypes (rs4680, rs4818, rs4633 and rs6269) and fibromyalgia included 60 367 participants (2713 ICD-9 diagnosed fibromyalgia) and found no association.57 They have also been refuted in other European CWP samples21 58 and a large candidate gene study of fibromyalgia.59 However, we identified rs165599, located at 3’-UTR of COMT, associated with CWP in the discovery sample but not in the meta-analysis or any of the replication cohorts. This variant is not in LD with previously studied COMT SNPs rs4680, rs4818, rs4633 and rs6269, and was found not to be associated with chronic musculoskeletal pain including CWP neither when studied as a single SNP nor as a part of a haploblock.60–62 Several explanations of our non-replication of COMT locus are possible. First, there was lower power pertaining to overall meta-analysis, which was estimated at 48% based on the effect size observed in the discovery sample (n=249 843), replication sample size (n=43 080) and the number of tests conducted (n=3). Our meta-analysis did have 90% power to detect a relative risk as small as 1.04 but the estimated COMT effect was only 1.012 (beta=0.0027±0.004; OR=1.012, 95% CI=0.97 to 1.05). However, our replication sample size was larger than many of the earlier studies that reported the association between COMT and CWP.20 63 Second, we observed a tendency towards non-significance for the COMT locus in the sensitivity GWAS due to the exclusion of participants with non-musculoskeletal pain from the control group suggesting that COMT predisposes to chronic pain in general. Finally, genetic factors underlying chronic pain and psychiatric comorbidity (e.g. depression and neuroticism) are known to be shared.64 However, previous GWAS on chronic pain,28 65 66 depression67 and neuroticism68 have failed to detect an association with COMT. Thus, if there is a role of COMT in CWP, it is likely minimal.
Epidemiological studies have consistently reported higher BMI to be associated with an increased risk of CWP.10 11 69 Our analysis showed significantly higher BMI in CWP cases compared with controls (p<0.0001) in all cohorts except RS-3. In line with this, we observed a positive genetic overlap between BMI and CWP independent of genetic confounders. Similarly, genetically independent pairwise genetic correlation for depressive symptoms, age of first birth and years of schooling was seen with CWP. These findings indicate the presence of shared molecular pathways underlying these traits.
Functional analysis showed that FUMA mapped genes differentially expressed in skeletal muscle, several areas of the CNS, pancreas, whole blood and heart tissues. These findings suggest the involvement of nervous, musculoskeletal and neuroendocrine systems in CWP. These physiological systems have been implicated in fibromyalgia by previous studies.70–72 Evidence suggests that both peripheral and central pain mechanisms influence CWP.73 74 We observed overall stronger enrichment for differentially expressed gene sets in skeletal muscle than other GTEx tissues. Also, skeletal muscle and DRG eQTLs colocalise with the RNF123 locus. These findings suggest a substantial involvement of peripheral pain mechanisms in CWP.
The study has limitations. The case definition of CWP depends on self-report together with exclusion of other conditions with symptoms leading to chronic pain.75 A clinical diagnosis of CWP would have been infeasible in a sample this large. Also, we used common SNPs to estimate the heritability of CWP, so the contribution of other variants in the heritability estimated remains unknown. The phenotype definition used in this study to estimate SNP heritability has differed from the Kato et al 13 study, where a modulated American College of Rheumatology76 criteria based on self-report was used to estimate broad-sense heritability. However, using UKB samples, a study reported the SNP heritability of pain all over the body, regardless of chronicity, on the liability scale was 0.31±0.072.64 We found a similar but slightly higher estimate for CWP (0.33±0.0004), suggesting our definition is meaningful and CWP is a trait of high genetic influence. Finally, our findings cannot be generalisable to ancestry other than northern Europeans (online supplemental text).
In summary, this study identified a novel association for CWP in the RNF123 locus and suggested the role of calcium regulation, by the involvement of the CDHR4, CACNA2D2 and ATP2C1 genes. The association of the COMT locus with CWP was not replicated, suggesting a small influence, if any. We found evidence that the epidemiological association of BMI and CWP is at least in part genetically mediated. Finally, our results suggest a profound role of peripheral mechanisms in the pathogenesis of CWP.
Acknowledgments
The authors would like to thank all the participants of UK Biobank, The Nord-Trøndelag Health Survey, English Longitudinal Study of Aging, TwinsUK and Rotterdam study I, II and III. The contribution of inhabitants, general practitioners and pharmacists of the Ommoord district to the Rotterdam Study is gratefully acknowledged. The UK Biobank study was approved by the National Health Service National Research Ethics Service (ref. 11/NW/0382) and all participants provided written informed consent. Genome-wide association analysis was performed using the UK Biobank resource under project number 18219. The English Longitudinal Study of Ageing is jointly run by University College London, Institute for Fiscal Studies, University of Manchester and National Centre for Social Research. Genetic analyses have been carried out by UCL Genomics and funded by the Economic and Social Research Council and the National Institute on Aging. Data governance was provided by the METADAC data access committee, funded by ESRC, Welcome and MRC (2015-2018: Grant Number MR/N01104X/1 2018-2020: Grant Number ES/S008349/1). TwinsUK is funded by the Wellcome Trust, Medical Research Council, European Union, Chronic Disease Research Foundation (CDRF), Zoe Global Ltd and the National Institute for Health Research (NIHR)-funded BioResource, Clinical Research Facility and Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London. The Nord-Trøndelag Health Study (The HUNT Study) is a collaboration between HUNT Research Centre (Faculty of Medicine and Health Sciences, NTNU, Norwegian University of Science and Technology), Trøndelag County Council, Central Norway Regional Health Authority and the Norwegian Institute of Public Health. The genotyping was financed by the National Institute of Health (NIH), University of Michigan, The Norwegian Research Council and Central Norway Regional Health Authority and the Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology (NTNU). The genotype quality control and imputation has been conducted by the K.G. Jebsen Center for Genetic Epidemiology, Department of Public Health and Nursing, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology (NTNU). The Rotterdam Study is supported by the Erasmus MC University Medical Center and Erasmus University Rotterdam; The Netherlands Organization for Scientific Research (NWO); The Netherlands Organization for Health Research and Development (ZonMw); the Research Institute for Diseases in the Elderly (RIDE); The Netherlands Genomics Initiative (NGI); the Ministry of Education, Culture and Science; the Ministry of Health, Welfare and Sports; the European Commission (DG XII) and the Municipality of Rotterdam.
Footnotes
Handling editor: Josef S Smolen
Collaborators: Members of HUNT All-In Pain: Amy E Martinsen (1. K. G. Jebsen Center for Genetic Epidemiology, Department of Public Health and Nursing, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology, Trondheim, Norway; 2. Department of Research, Innovation and Education, Division of Clinical Neuroscience, Oslo University Hospital, Oslo, Norway; 3. Institute of Clinical Medicine, Faculty of Medicine, University of Oslo, Oslo, Norway.), Anne Heidi Skogholt (K. G. Jebsen Center for Genetic Epidemiology, Department of Public Health and Nursing, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology, Trondheim, Norway.), Ben Brumpton (K. G. Jebsen Center for Genetic Epidemiology, Department of Public Health and Nursing, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology, Trondheim, Norway.), Cristen Willer (Department of Internal Medicine, Division of Cardiovascular Medicine, University of Michigan, Ann Arbor, 48109, MI, USA.), Ingrid Heuch (Department of Research, Innovation and Education, Division of Clinical Neuroscience, Oslo University Hospital, Oslo, Norway.), Ingunn Mundal (Department of Health Science, Molde University College, Molde, Norway.), Jonas Bille Nielsen (1. K. G. Jebsen Center for Genetic Epidemiology, Department of Public Health and Nursing, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology, Trondheim, Norway; 2. Department of Internal Medicine, Division of Cardiovascular Medicine, University of Michigan, Ann Arbor, 48109, MI, USA; 3. Department of Epidemiology Research, Statens Serum Institut, Copenhagen, Denmark.), Kjersti Storheim (1. Research and Communication Unit for Musculoskeletal Health (FORMI), Department of Research, Innovation and Education, Division of Clinical Neuroscience, Oslo University Hospital, Oslo, Norway; 2. Faculty of Health Sciences, Department of physiotherapy, Oslo Metropolitan University, Oslo, Norway.), Kristian Bernhard Nilsen (1. Department of Neuromedicine and Movement Science, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology (NTNU), Trondheim, Norway. 2. Department of Neurology, Oslo University Hospital, Oslo, Norway.), Lars Fritsche (Center for Statistical Genetics, Department of Biostatistics, University of Michigan, Ann Arbor, 48109, MI, USA.), Laurent F. Thomas (1. K. G. Jebsen Center for Genetic Epidemiology, Department of Public Health and Nursing, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology, Trondheim, Norway; 2. Department of Clinical and Molecular Medicine, Norwegian University of Science and Technology, Trondheim, Norway.), Linda M Pedersen (Department of Research, Innovation and Education, Division of Clinical Neuroscience, Oslo University Hospital, Oslo, Norway.), Maiken E Gabrielsen (K. G. Jebsen Center for Genetic Epidemiology, Department of Public Health and Nursing, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology, Trondheim, Norway.), Marianne Bakke Johnsen (1. K. G. Jebsen Center for Genetic Epidemiology, Department of Public Health and Nursing, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology, Trondheim, Norway; 2. Institute of Clinical Medicine, Faculty of Medicine, University of Oslo, Oslo, Norway; 3. Research and Communication Unit for Musculoskeletal Health (FORMI), Department of Research, Innovation and Education, Division of Clinical Neuroscience, Oslo University Hospital, Oslo, Norway.), Marie Udnesseter Lie (1. Institute of Clinical Medicine, Faculty of Medicine, University of Oslo, Oslo, Norway; 2. Research and Communication Unit for Musculoskeletal Health (FORMI), Department of Research, Innovation and Education, Division of Clinical Neuroscience, Oslo University Hospital, Oslo, Norway.), Oddgeir Holmen (HUNT Research Center, Department of Public Health and Nursing, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology, Trondheim, Norway.), Synne Øien Stensland (1. Research and Communication Unit for Musculoskeletal Health (FORMI), Department of Research, Innovation and Education, Division of Clinical Neuroscience, Oslo University Hospital, Oslo, Norway; 2. NKVTS, Norwegian Centre for Violence and Traumatic Stress Studies.), Wei Zhou (1. Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI, USA. 2. Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, Massachusetts, USA.)
Contributors: MSR prepared the manuscript. Discovery GWAS: MSR, MBF and FMKW designed the study. MSR, YAT, MBF and FMKW developed phenotype definition. MSR performed data management and GWAS in the discovery cohorts. Replication: MSR, MBF and FMKW developed replication protocol and coordinated the study; MSR, BSW, JAW, MBF and FMKW designed the study; BSW, SB, KHa, EAF, KHv, JAZ, HUNT All-in Pain authors, SOCC, JBvM and FMKW collected replication cohort’s data; MSR, BSW, SB, KHv, JAZ, SOCC and JBvM were involved data management; MSR, BSW, SB, KHa, EAF, JAZ, HUNT All-in Pain authors, MBF and FMKW developed phenotype definition; MSR, BSW, SB, SOCC and JBvM analysed the data. MSR performed replication meta-analysis. Post-GWAS bioinformatics: MSR, YAT and SZS performed post-GWAS bioinformatics analysis. MBF, YSA and FMKW provided the statistical and bioinformatics consultation in the project. MSR, KHa, SOCC, YAT, SZS, YSA, JBvM, MBF and FMKW interpreted the findings. FMKW was responsible for the overall supervision of the project. All authors commented on the manuscript and agreed on the final version to be published.
Funding: MSR received funding from the European Union’s Horizon 2020 research and innovation program IMforFUTURE, under H2020-MSCA-ITN grant agreement number 721815. The work of YAT was supported by PolyOmica and by the Russian Foundation for Basic Research (project 19-015-00151). The work of SZS was supported by the Russian Ministry of Science and Education under the 5-100 Excellence Programme and by the Ministry of Education and Science of the RF via the Institute of Cytology and Genetics SB RAS (project number 0259-2021-0009/AAAA-A17-117092070032-4). The work of YSA was supported by PolyOmica.
Competing interests: YSA is co-owner of Maatschap PolyOmica and PolyKnomics BV, private organisations, providing services, research and development in the field of computational and statistical, quantitative and computational (gen)omics.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Contributor Information
Collaborators: HUNT All-In Pain, Amy E Martinsen, Anne Heidi Skogholt, Ben Brumpton, Ingrid Heuch, Ingunn Mundal, Jonas Bille Nielsen, Kjersti Storheim, Kristian Bernhard Nilsen, Lars Fritsche, Laurent F Thomas, Linda M Pedersen, Maiken E Gabrielsen, Marianne Bakke Johnsen, Marie Udnesseter Lie, Oddgeir Holmen, Synne Øien Stensland, Wei Zhou, and Cristen Willer
Data availability statement
Summary statistics from our discovery and sensitivity GWAS was deposited at Zenodo (https://doi.org/10.5281/zenodo.4459546). Other data relevant to the study are included in the article or uploaded as online supplementary information.
Ethics statements
Patient consent for publication
Not required.
Ethics approval
Study-specific ethical approval was provided by the local ethics committees. All study participants provided written informed consent. Details are provided in the online supplementary material.
References
- 1.Häuser W, Ablin J, Fitzcharles M-A, et al. Fibromyalgia. Nat Rev Dis Primers 2015;1:15022. 10.1038/nrdp.2015.22 [DOI] [PubMed] [Google Scholar]
- 2.Wolfe F, Clauw DJ, Fitzcharles M-A, et al. 2016 revisions to the 2010/2011 fibromyalgia diagnostic criteria. Semin Arthritis Rheum 2016;46:319–29. 10.1016/j.semarthrit.2016.08.012 [DOI] [PubMed] [Google Scholar]
- 3.Shipley M. Chronic widespread pain and fibromyalgia syndrome. Medicine 2018;46:252–5. 10.1016/j.mpmed.2018.01.009 [DOI] [Google Scholar]
- 4.Fayaz A, Croft P, Langford RM, et al. Prevalence of chronic pain in the UK: a systematic review and meta-analysis of population studies. BMJ Open 2016;6:e010364. 10.1136/bmjopen-2015-010364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mansfield KE, Sim J, Jordan JL, et al. A systematic review and meta-analysis of the prevalence of chronic widespread pain in the general population. Pain 2016;157:55–64. 10.1097/j.pain.0000000000000314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boonen A, van den Heuvel R, van Tubergen A, et al. Large differences in cost of illness and wellbeing between patients with fibromyalgia, chronic low back pain, or ankylosing spondylitis. Ann Rheum Dis 2005;64:396–402. 10.1136/ard.2003.019711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Macfarlane GJ, Barnish MS, Jones GT. Persons with chronic widespread pain experience excess mortality: longitudinal results from UK Biobank and meta-analysis. Ann Rheum Dis 2017;76:1815–22. 10.1136/annrheumdis-2017-211476 [DOI] [PubMed] [Google Scholar]
- 8.Kvalheim S, Sandven I, Hagen K, et al. Smoking as a risk factor for chronic musculoskeletal complaints is influenced by age. The HUNT study. Pain 2013;154:1073–9. 10.1016/j.pain.2013.03.015 [DOI] [PubMed] [Google Scholar]
- 9.Kvalheim S, Sandvik L, Winsvold B, et al. Early menarche and chronic widespread musculoskeletal complaints--Results from the HUNT study. Eur J Pain 2016;20:458–64. 10.1002/ejp.747 [DOI] [PubMed] [Google Scholar]
- 10.Mundal I, Gråwe RW, Bjørngaard JH, et al. Prevalence and long-term predictors of persistent chronic widespread pain in the general population in an 11-year prospective study: the HUNT study. BMC Musculoskelet Disord 2014;15:213. 10.1186/1471-2474-15-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mundal I, Gråwe RW, Bjørngaard JH, et al. Psychosocial factors and risk of chronic widespread pain: an 11-year follow-up study--the HUNT study. Pain 2014;155:1555–61. 10.1016/j.pain.2014.04.033 [DOI] [PubMed] [Google Scholar]
- 12.Wright LJ, Schur E, Noonan C, et al. Chronic pain, overweight, and obesity: findings from a community-based twin registry. J Pain 2010;11:628–35. 10.1016/j.jpain.2009.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kato K, Sullivan PF, Evengård B, et al. Importance of genetic influences on chronic widespread pain. Arthritis Rheum 2006;54:1682–6. 10.1002/art.21798 [DOI] [PubMed] [Google Scholar]
- 14.Kerr JI, Burri A. Genetic and epigenetic epidemiology of chronic widespread pain. J Pain Res 2017;10:2021–9. 10.2147/JPR.S143869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peters MJ, Broer L, Willemen HLDM, et al. Genome-wide association study meta-analysis of chronic widespread pain: evidence for involvement of the 5p15.2 region. Ann Rheum Dis 2013;72:427–36. 10.1136/annrheumdis-2012-201742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bouhouche A, Benomar A, Bouslam N, et al. Mutation in the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (CCT5) gene causes autosomal recessive mutilating sensory neuropathy with spastic paraplegia. J Med Genet 2006;43:441–3. 10.1136/jmg.2005.039230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawson VH, Grewal J, Hackshaw KV, et al. Fibromyalgia syndrome and small fiber, early or mild sensory polyneuropathy. Muscle Nerve 2018;58:625–30. 10.1002/mus.26131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vehof J, Zavos HMS, Lachance G, et al. Shared genetic factors underlie chronic pain syndromes. Pain 2014;155:1562–8. 10.1016/j.pain.2014.05.002 [DOI] [PubMed] [Google Scholar]
- 19.Tsepilov YA, Freidin MB, Shadrina AS. Analysis of genetically independent phenotypes identifies shared genetic factors associated with chronic musculoskeletal pain at different anatomic sites. bioRxiv 2019;810283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gürsoy S, Erdal E, Herken H, et al. Significance of catechol-O-methyltransferase gene polymorphism in fibromyalgia syndrome. Rheumatol Int 2003;23:104–7. 10.1007/s00296-002-0260-5 [DOI] [PubMed] [Google Scholar]
- 21.Hagen K, Pettersen E, Stovner LJ, et al. No association between chronic musculoskeletal complaints and Val158Met polymorphism in the catechol-O-methyltransferase gene. The HUNT study. BMC Musculoskelet Disord 2006;7:40. 10.1186/1471-2474-7-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diatchenko L, Nackley AG, Slade GD, et al. Catechol-O-methyltransferase gene polymorphisms are associated with multiple pain-evoking stimuli. Pain 2006;125:216–24. 10.1016/j.pain.2006.05.024 [DOI] [PubMed] [Google Scholar]
- 23.Smith SB, Parisien M, Bair E, et al. Genome-Wide association reveals contribution of MRAS to painful temporomandibular disorder in males. Pain 2019;160:579–91. 10.1097/j.pain.0000000000001438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loh P-R, Tucker G, Bulik-Sullivan BK, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet 2015;47:284–90. 10.1038/ng.3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang J, Ferreira T, Morris AP, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet 2012;44:369–75. 10.1038/ng.2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willer CJ, Li Y, Abecasis GR. Metal: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010;26:2190–1. 10.1093/bioinformatics/btq340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bulik-Sullivan BK, Loh P-R, Finucane HK, et al. Ld score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 2015;47:291–5. 10.1038/ng.3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freidin MB, Tsepilov YA, Palmer M, et al. Insight into the genetic architecture of back pain and its risk factors from a study of 509,000 individuals. Pain 2019;160:1361–73. 10.1097/j.pain.0000000000001514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watanabe K, Taskesen E, van Bochoven A, et al. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 2017;8:1826. 10.1038/s41467-017-01261-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.GTEx Consortium . The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020;369:1318–30. 10.1126/science.aaz1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parisien M, Khoury S, Chabot-Doré A-J, et al. Effect of human genetic variability on gene expression in dorsal root ganglia and association with pain phenotypes. Cell Rep 2017;19:1940–52. 10.1016/j.celrep.2017.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carvalho-Silva D, Pierleoni A, Pignatelli M, et al. Open targets platform: new developments and updates two years on. Nucleic Acids Res 2019;47:D1056–65. 10.1093/nar/gky1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heberle H, Meirelles GV, da Silva FR, et al. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics 2015;16:169. 10.1186/s12859-015-0611-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamura T, Hara T, Matsumoto M, et al. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol 2004;6:1229–35. 10.1038/ncb1194 [DOI] [PubMed] [Google Scholar]
- 35.Wang S, Yang Y-K, Chen T, et al. RNF123 has an E3 ligase-independent function in RIG-I-like receptor-mediated antiviral signaling. EMBO Rep 2016;17:1155–68. 10.15252/embr.201541703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnston KJA, Adams MJ, Nicholl BI, et al. Genome-Wide association study of multisite chronic pain in UK Biobank. PLoS Genet 2019;15:e1008164. 10.1371/journal.pgen.1008164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vinik A, Rosenstock J, Sharma U, et al. Efficacy and safety of mirogabalin (DS-5565) for the treatment of diabetic peripheral neuropathic pain: a randomized, double-blind, placebo- and active comparator-controlled, adaptive proof-of-concept phase 2 study. Diabetes Care 2014;37:3253–61. 10.2337/dc14-1044 [DOI] [PubMed] [Google Scholar]
- 38.Emami Riedmaier A, Fisel P, Nies AT, et al. Metformin and cancer: from the old medicine cabinet to pharmacological pitfalls and prospects. Trends Pharmacol Sci 2013;34:126–35. 10.1016/j.tips.2012.11.005 [DOI] [PubMed] [Google Scholar]
- 39.Sanquer S, Maison P, Tomkiewicz C, et al. Expression of inosine monophosphate dehydrogenase type I and type II after mycophenolate mofetil treatment: a 2-year follow-up in kidney transplantation. Clin Pharmacol Ther 2008;83:328–35. 10.1038/sj.clpt.6100300 [DOI] [PubMed] [Google Scholar]
- 40.Patel R, Dickenson AH. Mechanisms of the gabapentinoids and α 2 δ-1 calcium channel subunit in neuropathic pain. Pharmacol Res Perspect 2016;4:e00205. 10.1002/prp2.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Derry S, Cording M, Wiffen PJ, et al. Pregabalin for pain in fibromyalgia in adults. Cochrane Database Syst Rev 2016;9:Cd011790. 10.1002/14651858.CD011790.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cooper TE, Derry S, Wiffen PJ, et al. Gabapentin for fibromyalgia pain in adults. Cochrane Database Syst Rev 2017;56:CD012188. 10.1002/14651858.CD012188.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sotomayor M, Gaudet R, Corey DP. Sorting out a promiscuous superfamily: towards cadherin connectomics. Trends Cell Biol 2014;24:524–36. 10.1016/j.tcb.2014.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cailliez F, Lavery R. Cadherin mechanics and complexation: the importance of calcium binding. Biophys J 2005;89:3895–903. 10.1529/biophysj.105.067322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Micaroni M, Giacchetti G, Plebani R, et al. ATP2C1 gene mutations in Hailey-Hailey disease and possible roles of SPCA1 isoforms in membrane trafficking. Cell Death Dis 2016;7:e2259. 10.1038/cddis.2016.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sudbrak R, Brown J, Dobson-Stone C, et al. Hailey-Hailey disease is caused by mutations in ATP2C1 encoding a novel Ca(2+) pump. Hum Mol Genet 2000;9:1131–40. 10.1093/hmg/9.7.1131 [DOI] [PubMed] [Google Scholar]
- 47.Kollman N, Bass J. Generalized familial benign chronic pemphigus (Hailey-Hailey disease) treated successfully with low-dose naltrexone. JAAD Case Rep 2018;4:725–7. 10.1016/j.jdcr.2018.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Albers LN, Arbiser JL, Feldman RJ. Treatment of Hailey-Hailey disease with low-dose naltrexone. JAMA Dermatol 2017;153:1018–20. 10.1001/jamadermatol.2017.2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cabanas H, Muraki K, Staines D, et al. Naltrexone restores impaired transient receptor potential melastatin 3 ion channel function in natural killer cells from myalgic Encephalomyelitis/Chronic fatigue syndrome patients. Front Immunol 2019;10:2545. 10.3389/fimmu.2019.02545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park J, Luo ZD. Calcium channel functions in pain processing. Channels 2010;4:510–7. 10.4161/chan.4.6.12869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bourinet E, Altier C, Hildebrand ME, et al. Calcium-permeable ion channels in pain signaling. Physiol Rev 2014;94:81–140. 10.1152/physrev.00023.2013 [DOI] [PubMed] [Google Scholar]
- 52.Younger J, Noor N, McCue R, et al. Low-dose naltrexone for the treatment of fibromyalgia: findings of a small, randomized, double-blind, placebo-controlled, counterbalanced, crossover trial assessing daily pain levels. Arthritis Rheum 2013;65:529–38. 10.1002/art.37734 [DOI] [PubMed] [Google Scholar]
- 53.Mogil JS. Pain genetics: past, present and future. Trends Genet 2012;28:258–66. 10.1016/j.tig.2012.02.004 [DOI] [PubMed] [Google Scholar]
- 54.edKambur O, Männistö PT. Catechol-O-Methyltransferase and pain. Int Rev Neurobiol 2010;95:227–79. 10.1016/B978-0-12-381326-8.00010-7 [DOI] [PubMed] [Google Scholar]
- 55.Park D-J, Lee S-S. New insights into the genetics of fibromyalgia. Korean J Intern Med 2017;32:984–95. 10.3904/kjim.2016.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang L, Zhu J, Chen Y, et al. Meta-Analysis reveals a lack of association between a common catechol-O-methyltransferase (COMT) polymorphism val¹⁵⁸met and fibromyalgia. Int J Clin Exp Pathol 2014;7:8489–97. [PMC free article] [PubMed] [Google Scholar]
- 57.Lee C, Liptan G, Kantorovich S, et al. Association of Catechol-O-methyltransferase single nucleotide polymorphisms, ethnicity, and sex in a large cohort of fibromyalgia patients. BMC Rheumatol 2018;2:38. 10.1186/s41927-018-0045-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nicholl BI, Holliday KL, Macfarlane GJ, et al. No evidence for a role of the catechol-O-methyltransferase pain sensitivity haplotypes in chronic widespread pain. Ann Rheum Dis 2010;69:2009–12. 10.1136/ard.2009.126086 [DOI] [PubMed] [Google Scholar]
- 59.Smith SB, Maixner DW, Fillingim RB, et al. Large candidate gene association study reveals genetic risk factors and therapeutic targets for fibromyalgia. Arthritis Rheum 2012;64:584–93. 10.1002/art.33338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vargas-Alarcón G, Fragoso J-M, Cruz-Robles D, et al. Catechol-O-Methyltransferase gene haplotypes in Mexican and Spanish patients with fibromyalgia. Arthritis Res Ther 2007;9:R110. 10.1186/ar2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hocking LJ, Smith BH, Jones GT, et al. Genetic variation in the beta2-adrenergic receptor but not catecholamine-O-methyltransferase predisposes to chronic pain: results from the 1958 British birth cohort study. Pain 2010;149:143–51. 10.1016/j.pain.2010.01.023 [DOI] [PubMed] [Google Scholar]
- 62.Diatchenko L, Slade GD, Nackley AG, et al. Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Hum Mol Genet 2005;14:135–43. 10.1093/hmg/ddi013 [DOI] [PubMed] [Google Scholar]
- 63.Tammimäki A, Männistö PT. Catechol-O-Methyltransferase gene polymorphism and chronic human pain: a systematic review and meta-analysis. Pharmacogenet Genomics 2012;22:673–91. 10.1097/FPC.0b013e3283560c46 [DOI] [PubMed] [Google Scholar]
- 64.Meng W, Adams MJ, Reel P, et al. Genetic correlations between pain phenotypes and depression and neuroticism. Eur J Hum Genet 2020;28:358–66. 10.1038/s41431-019-0530-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meng W, Chan BW, Harris C, et al. A genome-wide association study finds genetic variants associated with neck or shoulder pain in UK Biobank. Hum Mol Genet 2020;29:1396–404. 10.1093/hmg/ddaa058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meng W, Adams MJ, Palmer CNA, et al. Genome-wide association study of knee pain identifies associations with GDF5 and COL27A1 in UK Biobank. Commun Biol 2019;2:321. 10.1038/s42003-019-0568-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Howard DM, Adams MJ, Clarke T-K, et al. Genome-Wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci 2019;22:343–52. 10.1038/s41593-018-0326-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nagel M, Jansen PR, Stringer S, et al. Meta-Analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat Genet 2018;50:920–7. 10.1038/s41588-018-0151-7 [DOI] [PubMed] [Google Scholar]
- 69.Mork PJ, Vasseljen O, Nilsen TIL. Association between physical exercise, body mass index, and risk of fibromyalgia: longitudinal data from the Norwegian Nord-Trøndelag health study. Arthritis Care Res 2010;62:611–7. 10.1002/acr.20118 [DOI] [PubMed] [Google Scholar]
- 70.Olsen NJ, Park JH. Skeletal muscle abnormalities in patients with fibromyalgia. Am J Med Sci 1998;315:351–8. 10.1097/00000441-199806000-00003 [DOI] [PubMed] [Google Scholar]
- 71.Staud R. Autonomic dysfunction in fibromyalgia syndrome: postural orthostatic tachycardia. Curr Rheumatol Rep 2008;10:463–6. 10.1007/s11926-008-0076-8 [DOI] [PubMed] [Google Scholar]
- 72.Furlan R, Colombo S, Perego F, et al. Abnormalities of cardiovascular neural control and reduced orthostatic tolerance in patients with primary fibromyalgia. J Rheumatol 2005;32:1787–93. [PubMed] [Google Scholar]
- 73.Sluka KA, Clauw DJ. Neurobiology of fibromyalgia and chronic widespread pain. Neuroscience 2016;338:114–29. 10.1016/j.neuroscience.2016.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Staud R. Peripheral pain mechanisms in chronic widespread pain. Best Pract Res Clin Rheumatol 2011;25:155–64. 10.1016/j.berh.2010.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Häuser W, Perrot S, Sommer C, et al. Diagnostic confounders of chronic widespread pain: not always fibromyalgia. Pain Rep 2017;2:e598. 10.1097/PR9.0000000000000598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wolfe F, Smythe HA, Yunus MB, et al. The American College of rheumatology 1990 criteria for the classification of fibromyalgia. Report of the multicenter criteria Committee. Arthritis Rheum 1990;33:160–72. 10.1002/art.1780330203 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
annrheumdis-2020-219624supp001.pdf (1MB, pdf)
Data Availability Statement
Summary statistics from our discovery and sensitivity GWAS was deposited at Zenodo (https://doi.org/10.5281/zenodo.4459546). Other data relevant to the study are included in the article or uploaded as online supplementary information.