

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus responsible for causing the COVID-19 pandemic, can be detected in untreated wastewater. Wastewater surveillance of SARS-CoV-2 complements clinical data by offering earlier community-level detection, removing underlying factors such as access to healthcare, sampling asymptomatic patients, and reaching a greater population. Here, we compare 24-hour composite samples from the influents of two different wastewater treatment plants (WWTPs) in South Carolina, USA: Columbia and Rock Hill. The sampling intervals span the months of July 2020 and January 2021, which cover the first and second waves of elevated SARS-CoV-2 transmission and COVID-19 clinical cases in these regions. We identify four signature mutations in the surface glycoprotein (spike) gene that are associated with the following variants of interest or concern, VOI or VOC (listed in parenthesis): S477N (B.1.526, Iota), T478K (B.1.617.2, Delta), D614G (present in all VOC as of May 2021), and H655Y (P.1, Gamma). The N501Y mutation, which is associated with three variants of concern, was identified in samples from July 2020, but not detected in January 2021 samples. Comparison of mutations identified in viral sequence databases such as NCBI Virus and GISAID indicated that wastewater sampling detected mutations that were present in South Carolina, but not reflected in the clinical data deposited into databases.
Keywords: Wastewater-based epidemiology, SARS-CoV-2, Variant detection, Sewage surveillance
Graphical abstract

1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is responsible for the COVID-19 pandemic declared by the World Health Organization in March 2020 (Tedros, 2020). The SARS-CoV-2 pandemic has resulted in both loss of life and an economic downturn (US Census Bureau, 2021). Although the virus is predominantly transmitted by bioaerosols (Guzman, 2021), fecal shedding of SARS-CoV-2 has been established from both symptomatic and asymptomatic patients (Zhang et al., 2020), and there have been multiple reports of the detection of SARS-CoV-2 globally in both treated and untreated sewage (Crits-Christoph et al., 2020; Kumar et al., 2020; La Rosa et al., 2020; Westhaus et al., 2021). Wastewater sampling has emerged as a useful tool to aid policy makers in making decisions about stay-at-home orders and other measures to mitigate viral spread (McClary, 2021).
Advantages of SARS-CoV-2 wastewater surveillance include the following: (1) asymptomatic cases are captured in the data (Wu et al., 2020), (2) wastewater trends precede clinical data by as much as 10 days (Wu et al., 2020), and (3) wastewater data tracks infection trends independently from clinical data, without reflecting healthcare access and choices (“National Wastewater Surveillance System (NWSS) – a new public health tool to understand COVID-19 spread in a community|CDC”, 2021). However, the information provided by wastewater surveillance goes beyond tracking case counts: recent studies have shown that wastewater sampling can reveal which viral variants may be present in a specific location (Fontenele et al., 2021; Martin et al., 2020) as well as signature mutations that match clinical sequences or those not yet present in databases (Crits-Christoph et al., 2020; Jahn et al., 2021; Nemudryi et al., 2020). In particular, three variants, B.1.1.7 (Abbott, 2021), B.1.351 (Tegally et al., 2020), and P.1 (Candido et al., 2021) are considered concerning due to their potential for higher rates of transmission. These correspond to NextStrain clades 20I, 20H and 20J, respectively (Hadfield et al., 2018). These three variants of concern (Centers for Disease Control and Prevention, 2021), commonly referred to as the UK (World Health Organization label Alpha), South African (Beta), and Brazilian (Gamma) variants, share several mutations, such as the spike protein mutation from asparagine to tyrosine (N501Y). The N501Y mutation is of functional significance because it is located in the receptor-binding domain of the spike protein (Tegally et al., 2020). This domain binds to the human receptor that is thought to mediate viral entry into the cell (angiotensin-converting enzyme 2, or ACE2). The mutation from asparagine to tyrosine has been shown in mouse models to enhance binding affinity to the ACE2 receptor (Gu et al., 2020).
In this study, we sequence SARS-CoV-2 amplified from wastewater influent samples from two wastewater treatment plants (WWTPs) in the state of South Carolina, USA: Rock Hill and Columbia. The Rock Hill WWTP is located in York County on the northern border, whereas the Columbia WWTP is located in Richland County in the central area of the state. The sampling intervals span the months of July 2020 and January 2021, which cover the first and second waves of elevated SARS-CoV-2 transmission and COVID-19 clinical cases in these regions (Fig. 1 ). We report the detection of the N501Y mutation in wastewater as early as July 2020, as well as other mutations, some of which are present in variants of interest or concern. Many of these mutations, although reported in clinical data from other states or in South Carolina at other times, were not present in clinical sequences collected from patients in South Carolina during the corresponding intervals and deposited in the Global Initiative on Sharing All Influenza Data, or GISAID (Shu and McCauley, 2017), repository. This study affirms the value of wastewater surveillance, even from a limited number of WWTPs, as part of a statewide infectious disease mitigation program.
Fig. 1.

COVID-19 clinical cases peaked in Columbia (orange) and Rock Hill (blue), South Carolina, USA during July 2020 and January 2021. Stacked area chart depicting the seven-day moving average of clinical COVID-19 case counts reported for the USA postal zip codes served by the Columbia and Rock Hill WWTPs. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2. Materials and methods
2.1. Wastewater sampling
The Columbia and Rock Hill WWTPs are both secondary (activated sludge) WWTPs that treat municipal wastewater with 6% and 3% of total flow, respectively, permitted from industry. The Columbia WWTP serves a population of 363,714, whereas the Rock Hill WWTP serves 264,117 people. The monthly average flow of Columbia WWTP is 45 MGD. The monthly average flow of Rock Hill WWTP is 12 MGD.
One liter 24-hour composite wastewater samples were collected twice a week at the influent site of both the Columbia and Rock Hill WWTPs and transported on ice to the laboratory at the University of South Carolina where they were immediately processed. One mL of bovine respiratory syncytial virus (BRSV) vaccine (~80 million copies/mL) (INFORCE 3®) was added to 1 L of wastewater prior to concentration in order to quantify processing and viral extraction efficiency. The average BRSV viral recovery was 4-5%. The samples were then homogenized for 10 min using laboratory blenders and 250 mL of homogenized wastewater was decanted into centrifuge bottles. The samples were centrifuged using an Avanti® J-E Centrifuge (Beckman Coulter Lifesciences, Indianapolis, Indiana) with a JS-5.3 rotor for 30 min at 4577g without braking. The pellets were stored at −80 °C and 50 mL of the supernatants were concentrated to 400 μL using Milipore Amicon 30 kDa ultrafilters.
2.2. RNA extraction, library preparation, and sequencing
RNA was extracted from 200 μL of the concentrated supernatant using the Qiagen AllPrep PowerViral extraction kit as per the manufacturer's instructions and eluted in 51 μL of RNase-free water and stored at −80 °C. Sequencing libraries were prepared following the Oxford Nanopore Technologies (ONT) Eco PCR tiling of SARS-CoV-2 with native barcoding based off the protocol developed by the ARTIC network (Quick, 2020). Briefly, total RNA was transcribed into cDNA using the LunaScript® RT SuperMix Kit (New England Biolabs, Ipswich, MA). The resulting products were amplified by 40 cycles of PCR using two different primer pools (V3 design) to create 400 bp amplicons spanning the entire SARS-CoV-2 genome. The PCR products were cleaned with a 1:1 ratio of SPRISelect beads (Beckman Coulter Lifesciences, Indianapolis, IN) to sample rather than a 1:10 dilution of the PCR products as described in the protocol. The PCR products were then end-prepped using the NEBnext® Ultra™ II End Repair/dA-Tailing Module (New England Biolabs, Ipswich, MA). Sequencing barcodes and adapters (Oxford Nanopore Technologies, Oxford, UK) were sequentially ligated, and all remaining bead cleanups were performed using SPRIselect beads (Beckman Coulter Lifesciences, Indianapolis, IN). The final libraries were loaded onto two separate R9 version flow cells (Oxford Nanopore Technologies, Oxford, UK) and sequenced using a GridION X5.
Columbia and Rock Hill WWTP influent samples from July 2020 and one sample from June 28, 2020, were sequenced together on an R9 flow cell. 3.39 M reads (1.5 Gb) were analyzed with an average quality score (Qscore) of 12.72 and an average sequence length of 568 bp. Columbia and Rock Hill influent samples from January 2021, were sequenced together on an R9 flow cell. 2.58 M reads (2.1 Gb) were analyzed with an average quality score of 12.64 and an average read length of 525 bp. For a full list of samples and viral copy numbers ascertained by quantitative PCR (Supplementary Methods), see Supplementary Table S1.
2.3. Data analysis
Sequencing data processing was performed according to the ARTIC network nCoV-2019 novel coronavirus bioinformatics protocol (Loman et al., 2020). Basecalling and demultiplexing were performed within MinKNOW using the high-accuracy model of Guppy version 4.2.3 developed by Oxford Nanopore Technologies (ONT). The minimum barcode score was set to 40 and the dual barcoding option was applied. Reads were filtered using a Qscore threshold of 7 and reads outside of the length range of 400-700 bp were omitted to eliminate chimeric reads. Lastly, filtered reads were mapped to the SARS-CoV-2 genome (accession MN908947.3) using minimap (Li, 2018) within the artic minion command with normalization option enabled (--normalize 200). Variant calling was performed with inStrain (Olm et al., 2021) with the options -c 2 (minimum coverage of 2) and –pairing_filter all_reads (in order to accepted non-paired reads). Mutations identified within primer-binding regions and in problematic sites identified by De Maio and colleagues (De Maio et al., 2020; Maio et al., 2020) were removed and are not presented in Table 2, Table 3. InStrain results for the Columbia and Rock Hill WWTP samples are presented in Supplementary Datasets S1 and S2.
Table 2.
Non-synonymous mutations with >100× position coverage that are present in January 2021, but absent in July 2020. RdRp = RNA-dependent RNA polymerase. The entirety of the GISAID database was searched on June 17, 2021. Acknowledgement for contributions to GISAID are included in Supplementary Dataset S8.
| Nucleotide | Mutation in product | Coverage | SRA counts | GISAID | GISAID filtereda |
|---|---|---|---|---|---|
| Orf1ab polyprotein | |||||
| 6032 | NSP3_A1105T | 129 | 0 | 797 | 5 |
| 8083 | NSP3_M1788I | 116 | 120 | >10 k | 128 |
| 9928 | NSP4_M458I | 163 | 42 | 8321 | 61 |
| 11,085 | NSP6_Y38F | 124 | 0 | 218 | 1 |
| 12,986 | NSP9_M101V | 106 | 0 | 430 | 1 |
| 14,805 | RdRp_T455I | 102 | 0b | 0b | 0 |
| 16,175 | RdRp_L912F | 138 | 0 | 0 | 0 |
| Other | |||||
| 21,707 | Surface glycoprotein | 217 | 51 | 4923 | 1 |
| 27,964 | ORF8 protein | 107 | 2544 | 0c | 0c |
Location filtered by South Carolina and Collection date between January 1 and January 31, 2021, inclusive.
Absent from database search results, but present in the literature (Bui et al., 2021; Zeng et al., 2020).
GISAID not searchable by ORF8 substitutions.
Table 3.
Spike gene mutations identified in this work and associated references. NCBI SRA and GISAID were searched on June 16, 2021. High-confidence mutations are located in positions with at least 100× coverage, whereas medium-confidence mutations are located in positions with at least 50× coverage. Literature references are not comprehensive. Acknowledgement for contributions from GISAID are included in Supplementary Dataset S7.
| Nucleotide position | Mutation | Sample set | References | SRA Counts | GISAID | GISAID filtereda | |
|---|---|---|---|---|---|---|---|
| High-confidence, >100× coverage | |||||||
| 21,707 | H49Y | January 2021 | (Armero et al., 2021; Phan, 2020; Sixto-López et al., 2021) | 51 | 4919 | 1 | |
| 23,063 | N501Y | July 2020 | (Abbott, 2021; Candido et al., 2021; Tegally et al., 2020) | 49 | >10 k | 0b | |
| 24,319 | Q920K | July 2020 | (Armero et al., 2021) | 0 | 2 | 0 | |
| Medium-confidence, >50× coverage | |||||||
| 21,614 | L18F | January 2021 | (Grabowski et al., 2021) | 59 | >10 k | 4 | |
| 23,525 | H655Y | July 2020 | NA | 35 | 69 | 0 | |
| 24,007 | L822F | January 2021 | (Ansori et al., 2020; Guruprasad, 2021) | 52 | 1523 | 1 | |
| 24,023 | L858F | January 2021 | NA | 0 | 197 | 0 | |
| 24,026 | S884F | January 2021 | (Aktas, 2020; Pérez-Cataluña et al., 2021; Sharma et al., 2020) | 0 | 438 | 0 | |
| 24,134 | S939F | January 2021 | (Guruprasad, 2021; Laha et al., 2020; Oliva et al., 2021) | 119 | 6254 | 0 | |
| 24,213 | V1228L | January 2021 | (Aktas, 2020; Baruah and Bose, 2020; Guruprasad, 2021) | 45 | 4633 | 11 | |
| 24,733 | H1058Y | July 2020 | NA | 0 | 285 | 1 | |
Location filtered by South Carolina and Collection date between January 1 and January 31, 2021, and/or June 28, 2020 and July 31, 2020, depending on the sample sets where the mutation was observed.
Filtering by Collection Date of June 28 to July 31, 2020, but unrestricted location, resulted in 34 viruses with the N501Y mutation, 26 of which originated from samples in the USA.
In addition to performing inStrain analysis for all barcodes, each of which corresponded to a 24-hour composite influent sample collected from either Columbia or Rock Hill WWTPs, we also performed a combined analysis of Columbia and Rock Hill during each month (July 2020 or January 2021). This method allowed us to increase the sequencing depth by including unclassified reads with incomplete barcode ligation. Since samples from July 2020 were sequenced separately from January 2021, we were still able to compare WWTP samples in time, although geographic information was not separated. The combined analysis for July 2020 and January 2021 is presented in Supplementary Datasets S3 and S4, respectively.
3. Results and discussion
3.1. Detection of signature mutations of SARS-CoV-2 variants of concern from wastewater
Due to the composite nature of wastewater, in which viral fragments from different sources are combined, it is not possible at this time to determine whether mutations observed from the same sample correspond to the same viral genome. Also, viral fragments of different sizes may have stability differences in wastewater. Therefore, the observation of signature mutations in wastewater is insufficient to determine the presence of a Variant of Concern (VOC) (Centers for Disease Control and Prevention, 2021) within a community. Nevertheless, the observation of signature mutations from wastewater is an early warning of the potential presence of a VOC within a community and is thus still valuable.
Mutational profiles observed in the surface glycoprotein (spike) gene in Columbia and Rock Hill WWTP influent samples during the month of January 2021 are presented in Fig. 2 . Samples with less than 200× average depth and 70% SARS-CoV-2 genome coverage were excluded, hence samples from July 2020 are not presented due to depth and coverage below these thresholds (Supplementary Table S2). The average sequencing depth and SARS-CoV-2 genome coverage for samples from the Columbia WWTP in January 2021 was 314× and 92.9%, whereas the average depth and coverage for Rock Hill WWTP samples was 249× and 85.8%. A complete list of the sequencing depth and coverage for each sample is provided in Supplementary Table S2. Four mutations in the spike gene were detected that are present in one or more variants of interest (VOI) or variants of concern (VOC) (Centers for Disease Control and Prevention, 2021), which are listed in parenthesis: S477N (B.1.526, Iota), T478K (B.1.617.2, Delta), D614G (present in all VOC as of May 2021), H655Y (P.1, Gamma). A full list of mutations in the spike protein for each variant is provided at CoVariants.org (Hodcroft, 2021) and in Table 1 . Mutations were only considered with at least 10 reads corresponding to the divergent nucleotide and at least 50× coverage in the nucleotide position. Including the mutations found in VOI and VOC, 16 mutations in the spike gene were identified from all Rock Hill samples and 34 from Columbia samples. A full list of mutations across the entire SARS-CoV-2 genome is included in Supplementary Datasets S1 and S2 for both Columbia and Rock Hill WWTPs in July 2020 and January 2021 samples. Table 1 illustrates the value of sequencing SARS-CoV-2 from wastewater in addition to clinical samples (represented by sequences deposited in GISAID): mutations detected in VOI/VOC that were already known to be present in South Carolina by clinical sequencing data were corroborated by wastewater data, but more importantly, potential VOI/VOC were flagged by the mutations detected in wastewater that were absent from the clinical sequences in GISAID collected in January 2021.
Fig. 2.
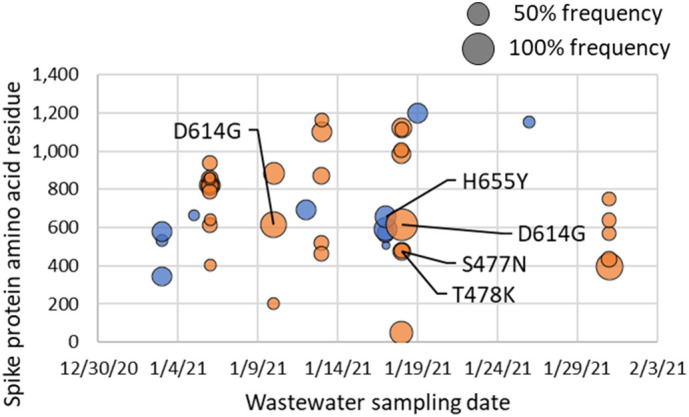
Spike gene mutations from the Wuhan SARS-CoV-2 reference genome (accession MN908947.3) detected in Columbia (orange) and Rock Hill (blue) WWTP influents during the month of January 2021. Bubble sizes represent the variant nucleotide frequency. Data labels are shown only for mutations identified in one or more variants of concern, but all detected mutations, both synonymous and nonsynonymous, are plotted (Supplementary Datasets S1 and S2). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 1.
Comparison of signature mutations detected in wastewater to sequenced Variants of Interest (VOI) or Variants of Concern (VOC) from clinical samples collected during January 2021 in South Carolina. Substitutions in bold were detected in WWTP influent samples. Clinical sequence data was taken from GISAID, and acknowledgements are included Supplementary Dataset S5. Spike protein substitutions were taken from the Center for Disease Control (Centers for Disease Control and Prevention, 2021). Substitutions shown in parenthesis indicate that the substitution is only observed in some sequences.
| Variant Pango lineage/WHO designation | Number of clinical Sequences (GISAID) | Collection date range | Spike Mutations | WWTP and collection date |
|---|---|---|---|---|
| B.1.351/Beta (VOC) | 8 | 01-08-21 to 01-26-21 |
D80A, D215G, 241del, 242del, 243del, K417N, E484K, N501Y, D614G, A701V | D614G: Columbia 01-10-21 and 01-18-21 |
| B.1.427, B.1.429/Epsilon (VOC) | 35 | 01-03-21 to 01-29-21 |
L452R, D614G; S13I, W152C, L452R, D614G | D614G: Columbia 01-10-21 and 01-18-21 |
| P.1/Gamma (VOC) | 0a | N/A | L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I | H655Y: Rock Hill 01-17-21, D614G: Columbia 01-10-21 and 01-18-21 |
| B.1.617.2/Delta (VOI) | 0b | N/A | Spike: T19R, (G142D), 156del, 157del, R158G, L452R, T478K, D614G, P681R, D950N | T478K: Columbia 01-18-21 |
| B.1.525/Eta (VOI) | 1 | 01-25-21 | A67V, 69del, 70del, 144del, E484K, D614G, Q677H, F888L | D614G: Columbia 01-10-21 and 01-18-21 |
| B.1.526/Iota (VOI) | 1 | (L5F*), T95I, D253G, (S477N*), (E484K*), D614G, (A701V*) | S477N: Columbia 01-18-21 |
First sequence for this variant in GISAID was submitted from a sample collected on 02-24-21.
First sequence for this variant in GISAID was submitted from a sample collected on 05-05-21.
3.2. Overlap in the mutational profiles of SARS-CoV-2 between Columbia and Rock Hill
We also aimed to determine similarities in SARS-CoV-2 mutations between Columbia and Rock Hill WWTP influents. Due to the low sequencing depth for barcoded samples in July 2020 (Supplementary Table S2), especially for Rock Hill, we avoided comparisons for July 2020 samples between Columbia and Rock Hill. In January 2021, we identified 85 shared locations in the SARS-CoV-2 genome where a mutation occurred. The Orf1ab gene occupies most of the SARS-CoV-2 genome length (71%). Similarly, we found that the largest percentage of shared mutational locations occurred in this gene. However, 26% of the shared locations were in the nucleocapsid phosphoprotein (N) gene, which constitutes only 4% of the genome length. We further investigated the shared mutations in this gene (Supplementary Table S3). In this analysis, we included low-frequency mutations with no coverage threshold, but also validated the mutations by comparison to sequence databases (Supplementary Table S3). Another study reported that the nucleocapsid is one of the SARS-CoV-2 proteome components with a high rate of mutation (Vilar and Isom, 2021). In particular, R203K and G204R both had a mutation rate of 0.22, indicating that they were present in 22% of all sequences as of December 2020 (Vilar and Isom, 2021). Similarly, S194 and M234 showed high rates of mutation (Vilar and Isom, 2021). Consistent with this study, we identified R203K, G204R, S194L, and M234I in the nucleocapsid protein as shared mutations between Columbia and Rock Hill WWTP influents in January 2021 (Supplementary Table S3). Therefore, mutations observed across multiple WWTP sites can reflect global mutation trends, which can be of value in identifying and investigating the spread of new, potentially more infectious strains of SARS-CoV-2.
3.3. Temporal trends of SARS-CoV-2 mutations observed in wastewater collected in January 2021
There was very little temporal overlap in the detected mutations for either the Columbia WWTP influent samples or the Rock Hill WWTP influent samples (Fig. 2), which may reflect the dynamic environment of wastewater in terms of the sewershed population served that is dependent on travel in and out of the community, as well as the wastewater conditions such as flowrate and types of contaminants. Detection of viral mutations in wastewater surveillance efforts may be sensitive to wastewater components such as salts, lipids, and urate, which can affect PCR efficiency (Farkas et al., 2020; Schrader et al., 2012). A synonymous guanine to adenine substitution at nucleotide position 25,297 (residue 1245) was detected in the spike gene in January 5 and 17 samples from the Rock Hill WWTP. A consensus adenine to guanine substitution in spike gene nucleotide position 23,403, corresponding to D614G was detected on January 10 and 18 in the Columbia WWTP influent. Only one mutation in the spike gene was shared between Columbia and Rock Hill: a synonymous adenine substitution for a thymine in the reference sequence at nucleotide position 23,269 (corresponding to amino acid 569 in the spike protein). Comparison of both high-confidence (>100× coverage) and medium-confidence (>50× coverage) divergent sites found in the Columbia WWTP samples from January 6 and 31 of 2021 indicated no shared mutations in the spike gene, despite comparable coverage. Across the entire SARS-CoV-2 genome, only five shared mutations out of 41 total with at least 100× coverage were found when comparing the Columbia WWTP influent sample from January 31 to the Columbia sample from January 6, 2021. Even when expanding the comparison to include mutations with less than 100× coverage in the January 6 sample, no additional shared mutations were identified. The five shared mutations were located in the region of the genome encoding the Orf1ab gene.
Out of the 34 divergent sites identified within the Columbia sample set (Supplementary Dataset S1), 31 were observed with less than 50% variant or consensus frequencies and all 16 divergent sites detected in the Rock Hill sample set were less than 50% frequency. In other words, the majority of the reads at positions with more than one detected nucleotide were consistent with the reference sequence. A notable exception is the D614G mutation in the spike protein observed in both the January 10 and 18 samples, where 100% of the reads aligned to the D614G variant, which is consistent with the global trend and evidence that the D614G mutation increases infectivity (Korber et al., 2020). The synonymous mutation of cytosine to thymine at position 22,747 (residue 395) in the genome was also a majority in the January 31 sample. The mutation H49Y was detected in the January 18 Columbia sample at 50% frequency. This mutation has been reported previously in several clinical samples (Armero et al., 2021; Phan, 2020; Sixto-López et al., 2021). The H49Y mutation has been associated with enhanced cell entry (Ozono et al., 2020), although at present it is not considered a variant of interest or concern.
3.4. Comparison of wastewater sequence diversity between July 2020 and January 2021
Peak case counts of SARS-CoV-2 in South Carolina were lower in July 2020 (2366 new cases on July 18, 2020) compared to January 2021 (7678 new cases reported on January 7) (CDC Case Surveillance Task Force, 2021), resulting in lower concentrations in wastewater (Supplementary Table S1) and this may have contributed to difficulty in amplification and sequencing. Combined analysis of both Columbia and Rock Hill sites with normalization resulted in an average sequencing depth of 415× for January 2021 and 300× for July 2020. Coverage of the SARS-CoV-2 genome was 99.8% for the combined analysis of January 2021 and 91.1% for July 2020. Variant calling with inStrain (Olm et al., 2021) detected 40 and 27 divergent sites with at least 100× position coverage and at least 10 divergent reads in July 2020 and January 2021, respectively. Fig. 3 summarizes the overlap in non-synonymous mutations between July 2020 and January 2021. Seven (four non-synonymous) mutations with at least 100× position coverage in the spike gene were detected in July 2020, whereas only two mutations (one nonsynonymous) were detected in January 2021. However, with a threshold of at least 50× position coverage, 11 mutations were detected in January 2021, and 10 were detected from the July 2020 sample set. Coverage of the spike gene in both July 2020 and January 2021 sequencing runs is presented in Supplementary Fig. S1. The July 2020 sequencing run experienced a region of amplicon dropout from 22,013 to 22,291 bp, which prohibited variant-calling in this area of the genome.
Fig. 3.

Non-synonymous shared or unique mutations detected in wastewater from July 2020 and January 2021. Each reported mutation had at least 100× coverage in its position and at least 10 divergent reads. RdRp = RNA-dependent RNA polymerase, 3'-to-5' = 3'-to-5' exonuclease.
In July 2020, the N501Y mutation of the spike gene was observed at 215× coverage. This mutation was not observed in the January 2021 combined analysis, although the SARS-CoV-2 genome had similar overall coverage (~400×) in this region to the July 2020 combined analysis. The N501Y mutation was not detected in the negative control for the July 2020 sequencing run. Globally, 34 sequences collected from June 28 to July 31, 2020, were deposited into both NCBI Virus and GISAID with the N501Y mutation. However, no sequences with the N501Y mutation were found from South Carolina in GISAID during this time frame, although there was an earlier reported observation of this mutation in May 2020. Twenty-six of the sequences with the N501Y mutation in the July time frame were from the USA (Table 2 ), and all except one were collected from patients in Texas. The N501Y observation in wastewater in South Carolina during July 2020 suggests a possible transmission event of a variant with the N501Y mutation between these two states, although it is also possible that there were already variants with the N501Y mutation present in South Carolina that were not captured in GISAID.
Both the evolution of SARS-CoV-2 as well as its geographic distribution are of interest in controlling the pandemic. Only 590 sequences with a collection date between January 1 and January 31, 2021 from the state of South Carolina were deposited in GISAID (Shu and McCauley, 2017) (acknowledgement included in Supplementary Dataset S5) as of May 26, 2021, despite 134,638 reported cases (CDC Case Surveillance Task Force, 2021). Similarly, only 377 nucleotide sequences from South Carolina were deposited in NCBI Virus (Hatcher et al., 2017) in the SARS-CoV-2 Data Hub with the same collection period. This disparity illustrates the value of wastewater sequencing, where this study alone includes viral sequences from approximately 600,000 people. Table 2 reports nonsynonymous mutations that were detected in the combined analysis of Columbia and Rock Hill WWTP influent samples in January 2021 but were absent in July 2020. Nine nonsynonymous mutations were present in January 2021 samples but absent from July 2020, and seven of these mutations were located in the Orf1b (Fig. 3 and Table 2). Mutations in the spike gene from July 2020 and January 2021 combined analysis of Columbia and Rock Hill are presented in Table 3 . To corroborate our findings, we searched the entirety of NCBI Virus (Hatcher et al., 2017) and GISAID (Shu and McCauley, 2017), as well as the literature (Table 2), for reports of the same mutations in SARS-CoV-2. We also filtered GISAID results by location and collection date to ascertain whether the detected mutations were present in data collected from South Carolina within the appropriate month. Notably, in many instances although the mutation was present in the entire sequence database, the mutation was not detected in sequences collected from South Carolina. This further supports the power of wastewater surveillance in capturing the sequence space of SARS-CoV-2 compared to clinical sampling alone.
4. Conclusions
We demonstrate here the value of amplifying and sequencing SARS-CoV-2 from wastewater to capture the sequence-space of mutations in the virus. We detected mutations reported in clinical data, including those in variants of concern, such as the N501Y mutation in the surface glycoprotein. Even during a single month, wastewater samples indicated a high degree of sequence diversity in the SARS-CoV-2 genome, with a total of 77 unique mutations (including both synonymous and nonsynonymous) in positions of at least 100× coverage detected in July 2020 samples and 230 mutations in January 2021. Wastewater samples from Columbia and Rock Hill alone captured SARS-CoV-2 sequence diversity that was absent in clinical samples from the entire state of South Carolina deposited into sequence databases like NCBI Virus and GISAID. We validated the observed mutations in SARS-CoV-2 samples from wastewater to the entirety of NCBI Virus and GISAID, as well as the literature, demonstrating that although many of the observed mutations were not detected in clinical samples collected during July 2020 or January 2021 from South Carolina, they have been observed elsewhere.
Although we successfully detected signature mutations present in variants of interest or concern in wastewater, we note that detection of SARS-CoV-2 in wastewater has its limitations compared to clinical sampling. The major obstacle is that amplicons are from a mixed pool of individuals and thus it is unlikely that mutations on different amplicons or even the same amplicon can be associated with a single genome or variant. However, as we have shown in this work, signature mutations associated with specific variants can still be detected and thus complement clinical datasets, which will always be limited to the number of tested patients, who are mostly symptomatic. Clinical sequences are more useful in determining transmission events that corroborates contact tracing and further informs disease-mitigation strategies (Walker et al., 2021), but wastewater surveillance can offer easier and faster detection of the presence of variants of interest or concern in a community, in addition to monitoring broader evolutionary trends at the population-level.
Data availability
Supplementary datasets are available through Mendeley Data: Swift, Candice (2021), “Community-level SARS-CoV-2 sequence diversity revealed by wastewater sampling”, Mendeley Data, V1, doi: 10.17632/ng8kd9wszx.1. Sequencing data in BAM format has been submitted to the National Center for Biotechnology Information Sequencing Read Archive (NCBI SRA) and is available at BioProject accession PRJNA745177.
CRediT authorship contribution statement
Candice L. Swift: Conceptualization, Investigation, Visualization, Writing – original draft. Mirza Isanovic: Investigation, Writing – review & editing. Karlen E. Correa Velez: Investigation, Writing – review & editing. R. Sean Norman: Conceptualization, Supervision, Funding acquisition, Visualization, Writing – review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We acknowledge the following funding sources: Center for Disease Control and Prevention #75D-301-18C-02903 and South Carolina Department of Health and Environmental Control (SCDHEC) #EQ-0-654. We are grateful to South Carolina utilities directors and operators, as well as SCDHEC for their contributions to the wastewater sampling and transportation that enabled this work. We gratefully acknowledge Authors from the Originating laboratories responsible for obtaining the specimens, as well as the Submitting laboratories where the genome data were generated and shared via GISAID, on which this research is based. A full list of Originating and Submitting laboratories referenced in this work is available in Supplementary Datasets S5-S8.
Editor: Warish Ahmed
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.scitotenv.2021.149691.
Appendix A. Supplementary data
Supplementary material
References
- Abbott S. 2021. Estimated Transmissibility and Severity of Novel SRS-CoV2 Variant of Concern 202012/01 in England. [Google Scholar]
- Aktas E. Bioinformatic Analysis Reveals That Some Mutations May Affect On Both Spike Structure Damage and Ligand Binding Site. bioRxiv. 2020 doi: 10.1101/2020.08.10.244632. [DOI] [Google Scholar]
- Ansori A.N.M., Kharisma V.D., Muttaqin S.S., Antonius Y., Parikesit A.A. Genetic variant of SARS-CoV-2 isolates in Indonesia: Spike glycoprotein gene. J. Pure Appl. Microbiol. 2020;14:971–978. doi: 10.22207/JPAM.14.SPL1.35. [DOI] [Google Scholar]
- Armero A., Berthet N., Avarre J.C. Intra-host diversity of sars-cov-2 should not be neglected: case of the state of Victoria, Australia. Viruses. 2021;13:1–15. doi: 10.3390/v13010133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruah V., Bose S. Immunoinformatics-aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019-nCoV. J. Med. Virol. 2020;92:495–500. doi: 10.1002/jmv.25698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui N.N., Lin Y.T., Huang S.H., Lin C.W. The extent of molecular variation in novel SARS-CoV-2 after the six-month global spread. Infect. Genet. Evol. 2021;91 doi: 10.1016/j.meegid.2021.104800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candido S., Mishra S., Crispim M.A.E., Sales F.C., Jesus J.G.De, Andrade P.S., Coletti T.M., Giulia M., Kraemer M.U., Jr N.G., Camilo C.C. Genomics and Epidemiology of a Novel SARS-CoV-2 Lineage in Manaus, Brazil. 2021. pp. 1–45. [Google Scholar]
- CDC Case Surveillance Task Force COVID-19 case surveillance public use data | data | Centers for Disease Control and Prevention [WWW document] 2021. https://data.cdc.gov/Case-Surveillance/COVID-19-Case-Surveillance-Public-Use-Data-with-Ge/n8mc-b4w4 URL. (accessed 5.26.21)
- Centers for Disease Control and Prevention . CDC; 2021. SARS-CoV-2 Variant Classifications and Definitions [WWW Document]https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html?CDC_AA_refVal=https%3A%2F%2Fwww.cdc.gov%2Fcoronavirus%2F2019-ncov%2Fcases-updates%2Fvariant-surveillance%2Fvariant-info.html URL. (accessed 5.26.21) [Google Scholar]
- Crits-Christoph A., Kantor R.S., Olm M.R., Whitney O.N., Al-Shayeb B., Lou Y.C., Flamholz A., Kennedy L.C., Greenwald H., Hinkle A., Hetzel J., Spitzer S., Koble J., Tan A., Hyde F., Schroth G., Kuersten S., Banfield J.F., Nelson K.L. Genome Sequencing of Sewage Detects Regionally Prevalent SARS-CoV-2 Variants. Vol. 12. 2020. pp. 1–9. medRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maio N., Walker C., Borges R., Weilguny L., Slodkowicz G., Goldman N. Masking strategies for SARS-CoV-2 alignments - Novel 2019 coronavirus / software and tools - Virological [WWW document] 2020. https://virological.org/t/masking-strategies-for-sars-cov-2-alignments/480 URL. (accessed 6.21.21)
- Farkas K., Hillary L.S., Malham S.K., McDonald J.E., Jones D.L. Wastewater and public health: the potential of wastewater surveillance for monitoring COVID-19. Curr. Opin. Environ. Sci. Health. 2020;17:14–20. doi: 10.1016/j.coesh.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenele R.S., Kraberger S., Hadfield J., Driver E.M., Bowes D., Holland L.A., Faleye T.O.C., Adhikari S., Kumar R., Inchausti R., Holmes W.K., Deitrick S., Brown P., Duty D., Smith T., Bhatnagar A., Yeager R.A., Holm R.H., Hoogesteijn von Reitzenstein N., Wheeler E., Dixon K., Constantine T., Wilson M.A., Lim E.S., Jiang X., Halden R.U., Scotch M., Varsani A. High-throughput sequencing of SARS-CoV-2 in wastewater provides insights into circulating variants. Prepr. Serv. Health Sci. 2021 doi: 10.1101/2021.01.22.21250320. medRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski F., Kochańczyk M., Lipniacki T. v-L18F substrain of SARS-CoV-2 VOC-202012/01 is rapidly spreading in England. medRxiv 2021.02.07.21251262. 2021 [Google Scholar]
- Gu H., Chen Q., Yang G., He L., Fan H., Deng Y., Wang Y., Teng Y., Zhao Z., Cui Y., Li Yuchang, Li X., Li J., Zhang N., Yang Xiaolan, Chen S., Guo Y., Zhao G., Wang X., Luo D., Wang H., Yang Xiao, Li Yan, Han G., He Y., Zhou X., Geng S., Sheng X., Jiang S., Sun S., Qin C., Zhou Y. 2020. Adaptation of SARS-CoV-2 in BALB/c Mice for Testing Vaccine Efficacy 1607, 1603–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruprasad L. 2021. Human SARS CoV-2 spike protein mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman M.I. An overview of the effect of bioaerosol size in coronavirus disease 2019 transmission. Int. J. Health Plann. Manag. 2021;36:257–266. doi: 10.1002/hpm.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield J., Megill C., Bell S.M., Huddleston J., Potter B., Callender C., Sagulenko P., Bedford T., Neher R.A. NextStrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34:4121–4123. doi: 10.1093/bioinformatics/bty407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher E.L., Zhdanov S.A., Bao Y., Blinkova O., Nawrocki E.P., Ostapchuck Y., Schaffer A.A., Rodney Brister J. Virus variation resource-improved response to emergent viral outbreaks. Nucleic Acids Res. 2017;45:D482–D490. doi: 10.1093/nar/gkw1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodcroft E.B. CoVariants: SARS-CoV-2 mutations and variants of interest [WWW document] 2021. https://covariants.org/ URL. (accessed 5.26.21)
- Jahn K., Dreifuss D., Topolsky I., Kull A., Ganesanandamoorthy P., Fernandez-Cassi X., Bänziger C., Stachler E., Fuhrmann L., Philipp Jablonski K., Chen C., Aquino C., Stadler T., Ort C., Kohn T., Julian T.R., Beerenwinkel N. 2021. Detection of SARS-CoV-2 Variants in Switzerland by Genomic Analysis of Wastewater Samples. medRxiv 2021.01.08.21249379. [Google Scholar]
- Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E.E., Bhattacharya T., Foley B., Hastie K.M., Parker M.D., Partridge D.G., Evans C.M., Freeman T.M., de Silva T.I., Angyal A., Brown R.L., Carrilero L., Green L.R., Groves D.C., Johnson K.J., Keeley A.J., Lindsey B.B., Parsons P.J., Raza M., Rowland-Jones S., Smith N., Tucker R.M., Wang D., Wyles M.D., McDanal C., Perez L.G., Tang H., Moon-Walker A., Whelan S.P., LaBranche C.C., Saphire E.O., Montefiori D.C. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827.e19. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M., Patel A.K., Shah A.V., Raval J., Rajpara N., Joshi M., Joshi C.G. First proof of the capability of wastewater surveillance for COVID-19 in India through detection of genetic material of SARS-CoV-2. Sci. Total Environ. 2020;746 doi: 10.1016/j.scitotenv.2020.141326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laha S., Chakraborty J., Das S., Manna S.K., Biswas S., Chatterjee R. Characterizations of SARS-CoV-2 mutational profile, spike protein stability and viral transmission. Infect. Genet. Evol. 2020;85 doi: 10.1016/j.meegid.2020.104445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rosa G., Iaconelli M., Mancini P., Bonanno Ferraro G., Veneri C., Bonadonna L., Lucentini L., Suffredini E. First detection of SARS-CoV-2 in untreated wastewaters in Italy. Sci. Total Environ. 2020;736 doi: 10.1016/j.scitotenv.2020.139652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman N., Rowe W., Rambaut A. nCoV-2019 novel coronavirus bioinformatics protocol [WWW Document] ARTIC Netw. 2020 https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html [Google Scholar]
- Maio N.D., W.C, Borges R., Weilguny L., Slodkowicz G., Goldman N. Virological.org; 2020. Issues with SARS-CoV-2 Sequencing Data - Novel 2019 Coronavirus / nCoV-2019 Genomic Epidemiology - Virological [WWW Document]https://virological.org/t/issues-with-sars-cov-2-sequencing-data/473 URL. (accessed 6.16.21) [Google Scholar]
- Martin J., Klapsa D., Wilton T., Zambon M., Bentley E., Bujaki E., Fritzsche M., Mate R., Majumdar M. Tracking SARS-CoV-2 in sewage: evidence of changes in virus variant predominance during COVID-19 pandemic. Viruses. 2020;12:1144. doi: 10.3390/v12101144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClary J.S. SARS-CoV-2 Wastewater Surveillance for Public Health Action: Connecting Perspectives From Wastewater Researchers and Public Health Officials During a Global Pandemic. 2021. pp. 1–21. Preprint. [DOI] [Google Scholar]
- National Wastewater Surveillance System (NWSS) A new public health tool to understand COVID-19 spread in a community | CDC [WWW document] 2021. https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/wastewater-surveillance.html URL. (accessed 4.20.21)
- Nemudryi A., Nemudraia A., Wiegand T., Surya K., Buyukyoruk M., Cicha C., Vanderwood K.K., Wilkinson R., Wiedenheft B. Temporal detection and phylogenetic assessment of SARS-CoV-2 in municipal wastewater. Cell Rep. Med. 2020;1 doi: 10.1016/j.xcrm.2020.100098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva R., Shaikh A.R., Petta A., Vangone A., Cavallo L. D936y and other mutations in the fusion core of the sars-cov-2 spike protein heptad repeat 1: Frequency, geographical distribution, and structural effect. Molecules. 2021;26 doi: 10.3390/molecules26092622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olm M.R., Crits-Christoph A., Bouma-Gregson K., Firek B.A., Morowitz M.J., Banfield J.F. inStrain profiles population microdiversity from metagenomic data and sensitively detects shared microbial strains. Nat. Biotechnol. 2021 doi: 10.1038/s41587-020-00797-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozono S., Zhang Y., Ode H., Tan T.S., Imai K., Miyoshi K., Kishigami S., Ueno T., Iwatani Y., Suzuki T., Tokunaga K. 2020. Naturally Mutated Spike Proteins of SARS-CoV-2 Variants Show Differential Levels of Cell Entry. [DOI] [Google Scholar]
- Pérez-Cataluña A., Chiner-Oms Á., Cuevas-Ferrando E., Díaz-Reolid A., Falcó I., Randazzo W., Girón-Guzmán I., Allende A., Bracho M.A., Comas I., Sánchez G. Detection of genomic variants of sars-cov-2 circulating in wastewater by high-throughput sequencing. medRxiv 2021.02.08.21251355. 2021 [Google Scholar]
- Phan T. Genetic diversity and evolution of SARS-CoV-2. Infect. Genet. Evol. 2020;81 doi: 10.1016/j.meegid.2020.104260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick J. nCoV-2019 sequencing protocol v3 (LoCost) [WWW document] 2020. https://www.protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bh42j8ye URL. (accessed 5.18.21)
- Schrader C., Schielke A., Ellerbroek L., Johne R. PCR inhibitors – occurrence, properties and removal. J. Appl. Microbiol. 2012;113:1014–1026. doi: 10.1111/J.1365-2672.2012.05384.X. [DOI] [PubMed] [Google Scholar]
- Sharma S., Dash P.K., Sharma S.K., Srivastava A., Kumar J.S., Karothia B.S., Chelvam K.T., Singh S., Gupta A., Yadav R.G., Yadav R., Greshma T.S., Kushwah P., Bhushan R., Nagar D.P., Nandan M., Kumar S., Thavaselvam D., Dubey D.K. Emergence and expansion of highly infectious spike:D614G mutant SARS-CoV-2 in central India (preprint). bioRxiv 2020.09.15.297846. 2020 doi: 10.1101/2020.09.15.297846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y., McCauley J. GISAID: global initiative on sharing all influenza data – from vision to reality. Eurosurveillance. 2017;22:2–4. doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixto-López Y., Correa-Basurto J., Bello M., Landeros-Rivera B., Garzón-Tiznado J.A., Montaño S. Structural insights into SARS-CoV-2 spike protein and its natural mutants found in mexican population. Sci. Rep. 2021;11:1–16. doi: 10.1038/s41598-021-84053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedros A.G. WHO Dir. Gen. Speeches. 2020. WHO Director-General’s opening remarks at the media briefing on COVID-19 - 11 March 2020. [Google Scholar]
- Tegally H., Wilkinson E., Giovanetti M., Iranzadeh A., Fonseca V., Giandhari J., Doolabh D., Pillay S., San E.J., Msomi N., Mlisana K., von Gottberg A., Walaza S., Allam M., Ismail A., Mohale T., Glass A.J., Engelbrecht S., van Zyl G., Preiser W., Petruccione F., Sigal A., Hardie D., Marais G., Hsiao M., Korsman S., Davies M.A., Tyers L., Mudau I., York D., Maslo C., Goedhals D., Abrahams S., Laguda-Akingba O., Alisoltani-Dehkordi A., Godzik A., Wibmer C.K., Sewell B.T., Lourenço J., Alcantara L.C.J., Kosakovsky Pond S.L., Weaver S., Martin D., Lessells R.J., Bhiman J.N., Williamson C., de Oliveira T. Emergence and Rapid Spread of a New Severe Acute Respiratory Syndrome-related Coronavirus 2 (SARS-CoV-2) Lineage With Multiple Spike Mutations in South Africa. 2020. p. 2. medRxiv. [DOI] [Google Scholar]
- US Census Bureau . 2021. Initial Impact of COVID-19 on U.S. Economy More Widespread Than on Mortality. [Google Scholar]
- Vilar S., Isom D.G. One year of SARS-CoV-2: how much has the virus changed? Biology (Basel) 2021;10:1–18. doi: 10.3390/biology10020091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker A., Houwaart T., Finzer P., Ehlkes L., Tyshaieva A., Damagnez M., Strelow D., Duplessis A., Nicolai J., Wienemann T., Tamayo T., Kohns Vasconcelos M., Hülse L., Hoffmann K., Lübke N., Hauka S., Andree M., Däumer M.P., Thielen A., Kolbe-Busch S., Göbels K., Zotz R., Pfeffer K., Timm J., Dilthey A.T. 2021. Characterization of SARS-CoV-2 Genetic Structure and Infection Clusters in a Large German City Based on Integrated Genomic Surveillance, Outbreak Analysis, and Contact Tracing. medRxiv 2021.02.13.21251678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhaus S., Weber F.A., Schiwy S., Linnemann V., Brinkmann M., Widera M., Greve C., Janke A., Hollert H., Wintgens T., Ciesek S. Detection of SARS-CoV-2 in raw and treated wastewater in Germany – suitability for COVID-19 surveillance and potential transmission risks. Sci. Total Environ. 2021;751 doi: 10.1016/j.scitotenv.2020.141750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Xiao A., Zhang J., Moniz K., Endo N., Armas F., Bonneau R., Brown M., Bushman M., Chai P., Duvallet C., Erickson T., Foppe K., Ghaeli N., Gu X., Hanage W., Huang K., Lee W.L., Matus M., McElroy K., Nagler J., Rhode S., Santillana M., Tucker J., Wuertz S., Zhao S., Thompson J., Alm E. SARS-CoV-2 titers in wastewater foreshadow dynamics and clinical presentation of new COVID-19 cases. Prepr. Serv. Health Sci. 2020;1–44 doi: 10.1101/2020.06.15.20117747. medRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H.L., Dichio V., Horta E.R., Thorell K., Aurell E. Global analysis of more than 50,000 SARS-CoV-2 genomes reveals epistasis between eight viral genes. Proc. Natl. Acad. Sci. U. S. A. 2020;117:31519–31526. doi: 10.1073/pnas.2012331117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Du R., Li B., Zheng X., Yang X., Hu B., Wang Y., Xiao G., Yan B., Shi Z., Zhou P. 2020. Molecular and Serological Investigation of 2019- nCoV Infected Patients: Implication of Multiple Shedding Routes. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Data Availability Statement
Supplementary datasets are available through Mendeley Data: Swift, Candice (2021), “Community-level SARS-CoV-2 sequence diversity revealed by wastewater sampling”, Mendeley Data, V1, doi: 10.17632/ng8kd9wszx.1. Sequencing data in BAM format has been submitted to the National Center for Biotechnology Information Sequencing Read Archive (NCBI SRA) and is available at BioProject accession PRJNA745177.