

Abstract
Different tetrahydrobenzo[b]thiophene derivatives were explored as new tubulin polymerization destabilizers to arrest tumor cell mitosis. A series of compounds incorporating the tetrahydrobenzo[b]thiophene scaffold were synthesized, and their biological activities were investigated. The cytotoxicity of each of the synthesized compounds was assessed against a range of cell lines. Specifically, the benzyl urea tetrahydrobenzo[b]thiophene derivative, 1-benzyl-3-(3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)urea (BU17), was identified as the most potent compound with broad-spectrum antitumor activity against several cancer cell lines. The potential mechanism(s) of action were investigated where dose-dependent G2/M accumulation and A549 cell cycle arrest were detected. Additionally, A549 cells treated with BU17 expressed enhanced levels of caspase 3 and 9, indicating the induction of apoptosis. Furthermore, it was found that BU17 inhibits WEE1 kinase and targets tubulin by blocking its polymerization. BU17 was also formulated into PLGA nanoparticles, and it was demonstrated that BU17-loaded nanoparticles could significantly enhance antitumor activity compared to the soluble counterpart.
Keywords: Tetrahydrobenzo[b]thiophenes, Antimitotic drugs, Tubulin polymerization, Microtubule assembly, Cell cycle arrest, Caspase activity, Nanoparticles
Graphical Abstract
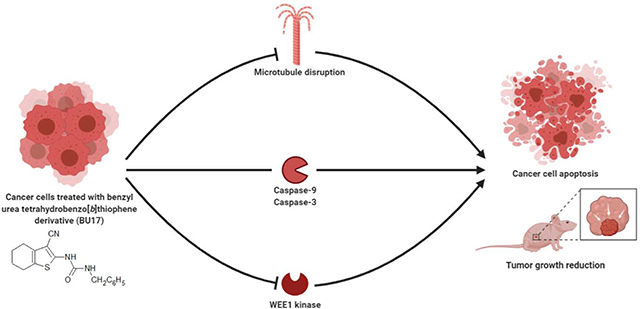
Different types of tetrahydrobenzo[b]thiophen derivatives have been synthesized to investigate their antitumor activity. 1-benzyl-3-(3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)urea was identified as the most potent compound with broad-spectrum antitumor activity, demonstrating strong apoptotic activities (inducing high expression of caspase-3 and −9) and antiproliferative activities through the inhibition of both tubulin polymerization and WEE1 kinase.
1. Introduction
Cancer is one of the deadliest diseases in modern history, with many types being difficult to eradicate. In 2018, approximately 17 million people were diagnosed with cancer, and 9.5 million deaths from cancer were reported worldwide [1]. Cancer can be linked to overactive positive cell cycle regulators, and it was found that altered expression and activity of cell cycle-related proteins cause dysregulated proliferation of cancer cells [2]. New targeted chemotherapeutic agents are being developed and explored to specifically kill cancer cells [3, 4]. In particular, antimitotic drugs that target microtubules and inhibit mitosis have been proposed as targeted antiproliferative drug candidates [5–7]. For example, vinca alkaloids (e.g. Vincristine Sulfate®, Pfizer) and taxanes (e.g. paclitaxel (PTX), Taxol®, Bristol Myers Squibb) are among the most effective cancer treatments that affect the dynamics of microtubules leading to cell death [8, 9]. However, these treatments possess drawbacks such as: complicated manufacturing processes; cancers can often develop resistance to these drugs; and these drugs are often associated with certain adverse effects (e.g., neuropathy and leukopenia) [10–13].
Many heterocyclic compounds bearing thiophene (e.g. AZD-7762, OSI-930, and PF-4989216) have been known to target cancer-specific proteins leading to inhibition or activation of specific signaling pathways in cancer cells [14–23]. In addition, several N-(3-cyano-4,5,6,7-tetrahydro-1-benzothien-2-yl)amides were reported as potent and selective JNK2 and JNK3 inhibitors [24]. Another thiophene bearing drug, nocodazole, has been shown to have antimitotic effects by binding to tubulin and arresting the cell cycle at the G2/M phase [25, 26]. Some thiophene-bearing antimitotic compounds such as ELR-510444 have been advanced to phase II clinical trials [27]. Additionally, other benzothiophene derivatives were found to have antitumor properties through their capacity to destabilize tubulin polymerization [28–30]. Recently, we reported a collection of benzylamines, benzamides, and ureas with the cyclohepta[b]thiophene scaffold, and 1-benzyl-3-(3-cyano-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophen-2-yl)urea was identified as a tubulin polymerization inhibitor and promising antiproliferative agent [31]. In our current work, another set of thiophene featuring compounds was synthesized using a tetrahydrobenzo[b]thiophene scaffold, with the aim of developing more potent anticancer compounds.
One of the efficient approaches to delivering drugs to tumors is through nanoparticles. Polymeric nanoparticles enhance the delivery of hydrophobic drugs, provide sustained drug release, and achieve enhanced drug delivery to the tumor site through substantial improvement in the drug accumulation and retention effect [32–35]. In this work, a series of benzylamines, benzamides and urea derivatives containing the tetrahydrobenzo[b]thiophene scaffold were fabricated and evaluated in vitro, and the lead compound was advanced to an in vivo study for testing its anticancer efficacy; with the mechanism of action of the most promising derivative also being explored. In addition, a nanoparticle-based formulation was developed for the most promising derivative to improve drug efficacy and safety.
2. Results and Discussion
Chemistry and properties of synthesized compounds.
In this work, the 6-membered ring tetrahydrobenzo[b]thiophene was utilized as a scaffold for ring optimization as follow-up research to our previous study [31]. Our target compounds were generated as illustrated in Figure 1. Fabrication of 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (compound 1) was achieved by reacting sulfur, malononitrile, diethylamine, and cyclohexanone in ethanol overnight [36]. Benzamides (compounds BZ02-BZ05) were synthesized via condensation of compound 1 and the appropriate acid chloride in pyridine at 80 °C. Condensation of compound 1 with the corresponding benzyl bromide in dimethylformamide gave the targeted benzylamine derivatives BZA06-BZA11. The synthesis of urea derivatives UD12-UD19 was achieved by adding the suitable aryl isocyanate to the amine 1. Properties and purity of the synthesized compounds were also studied, see the Supporting Information.
Figure 1. Fabrication scheme of tetrahyrdrobenzo[b]thiophene derivatives from 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (compound 1).
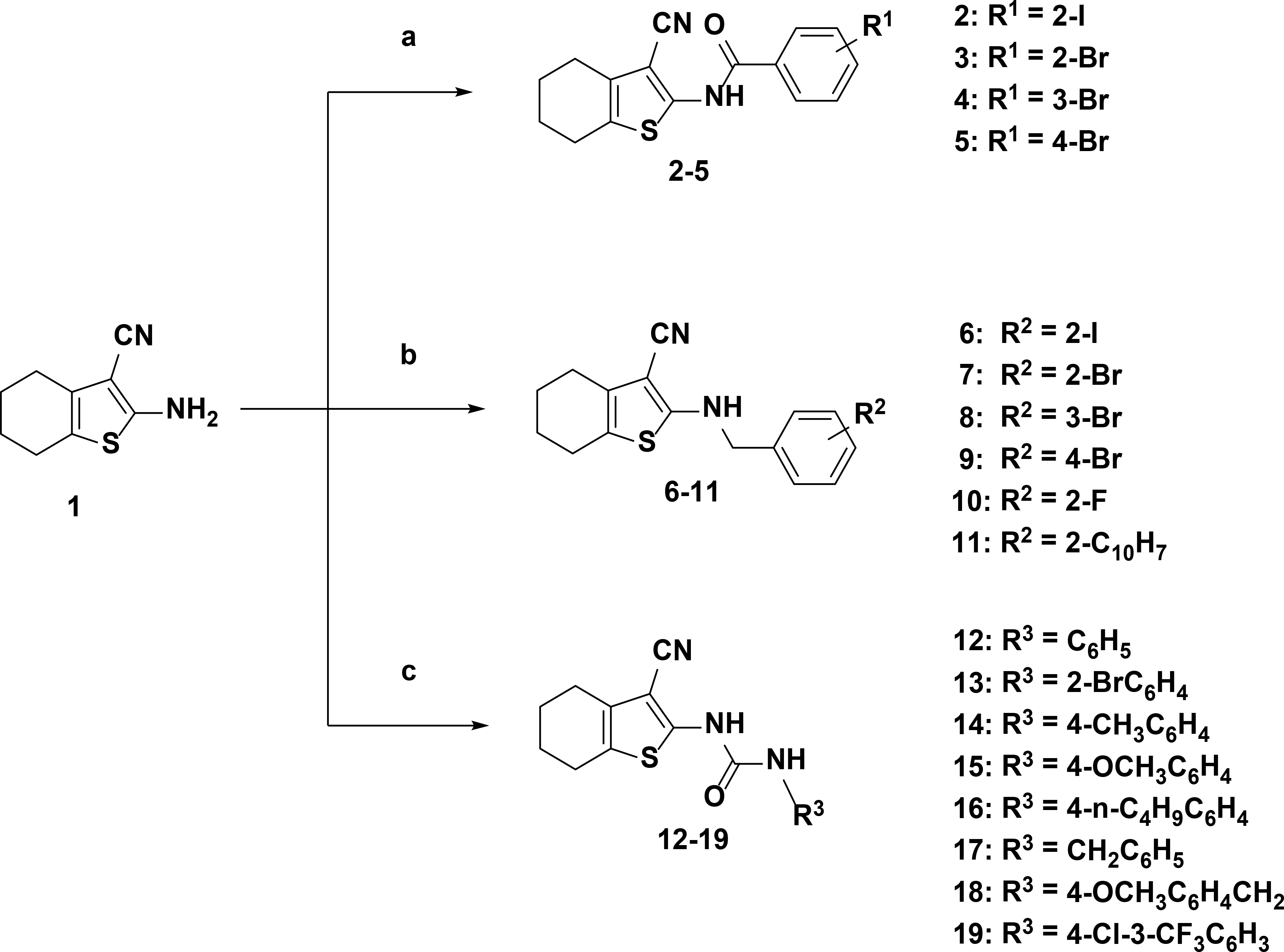
(a) Acid chlorides and pyridine based synthesis pathway; (b) benzyl bromides and dimethylformamide based synthesis pathway; (c) aryl isocyanates, dimethylformamide, and pyridine based pathway.
Cell viability testing with A549.
Several reports have highlighted the anticancer activity of thiophene-based derivatives [37]. For example, researchers have demonstrated the fabrication of a series of thiophene derivatives, with their anticancer properties being assessed using a wide range different of tumor cell types. Findings from these studies indicated that thiophene derivatives were effective against several tumor cell lines and had substantial antitumor effects against the non-small cell lung cancer cell line, A549, and the colorectal cancer cell line, CT26 [38–41]. In our study, the preliminary anticancer effect of the synthesized compounds was evaluated using 5 concentrations (100, 50, 25, 10, 1, 0.1 μM) against the A549 cell line using the MTS method. Table 1 shows the inhibitory concentrations, which produced 50% cell viability (IC50), of the investigated products. It was found that 2-iodobenzamide (BZ02) displayed enhanced inhibitory activity against the A549 cell line (IC50= 6.10 μM) when compared to other benzamides. Replacement of the iodide in compound 2 with bromide (i.e., BZ03) greatly reduced the inhibitory activity. In addition, the reduction of the benzamide (BZ02) into benzylamine (i.e., BZA06) resulted in an IC50 value of 30 μM. It was also found that the benzylamine (BZA09, IC50= 2.73 μM) was more potent than the corresponding benzamide (BZ05, IC50= 9.49 μM). Among the urea derivatives, the ring deactivated compounds UD13 and UD19 demonstrated considerably greater antiproliferative activity (IC50= 12.19 and 7.2 μM; respectively) than the ring activated derivatives UD14-UD16 and UD18 (IC50 > 50 μM). Furthermore, compound BU17, bearing the benzyl group with higher flexibility than the phenyl group in compound UD12, exhibited a strong impact on cell viability with an IC50 value of 9.00 μM versus IC50 > 50 μM for compound UD12.
Table 1.
Cytotoxic potency (IC50) of all synthesized compounds upon culture with A549
| Compound No. | R | IC50 (μM) |
|---|---|---|
| BZ02 | 2-I | 6.10 ± 0.39 |
| BZ03 | 2-Br | >50 |
| BZ04 | 3-Br | >50 |
| BZ05 | 4-Br | 9.49 ± 1.32 |
| BZA06 | 2-I | 30.7 ± 0.07 |
| BZA07 | 2-Br | >50 |
| BZA08 | 3-Br | >50 |
| BZA09 | 4-Br | 2.73 ± 0.81 |
| BZA10 | 2-F | >50 |
| BZA11 | 2-C10H7 | >50 |
| BU12 | C6H5 | >50 |
| BU13 | 2-BrC6H4 | 12.19 ± 1.43 |
| BU14 | 4-CH3C6H4 | >50 |
| BU15 | 4-OCH3C6H4 | >50 |
| BU16 | 4-n-C4H9C6H4 | >50 |
| BU17 | C6H5CH2 | 9.00 ± 0.94 |
| BU18 | 4-OCH3C6H4CH2 | >50 |
| BU19 | 4-Cl-3-CF3 | 7.2 ± 0.61 |
Cell viability assay with a panel of 60 cell lines (single-dose study).
Based on the initial cell viability data with A549 cells, 16 of the synthesized compounds (BZ02-BZ05, BZA06-BZA10, and UD13-UD19) were advanced to preliminary studies investigating their antiproliferative activity at 10 μM concentration against 60 human cancer cell lines at the National Cancer Institute (NCI). Percentage growth inhibition (%GI) of these cell lines incubated with benzamides, benzylamines, and ureas are illustrated in Tables 2 and 3 while the mean % GI of the tested compounds is illustrated in Figure 2. It was found that compounds BZ02 and BZ05 displayed enhanced inhibitory effects in the vast majority of cell lines especially leukemia, colon, and CNS cancer cells. However, it was noticed that compound BZ02 was poorly cytotoxic towards prostate cancer cell lines. Interestingly, two benzamide compounds (BZ03 and BZ04) and a benzylamine compound (BZA06) exhibited selective inhibition towards MDA-MB-435 (a breast cancer cell line) and K-562 (an erythroleukemic cell line). Surprisingly, benzyl urea analog (compound BU17) displayed potent growth inhibition of most of the 60 cancer cell lines tested.
Table 2.
Percent growth inhibition (%GI) of 60 cancer cell lines treated with benzamide and benzylamine derivatives (10 μM).
| Cell line | %GI | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| BZ02 | BZ03 | BZ04 | BZ05 | BZA06 | BZA07 | BZA08 | BZA09 | BZA10 | |
| Leukemia | |||||||||
| CCRF-CEM | 76.33 | 19.72 | 36.49 | 79.57 | - | - | - | - | - |
| HL-60(TB) | 87.4 | 27.46 | 37.56 | 85.91 | - | - | - | - | - |
| K-562 | 85.19 | 72.39 | 77.52 | 84.07 | 15.55 | - | 10.78 | 11.75 | 14.05 |
| MOLT-4 | 65.12 | 36.49 | 47.66 | 67.49 | 17.25 | - | 16.24 | 12.95 | - |
| RPMI-8226 | 53.55 | 14.68 | 22.18 | 66.74 | 13.38 | - | - | - | - |
| SR | 61.36 | 42.76 | 68.07 | 49.75 | 19.52 | - | - | - | - |
| Non-Small Cell Lung Cancer | |||||||||
| A549/ATCC | 46.4 | 27.68 | 24.84 | 46.43 | 12.15 | - | - | - | - |
| EKVX | 51.5 | 12.99 | 63.01 | 23.69 | - | - | - | - | |
| HOP-62 | 57.84 | 16.73 | 53.6 | 14.21 | - | - | - | - | |
| HOP-92 | 28.4 | 13.83 | 18.55 | 17.72 | - | - | - | - | |
| NCI-H226 | 58.34 | 25.68 | 10.19 | 55.83 | 26.26 | - | - | - | 11.91 |
| NCI-H23 | 26.57 | - | 34.98 | 16.10 | - | - | - | - | |
| NCI-H322M | 11.49 | 20.9 | 47.9 | 15.80 | - | - | - | - | |
| NCI-H460 | 82.15 | 12.41 | 24.74 | 85.65 | - | - | - | - | - |
| NCI-H522 | 75.37 | 25.84 | 24.1 | 77.43 | 22.07 | - | - | - | 15.92 |
| Colon Cancer | |||||||||
| COLO 205 | NT | NT | - | NT | - | NT | NT | - | NT |
| HCC-2998 | 28.42 | 13.12 | - | 31.4 | - | - | - | - | - |
| HCT-116 | 73.63 | 12.01 | 25.15 | 77.73 | 14.07 | - | - | - | - |
| HCT-15 | 78.52 | 56.3 | 54.17 | 77.76 | - | - | - | - | - |
| HT29 | 82.56 | 55.47 | 40.53 | 84.22 | - | - | - | - | - |
| KM 12 | 65.85 | 37.22 | 30.46 | 74.73 | - | - | - | - | - |
| SW-620 | 79.04 | 54.5 | 49.51 | 80.79 | - | - | - | - | - |
| CNS Cancer | |||||||||
| SF-268 | 41.93 | - | - | 38.3 | - | - | - | - | - |
| SF-295 | 78.92 | 19.76 | 19.17 | 78.27 | - | - | - | - | - |
| SF-539 | 77.06 | 12.72 | 13.44 | 89.47 | - | - | - | - | - |
| SNB-19 | 60.84 | 17.86 | 18.92 | 61.49 | - | - | - | - | - |
| SNB-75 | 83.26 | 20.09 | 15.59 | 86.38 | 21.81 | - | - | - | - |
| U251 | 70.34 | 19.8 | 11.84 | 68.85 | 12.57 | - | - | - | - |
| Melanoma | |||||||||
| LOX IMVI | 63.17 | 26.13 | 20.01 | 61.34 | 11.70 | - | 16.38 | - | 11.64 |
| MALME-3M | 39.06 | 28.33 | 37.58 | 50.76 | 13.81 | - | - | - | - |
| M14 | 83.72 | 34.98 | 27.84 | 82.7 | - | - | - | - | - |
| MDA-MB-435 | L | 97.05 | 89.22 | L | - | - | - | - | - |
| SK-MEL-2 | 73.59 | 45.23 | - | 62.98 | - | - | - | - | - |
| SK-MEL-28 | 35.92 | 20.21 | 13.75 | 40.95 | - | - | - | - | - |
| SK-MEL-5 | 80.76 | 24.01 | 10.23 | 90.4 | - | - | - | - | - |
| UACC-257 | 39.3 | - | 16.56 | 33.88 | - | - | - | - | - |
| UACC-62 | 61.95 | 37.55 | 28.29 | 70.91 | 32.02 | 15.75 | 29.36 | 18.1 | 20.59 |
| Ovarian Cancer | |||||||||
| IGORV1 | 49.41 | 11.93 | 32.63 | 46.28 | 38.21 | - | 13.16 | - | - |
| OVCAR-3 | 83.34 | 14.14 | - | 83.23 | - | - | - | - | - |
| OVCAR-4 | 20.08 | - | 24.91 | 30.35 | - | - | - | 11.68 | - |
| OVCAR-5 | 29.21 | - | - | 44.26 | - | - | - | - | - |
| OVCAR-8 | 48.56 | - | 18.11 | 35.45 | - | - | - | - | - |
| NCI/ADR-RES | 86.76 | 52.06 | 23.19 | 83.39 | - | - | - | - | - |
| SK-OV-3 | NT | NT | 14.83 | NT | - | NT | NT | NT | NT |
| Renal Cancer | |||||||||
| 786–0 | 47.83 | 5.79 | 17.38 | 53.92 | - | - | - | 12.79 | - |
| A498 | NT | NT | NT | NT | - | NT | NT | - | NT |
| ACHN | 49.06 | - | 24.37 | 52.42 | 35.95 | - | - | - | - |
| CAKI-1 | 54.98 | 23.26 | 26.99 | 45.74 | - | 15.26 | 15.91 | - | 12.51 |
| RXF 393 | L | 14.86 | - | 86.29 | 12.39 | 11.89 | - | - | - |
| SN12C | 46.52 | - | 16.06 | 54.34 | - | - | - | - | - |
| TK-10 | - | - | 20.84 | 27.26 | 33.20 | - | - | - | - |
| UO-31 | 41.93 | 15.8 | 17.7 | 36.95 | - | 28.63 | 19.67 | - | 13.18 |
| Prostate Cancer | |||||||||
| PC-3 | 52.93 | - | - | 71.01 | - | - | - | - | - |
| DU-145 | 37.15 | - | - | 66.66 | - | - | - | - | - |
| Breast Cancer | |||||||||
| MCF-7 | 84.96 | 50.04 | 48.17 | 86.86 | 18.22 | - | 17.1 | - | - |
| MDA-MB-231/ATCC | 45.33 | 21.13 | 17.82 | 55.09 | 18.80 | - | - | - | - |
| HS 578T | 67.9 | 25.19 | 19.27 | 60.89 | - | - | - | - | - |
| BT-549 | 47.79 | 16.82 | - | 47.82 | - | - | - | - | - |
| T-47D | 38.56 | - | 22.08 | 52.8 | 25.85 | - | - | 29.48 | 15.53 |
| MDA-MB-468 | 49.97 | 33.4 | 40.7 | 80.47 | 13.55 | - | - | - | - |
(-): GI < 10%
(L): compound was lethal
(NT): not tested
Table 3.
Percent growth inhibition (%GI) of 60 cancer cell lines treated with benzyl urea derivatives (10 μM).
| Cell line | %GI | ||||||
|---|---|---|---|---|---|---|---|
| BU13 | BU14 | BU15 | BU16 | BU17 | BU18 | BU19 | |
| Leukemia | |||||||
| CCRF-CEM | - | - | - | - | 83.37 | NT | NT |
| HL-60(TB) | - | 13.86 | 24.51 | - | L | - | 25.42 |
| K-562 | - | - | 14.91 | 34.19 | 86.02 | - | 18.27 |
| MOLT-4 | - | - | - | 14.35 | 82.29 | - | - |
| RPMI-8226 | - | - | - | - | 82.34 | - | 18.96 |
| SR | 22.13 | - | - | 62.19 | 76.75 | 13.81 | - |
| Non-Small Cell Lung Cancer | |||||||
| A549/ATCC | - | - | - | 15.6 | 71.45 | - | - |
| EKVX | - | - | - | - | 42.23 | - | 19.05 |
| HOP-62 | - | - | - | - | 59.79 | - | 10.99 |
| HOP-92 | - | - | - | - | 58.03 | - | 18.72 |
| NCI-H226 | - | - | - | 19.08 | 70.53 | - | - |
| NCI-H23 | - | - | - | - | 70.97 | - | - |
| NCI-H322M | - | - | - | 21.81 | 57.82 | - | - |
| NCI-H460 | - | - | - | 14.88 | 97.38 | - | - |
| NCI-H522 | - | - | 16.63 | 19.81 | 78.07 | - | - |
| Colon Cancer | |||||||
| COLO 205 | NT | - | NT | NT | 89.93 | - | - |
| HCC-2998 | - | - | - | - | 77.56 | - | - |
| HCT-116 | - | - | - | 15.56 | 87.78 | - | - |
| HCT-15 | - | - | - | 17.04 | 79.73 | - | - |
| HT29 | - | - | - | 15.54 | 87.21 | - | - |
| KM 12 | - | - | - | 13.47 | 79.5 | - | - |
| SW-620 | - | - | - | - | 76.02 | - | - |
| CNS Cancer | |||||||
| SF-268 | - | - | - | - | 53.08 | - | - |
| SF-295 | - | - | - | - | 79.02 | - | - |
| SF-539 | - | - | - | - | L | - | - |
| SNB-19 | - | - | - | - | 65.89 | - | - |
| SNB-75 | - | - | - | - | 73.01 | - | 17.46 |
| U251 | - | - | - | - | 73.9 | - | - |
| Melanoma | |||||||
| LOX IMVI | - | - | - | 42.98 | 75.96 | - | - |
| MALME-3M | 10.28 | - | - | - | 52.48 | - | - |
| M14 | - | - | - | 14.62 | 89.36 | - | - |
| MDA-MB-435 | - | - | - | 25.09 | L | 14.91 | - |
| SK-MEL-2 | - | - | - | - | 54.43 | - | - |
| SK-MEL-28 | - | - | - | - | 35.09 | - | - |
| SK-MEL-5 | - | - | - | 20.49 | 86.65 | - | - |
| UACC-257 | - | - | - | - | 32.02 | - | - |
| UACC-62 | 12.92 | 25.77 | 25.07 | 57.08 | 60.66 | - | - |
| Ovarian Cancer | |||||||
| IGORV1 | - | - | - | - | 71.19 | - | - |
| OVCAR-3 | - | - | - | - | L | - | - |
| OVCAR-4 | - | - | - | - | 51.58 | - | - |
| OVCAR-5 | - | - | - | 87.10 | 54.00 | - | - |
| OVCAR-8 | - | - | - | - | 73.19 | - | - |
| NCI/ADR-RES | - | - | - | 87.38 | 88.86 | - | - |
| SK-OV-3 | NT | NT | NT | NT | 64.89 | - | - |
| Renal Cancer | |||||||
| 786–0 | - | - | - | 86.31 | 64.39 | - | - |
| A498 | NT | NT | NT | NT | NT | 77.36 | NT |
| ACHN | - | - | - | 83.62 | 53.81 | - | - |
| CAKI-1 | - | - | - | 69.00 | 61.90 | 80.81 | - |
| RXF 393 | 85.22 | - | - | - | 99.98 | NT | 80.16 |
| SN12C | - | - | - | - | 64.72 | - | - |
| TK-10 | - | - | - | 81.60 | 50.93 | 74.58 | - |
| UO-31 | 78.13 | 77.19 | 79.71 | 80.65 | 52.84 | - | 88.27 |
| Prostate Cancer | |||||||
| PC-3 | - | - | - | 82.06 | 73.05 | - | - |
| DU-145 | - | - | - | - | 89.93 | - | - |
| Breast Cancer | |||||||
| MCF-7 | - | - | - | - | 80.49 | - | - |
| MDA-MB-231/ATCC | - | - | - | 82.19 | 81.16 | - | - |
| HS 578T | - | - | - | - | 90.36 | - | 87.78 |
| BT-549 | - | - | - | - | 75.92 | - | - |
| T-47D | - | - | 87.21 | - | 60.02 | - | 78.38 |
| MDA-MB-468 | - | - | - | - | L | - | - |
(-): GI < 10%
(L): compound was lethal
(NT): not tested
Figure 2.
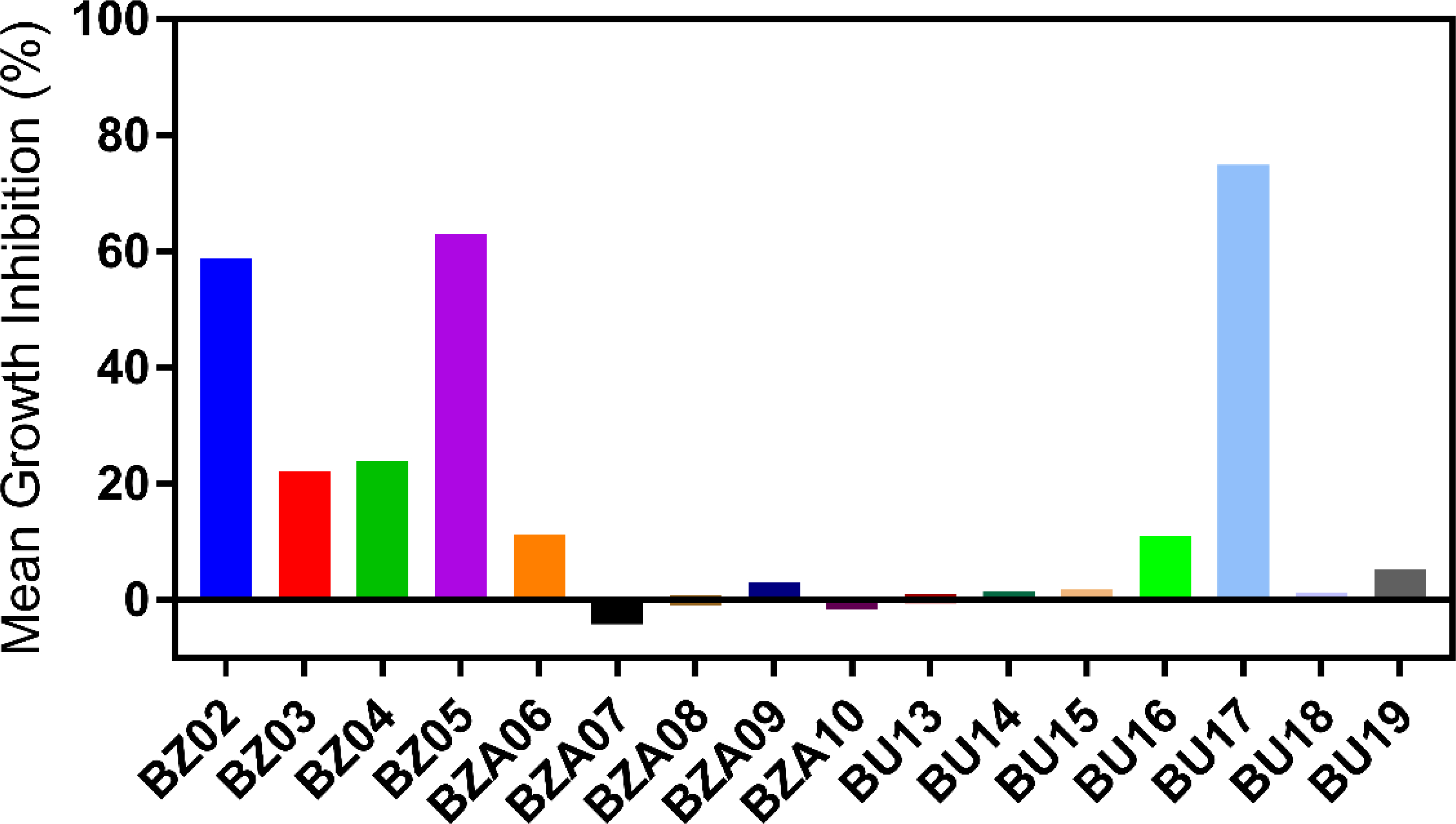
Percent growth inhibition (%GI) of the synthesized compounds tested on 60 cancer cell lines at the NCI.
Cell viability assay with a panel of 60 cell lines (multidose study).
Based on the single-dose study, BU17 was selected as a lead compound and advanced to subsequent studies where five increasing concentrations were tested against 60 cancer cell lines. In this experiment, the concentration of BU17 needed to inhibit cell growth by 50% (GI50), the concentration of BU17 needed for total growth inhibition (TGI), and the concentration of BU17 required for 50% lethality (LC50) were identified. The three parameters are shown in Tables 4 and 5. The results showed that compound BU17 displayed potent antitumor properties against the majority of tested cancer cell lines although the GI50 against SK-MEL-2, SK-MEL-28, and UACC-257 (28.5, 30.4, and 50.5 μM, respectively) indicated moderate antitumor activity. To elaborate, compound BU17 inhibited the growth of the majority of tested cancer cell lines at sub-micromolar concentrations (0.201–0.98 μM). In addition, BU17 exhibited GI50 concentrations against EKVX, HOP-62, HOP-92, NCI-H226, NCI-H23, HCC-2998, SF-268, OVCAR-4, OVCAR-5, OVCAR-8, SK-OV-3, SN 12C, TK-10, HS 578T, and BT-549 cell lines that ranged from 1.03 – 12.04 μM. Interestingly, it was found that compound BU17 exhibited superior antiproliferative activity to the antimitotic drug nocodazole against OVACAR-4, OVACAR-5, CAKI-1, and T47D demonstrating GI50 values of 3.46, 2.07, 0.687 and 1.44 μM versus 22.28, 20.75, 1.109 and 81.28 μM, respectively. In terms of the drug lethality, compound BU17 exhibited LC50 values >100 μM against all the tested cancer cell lines.
Table 4.
GI50 values (μM) generated for BU17 and nocodazole upon screening NCI-60 cell lines
| GI50 | GI50 | ||||
|---|---|---|---|---|---|
| Cell Lines | BU17 | Nocodazole | Cell Lines | BU17 | Nocodazole |
| Leukemia | Melanoma | ||||
| CCRF-CEM | 0.521 | 0.014 | M14 | 0.439 | 0.013 |
| HL-60(TB) | 0.246 | 0.011 | MDA-MB-435 | 0.235 | 0.011 |
| K-562 | 0.442 | 0.011 | SK-MEL-2 | 28.5 | 0.019 |
| MOLT-4 | 0.509 | 0.020 | SK-MEL-28 | 30.4 | 0.095 |
| RPMI-8226 | 0.719 | 0.013 | SK-MEL-5 | 0.467 | 0.018 |
| SR | 0.334 | 0.015 | UACC-257 | 50.5 | 0.148 |
| Non-Small Cell Lung Cancer | UACC-62 | 0.552 | 0.018 | ||
| A549/ATCC | 0.652 | 0.018 | Ovarian Cancer | ||
| EKVX | 2.45 | 0.039 | IGROV1 | 0.832 | 0.024 |
| HOP-62 | 2.93 | 0.028 | OVCAR-3 | 0.351 | 0.017 |
| HOP-92 | 5.88 | 0.292 | OVCAR-4 | 3.46 | 22.28 |
| NCI-H226 | 2.26 | 0.062 | OVCAR-5 | 2.07 | 20.75 |
| NCI-H23 | 3.38 | 0.016 | OVCAR-8 | 2.02 | 0.029 |
| NCI-H322M | 0.764 | 0.023 | NCI/ADR-RES | 0.478 | 0.014 |
| NCI-H460 | 0.453 | 0.017 | SK-OV-3 | 9.20 | 0.017 |
| NCI-H522 | 0.951 | 0.012 | Renal Cancer | ||
| Colon Cancer | 786–0 | 0.982 | 0.078 | ||
| COLO | 0.578 | 0.019 | A498 | 0.277 | 0.019 |
| HCC-2998 | 2.30 | 0.038 | ACHN | 0.691 | 0.225 |
| HCT-116 | 0.525 | 0.013 | CAKI-1 | 0.687 | 1.109 |
| HCT-15 | 0.461 | 0.019 | RXF 393 | 0.724 | 0.070 |
| HT29 | 0.408 | 0.013 | SN 12C | 1.03 | 0.073 |
| KM12 | 0.440 | 0.013 | TK-10 | 12.4 | 2.951 |
| SW-620 | 0.433 | 0.013 | UO-31 | 0.865 | 0.058 |
| CNS Cancer | Prostate Cancer | ||||
| SF-268 | 2.56 | 0.089 | PC-3 | 0.709 | 0.020 |
| SF-295 | 0.777 | 0.012 | DU-145 | 0.858 | 0.014 |
| SF-539 | 0.441 | 0.017 | Breast Cancer | ||
| SNB-19 | 0.778 | 0.086 | MCF-7 | 0.374 | 0.050 |
| SNB-75 | 0.201 | 0.015 | MDA-MB-468 | 0.500 | NT |
| U251 | 0.742 | 0.040 | HS 578T | 1.44 | 0.016 |
| Melanoma | T-47D | 1.44 | 81.28 | ||
| LOX IMVI | 0.699 | 0.021 | MDA-MB-231/ATCC | 1.44 | 0.171 |
| MALME-3M | 0.818 | 0.027 | |||
Table 5.
TGI and LC50 values (μM) for BU17 and nocodazole upon screening NCI-60 cell lines
| BU17 | Nocodazole | BU17 | Nocodazole | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Cell Lines | TGI | LC50 | TGI | LC50 | Cell Lines | TGI | LC50 | TGI | LC50 |
| Leukemia | Melanoma | ||||||||
| CCRF-CEM | >100 | >100 | 19.9 | 100 | M14 | >100 | >100 | 5.01 | >100 |
| HL-60(TB) | 0.759 | >100 | 0.05 | 79.43 | MDA-MB-435 | 0.651 | >100 | 0.1 | 1.58 |
| K-562 | >100 | >100 | 39.8 | 79.43 | SK-MEL-2 | >100 | >100 | 12.59 | >100 |
| MOLT-4 | >100 | >100 | 12.58 | 79.43 | SK-MEL-28 | >100 | >100 | 63.09 | >100 |
| RPMI-8226 | >100 | >100 | 6.31 | >100 | SK-MEL-5 | 9.71 | >100 | 6.31 | 50.11 |
| SR | >100 | >100 | 6.31 | 79.43 | UACC-257 | >100 | >100 | 100 | >100 |
| Non-Small Cell Lung Cancer | UACC-62 | >100 | >100 | 63.09 | >100 | ||||
| A549/ATCC | >100 | >100 | 50.12 | 100 | Ovarian Cancer | ||||
| EKVX | >100 | >100 | 31.62 | >100 | IGROV1 | >100 | >100 | 39.81 | >100 |
| HOP-62 | >100 | >100 | 100 | >100 | OVCAR-3 | 2.05 | >100 | 100 | 79.43 |
| HOP-92 | 56.0 | >100 | 7.94 | >100 | OVCAR-4 | >100 | >100 | 63.09 | >100 |
| NCI-H226 | 93.4 | >100 | 1.26 | >100 | OVCAR-5 | >100 | >100 | 100 | >100 |
| NCI-H23 | >100 | >100 | 0.63 | 63.09 | OVCAR-8 | >100 | >100 | 15.8 | >100 |
| NCI-H322M | >100 | >100 | 79.43 | >100 | NCI/ADR-RES | >100 | >100 | 0.16 | >100 |
| NCI-H460 | >100 | >100 | 10 | 79.43 | SK-OV-3 | >100 | >100 | 10.00 | >100 |
| NCI-H522 | >100 | >100 | 0.039 | >100 | Renal Cancer | ||||
| Colon Cancer | 786–0 | >100 | >100 | 31.62 | 79.43 | ||||
| COLO | 8.34 | >100 | 1.99 | 25.11 | A498 | 1.70 | >100 | 1.58 | 79.43 |
| HCC-2998 | >100 | >100 | 12.56 | >100 | ACHN | >100 | >100 | 79.43 | >100 |
| HCT-116 | >100 | >100 | 15.84 | >100 | CAKI-1 | >100 | >100 | 63.09 | >100 |
| HCT-15 | >100 | >100 | 2.51 | >100 | RXF 393 | 65.3 | >100 | 6.31 | 79.43 |
| HT29 | >100 | >100 | 1.00 | 25.11 | SN 12C | >100 | >100 | 31.62 | >100 |
| KM12 | >100 | >100 | 39.81 | >100 | TK-10 | >100 | >100 | 63.09 | >100 |
| SW-620 | >100 | >100 | 100 | >100 | UO-31 | >100 | >100 | 39.81 | 79.43 |
| CNS Cancer | Prostate Cancer | ||||||||
| SF-268 | >100 | >100 | 63.09 | >100 | PC-3 | >100 | >100 | 15.84 | >100 |
| SF-295 | 91.7 | >100 | 0.03 | 63.09 | DU-145 | 11.2 | >100 | 1.58 | 79.43 |
| SF-539 | 10.3 | >100 | 0.501 | >100 | Breast Cancer | ||||
| SNB-19 | >100 | >100 | 39.81 | >100 | MCF-7 | >100 | >100 | 50.12 | >100 |
| SNB-75 | 34.0 | >100 | 63.09 | >100 | MDA-MB-468 | 7.14 | >100 | 79.43 | >100 |
| U251 | 66.7 | >100 | 1.99 | 50.12 | HS 578T | >100 | >100 | 79.43 | >100 |
| Melanoma | T-47D | >100 | >100 | 100 | >100 | ||||
| LOX IMVI | >100 | >100 | 39.81 | >100 | MDA-MB-231/ATCC | >100 | >100 | 100 | >100 |
| MALME-3M | >100 | >100 | 39.81 | >100 | |||||
Cell cycle analysis.
To investigate the mechanism by which the BU17 exerts its antiproliferative activity, analysis of the cell cycle of A549 cells incubated with increasing concentrations of compound BU17 (1, 5, 10, 50 μM) was performed. Cells were incubated with the treatment for 24 h, then the analysis was carried out to identify the percentage of cells in each cell cycle phase and before a significant number of A549 cells underwent the apoptosis (Figure 3 and Table 6). In this experiment, it was found that BU17 shifted the cell population from G0/G1 and S phases to the G2/M phase in a concentration-dependent manner.
Figure 3.
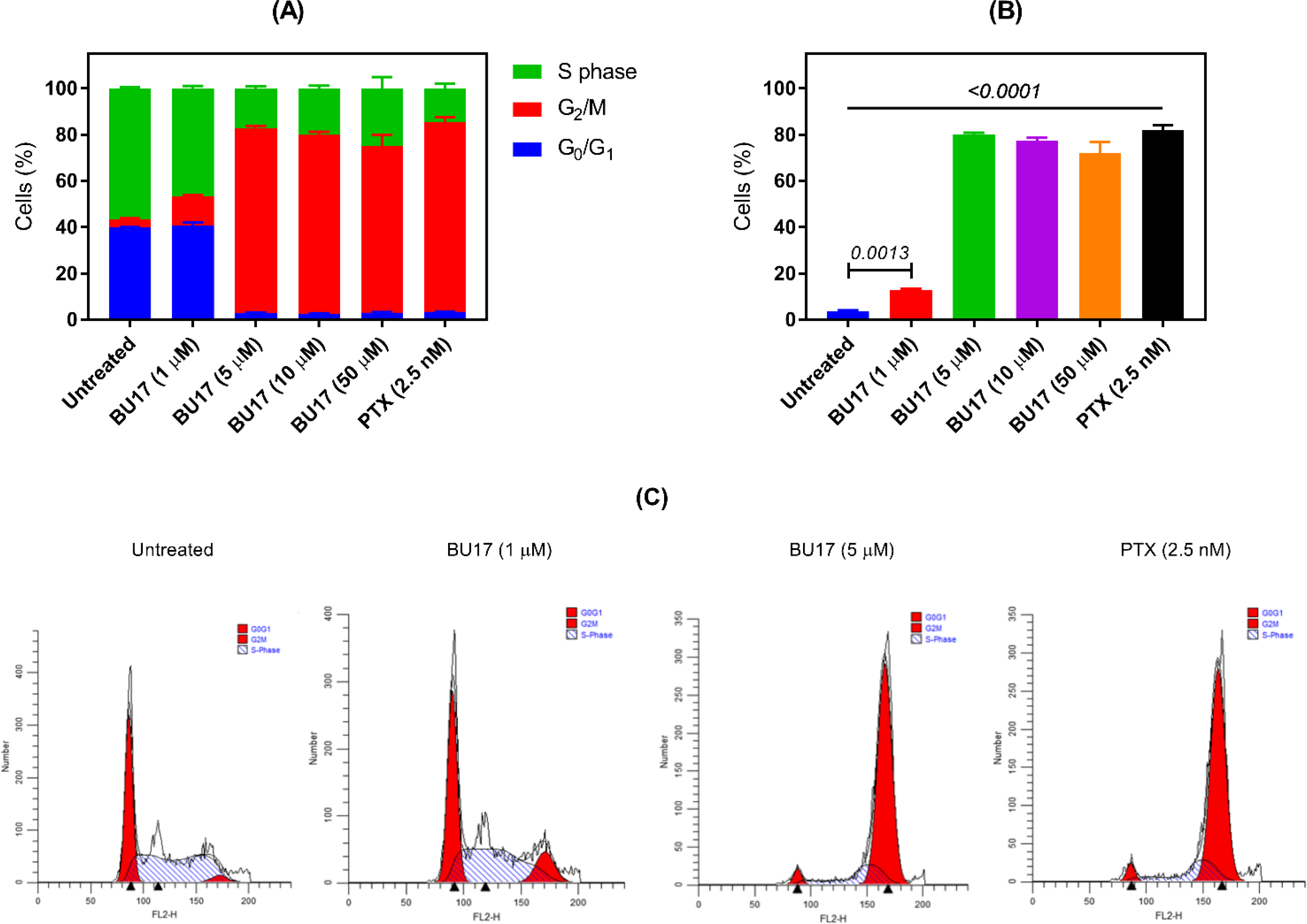
Analysis of the cell cycle of A549 cells treated with either BU17 or PTX for 24 h. (A) Cell cycle phase distribution of variously treated A549 cells. (B) Proportion (%) of A549 cells in the G2/M phase. (C) Histograms indicating the proportion of A549 cells in each cell cycle phase after incubation with either BU17 or PTX.
Table 6.
Cell cycle analysis (by phases, expressed as %) of A549 cells incubated with either BU17 or PTX.
| Groups | G0/G1 Phase | G2/M Phase | S Phase |
|---|---|---|---|
| Untreated cells | 39.78 ±0.23 | 3.57 ± 0.50 | 56.65 ± 0.53 |
| BU17 (1 μM) | 40.59 ± 1.51 | 12.86 ± 0.58 | 46.54 ± 1.05 |
| BU17 (5 μM) | 2.79 ± 0.22 | 80.02 ± 0.78 | 17.19 ± 0.94 |
| BU17 (10 μM) | 2.58 ± 0.28 | 77.34 ± 1.25 | 20.08 ± 1.10 |
| BU17 (50 μM) | 2.99 ± 0.27 | 71.99 ± 4.88 | 25.01 ± 4.88 |
| PTX (2.5 nM) | 3.41 ± 0.18 | 81.97 ± 2.06 | 14.62 ± 1.99 |
Apoptosis detection.
To investigate the potential anticancer mechanism(s) of action of BU17, A549 cells were treated with BU17, and apoptosis was assessed by staining with annexin V/propidium iodide (Figure 4A–C). Untreated cells showed 0.24% cell debris/necrotic cells in Q1, 0.0111% late apoptotic cells in Q2, 0.73% in the early apoptosis phase in Q3, and 99.0% live cells in Q4. In contrast, cells treated with 10 μM of BU17 displayed 2.65% cell debris in Q1, 4.39% late apoptotic cells in Q2, 10.9% in early apoptosis Q3, and 82.1% live cells in Q4. Therefore, compound BU17 substantially induced apoptosis in the A549 cells.
Figure 4.
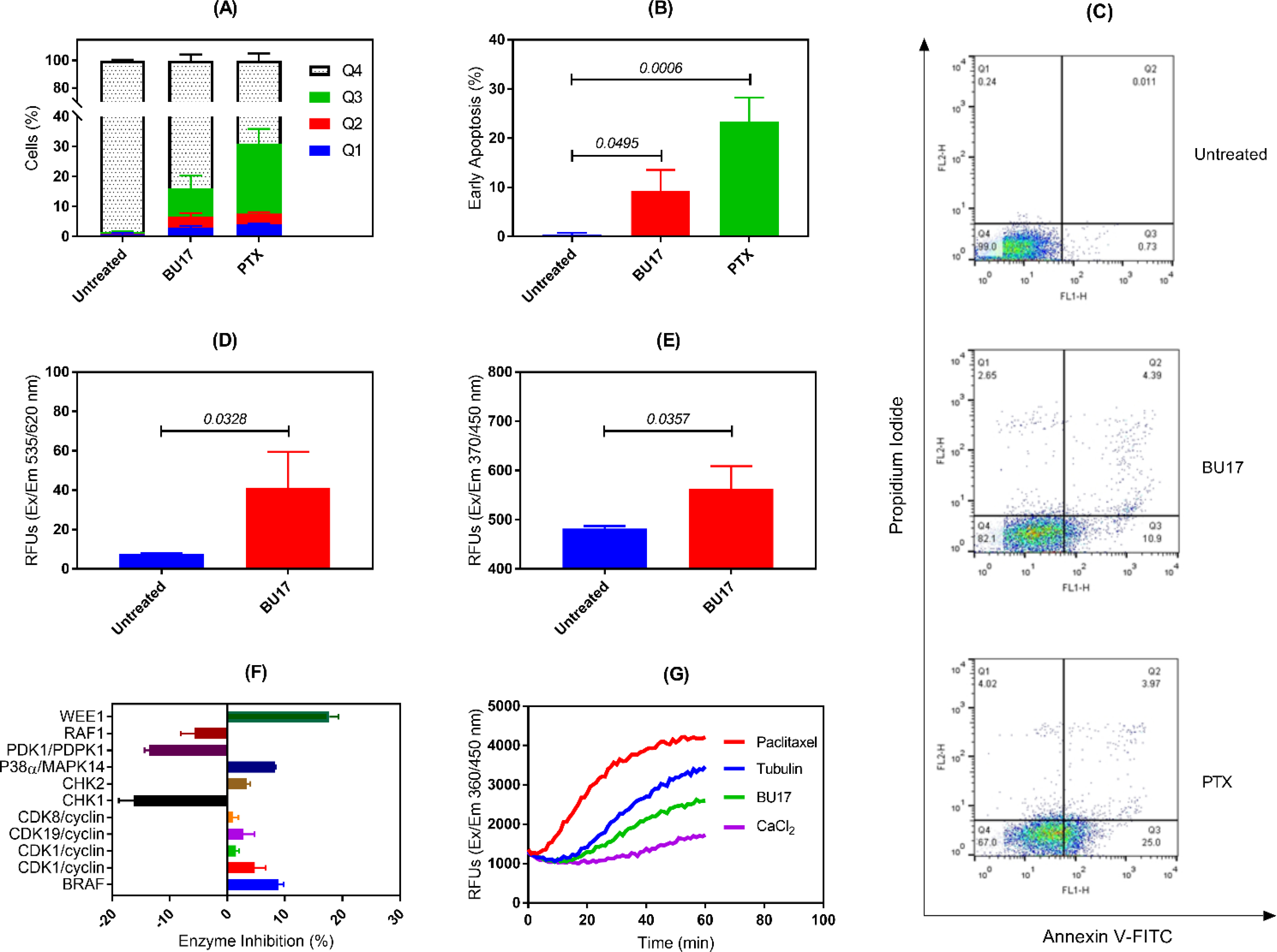
Summary of key mechanistic studies of BU17. (A) Percentages of A549 cells (treated as indicated) in necrotic cells (Q1), late apoptotic cells (Q2), early apoptotic cells (Q3), and live cells (Q4). (B) Detection of early apoptosis in the treated A549 cells as indicated. (C) Dot-plots of A549 cells treated as indicated. (D) Detection of caspase 3 expressions in A549 cells incubated with BU17. (E) Detection of caspase 9 expressions in A549 cells incubated with BU17. (F) Percentage enzymatic inhibition of BU17 against a panel of 11 kinases. (G) Effect of BU17 on tubulin polymerization rate. BU17 was used at 10 μM and PTX was used at 5 nM for all experiments.
Caspase 3 and 9 activity.
In this experiment, the expression of caspase 3 and 9 was measured in A549 cells treated with BU17 (10 μM) to detect apoptosis. The results indicated significantly higher levels of caspase 3 and 9 in A549 cells incubated with BU17 compared to untreated cells (Figure 4D and 4E). This suggests that BU17 triggered apoptosis via intrinsic pathways as evidenced by the high levels of caspase 9 in the treated cells.
JNK1 and JNK2 pathways.
Due to the similar structures of BU17 and the JNK inhibitors VIa and VIb (Figure 1), BU17 was tested for its capacity to inhibit JNK1 and JNK2. Results showed that compound BU17 had no inhibitory activity towards JNK1 and JNK2 enzymes. Specifically, the percentage of enzyme inhibition of compound BU17 on both enzymes was not different from the control group (DMSO) (percentage of enzyme inhibition of JNK1 and JNK2 relative to DMSO control was −7.87 and −1.67, respectively). This suggests that there could be other mechanisms of action(s) by which compound BU17 exerts its antitumor activity.
Activity of kinases.
BU17 was tested against 11 kinases that could be involved in cell cycle arrest. Based on the results, compound BU17 displayed moderate inhibitory activity against WEE1 (Figure 4F); suggesting an enhanced activity towards programmed cell death in response to DNA damage. Since the tested compound demonstrated very weak inhibitory activity towards the other tested kinases (>10%), it might cause cell cycle arrest by targeting different mechanisms.
Microtubule disruption.
According to the data obtained from the cell cycle experiment, BU17 was found to shift the cell population to G2/M. It has been widely shown that drugs with antimitotic activity due to G2/M phase arrest can also disrupt microtubule formation [8, 42]. Therefore, the impact of BU17 on tubulin polymerization rate was studied. Results displayed that BU17 substantially slowed down the tubulin polymerization rate and, thus, disrupted microtubule assembly (Figure 4G). Furthermore, the effect of BU17 on tubulin polymerization was opposite to PTX which is known to promote the tubulin polymerization rate.
Properties of nanoparticles.
The nanoparticles (loaded with BU17) were successfully prepared using a biodegradable and biocompatible polymer poly(lactic-co-glycolic acid) (PLGA; 50:50) with the surfactant D-α-tocopherol polyethylene glycol 1000 succinate, which inhibits the efflux transporter, P-glycoprotein (Figure 5A). The average particle diameter was 183.9 ± 4.09 nm with a marginal size distribution, and they displayed an average zeta potential of −40.73 mV (Figure 5B). In addition, scanning electron microscope images showed spherical nanoparticles with smooth surfaces (Figure 5C).
Figure 5.
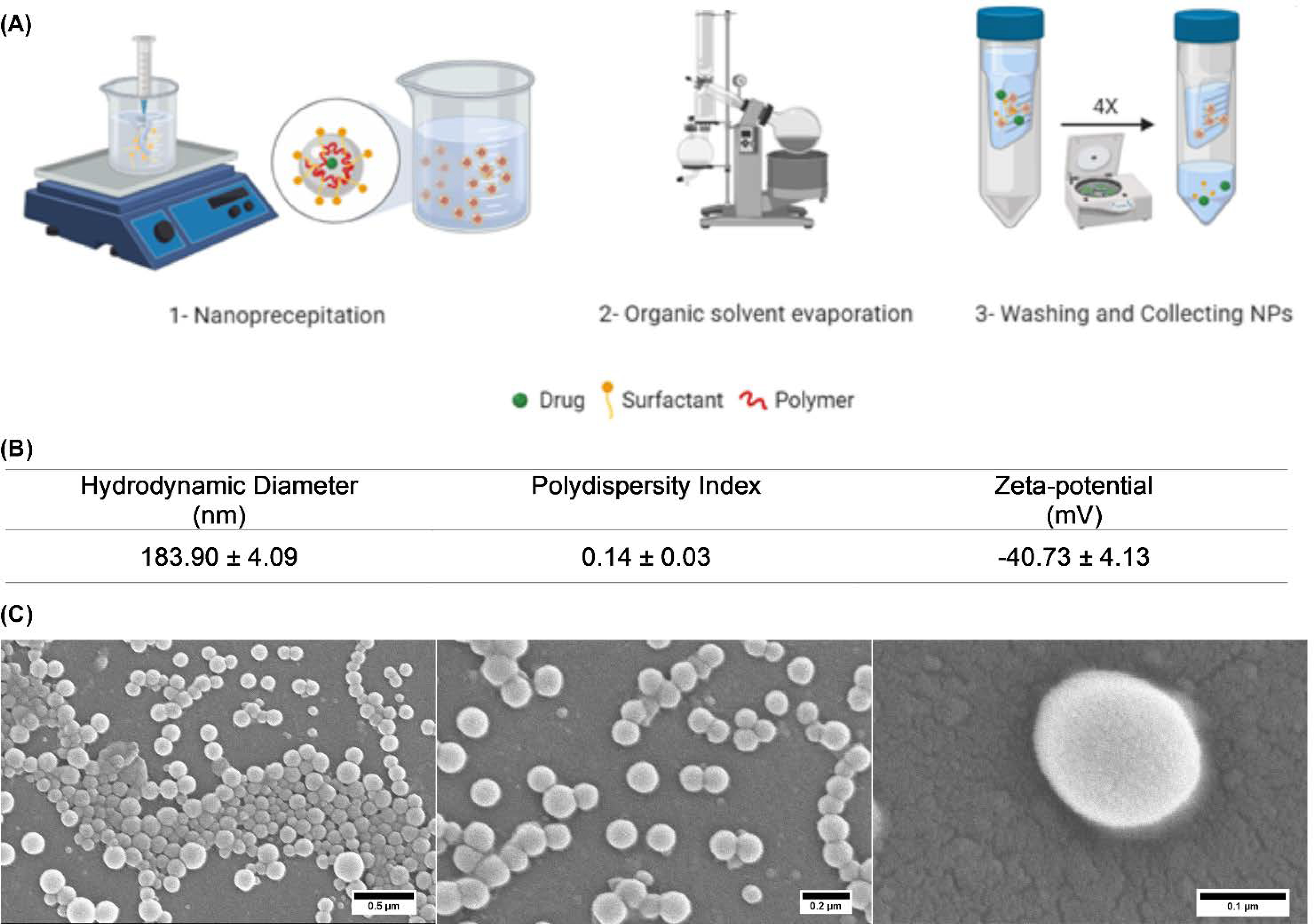
Fabrication and properties of BU17-loaded PLGA nanoparticles. (A) Diagram outlining preparation of BU17-loaded PLGA nanoparticles. (B) Characterization of the formulated nanoparticles by Zetasizer NanoZS. (C) Scanning electron microscope images of BU17-loaded PLGA nanoparticles.
Cell viability assay of compound BU17 in solution and nanoparticles.
To evaluate the antitumor activity of compound BU17 and its nanoparticle formulation prior to in vivo testing, cell viability testing of BU17, and its nanoparticles against CT26 murine colon carcinoma was performed adopting the MTS method in vitro (Figure 8). The drug-loaded nanoparticles displayed a significant decrease in cell viability (IC50 = 0.08 μM) in comparison to the BU17 in solution (IC50 = 1.52 μM) which means enhanced efficacy and delivery of BU17 when formulated into nanoparticles (Figure 6A).
Figure 6.
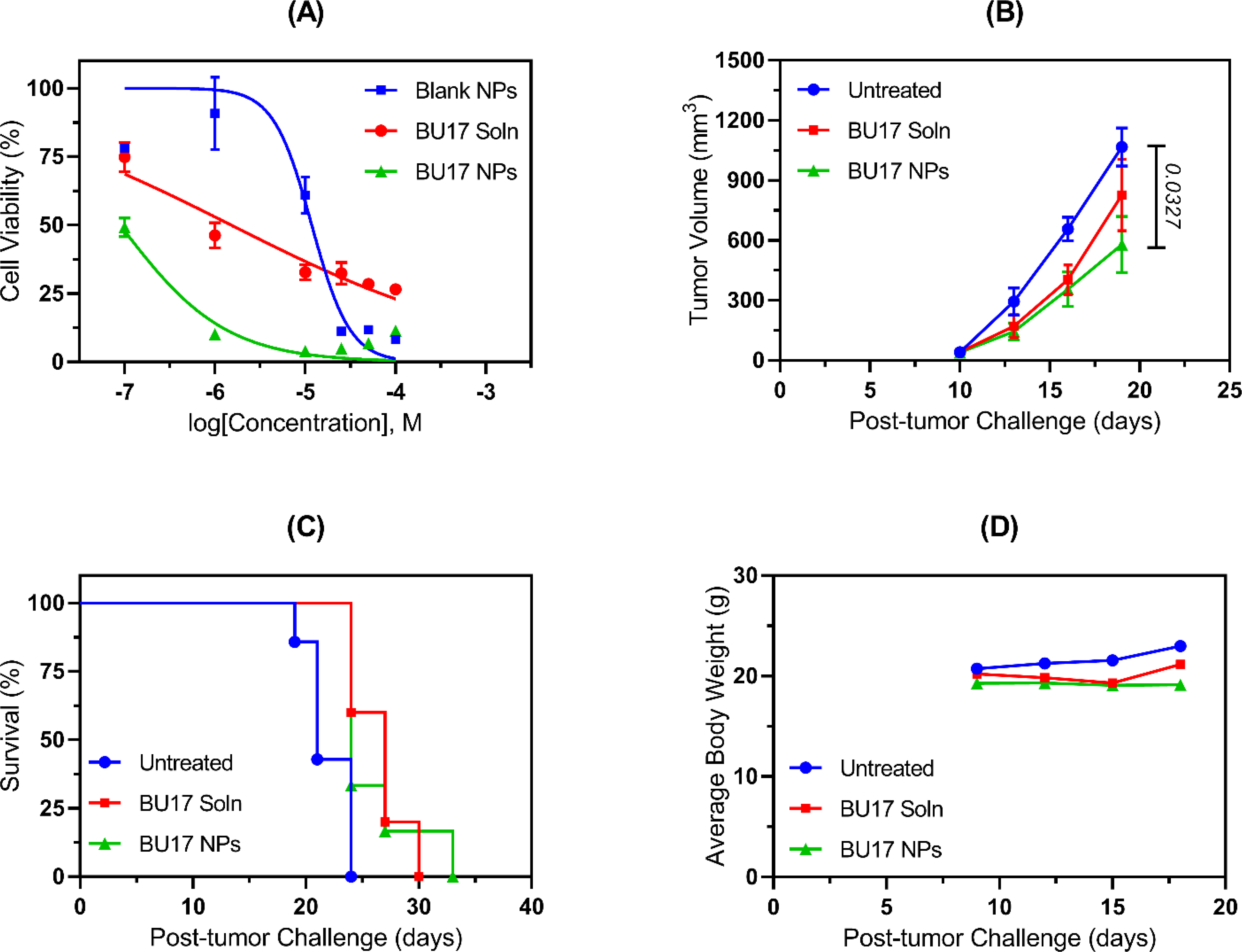
In vitro and in vivo antitumor efficacy of BU17 in solution (BU17 Soln) vs BU17-loaded nanoparticles (BU17 NPs). (A) Dose-response curves resulting from CT26 cells being incubated in vitro with either blank nanoparticles (IC50 11.84 μM), BU17 solution (IC50 1.52 μM), or BU17 nanoparticles (IC50 0.08 μM). (B) Tumor progression curve for mice inoculated with CT26 cells and administered with the designated treatment. (C) Mice survival curves where mice were euthanized when the tumor size exceeded 20 mm at the largest diameter or 10 mm in height. Survival analysis was initially performed by the Log-rank (Mantel-Cox) test using GraphPad Prism 8, and subsequent statistical analysis was carried out by pairwise comparisons where data were analyzed using Log-rank test (Tukey-Kramer adjusted) using SAS 9.4 software. (D) Mice body weight measurements over time.
In vivo antitumor efficacy.
Although the majority of the in vitro studies were performed with non-small cell lung cancer cell line A549, the in vivo study was carried out in a colon cancer cell line (CT26) to further confirm the broad-spectrum antitumor activity of BU17 that was observed in the NCI-60 screening assay. Colon cancer is one of the most common and most deadly forms of cancer. Thus, compound BU17 in soluble form was tested for its in vivo antitumor activity and compared to BU17-loaded nanoparticles in a CT26 tumor model (colon carcinoma cells). Both treatments demonstrated enhanced inhibition of tumor progression in comparison to the untreated group (Figure 6B). However, only mice treated with BU17-loaded nanoparticles had smaller tumor volumes that were significantly lower than untreated control mice (P=0.0327). Also, the mixed-effects model analysis of longitudinal data of the tumor growth of mice treated with BU17-loaded nanoparticles indicated that there was a significant decrease in the tumor growth in comparison to the untreated group (P=0.0144). In terms of the mouse survival study, soluble BU17 and BU17-loaded nanoparticle treated mice had significantly longer survival than untreated mice (P-values were 0.0430 and 0.0398, respectively) (Figure 6C). All mice displayed no significant changes in body weight except for one mouse treated with soluble BU17 and was euthanized on day 30 due to substantial loss in its body weight (>20% of its weight on the first day of the animal study) (Figure 6D).
3. Conclusion
This study documented the fabrication and antiproliferative potency of benzamides, benzylamines, and ureas harboring a tetrahydrobenzo[b]thiophene scaffold. The study revealed the benzyl urea derivative, BU17, to be a promising and broad-spectrum antiproliferative agent that targets rapidly growing cancer cells by inhibiting tubulin polymerization. The potential mechanism of action was confirmed by in vitro destabilization of tubulin polymerization, the arrest of the cell cycle at the G2/M phase, and early apoptosis detection. A polymeric nanoparticle formulation of BU17 was fabricated and tested in vivo as an anticancer formulation in the CT26 murine tumor model. The formulated compound BU17 was superior to the free drug with respect to tumor growth inhibition and mice weight stability, the former of which might be attributed to improved drug delivery by the nanoparticles to the tumor. In the future, we plan to define the tubulin-binding sites of BU17; and different benzyl urea derivatives will be explored in structure-activity relationship studies. In addition, in vivo testing with human xenograft of compound BU17 and its nanoparticles will be studied.
Methods
Synthesis and characterization of 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (compound 1), benzamides (compounds BZ02-BZ05), benzylamines (compounds BZA06-BZA11), and benzyl urea derivatives (compounds BU12-BU19).
Chemical reagents and organic solvents were obtained from Sigma-Aldrich (St. Louis, MI) and used without further purification. The synthesis was performed following previously reported protocols [31, 36].
Materials for in vitro screening:
The cell lines A549 and CT26 were purchased from American Type Culture Collection (Manassas, VA). Both cell lines were cultured in RPMI-1640 medium supplemented with 1% Pen-Strep (100 U/mL, Gibco, Gaithersburg, MD) and 10% FBS (Atlanta Biologicals, Lawrenceville, GA). In this work, cells were maintained in a 5% CO2 cell culture incubator set at 37 °C. CellTiter 96 aqueous MTS reagent powder (MTS, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was purchased from Promega Corporation (San Luis Obispo, CA) while NP-40 Surfact-Amps detergent solution and propidium iodide were purchased from Thermo Fisher Scientific (Waltham, MA). RNAase A solution was obtained from Invitrogen (Thermo Fisher Scientific, Carlsbad, CA). Annexin V Apoptosis Detection Kit was also purchased from Thermo Fisher Scientific (Waltham, MA).
Cell viability testing using MTS assay.
In this experiment, A549 cancer cells were seeded in a 96-well plate at 2 × 103 cells in 100 μL of medium per well [31]. After 24 h of incubation, cells were incubated with the treatments for 48 h. This was followed by adding the MTS reagent (according to the manufacturer’s instructions), and cell viability (%) was calculated based on the absorbance data measured at 490 nm using Spectramax Plus Spectrophotometer (Molecular Devices, San Jose, CA).
Cell viability testing by the National Cancer Institute (NCI).
Based on the preliminary data obtained from the MTS assay with A549 tumor cells, compounds that exhibited strong antitumor activity were advanced for further testing at the NCI to detect potential anticancer activity using a diverse range of cell lines. Cell viability testing of a panel of 60 human cancer cell lines was performed at the National Cancer Institute, Bethesda, Maryland, USA, following standard NCI-60 methodology. Cell lines representing different types of cancers were included in the in vitro test. According to the NCI-60 five-dose screening method, three dose-response parameters were calculated as follows: (i) Growth inhibition of 50% (GI50) was calculated from [(Ti-Tz)/(C-Tz)] × 100 = 50, which is the drug concentration resulting in a 50% reduction in the net protein increase (as measured by SRB staining) compared to control cells (C = control growth; Ti = test growth in the presence of drug at five concentration levels; Tz = cell population for each cell line at time zero (the time of drug addition)), (ii) The drug concentration resulting in total growth inhibition (TGI) is calculated from Ti = Tz, and (iii) The LC50 (concentration of drug resulting in a 50% reduction in the measured protein at the end of the drug treatment as compared to that at the beginning) indicating a net loss of cells following treatment is calculated from [(Ti-Tz)/Tz] × 100 = −50 [43].
Cell cycle analysis.
A549 cells were seeded in a 6-well plate at density 150 × 103 cells/well for 24 h. Cells were then incubated with the different concentrations of treatments for 36 h. Subsequently, cell cycle reagents (e.g. NP40 and propidium iodide, Thermo Fisher Scientific, Waltham, MA) were added as previously described [31]. Cell samples were finally acquired using a FACScan flow cytometer (BD Biosciences, San Jose, CA), and the data files were analyzed using ModFit LT and FlowJo software.
Apoptosis assay.
In a 6-well plate, A549 cells were seeded at a density of 150 × 103 cells/well for 24 h. Cells were then treated with the designated treatment for 48 h. After incubation with the treatments, cells were stained with annexin V and propidium iodide (Thermo Fisher Scientific, Waltham, MA). The stepwise description of this assay protocol is reported in our previously published work [31]. In this study, PTX and BU17 were tested at concentrations of 5 nM and 10 μM (selected based on the A549 MTS assay data, IC50 = 9 μM), respectively. Finally, samples were measured using the FACScan flow cytometer and the data were analyzed using FlowJo software.
Caspase activity assay.
In this experiment, the activity of two caspases (3 and 9) was evaluated as previously described [31]. Briefly, A549 cells were incubated with the treatment (10 μM of BU17, which was selected based on the MTS assay data obtained from the A549 cell line, IC50 = 9 μM) for 48 h, then caspase kit reagents were added (as per manufacturer’s instructions, Abcam, Burlingame, CA) and the fluorescence was measured using a spectrophotometer (Spectramax M5 Microplate Reader, Molecular Devices).
JNK1 and JNK2 inhibition assay.
Reaction Biology Corporation Kinase HotSpot (Malvern, PA) tested the inhibitory activity of compound BU17 towards JNK1 and JNK2 at a single concentration (10 μM, selected based on the MTS assay data obtained from the A549 cell line, IC50 = 9 μM) in duplicate mode, and reactions were carried out at 10 μM ATP.
Panel of 11 kinases.
Reaction Biology Corporation Kinase HotSpot tested the inhibitory activity of BU17 towards 11 protein kinases (BRAF, CDK1/cyclin A, CDK1/cyclin B, CDK19/cyclin C, CDK8/cyclin C, CHK1, CHK2, P38α/MAPK14, PDK1/PDPK1, RAF1 and WEE1) at a single concentration (10 μM, selected based on the MTS assay data obtained from the A549 cell line, IC50 = 9 μM) in duplicate mode and reactions were carried out at 10 μM ATP.
Tubulin polymerization assay.
This assay was performed to study the effect of BU17 on the microtubule formation and, hence, the cell division process. In brief, the BU17 solution (3 μM) was tested by incubation with tubulin, and the fluorescence was measured over time as previously described [31]. In this experiment, PTX and CaCl2 were used as assay controls. Both BU17 and PTX were tested at a final concentration of 3 μM while CaCl2 was used at a concentration of 75 μM, as per the manufacturer’s protocol (Cytoskeleton Inc., Denver, CO).
Formulation of compound BU17 into nanoparticles.
Using a nanoprecipitation technique, nanoparticles encapsulating compound BU17 were prepared. Briefly, 1 mg of compound BU17 and 30 mg of Resomer® RG 502 H, Poly(D,L-lactide-co-glycolide) were dissolved in 950 μL acetone and 50 μL of ethanol. The organic phase was transferred to a 3 mL syringe with a 26 G/0.5 needle, and the tip was submerged in a 30 mL stirring aqueous solution of 0.1 % w/v TPGS (Sigma-Aldrich). Next, the suspension was transferred to a rotary evaporator (Heidolph, Wood Dale, IL) 50 mbar, 90 rpm for 2 h to evaporate the organic solvents. Amicon ultra-15 centrifugal filter units (Mw cutoff = 100 kDa, EMD Millipore, Burlington, MA) and Eppendorf centrifuge 5804 R (Eppendorf, Hamburg, Germany) were used to wash and collect the formed nanoparticles at 600 ×g for 30 min 4 times. To evaluate the shape and size of the nanoparticles, a drop of diluted nanoparticle suspension was allowed to dry overnight on an aluminum SEM stub then coated with gold and palladium, and images were taken using a Hitachi S-4800 SEM (Hitachi High-Technologies, Tokyo, Japan). To measure the size, size distribution, and surface charge of the prepared nanoparticles, 10 μL of the nanoparticles was diluted with 990 μL of nanopure water and measured using a Zetasizer Nano-ZS particle analyzer (Malvern Panalytical, Malvern, United Kingdom). In addition, the drug content was estimated by HPLC-ultraviolet after dissolving the nanoparticles in a mixture of water and acetonitrile, using reversed-phase 5 μm C18 column (6 × 15 mm) and a mobile phase of acetonitrile:water (60:40, v/v) set at a flow rate of 1 mL/ min. Compound BU17 was detected by a UV detector at λ 295 nm after 6.25 min.
Cell viability testing of compound BU17 in solution vs nanoparticles.
The murine CT26 cells were seeded in 96-well plates at a density of 2 X 103 cells/ well for 24 h. Cells were incubated with increasing concentrations (0.1, 1, 10, 25, 50, 50, 100 μM) of BU17 either in nanoparticles or soluble form. Then, the MTS assay was performed as described in the above section.
In vivo antitumor efficacy.
In this study, 6–8 weeks old female immunocompetent BALB/cJ mice were inoculated with syngeneic CT26 cells at a density of 1 × 106 cells/mouse subcutaneously in the right flank. After one week, tumor volumes were measured, and the mice were randomized into 3 groups. Mice in the first group (n=5) were treated with 150 μg of BU17 in 10% (v/v) Tween-80 solution per mice. Also, 150 μg of compound BU17-loaded nanoparticles were injected into the second group (n=6), and no treatment for the last group (n=7). The retro-orbital venous sinus injection was adopted as an intravenous route of treatment administration for 10 successive days. The mice weights were measured daily, and the tumor volumes were calculated every 2 days starting from day 8 post-tumor challenge by measuring the tumor diameters (D, mm) and height (H, mm) using a digital caliper as displayed in Equation (1). Mice were euthanized once the tumor diameter exceeded 20 mm or the tumor height exceeded 10 mm. This animal study was approved by the University of Iowa Institutional Animal Care and Use Committee (IACUC).
| Equation 1: |
Statistical analysis.
Data obtained from cell cycle analysis and apoptosis detection experiments were initially analyzed by one-way analysis of variance (ANOVA) using the F-test and followed by Tukey’s multiple comparison test to compare all pairs of treatments. An unpaired two-tailed t-test was used to compare data obtained from the caspase 3 and 9 activity experiment. Data of the tumor progression and body weight were analyzed with a linear mixed-effects model using SAS PROC MIXED (SAS Institute Inc., Cary, NC) [44]. Comparison of survival curves was initially performed by the Log-rank (Mantel-Cox) test using GraphPad Prism 8 (GraphPad Software, La Jolla, CA), and further statistical analysis was carried out by pairwise comparisons, and data were analyzed using Log-rank test (Tukey-Kramer adjusted) using SAS 9.4 software [45]. In all tests, differences were considered statistically significant when P<0.05.
Supplementary Material
Acknowledgments
We thank Dr. Azza R. Maarouf and Dr. Ghada S. Hassan (Mansoura University) for their discussions on the science in this manuscript. We would like to express our sincere gratitude to the National Cancer Institute (NCI, Bethesda, MD) for the NCI-60 screening of the new compounds and P30 CA086862 for support. We acknowledge the Flow Cytometry Facility, which is a Carver College of Medicine/Holden Comprehensive Cancer Center core research facility at the University of Iowa. The facility is funded through user fees and the generous financial support of the Carver College of Medicine, Holden Comprehensive Cancer Center, and Iowa City Veteran’s Administration Medical Center. We acknowledge the Lyle and Sharon Bighley Chair to A.K.S. for support.
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information
Properties and purity of the synthesized compounds.
References
- [1].Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA Cancer J Clin 68(6) (2018) 394–424. [DOI] [PubMed] [Google Scholar]
- [2].Otto T, Sicinski P, Cell cycle proteins as promising targets in cancer therapy, Nat Rev Cancer 17(2) (2017) 93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vanneman M, Dranoff G, Combining immunotherapy and targeted therapies in cancer treatment, Nat Rev Cancer 12(4) (2012) 237–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Marin JJ, Romero MR, Blazquez AG, Herraez E, Keck E, Briz O, Importance and limitations of chemotherapy among the available treatments for gastrointestinal tumours, Anticancer Agents Med Chem 9(2) (2009) 162–84. [DOI] [PubMed] [Google Scholar]
- [5].Jordan MA, Wilson L, Microtubules as a target for anticancer drugs, Nat Rev Cancer 4(4) (2004) 253–65. [DOI] [PubMed] [Google Scholar]
- [6].Nogales E, Structural Insights into Microtubule Function, Annual Review of Biophysics and Biomolecular Structure 30(1) (2001) 397–420. [DOI] [PubMed] [Google Scholar]
- [7].Prota AE, Bargsten K, Zurwerra D, Field JJ, Diaz JF, Altmann KH, Steinmetz MO, Molecular mechanism of action of microtubule-stabilizing anticancer agents, Science 339(6119) (2013) 587–90. [DOI] [PubMed] [Google Scholar]
- [8].Bates D, Eastman A, Microtubule destabilising agents: far more than just antimitotic anticancer drugs, Br J Clin Pharmacol 83(2) (2017) 255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lee CT, Huang YW, Yang CH, Huang KS, Drug delivery systems and combination therapy by using vinca alkaloids, Curr Top Med Chem 15(15) (2015) 1491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cavaletti G, Cavalletti E, Oggioni N, Sottani C, Minoia C, D’Incalci M, Zucchetti M, Marmiroli P, Tredici G, Distribution of paclitaxel within the nervous system of the rat after repeated intravenous administration, Neurotoxicology 21(3) (2000) 389–93. [PubMed] [Google Scholar]
- [11].Zajaczkowska R, Kocot-Kepska M, Leppert W, Wrzosek A, Mika J, Wordliczek J, Mechanisms of Chemotherapy-Induced Peripheral Neuropathy, Int J Mol Sci 20(6) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dumontet C, Sikic BI, Mechanisms of action of and resistance to antitubulin agents: microtubule dynamics, drug transport, and cell death, J Clin Oncol 17(3) (1999) 1061–70. [DOI] [PubMed] [Google Scholar]
- [13].Gao P, Rush BD, Pfund WP, Huang T, Bauer JM, Morozowich W, Kuo MS, Hageman MJ, Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability, J Pharm Sci 92(12) (2003) 2386–98. [DOI] [PubMed] [Google Scholar]
- [14].Patch RJ, Baumann CA, Liu J, Gibbs AC, Ott H, Lattanze J, Player MR, Identification of 2-acylaminothiophene-3-carboxamides as potent inhibitors of FLT3, Bioorg Med Chem Lett 16(12) (2006) 3282–6. [DOI] [PubMed] [Google Scholar]
- [15].Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, Green S, Haye HR, Horn CL, Janetka JW, Liu D, Mouchet E, Ready S, Rosenthal JL, Queva C, Schwartz GK, Taylor KJ, Tse AN, Walker GE, White AM, AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies, Mol Cancer Ther 7(9) (2008) 2955–66. [DOI] [PubMed] [Google Scholar]
- [16].Gui X, Yang H, Li T, Tan X, Shi P, Li M, Du F, Chen ZJ, Autophagy induction via STING trafficking is a primordial function of the cGAS pathway, Nature 567(7747) (2019) 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dumble M, Crouthamel MC, Zhang SY, Schaber M, Levy D, Robell K, Liu Q, Figueroa DJ, Minthorn EA, Seefeld MA, Rouse MB, Rabindran SK, Heerding DA, Kumar R, Discovery of novel AKT inhibitors with enhanced anti-tumor effects in combination with the MEK inhibitor, PLoS One 9(6) (2014) e100880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Walls M, Baxi SM, Mehta PP, Liu KK, Zhu J, Estrella H, Li C, Zientek M, Zong Q, Smeal T, Yin MJ, Targeting small cell lung cancer harboring PIK3CA mutation with a selective oral PI3K inhibitor PF-4989216, Clin Cancer Res 20(3) (2014) 631–43. [DOI] [PubMed] [Google Scholar]
- [19].Wu CP, Murakami M, Hsiao SH, Chou AW, Li YQ, Huang YH, Hung TH, Ambudkar SV, Overexpression of ATP-Binding Cassette Subfamily G Member 2 Confers Resistance to Phosphatidylinositol 3-Kinase Inhibitor PF-4989216 in Cancer Cells, Mol Pharm 14(7) (2017) 2368–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Garton AJ, Crew AP, Franklin M, Cooke AR, Wynne GM, Castaldo L, Kahler J, Winski SL, Franks A, Brown EN, Bittner MA, Keily JF, Briner P, Hidden C, Srebernak MC, Pirrit C, O’Connor M, Chan A, Vulevic B, Henninger D, Hart K, Sennello R, Li AH, Zhang T, Richardson F, Emerson DL, Castelhano AL, Arnold LD, Gibson NW, OSI-930: a novel selective inhibitor of Kit and kinase insert domain receptor tyrosine kinases with antitumor activity in mouse xenograft models, Cancer Res 66(2) (2006) 1015–24. [DOI] [PubMed] [Google Scholar]
- [21].Dallemagne P, Khanh LP, Alsaidi A, Renault O, Varlet I, Collot V, Bureau R, Rault S, Synthesis and biological evaluation of cyclopenta[c]thiophene related compounds as new antitumor agents, Bioorg Med Chem 10(7) (2002) 2185–91. [DOI] [PubMed] [Google Scholar]
- [22].Zhang HZ, Kasibhatla S, Kuemmerle J, Kemnitzer W, Ollis-Mason K, Qiu L, Crogan-Grundy C, Tseng B, Drewe J, Cai SX, Discovery and structure-activity relationship of 3-aryl-5-aryl-1,2,4-oxadiazoles as a new series of apoptosis inducers and potential anticancer agents, J Med Chem 48(16) (2005) 5215–23. [DOI] [PubMed] [Google Scholar]
- [23].Lambert M, Jambon S, Depauw S, David-Cordonnier MH, Targeting Transcription Factors for Cancer Treatment, Molecules 23(6) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Angell RM, Atkinson FL, Brown MJ, Chuang TT, Christopher JA, Cichy-Knight M, Dunn AK, Hightower KE, Malkakorpi S, Musgrave JR, Neu M, Rowland P, Shea RL, Smith JL, Somers DO, Thomas SA, Thompson G, Wang R, N-(3-Cyano-4,5,6,7-tetrahydro-1-benzothien-2-yl)amides as potent, selective, inhibitors of JNK2 and JNK3, Bioorg Med Chem Lett 17(5) (2007) 1296–301. [DOI] [PubMed] [Google Scholar]
- [25].Xu K, Schwarz PM, Ludueña RF, Interaction of nocodazole with tubulin isotypes, Drug Development Research 55(2) (2002) 91–96. [Google Scholar]
- [26].Feng R, Li S, Lu C, Andreas C, Stolz DB, Mapara MY, Lentzsch S, Targeting the microtubular network as a new antimyeloma strategy, Mol Cancer Ther 10(10) (2011) 1886–96. [DOI] [PubMed] [Google Scholar]
- [27].Risinger AL, Westbrook CD, Encinas A, Mulbaier M, Schultes CM, Wawro S, Lewis JD, Janssen B, Giles FJ, Mooberry SL, ELR510444, a novel microtubule disruptor with multiple mechanisms of action, J Pharmacol Exp Ther 336(3) (2011) 652–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Romagnoli R, Baraldi PG, Brancale A, Ricci A, Hamel E, Bortolozzi R, Basso G, Viola G, Convergent Synthesis and Biological Evaluation of 2-Amino-4-(3′,4′,5′-trimethoxyphenyl)-5-aryl Thiazoles as Microtubule Targeting Agents, Journal of Medicinal Chemistry 54(14) (2011) 5144–5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Romagnoli R, Baraldi PG, Carrion MD, Cara CL, Preti D, Fruttarolo F, Pavani MG, Tabrizi MA, Tolomeo M, Grimaudo S, Di Cristina A, Balzarini J, Hadfield JA, Brancale A, Hamel E, Synthesis and Biological Evaluation of 2- and 3-Aminobenzo[b]thiophene Derivatives as Antimitotic Agents and Inhibitors of Tubulin Polymerization, Journal of Medicinal Chemistry 50(9) (2007) 2273–2277. [DOI] [PubMed] [Google Scholar]
- [30].Simoni D, Romagnoli R, Baruchello R, Rondanin R, Grisolia G, Eleopra M, Rizzi M, Tolomeo M, Giannini G, Alloatti D, Castorina M, Marcellini M, Pisano C, Novel A-Ring and B-Ring Modified Combretastatin A-4 (CA-4) Analogues Endowed with Interesting Cytotoxic Activity, Journal of Medicinal Chemistry 51(19) (2008) 6211–6215. [DOI] [PubMed] [Google Scholar]
- [31].Abdel-Rahman SA, El-Damasy AK, Hassan GS, Wafa EI, Geary SM, Maarouf AR, Salem AK, Cyclohepta[b]thiophenes as Potential Antiproliferative Agents: Design, Synthesis, In Vitro, and In Vivo Anticancer Evaluation, ACS Pharmacol Transl Sci 3(5) (2020) 965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Byrne JD, Betancourt T, Brannon-Peppas L, Active targeting schemes for nanoparticle systems in cancer therapeutics, Adv Drug Deliv Rev 60(15) (2008) 1615–26. [DOI] [PubMed] [Google Scholar]
- [33].Maeda H, Wu J, Sawa T, Matsumura Y, Hori K, Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review, J Control Release 65(1–2) (2000) 271–84. [DOI] [PubMed] [Google Scholar]
- [34].Xu L, Xu S, Wang H, Zhang J, Chen Z, Pan L, Wang J, Wei X, Xie H, Zhou L, Zheng S, Xu X, Enhancing the Efficacy and Safety of Doxorubicin against Hepatocellular Carcinoma through a Modular Assembly Approach: The Combination of Polymeric Prodrug Design, Nanoparticle Encapsulation, and Cancer Cell-Specific Drug Targeting, ACS Appl Mater Interfaces 10(4) (2018) 3229–3240. [DOI] [PubMed] [Google Scholar]
- [35].Ebeid K, Meng X, Thiel KW, Do AV, Geary SM, Morris AS, Pham EL, Wongrakpanich A, Chhonker YS, Murry DJ, Leslie KK, Salem AK, Synthetically lethal nanoparticles for treatment of endometrial cancer, Nat Nanotechnol 13(1) (2018) 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gewald K, Schinke E, Böttcher H, Heterocyclen aus CH-aciden Nitrilen, VIII. 2-Amino-thiophene aus methylenaktiven Nitrilen, Carbonylverbindungen und Schwefel, Chemische Berichte 99(1) (1966) 94–100. [Google Scholar]
- [37].Archna S. Pathania, Chawla PA, Thiophene-based derivatives as anticancer agents: An overview on decade’s work, Bioorg Chem 101 (2020) 104026. [DOI] [PubMed] [Google Scholar]
- [38].Sztanke M, Rzymowska J, Sztanke K, Synthesis, structure elucidation and identification of antiproliferative activities of a novel class of thiophene bioisosteres bearing the privileged 7,8-dihydroimidazo[2,1-c][1,2,4]triazin-4(6H)-one scaffold, Bioorg Med Chem 23(13) (2015) 3448–56. [DOI] [PubMed] [Google Scholar]
- [39].Shah R, Verma PK, Synthesis of thiophene derivatives and their anti-microbial, antioxidant, anticorrosion and anticancer activity, BMC Chemistry 13(1) (2019) 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Megally Abdo NY, Samir EM, Mohareb RM, Synthesis and evaluation of novel 4H-pyrazole and thiophene derivatives derived from chalcone as potential anti-proliferative agents, Pim-1 kinase inhibitors, and PAINS, Journal of Heterocyclic Chemistry 57(4) (2020) 1993–2009. [Google Scholar]
- [41].Bhattarai DP, Kim BS, NIR-Triggered Hyperthermal Effect of Polythiophene Nanoparticles Synthesized by Surfactant-Free Oxidative Polymerization Method on Colorectal Carcinoma Cells, Cells 9(9) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mohan R, Banerjee M, Ray A, Manna T, Wilson L, Owa T, Bhattacharyya B, Panda D, Antimitotic sulfonamides inhibit microtubule assembly dynamics and cancer cell proliferation, Biochemistry 45(17) (2006) 5440–9. [DOI] [PubMed] [Google Scholar]
- [43].NCI-60 Screening Methodology. <https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm>, 2015.
- [44].Wafa EI, Geary SM, Ross KA, Goodman JT, Narasimhan B, Salem AK, Pentaerythritol-based lipid A bolsters the antitumor efficacy of a polyanhydride particle-based cancer vaccine, Nanomedicine 21 (2019) 102055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wafa EI, Geary SM, Ross KA, Goodman JT, Narasimhan B, Salem AK, Single Dose of a Polyanhydride Particle-Based Vaccine Generates Potent Antigen-Specific Antitumor Immune Responses, J Pharmacol Exp Ther 370(3) (2019) 855–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.