

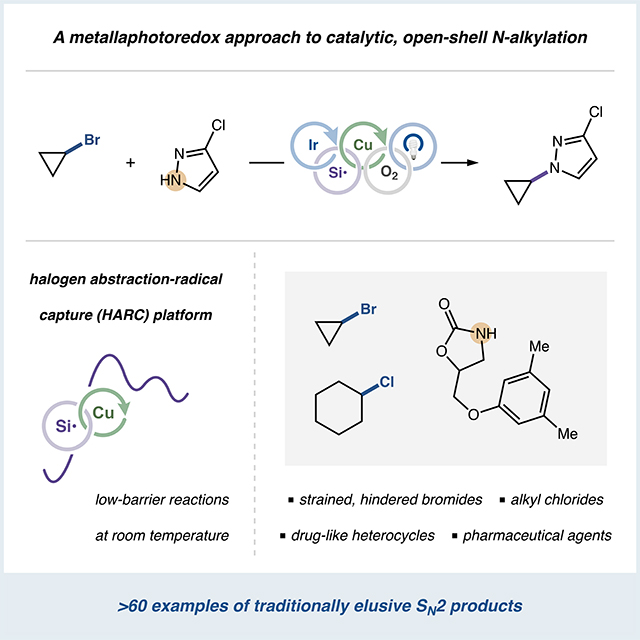
SUMMARY
The catalytic union of amides, sulfonamides, anilines, imines or N-heterocycles with a broad spectrum of electronically and sterically diverse alkyl bromides has been achieved via a visible light-induced metallaphotoredox platform. The use of a halogen abstraction–radical capture (HARC) mechanism allows for room temperature coupling of C(sp3)-bromides using simple Cu(II) salts, effectively bypassing the prohibitively high barriers typically associated with thermally-induced SN2 or SN1 N-alkylation. This regio- and chemoselective protocol is compatible with >10 classes of medicinally-relevant N-nucleophiles, including established pharmaceutical agents, in addition to structurally diverse primary, secondary and tertiary alkyl bromides. Furthermore, the capacity of HARC methodologies to engage conventionally inert coupling partners is highlighted via the union of N-nucleophiles with cyclopropyl bromides and unactivated alkyl chlorides, substrates that are incompatible with nucleophilic substitution pathways. Preliminary mechanistic experiments validate the dual catalytic, open-shell nature of this platform, which enables reactivity previously unattainable in traditional halide-based N-alkylation systems.
Keywords: photoredox, copper catalysis, N-alkylation, halogen abstraction, cyclopropylation, organochloride activation
Graphical Abstract
eTOC Blurb
Traditional substitution reactions between nitrogen nucleophiles and alkyl halides feature well-established, substrate-dependent limitations and competing reaction pathways under thermally-induced conditions. Herein, we report that a metallaphotoredox approach, utilizing a halogen abstraction-radical capture (HARC) mechanism, provides a valuable alternative to conventional N-alkylation. This visible light-induced, copper-catalyzed protocol is successful for coupling >10 classes of N-nucleophiles with diverse primary, secondary or tertiary alkyl bromides. Moreover, this open-shell platform alleviates outstanding N-alkylation challenges regarding regioselectivity, direct cyclopropylation and secondary alkyl chloride functionalization.
INTRODUCTION
The societal need to discover and produce pharmaceuticals, agrochemicals and functional materials has long established a demand for new C–N bond-forming reactions that are successful across diverse combinations of complex substrates.1–3 Indeed, over the last two decades, palladium-, nickel- and copper-catalyzed protocols such as Buchwald-Hartwig,4,5 Ullmann-Goldberg6 and Chan-Evans-Lam7 C(sp2)–N cross-couplings have emerged as mainstay strategies to generically access versatile N-aryl structural motifs (Scheme 1).8,9 In contrast, transition metal-mediated couplings affording C(sp3)–N bonds are comparatively underdeveloped as broadly applicable synthetic technologies,10 despite growing recognition of the importance of C(sp3)-incorporation when designing new drugs and agrochemicals.11–13 Instead, practical approaches to forging C(sp3)–N bonds remain generally limited to classical methods, including Mitsunobu substitution,14 Curtius rearrangements,15 olefin hydroamination16,17 or reductive amination.18
Scheme 1. Catalytic N-alkylation via HARC coupling of alkyl bromides.

Among the traditional N-alkylation strategies, direct SN2 or SN1 substitution of alkyl halides with N-nucleophiles remains the most adopted technology for generating alkylamine-bearing biomedical agents19,20 and among the most frequently utilized transformations across all chemical industries.21,22 Despite the long-standing popularity of this closed-shell N-alkylation pathway, substitution reactions typically face substantial halide-dependent activation barriers, which renders numerous substrates (in particular strained or hindered halides) inert to functionalization (Scheme 1).21,22 Even for reactive halides, elevated barriers for substitution result in high-temperature requirements that impose broad limitations, including low selectivity for a single constitutional isomer (or regioisomer), non-controlled overalkylation events and competing elimination pathways.21,22 While in some cases, metal-mediated variants of halide N-alkylation have removed these limitations,23,24 there remains an outstanding need to identify new, robust mechanisms that allow such couplings across an expansive range of complex halide and N-heterocyclic systems while employing ambient, low-temperature conditions.
Within the select C(sp3)–N cross-coupling methods already disclosed, copper catalysis has increasingly been featured as a key component for enhancing efficiency and scope, exploiting the well-precedented capacity for high-valent Cu(III) states to induce carbon–heteroatom bond formation via reductive elimination.25,26 Buchwald, Hartwig, König, Watson and others have elegantly engaged several abundant alkyl feedstocks in such protocols, including olefins,27,28 boronic acids and esters,29–32 hydrocarbons (activated in situ via hydrogen atom transfer)33,34 and carboxylic acids (typically deployed as redox-active electrophiles).35–38 However, platforms for copper-catalyzed N-alkylation using alkyl halides, which are readily available and prototypical SN2 electrophiles, are currently restricted by limited mechanistic approaches and reduced generality in substrate scope. Within this area, the groups of Fu and Peters have made notable advances by designing several distinct organohalide-based N-alkylation platforms, primarily using transiently-generated Cu(I)-amido intermediates for aliphatic radical generation via single-electron transfer (SET).39–43 In these systems, successful activation of both coupling partners by a single (often photoactive) copper complex has frequently enabled N-alkylation using readily reducible electrophiles (i.e., iodides and α-halocarbonyls), with additional examples demonstrated using aliphatic bromides in tandem with amide- or indole-derived nucleophiles.39–43 Ultimately, these reports from Fu and Peters have conclusively established that merging open-shell and copper-mediated activation modes can indeed facilitate catalytic C(sp3)–N bond formation.44
Over the past decade, metallaphotoredox catalysis has emerged as a powerful platform to employ open-shell intermediates in concert with transition metal-catalyzed cross-coupling under mild conditions.45,46 This approach has recently been combined with copper catalysis,47 in particular for forging traditionally elusive C–CF348–52 and C–N35–37 bonds. In the context of organohalide functionalization, such strategies have most notably enabled Ullmann-Goldberg couplings of diverse (hetero)aryl bromides at room temperature using a previously unreported mechanistic approach.53 This transformation utilizes photoredox-generated silyl radicals, which rapidly convert aryl bromides to radical intermediates via halogen atom transfer, to facilitate ambient or low-temperature coupling by replacing sluggish Cu(I) oxidative addition with a kinetically facile halogen abstraction-radical capture (HARC) sequence.54–56 Based on this new mechanism for C–N coupling, we hypothesized that an underexplored alkyl-HARC regime could generically provide aliphatic radicals from the corresponding bromides, ultimately enabling low-barrier N-alkylation via well-established copper-catalyzed C(sp3)–N bond formation (Scheme 1). Specifically, using these previously unexplored principles for N-alkylation, we anticipated that independent steps for halide activation (by photoredox-mediated halogen abstraction) and nucleophile activation (by copper ligation) would confer widespread structural and electronic generality with respect to each coupling partner, thereby addressing key limitations in halide-based catalytic C(sp3)–N coupling. Herein, we report the successful design and execution of this dual copper/photoredox N-alkylation platform, which demonstrates reactivity and selectivity surpassing that of traditional substitution due to synergistic copper and silyl radical-based activation modes.
RESULTS AND DISCUSSION
HARC N-alkylation of 3-chloroindazole using alkyl bromides
Our initial studies began by investigating the alkylation of a medicinally-relevant 1H-indazole model N-nucleophile with bromocyclohexane (Figure 1). Gratifyingly, following optimization efforts (see Figures S1–S21), 93% yield of product 1 was obtained after 4 hours of blue light irradiation (450 nm) using Ir(III) photocatalyst Ir[dF(CF3)ppy]2[4,4′-d(CF3)bpy]PF6 ([Ir-1]), commercial copper precatalyst Cu(TMHD)2, tris(trimethylsilyl)silanol (supersilanol) as a silyl radical precursor and acetonitrile as solvent (entry 1). Several elements proved critical for optimal reactivity, including a vent needle for air incorporation, the Integrated Photoreactor (IPR) as a standardized device for reaction vial irradiation,57 LiOt-Bu as base and water as an additive.58–60 Anhydrous systems were consistently detrimental (entry 2), even under conditions closely mimicking the previously reported HARC-Ullmann-Goldberg reaction (i.e. 1,1,3,3-tetramethylguanidine as base, entry 3).53 Control reactions verified the importance of oxygen for catalytic turnover (entry 4), in addition to the superior performance of high-intensity IPR setups, as weaker blue light sources (i.e., Kessil lamps) were generally inadequate for robust product formation (entry 5). As expected, base, photocatalyst, copper and supersilanol were all required for substantial reactivity (entries 6–9), consistent with the desired HARC mechanism that should require both photocatalytic halogen atom abstraction and copper-mediated bond formation to achieve coupling (see Supplemental Information and Figures S26–S27 for additional control experiments and perturbation studies, which confirm the robustness of the optimized conditions).
Figure 1. Control reactions of optimized HARC N-alkylation conditions.

Reactions performed with (TMS)3SiOH (2.5 equiv), LiOt-Bu (3.0 equiv), and H2O (10 equiv) in MeCN (0.1 M), under IPR irradiation, unless otherwise indicated. aYields determined by 1H NMR analysis. See Supplemental Information for specific experimental details. TMHD, 2,2,6,6-tetramethyl-3,5-heptanedionate; IPR, Integrated Photoreactor; TMG, 1,1,3,3-tetramethylguanidine.
With optimized conditions in hand, we next sought to examine the scope of bromides amenable to coupling (Figure 2). Strained cyclic bromides, which are often sluggish SN1 or SN2 partners,21,22 were effective substrates, delivering N-cyclobutyl and N-azetidinyl products in high yield (2 and 3, 87% and 90% yield, respectively). Larger cyclic bromides containing pyrrolidine (4, 55% yield), tetrahydropyran (5, 71% yield) or cycloheptane (6, 60% yield) cores could also be successfully functionalized. Acyclic bromides were also readily implemented in this protocol (7–9, 54–75% yield), demonstrating complementarity to SN2 methods via reactivity with neopentyl substrates.21,22 For more complex secondary cases, spirocyclic and bridged bicyclic frameworks (10–12, 47–92% yield), an adamantyl system (13, 54% yield), a biologically-relevant sterol scaffold (14, 56% yield) and a substrate bearing heterocyclic functionality (15, 87% yield) were all broadly competent for alkyl-HARC coupling.
Figure 2. Alkyl bromide scope for copper-HARC N-alkylation.

Isolated yields unless otherwise indicated. r.r. >20:1 in all cases. Reactions generally performed under air with photocatalyst [Ir-1] (0.8 mol%), Cu(TMHD)2 (20–30 mol%), (TMS)3SiOH (2.5 equiv), LiOt-Bu (3.0 equiv), H2O (10 equiv), N-nucleophile (0.25 mmol, 1.0 equiv) and alkyl bromide (2.5 equiv) in MeCN (0.1 M) under IPR irradiation (450 nm) for 4 h. See Supplemental Information for specific experimental details. ad.r. 4:1. b0.05 mmol scale; toluene/MeCN (7:3, 0.1 M) as solvent; d.r. 1:1; yield determined by 1H NMR analysis. c(TMS)3SiNHAd as silyl radical source. d60 mol% Cu(TMHD)2. eIsolated yield from five combined 0.05 mmol scale reactions (0.25 mmol total) due to reduced yields (>5% loss of yield) on typical 0.25 mmol scale. fIr[dF(CF3)ppy]2(dtbbpy)PF6 [Ir-2] as photocatalyst. g3.5 equiv. (TMS)3SiNHAd used. Ad, 1-adamantyl.
To further illustrate the generality of this platform, we also evaluated a series of tertiary bromide electrophiles, which are typically challenging substrates in both metal-free and transition metal-catalyzed N-alkylations.21–24 Although supersilanol was insufficient for accomplishing these couplings, we were delighted to find that the recently disclosed aminosilane (TMS)3SiNHAd, which furnishes a more nucleophilic silyl radical with greater kinetic capacity for halogen atom abstraction,61 was competent for accessing such tertiary N-alkyl products. Numerous scaffolds reacted efficiently under these conditions, including derivatives of adamantane (16 and 17, 63% and 62% yield, respectively), bicyclo[2.2.2]octane (18 and 19, 57% and 51% yield, respectively), bicyclo[1.1.1]pentane (20, 64% yield) and cubane (21, 51% yield).62 Evidently, this method can be readily applied to the formation of rigid bioisosteric alkyamines, structures of high value for medicinal chemistry programs which currently suffer from limited (or non-existent) synthetic approaches via modular cross-coupling techniques.63
Evaluation of N-nucleophile and pharmaceutical agent scope
We then turned our attention to the scope of N-nucleophiles suitable for this transformation (Figure 3). Using a strained model halide substrate, a variety of indazoles (22–24, 66–86% yield) and pyrazoles (25–27, 57–90% yield) were alkylated in high efficiency. Consistent with previous copper metallaphotoredox studies,35 this method delivers all products as single regioisomers, an elusive selectivity trend for traditional nucleophilic substitution approaches.21,22,64,65 For heterocycles featuring one reactive site, azaindoles (28–30, 54–85% yield), indoles (31–33, 65–68% yield), pyrrole (34, 64% yield) and carbazoles (35 and 36, 58% and 75% yield, respectively) were readily functionalized, as were benzamide N-acyl partners bearing either electron-rich or electron-deficient arene frameworks (37–39, 49–78% yield). Furthermore, sulfonamide (40, 74% yield), carbamate (41, 71% yield), aniline (42, 58% yield), oxindole (43, 91% yield) and dihydroquinolone nucleophiles (44, 56% yield) were also successfully coupled. Remarkably, exquisite chemoselectivity was observed for bromide abstraction in all cases, leaving aryl or alkyl chloride functionality intact and available for subsequent synthetic manipulation. Notably, benzophenone imine (45, 82% yield) could also be utilized for accessing derivatives of primary amines, motifs typically inaccessible via substitution due to predominant overalkylation when using nucleophiles (such as ammonia) containing several labile N–H bonds21,22,24 In total, 13 diverse nucleophile classes were amenable to coupling, demonstrating the generality of alkyl-HARC mechanisms with independent silyl radical and copper-based activation steps (for additional examples and current limitations, see Figure S30).66
Figure 3. N-nucleophile scope for copper-HARC N-alkylation.

Isolated yields unless otherwise indicated. See Figure 2 for general conditions and Supplemental Information for specific experimental details. aDBN (1.0 equiv) used as additive. b0.5 equiv. LiOt-Bu used. cYield determined by 1H NMR analysis. DBN, 1,5-diazabicyclo[4.3.0]non-5-ene.
To survey the utility of this method in drug discovery settings, we also employed this alkyl-HARC protocol to functionalize various commercial N-nucleophilic pharmaceutical agents (Figure 4). Celebrex (46, 87%), Navoban (47, 52% yield), Skelaxin (48, 87% yield) and Dogmatil (49, 73% yield) were all expediently converted to complex N-alkyl analogs using various bromide coupling partners, with basic and oxidation-prone67 tertiary amines notably tolerated in several cases.68 Given the prevalence of heterocyclic N-nucleophiles in drug candidates, this platform should provide medicinal chemists facile access to diverse pharmaceutical analogs via modular, late-stage N-alkylation.
Figure 4. Late-stage N-alkylation of pharmaceutical agents.

All yields are isolated. See Supplemental Information for specific experimental details.
Extension of HARC methodology to cyclopropyl and chloride electrophiles
Exploiting this complementary N-alkylation protocol, we further sought to couple halides broadly considered inert within SN1 or SN2 substitution platforms, initially targeting the generation of heterocyclic cyclopropylamine derivatives. These motifs, although pharmaceutically valuable,69 are challenging to reliably access late-stage due to the generally inert nature of cyclopropane electrophiles to nucleophilic displacement,35,70–74 instead necessitating the use of preformed organometallic derivatives.75–79 Gratifyingly, alkyl-HARC cyclopropylation employing commercial and bench-stable bromocyclopropane was indeed achievable for various N-nucleophiles with good to excellent efficiency (50–59, 48–85% yield, Figure 5). Moreover, this approach was applicable to more complex structures, providing Skelaxin (60, 84% yield) and Celebrex (61, 51% yield) analogs in expedient fashion. This accomplishment highlights the unique capabilities of HARC-based N-alkylations, permitting single-step syntheses of elusive drug-like cyclopropylamines.
Figure 5. Direct cyclopropylation of N-nucleophiles using bromocyclopropane.

Isolated yields unless otherwise indicated. See Supplemental Information for specific experimental details. ar.r. >20:1. bYield determined by 1H NMR analysis.
Additionally, we anticipated that (TMS)3SiNHAd, originally designed by our laboratory for organochloride abstraction,61 could activate typically unreactive non-primary alkyl chlorides towards HARC N-alkylation (Figure 6). We were pleased to find that, under modified conditions, conversion of secondary alkyl chlorides to the corresponding radicals using (TMS)3SiNHAd provided facile N-alkylation reactivity (62 and 1, 52% and 50% yield, respectively). The significance of this discovery was further verified via rapid generation of trans-amino alcohol 63 (40% yield) for which the necessary bromohydrin partner is unavailable in gram-scale quantities from chemical vendors, thus demonstrating single-step entry into chemical space unattainable from commercially-available bromides (for control reactions verifying the mechanism that enables formation of 63, see Figure S28). The chlorocyclohexanol stereoisomeric mixture employed ultimately furnished 63 in a diastereoconvergent fashion, a notable consequence of exploiting copper-mediated bond formation as an elementary step to facilitate N-alkylation. We expect this new metallaphotoredox N-alkylation strategy to enable a variety of previously unachievable chloride substitution reactions, and further expansion of this variant of the copper-HARC protocol is currently underway.
Figure 6. Extension of HARC coupling to unactivated alkyl chlorides.

All yields are isolated. r.r. >20:1 in all cases. aInitial alkyl chloride d.r. 1:1. See Supplemental Information for specific experimental details.
Mechanistic proposal and comparisons to traditional substitution methods
Following our studies on the applicability of this HARC N-alkylation to various synthetic contexts, we also sought to investigate the mechanism of coupling. In particular, we set out to verify that the achievements of this platform, with respect to the breadth of scope, are in fact complimentary to those possible within established substitution conditions, and that the HARC design principle is utilized to accomplish these advances. We started by identifying products 1, 18 and 50 as representative examples for comparing our metallaphotoredox method against prototypical substitution approaches. Previous reports indicate that these adducts are formed in trace or modest yield (with moderate N1-regioselectivity) under traditional substitution conditions when using 3-chloroindazole and the corresponding bromide electrophiles (Figure 7),35 presumably due to prohibitively high kinetic barriers for halide displacement which cannot be overcome using thermal activation. Consequently, efficient and regioselective generation of 1, 18 and 50 under HARC N-alkylation conditions (57–93% yield, see Figures 1, 2, 5 and 7) signifies that this new platform will indeed enable reactivity not attainable from standard two-electron processes, implying that alternative reaction pathways must be operative. Furthermore, when subjecting 3-chloroindazole and bromocyclohexane to the optimized HARC-coupling conditions using only ambient light as a photon source, product 1 was not detected (Figure 7), eliminating the possibility of background non-photonic substitution pathways (these trends were consistent across all 13 classes of nucleophiles subjected to coupling, see Figure S29). Collectively, these results indicate both that unique mechanisms beyond those of traditional nucleophilic substitution are responsible for the robust performance observed in HARC N-alkylation reactions, and that such N-alkyl products are otherwise inaccessible when using conditions designed to best facilitate classical SN2 or SN1 processes.
Figure 7. Comparisons with traditional substitution approaches and preliminary mechanistic experiments.

All products formed using HARC conditions displayed r.r. >20:1. See reference 35, indicated main text Figures, or Figures S22–S25 and S29 for additional experimental details. TEMPO, 2,2,6,6-tetramethylpiperidin-1-yl)oxyl.
To validate the hypothesized open-shell nature of this transformation, we further subjected 3-chloroindazole to coupling conditions designed to indicate the presence of radical intermediates. When using (bromomethyl)cyclopropane as the halide coupling partner, no direct coupling was observed, with N-homoallyl indazole 64 instead generated as the primary product in 33% yield (Figure 7). Given the rapid rate for unimolecular ring-opening of the cyclopropylmethyl radical (k = 7.8 × 107 s−1 at 20 °C)80 to generate homoallylic isomers, this outcome is consistent with the intermediacy of alkyl radicals (presumably generated via halogen atom transfer, as supersilanol is required for detectable product formation, see Figure 1) in the HARC N-alkylation protocol. However, to further distinguish between the possibilities of radical ring-opening and copper-mediated β-carbon elimination (which would not necessarily proceed via open-shell intermediates),81 TEMPO-trapping studies were also conducted. When adding the persistent radical 2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) to the standard bromocyclohexane coupling reaction, product 1 was not detected, and the major species observed from bromocyclohexane conversion was TEMPO-trapped adduct 65 (Figure 7). Given that adduct 65 is well-established to be forged via radical-radical coupling of TEMPO with the transient cyclohexyl radical,82 these results, in tandem with ring-opening studies and control experiments (Figures 1, 7 and S22–S25), denote the anticipated open-shell pathways needed to enable HARC-type coupling.
Based on the above findings and preceding literature regarding the behavior of copper complexes in systems containing open-shell intermediates, a plausible mechanism and summary of optimal conditions for copper-HARC N-alkylation is detailed in Scheme 2. Initial photoexcitation of Ir(III) photocatalyst [Ir-1] with blue light from IPR irradiation and subsequent intersystem crossing would generate the long-lived triplet excited state II (lifetime τ = 280 ns), a potent single-electron oxidant (E1/2red[*IrIII/IrII] = +1.65 V vs. SCE in MeCN).83 Consistent with previous aerobic photocatalytic transformations featuring silyl radical activation of halides,53 ensuing SET between complex II and a silyl radical precursor such as supersilanol (IV; Epa [IV/IV+•] = +1.54 V vs. SCE in MeCN)48 would afford reduced Ir(II) photocatalyst III and, upon deprotonation and radical Brook rearrangement, silicon-centered radical V. This nucleophilic silyl radical can perform facile halogen atom abstraction, converting alkyl bromide VI to aliphatic radical intermediate VII.84 Concurrently, reduced Ir(II) complex III (E1/2red[IrIII/IrII] = −0.79 V vs. SCE in MeCN)83 can be re-oxidized by molecular oxygen to regenerate ground-state Ir(III) photocatalyst [Ir-1]. Independently, Cu(I) catalyst VIII, N-nucleophile IX and base would combine to produce anionic Cu(I)-amido complex X, eventually affording neutral Cu(II)-amido intermediate XI following SET with oxygen. Such copper complexes are well-documented to furnish C(sp3)–N bonds upon encountering alkyl radicals,33–36,44 presumably beginning with capture of radical VII (predicted to proceed at near-diffusion rates)85–87 to afford HARC-generated Cu(III)-alkyl complex XII. Subsequent reductive elimination from XII would deliver N-alkyl product XIII while regenerating Cu(I) catalyst VIII, ultimately closing both catalytic cycles.88–92 Unique to this platform is the decoupled nature of halide activation (by photocatalytic halogen atom transfer) and nucleophile activation (by copper catalysis). This design principle is evidently responsible for conferring generality (with respect to scope) across both coupling partners, and for offering reactivity and selectivity surpassing that of previously reported halide-based N-alkylation systems.
Scheme 2. Plausible mechanism and summary of conditions for copper-HARC N-alkylation.

Conclusion
In summary, an underexplored halogen abstraction-radical capture (HARC) strategy has been applied to the copper metallaphotoredox alkylation of N-nucleophiles using alkyl halides as convenient electrophilic partners. This coupling method requires no substrate preactivation, operates under mild aerobic conditions, and provides reactivity with a broad range of alkyl bromides and N-heterocycles, including those typically recalcitrant within thermally-induced SN1 or SN2 settings. The demonstrated late-stage utility of this method, as well as the compatibility of substrates traditionally inert to substitution (such as bromocyclopropane and various alkyl chlorides), makes this technology particularly promising for exploring diverse sp3-rich chemical space in medicinal chemistry settings. We anticipate that HARC coupling mechanisms, including those demonstrated for N-alkylation, will continue to be developed and deployed for complex molecule synthesis, ideally providing synthetic chemists access to elusive alkylamines (or other motifs) that cannot be generated through conventional approaches.
EXPERIMENTAL PROCEDURES
General procedure for HARC N-alkylation of indazoles and pyrazoles
To an oven-dried 40 mL vial equipped with a Teflon stir bar was added indazole or pyrazole nucleophile (0.25 mmol, 1.0 equiv.), Ir[dF(CF3)ppy]2[4,4′-d(CF3)bpy]PF6 ([Ir-1], 2.3 mg, 2.0 μmol, 0.008 equiv.), bis(2,2,6,6-tetramethyl-3,5-heptanedionato)copper(II) (Cu(TMHD)2, 22–32 mg, 0.05–0.075 mmol, 0.2–0.3 equiv.), LiOt-Bu (60 mg, 0.75 mmol, 3.0 equiv.), MeCN (2.5 mL, 0.1 M) and water (45 μL, 2.5 mmol, 10 equiv.). The resulting solution was stirred for 1–2 minutes under air to ensure complete ligation of the nucleophile to the copper precatalyst. Following this complexation period, alkyl halide (0.625 mmol, 2.5 equiv.) and (TMS)3SiOH (165 mg, 0.625 mmol, 2.5 equiv.) were added to the mixture, after which the vial was capped and an 18G vent needle was inserted through the Teflon-lined septum. The reaction mixture was subsequently stirred under air within the Integrated Photoreactor (450 nm irradiation) for 4 hours. After 4 hours, the reaction mixture was diluted with EtOAc (5 mL), followed by the addition of KF on alumina (40 wt. % from Sigma-Aldrich, 1.0 g) and tetrabutylammonium bromide (500 mg) to the vial. This suspension was stirred under air for 2–24 hours, then filtered into a separatory funnel, using an additional 25 mL EtOAc wash to ensure complete transfer from the vial. The organic layer was subsequently washed with saturated Na2CO3 (10 mL), water (10 mL) and brine (10 mL), and the collected aqueous layer was extracted with EtOAc (10 mL). The combined organics were dried over MgSO4 and concentrated in vacuo to obtain the crude product. This residue was then purified by silica gel chromatography to afford the desired N-alkylated product.
General procedure for HARC N-alkylation of other N-nucleophiles
To an oven-dried 40 mL vial equipped with a Teflon stir bar was added N-nucleophile (0.25 mmol, 1.0 equiv.), Ir[dF(CF3)ppy]2[4,4′-d(CF3)bpy]PF6 ([Ir-1], 2.3 mg, 2.0 μmol, 0.008 equiv.), MeCN (2.5 mL, 0.1 M) and 1,5-diazabicyclo[4.3.0]non-5-ene (DBN, 31 μL, 0.25 mmol, 1.0 equiv.) The resulting homogeneous solution was stirred for 5 minutes, after which LiOt-Bu (60 mg, 0.75 mmol, 3.0 equiv.) and water (45 μL, 2.5 mmol, 10 equiv.) were added to the vial. This suspension was then sonicated under air for 1 minute until the mixture became homogeneous. Cu(TMHD)2 (22–32 mg, 0.05–0.075 mmol, 0.2–0.3 equiv.) was then added to the vial, and the solution was stirred for 1–2 minutes under air to ensure complete ligation of the nucleophile to the copper precatalyst. Following this complexation period, alkyl halide (0.625 mmol, 2.5 equiv.) and (TMS)3SiOH (165 mg, 0.625 mmol, 2.5 equiv.) were added to the mixture, after which the vial was capped and an 18G vent needle was inserted through the Teflon-lined septum. The reaction mixture was subsequently stirred under air within the Integrated Photoreactor (450 nm irradiation) for 4 hours. After 4 hours, the reaction mixture was diluted with EtOAc (5 mL), followed by the addition of KF on alumina (40 wt. %, 1.0 g) and tetrabutylammonium bromide (500 mg) to the vial. This suspension was stirred under air for 2–24 hours, then filtered into a separatory funnel, using an additional 25 mL EtOAc wash to ensure complete transfer from the vial. The organic layer was subsequently washed with saturated Na2CO3 (10 mL), water (10 mL) and brine (10 mL), and the collected aqueous layer was extracted with EtOAc (10 mL). The combined organics were dried over MgSO4 and concentrated in vacuo to obtain the crude product. This residue was then purified by silica gel chromatography to afford the desired N-alkylated product.
Other experimental details and examples (Figures S1–S31), as well as characterization data (Figures S32–S184), can be found in the Supplemental Information.
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, David W. C. MacMillan (dmacmill@princeton.edu).
Materials availability
This study did not involve the design of unique reagents or catalysts for chemical synthesis.
Data and code availability
There is no dataset or code associated with this publication. All relevant procedures and experimental data are provided in the Supplemental Information.
Supplementary Material
The bigger picture.
Alkylamines, and their N-heterocyclic derivatives, are key structural features in molecules central to the medical, agroscience and materials science industries. Although N-alkylation using nitrogen nucleophiles and halides has historically been accomplished via nucleophilic substitution, this process is frequently inefficient and poorly selective under thermal activation. We report a kinetically facile, room temperature N-alkylation achieved via underexplored halogen abstraction-radical capture pathways mediated by independent photoredox and copper catalytic cycles. With broad scope and late-stage applicability, even for traditionally inert cyclopropane and chloride electrophiles, this protocol offers new and efficient strategies for modern amine synthesis. These developments can ideally provide medicinal chemists with single-step access to valuable sp3-rich chemical diversity in drug discovery, including alkylamine motifs unattainable via conventional N-alkylation methods.
Highlights.
General, room temperature N-alkylation via copper metallaphotoredox catalysis
Broad reactivity across diverse alkyl bromides, N-heterocycles and pharmaceuticals
Convenient approach to N-cyclopropylation using easily-handled bromocyclopropane
Readily extended to functionalization of unactivated secondary alkyl chlorides
ACKNOWLEDGMENTS
The authors are grateful for financial support provided by the National Institute of General Medical Sciences (NIGMS), the NIH (under Award R35GM134897-01), the Princeton Catalysis Initiative, and kind gifts from Merck, Janssen, BMS, Genentech, Celgene and Pfizer. N. W. D. acknowledges Princeton University, N. Brink and the Brink Family for a Brink Graduate Fellowship and acknowledges Princeton University, E. Taylor and the Taylor family for an Edward C. Taylor Fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIGMS. The authors thank M. N. Lavagnino, Y. Liang, W. Liu, H. A. Sakai and X. Zhang for helpful scientific discussions.
Footnotes
DECLARATION OF INTERESTS
D.W.C.M. declares a competing financial interest with respect to the Integrated Photoreactor.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and Figures S1–S184 and can be found with this article online at https://doi.org/10.1016/j.chempr.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES AND NOTES
- 1.Vitaku E; Smith DT; Njardarson JT (2014). Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- 2.Dugger RW; Ragan JA; Ripin DHB (2005). Survey of GMP Bulk Reactions Run in a Research Facility between 1985 and 2002. Org. Process Res. Dev 9, 253–258. [Google Scholar]
- 3.Froidevaux V; Negrell C; Caillol S; Pascault J-P; Boutevin B (2016). Biobased Amines: From Synthesis to Polymers; Present and Future. Chem. Rev 116, 14181–14224. [DOI] [PubMed] [Google Scholar]
- 4.Ruiz-Castillo P; Buchwald SL (2016). Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hazari N; Melvin PR; Beromi MM (2017). Well-defined nickel and palladium precatalysts for cross-coupling. Nat. Rev. Chem 1, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sambiago C; Marsden SP; Blacker AJ; McGowan PC (2014). Copper catalyzed Ullmann type chemistry: from mechanistic aspects to modern development. Chem. Soc. Rev 43, 3525–3550. [DOI] [PubMed] [Google Scholar]
- 7.West MJ; Fyfe JWB; Vantourout JC; Watson AJB (2019). Mechanistic Development and Recent Applications of the Chan-Lam Amination. Chem. Rev 119, 12491–12523. [DOI] [PubMed] [Google Scholar]
- 8.For direct comparisons of various metal-catalyzed C(sp2)–N coupling methodologies, see: Beletskaya IP; Cheprakov AV (2012). The Complimentary Competitors: Palladium and Copper in C–N Cross-Coupling Reactions. Organometallics 31, 7753–7808.
- 9.Bariwal J; Van der Eycken E (2013). C–N bond forming cross-coupling reactions: an overview. Chem. Soc. Rev 42, 9283–9303. [DOI] [PubMed] [Google Scholar]
- 10.Trowbridge A; Walton SM; Gaunt MJ (2020). New Strategies for the Transition-Metal Catalyzed Synthesis of Aliphatic Amines. Chem. Rev 120, 2613–2692. [DOI] [PubMed] [Google Scholar]
- 11.For reviews on the enhanced industrial performance of sp3-rich compounds, see Refs. 12–13, and: Lovering F; Bikker J; Humblet C (2009). Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 52, 6752–6756.
- 12.Foley DJ; Craven PGE; Collins PM; Doveston RG; Aimon A; Talon R; Churcher I; von Delft F; Marsden SP; Nelson A (2017). Synthesis and Demonstration of the Biological Relevance of sp3-rich Scaffolds Distantly Related to Natural Product Frameworks. Chem. Eur. J 23, 15227–15232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeschke P (2018). Current status of chirality in agrochemicals. Pest Manag. Sci 74, 2389–2404. [DOI] [PubMed] [Google Scholar]
- 14.Fletcher S (2015). The Mitsunobu reaction in the 21st century. Org. Chem. Front 2, 739–752. [Google Scholar]
- 15.Ghosh AK; Brindisi M; Sarkar A (2018). The Curtius Rearrangement: Applications in Modern Drug Discovery and Medicinal Chemistry. ChemMedChem 13, 2351–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Müller TE; Hultzsch KC; Yus M; Foubelo F; Tada M (2008). Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem. Rev 108, 3795–3892. [DOI] [PubMed] [Google Scholar]
- 17.Huang L; Arndt M; Gooßen K; Heydt H; Gooßen LJ (2015). Late Transition Metal-Catalyzed Hydroamination and Hydroamidation. Chem. Rev 115, 2596–2697. [DOI] [PubMed] [Google Scholar]
- 18.Abdel-Magid AF; Mehrman SJ (2006). A Review on the Use of Sodium Triacetoxyborohydride in the Reductive Amination of Ketones and Aldehydes. Org. Process Res. Dev 10, 971–1031. [Google Scholar]
- 19.Brown DG; Boström J (2016). Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem 59, 4443–4458. [DOI] [PubMed] [Google Scholar]
- 20.Carey JS; Laffan D; Thomson C; Williams MT (2006). Analysis of the reactions used for the preparation of drug candidate molecules. Org. Biomol. Chem 4, 2337–2347. [DOI] [PubMed] [Google Scholar]
- 21.For discussions of the fundamental importance and aspects of N-alkylation via halide substitution, see Ref. 22, and: March J (1985). Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (Wiley; ).
- 22.McMurry J (2012). Organic Chemistry (Thomson Brooks/Cole; ). [Google Scholar]
- 23.For a general review of metal-mediated substitution reactions, see: Fu GC (2017). Transition-Metal Catalysis of Nucleophilic Substitution Reactions: A Radical Alternative to SN1 and SN2 Processes. ACS Cent. Sci 3, 692–700.
- 24.Peacock DM; Roos CB; Hartwig JF (2016). Palladium-Catalyzed Cross Coupling of Secondary and Tertiary Alkyl Bromides with a Nitrogen Nucleophile. ACS Cent. Sci 2, 647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casitas A; Ribas X (2013). The role of organometallic copper(III) complexes in homogeneous catalysis. Chem. Sci 4, 2301–2318. [Google Scholar]
- 26.Hickman AJ; Sanford MS (2012). High-valent organometallic copper and palladium in catalysis. Nature 484, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu S; Niljianskul N; Buchwald SL (2013). Enantio- and Regioselective CuH-Catalyzed Hydroamination of Alkenes. J. Am. Chem. Soc 135, 15746–15749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu RY; Buchwald SL (2020). CuH-Catalyzed Olefin Functionalization: From Hydroamination to Carbonyl Addition. Acc. Chem. Res 53, 1229–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rucker RP; Whittaker AM; Dang H; Lalic G (2012). Synthesis of Tertiary Alkyl Amines from Terminal Alkenes: Copper-Catalyzed Amination of Alkyl Boranes. J. Am. Chem. Soc 134, 6571–6574. [DOI] [PubMed] [Google Scholar]
- 30.Sueki S; Kuninobu Y (2013). Copper-Catalyzed N- and O-Alkylation of Amines and Phenols using Alkylborane Reagents. Org. Lett 15, 1544–1547. [DOI] [PubMed] [Google Scholar]
- 31.Rossi SA; Shimkin KW; Xu Q; Mori-Guiroz LM; Watson DA (2013). Selective Formation of Secondary Amides via the Copper-Catalyzed Cross-Coupling of Alkylboronic Acids with Primary Amides. Org. Lett 15, 2314–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mori-Quiroz LM; Shimkin KW; Rezazadeh S; Kozlowski RA; Watson DA (2016). Copper-Catalyzed Amidation of Primary and Secondary Alkyl Boronic Esters. Chem. Eur. J 22, 15654–15658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran BL; Li B; Driess M; Hartwig JF (2014). Copper-Catalyzed Intermolecular Amidation and Imidation of Unactivated Alkanes. J. Am. Chem. Soc 136, 2555–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng Y-W; Narobe R; Donabauer K; Yakubov S; König B (2020). Copper(II)-Photocatalyzed N–H Alkylation with Alkanes. ACS Catal 10, 8582–8589. [Google Scholar]
- 35.Liang Y; Zhang X; MacMillan DWC (2018). Decarboxylative sp3 C–N coupling via dual copper and photoredox catalysis. Nature 559, 83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X; Smith RT; Le C; McCarver SJ; Shireman BT; Carruthers NI; MacMillan DWC (2020). Copper-mediated synthesis of drug-like bicyclopentanes. Nature 580, 220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mao R; Frey A; Balon J; Hu X (2018). Decarboxylative C(sp3)–N cross-coupling via synergetic photoredox and copper catalysis. Nat. Catal 1, 120–126. [Google Scholar]
- 38.Zhao W; Wurz RP; Peters JC; Fu GC (2017). Photoinduced, Copper-Catalyzed Decarboxylative C–N Coupling to Generate Protected Amines: An Alternative to the Curtius Rearrangement. J. Am. Chem. Soc 139, 12153–12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bissember AC; Lundgren RJ; Creutz SE; Peters JC; Fu GC (2013). Transition Metal-Catalyzed Alkylations of Amines with Alkyl Halides: Photoinduced, Copper-Catalyzed Couplings of Carbazoles. Angew. Chem., Int. Ed 52, 5129–5133. [DOI] [PubMed] [Google Scholar]
- 40.Do H-Q; Bachman S; Bissember AC; Peters JC; Fu GC (2014). Photoinduced, Copper-Catalyzed Alkylation of Amides with Unactivated Secondary Alkyl Halides at Room Temperature. J. Am. Chem. Soc 136, 2162–2167. [DOI] [PubMed] [Google Scholar]
- 41.Kainz QM; Matier CD; Bartoszewicz A; Zultanski SL; Peters JC; Fu GC (2016). Asymmetric copper-catalyzed C–N cross-couplings induced by visible light. Science 351, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahn JM; Peters JC; Fu GC (2017). Design of a Photoredox Catalyst that Enables the Direct Synthesis of Carbamate-Protected Primary Amines via Photoinduced, Copper-Catalyzed N-Alkylation Reactions of Unactivated Secondary Halides. J. Am. Chem. Soc 139, 18101–18106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bartoszewicz A; Matier CD; Fu GC (2019). Enantioconvergent Alkylations of Amines by Alkyl Electrophiles: Copper-Catalyzed Nucleophilic Substitutions of Racemic α-Halolactams by Indoles. J. Am. Chem. Soc 141, 14864–14869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.For further mechanistic details related to photoinduced C(sp3)–N couplings using copper photoreductants, see: Ahn JM; Ratani TS; Hannoun KI; Fu GC; Peters JC (2017). Photoinduced, Copper-Catalyzed Alkylation of Amines: A Mechanistic Study of the Cross-Coupling of Carbazole with Alkyl Bromides. J. Am. Chem. Soc 139, 12716–12723.
- 45.For a general review of metallaphotoredox cross-couplings, see: Twilton J; Le CC; Zhang P; Shaw MH; Evans RW; MacMillan DWC (2017). The Merger of Transition Metal and Photocatalysis. Nat. Rev. Chem 1, 1–18.
- 46.Levin MD; Kim S; Toste FD (2016). Photoredox Catalysis Unlocks Single-Electron Elementary Steps in Transition Metal Catalyzed Cross-Coupling. ACS Cent. Sci 2, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hossain A; Bhattacharyya A; Reiser O (2019). Copper’s rapid ascent in visible-light photoredox catalysis. Science 364, 1–11. [DOI] [PubMed] [Google Scholar]
- 48.Le C; Chen TQ; Liang T; Zhang P; MacMillan DWC (2018). A radical approach to the copper oxidative addition problem: Trifluoromethylation of bromoarenes. Science 360, 1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao X; MacMillan DWC (2020). Metallaphotoredox Perfluoroalkylation of Organobromides. J. Am. Chem. Soc 142, 19480–19486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kautzky JA; Wang T; Evans RW; MacMillan DWC (2018). Decarboxylative Trifluoromethylation of Aliphatic Carboxylic Acids. J. Am. Chem. Soc 140, 6522–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kornfilt DJP; MacMillan DWC (2019). Copper-Catalyzed Trifluoromethylation of Alkyl Bromides. J. Am. Chem. Soc 141, 6853–6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sarver PJ; Bacauanu V; Schultz DM; DiRocco DA; Lam Y-h.; Sherer EC; MacMillan DWC (2020). The merger of decatungstate and copper catalysis to enable aliphatic C(sp3)–H trifluoromethylation. Nat. Chem 12, 459–467. [DOI] [PubMed] [Google Scholar]
- 53.Lavagnino MN; Liang T; MacMillan DWC (2020). HARC as an open-shell strategy to bypass oxidative addition in Ullmann-Goldberg couplings. Proc. Natl. Acad. Sci. U. S. A 117, 21058–21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.For kinetics and applications of silyl radicals in halogen atom abstraction processes, see Refs. 55–56, and: Chatgilialoglu C (1995). Structural and Chemical Properties of Silyl Radicals. Chem. Rev. 95, 1229–1251.
- 55.Chatgilialoglu C (1992). Organosilanes as radical-based reducing agents in synthesis. Acc. Chem. Res. 25, 188–194. [Google Scholar]
- 56.Chatgilialoglu C (2004). Organosilanes in Radical Chemistry (Wiley; ). [Google Scholar]
- 57.Le CC; Wismer MK; Shi Z-C; Zhang R; Conway DV; Li G; Vachal P; Davies IW; MacMillan DWC (2017). A General Small-Scale Reactor to Enable Standardization and Acceleration of Photocatalytic Reactions. ACS Cent. Sci 3, 647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Although the advantageous role of water is still under investigation, several possibilities are consistent with preceding reports, including a) enhanced thermodynamic favorability for SET oxidation of supersilanol via hydrogen bonding-mediated proton-coupled electron transfer (PCET), b) enhanced rates of superoxide disproportionation, suppressing formation of deleterious alcohol and ketone byproducts derived from the alkyl radical (see Figure S3), and c) enhanced propensity for transmetalation/ligand exchange via modulation of the copper coordination sphere, thereby facilitating formation of the required alkyl Cu(III)–amido intermediate. For further discussions of each possibility, see Refs. 59–60, and: Gentry EC; Knowles RR (2016). Synthetic Applications of Proton-Coupled Electron Transfer. Acc. Chem. Res 49, 1546–1556.
- 59.Che Y; Tsushima M; Matsumoto F; Okajima T; Tokuda K; Ohsaka T (1996). Water-Induced Disproportionation of Superoxide Ion in Aprotic Solvents. J. Phys. Chem 100, 20134–20137. [Google Scholar]
- 60.Su X-L; Ye L; Chen J-J; Liu X-D; Jiang S-P; Wang F-L; Liu L; Yang C-J; Chang X-Y; Li Z-L; Gu Q,-S; Liu X-Y (2020). Copper-Catalyzed Enantioconvergent Cross-Coupling of Racemic Alkyl Bromides with Azole C(sp2)–H Bonds. Angew. Chem., Int. Ed 60, 380–384. [DOI] [PubMed] [Google Scholar]
- 61.Sakai HA; Liu W; Le CC; MacMillan DWC (2020). Cross-Electrophile Coupling of Unactivated Alkyl Chlorides. J. Am. Chem. Soc 142, 11691–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Although copper catalyst turnover numbers (TONs) reported in Figure 2 range from <1 to 4.6, higher TONs (>10) can typically be achieved at lower loadings of the copper catalyst, consistently accompanied by a small decrease in yield of N-alkylated product (see Figure S21). Given the inexpensive and earth-abundant nature of copper as a metal for homogeneous catalysis, higher loadings (>20 mol%) were employed to maximize yield in all cases, despite the reduction in TONs. For further discussions regarding the considerations of copper as a non-precious metal for catalysis, see: Liu X; Zheng H; Sun Z; Han A; Du P (2015). Earth-Abundant Copper-Based Bifunctional Electrocatalyst for Both Catalytic Hydrogen Production and Water Oxidation. ACS Catal 5, 1530–1538.
- 63.Locke GM; Bernhard SSR; Senge MO (2019). Nonconjugated Hydrocarbons as Rigid-Linear Motifs: Isosteres for Material Sciences and Bioorganic and Medicinal Chemistry. Chem. Eur. J 25, 4590–4647. [DOI] [PubMed] [Google Scholar]
- 64.For additional examples of poorly regioselective N-alkylations of azoles using organohalides, see Ref. 65, and: Schmidt A; Beutler A; Snovydovych B (2008). Recent Advances in the Chemistry of Indazoles. Eur. J. Org. Chem 2008, 4073–4095.
- 65.Teixeira FC; Ramos H; Antunes IF; Curto MJM; Duarte MT; Bento I (2006). Synthesis and Structural Characterization of 1- and 2-Substituted Indazoles: Ester and Carboxylic Acid Derivatives. Molecules 11, 867–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Several classes of substrates, including alkylamines, (benz)imidazoles, (benzo)triazoles, pyridones, α-bromocarbonyls, tert-butyl bromide and sterically encumbered secondary acyclic bromides have thus far been recalcitrant within HARC N-alkylation protocols. For specific examples and reaction conditions, see Figure S30.
- 67.Smith JRL; Mashender D (1976). Amine oxidation. Part IX. The electrochemical oxidation of some tertiary amines: the effect of structure on reactivity. J. Chem. Soc., Perkin Trans 2, 47–51. [Google Scholar]
- 68.For examples of the challenges typically posed by tertiary amines in metallaphotoredox cross-couplings, see: Dombrowski AW; Gesmundo NJ; Aguirre AL; Sarris KA; Young JM; Bogdan AR; Martin MC; Gedeon S; Wang Y (2020). Expanding the Medicinal Chemist Toolbox: Comparing Seven C(sp2)–C(sp3) Cross-Coupling Methods by Library Synthesis. ACS Med. Chem. Lett 11, 597–604.
- 69.For examples of the medicinally-relevant geometric, metabolic and permeation properties associated with cyclopropylamines, see: Talele TT (2016). The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem 59, 8712–8756.
- 70.For additional examples of ineffective (<15% yield) substitution reactions involving related heterocyclic N-nucleophiles and cyclopropyl halides, see Ref. 71, and: Kaur N; Monga V; Jain R (2004). Facile one-step synthesis of N-α-Boc-1-alkyl-L-histidines. Tetrahedron Lett 45, 6883–6885.
- 71.Lambertucci C; Antonini I; Buccioni M; Dal Ben D; Kachare DD; Volpini R; Klotz K-N; Cristalli G (2009). 8-Bromo-9-alkyl adenine derivatives as tools for developing new adenosine A2A and A2B receptors ligands. Bioorg. Med. Chem 17, 2812–2822. [DOI] [PubMed] [Google Scholar]
- 72.For alternative approaches featuring formal nucleophilic substitutions of acyl-substituted bromocyclopropanes that circumvent SN2 pathways via initial dehydrobromination, see Refs. 73–74, and: Alnasleh BK; Sherrill WM; Rubina M; Banning J; Rubin M (2009). Highly Diastereoselective Formal Nucleophilic Substitution of Bromocyclopropanes. J. Am. Chem. Soc 131, 6906–6907.
- 73.Prosser AR; Banning JE; Rubina M; Rubin M (2010). Formal Nucleophilic Substitution of Bromocyclopropanes with Amides en route to Conformationally Constrained β-Amino Acid Derivatives. Org. Lett 12, 3968–3971. [DOI] [PubMed] [Google Scholar]
- 74.Ryabchuk P; Rubina M; Xu J; Rubin M (2012). Formal Nucleophilic Substitution of Bromocyclopropanes with Azoles. Org. Lett 14, 1752–1755. [DOI] [PubMed] [Google Scholar]
- 75.For select copper-mediated N-cyclopropylation platforms (utilized for multiple substrates) that feature transmetalation with cyclopropyl organometallics, see Refs. 76–79, and: Gagnon A; St-Onge M; Little K; Duplessis M; Barabé F (2007). Direct N-Cyclopropanation of Cyclic Amides and Azoles Employing a Cyclopropylbismuth Reagent. J. Am. Chem. Soc 129, 44–45.
- 76.Bénard S; Neuville L; Zhu J (2008). Copper-Mediated N-Cyclopropanation of Azoles, Amides, and Sulfonamides by Cyclopropylboronic Acid. J. Org. Chem 73, 6441–6444. [DOI] [PubMed] [Google Scholar]
- 77.Tsuritani T; Strotman NA; Yamamoto Y; Kawasaki M; Yasuda N; Mase T (2008). N-Cyclopropanation of Indoles and Cyclic Amides with Copper(II) Reagent. Org. Lett 10, 1653–1655. [DOI] [PubMed] [Google Scholar]
- 78.Bénard S; Neuville L; Zhu J (2010). Copper-promoted N-cyclopropylation of anilines and amines by cyclopropylboronic acid. Chem. Commun 46, 3393–3395. [DOI] [PubMed] [Google Scholar]
- 79.Derosa J; O’Duill ML; Holcomb M; Boulous MN; Patman RL; Wang F; Tran-Dubé M; McAlpine I; Engle KM (2018). Copper-Catalyzed Chan-Lam Cyclopropylation of Phenols and Azaheterocycles. J. Org. Chem 83, 3417–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Newcomb M (2012). Radical Kinetics and Clocks. In Encyclopedia of Radicals in Chemistry, Biology and Materials, Chatgilialoglu C and Studer A, eds. (Wiley; ), pp. 107–124. [Google Scholar]
- 81.Kang Y-W; Cho YJ; Ko K-Y; Jang H-Y (2015). Copper-catalyzed carbon-carbon bond cleavage of primary propargyl alcohols: β-carbon elimination of hemiaminal intermediates. Catal. Sci. Technol 5, 3931–3934. [Google Scholar]
- 82.Yi J; Badir SO; Alam R; Molander GA (2019) Photoredox-Catalyzed Multicomponent Petasis Reaction with Alkyltrifluoroborates. J. Org. Chem 21, 4853–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Choi GC; Zhu Q; Miller DC; Gu CJ; Knowles RR (2016). Catalytic alkylation of remote C–H bonds enabled by proton-coupled electron transfer. Nature 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.For use of tris(trimethylsilyl)silanol in alkyl bromide halogen atom abstraction following radical Brook rearrangement, see: Smith RT; Zhang X; Rincón JA; Agejas J; Mateos C; Barberis M; García-Cerada S; de Frutos O; MacMillan DWC (2018). Metallaphotoredox-Catalyzed Cross-Electrophile Csp3–Csp3 Coupling of Aliphatic Bromides. J. Am. Chem. Soc 140, 17433–17438.
- 85.For discussions of the kinetics associated with formation/decomposition of putative alkylcopper(III) intermediates formed via alkyl radical capture, see Refs. 86–87, and: Kochi JK; Bemis A; Jenkins CL (1968). Mechanism of Electron Transfer Oxidation of Alkyl Radicals by Copper(II) Complexes. J. Am. Chem. Soc 90, 4616–4625.
- 86.Jenkins CL; Kochi JK (1972). Homolytic and ionic mechanisms in the ligand-transfer oxidation of alkyl radicals by copper(II) halides and pseudohalides. J. Am. Chem. Soc 94, 856–865. [Google Scholar]
- 87.Freiberg M; Meyerstein D (1980). Reactions of aliphatic free radicals with copper cations in aqueous solution. Part 2.–Reactions with cupric ions: a pulse radiolysis study. J. Chem. Soc. Faraday Trans 1, 1825–1837. [Google Scholar]
- 88.Although the proposed mechanism is favored for coupling of nucleophilic radicals such as VII, we also recognize the possibility of an alternative pathway, wherein C–N bond formation is achieved via outer-sphere group transfer (or copper-mediated radical-radical coupling) from Cu(II)-amido complex XI (formally a Cu-stabilized amido radical) to alkyl radical intermediate VII, ultimately furnishing product XIII and regenerating catalyst VIII in parallel with the radical capture/reductive elimination pathway depicted in Scheme 2. For further discussion of both mechanistic possibilities and examples of outer-sphere bond formation involving Cu(II) complexes and radical intermediates, see Refs. 89–92.
- 89.Qi X; Zhu L; Bai R; Lan Y (2017). Stabilization of Two Radicals with One Metal: A Stepwise Coupling Model for Copper-Catalyzed Radical-Radical Cross-Coupling. Sci. Rep 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li S-J; Lan Y (2020). Is Cu(III) a necessary intermediate in Cu-mediated coupling reactions? A mechanistic point of view. Chem. Commun 56, 6609–6619. [DOI] [PubMed] [Google Scholar]
- 91.Zhang Q; Wang T; Zhang X; Tong S; Wu Y-D; Wang M-X (2019). Radical Reactivity, Catalysis, and Reaction Mechanism of Arylcopper(II) Compounds: The Missing Link in Organocopper Chemistry. J. Am. Chem. Soc 141, 18341–18348. [DOI] [PubMed] [Google Scholar]
- 92.Zheng C; Lu F; Lu H; Xin J; Deng Y; Yang D; Wang S; Huang Z; Gao M; Lei A (2018). Copper-catalyzed selective radical-radical cross-coupling for C–S bond formation: an access to α-alkylthionitriles. Chem. Commun 54, 5574–5577. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
There is no dataset or code associated with this publication. All relevant procedures and experimental data are provided in the Supplemental Information.