

Abstract
Native mass spectrometry analysis of membrane proteins has yielded many useful insights in recent years with respect to membrane protein-lipid interactions, including identifying specific interactions and even measuring binding affinities based on observed abundances of lipid-bound ions after collision-induced dissociation (CID). However, the behavior of non-covalent complexes subjected to extensive CID can in principle be affected by numerous factors related to gas-phase chemistry, including gas-phase basicity (GB) and acidity, shared-proton bonds, and other factors. A recent report from our group showed that common lipids span a wide range of GB values. Notably, phosphatidylcholine (PC) and sphingomyelin lipids are more basic than arginine, suggesting they may strip charge upon dissociation in positive ion mode, while phosphoserine lipids are slightly less basic than arginine and may form especially strong shared-proton bonds. Here, we use CID to probe the strength of non-specific gas-phase interactions between lipid head groups and several soluble proteins, used to deliberately avoid possible physiological protein-lipid interactions. The strengths of the protein-head group interactions follow the trend predicted based solely on lipid and amino acid GBs: phosphoserine (PS) head group forms the strongest bonds with these proteins and out-competes the other head groups studied, while glycerophosphocholine (GPC) head groups form the weakest interactions and dissociate carrying away a positive charge. These results indicate that gas-phase thermochemistry can play an important role in determining which head groups remain bound to protein ions with native-like structures and charge states in positive ion mode upon extensive collisional activation.
Keywords: lipids, native mass spectrometry, thermochemistry, gas-phase basicity, gas-phase ion energetics, collision induced dissociation
Graphical Abstract
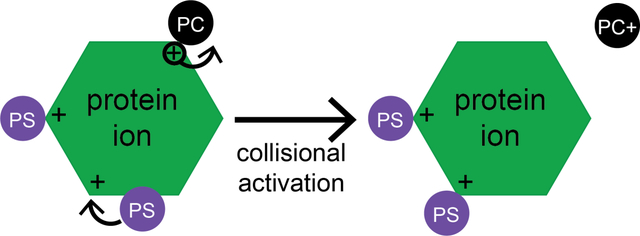
Introduction
Native mass spectrometry can be a powerful tool in determining the composition, stoichiometry, and structure of biochemical analytes, many of which can be challenging to accurately and precisely characterize using other techniques. As native mass spectrometry (MS) instrumentation and sample preparation methods have improved, a wider range of biochemical targets have come under investigation [1, 2], including native membrane protein-lipid and protein-drug interactions [3–11]. Many membrane proteins can be solubilized using local environments that mimic the lipid membrane, such as detergent micelles [12–14], or lipoprotein Nanodiscs [15–17], and are amenable to native electrospray ionization (ESI) [2, 18]. In parallel, many protein-drug complexes are stable to electrospray ionization, and their stoichiometry and structure can be probed by native ion mobility-mass spectrometry (IM-MS), Collision-Induced Unfolding, and other gas-phase methods. Seminal work from the Robinson [6, 12, 19–22], Laganowsky [23–28], Klassen [29–32], and Ruotolo [33–35] groups and many others has demonstrated the power and utility of these approaches.
Ideally, the stoichiometry, binding sites, and apparent binding strengths of protein-lipid, protein-drug, and other biochemical interactions studied using native (IM-)MS are a direct reflection of the condensed phase. However, because the dielectric permittivity of the vacuum environment is much lower than in solution, and water molecules and other co-solutes that contribute to biomolecular structure and stability are largely absent in the biomolecular ions produced by electrospray ionization (ESI), care must be taken to ensure that the observed ions do not undergo unwanted changes in structure during the native (IM-)MS experiment [36–38]. Numerous experiments and computations have shown that biomolecular ions can become kinetically trapped (see Figure 1) in native-like conformations during ESI under carefully controlled instrumental conditions [37, 39–43]. Typically, micron- or submicron-sized ESI emitters (nanoelectrospray ionization, “nESI”) and low-micromolar (or lower) concentrations of analytes and non-specific cosolutes (such as metal ions or other potential adducts) are used as well to limit condensation of unwanted adducts onto and evaporation-induced oligomerization of biomolecules during the ESI process [44–48].
Figure 1.
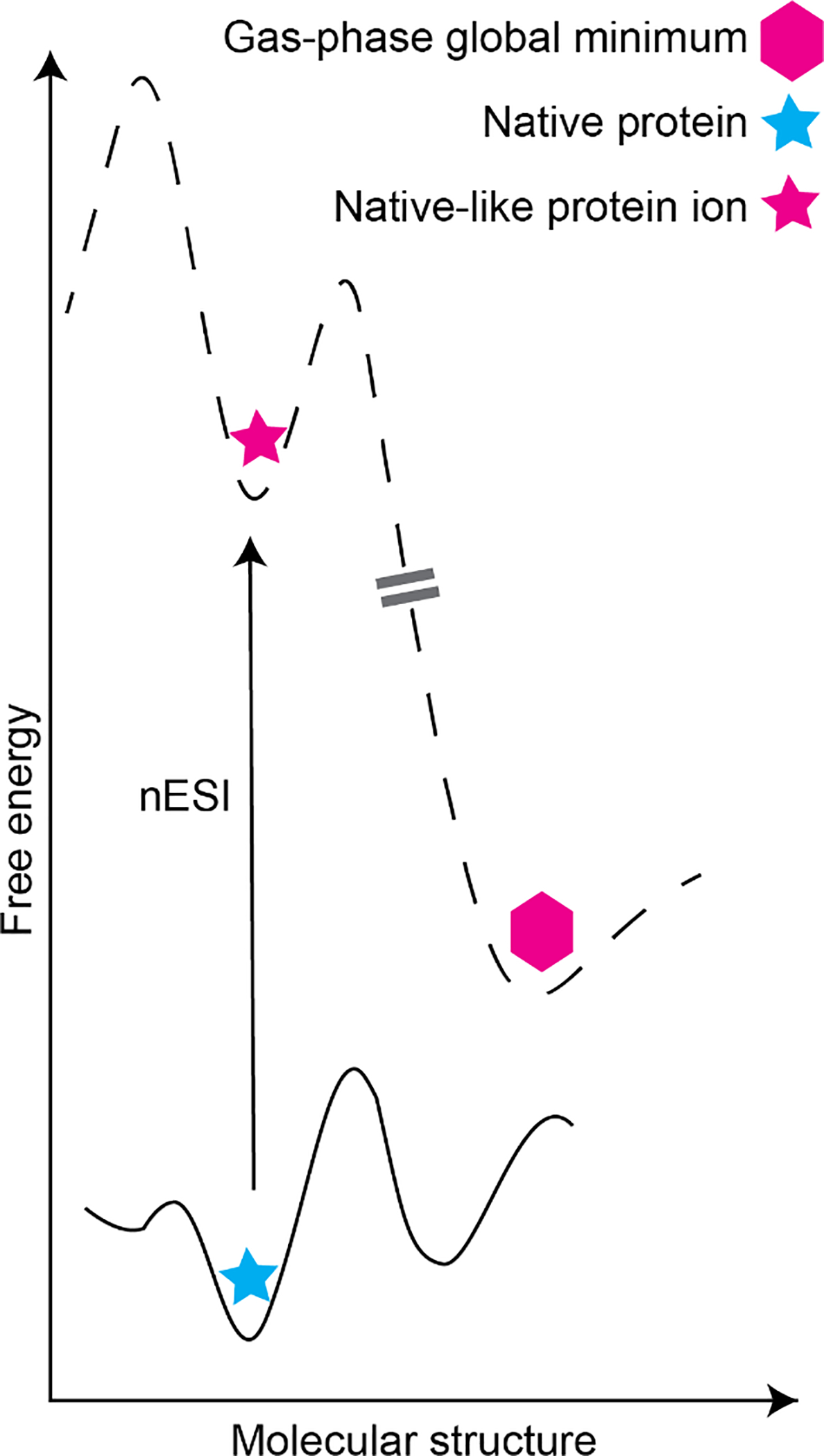
Schematic potential energy surface for native protein-adduct complex in aqueous solution (bottom; blue star represents global minimum-energy structure) and after transfer to the gas phase environment using nESI, in which energy is imparted to desolvate the protein ion (top). Note that relative energy barrier heights for adduct migration, protein unfolding, and other processes can differ between the solution and gas phase due to the absence of water and other cosolutes in the latter environment. Double-dashed line indicates variety of possible pathways to the gas-phase global minimum structure (pink hexagon), which may differ substantially from the kinetically trapped native-like protein ion (pink star).
In the past decade, several groups have demonstrated excellent agreement between binding behavior in solution and results inferred from native (IM-)MS experiments [10, 12, 24, 27, 28, 49]. For example, Klassen’s “Catch-and-Release” native MS experiments [30, 32, 50] have indicated similar ganglioside preferences of peripheral bacterial toxin proteins to those known from condensed-phase techniques. The Robinson and Laganowsky groups and others have also determined phospholipid binding preferences of both bacterial and human transmembrane proteins and found them to agree very well with condensed-phase results [10, 12, 19, 21, 22, 25, 27, 28, 49, 51–53]. Laganowsky has developed instrumentation for studying protein-lipid interactions as a function of solution temperature and demonstrated that lipid binding thermodynamics inferred from abundances of gas-phase protein-lipid complex ions are in agreement with those determined in the condensed phase [23, 24, 27].
Crucial to the accuracy of these types of experiments are the preservation of condensed-phase structures and interactions during the nanoESI process without introducing new, artifactual ones. Two ways in which these artifacts can in principle be introduced are: 1) adduction of non-specific cosolutes to the biomolecular ion during the nanoESI process due to droplet condensation (“non-specific adduction”) [54] and 2) changes in the structure or location of a specific binding site due to heating and relaxation of the complex ion in the gas-phase environment, where the potential energy surface may be different from that in solution (“adduct migration”). Evidence for non-specific adduction is readily observed as formation of oligomers and complexes known to be absent in solution, often as a result of using large ESI emitters and/or high biomolecule concentrations that statistically place multiple analytes within each initial electrospray droplet [44, 45]. This effect can even be exploited to displace metal cations from proteins during the nanoESI process in methods such as “buffer loading” [54] or to produce salt cluster ions for mass calibration [55]. Much less studied is the phenomenon of adduct migration, although much evidence supports the lability of charged species such as protons to migrate on the surface of native-like protein ions, which can result in unfolding and/or dissociation of the ions upon gas-phase activation [56–62]. Infrared photodissociation spectroscopy studies further indicate that protons can move from basic sites on organic ions that are favorable in solution to other basic sites that are more favorable in the gas phase during the ESI process [61, 63–66]. Because hydrogen bonds, shared-proton bonds, and other polar interactions can have very different relative stabilities in the gas phase versus in solution [65, 67], thermochemical barriers for disruption of non-covalent interactions can be dramatically different in the aqueous and gas-phase environment as illustrated in Figure 1 [59]. For example, hydrochloric acid is a much stronger acid in aqueous solution than phosphoric acid, but their relative acidities are reversed in the gas phase [68]. Similarly, carboxylic acid groups are poor bases in aqueous solution but relatively strong bases in the gas phase [65, 67].
The method most commonly used to dissociate unwanted adducts, such as detergent molecules, from protein-lipid complexes in native (IM-)MS studies, Collision-Induced Dissociation (CID), is a “slow heating” method [13, 69–71]. Dissociation in CID occurs on the many-microsecond to millisecond timescale [72, 73]. It is plausible that, under some experimental conditions, lipids initially bound at the surface of a membrane protein can undergo a change in binding geometry or migrate along the protein surface prior to complete dissociation. Whether these processes occur under a given set of experimental conditions inherently depends on the gas-phase potential energy surface for the protein-lipid interaction and the timescale of the dissociation process.
Here, we use a recently-demonstrated method for experimentally measuring gas-phase binding entropy and enthalpy barriers to determine gas-phase dissociation barrier thermochemistry of several common lipid head groups non-specifically bound to ions of soluble proteins in positive ion mode [73]. Relative gas-phase binding affinities to these protein ions are found to agree very well with expectations based on measured and computed gas-phase basicities of the lipid head groups [74]. This strategy is intended to circumvent ambiguities in interpretation that could arise from the presence of competing specific interactions and is used as a benchmark for evidence of non-specific lipid binding. Finally, we discuss implications of these results for identifying and determining the strength of physiologically relevant protein-lipid interactions using existing native (IM-)MS strategies.
Materials and Methods
Sample preparation.
Glycerophosphorylcholine, phosphorylcholine, phosphorylethanolamine, glycerol 1-phosphate, phosphoserine, ubiquitin, lysozyme, and transferrin were purchased from Millipore Sigma (Saint Louis, MO, USA). Lyophilized proteins were reconstituted in ultrapure 18 MΩ cm water and buffer-exchanged into 200 mM ammonium acetate, pH 7–7.5. Lipid head groups were dissolved in 200 mM ammonium acetate, pH 7–7.5. For experiments with ubiquitin and lysozyme, protein and lipid head group solutions were combined such that the final concentrations of protein and head group were 10 μM and 100 μM, respectively, while for experiments with transferrin the final concentrations were 5 μM protein and either 500 μM head group or, for competitive binding experiments, 500 μM of each head group under investigation.
Native IM-MS and CID.
All mass spectrometry experiments were performed in triplicate (transferrin experiments) or quadruplicate (all other experiments) on separate days in “Sensitivity” mode using a Synapt G2-Si (Waters Corp., Milford, MA, USA) with a nESI source. nESI emitters were pulled from 0.78 mm i.d. borosilicate capillaries to a final i.d. of ~1 μm using a Flaming-Brown P-97 micropipette puller (Sutter Instrument, Novato, CA, USA). Emitters were loaded with 3–5 μL of sample, and 0.7–1.1 kV was applied to a platinum wire in electrical contact with the solution to initiate electrospray. The source was held at ambient temperature, the sampling cone voltage was set to 25 V, and nitrogen, helium, and argon gas flow rates were 50, 100, and 5 mL/min, respectively. An ion mobility traveling wave velocity of 500 m/s was used for experiments with ubiquitin and lysozyme, a velocity of 400 m/s was used for experiments with transferrin, and a wave height of 20 V was used for all experiments. For CID experiments with ubiquitin and lysozyme, the singly-adducted state was isolated for a given charge state using a 32k quadrupole with the LM resolution set to 12. Dissociation was achieved by increasing the Trap CE in 1 V increments, beginning at the threshold for observable dissociation and continuing until the precursor was fully dissociated or significant covalent bond fragmentation was observed. For transferrin, due to the higher charge states present, there was insufficient resolution to completely isolate the singly-adducted state. Instead, an entire charge state was isolated with the LM resolution set to 4 and CID performed by increasing the Trap CE in 10 V increments from 10 V to 100 V. Additional mass spectra for transferrin were acquired without isolation at Trap CE values of 10, 50, 70, and 100 V.
Data analysis.
CID data for lipid head group-bound ubiquitin and lysozyme were analyzed in a similar manner to that described in our previous publication [73], (see also Supp. Info. for a brief description). Briefly, arrival time distributions for precursor and product ions were extracted using TwimExtract [75] and integrated in Igor Pro v. 6.3 (WaveMetrics, Portland, OR, USA).
For experiments with transferrin, native mass spectra were deconvolved using UniDec [76], and the Gabor Transform method in iFAMS (for “Double FT” analysis) [77, 78], to determine mass and charge state distributions as well as total lipid head group adduction. UniDec input parameters were: charge state range 10–25+, mass range 78–84 kDa, and peak full-width-at-half-maximum (fwhm) 3.0. Deconvolved mass spectra were analyzed further using Igor Pro v. 6.3 and iFAMS v. 5.3 [77, 79].
Results and Discussion
By probing the energetics of non-specific lipid head group binding, a baseline for what might be expected in the case of lipid-protein interactions governed by gas-phase, as opposed to condensed-phase, chemistry can be determined. To do this, we employ our recently introduced method for determining activation energies for protein and protein complex CID to determine dissociation barrier thermochemistry of lipid head groups non-specifically adducted to soluble proteins.
Several lipid head groups representing common phospholipids were studied. Structures of phosphorylethanolamine (PE), sodium glycerol 1-phosphate (PG), phosphoserine (PS), phosphorylcholine (PC), and glycerophosphorylcholine (GPC) are shown in Figure 2. GPC was studied in addition to PC due to the prevalence of calcium as a contaminant in PC solutions; calcium adduction was found to decrease spectral quality and complicate analysis. Ubiquitin (Ubq, 8.6 kDa) and lysozyme (LZ, 14 kDa) were used as model soluble proteins with no known native phospholipid interactions. Native mass spectra of each protein with no head group present were acquired and are shown in Figure S1. The most abundant native-like charge state was chosen for subsequent CID experiments; 5+ for Ubq and 7+ for LZ. Additional solutions of each lipid head group with Ubq or LZ protein were prepared in 10:1 head group:protein molar ratios. Statistically, at these lipid head group concentrations, multiple lipid head groups are present in each electrospray droplet, resulting in evaporation-induced, non-specific adduction of one or more lipid head groups onto protein ions present in the same droplet. The singly-adducted state of each target ion complex was isolated and subsequently dissociated in the Trap using CID over a range of collision energies.
Figure 2.
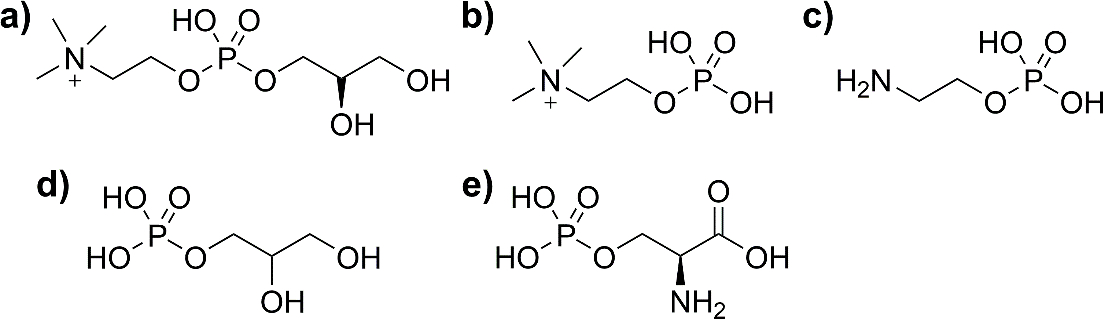
Structures of lipid head groups (a) glycerophosphorylcholine, GPC; (b) phosphorylcholine, PC; (c) phosphorylethanolamine, PE; (d) glycerol 1-phosphate, PG; and (e) phosphoserine, PS. Protonation states shown indicate those observed upon gas-phase dissociation from complexes with proteins in positive ion mode.
Mass spectra of lipid head group binding.
For Ubq, representative mass spectra for GPC, PS, PC, PE, and PG head groups, are shown in Figure 3. (A nESI spectrum of Ubq with no lipid head group present is shown in Figure S1 for comparison. Replicate data with lipid head groups are shown in Figures S2–S6, illustrating the level of reproducibility of the experiments on different days using different nESI capillaries.) Despite the apparent variability in the extent of head group binding between replicates, which is attributed to the use of different nESI capillaries, GPC and PC consistently display the lowest extent of binding, with up to two bound to Ubq5+ (Figures 3a, 3d, S2, and S3, respectively); PS exhibits the greatest extent of binding, with up to twelve bound to Ubq5+ (Figures 3m, and S4); and PE and PG fall between these extremes (Figures 3g, 3j, S5, and S6). Interestingly, GPC acts as a mild charge reducing reagent under the same instrumental conditions, increasing the relative abundance of Ubq4+ in the raw nESI spectrum as compared to all other head groups studied.
Figure 3.
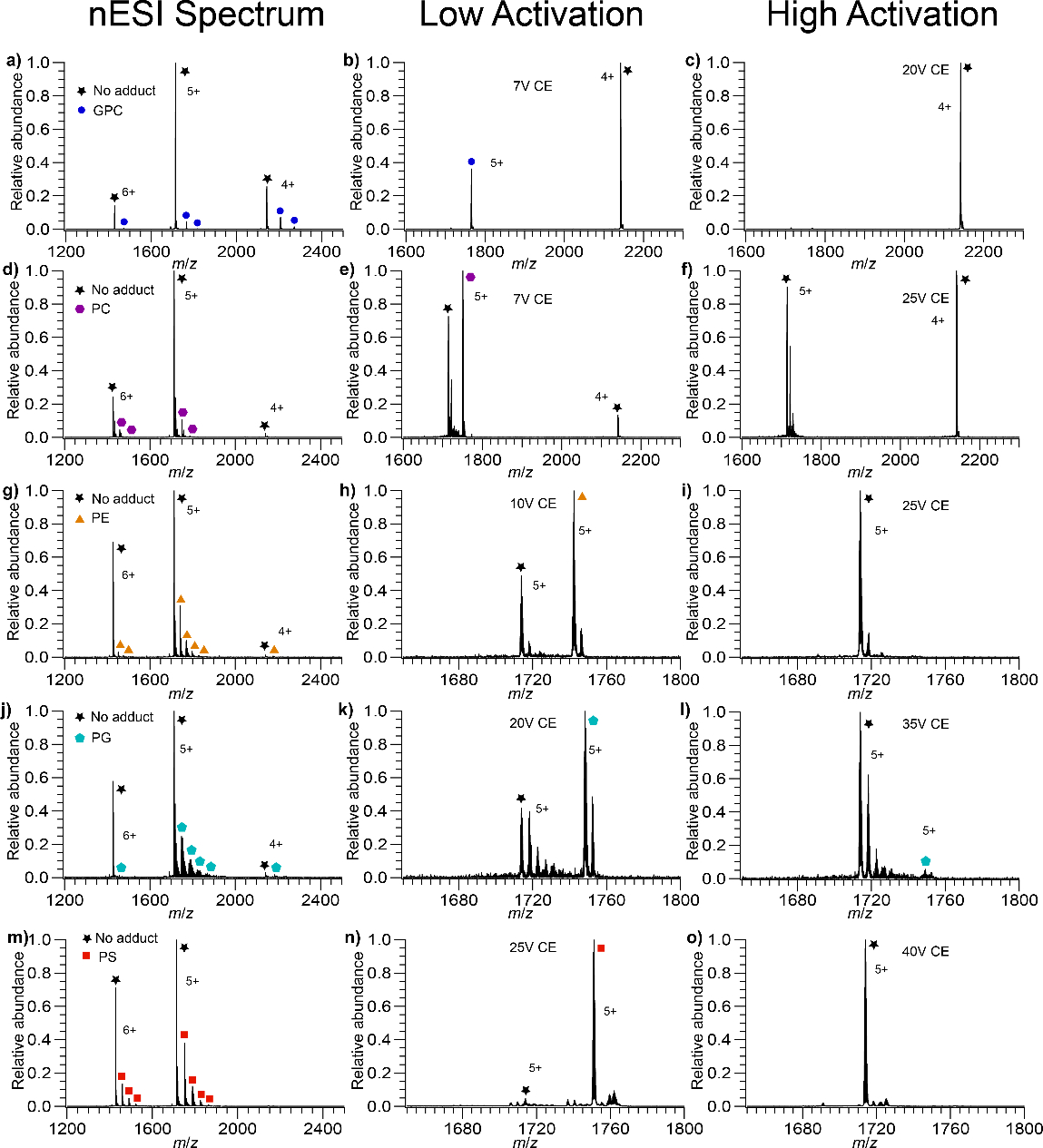
Mass spectra of GPC, PS, PC, PG, and PE bound to Ubq. (a) Ubq with GPC bound, (b) isolated Ubq5+ with one GPC bound at low activation, (c) isolated Ubq5+ with one GPC bound at high activation, (d) Ubq with PC bound, (e) isolated Ubq5+ with one PC bound at low activation, (f) isolated Ubq5+ with one PC bound at high activation, (g) Ubq with PE bound, (h) isolated Ubq5+ with one PE bound at low activation, (i) isolated Ubq5+ with one PE bound at high activation, (j) Ubq with PG bound, (k) isolated Ubq5+ with one PG bound at low activation, (l) isolated Ubq5+ with one PG bound at high activation, (m) Ubq with PS bound, (n) isolated Ubq5+ with one PS bound at low activation, (o) isolated Ubq5+ with one PS bound at high activation. In the isolated spectra at low activation there is a small amount of Ubq with no adducts present, due to a combination of imperfect mass selection (most prominent in the PC spectra) as well as dissociation due to the isolation (most prevalent in the PE and PG spectra). Data from three other replicates for each head group, acquired using different nESI capillaries, are shown in the Supporting Information (Figures S2–S6).
Analogous mass spectra for LZ with each head group are shown in Figures 4 and S7–S11. (A nESI spectrum of LZ with no lipid head group present is shown in Figure S1 for comparison.) Overall, the level of adduction of head group and the ease with which they can be dissociated from LZ follows the same trends as for Ubq. PS, PG, and PE typically adduct to a greater extent than do PC and GPC, and GPC has a slight charge-reducing effect, in this case increasing the relative abundance of LZ6+ in the raw nESI mass spectra. Given the variation in the extent of lipid head group binding observed between the replicates, this overall agreement between the Ubq and LZ data suggests that these trends are relatively robust toward differences in the nESI process (such as initial droplet size or heating experienced during the nESI process) occurring between different nESI capillaries.
Figure 4.
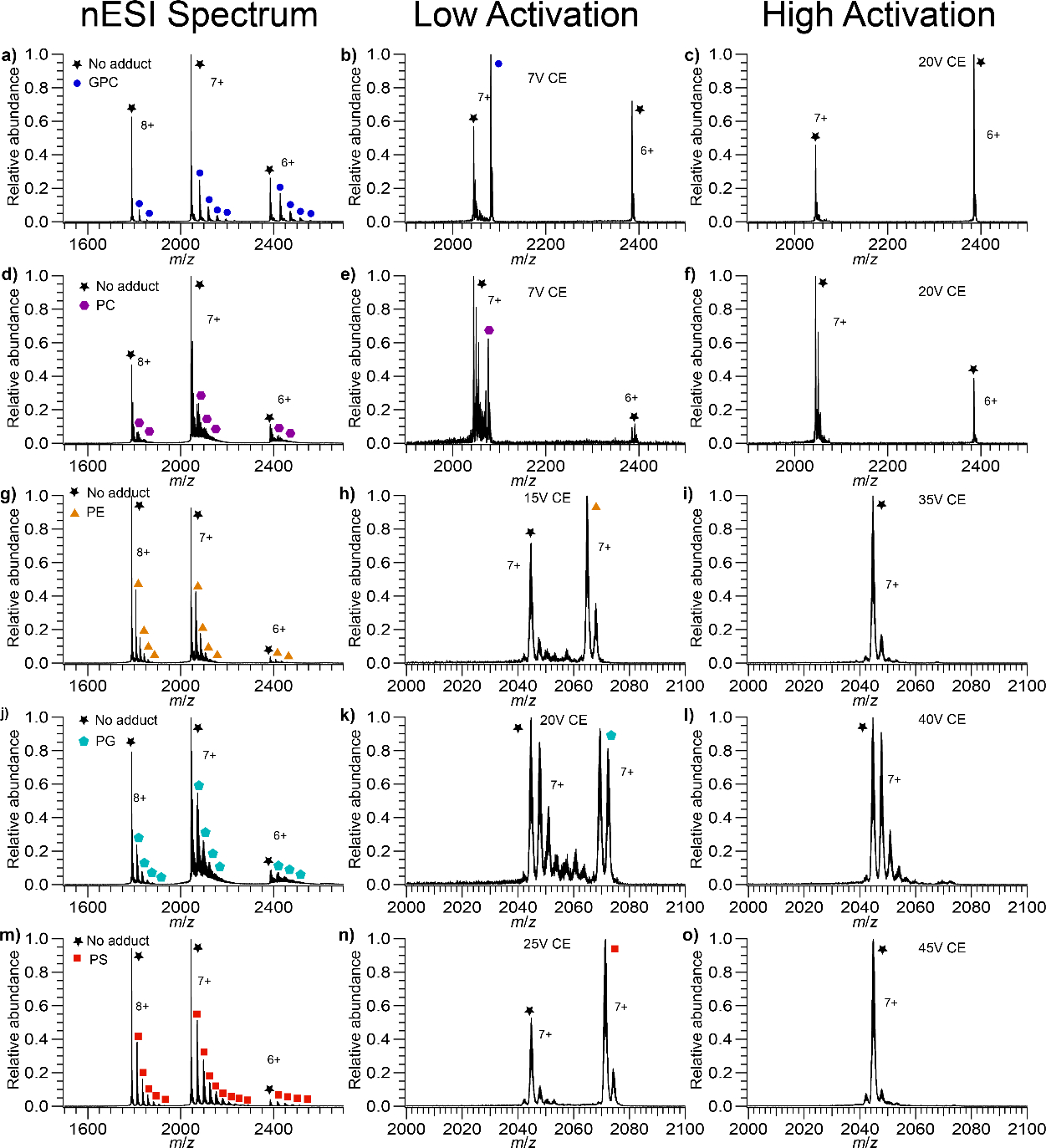
Mass spectra of GPC, PC, PE, PG, and PS bound to LZ. (a) LZ with GPC bound (b) isolated LZ7+ with GPC bound at low activation (c) isolated LZ7+ with GPC bound at high activation (d) LZ with PC bound (e) isolated LZ7+ with PC bound at low activation (f) isolated LZ7+ with PC bound at high activation (g) LZ with PE bound (h) isolated LZ7+ with PE bound at low activation (i) isolated LZ7+ with PE bound at high activation (j) LZ with PG bound (k) isolated LZ7+ with PG bound at low activation (l) isolated LZ7+ with PG bound at high activation (m) LZ with PS bound (n) isolated LZ7+ with PS bound at low activation (o) isolated LZ7+ with PS bound at high activation. PS binds the most extensively, PC the least, with GPC, PE, and PG falling in between. In the isolated spectra at low activation there is some LZ with no adducts, due to a combination of imperfect isolation and dissociation due to the isolation.
CID of protein-head group complexes: proton abstraction.
Thermochemistry of the protein-lipid head group interactions was probed using CID and compared to expectations based on the gas-phase basicity (−ΔG° of protonation at 298 K) of each head group. Our hypothesis was that PC and GPC should be most likely to abstract a proton from Ubq or LZ ions due to the very high GB of the PC head group (exceeding that of the most basic amino acid, arginine), whereas less proton abstraction should be observed for the other, lower-basicity head groups. For each protein and head group combination, CID was performed by scanning the collision voltage in 1 V increments from a minimally-activating voltage to one sufficient to cause complete head group dissociation but not observable covalent bond fragmentation. Isolation mass spectra for the singly-adducted state of each protein are shown in the middle column of Figures 3 (Ubq), 4 (LZ), and S2–S11 (both Ubq and LZ). To account for the presence of some non-adducted Ubq and LZ observed after isolation, the raw fraction of observed precursor in subsequent CID kinetics experiments was divided by this initial fraction of dissociated precursor observed upon isolation. The right-hand columns of Figures 3, 4, and S2–S11 show isolated mass spectra at high activation, indicating that there is a single CID product in all cases except PC. While PE, PG, and PS all dissociate exclusively as neutral species, GPC dissociates as a cation, and PC dissociates predominantly as a cation and to a lesser extent as a neutral. These results agree with those predicted by comparison of GB values of the lipid head groups and basic amino acids [74].
CID of protein-head group complexes: dissociation energies.
In addition to the charge-abstraction capabilities of each head group, the fraction of lipid head group-bound protein ions surviving activation was also studied as a function of CID voltage and compared to predictions based on GB. Seminal experimental and computational results from Johnson and coworkers [80] showed that gas-phase bases with similar GB tend to form stronger shared-proton bonds than do those with very different proton binding energies. Arginine residues are highly basic in the gas phase (GB 1006.6 kJ/mol [67]) and are plentiful at the surface of many native proteins. Thus, it is expected that these residues will carry the majority of positive charges upon nESI. Because PC and GPC have quaternary ammonium groups, they cannot share a proton via these functional groups with residues on the protein surface, thus any shared-proton interactions involving these head groups and a protonated amino acid likely involve the phosphate group. Such an interaction should be neutral, because the positive charge is held by the quaternary ammonium group, in these cases. By contrast, PE and PS have highly basic primary amine groups that can form strong shared-proton interactions with protonated amino acids, and the charge in such interactions is localized there. PG has relatively low basicity phosphoric acid and glycerol groups, thus it can form either neutral or net positively-charged interactions with protonated amino acids. Based on their relative GB and these chemical constraints, we therefore hypothesized that PC and GPC should form relatively weak interactions with protonated protein ions, because the interaction site is likely neutral, whereas PE, PS, and PG can form stronger, net positively-charged, shared-proton bonds with the protein. We further expected that PE and PS should form stronger interactions than PG, based on the higher computed GB of these two head groups (~950 kJ/mol, much closer to that of arginine) as compared to PG (~906 kJ/mol) [74].
For both Ubq and LZ, in each set of trials, the same ranking of interaction strength is observed experimentally, that is, GPC dissociates at the lowest CID voltage, followed closely by PC, then PE, PG, and finally PS (Figures 5 and 6). This ordering is almost identical to the ordering expected based on the above arguments, except that PG binds more tightly than expected. For both proteins, PC and PG adduct mass spectra indicate more concomitant sodium adduction than observed for the other head groups. Increased salt adduction can lead to increases in measured CID energies [81], but the degree of sodiation in the nESI mass spectra did not appear to affect the dissociation behavior of the lipid head groups studied here for either protein, indicating that any effect of increased sodium adduction is likely small. Instead, since the non-specific interactions studied here are formed in the latter stages of the ESI process and may be concurrent with charging of the protein, it is possible that the presence of a significant amount of PG causes alternative charge configurations, i.e., protonation at sites other than arginine, to become favored. This possibility of solution additives affecting the location but not the number of charge sites is intriguing; further studies are necessary to confirm this possibility but are beyond the scope of this report.
Figure 5.
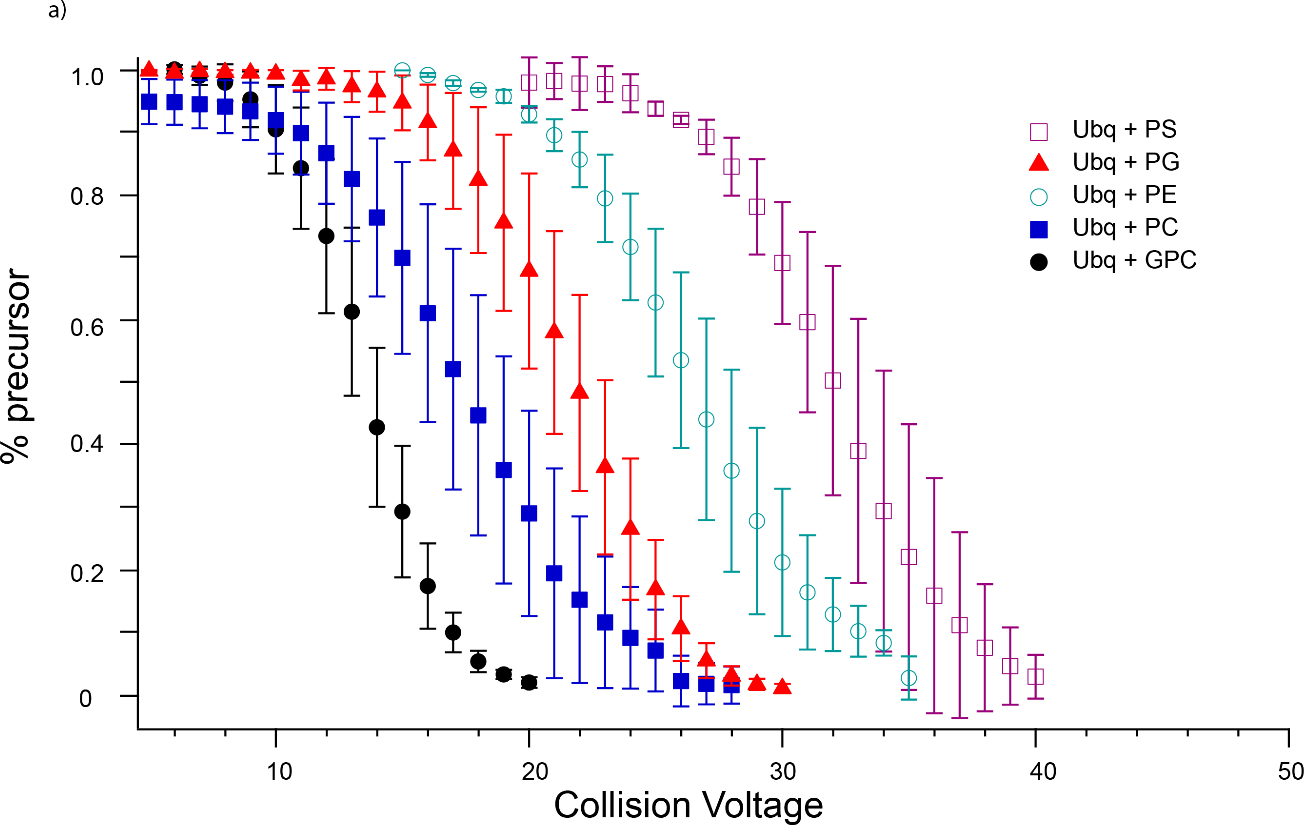
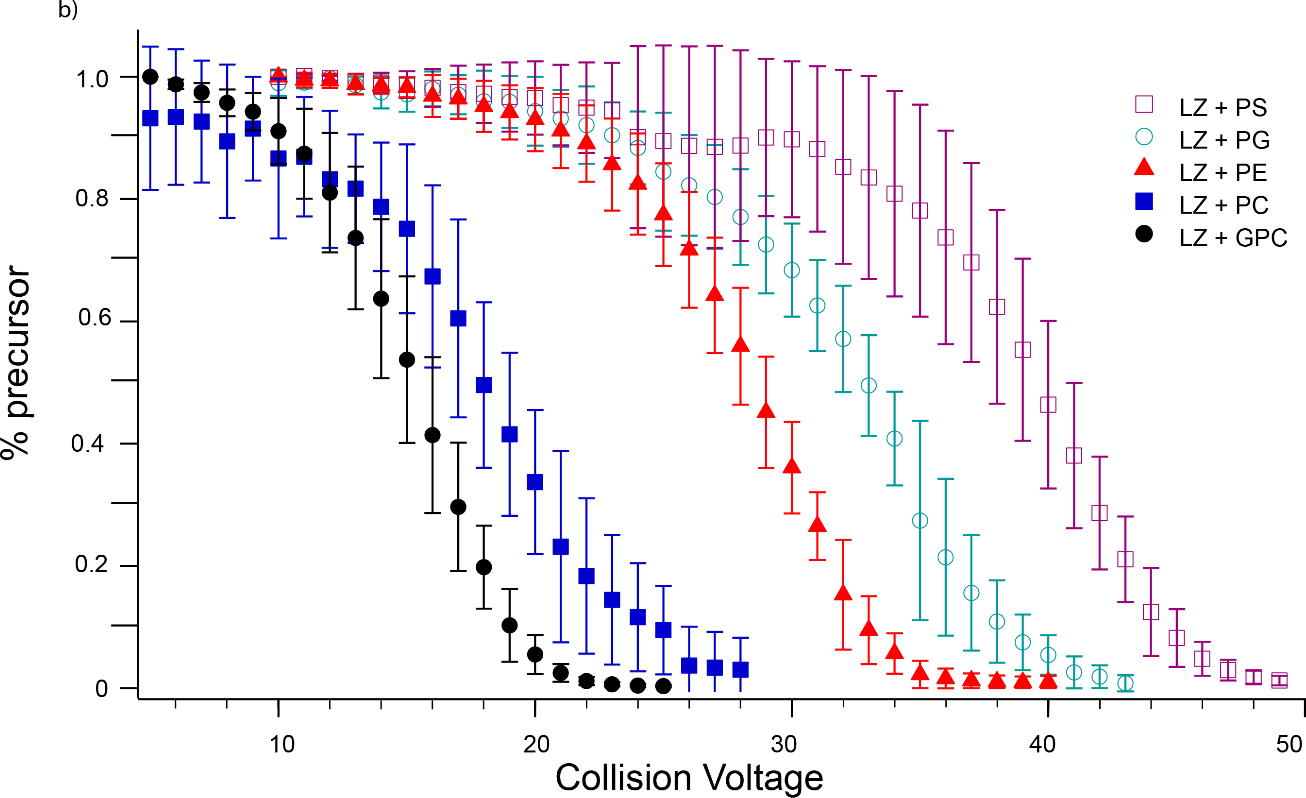
Breakdown curves for dissociation of lipid head groups from (a) Ubq5+ (b) LZ7+. For both proteins, the same trend in lipid head group binding strength is observed, namely PS > PG > PE > PC > GPC. Some dissociation of GPC from Ubq was observed upon isolation, as shown in (a); this was subtracted from CID-induced dissociation in determining barrier thermochemistry values.
Figure 6.
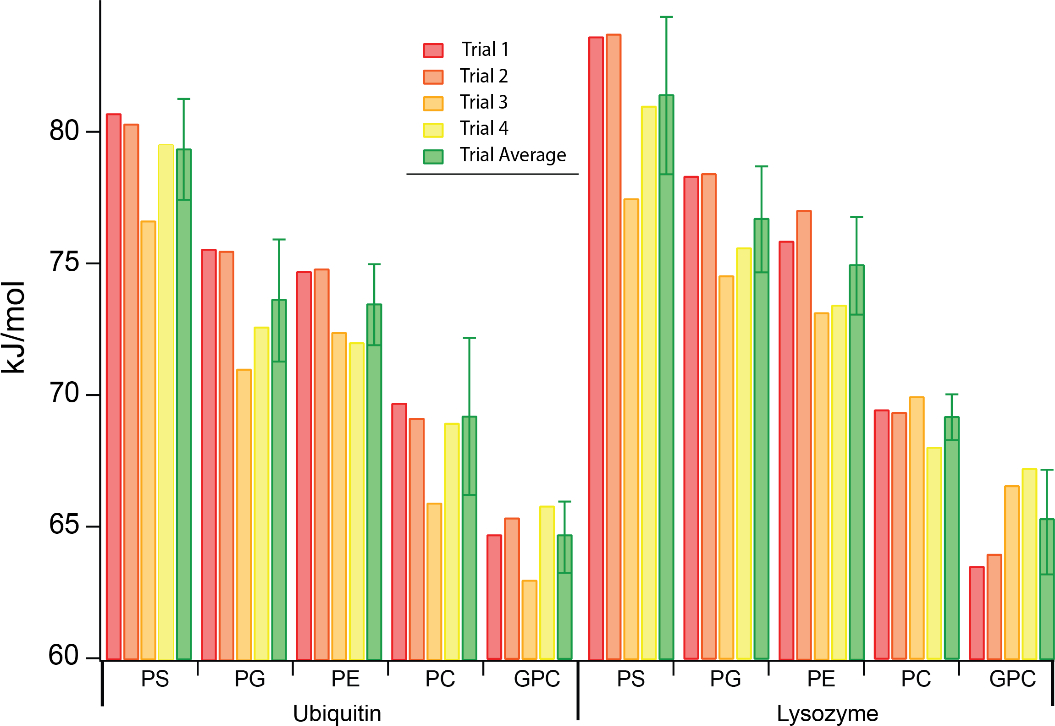
Gibbs free energies of dissociation for lipid head groups bound to Ubq and LZ in CID experiments, along with their average and standard deviation across four replicate experiments performed on different days with different nESI capillaries.
Activation energies for protein-head group CID.
Activation energies were determined using our previously-introduced method for each protein and head group. Kinetic plots as a function of reciprocal effective temperature for Ubq and LZ are shown in Figures S12 and S13, respectively. ΔG‡ for each head group and protein are illustrated in Figure 6, and ΔG‡, ΔH‡, and ΔS‡ values are shown in Table S1. (For precursor ions that exhibited some dissociation upon isolation, this dissociation was subtracted from that produced by subsequent CID activation in deriving barrier thermochemistry values.) Despite variability in the extent of lipid head group binding between replicates in the nESI mass spectra, ΔG‡ values fall between 65 and 81 kJ/mol for all head groups and both proteins and follow the same trend as the midpoint CID voltages of the breakdown curves, i.e., PS > PG > PE > PC > GPC. Good reproducibility was observed between all 4 trials, with a standard deviation of ~1–3 kJ/mol (Figure 6; and Table S1). The highest ΔH‡ measured for both proteins are for GPC and PC (Table S1), likely due to the presence of a reverse Coulomb barrier, as these two lipid head groups dissociate bearing a positive charge.
GPC and PC also have the highest ΔS‡ values, which may be indicative of rearrangement to a relatively large ensemble of conformations following abstraction of charge by GPC/PC. Thus, activation energetics measured by this method agree with trends inferred from breakdown curve data and have the unique further advantage of uncovering subtle differences in the CID process for different head groups, as indicated by differences in ΔS‡ and ΔH‡ that may not be obvious from the appearance of the breakdown curves or the 50% dissociation voltage alone.
Non-specific binding to a larger soluble protein: transferrin.
We also investigated non-specific lipid head group binding to a much larger soluble protein, transferrin (TF, 80 kDa), chosen for its homogenous mass distribution, which facilitates assignment of mass spectral peaks in comparison to other large proteins with multiple isoforms (Figure S14). Mass spectra were first collected under identical instrumental conditions without isolation to assess the overall extent of lipid head group adduction. Figure 7 shows mass spectra for TF with either PS, PE, PG, or GPC head groups bound. (Extensive calcium binding was observed for this protein with PC head group due to the presence of calcium in the PC reagent; PC binding to TF was not explored further due to the very low mass spectral resolution obtained due to this calcium interference.) At a low level of activation (Figure 7a), individual adducts were not resolved for any of the head groups. Increasing the Trap CE to 70 V clearly resolves adducts for all head groups except PG, which is partially resolved (Figure 7c–f). PG is anionic, and sodium counter-ions present in the PG sample adduct to this protein and decrease mass spectral resolution. The extent of binding under these conditions follows the order PS > PE ~ PG > GPC, in agreement with that observed for Ubq and LZ. In addition, at 70 V Trap CE, GPC strips charge from TF, shifting the charge state distribution lower (Figure 7b and S17). This, again, agrees with predictions based solely on the GB of this head group and is in line with the above observations of GPC dissociating with a charge from Ubq and LZ.
Figure 7.
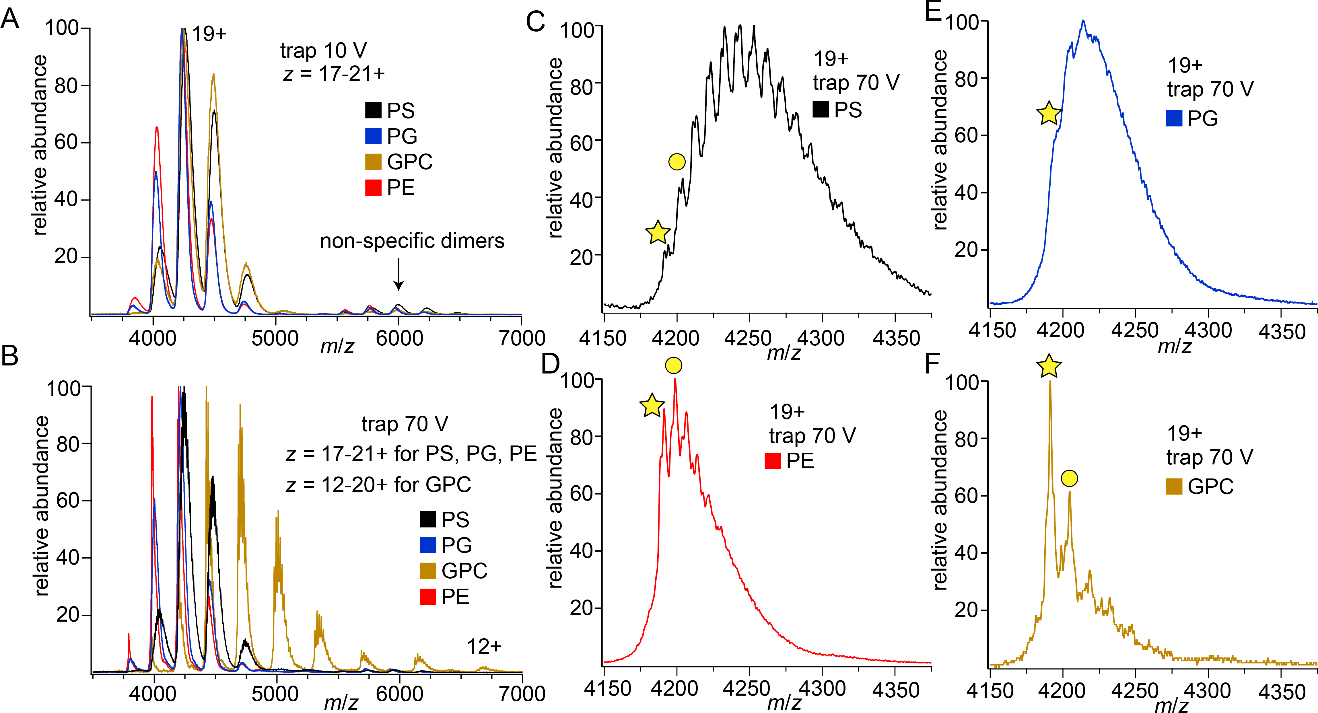
Mass spectra showing lipid head group adduction to TF at (a) low, 10 V and (b) moderate, 70 V levels of Trap CE. With moderate activation GPC has extensively stripped charge from TF. The other panel show TF19+ at 70 V Trap CE for each head group: (c) PS (d) PE (e) PG (f) GPC. Stars identify TF19+ with no adducts, while circles mark the first resolvable head group adduct. Significantly more PS adducts are observed than for the other head groups.
Deconvolved mass spectra at several Trap CE values reveal the relative strength of binding of the lipid head groups to TF (Figure S16), confirming that PS requires the most activation to dissociate. Interestingly, while GPC dissociates readily, the overall number of GPC adducts remains equal to or perhaps slightly higher than the number of PE or PG adducts when the whole charge state distribution is examined (Figure S16b–d). This is attributed to the charge stripping effect of GPC, that is, as GPC adducts dissociate and lower the charge of the remaining protein ion, the Coulombic contribution to the barrier for further dissociation of GPC adducts decreases, allowing some GPC adduction to persist even at high levels of activation. Overall, the results for TF are in excellent agreement with those for Ubq and LZ and further confirm the essential role of GB in lipid head group binding energetics for these proteins.
Competitive head group binding to transferrin.
Ideally, native MS can be used to accurately determine the relative binding strength of lipids to membrane proteins based on the relative abundances observed of different lipid adducts in native mass spectra. In most cases, this is currently done by embedding membrane proteins in detergent micelles or Nanodiscs with at least two different lipid types, and inferring from CID experiments which lipids remain bound after most are dissociated from the initial complexes [13, 17, 22, 24, 82]. Based on the above results for Ubq and LZ, we hypothesized that, for non-specific complexes of TF with adducted lipid head groups formed by nESI from mixtures of the above head groups, PS would form the strongest interactions with TF and survive the greatest level of collisional activation. Figure 8 illustrates competitive head group binding experiments in which identical concentrations (500 μM) of PS and another head group (GPC, PE, and PG) were mixed with TF in ammonium acetate solution and ionized using nESI. At the lowest level of gas-phase activation (Trap potential 20 V), deconvolved mass spectra exhibited a broad, poorly-resolved “hump” for each head group combination, consistent with extensive adduction.
Figure 8.
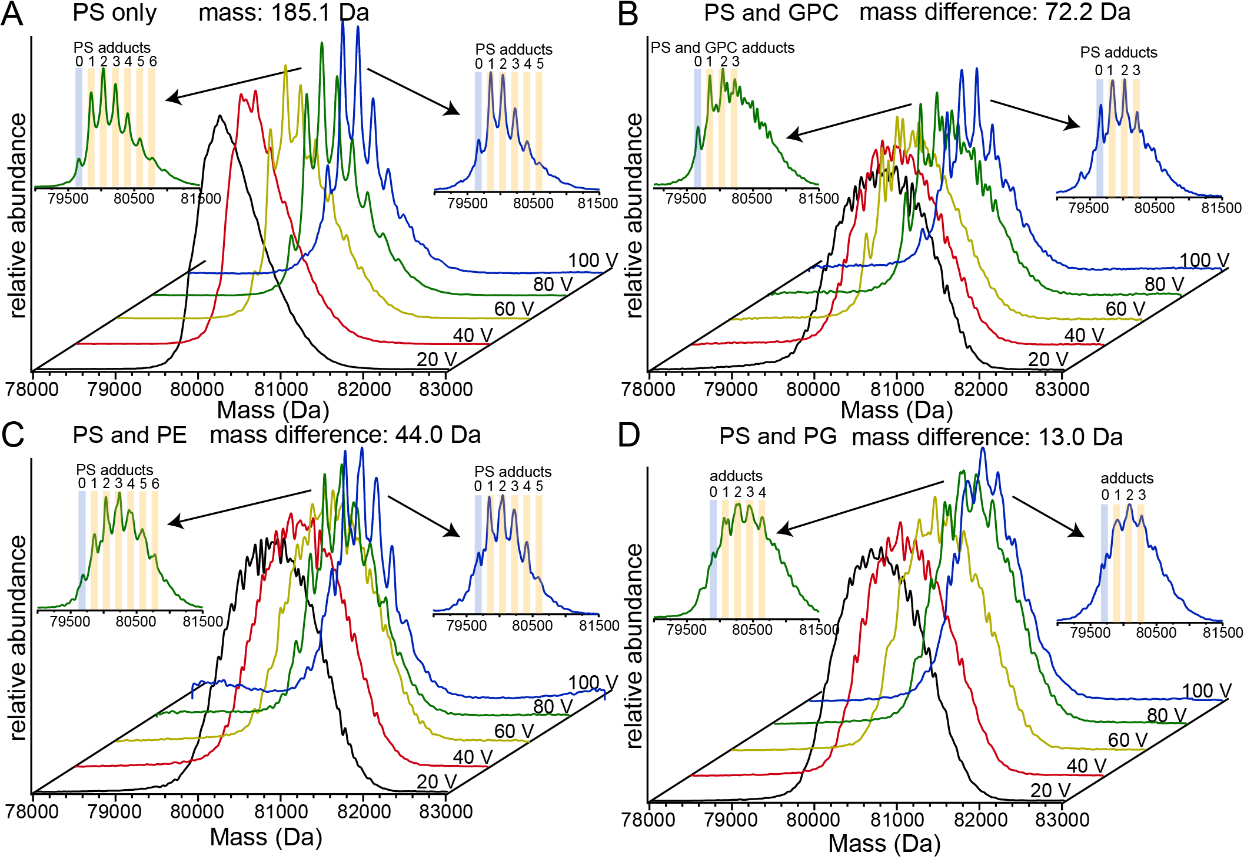
Deconvolved mass spectra of lipid head group binding to transferrin at different levels of collisional activation. (a) PS only (b) competitive binding between PS and GPC (c) competitive binding between PS and PE (d) competitive binding between PS and PG.
Because the resolution of these low-activation TF-adduct mass spectra was poor, the breadth of the deconvolved mass spectra (defined at 10% of maximum intensity) was instead used to characterize the maximum possible extent of binding of each different head group. For PS alone, the deconvolved mass spectrum is ~2,000 Da wide (indicating a maximum of ~11 PS head groups adducted), whereas each of the PS/GPC, PS/PE, and PS/PG mixed head group spectra is ~2,500 Da wide. For each of these competing head group mass spectra, this spectral width indicates a maximum of ~13.5 PS or ~10 GPC, ~21 PE, or ~16 PG adducts, respectively, or combinations of these head groups with fewer of each than these maximal numbers. Upon more extensive collisional activation, the observed width of the deconvolved mass spectra decreases in each case, until at 100 V, moderately well-resolved adduct peaks with a spacing consistent with PS (measured spacing 184 ± 2 Da, as compared to molecular weight 185.07 Da), as well as a spectral width of ~1,500 Da, are observed in each case. This width is consistent with a maximum of ~8 PS, ~6 GPC, 12.5 PE, or ~9 PG adducts. Due to the large mass difference between the PS and GPC head groups (185 and 257 Da, respectively), all of the moderately-well resolved peaks in the PS and PS/GPC spectra at 100 V collisional activation could be unambiguously assigned to PS adducts, and no distinct GPC adduct complexes were clearly resolved. This result indicates that PS binds more strongly to TF than does GPC, consistent with expectations based on the Ubq and LZ experiments described above. The lower mass spectral resolution obtained with PS/PE and PS/PG at 100 V activation, by contrast, likely indicates the presence of both head group types, due to the smaller mass differences between these head groups. For PS/PE, mass spacings consistent with both PS (measured spacing 180 Da ± 10 Da) and PE (measured spacing 144 ± 4 Da, as compared to PE molecular weight 141.06 Da) were both identified by “Double Fourier Transform” of the mass spectrum [78, 79], with the PS peak larger than the PE peak. For PS/PG, mass spacings of 183 ± 4 Da and 175 ± 4 Da were observed by Double Fourier Transform of the mass spectrum, similar to the molecular weights of PS and PG (185.07 Da and 172.07 Da, respectively), with both peaks having similar intensities. Overall, these results are highly consistent with expectations based on the Ubq and LZ results described above as well as head group GB values, indicating a level of adduction after 100 V activation in the order PS ~ PG > PE > GPC. These results illustrate that, at high levels of activation used to remove most head groups, gas-phase binding thermochemistry can play a large role in determining which head groups are robust to dissociation.
Conclusions
Non-specific binding of several model lipid head groups to soluble proteins in positive ion mode was investigated with native MS and CID, including the propensity of these head groups to remove charge upon dissociation and dissociation barrier thermochemistry. PC and GPC head groups have the greatest tendency to remove charge from the proteins studied upon CID, and PS, PE, and PG head groups dissociate exclusively as neutral molecules, in agreement with expectations based solely on the relative gas-phase basicities (GB) of these head groups and those of amino acid residues [67, 74]. PS head group has the highest barrier for dissociation from the proteins studied, followed by PG, PE, PC, and GPC, and these results are robust to variability in the overall extent of head group binding across replicates due to use of different nESI capillaries. These results are largely in agreement with expectations based on relative GB values, in combination with the fact that PC and GPC cannot participate in shared-proton interactions at their permanent charge sites due to the presence of the quaternary ammonium group. However, PG, which might not be expected to form a strong interaction based on its relatively low GB, in fact forms the second strongest interaction.
ΔG‡ values for dissociation of each head group are remarkably similar for Ubq and LZ (<4% difference for the same head group, as compared to a difference of ~20% between the most tightly and most weakly bound head groups) and follow identical trends. Intriguingly, the range of ΔG‡ is relatively narrow (~65–82 kJ/mol) despite a wider range of ΔH‡ (105–159 kJ/mol for Ubq and 84–117 kJ/mol for LZ), indicating a large degree of enthalpy-entropy compensation. In agreement with results for Ubq and LZ, competitive binding experiments for the same lipid head groups to TF, a much larger protein, show that PS forms the strongest non-specific bonds to TF and that the presence of GPC adducts on TF leads to significant charge stripping upon CID. Together, the above results suggest a signature for non-specific binding of lipids to proteins with native-like structures and charge states in positive ion mode: prevalent charge stripping by lipids with phosphocholine head groups (such as phosphatidylcholines and sphingomyelins) upon CID and lipid binding strengths in the order PS > PG > PE > PC.
Adduct migration and evaporation-induced non-specific adduction are both phenomena that must be considered in designing and interpreting native MS experiments to identify physiologically relevant protein adducts such as drug targets, lipids, or cofactors. Because the potential energy surface in the gas phase can be very different from that in solution, slow-heating methods such as CID can in principle result in dissociation behavior that reflects the gas-phase potential energy surface more than that in solution, especially when activation is very extensive. Whether this occurs inherently depends on relative barriers for dissociation as well as the internal temperature of the ions achieved during CID and the timescale of the experiment. The above results provide a benchmark for assessing whether signatures of dissociation behavior dominated by gas-phase thermochemical properties, possibly after adduct migration, may be present in native MS-based lipid binding experiments. In combination with the ion heating/cooling and CID kinetics modeling described here, these results also provide a model for examining effects of gas-phase enthalpy and entropy in protein-ligand interactions using native MS. Furthermore, PC and GPC head groups behave similarly to other reagents used for charge reduction in native MS experiments in positive ion mode [83–85], suggesting they could be useful in this capacity. PS head group, in contrast, may adduct strongly to many native protein ions, potentially enabling deliberate alteration of gas-phase unfolding and dissociation pathways, as observed recently for some covalent protein adducts [86].
Supplementary Material
Highlights.
Phosphoserine, phosphoglycerol, phosphoethanolamine, and (glycero)phosphocholine head groups are deliberately adducted to native-like soluble protein ions
Gas-phase dissociation barrier energetics and charge abstraction behavior upon collisional activation follow expectations based on head group gas-phase basicities and chemical structure
Robust signatures of gas-phase thermochemistry-dominated head group dissociation are identified
Acknowledgements
This work was funded by the National Institute of Health (award R21AI125804). The authors thank Prof. Joseph A. Loo for helpful conversations.
Footnotes
Conflict of Interest Statement
The authors declare no conflict of interest.
Online Supplementary Materials
Additional information about theoretical methods, replicate mass spectrometry data, and barrier thermochemistry tables are available online free of charge at https://journals.elsevier.com/international-journal-of-mass-spectrometry.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Leney AC, Heck AJR, Native Mass Spectrometry: What is in the Name?, J. Am. Soc. Mass Spectrom 28 (2017) 5–13. [DOI] [PubMed] [Google Scholar]
- 2.Marcoux J, Robinson Carol V., Twenty Years of Gas Phase Structural Biology, Structure 21 (2013) 1541–1550. [DOI] [PubMed] [Google Scholar]
- 3.Zhou M, Morgner N, Barrera NP, Politis A, Isaacson SC, Matak-Vinkovic D, Murata T, Bernal RA, Stock D, Robinson CV, Mass Spectrometry of Intact V-Type ATPases Reveals Bound Lipids and the Effects of Nucleotide Binding, Science 334 (2011) 380–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Spoel D, Marklund EG, Larsson DSD, Caleman C, Proteins, Lipids, and Water in the Gas Phase, Macromol. Biosci 11 (2011) 50–59. [DOI] [PubMed] [Google Scholar]
- 5.Yeagle PL, Non-covalent binding of membrane lipids to membrane proteins, Biochim. Biophys. Acta-Biomembr 1838 (2014) 1548–1559. [DOI] [PubMed] [Google Scholar]
- 6.Laganowsky A, Reading E, Allison TM, Ulmschneider MB, Degiacomi MT, Baldwin AJ, Robinson CV, Membrane proteins bind lipids selectively to modulate their structure and function, Nature 510 (2014) 172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poveda JA, Giudici AM, Renart ML, Molina ML, Montoya E, Fernández-Carvajal A, Fernández-Ballester G, Encinar JA, González-Ros JM, Lipid modulation of ion channels through specific binding sites, Biochim. Biophys. Acta 1838 (2014) 1560–1567. [DOI] [PubMed] [Google Scholar]
- 8.Bechara C, Noll A, Morgner N, Degiacomi MT, Tampe R, Robinson CV, A Subset of Annular Lipids is Linked to the Flippase Activity of an ABC Transporter, Biophys. J 108 (2015) 202a. [DOI] [PubMed] [Google Scholar]
- 9.Pliotas C, Dahl ACE, Rasmussen T, Mahendran KR, Smith TK, Marius P, Gault J, Banda T, Rasmussen A, Miller S, Robinson CV, Bayley H, Sansom MSP, Booth IR, Naismith JH, The role of lipids in mechanosensation, Nat. Struct. Mol. Biol 22 (2015) 991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saliba AE, Vonkova I, Gavin AC, The systematic analysis of protein-lipid interactions comes of age, Nat. Rev. Mol. Cell. Biol 16 (2015) 753–761. [DOI] [PubMed] [Google Scholar]
- 11.Hooker BS, Bigelow DJ, Lin CT, Methods for mapping of interaction networks involving membrane proteins, Biochem. Biophys. Res. Commun 363 (2007) 457–461. [DOI] [PubMed] [Google Scholar]
- 12.Barrera NP, Di Bartolo N, Booth PJ, Robinson CV, Micelles protect membrane complexes from solution to vacuum, Science 321 (2008) 243–246. [DOI] [PubMed] [Google Scholar]
- 13.Landreh M, Liko I, Uzdavinys P, Coincon M, Hopper JTS, Drew D, Robinson CV, Controlling release, unfolding and dissociation of membrane protein complexes in the gas phase through collisional cooling, Chem. Commun 51 (2015) 15582–15584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson JW, Rolland AD, Klausen GM, Prell JS, Ion Mobility-Mass Spectrometry Reveals That α-Hemolysin from Staphylococcus aureus Simultaneously Forms Hexameric and Heptameric Complexes in Detergent Micelle Solutions, Anal. Chem 91 (2019) 10204–10211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walker LR, Marzluff EM, Townsend JA, Resager WC, Marty MT, Native Mass Spectrometry of Antimicrobial Peptides in Lipid Nanodiscs Elucidates Complex Assembly, Anal. Chem 91 (2019) 9284–9291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marty MT, Wilcox KC, Klein WL, Sligar SG, Nanodisc-solubilized membrane protein library reflects the membrane proteome, Anal. Bioanal. Chem 405 (2013) 4009–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marty MT, Hoi KK, Gault J, Robinson CV, Probing the Lipid Annular Belt by Gas-Phase Dissociation of Membrane Proteins in Nanodiscs, Angew. Chem.-Int. Edit 55 (2016) 550–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laganowsky A, Reading E, Hopper JTS, Robinson CV, Mass Spectrometry of Intact Membrane Protein Complexes, Nat. Protoc 8 (2013) 639–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrera NP, Isaacson SC, Zhou M, Bavro VN, Welch A, Schaedler TA, Seeger MA, Miguel RN, Korkhov VM, van Veen HW, Venter H, Walmsley AR, Tate CG, Robinson CV, Mass Spectrometry of Membrane Transporters Reveals Subunit Stoichiometry and Interactions, Nat. Methods 6 (2009) 585–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrera NP, Robinson CV, Advances in the Mass Spectrometry of Membrane Proteins: From Individual Proteins to Intact Complexes, Annu. Rev. Biochem 80 (2011) 247–271. [DOI] [PubMed] [Google Scholar]
- 21.Bechara C, Noell A, Morgner N, Degiacomi MT, Tampe R, Robinson CV, A Subset of Annular Lipids is Linked to the Flippase Activity of an ABC Transporter, Nat. Chem 7 (2015) 255–262. [DOI] [PubMed] [Google Scholar]
- 22.Bolla JR, Corey RA, Sahin C, Gault J, Hummer A, Hopper JTS, Lane DP, Drew D, Allison TM, Stansfeld PJ, Robinson CV, Landreh M, A Mass-Spectrometry-Based Approach to Distinguish Annular and Specific Lipid Binding to Membrane Proteins, Angew. Chem.-Int. Edit 59 (2020) 3523–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allison TM, Reading E, Liko I, Baldwin AJ, Laganowsky A, Robinson CV, Quantifying the stabilizing effects of protein–ligand interactions in the gas phase, Nat. Commun 6 (2015) 8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cong X, Liu Y, Liu W, Liang X, Russell DH, Laganowsky A, Determining Membrane Protein–Lipid Binding Thermodynamics Using Native Mass Spectrometry, J. Am. Chem. Soc 138 (2016) 4346–4349. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Cong X, Liu W, Laganowsky A, Characterization of Membrane Protein–Lipid Interactions by Mass Spectrometry Ion Mobility Mass Spectrometry, J. Am. Soc. Mass Spectrom 28 (2017) 579–586. [DOI] [PubMed] [Google Scholar]
- 26.Reading E, Walton Troy A., Liko I, Marty Michael T., Laganowsky A, Rees Douglas C., Robinson Carol V., The Effect of Detergent, Temperature, and Lipid on the Oligomeric State of MscL Constructs: Insights from Mass Spectrometry, Chem. Biol 22 (2015) 593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patrick JW, Boone CD, Liu W, Conover GM, Liu Y, Cong X, Laganowsky A, Allostery revealed within lipid binding events to membrane proteins, Proc. Natl. Acad. Sci. U. S. A 115 (2018) 2976–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y, LoCaste CE, Liu W, Poltash ML, Russell DH, Laganowsky A, Selective binding of a toxin and phosphatidylinositides to a mammalian potassium channel, Nat. Commun 10 (2019) 1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kitova EN, El-Hawiet A, Schnier PD, Klassen JS, Reliable Determinations of Protein–Ligand Interactions by Direct ESI-MS Measurements. Are We There Yet?, J. Am. Soc. Mass Spectrom 23 (2012) 431–441. [DOI] [PubMed] [Google Scholar]
- 30.Li J, Han L, Li J, Kitova EN, Xiong ZJ, Privé GG, Klassen JS, Detecting Protein–Glycolipid Interactions Using CaR-ESI-MS and Model Membranes: Comparison of Pre-loaded and Passively Loaded Picodiscs, J. Am. Soc. Mass Spectrom 29 (2018) 1493–1504. [DOI] [PubMed] [Google Scholar]
- 31.Sun J, Kitova EN, Klassen JS, Method for Stabilizing Protein−Ligand Complexes in Nanoelectrospray Ionization Mass Spectrometry, Anal. Chem 79 (2007) 416–425. [DOI] [PubMed] [Google Scholar]
- 32.Zhang YX, Liu L, Daneshfar R, Kitova EN, Li CS, Jia F, Cairo CW, Klassen JS, Protein-Glycosphingolipid Interactions Revealed Using Catch-and-Release Mass Spectrometry, Anal. Chem 84 (2012) 7618–7621. [DOI] [PubMed] [Google Scholar]
- 33.Eschweiler JD, Kerr R, Rabuck-Gibbons J, Ruotolo BT, Sizing Up Protein–Ligand Complexes: The Rise of Structural Mass Spectrometry Approaches in the Pharmaceutical Sciences, Annu. Rev. Anal. Chem 10 (2017) 25–44. [DOI] [PubMed] [Google Scholar]
- 34.Hyung S-J, Robinson CV, Ruotolo BT, Gas-Phase Unfolding and Disassembly Reveals Stability Differences in Ligand-Bound Multiprotein Complexes, Chem. Biol 16 (2009) 382–390. [DOI] [PubMed] [Google Scholar]
- 35.Niu S, Rabuck JN, Ruotolo BT, Ion mobility-mass spectrometry of intact protein–ligand complexes for pharmaceutical drug discovery and development, Curr. Opin. Chem. Biol 17 (2013) 809–817. [DOI] [PubMed] [Google Scholar]
- 36.Vahidi S, Stocks BB, Konermann L, Partially Disordered Proteins Studied by Ion Mobility-Mass Spectrometry: Implications for the Preservation of Solution Phase Structure in the Gas Phase, Anal. Chem 85 (2013) 10471–10478. [DOI] [PubMed] [Google Scholar]
- 37.Rolland AD, Prell JS, Computational insights into compaction of gas-phase protein and protein complex ions in native ion mobility-mass spectrometry, Trends Anal. Chem 116 (2019) 282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin V, Konermann L, Probing the Effects of Heterogeneous Oxidative Modifications on the Stability of Cytochrome c in Solution and in the Gas Phase, J. Am. Soc. Mass Spectrom 32 (2021) 73–83. [DOI] [PubMed] [Google Scholar]
- 39.Silveira JA, Fort KL, Kim D, Servage KA, Pierson NA, Clemmer DE, Russell DH, From Solution to the Gas Phase: Stepwise Dehydration and Kinetic Trapping of Substance P Reveals the Origin of Peptide Conformations, J. Am. Chem. Soc 135 (2013) 19147–19153. [DOI] [PubMed] [Google Scholar]
- 40.Servage KA, Silveira JA, Fort KL, Russell DH, From Solution to Gas Phase: The Implications of Intramolecular Interactions on the Evaporative Dynamics of Substance P During Electrospray Ionization, J. Phys. Chem. B 119 (2015) 4693–4698. [DOI] [PubMed] [Google Scholar]
- 41.Zhu Y, Roy HA, Cunningham NA, Strobehn SF, Gao J, Munshi MU, Berden G, Oomens J, Rodgers MT, IRMPD Action Spectroscopy ER -CID Experiments, and Theoretical Studies of Sodium Cationized Thymidine and 5-Methyluridine: Kinetic Trapping During the ESI Desolvation Process Preserves the Solution Structure of [Thd+Na], J. Am. Soc. Mass Spectrom 28 (2017) 2423–2437. [DOI] [PubMed] [Google Scholar]
- 42.Hamdy OM, Julian RR, Reflections on charge state distributions, protein structure, and the mystical mechanism of electrospray ionization, J Am Soc Mass Spectrom 23 (2012) 1–6. [DOI] [PubMed] [Google Scholar]
- 43.Breuker K, McLafferty FW, Stepwise evolution of protein native structure with electrospray into the gas phase, 10−12 to 102 s, Proc. Natl. Acad. Sci. U. S. A 105 (2008) 18145–18152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang W, Kitova EN, Klassen JS, Nonspecific Protein−Carbohydrate Complexes Produced by Nanoelectrospray Ionization. Factors Influencing Their Formation and Stability, Anal. Chem 77 (2005) 3060–3071. [DOI] [PubMed] [Google Scholar]
- 45.Wheeler LC, Donor MT, Prell JS, Harms MJ, Multiple Evolutionary Origins of Ubiquitous Cu2+ and Zn2+ Binding in the S100 Protein Family, PLOS ONE 11 (2016) e0164740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Susa AC, Xia Z, Williams ER, Small Emitter Tips for Native Mass Spectrometry of Proteins and Protein Complexes from Nonvolatile Buffers That Mimic the Intracellular Environment, Anal. Chem 89 (2017) 3116–3122. [DOI] [PubMed] [Google Scholar]
- 47.Susa AC, Lippens JL, Xia Z, Loo JA, Campuzano IDG, Williams ER, Submicrometer Emitter ESI Tips for Native Mass Spectrometry of Membrane Proteins in Ionic and Nonionic Detergents, J. Am. Soc. Mass Spectrom 29 (2018) 203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reardon PN, Jara KA, Rolland AD, Smith DA, Hoang HTM, Prell JS, Barbar EJ, The dynein light chain 8 (LC8) binds predominantly “in-register” to a multivalent intrinsically disordered partner, J. Biol. Chem 295 (2020) 4912–4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marcoux J, Wang SC, Politis A, Reading E, Ma J, Biggin PC, Zhou M, Tao H, Zhang Q, Chang G, Morgner N, Robinson CV, Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 9704–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leney AC, Fan X, Kitova EN, Klassen JS, Nanodiscs and Electrospray Ionization Mass Spectrometry: A Tool for Screening Glycolipids Against Proteins, Anal. Chem 86 (2014) 5271–5277. [DOI] [PubMed] [Google Scholar]
- 51.Harvey SR, Liu Y, Liu W, Wysocki VH, Laganowsky A, Surface induced dissociation as a tool to study membrane protein complexes, Chem. Commun 53 (2017) 3106–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chorev DS, Tang H, Rouse SL, Bolla JR, von Kügelgen A, Baker LA, Wu D, Gault J, Grünewald K, Bharat TAM, Matthews SJ, Robinson CV, The use of sonicated lipid vesicles for mass spectrometry of membrane protein complexes, Nat. Protoc 15 (2020) 1690–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chorev DS, Baker LA, Wu D, Beilsten-Edmands V, Rouse SL, Zeev-Ben-Mordehai T, Jiko C, Samsudin F, Gerle C, Khalid S, Stewart AG, Matthews SJ, Grünewald K, Robinson CV, Protein assemblies ejected directly from native membranes yield complexes for mass spectrometry, Science 362 (2018) 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iavarone AT, Udekwu OA, Williams ER, Buffer loading for counteracting metal salt-induced signal suppression in electrospray ionization, Anal. Chem 76 (2004) 3944–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anacleto JF, Pleasance S, Boyd RK, Calibration of ion spray mass spectra using cluster ions, Org. Mass Spectrom 27 (1992) 660–666. [Google Scholar]
- 56.Donor MT, Ewing SA, Zenaidee MA, Donald WA, Prell JS, Extended Protein Ions Are Formed by the Chain Ejection Model in Chemical Supercharging Electrospray Ionization, Anal. Chem 89 (2017) 5107–5114. [DOI] [PubMed] [Google Scholar]
- 57.Jones JL, Dongre AR, Somogyi A, Wysocki VH, Sequence Dependence of Peptide Fragmentation Efficiency Curves Determined by Electrospray Ionization/Surface-Induced Dissociation Mass Spectrometry, J. Am. Chem. Soc 116 (1994) 8368–8369. [Google Scholar]
- 58.Wysocki VH, Tsaprailis G, Smith LL, Breci LA, Mobile and localized protons: a framework for understanding peptide dissociation, J. Mass Spectrom 35 (2000) 1399–1406. [DOI] [PubMed] [Google Scholar]
- 59.Shelimov KB, Clemmer DE, Hudgins RR, Jarrold MF, Protein Structure in Vacuo: Gas-Phase Conformations of BPTI and Cytochrome c, J. Am. Chem. Soc 119 (1997) 2240–2248. [Google Scholar]
- 60.Paizs B, Suhai S, Fragmentation pathways of protonated peptides, Mass Spectrom. Rev 24 (2005) 508–548. [DOI] [PubMed] [Google Scholar]
- 61.Polfer NC, Oomens J, Suhai S, Paizs B, Infrared Spectroscopy and Theoretical Studies on Gas-Phase Protonated Leu-enkephalin and Its Fragments: Direct Experimental Evidence for the Mobile Proton, J. Am. Chem. Soc 129 (2007) 5887–5897. [DOI] [PubMed] [Google Scholar]
- 62.Wyttenbach T, Paizs B, Barran P, Breci L, Liu D, Suhai S, Wysocki VH, Bowers MT, The Effect of the Initial Water of Hydration on the Energetics, Structures, and H/D Exchange Mechanism of a Family of Pentapeptides: An Experimental and Theoretical Study, J. Am. Chem. Soc 125 (2003) 13768–13775. [DOI] [PubMed] [Google Scholar]
- 63.Bush MF, O’Brien JT, Prell JS, Saykally RJ, Williams ER, Infrared Spectroscopy of Cationized Arginine in the Gas Phase: Direct Evidence for the Transition from Nonzwitterionic to Zwitterionic Structure, J. Am. Chem. Soc 129 (2007) 1612–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oh H-B, Lin C, Hwang HY, Zhai H, Breuker K, Zabrouskov V, Carpenter BK, McLafferty FW, Infrared Photodissociation Spectroscopy of Electrosprayed Ions in a Fourier Transform Mass Spectrometer, J. Am. Chem. Soc 127 (2005) 4076–4083. [DOI] [PubMed] [Google Scholar]
- 65.Chang TM, Prell JS, Warrick ER, Williams ER, Where’s the Charge? Protonation Sites in Gaseous Ions Change with Hydration, J. Am. Chem. Soc 134 (2012) 15805–15813. [DOI] [PubMed] [Google Scholar]
- 66.Steill JD, Oomens J, Gas-Phase Deprotonation of p-Hydroxybenzoic Acid Investigated by IR Spectroscopy: Solution-Phase Structure Is Retained upon ESI, J. Am. Chem. Soc 131 (2009) 13570–13571. [DOI] [PubMed] [Google Scholar]
- 67.Hunter EPL, Lias SG, Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update, J. Phys. Chem. Ref. Data 27 (1998) 413–656. [Google Scholar]
- 68.Morris RA, Knighton WB, Viggiano AA, Hoffman BC, Schaefer Iii HF, The gas-phase acidity of H3PO4, J. Chem. Phys 106 (1997) 3545–3547. [Google Scholar]
- 69.Beardsley RL, Jones CM, Galhena AS, Wysocki VH, Noncovalent Protein Tetramers and Pentamers with “n” Charges Yield Monomers with n/4 and n/5 Charges, Anal. Chem 81 (2009) 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benesch JLP, Aquilina JA, Ruotolo BT, Sobott F, Robinson CV, Tandem Mass Spectrometry Reveals the Quaternary Organization of Macromolecular Assemblies, Chem. Biol 13 (2006) 597–605. [DOI] [PubMed] [Google Scholar]
- 71.Wanasundara SN, Thachuk M, Toward an Improved Understanding of the Dissociation Mechanism of Gas Phase Protein Complexes, J. Phys. Chem. B 114 (2010) 11646–11653. [DOI] [PubMed] [Google Scholar]
- 72.Gordon SM, Reid NW, An investigation of the kinetic shift in mass spectrometry, Int. J. Mass Spectrom. Ion Phys 18 (1975) 379–391. [Google Scholar]
- 73.Donor MT, Shepherd SO, Prell JS, Rapid Determination of Activation Energies for Gas-Phase Protein Unfolding and Dissociation in a Q-IM-ToF Mass Spectrometer, J. Am. Soc. Mass Spectrom 31 (2020) 602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miller ZM, Zhang JD, Donald WA, Prell JS, Gas-Phase Protonation Thermodynamics of Biological Lipids: Experiment, Theory, and Implications, Anal. Chem 92 (2020) 10365–10374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Haynes SE, Polasky DA, Dixit SM, Majmudar JD, Neeson K, Ruotolo BT, Martin BR, Variable-Velocity Traveling-Wave Ion Mobility Separation Enhancing Peak Capacity for Data-Independent Acquisition Proteomics, Anal. Chem 89 (2017) 5669–5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marty MT, Baldwin AJ, Marklund EG, Hochberg GKA, Benesch JLP, Robinson CV, Bayesian Deconvolution of Mass and Ion Mobility Spectra: From Binary Interactions to Polydisperse Ensembles, Anal. Chem 87 (2015) 4370–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cleary SP, Thompson AM, Prell JS, Fourier Analysis Method for Analyzing Highly Congested Mass Spectra of Ion Populations with Repeated Subunits, Anal. Chem 88 (2016) 6205–6213. [DOI] [PubMed] [Google Scholar]
- 78.Hoi KK, Robinson CV, Marty MT, Unraveling the Composition and Behavior of Heterogeneous Lipid Nanodiscs by Mass Spectrometry, Anal. Chem 88 (2016) 6199–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cleary SP, Prell JS, Distinct classes of multi-subunit heterogeneity: analysis using Fourier Transform methods and native mass spectrometry, Analyst 145 (2020) 4688–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roscioli JR, McCunn LR, Johnson MA, Quantum Structure of the Intermolecular Proton Bond, Science 316 (2007) 249. [DOI] [PubMed] [Google Scholar]
- 81.Freeke J, Robinson CV, Ruotolo BT, Residual counter ions can stabilise a large protein complex in the gas phase, Int. J. Mass Spectrom 298 (2010) 91–98. [Google Scholar]
- 82.Landreh M, Costeira-Paulo J, Gault J, Marklund EG, Robinson CV, Effects of Detergent Micelles on Lipid Binding to Proteins in Electrospray Ionization Mass Spectrometry, Anal. Chem 89 (2017) 7425–7430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kaldmäe M, Österlund N, Lianoudaki D, Sahin C, Bergman P, Nyman T, Kronqvist N, Ilag LL, Allison TM, Marklund EG, Landreh M, Gas-Phase Collisions with Trimethylamine-N-Oxide Enable Activation-Controlled Protein Ion Charge Reduction, J Am Soc Mass Spectrom 30 (2019) 1385–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Townsend JA, Keener JE, Miller ZM, Prell JS, Marty MT, Imidazole Derivatives Improve Charge Reduction and Stabilization for Native Mass Spectrometry, Anal. Chem 91 (2019) 14765–14772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lemaire D, Marie G, Serani L, Laprévote O, Stabilization of Gas-Phase Noncovalent Macromolecular Complexes in Electrospray Mass Spectrometry Using Aqueous Triethylammonium Bicarbonate Buffer, Anal. Chem 73 (2001) 1699–1706. [DOI] [PubMed] [Google Scholar]
- 86.Polasky DA, Dixit SM, Keating MF, Gadkari VV, Andrews PC, Ruotolo BT, Pervasive Charge Solvation Permeates Native-like Protein Ions and Dramatically Influences Top-down Sequencing Data, J. Am. Chem. Soc 142 (2020) 6750–6760. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.