

Abstract
Progressive accumulation of aggregation-prone proteins, amyloid-β (Aβ) and hyperphosphorylated tau (p-tau), are the defining hallmarks of Alzheimer’s disease (AD). The mechanisms by which Aβ and p-tau are transmitted throughout the diseased brain are not yet completely understood. Interest in exosome research has grown dramatically over the past few years, specifically due to their potential role as biomarkers for staging of neurodegenerative diseases, including AD. Despite their diagnostic utility, the pathogenic potential of exosomes has yet to be fully elucidated. In this study, we use a series of recombinant tau antibodies to characterize a new model of human tau in vivo. Exosome suspensions derived from neuronally-differentiated, human induced pluripotent stem cells that express the repeat domain of tau P301L and V337M mutations (NiPSCEs) were injected into the wild-type mouse brain and pathological changes were characterized by immunostaining at one- (1 m) and two-month (2 m) post-injection. We found that tau inclusions were present throughout the brain at 2m post-injection, which were detectable using antibodies raised against full-length tau (K9JA) and misfolded tau (MC1). Furthermore, we found that phosphorylated tau immunoreactivity was elevated 1m post-injection, which was surprisingly normalized after 2m. Finally, we observed extensive degeneration of neuronal dendrites in both ipsilateral and contralateral hippocampi in NiPSCE treated mice. In summary, we demonstrate that exosomes are sufficient to cause long-distance propagation of tau pathology and neurodegeneration in vivo. These novel findings support an active role of exosomes in AD pathogenesis.
Keywords: Alzheimer’s disease, exosomes, induced pluripotent stem cells, tau propagation
INTRODUCTION
Progressive accumulation of specific aggregation-prone proteins is the defining hallmark of many neurodegenerative diseases including Alzheimer’s disease (AD). AD is characterized by the accumulation of amyloid-β (Aβ) and hyperphosphorylated tau (p-tau) protein. P-tau exhibits reduced ability to bind and stabilize microtubules. Destabilized microtubules impair synaptic transmission, while the mis-localized p-tau aggregates form insoluble paired helical filaments (PHFs) that form neurofibrillary (tau) tangles. Although tau is known to be primarily intracellular, studies suggest that tau aggregates are secreted and can propagate from neuron to neuron in a prion-like manner under pathological conditions [1–3]. The spreading of tau in a “prion-like” manner has long been known to occur in human AD brains. First described by Braak and Braak [4], this spatially consistent pattern begins in the entorhinal cortex (EC), propagates to the hippocampus via the perforant pathway [5], and eventually culminates in the primary visual cortex. The mechanisms by which p-tau is transmitted throughout the diseased brain are not completely understood. Aβ can be trafficked transsynaptically between neurons suggests that tau and other aggregation-prone proteins can spread in a similar manner [6], potentially via neuronally-derived exosomes.
Exosomes are extracellular vesicles released by the fusion of endocytic multivesicular bodies with the plasma membrane [7]. Once released different, exosomes can mediate intercellular communication by fusing to recipient cells and depositing cargo into the intracellular space [8, 9]. Exosomes can activate intracellular signaling without ligand-receptor internalization or they can undergo endocytic mechanisms (phagocytosis or receptor-mediated endocytosis) to release their contents into the target cells. Alternatively, exosomes can be enzymatically perforated, resulting in the release of their content into the extracellular space. Subsequently, exosome cargo can bind to cell surface receptors on target cells to activate intracellular signaling [10].
Recent advances in the isolation and characterization of neuronally derived, plasma-based exosomes (NDEs) have enhanced their utility as diagnostic biomarkers [11–15]. We recently demonstrated that elevated levels of AD-related proteins contained within plasma NDEs, including p-tau and Aβ42, accurately predicted the conversion of mild cognitive impairment to AD [16]. Additional studies have demonstrated that plasma NDEs have the ability to differentiate between cognitively normal controls from patients with AD [11], frontotemporal dementia [17], Down syndrome [13], and acute brain injury [12]. Despite the diagnostic utility of plasma NDEs, the pathological consequence of NDE cargo proteins has yet to be fully determined. A number of in vitro models have been developed to study tau propagation; however, few animal models have been developed due to the lengthy time course required to observe tau propagation in the rodent brain [18].
Recently, we utilized a human neuronal model to characterize neuropathological changes of exosome cargo proteins in the normal mouse brain [19]. In our neuronal model, human induced pluripotent stem cells (hiPSCs)-derived neurons expressed the tau repeat domain (tau-RD) with P301L and V337M mutations fused to a YFP reporter (tau-RD-LM-YFP) [20]. Our previous work demonstrated that exosomes could cause tau deposition in mouse brain tissue.
However, the effects on widespread tau propagation were not addressed. In the current study, we use a series of recombinant tau antibodies to investigate and characterize a new model of human tau (h-tau) propagation in vivo.
METHODS
Generation of tau-RD-LM-YFP seeds from the conditioned media of human iPSC cells
Neuronal cultures derived from non-demented controls (NDC) were generated as previously described [21]. Neural stem cells (NSCs) were seeded at a density of 150,000 cells/cm2 on Matrigel-coated (70μg/mL) plastic cell culture dishes. NSCs were grown to 80% confluency, at which time neuronal differentiation was initiated through withdrawal of fibroblast growth factor (bFGF) from the NSC media (DMEM-12,1% N-2, 2%, B-27, Pen-Strep, 20 ng/mL bFGF). The iPSC line from which was NSCs were derived was described previously (see “NDC1” in [22]). Four-week-differentiated neuronal cultures were transduced with tau-RD-LM-YFP or YFP adenovirus vectors and incubated for 24 h. After the 24 h incubation, media was aspirated, cells were washed with PBS, and fresh media was applied to ensure removal of virus. Three days post-transduction, 2mL of fresh neuronal media was added to each well and the cells incubated for 3 additional days. Tau-RD-LM-YFP conditioned media was collected and centrifuged at 1000 RPM for 5 min at 4°C to remove cell debris.
Isolation of neuronally differentiated induced pluripotent stem cell-derived exosomes (NiPSCEs) from cell culture media
2mL of cell culture media were incubated with 0.4mL ExoQuick™-TC (EXOTCxxA-1, System Biosciences, Inc.) rotating overnight at 4°C. After centrifugation at 1500×g for 30 min at 4°C, supernatant was collected, and the resultant pellet was suspended in 300 μL of 1x PBS with protease and phosphatase inhibitor cocktail EDTA-free and stored at −80°C.
Characterization of exosomes preparations by size and western blot
Nanoparticle tracking analysis (NTA) was used to characterize NiPSCEs derived from cell culture media. NiPSCEs were pooled in 100 μl of 1x PBS, diluted 1:2,000 and visualized with a NanoSight LM10 instrument as described [16].
2μg of NiPSCEs was diluted with 2x Laemmli sample buffer (161–0737, BioRad) and resolved on 4–20% Mini-PROTEAN™ TGX™ Precast Protein Gel (456–1096, BioRad). Protein was transferred to polyvinylidene difluoride (PVDF) membranes using iBlot Semi-Dry transfer cell (IB25001, Invitrogen, ThermoFisher Scientific). Blots were blocked in 5% bovine serum albumin (BSA) in tris-buffered saline (TBS) with 0.1% Tween-20 (TBST) at room temperature for 1hr on a platform shaker. Primary antibodies were applied overnight at 4°C in blocking solution. The primary antibodies used were paired helical filament 1 (PHF1) (1:500; gift from Dr. Peter Davies); MC-1 (mouse anti-human tau; raised against the N-terminus (amino acids 7–9), and aa 313–322 in the third microtubule binding domain. MC1 does not react with FAC1 [23]; 1:100, gift from Dr. Peter Davies); and Purified Mouse Anti-Flotillin-1 (exosome surface marker; raised against mouse flotillin-1 aa. 312–428, cross reacts with human flotillin-1; 1:100, 610821, BD Transduction Laboratories, San Jose, CA). Following overnight incubation, blots were washed 3x in 1x TBST then incubated in horseradish peroxidase conjugated secondary antibodies (anti-mouse, 1:1000; Thermo-Fisher). Enhanced chemiluminescence (ECL) substrate was applied before visualization by GelDoc (BioRad). Blot analysis was performed on ImageLab 5.2.1 (BioRad).
Exosome injection, tissue preparation, immunohistochemistry, and image analysis
2μL (0.5μg) of exosome preparations was injected into the right hippocampus of wild-type, female, C57BL/6 mice (n=5/group, 3–4 month old). Female mice were exclusively utilized in this study because epidemiological reports in humans [24] and scientific reports in transgenic mice [23, 25] have demonstrated the risk of AD is higher in females than in males.
Briefly, mice were placed under anesthesia on a Koft stereotaxic apparatus and coordinates (hippocampus: AP −2.0 mm, lateral 1.5 mm, depth 1.3 mm) were determined using a Franklin and Paxinos Atlas, as previously described [26]. Exosome preparations were administered using a Hamilton syringe connected to a hydraulic system to inject the solution at a rate of 1 μl over 2 min as previously described [15, 16]. To allow diffusion of the solution into the brain tissue, the needle was left for an additional 5 min after the completion of the injection. Mice received unilateral injections (right side) to allow comparisons against the contralateral side. Additional controls were performed by injecting wild-type littermates with 1x PBS (n=5). Mice were 3–4 months at the time of the injection and maintained for 1m and 2m after exosome injection.
Mice were sacrificed following NIH guidelines. Briefly, all mice were deeply anesthetized with ketamine (100 mg/kg)/xylazine (10 mg/kg) and then decapitated following transcardial perfusion with 0.9% saline. Whole brains were post-fixed for 24 h in 4% phosphate-buffered paraformaldehyde (pH 7.4) at 4°C and 30μM-thick, coronal sections were cut on a freezing-sliding microtome. Mice sections were stored at −20°C in cryoprotectant solution containing 20% glycerol and 30% ethylene glycol in 0.1 m phosphate buffer until use.
Analysis of exosome-mediated tau propagation was performed in serially sectioned, free-floating and blind coded sections. Sections were incubated overnight at 4°C with, K9JA (rabbit anti-human total tau; raised against C-terminal of human tau, amino acids 243–441; 1:32,000; DAKO A-0024); PHF1 (mouse anti-phospho tau, S396/404; 1:500; gift from Dr. Peter Davies); and MC-1 (mouse anti-human tau; raised against the N-terminus (amino acids 7–9), and aa 313–322 in the third microtubule binding domain. MC1 does not react with FAC1 [23]; 1:100, gift from Dr. Peter Davies) followed by anti-rabbit or anti-mouse IgG1 secondary antibodies (1:100, Vector Laboratories, Inc.), and visualized with diaminobenzidine (DAB). The mounted slices were imaged via Leica DMi8 (Leica Microsystems) at 10x magnification and stitched together using Leica Application Suite X. Quantification of positively stained cells was performed using ImageJ (NIH) Cell Counter plug-in (Kurt De Vos, University of Sheffield).
Double-label immunofluorescence was conducted using sections immunolabeled overnight with antibodies against the dendritic marker microtubule-associated protein-2 (MAP2; 1:500, (AB5622), Millipore) and PHF1. Sections were visualized with FITC-tagged secondary antibody or the Tyramide Signal Amplification Direct (Red) system (1:100, NEL702001, Perkin Elmer), mounted under glass coverslips with anti-fading media (H-1000, Vector Laboratories), and imaged with the laser scanning confocal microscope (MRC1024, BioRad Laboratories).
Quantification of dendritic blebbing
Brain tissue from control and NiPSCE treated mice was fixed, sectioned and immunostained with MAP2 (1:500, AB5622, Millipore) and FITC-conjugated secondary antibody (anti-mouse, 1:100, FI200, Vector Laboratories); mounted under glass coverslips with anti-fading media (Vector Laboratories); and imaged with the laser scanning confocal microscope (MRC1024, BioRad Laboratories). To quantify dendritic blebbing, dendrites were traced and measured using ImageJ (NIH) and the number of blebs were counted. Data are reported as the average number of blebs/length of dendrite. Analysis of dendritic blebbing was performed under blinded conditions (one lab member imaged sections while a second member performed the blinded analysis).
Statistical analysis
All experiments were done blind by coding the specimen. Values in the figures are expressed as means±SEM. Statistical significance was determined between control, 1m and 2m mouse groups using one-way ANOVA with Bonferonni post-hoc test (GraphPad Prism 6, La Jolla, CA).
RESULTS
Human tau delivered by exosomes derived from human tau-RD-LM-YFP conditioned media propagated in normal mouse brain
We previously showed transfer of human tau to the recipient mouse neurons when exosomes derived from human iPSC neuronal cultures expressing tau-RD-LM-YFP (NiPSCE-tau-RD-LM-YFP) were injected into mouse brain [19]. Here, we expanded on our previous findings to investigate whether or not exosomes are sufficient to cause tau propagation in vivo. NTA and immunoblot were conducted to characterize exosome preparations prior to injection. NTA demonstrated the size distribution of the NiPSCE-tau-RD-LM-YFP (referred to as NiPSCE from this point on) preparation derived from tau-RD-LM-YFP conditioned media were similar in size and shape to our previous study [19] (Fig. 1A). We reported previously that tau-RD-LM-YFP derived exosomes contain tau, which was detectable with K9JA antibodies that recognize the C-terminus of human tau, amino acids 243–441. In order to determine whether or not these exosomes also contain modified forms of tau, we probed exosomes lysates with antibodies that recognize pathogenically phosphorylated (PHF1) and misfolded (MC-1) tau (Fig. 1B). Both forms of tau were detectable in exosomes but not in nonexosomal fractions. The non-exo fraction is defined as the supernatant fraction which was collected following exosome precipitation. The exosome surface marker flotilin-1 was used as a loading control during immunoblot.
Fig. 1.

Exosomes derived from neuronally-differentiated, human induced pluripotent stem cells (NiPSCEs) contain human tau species. Representative NTA plot of averaged size/concentration for exosomes derived from tau-RD-LM-YFP conditioned media from induced pluripotent stem cells (NiPSCEs) (A). Non-exosome (non-exo) and exosomal (exo) fractions were probed against the human recombinant tau protein using MC-1 and PHF1 antibodies. Representative western blots demonstrate that MC-1 and PHF1 immunoreactive tau is only detected in the exosomal fraction derived from NiPSCEs. Exosome surface marker, Flotillin-1, was used as a loading control (B).
Next, NiPSCE preparations were stereotactically injected into the right hippocampus of 3–4-month-old female C57/Bl6 mice. Female mice were used for this study due to the finding that female mice expressing mutated forms of tau display a more robust pathological profile than male counterparts [23, 25]. 1m or 2m post-injection mouse brains were sectioned and stained with a series of recombinant tau antibodies to characterize tau pathology across the whole mouse brain. A separate cohort of female C57/Bl6 mice was injected with 1x PBS as an injection control. At 2m post-injection, K9JA immunolabeling revealed propagation of h-tau ipsilateral and contralateral to the injection site in the normal mouse brain (Fig. 2A). Proximal to the injection site, K9JA immunoreactivity was observed primarily in the ipsilateral thalamic nuclear regions (TH) and contralateral CA1 (Fig. 2B). Widespread K9JA immunoreactivity was also observed in the hippocampi, TH, and piriform/entorhinal (Pir/EC) cortices (Fig. 2A, B) of 2m NiPSCE injected mice. These data demonstrate that exosome-associated h-tau can transfer and propagate in mouse neurons in vivo.
Fig. 2.
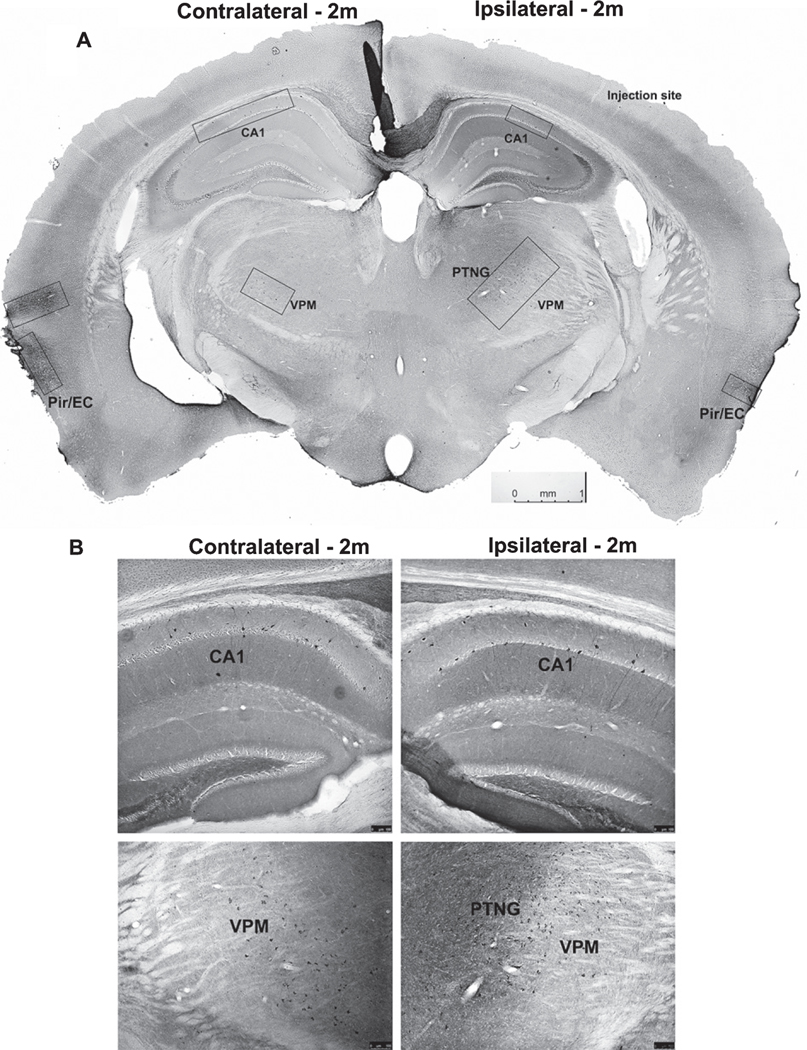
Widespread tau propagation is observed in normal mouse brain following exosome injection. Representative photomicrograph of coronal mouse section taken from a C57Bl/6 mouse injected with NiPSCEs and sacrificed at 2m post-injection. Sections were stained with the tau antibody, K9JA, and visualized with DAB. Size and location of boxes indicate varying density of K9JA immunoreactivity throughout the mouse brain (A). Representative photomicrographs of K9JA immunoreactivity in ipsilateral and contralateral hippocampi (top images), and TH brain regions (bottom images) of C57BL/6 mice injected with NiPSCEs and sacrificed at 2m post–injection (B). (n=5/timepoint, scale bar=100 μM).
We then examined if exosomes propagated pathological forms of tau in vivo. Immunohistochemical (Fig. 3A–C) and immunofluorescent (Fig. 4A, B) staining were conducted on control mice and NiPSCE injected mice using PHF1 anti-tau antibodies that recognize phosphorylation of tau serine residues (396 and 404). PHF1 labeling is routinely used to identify tau pathology in human AD brains and transgenic AD animal models. Here, we observed PHF1 immunoreactivity in the hippocampus, neocortex, Pir/EC and TH (Fig. 3B, C) on the ipsilateral side of the mouse brain. The nuclei of the TH include the posterior thalamic nuclear group (PTNG), ventral posterolateral thalamic nuclei (VPL), and ventral posteromedial thalamic nuclei (VPM). PHF1 immunoreactivity was not observed in the brains of control mice (Fig. 3A). Interestingly, a similar yet less pronounced pattern of PHF1 immunoreactivity was observed on the contralateral side of the mouse brain (Fig. 3B, C). These data suggest that NiPSCEs mediated long-distance propagation of human p-tau in various regions of the mouse brain. Double-labeling using immunofluorescence with PHF1 and the neuronal marker, MAP2 revealed a strong colocalization between the two markers in 1m NiPSCE injected mice (Fig. 4A). The colocalization of PHF1 and MAP2 was evident in neuronal cell bodies and to a lesser extent, in dendrites (Fig. 4B, Z-plane images of MAP2 and PHF1 colocalization can be found in Supplementary Figure 1).
Fig. 3.

PHF1 immunoreactivity reveals propagation of p-tau in normal mouse CNS. Representative photomicrographs of coronal mouse sections taken from a C57/Bl6 mouse injected with 1x PBS (control) (A) or NiPSCEs sacrificed at 1m (B) and 2m (C) post-injection, stained with p-tau antibody, PHF1, and visualized with DAB. The mounted slices were imaged via Leica DMi8 (Leica Microsystems) at 10x magnification and stitched together using Leica Application Suite X (LASx). All injection control mice were negative for human tau staining (A). Tau propagation, as measured by the transmission of PHF1, was detected in the hippocampus (CA1 and CA3); neocortex; piriform (Pir)/entorhinal cortex (EC); thalamic nuclei regions (TH) including post-thalamic nuclear group (PTNG), ventral posteromedial thalamic nuclei (VPM), ventral posterolateral thalamic nuclei; and zona incerta (ZI) (scale bar=1mm, n=5/timepoint). Brain regions that displayed PHF1 immunoreactivity were labeled on both hemispheres.
Fig. 4.

PHF1 immunoreactivity is significantly increased in injected mouse brains at 1m post-injection. Representative photomicrographs of the ipsilateral CA3 hippocampus (A) obtained from a NiPSCE injected mouse sacrificed at 1m post–injection and co-labeled with PHF1 and neural maker, MAP2. Colocalization of PHF1 and MAP2 was evident in neuronal cell bodies (insert, B, scale bar, 25 μm) and to a lesser extent, dendrites in the hippocampus of 1m NiPSCE injected mice (A). Increased PHF1 immunoreactivity was observed in the ipsilateral hippocampus of NiPSCE injected mice sacrificed at 1m post–injection as compared to mice sacrificed at 2m post–injection and control mice (C). No changes in PHF1 immunoreactivity in contralateral HP (D), ipsilateral TH (E), or contralateral TH (F) were detected between control versus 1m or 2m groups or between 1m and 2m groups. (n=5/timepoint; One way ANOVA with Bonferonni post-hoc test **p<0.01 versus control; ##p<0.01 versus 1m).
Next, we quantified the number of PHF1 positive (PHF1+) cells in the hippocampi and TH regions of NiPSCE injected mice and in control mice (Fig. 4C–F). The number of PHF1 +cells was significantly increased in the ipsilateral hippocampus, specifically in the CA1 and CA3 (Fig. 4C, p<0.001 versus control), of 1m NiPSCE injected mice as compared to control mice. Notably, there was a significant reduction in PHF1 immunoreactivity in the ipsilateral hippocampus at 2m post-injection as compared to 1m NiPSCE injected mice sacrificed (Fig. 4C, p<0.001 versus 1m).
MC-1 immunoreactivity was assessed in NiPSCE injected mice via immunostaining. MC-1 recognizes conformational tau epitopes which are closely associated with severe AD [27]. Quantification of MC-1 immunoreactivity revealed MC-1 immunoreactivity was significantly increased in the ipsilateral hippocampus and ipsilateral TH (Fig. 5) of mice sacrificed at 2m post-injection as compared to mice sacrificed at 1m post-injection (2m, 37.2±8.077 MC-1+cells versus 1m, 22.6±6.337 MC-1+cells; p<0.05; 2m, 85±18.49 MC-1+cells versus data not shown, 1m, 66.8±14.48 MC-1+cells, p<0.05) and compared to control mice (2m, 37.2±8.077 MC-1+cells versus control, 8.8±4.164 MC-1+cells; 2m, 85±18.49 MC-1+cells versus control, 16.6±10.75 MC-1+cells). No significant differences were detected in the contralateral hemisphere.
Fig. 5.

Deposits of pathogenically misfolded tau are present in the brain 2m post-injection. Representative photomicrograph of a MC-1 stained mouse section, no MC-1 immunoreactivity was observed in the brains of control mice (n=5/timepoint; scale bar, 100 μm). Representative photomicrographs of MC-1 immunoreactivity in the ipsilateral hippocampus and ipsilateral TH obtained from a control (A-B), 1m (C-D), and 2m (E-F) NiPSCE injected mice (n=5/timepoint; scale bar, 100 μm). Quantification of MC-1+cells (G-J) in NiPSCE injected mouse brains and controls shows that MC-1 immunoreactivity was significantly increased in the ipsilateral hippocampus (G), and ipsilateral TH (I) of mice sacrificed at 2m post-injection as compared to control mice. (n=5/group/timepoint; One-way ANOVA with Bonferonni post hoc test, *p<0.05 versus control).
Exosome associated-tau is toxic to recipient cells.
Finally, to determine the pathological consequences of exosome-associated tau, we quantified dendritic blebbing of hippocampal neurons in control and NiPSCE injected mice. Dendritic blebbing is a feature of apoptotic neurons that is present in the brains of tau overexpressing mice [28–30]. We found that injections of NiPSCEs increased dendritic blebbing of hippocampal neurons ipsilateral and contralateral to the injection site (Fig. 6). In total these data demonstrate that pathological forms of tau are packaged within exosomes and that exosomes are sufficient to cause tau propagation and neurodegeneration in the naïve mouse brain.
Fig. 6.

Exosome associated-tau is toxic to recipient cells. Brain sections from control and NiPSCE treated mice were immunolabeled with neuron-specific MAP2 antibodies. Hippocampal neurons ipsilateral (A) and contralateral (B) to the injection site were imaged using confocal microscopy (20X, scale bar = 10 μm). Dendritic blebbing was quantified as described in the Materials and Methods. C) Data is expressed as the mean number of blebs/length of dendrite (arbitrary units)±SEM. Data was analyzed with a 2-way ANOVA and Bonferonni post-hoc test. ***p<0.001. **p<0.01. n=17–35 dendrite per group.
DISCUSSION
Previous studies report that brain homogenates from mouse models of tauopathy [31], and homogenates from human tauopathies [32], can initiate tau pathology in the naïve mouse brain yet the specific mediators of tau seeding remain unknown. The present work demonstrates that exosomes are sufficient to seed pathological forms of tau resulting in brain-wide propagation. It remains unclear whether or not the tau pathology we observed in our model involved corruption of endogenously produced mouse tau, which will be addressed in future studies. Although this study proposes a pathogenic role for exosomes in tauopathies, tau seeding likely occurs by more than one mechanism. For instance, intracerebrally injected synthetic tau aggregates also initiate tau propagation in the mouse brain [33]. Therefore, we hypothesize that pathogenic tau seeding occurs by both exosome-independent and exosome-mediated processes.
Trans-synaptic spread of misfolded tau has been suggested to underlie tau propagation in vitro [3] and in vivo [18], although non-synaptic-mediated mechanisms have also been proposed [34]. A transgenic animal model harboring the same P301L mutation expressed by our cellular model demonstrated that tau pathology traverses neural networks in an age-dependent manner. Tau pathology reportedly began in the medial entorhinal cortex (MEC) and travelled through MEC axons to their respective terminals in the molecular layer of the dentate gyrus [18]. In this model, PHF1 immunoreactivity was not observed until 12m and was restricted to the MEC and dentate gyrus. Here, we observed widespread PHF1 immunoreactivity as early as 1m, particularly, in the CA1 and CA3 hippocampus, and in the piriform/entorhinal cortices of both hemispheres suggesting that tau propagation in our model could be network dependent. Further work will be needed to establish whether the propagation of exosome-associated tau and Aβ is solely dependent on synaptically connected pathways.
In our exosome-based model, the pattern of staining was starkly different between PHF1 and MC-1 at both time points. We hypothesized that pathogenically phosphorylated forms of tau would be present in the ipsilateral hippocampus 1m and 2m following exosome injections. Strikingly, although the number of PHF1 positive cells in ipsilateral hippocampi was increased after 1m, this was normalized in 2m samples. As demonstrated in Fig. 2, tau deposits were present in the ipsilateral hippocampus 2m post-injection; therefore, the loss of PHF1 immunoreactivity was not due to a lack of tau aggregates. While unexpected, these results are not unlike those obtained in a murine synucleinopathy model in which mice expressing A53T mutant human alpha-synuclein were found to have increased PHF1 immunoreactive tau at 4 and 8 months of age, which was normalized at 12 months [35]. Therefore, we hypothesize that PHF1 phosphorylation may be a transient phenomenon in some tauopathies. Alternatively, because there was an increase in misfolded tau (detected by the conformation specific antibody MC-1) in the ipsilateral hippocampus at 2m post-injection, the decrease in PHF1 between 1m and 2m samples may be the result of protein folding and/or aggregation masking the PHF1 epitope. This oddity will need to be addressed in future work.
Whether or not the uptake of tau contained within neuronally-derived exosomes is cell specific remains unknown. Here, we only determined neuronal cells were the recipients of exosome-associated tau. Similarly, Ahmed et al. developed an in vivo model of rapid tau propagation [36], where tau pathology was exclusive to neurons. However, recent work from Asai and colleagues demonstrated that pharmacological inhibition of exosome release, as well as ablation of microglia, decreases tau pathology in a mouse model of tauopathy [34]. The present work demonstrates that exosome-associated tau can also seed pathology and that exosomes are sufficient to initiate propagation in the naïve mouse brain. The mechanisms of post-injection propagation were not explored in this study and it remains unclear whether or not tau pathology is conveyed via microglia-dependent mechanisms, as proposed by Asai et. Al., or microglia-independent mechanism such as transsynaptic transport [37, 38]. We hypothesize that both these processes contributed to the post-injection pattern of tau pathology we observed in our animals, however, the relative contributions of synaptically trafficked tau versus microglia trafficked tau in our model remain unclear.
Lastly, we demonstrated that exosome-associated tau is toxic to recipient cells. Dendritic blebbing is a marker of neurodegeneration [39] that is present in rodent tauopathies [28, 40]. Here, we found extensive dendritic blebbing and dystrophic neurites in the brains of NiPSCE injected mice. Future studies will assess the long-term functional and neurobehavioral changes in mice following NiPSCE injection.
Our results may have therapeutic implications, suggesting that pathological tau species capable of long-distance propagation may not be removed by passive tau immunotherapy, which targets extracellular tau outside exosomes [41]. Anti-sense oligonucleotide against tau, another potential anti-tau therapy in development [42, 43], may possibly be able to reduce the exosomal tau. However, more research is needed to determine how pathological tau is sorted into the exosomes, so that these toxic species can be targeted.
In summary, we conclude that exosomes are sufficient to initiate tau propagation and are therefore may play a pathogenic role in AD. Future studies investigating the mechanisms that underlie tau packaging into exosomes and the uptake of exosome cargo by recipient cells may reveal new therapeutic targets for the treatment of tau-linked neurodegenerative disorders.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Yuan and Rissman laboratories for technical assistance and critical reading of this manuscript. We thank Dr. Peter Davies (Albert Einstein College of Medicine) for donating antibodies MC-1 and PHF1. This work was supported by NIA grants AG057459, AG057469, AG051848, and AG018440 to RAR, and VA Merit Awards BX003040 and BX004312 to RAR.
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18–0776r2).
SUPPLEMENTARY MATERIAL
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-180776.
REFERENCES
- [1].Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H, Raposo G (2004) Cells release prions in association with exosomes. Proc Natl Acad Sci U S A 101, 9683–9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ayers JI, Giasson BI, Borchelt DR (2018) Prion-like Spreading in Tauopathies. Biol Psychiatry 83, 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Frost B, Jacks RL, Diamond MI (2009) Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 284, 12845–12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259. [DOI] [PubMed] [Google Scholar]
- [5].Witter MP (2007) The perforant path: Projections from the entorhinal cortex to the dentate gyrus. Prog Brain Res 163, 43–61. [DOI] [PubMed] [Google Scholar]
- [6].Spires-Jones TL, Hyman BT (2014) The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 82, 756–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Edgar JR (2016) Q&A: What are exosomes, exactly? BMC Biol 14, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Simons M, Raposo G (2009) Exosomes–vesicular carriers for intercellular communication. Curr Opin Cell Biol 21, 575–581. [DOI] [PubMed] [Google Scholar]
- [9].Budnik V, Ruiz-Cañada C, Wendler F (2016) Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci 17, 160–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Urbanelli L, Magini A, Buratta S, Brozzi A, Sagini K, Polchi A, Tancini B, Emiliani C (2013) Signaling pathways in exosomes biogenesis, secretion and fate. Genes (Basel) 4, 152–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fiandaca MS, Kapogiannis D, Mapstone M, Boxer A, Eitan E, Schwartz JB, Abner EL, Petersen RC, Federoff HJ, Miller BL, Goetzl EJ (2015) Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement 11, 600–607 e601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Goetzl L, Merabova N, Darbinian N, Martirosyan D, Poletto E, Fugarolas K, Menkiti O (2018) Diagnostic potential of neural exosome cargo as biomarkers for acute brain injury. Ann Clin Transl Neurol 5, 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hamlett ED, Goetzl EJ, Ledreux A, Vasilevko V, Boger HA, LaRosa A, Clark D, Carroll SL, Carmona-Iragui M, Fortea J, Mufson EJ, Sabbagh M, Mohammed AH, Hartley D, Doran E, Lott IT, Granholm AC (2017) Neuronal exosomes reveal Alzheimer’s disease biomarkers in Down syndrome. Alzheimers Dement 13, 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Stern RA, Tripodis Y, Baugh CM, Fritts NG, Martin BM, Chaisson C, Cantu RC, Joyce JA, Shah S, Ikezu T, Zhang J, Gercel-Taylor C, Taylor DD (2016) Preliminary Study of plasma exosomal tau as a potential biomarker for chronic traumatic encephalopathy. J Alzheimers Dis 51, 1099–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ngolab J, Trinh I, Rockenstein E, Mante M, Florio J, Trejo M, Masliah D, Adame A, Masliah E, Rissman RA (2017) Brain-derived exosomes from dementia with Lewy bodies propagate α-synuclein pathology. Acta Neuropathol Commun 5, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Winston CN, Goetzl EJ, Akers JC, Carter BS, Rockenstein EM, Galasko D, Masliah E, Rissman RA (2016) Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement (Amst) 3, 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Goetzl EJ, Kapogiannis D, Schwartz JB, Lobach IV, Goetzl L, Abner EL, Jicha GA, Karydas AM, Boxer A, Miller BL (2016) Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J 30, 4141–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT (2012) Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73, 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Reilly P, Winston CN, Baron K, Trejo M, Rockenstein EM, Akers JC, Kfoury N, Diamond M, Masliah E, Rissman RA, Yuan SH (2017) Novel human neuronal tau model exhibiting neurofibrillary tangles and transcellular propagation. Neurobiol Dis 106, 222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Reilly P, Winston CN, Baron KR, Trejo M, Rockenstein EM, Akers JC, Kfoury N, Diamond M, Masliah E, Rissman RA, Yuan SH (2017) Novel human neuronal tau model exhibiting neurofibrillary tangles and transcellular propagation. Neurobiol Dis 106, 222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yuan SH, Martin J, Elia J, Flippin J, Paramban RI, Hefferan MP, Vidal JG, Mu Y, Killian RL, Israel MA, Emre N, Marsala S, Marsala M, Gage FH, Goldstein LS, Carson CT (2011) Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One 6, e17540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LS (2012) Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Turner RS (2001) Alzheimer’s disease in man and transgenic mice: Females at higher risk. Am J Pathol 158, 797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Barnes LL, Wilson RS, Bienias JL, Schneider JA, Evans DA, Bennett DA (2005) Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch Gen Psychiatry 62, 685–691. [DOI] [PubMed] [Google Scholar]
- [25].Carroll JC, Rosario ER, Kreimer S, Villamagna A, Gentzschein E, Stanczyk FZ, Pike CJ (2010) Sex differences in β-amyloid accumulation in 3xTg-AD mice: Role of neonatal sex steroid hormone exposure. Brain Res 1366, 233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Spencer B, Michael S, Shen J, Kosberg K, Rockenstein E, Patrick C, Adame A, Masliah E (2013) Lentivirus mediated delivery of neurosin promotes clearance of wild-type α-synuclein and reduces the pathology in an α-synuclein model of LBD. Mol Ther 21, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [27].Jicha GA, Bowser R, KazamI G, Davies P (1997) Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res 48, 128–132. [DOI] [PubMed] [Google Scholar]
- [28].Jaworski T, Lechat B, Demedts D, Gielis L, Devijver H, Borghgraef P, Duimel H, Verheyen F, Kugler S, Van Leuven F (2011) Dendritic degeneration, neurovascular defects, and inflammation precede neuronal loss in a mouse model for tau-mediated neurodegeneration. Am J Pathol 179, 2001–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Erturk A, Wang Y, Sheng M (2014) Local pruning of dendrites and spines by caspase-3-dependent and proteasome-limited mechanisms. J Neurosci 34, 16721688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Park JS, Bateman MC, Goldberg MP (1996) Rapid alterations in dendrite morphology during sublethal hypoxia or glutamate receptor activation. Neurobiol Dis 3, 215–227. [DOI] [PubMed] [Google Scholar]
- [31].Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 11, 909–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M, Ghetti B, Goedert M, Tolnay M (2013) Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A 110, 9535–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM (2013) Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 33, 1024–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kugler S, Ikezu T (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18, 1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Oaks AW, Frankfurt M, Finkelstein DI, Sidhu A (2013) Age-dependent effects of A53T alpha-synuclein on behavior and dopaminergic function. PLoS One 8, e60378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H, Parhizkar S, Ward MA, Cavallini A, Jackson S, Bose S, Clavaguera F, Tolnay M, Lavenir I, Goedert M, Hutton ML, O’Neill MJ (2014) A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: The pattern of spread is determined by connectivity, not proximity. Acta Neuropathol 127, 667–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang Y, Balaji V, Kaniyappan S, Kruger L, Irsen S, Tepper K, Chandupatla R, Maetzler W, Schneider A, Mandelkow E, Mandelkow EM (2017) The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener 12, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dujardin S, Lecolle K, Caillierez R, Begard S, Zommer N, Lachaud C, Carrier S, Dufour N, Auregan G, Winderickx J, Hantraye P, Deglon N, Colin M, Buee L (2014) Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: Relevance to sporadic tauopathies. Acta Neuropathol Commun 2, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kweon JH, Kim S, Lee SB (2017) The cellular basis of dendrite pathology in neurodegenerative diseases. BMB Rep 50, 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hoffmann NA, Dorostkar MM, Blumenstock S, Goedert M, Herms J (2013) Impaired plasticity of cortical dendritic spines in P301S tau transgenic mice. Acta Neuropathol Commun 1, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yanamandra K, Jiang H, Mahan TE, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM (2015) Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann Clin Transl Neurol 2, 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].DeVos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR, Schuler DR, Maloney SE, Wozniak DF, Rigo F, Bennett CF, Cirrito JR, Holtzman DM, Miller TM (2013) Antisense reduction of tau in adult mice protects against seizures. J Neurosci 33, 12887–12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, Chen G, Shen T, Tran H, Nichols B, Zanardi TA, Kordasiewicz HB, Swayze EE, Bennett CF, Diamond MI, Miller TM (2017) Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med 9, eaag0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.