


Abstract
Most coding genes in the human genome are annotated with multiple alternative transcripts. However, clear evidence for the functional relevance of the protein isoforms produced by these alternative transcripts is often hard to find. Alternative isoforms generated from tandem exon duplication-derived substitutions are an exception. These splice events are rare, but have important functional consequences. Here, we have catalogued the 236 tandem exon duplication-derived substitutions annotated in the GENCODE human reference set. We find that more than 90% of the events have a last common ancestor in teleost fish, so are at least 425 million years old, and twenty-one can be traced back to the Bilateria clade. Alternative isoforms generated from tandem exon duplication-derived substitutions also have significantly more clinical impact than other alternative isoforms. Tandem exon duplication-derived substitutions have >25 times as many pathogenic and likely pathogenic mutations as other alternative events. Tandem exon duplication-derived substitutions appear to have vital functional roles in the cell and may have played a prominent part in metazoan evolution.
INTRODUCTION
Alternative splicing of messenger RNA is predicted to occur in almost all multi-exon coding genes (1,2) and the human reference genome is annotated with an ever-expanding number of alternative protein coding transcripts (3–5). Alternative splicing has unequivocal support at the transcript level (6–8), although the vast majority of predicted alternative protein products evade detection at the protein level (9,10). The lack of peptides for alternative splice isoforms is a real biological phenomenon; we found that just 0.37% of all reliable peptides detected in large-scale proteomics experiments mapped to alternative splice isoforms (10). What exactly happens to the missing alternative isoforms and how many alternative splice isoforms are functional at the protein level are just two of many unresolved questions (11).
Alternative splicing events are generally classified by the mechanism of their generation. However, the final product of a coding gene is a protein isoform, so a protein-level classification makes more biological sense. At the protein level, there are just three basic types of alternative splicing, insertions, deletions and substitutions. Substitutions can be further broken down by whether they are homologous or non-homologous. Non-homologous exons can be incorporated into coding genes from various sources, from adjacent genes (12), from fragments of transposable elements (13,14), or they can be entirely novel exons (15). Homologous substitution events are produced by the alternative splicing of exons that have been duplicated in tandem, duplicated exons that are adjacent in sequence.
Substitutions at the protein level can be generated from a variety of mechanisms. This is determined by the position of the substitution; those substitutions internal to the sequence are produced from mutually exclusive splicing of exons (16,17). Mutually exclusive splicing (16) is one of the rarest splice events in the human genome (1). By strict definition, mutually exclusive splicing does not generate substitutions at the C-terminal or N-terminal; instead, amino acid substitutions at the C-terminal are generated through alternative poly-adenylation (poly(A)) or from exon skips that cause frameshifts, while N-terminal substitutions derive from alternative promoter usage.
After duplication, most tandem exons end up constitutively spliced within the same transcript, but a small number are alternatively spliced so that transcripts will include one or other (but not both) of the exons. These tandem duplicated exons produce alternative protein isoforms that have homologous regions. Unlike most novel alternative isoforms, alternative isoforms generated from tandem exon duplications can be functional right away since they initially share the same sequence as the main isoform. Evolution away from the initial exon sequence allows the new isoform to gain subtle differences in structure and function.
The most famous example of this is Drosophila gene Dscam1 (18) which has four sets of multiple tandem duplicated exons that are mutually exclusively spliced and can, theoretically, produce thousands of homologous protein isoforms (Figure 1). Curiously, while there are numerous examples of tandem duplicated exon events involving multiple mutually exclusively spliced internal homologous exons in Drosophila (e.g. mrp, 14–3–3zeta, Pfk), internal mutually exclusive splicing events in vertebrates are supposed to involve no more than two exons (19) because the mechanisms to allow splicing of multiple exons in a mutually exclusively manner in invertebrates do not exist in vertebrates (20).
Figure 1.
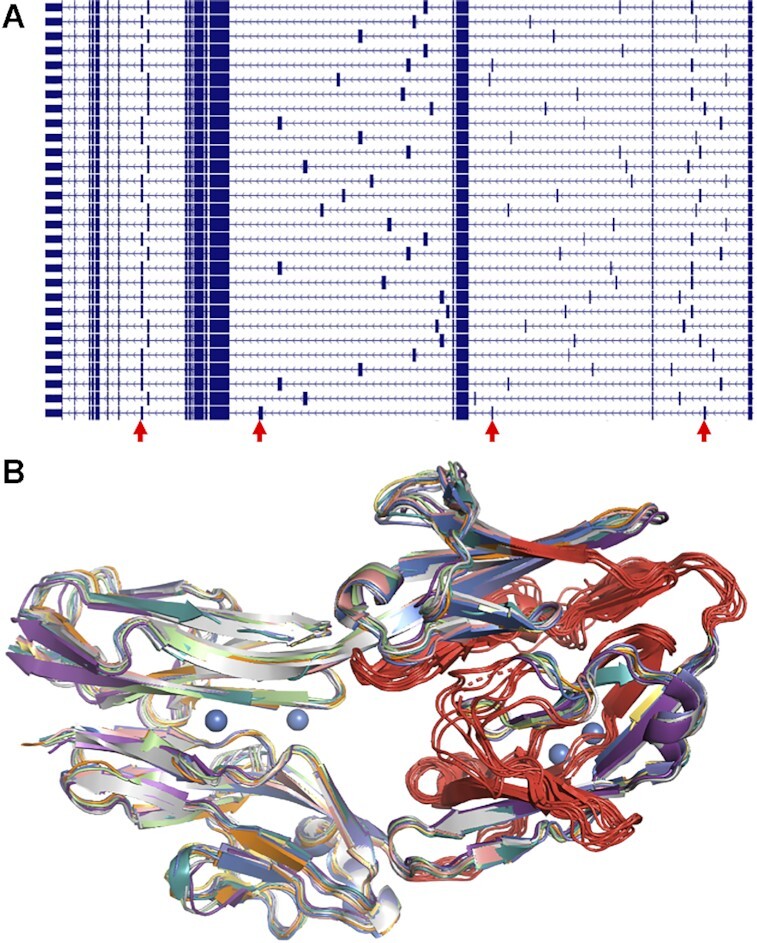
Mutually exclusively spliced tandem exon duplication-derived substitution in the Dscam1 gene. (A) A selection of Drosophila gene Dscam1 transcript models from the UCSC genome browser (21). Dscam1 has four separate sets of homologous exons - each transcript includes just one of the four sets of exons. Regions with the homologous exons are indicated by the arrows. (B) The ten crystalised structures from the PDB (22) of the first four immunoglobulin domains in Dscam1 protein (18) shown in cartoon format. The constant regions in the ten proteins are shown in different colours, while the homologous regions translated from the two 5′ tandem exon duplication-derived substitutions are shown in red. All 10 structures are highly similar despite the differences in sequence, but loop regions translated from the homologous exons have distinct backbones. The structures in all figures were represented using PyMol.
Tandem exon duplication-derived substitutions are not always mutually exclusively spliced, however. Tandemly duplicated 5′ CDS and alternative promoter usage produces homologous N-terminal sequences and 3′ CDS duplications can go on to generate homologous C-terminal sequences via alternative poly(A) use. The human gene set does have examples of multiple alternatively spliced tandem exon duplications that produce three or more N-terminals or C-terminals.
Kondrashov and Koonin (24) were the first to characterise alternatively spliced tandem duplicated exons. They found 50 pairs in distinct vertebrate and invertebrate species and suggested that these tandem exon duplications might have allowed alternative isoforms to have specialised functional roles. They also hypothesised that since exon duplication was common, it might be involved in as much as 10% of alternative splicing. Copley (25) found mutually exclusively spliced exon duplications that appeared to have arisen independently in three different ion channel families in human and Drosophila genes. This evidence of convergent evolution suggested that tandem exon duplications might provide advantages in certain protein families. Letunic et al. (26) suggested that tandem exon duplications might be responsible for 20% of alternative splicing events. However, with time it has become clear that most tandem duplicated exons are incorporated as part of the constitutive isoform.
Hatje and Kollmar (17,27) carried out two large-scale analyses of mutually exclusive splicing. The first, in Drosophila (17), found that mutually exclusively spliced exons were enriched in transmembrane transporters and ion channels. Most exons were also conserved across Drosophila species, though they speculated that mutually exclusive spliced exons might also have a role in speciation. In their second analysis (27), the authors predicted 629 clusters of mutually exclusively spliced exons in the human genome. A total of 42% of the 1399 mutually exclusively spliced exons from the 629 clusters were not annotated as coding in RefSeq. The authors used a strict definition of mutually exclusive splicing, but not all events involved tandem duplicated exons.
A recent study (30) of the functional properties of 143 homologous mutually exclusive spliced events in the human reference set (4) found that regions translated from these exons are enriched in surface-exposed residues and tend to cluster near protein functional sites. The authors suggest that these homologous substitution events may affect protein specificity and selectivity.
As part of a large-scale proteomics analysis of alternative splicing in the human reference set, we found that tandem exon duplication-derived substitutions were detected significantly more often at the protein level than would be expected (9,10,28). In fact, alternative isoforms generated from tandem duplicated homologous exons made up >10% of detected alternative proteins across a range of species (9). In general, most of the splice events detected in these proteomics experiments maintained their functional domain composition (10) and had considerable cross-species support (1,29).
In this study, we have catalogued and characterised the 236 tandem exon duplication-derived substitutions annotated in the current definition of the human gene set (4). We find that the vast majority of the homologous exons in these events are highly conserved and that 21 arose even before the separation of vertebrates and invertebrates, >670 million years ago. We detect more than a third of these splice events in proteomics experiments. Most importantly, we show that alternative exons generated by tandem duplication are highly enriched in pathogenic mutations, supporting the hypothesis that these tandem exon duplication-derived substitutions are a highly important class of splice event.
MATERIALS AND METHODS
Annotation databases
We used the GENCODE v33 human gene set (4) as the basis for the analyses. Homology searches were carried out against other vertebrate species using the Ensembl (31), RefSeq (5) and UniProtKB (32) annotations for those species. We checked for homology to the human tandem duplication events in the Flybase (33) and APPRIS (34) databases for Drosophila, and in the RefSeq and UniProtKB databases for other invertebrate species.
For the analysis of alternatively spliced exons we defined the main transcript variants from the GENCODE v33 human gene set using the APPRIS principal isoforms (34). APPRIS defines principal isoforms based on cross-species conservation and conserved protein features. Alternative exons in the GENCODE v33 human gene set were defined as all exons that did not overlap APPRIS principal exons.
Manual curation of annotated tandem exon duplications
We manually curated tandem exon duplication-derived substitutions in the GENCODE human gene set. The manual curation process took six years. The initial set was generated with a BLAST search (35) followed by a manual curation step. We translated all exon sequences that were longer than 30 nucleotides in Ensembl version 78 and used these to search for homology with BLAST v2.2.25. Homologous regions found in BLAST had to have an e-value of less than 0.005. We required each of the resulting potential homologous exons to occupy a position within the alternative transcript that was equivalent to the position of the query exon. In the manual curation step we discarded all read-through transcripts, where one of the alternative exons belonged to a paralogous neighbour gene or pseudogene. Our initial analysis yielded 129 alternative homologous regions.
No single method will detect all the tandem duplicated exon substitutions in the human gene set, so we added more examples to our set by manual annotation. We included additional cases of homologous exons that were identified while working with the APPRIS database and those found in studies carried out as part of the GENCODE consortium, including analyses of alternative splicing and gene models (11,14,29), analyses of UniProt and RefSeq annotations (36), analyses of proteomics data (28). We wrote a script to predict the effect of splice events at the protein level as part of the development of the TRIFID functional isoform predictor (37) and manually analysed those substitutions predicted to be homologous based on the similarity of their amino acid sequences. When we detected exons that were annotated in RefSeq, but not annotated in GENCODE, we brought them to the attention of the GENCODE manual annotators.
The final set of tandem exon duplication-derived substitution events were all annotated as coding in v33 of the GENCODE human reference set. The translated exons had to have a minimum of eight amino acid residues and had to either have been detected in the BLAST search or to have measurable evidence of homology. Within this last group, we included those events in which both exons mapped to the same functional domain or motif, or were most similar to the same known structure. We also included those exons that had evolved to the point at which the similarity was no longer apparent except for a small number of residues, as long as those residues completed a functional domain or similar motif (e.g. the C-terminals in the Plasma membrane calcium-transporting ATPases).
Determination of last common ancestor
We carried out a manual annotation of the age of each duplication event. The initial analysis of gene age was carried out with BLAST searches against the genomes of five distant vertebrate species, coelacanth (LatCha1) (38), fugu (Fugu4) (39), spotted gar (LepOcu1) (40), zebrafish (GRCz11) (41) and lamprey (Pmarinus7) (42). The taxa were retrieved from Ensembl v99 and we scanned each genome using TBLASTN with an e-value threshold of 0.1 and without low complexity filtering. When we found multiple hits in the same gene for each homologous region, we determined which hit was most similar to each region. To count as an evolutionarily conserved tandem duplication event, each homologous region had to be most similar to a different TBLASTN hit and the two orthologous exons detected in the searches had to be sequential and in the same gene.
After that we searched for the most distant homologue for each pair of tandem duplicated exons manually in three databases: UniProtKB, RefSeq and Flybase. Where two species had a pair of homologous exons at the same position, we did not automatically assume that they were the result of a common ancestral duplication event, because the two events could have been acquired independently. Instead, we examined each pair of homologous exons in detail, building multiple sequence alignments with Kalign (43) and Muscle (44). We determined whether the two exons had evolved from the same event or independently by comparing the similarity between the equivalent homologous regions in the two species and the between the homologous regions within the same species. To infer common ancestry, we required that similarity was higher between species than within species and that the splice sites were the same. Where the relationship was not clear, we generated a phylogenetic tree to determine the provenance of each substitution event.
When we searched for evidence of duplication prior to the Chordata phylum, we required that we had previously detected an orthologous event in sharks, rays or lamprey. We required the tandem duplicated exon event was detected in tunicates or lancelets in order to search for evidence of an evolutionary relationship with pairs of exons within the Bilateria clade.
We dated the origin of each tandem exon duplication-derived substitution to the last common ancestor (LCA) between human and the most distant species in which we found evidence for the substitution event. We estimated the ages from TimeTree (45), taking the average of seven recent analyses (46–52) and rounding to the nearest 5 million years.
Manual curation of other alternative exons
In order to produce a background set against which tandem exon duplication-derived substitutions could be compared, we curated a set of alternative exons. Alternative exons were defined as those GENCODE v33 exons tagged as alternative or minor by APPRIS, that were a minimum of 42 bases and that did not overlap with the coordinates of any exon from the APPRIS principal transcript. The exons were a minimum of 42 bases in order to detect orthologous exons in other species in BLAST searches. There were 12 019 alternative exons that passed these filters. If a splice event used more than one exon from the list, these exons were considered to be part of the same event. We pooled the 12 019 alternative exons into 10 599 splice events, 220 of which were tandem exon duplication-derived substitutions (the remainder had exons shorter than 42 bases).
We checked for homology with exons in fish by carrying out TBLASTN with an e-value threshold of 0.1 and without low complexity filtering searches against the same five vertebrate genomes as we used for the homologous exon search, coelacanth, fugu, spotted gar, zebrafish and lamprey, retrieved from Ensembl v99. If we found a hit, we analysed conservation manually against protein databases and confirmed homology with hits to regions from any fish species. As with the homologous exon analysis, we required evidence that each splicing event was conserved in the same gene in at least one fish species to include it in the list of splice events conserved in fish.
Proteomics
We analysed two large-scale proteomics analyses (53,54) for evidence of the expression of homologous exons. These experiments were carried out on multiple tissue types. We downloaded the data from ProteomeXchange (55) with identifiers PXD000561 and PXD010154.
We used the GENCODE v33 human reference set for the peptide search, excluding read-through genes (36) and setting aside experiments that did not use trypsin. We then mapped spectra from the two experiments to the reduced GENCODE v33 gene set using the Comet search engine (56). Comet was run with default parameters, allowing oxidation of methionines. Peptide spectrum matches (PSM) were post-processed using Percolator (57). Percolator posterior error probability (PEP) values were used to filter Comet PSMs. PSMs with PEP values of <0.001 were deemed to be valid. In addition, detected peptides were required to be fully tryptic and to have a maximum of one missed cleavage. Peptides that mapped to more than one gene were discarded.
Combining spectra from many different experiments will inevitably expand the false discovery rate (58). We were already using a conservative PEP score. To reduce false positive identifications further, each peptide had to be identified at least twice over the 1632 experiments. These steps will have eliminated many false positive matches; we found no peptides that mapped to olfactory receptors for example (59), but they will not have eliminated all of them. For example, we did identify PSMs for peptides that map to the alternative C-terminal of BLOC1S6, which are almost certainly false positives (58).
In order to detect evidence for a pair of homologous exons, we required that at least one valid peptide supported by at least two PSM from different experiments mapped to each homologous region.
RESULTS
We identified 236 tandem exon duplication-derived substitutions in 215 distinct coding genes across the whole GENCODE v33 human reference gene set. The manually curated set is available in Supplementary Table S1. The homologous regions range in size from the minimum eight amino acid residues (LMO1 and MYL6) to just over 250 residues (IKBIP, MASP1 and KIAA1958). Fourteen genes have more than one tandem exon duplication-derived substitution.
The set of tandem exon duplication-derived substitutions is made up of 39 substitutions at the N-terminal, 77 substitutions at the C-terminal, 119 internal substitutions and one whole protein swap (DUSP13). Most involve the swapping of a single homologous exon for another, but some events are more complex. G protein subunit alpha o1 (GNAO1), for example, produces two different proteins by swapping the two final coding exons for a pair of tandem duplicated exons (Figure 2). The two exons are always found together, so this is a single event. In ZFP64, coding exons 3, 4, 5 and part of 6 were duplicated to produce a new C-terminal. The resulting alternative transcript includes two copies of exon 3 and a truncated version of exon 6.
Figure 2.

Homologous duplicated regions in GNAO1, NECTIN2 and the protocadherin alpha locus (A) and (B). Complex homologous exons (gold) in GNAO1 and NECTIN2. Homologous exons are depicted in the gene model and as partial protein sequence alignment (homologous sequence in gold), and their position within the domain organization of the corresponding proteins is also shown. The regions corresponding to the trans-membrane helices in NECTIN2 are shown in red. (C) In the protocadherin alpha gene cluster fifteen tandem duplicated exons (in gold) are annotated as fifteen different genes. Each of the 15 distinct ‘genes’ generates a transcript made up of a single large tandemly duplicated 5′ CDS and the same three 3′ CDS. The 3′ CDS present in all 15 ‘genes’ code for the Cadherin tail domain.
Homology between the exons in GNAO1 transcripts is evident, but this is not always the case. The C-terminal swap in gene NECTIN2 (Figure 2) is also composed of multiple exons. Here the event almost certainly also arose by tandem duplication, but the homology is less clear. However, the region around the trans-membrane helix retains similarity, both C-terminals have a conserved PDZ binding motif, and HHPRED (60) searches show that the N-terminal sections in both tandem exon duplication-derived substitutions are significantly similar to the same set of PDB (22) structures, including structures 2k9y, an Ephrin type-A receptor, and 3j8f, a Capsid protein from a poliovirus receptor. NECTIN2 was previously named ‘Poliovirus receptor-related protein 2’.
Almost all homologous exons are found exclusively in pairs, but five genes are annotated with three sets of interchangeable tandem duplicated exons, either at the 5′ end (GCNT2 and KCNAB1) or at the 3′ end (LAMP2, TPM1 and TPM3). Outside of our set, there are also multiple 5′ tandem duplicated exons in the protocadherin and UDP glucuronosyltransferase family gene clusters, but these homologous exons are annotated as separate ‘genes’ so are not listed.
In contrast to Drosophila, however, there are no annotated internal (mutually exclusively spliced) tandem exon duplication-derived substitutions in the GENCODE reference set, as would be expected (20).
Gene and exon duplications
The distinct tandem exon duplication-derived substitutions were either generated from exon duplications or duplications of genes that already contained homologous exons. A total of 94 of the 236 tandem exon duplication-derived events (39.8%) were paralogues, so had been part of a gene duplication. The 94 substitutions arose from 38 unique tandem exon duplications, so 56 of the 236 substitution events were created from gene duplications rather than exon duplications. The family with most gene duplications is the sodium channel alpha subunit family, which has six family members that maintained the ancestral tandem exon duplication-derived substitution event (SCN1A, SCN2A, SCN3A, SCN5A, SCN8A y SCN9A).
Gene clusters
The work in this paper concentrates on tandem exon duplication-derived substitutions within the same gene. However, there are five ‘clusters’ outside of our set in which tandem exon duplications have been defined as distinct ‘genes’ (61). The protocadherin gamma cluster is made up of 21 separate ‘genes’. Each of these 21 ‘genes’ is composed of four coding exons, 3 of which are common to all 21 ‘genes’. The fourth exon in each ‘gene’ is one of 21 homologous 5′ coding exons that have been generated by tandem exon duplication. Similarly, the protocadherin alpha family is made up of 15 ‘genes’ that have three common downstream coding exons and a single unique homologous exon at the 5′ end (Figure 2C). The UDP glucuronosyltransferase family 1 member A cluster is made up of nine ‘genes’ with a similar organisation, but this time the main transcript of each ‘gene’ has four common 3′ coding exons in addition to the large upstream homologous exon that is unique to each of the ‘genes’. The unique amino terminus produced by each of these exons determines the substrate of this enzyme. The UDP glucuronosyltransferase family 2 member A cluster has two ‘genes’ with distinct 5′ homologous coding exons. Finally, the RNASE4/ANG cluster consists of two ‘genes’ with distinct homologous single coding exons and a shared 5′ UTR exon. These five clusters contain a total of 44 further tandem exon duplication-derived substitutions.
Comparison to previous studies
In the first study of tandem duplicated exons (24), Kondrashov and Koonin listed 50 events from a range of different species. Of the tandem exon duplication-derived substitutions in this paper, we might expect that GENCODE human reference set includes the 39 that are listed as being present in human or eutherian species. In fact, 36 events from Kondrashov and Koonin's list coincide with GENCODE v33 tandem exon duplication-derived substitutions (Figure 3).
Figure 3.
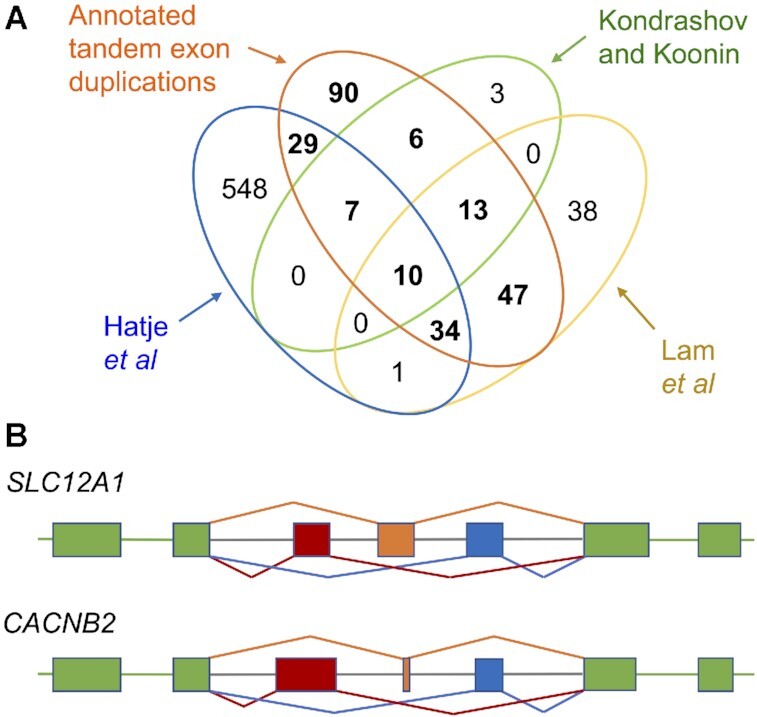
Overlap between tandem exon duplications and previous studies. (A) The intersection of the Kondrashov and Koonin (24), Hatje et al. (27), Lam et al. (30) analyses with the tandem exon duplication-derived substitutions annotated in GENCODE v33. The distribution of tandem exon duplication-derived substitutions is in bold. (B) The two clusters of multiple mutually exclusively spliced exons annotated in the human reference sets. Exons proportional to size, introns not to scale. Constitutive exons are shown in green, the three mutually exclusively spliced exons in each gene are shown in dark red, orange and blue. In SLC12A1 coding exons 2, 3, 4a/b/c, 5 and 6 are shown. The most recent duplication (last common ancestor with amphibians) is exon 4a (in dark red). In CACNB2 the coding exons are 5, 6, 7a/b/c, 8 and 9. The most recently evolved of these non-homologous exons (last common ancestor with amphibians) is exon 7c (blue).
Two more recent large-scale studies (27,30) concentrated on mutually exclusively spliced exons rather than events from tandem exon duplications. According to the strict definition of mutually exclusive splicing only one of each pair of exons can ever be spliced into the mature mRNA at any one time (16) and substitutions generated by alternative promoters and alternative poly(A) splicing events are excluded. Just over half of our tandem exon duplication-derived substitutions take place at the 3′ or 5′ end of the transcript, so not all are mutually exclusively spliced. Mutually exclusively spliced events can involve unrelated exons, so not all mutually exclusively spliced events involve tandem duplicated exons. However, there is an overlap between tandem exon duplication-derived substitutions and mutually exclusively spliced events.
Hatje et al. used RNA-seq data to predict and validate 629 clusters of distinct mutually exclusively spliced exons in the human genome (27). A total of 76 of the clusters in their study are identical to the tandem exon duplication-derived substitutions annotated in GENCODE v33 (Figure 3), while a further four overlap GENCODE-annotated events but are not identical. The remaining 549 Hatje et al. predicted mutually exclusive events fall into four main categories: clusters involving unannotated predicted exons (431 clusters), clusters involving exons that cannot possibly splice in a mutually exclusive manner (68), exon pairs that cDNA evidence suggests splice constitutively (35), and annotated exon pairs that are mutually exclusively spliced but not homologous (15). A more detailed breakdown of the Hatje et al. data set is available in the supplementary material.
Most of the 156 tandem exon duplication-derived substitutions annotated in GENCODE v33 that were not predicted by Hatje et al. are C-terminal and N-terminal substitution events, which were not considered mutually exclusive. However, 39 annotated mutually exclusively spliced tandem exon duplication-derived substitutions were not listed by Hatje et al.
The Lam et al. study (30) analysed 143 homologous mutually exclusive splicing events in 125 genes from the Ensembl human reference set (31). This study included those isoforms produced from alternative promoter and alternative poly(A) usage, but homologous exons were not allowed to occur together in the same transcript. This rule excluded examples in which minor transcripts included both duplicated exons (e.g. TPM1). The events themselves are not listed in the paper, but 89 of the 125 genes overlap with genes that have tandem exon duplication-derived substitutions, so as long as the Lam et al. study identified all the annotated tandem exon duplication-derived substitutions in these 89 genes, 104 of the events in the study will coincide with our tandem exon duplication-derived substitutions (Figure 3). The remaining 39 events in Lam et al either did not fit our definition of homologous exons or involved read-through transcripts that we did not include in our analysis because we believe they are unlikely to be translated into cellular proteins (36).
One curious result is that only 11 of the GENCODE v33 tandem exon duplication-derived substitutions are found in all three previous analyses (Figure 3): CACNA1A, DNM2, EYA4, MAPK8, MAPK14, MEF2D, P4HA1, SLC7A2, SLC25A3, SNAP25 and STX3. A total of 90 annotated tandem exon duplication-derived substitutions (38.1%) were not identified in any of the previous studies.
Do vertebrates have a mechanism that allows mutually exclusive splicing of multiple exons?
The analysis of previous studies also finds four tandem exon duplication-derived substitutions that are not annotated by GENCODE. Hatje et al. (27) identified three events, two of which are already annotated in RefSeq. Exons in CEPT1 and FAR1 are conserved in sharks and rays (gnathostoma), while the exon in SRPK1 has a last common ancestor with Styela clava (chordates). These are candidates to be added to the GENCODE reference set, as is the exon identified by Kondrashov and Koonin in SLC12A1.
The tandem exon duplication identified by Kondrashov and Koonin in SLC12A1 is also annotated in RefSeq and conserved at least in Xenopus tropicalis, so it is highly likely to be coding. What is surprising about this exon is that it forms a cluster of three with two other mutually exclusively spliced tandem duplicated exons (Figure 3), making this the only known case of homologous multiple mutually exclusively spliced exons in the human reference set.
Although Hatje et al. claimed to have found evidence of 69 multiple exon mutually exclusive spliced clusters in humans, almost half of their predictions are not even biologically possible (for example the supposed mutually exclusively spliced exons are in different genes, or have overlapping coordinates, see supplementary data for details). Another 34 have no supporting evidence except for a small number of RNA-Seq reads. However, one prediction, the mutually exclusively spliced exons in CACNB2, is supported by cDNA evidence and is annotated in both the GENCODE and RefSeq reference sets (Figure 3). As with SLC12A1, two exons are ancient and the third exon appears to be conserved in amphibians. However, the exons in the CACNB2 event are not homologous and are unlikely to have evolved via tandem duplication.
Cross-species conservation
We have previously noted that many homologous exons are highly conserved (10,28). Here, we found that both homologous exons are annotated in at least one fish genome in 215 of the 236 human tandem exon duplication-derived substitutions. This means that 91.1% of these events were already present in a common ancestor of mammals and ray-finned fish during, approximately 425 million years ago.
The proportion of orthologous tandem exon duplication-derived substitutions detected in fish is all the more remarkable because the vast majority of alternative exons are of much more recent evolutionary origin (28,62). In order to compare the conservation of homologous exons with other alternative exons, we took all alternative exons longer than 42 bases that did not overlap with exons from principal transcripts (a total of 12,018 alternative exons from 10,599 alternative events) and used BLAST to search for homology against the genomes of 5 fish species. We carried out manual analyses a posteriori for each exon to remove false positive hits and to ensure that all exons in a splice event were conserved in fish (see methods). If the splice event was annotated in at least one fish species, we determined that the alternative exon had its origin in the last common ancestor of humans and fish.
After manual analysis, we found that 615 of 10 599 alternative events (5.8%) had orthologues in at least one fish genome. Almost 30% of these (182) are tandem exon duplication-derived substitutions. Although we have analysed a large number of exons, this is only an estimate of the real numbers of alternative exons conserved in fish. The analysis is limited to coding exons that are 42 bases long and does not include exons that overlap principal exons (so excluding retained introns, and alternative 3′ and 5′ splice sites). Furthermore, some exons may have diverged to such an extent that they are no longer recognised by BLAST. For example, we failed to detect homology for nineteen tandem exon duplication-derived substitutions that we know are conserved in fish. One example was the homologous alternative exon in NRG1. Since we found no homology with any of the five fish species, it did not count as conserved in this analysis.
Tandem exon duplication-derived substitutions and mutually exclusive splicing
It has been suggested that mutually exclusively spliced exons are more conserved than other splice events (27). In order to test this theory, we used Muscle (44) to generate pairwise alignments between APPRIS principal isoforms and alternative isoforms encompassing the 10 599 alternative events with exons greater than 42 bases used in the conservation analysis. We classified each of the 10 599 alternative events by their effect on the protein sequence; whether the alternative exons generated tandem exon duplication-derived substitutions, in-frame insertions, or non-homologous internal, C-terminal or N-terminal substitutions.
It is possible to relate these protein-level events to classical splice event types. Tandem exon duplication-derived substitutions are generated from a mixture of alternative promoter events (those at the N-terminal), alternative poly(A) usage (C-terminal) and mutually exclusive exon splice events (internal). Internal substitutions also include those generated from mutually exclusively spliced exons that have no apparent homology. All non-homologous N-terminal substitutions are the result of alternative promoter usage.
Insertions are generated from cassette exons that are longer than 14 amino acids and that preserve the frame. Alternative events generated by skipping exons present in the principal variant do not have unique exons, so are not included in this analysis. Not all exon-skipping events generate insertions; many non-homologous C-terminal substitutions are generated by alternative poly(A) splicing, but out-of-frame insertions from exon skips will also generate distinct C-terminals. Alternative exons generated from alternative 5′ or 3′ splice sites or retained introns are not included in this analysis because these types of splice events overlap exons from the principal variant.
Between tandem exon duplication-derived and non-homologous substitutions, 226 events are generated from mutually exclusively spliced exons. We detected the equivalent event in fish for 110 of these (48.7%). By way of comparison, just 505 of the remaining 10 373 events were detected in fish (4.9%). So, mutually exclusively spliced events (both homologous and non-homologous) are substantially more conserved than other splice events in fish genomes.
However, the conservation signal comes almost entirely from the tandem exon duplication-derived exons. If we divide mutually exclusively spliced events into those that involve homologous exons and those that do not, equivalent events can be found in fish for 100 of the 111 events derived from tandem exon duplications (90.9%), compared to just 10 of the 115 internal substitutions that are not (Figure 4). In fact, mutually exclusively spliced events that involve non-homologous exons are as frequently conserved in fish (8.7%) as in-frame insertions (9.5%) and non-homologous N-terminal substitutions (8.3%). It is clear that it is mutually exclusive splice events involving tandem duplicated homologous exons that are conserved, and not the mutually exclusive splice events themselves.
Figure 4.

The percentage of splice events with an orthologue in fish The percentage of splice events in which orthologues of both exons are found in at least one fish species. For insertions, the inserted exons had to be conserved in fish and a sequence without the insertion had to be annotated in a fish species. Tandem exon duplication-derived substitutions (orange) are divided into three groups: ‘internal, homologous’; ‘N-terminal, homologous’ and ‘C-terminal, homologous’ in order to allow comparisons with non-homologous substitutions in equivalent positions (yellow).
In fact, events generated from tandem exon duplications are conserved between humans and fish regardless of the splicing mechanism. In addition to the 90.1% conservation rate for mutually exclusively spliced tandem exon duplications, 74.6% of the alternative poly(A) events and 80% of the alternative promoter events that involved homologous tandem exon duplications were conserved in both humans and fish. Cross species conservation of splice events is clearly related to tandem exon duplications and not to the mutually exclusive splicing process.
A total of 182 tandem exon duplication-derived substitutions could be traced back to a last common ancestor with fish (83.5%). Just 4.17% of non-tandem exon derived events were conserved in fish. Tandem exon duplication-derived substitutions are 20 times more likely than other splice events to have a last common ancestor with fish.
Most tandem duplicated exons are ancient
We carried out further manual analysis to determine the approximate date of the last common ancestor of all tandem exon-derived duplications in our set. Analysis of the conservation of tandem duplicated exon substitutions showed that a large majority of the substitutions (84.3%) were already present in the last common ancestor of humans and sharks (jawed vertebrates), and that almost two thirds (62.3%) of the events are shared between humans and lamprey (Figure 5). In addition, over a third of (36%) were present in the chordate phylum, at the end of the Proterozoic >560 million years ago.
Figure 5.

Age of last common ancestor and two tandem exon substitutions conserved in Bilaterian clade. (A) Last common ancestors of each tandem exon derived duplication event binned by age. Age of the last common ancestor was searched manually using FlyBase, RefSeq and UniProtKB. Approximate ages were estimated from TimeTree (45), see methods section. (B) The tandem exon duplication-derived substitution event in PRKCB mapped onto the structures of Protein kinase C beta (PDB: 3pfq, in blue) and Protein kinase C eta (PDB: 3txo in orange) in cartoon form. The homologous exons code for a C-terminal arm that wraps around the kinase domain. Residues conserved in both exons in vertebrates and invertebrates are shown in stick form in purple and most are hydrophobic residues that anchor the arm to the kinase domain. Residues conserved in one of the two homologous exons and not in the other are shown in stick form in orange and blue. These residues cluster in the helix in the top left-hand corner. (C) The tandem exon duplication-derived substitution event in RAB37 mapped onto the structure of Rab-26 (PDB: 2g6b, blue), showing just the surface of the protein. The residues that are identical in the two homologous regions are shown in red and again are mainly found in the interior. Residues that differ in the homologous region (yellow) form a surface patch that is likely to allow RAB37 isoforms to interact with different proteins.
Alternative exons conserved between vertebrate and invertebrate clades are rare. However, given that so many tandem exon duplication-derived substitutions appear to have their roots in the beginnings of the vertebrate subphylum, we surmised that some might be even older. Kondrashov and Koonin had found that the C-terminal duplication in human FBLN1 and C. elegans Fbl-1 arose in the last common ancestor of the two species. Hatje et al found four mutually exclusive events that coincided over the same region as events in orthologous genes in Drosophila, but they did not investigate whether the events were orthologous (see supplementary materials).
We interrogated the Drosophila genome (33) with the 215 tandem exon duplication-derived substitutions with vertebrate conservation to see whether they had last common ancestors that pre-dated the split between human and Drosophila. Thirteen events had orthologues in Drosophila, though all but three have already been reported. Nine events came from three families, the events in the ATPase membrane Ca2+ transporter family, genes ATP2B1, ATP2B2, ATP2B3 and ATP2B4 (29), the N-terminal event in the tropomyosin family, present in genes TPM1, TPM3, TPM4 (63), and one event in the actinin family, specifically the event present in both ACTN2 and ACTN4 (64). The other four events came from FOXP1 (65), PNPLA6, PPP1R12B and RAB37 (Figure 5).
There was evidence of a last common ancestor between vertebrates and chordate species (tunicates or lancelets) for 85 tandem duplications. We searched for orthologues of these 85 duplications across the bilaterian clade. We found equivalent exons in multiple species for nine more tandem exon duplication-derived events: ATE1 (in common octopus, for example), FBLN1 (C. elegans), OGDH (Pacific oyster), P4HA1 (C. remanei), PRKCB (crown of thorns starfish, Figure 5) PRKG1 (Taurus scarab) and STX2 and STX3 (Daphnia Magna). We found equivalent exons in multiple species for the event in gene ACOX1 too, but despite having similar conserved residues, the phylogenetic tree suggested that the event had evolved three times, once in chordates, once in echinoderms and once in molluscs.
In total, we found evidence that 21 tandem exon duplication-derived substitutions (from 14 families) appeared before the split between vertebrates and invertebrates, so are at least 670 million years old. The same number of events have arisen since the split between tetrapods and ray-finned fish, 425 million years ago.
Tandem exon duplication-derived substitutions are also enriched in proteomics studies
We analysed the data from two large-scale proteomics data sets for evidence of expression of homologous exons at the protein level (see methods). In order to identify peptides, we required at least two valid peptide-spectrum matches and to identify a gene, we required at least two reliably identified non-overlapping peptides. We detected 11,928 coding genes across the two analyses. We identified those alternative splicing events that had one or more validated peptides that mapped uniquely to each side of the splice event (9), a total of 628 splice events across 522 genes. There was evidence for the translation of both exons in 81 of the tandem exon duplication-derived substitutions (34.3% of all annotated events). There was also peptide evidence for 16 of the 44 tandem exon duplication-derived substitutions annotated in the PCDH and UGT ‘gene’ clusters (36.4%), though these did not count as splice variants because they are currently annotated as separate genes.
How significant is the enrichment in tandem exon duplication-derived substitutions?
To compare the proportion of each type of alternative event detected in the proteomics analysis, we used the alternative events from exons longer than 42 bases from the conservation analysis and matched the splice events detected in the proteomics experiments with those from the conservation analysis. The results can be seen in Figure 6A. We detected peptides for 37.2% of tandem exon duplication-derived substitutions that were at least 42 bases long and 4.7% of in-frame insertions longer than 42 bases. No other splice event had a detection rate higher than 3%. Overall, we detected peptides for just 1.8% of events other than tandem exon duplication-derived substitutions.
Figure 6.
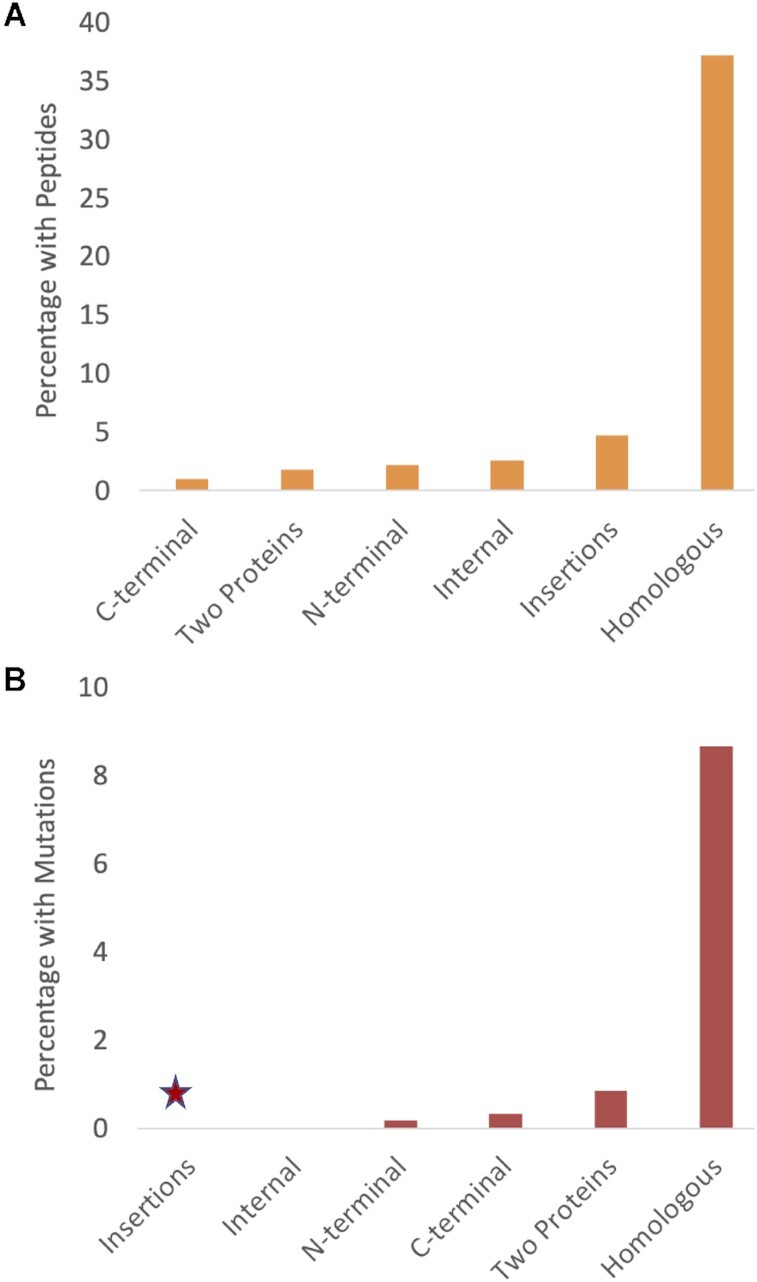
Percentage of alternative events detected in proteomics experiments and with pathogenic mutations. (A) The percentage of alternative splice events for which we detected peptides in proteomics experiments. (B) The percentage of splice events in which pathogenic or likely pathogenic mutations from ClinVar mapped to both alternative and principal exons. Insertions are marked with an asterisk because it was not possible to interrogate insertions for clinically relevant mutations that mapped to both exons.
Fisher's exact tests show that tandem exon duplication-derived substitutions are significantly more frequently detected in proteomics experiments (P-value of P < 0.00001 against all other event types). Indeed, these events were detected in proteomics experiments >20 times as frequently as all other splice events.
Even among the 615 conserved exons, we detected significantly more tandem duplication events than we would expect. We found peptides for 72 of 182 conserved tandem duplication events and for 60 of 433 other conserved events, proportionally three times as many conserved tandem exon duplication events. Again, the difference is highly significant (P-value < 0.00001).
The clinical importance of tandem exon duplication-derived substitutions
Hatje et al. (27) compared mutations in mutually exclusively spliced exons against those in all other exons. They found that both mutually exclusively spliced exons and cassette exons were significantly enriched in pathogenic mutations, which was a remarkable result. However, the result was erroneous because the authors miscalculated the number of background annotated coding exons in the human reference set by a whole order of magnitude. If they had used the correct number of annotated coding exons, both mutually exclusively spliced exons and cassette exons would have been significantly depleted in pathogenic mutations (see supplementary material).
In any case, the correct comparison is between the mutually exclusively spliced exons and non-mutually exclusively spliced exons from the same genes, rather than exons from the whole reference set. Here, we first tested whether genes with annotated tandem duplicated exon substitutions had proportionally more pathogenic mutations than other coding genes, and then we compared alternatively spliced tandem duplicated exons against all other exons from the same genes. We used the Ensembl Variant Effect Predictor (66) to map mutations in the ClinVar database (version 14th of November 2020, 67) to all exons in v33 of the GENCODE gene set. There were ClinVar entries for 10 879 coding genes (54.8% of annotated coding genes), and for 165 of the 215 genes with tandem exon duplication-derived substitutions (76.7%). Genes with tandem exon duplication-derived substitutions are enriched in the ClinVar database and this enrichment is significant (Fisher's exact test, P < 0.00001).
In order to determine whether alternatively spliced tandem duplicated exons are significantly enriched in pathogenic mutations, we concentrated solely on those genes with tandem exon duplication-derived substitutions. We compared the rate of pathogenic mutations in tandem duplicated exons with all other exons from the main transcript of each gene. We also extended the range of each exon by 5 bases at the 3′ end and 3 bases at the 5′ end to allow for intronic mutations that affected splicing. Main transcripts were determined using the APPRIS principal isoforms (34). Pathogenic ClinVar mutations mapped to 47 of the 505 tandem duplicated exons (9.3% of tandem duplicated exons) and to 840 of the 4218 coding exons from the main transcript (19.9%) across the 215 genes. This is despite the fact that tandem duplicated exons are, on average, 50% longer than exons from the principal transcripts. Fisher's exact tests show that, rather than being significantly enriched, alternatively spliced tandem duplicated exons are actually significantly depleted in pathogenic mutations.
The explanation for the depletion is simple. ClinVar pathogenic mutations map overwhelmingly to the main isoforms in each gene rather than from alternative variants; 98.8% of ‘Pathogenic’ mutations and 98.4% of ‘Likely pathogenic’ mutations map to exons that are part of APPRIS principal variants. In fact, ClinVar pathogenic mutations map to alternative exons in just 278 genes. This should not be surprising as we have already shown that principal protein isoforms are more highly expressed than alternative isoforms (68).
To determine whether tandem exon duplication-derived substitutions are more enriched in pathogenic mutations than other alternative splice events, we required ClinVar pathogenic or likely pathogenic mutations to map to both exons in a splice event. This guarantees that one of the mutations is in an alternative exon. To account for the enrichment in ClinVar mutations in genes with tandem exon duplication-derived substitutions, we used the intersection of the 10,599 events with non-overlapping alternative exons longer than 42 bases from the conservation analysis and the 10 879 genes that are annotated with ClinVar variants. We excluded insertions because it is impossible to map variants to both exons in a splice event. This left us with 6053 events, 173 of which were tandem exon substitution events.
There were just 32 events in which pathogenic or likely pathogenic mutations mapped to both the principal and alternative exons and 14 of the 32 events involved tandem duplicated exons, 8.1% of the 173 tandem exon substitutions (Figure 6B). Meanwhile, pathogenic or likely pathogenic mutations mapped to both exons in an event in just 18 of 5880 C-terminal swaps, N-terminal swaps, internal mutually exclusively spliced non-homologous swaps and genes with two distinct protein sequences (Figure 6B), just 0.3%. The most interesting result was that four of these 18 events involved the swap of one Pfam domain for a completely different functional domain. These events are extremely rare in the human genome.
Proportionally we found 27 times as many pathogenic and likely pathogenic mutations in tandem duplicated exon substitutions as we did in all other alternative events. This remarkable result is clearly significant (Fisher's exact test, P < 0.00001). Part of the reason for their importance will be that tandem duplicated exons in alternative events are more conserved, but these events were significantly enriched in pathogenic and likely pathogenic mutations even when we take this into account. In total, 394 of the 615 alternative events conserved in fish are in genes that have mutations in ClinVar, including 152 conserved tandem exon duplication events and just 242 other conserved alternative events. Thirteen of the 152 tandem exon duplication events had pathogenic and likely pathogenic mutations in both exons (8.6%), but just four of the 18 other splice events with pathogenic and likely pathogenic mutations in both exons were conserved in fish, and 4 of 242 is just 1.7%. Tandem exon duplication derived events have five times as many pathogenic mutations, even when conserved events are taken into account. Despite the tiny numbers involved, the difference again is significant (Fisher's exact test, P = 0.0016).
Three of the tandem exon duplication-derived substitutions (from genes OTOF, SCN2A and KRAS) in which pathogenic or likely pathogenic mutations map to both exons are detailed in Figure 7.
Figure 7.

Tandem duplicated exons and ClinVar pathogenic mutations. (A) View of the gene OTOF from the UCSC Genome Browser, showing coding exons (wide blue rectangles), non-coding exons (narrower blue rectangles) and introns (blue lines with arrows). Mapped ClinVar mutations are shown below the transcripts; pathogenic (P) and likely pathogenic (LP) are shown as red spots, variants of unknown significance (VUS) are dark blue, likely benign (LB) and benign (B) as green and others (OTH) as grey. The larger the spots, the larger the mutation; most mutations are single nucleotide variations. (B) Close up view of two of the transcripts from OTOF, showing just the homologous 3′ coding exons; pathogenic and likely pathogenic mutations map to both exons. (C) Close up view of SCN2A transcripts in the UCSC Genome Browser, showing homologous mutually exclusively spliced coding exons; pathogenic and likely pathogenic mutations map to both exons. (D) Close up view of KRAS transcripts in the UCSC Genome Browser, showing homologous 3′ coding exons; pathogenic and likely pathogenic mutations map to one exon, a likely pathogenic mutation maps to the other. (E) Close up of one of the residues affected by pathogenic mutations in splice isoform KRAS4B (PDB: 6ms9). The mutation changes aspartate residue 153 (shown in red) to a glycine. The aspartate plays an important role in the structure of KRAS4B, forming a salt bridge with arginine 149 (orange and blue sticks), which in turns forms hydrogen bonds with the asparagine at residue 26 (purple and red sticks). The equivalent residue to aspartate 153 in isoform KRAS4A is a glutamate, and glutamate also forms salt bridges with arginine. The likely pathogenic mutation in KRAS4A, shown in section D, changes the glutamate to valine. Neither valine nor glycine form salt bridges. (F) Close up of the second residue affected by the pathogenic mutation in splice isoform KRAS4B (PDB: 6ms9). The mutation changes phenylalanine residue 156 (shown in red) to a valine (a much smaller hydrophobic amino acid). The phenylalanine nestles in a highly hydrophobic pocket (hydrophobic residues shown here as blue sticks), which is crucial for maintaining the structure (and therefore the function) of KRAS4B. Both KRAS4A and KRAS4B have a phenylalanine residue in this position.
Fibroblast growth factor receptor 2
Of the 15 cases in which clinically relevant mutations mapped to both tandem duplicated exons, the pathologies linked to mutations were different only for two of the events. One of these was the event in FGFR2. The two fibroblast growth factor receptor isoforms (FGFR2b and FGFR2c) differ in tissue expression and in the specificity of their ligands. FGFR2c is expressed in the epithelium and is a receptor for four fibroblast growth factor subfamilies, FGF1, FGF4, FGF8 and FGF9 (69), while FGFR2b is specific to the mesenchyme and only binds ligands from the FGF7 subfamily (70).
The tandem exon duplication-derived substitution in the FGFR family (FGFR1, FGFR2 and FGFR3) affects the third immunoglobulin receptor domain, the domain that is mainly responsible for FGF binding specificity (70). The exon codes for the C-terminal half of the domain and differences between the two isoforms principally affect the two loops in contact with the FGF ligands (Figure 8). Mutations to the exon specific to FGFR2c-specific exon are related to 35 different development abnormalities including Crouzon syndrome, Jackson-Weiss syndrome and Pfeiffer syndrome. The mutation to the FGFR2b exon, a change from serine to cysteine at residue 320 (S320C) is related to lung squamous cell cancer (71). The serine is adjacent to the FGF binding site (Figure 8) and one possibility is that the change to cysteine allows the residue to make a hydrogen bond with a glutamate 112 on the FGF ligand, possibly via a bridge with a water molecule, thus making the FGF-FGFR2b interaction even more stable.
Figure 8.
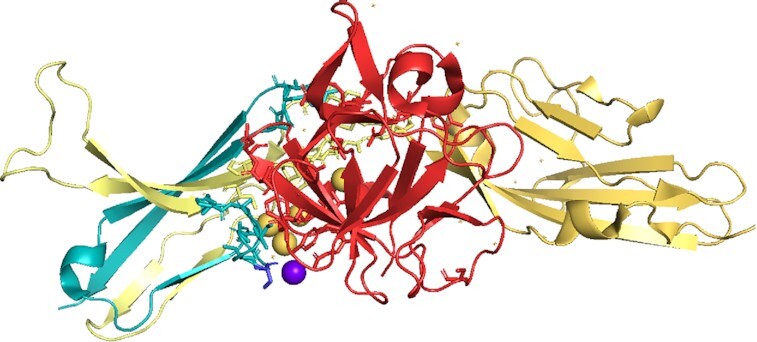
The tandem exon duplication-derived substitution in FGFR2 determines FGF binding specificity Ligand FGF10 bound to isoform FGFR2b in the PDB structure 1nun. FGF10 is in red, the constant part of FGFR2b in yellow and the tandem duplicated exon that generates half of domain 3 in teal. Residues involved in the interaction between receptor and ligand are shown in stick form. Waters that form part of hydrogen bonds are shown as spheres. Residue S320 (shown as a blue stick) has been associated with lung squamous cell cancer (71). The residue is very close to the binding site with FGF10. It is possible that the mutation to cysteine might allow it to form a hydrogen bond with a glutamate in FGF10 via a water molecule (purple sphere), thus strengthening the binding between FGFR2b and FGF10.
Curiously, the use of tandem exon duplications to modulate FGF binding specificity in fibroblast growth factor receptors seems to confer certain advantages because this pattern is not restricted to vertebrates. Arthropod species have multiple FGF ligands too, including homologues of the FGF7 subfamily, and species from Bombus to Anopheles also generate distinct alternative isoforms for FGF receptors from homologous exons, though Drosophila species seem to have lost the duplicated exon. The last common ancestor of the arthropod event can be traced back at least 350 million years. However, in arthropods it is the first exon in the third immunoglobulin receptor domain that is duplicated rather than the second exon as in vertebrates. The substitution in arthropods affects the N-terminal half of this domain and the linker (the yellow region in the third domain in Figure 8).
DISCUSSION
Tandem exon duplication-derived substitutions are a rare but strikingly important class of alternative splicing events. They provide genes with two identical copies of a functionally important protein, and with time these two proteins can evolve subtly different roles. Here, we have manually curated an exhaustive collection of alternatively spliced tandem duplicated exons from the human reference set. Alternative isoforms generated from these events are highly likely to be functionally important and we have added them to the functionally important isoforms already listed in the APPRIS database (34).
There are just 236 tandem exon duplication-derived substitutions in this set, many fewer than the ∼80 000 splice events currently annotated in the human reference gene set, yet they are highly conserved. We found that more than 90% arose from a common ancestor that predates the separation of tetrapods and fish, >425 million years ago. More than one in eleven were already present in the last common ancestor of invertebrates, >670 million years ago. These would be among the oldest splice events yet recorded.
As a control, we also manually curated the set of all annotated splice events involving alternative exons longer than 42 bases that had a last common ancestor with ray-finned fish. Fewer than 5% of these events were conserved in fish genomes. Tandem exon duplication-derived substitutions were more than 20 times more likely to be conserved in fish than other alternative splice events.
Tandem exon duplication-derived substitutions are not just ancient, they are also highly expressed. Proportionally, we detected peptide evidence for more than 20 times as many alternative isoforms generated from these homologous substitution events as for all other splice event types. We have also found that many of the conserved protein isoforms generated from these events are tissue specific; tandem exon duplication-derived substitutions make up a third of all events with tissue-specific differences at the protein level, and initial results indicate that they may have played an important role in the evolution of tissues (28).
The most important result is that tandem exon duplication-derived substitutions are remarkably enriched in pathogenic mutations. Although ClinVar (67) pathogenic mutations rarely map to alternative exons, 15 of the 35 events in which pathogenic or likely pathogenic mutations mapped to both exons in a splice event were tandem exon duplication-derived substitutions. Proportionally there were more than 27 times as many mutations affecting both exons in tandem exon duplication-derived substitutions as all other event types. Even with so few pathogenic mutations mapping to alternative exons, tandem exon duplication-derived substitution are significantly enriched in pathogenic and likely pathogenic mutations, suggesting that many tandem exon duplication-derived substitution events are likely to be clinically relevant. Even when we took the enhanced conservation of tandem exon duplication events into account, these tandem exon derived substitutions still had significantly more pathogenic mutations than all other alternative splice event types. This suggests that the reason that tandem exon substitution events are more conserved and have significantly more pathogenic mutations than other types of splice events is because they are more functionally important than other types of splice events.
Our set of tandem exon duplication-derived substitutions is not yet complete. There are at least five homologous exons in the human genome that have yet to be annotated in the GENCODE human gene set. As well as the exons in CEPT1, FAR1, SRPK1 and SLC12A1 that were identified in other studies, RefSeq annotates a homologous 5′ CDS that is conserved in sharks in gene CYRIB that none of the three previous studies reported. TTN, the largest coding gene in the human genome, also generates alternative isoforms from homologous exons, but we have not included TTN in the set because its size makes it difficult to analyse. There are also 44 events currently annotated as separate coding genes in five ‘gene clusters’. These events are also functionally important; we find peptide evidence for 36% of them. There is no logical reason for continuing to annotate the protocadherin and UDP glucuronosyltransferase family transcripts as distinct genes. These alternative transcripts produce proteins with different (but similar) N-terminals by virtue of having different promoters. The reason that they are annotated as separate genes is purely historical. Early researchers related the protocadherin families to immunoglobulins and T-cell receptors (72,73), even though the splicing mechanism is no different from PLEC, KCNAB1, FRMD4A, or many other genes that have multiple alternative promoters. No-one would suggest that the transcripts in these genes, or in genes ABR, MAST4 or UTRN, were clusters of different genes.
The three apparently mutually exclusively spliced exons in SLC12A1 and CACNB1 are intriguing. In both cases cross-species conservation suggests that all three exons are functionally important, the cDNA evidence supports mutually exclusive splicing of the exons, and they cannot be spliced together because that would cause a frameshift and lead to NMD. This would suggest that vertebrates might well have a mechanism to deal with multiple mutually exclusively spliced exons after all, though perhaps not as well developed as that of Drosophila (20).
The fact that these homologous alternatively spliced exons are both rare and highly conserved suggests that either exon duplication happens infrequently or that exon duplication is rarely fixed as part of alternative splicing events. In fact, we know that exon duplication in the human genome is a common occurrence (26), so it must be the fixing of duplicated exons as part of alternative splice events that is highly infrequent.
Lam et al. (30) made a start on the functional characterisation of isoforms derived from tandem exon duplications, but we still have little idea of the functional role of most of the isoforms generated from these events. The high level of translation and the remarkable enrichment in pathogenic mutations in these ancient alternatively spliced exons demonstrates that tandem exon duplication-derived substitutions have important roles in the cell. Many are also likely to have clinical relevance, highlighting the importance of investigating both principal and alternative exons if working with these genes.
We believe that the annotation of a complete set of tandem exon duplication-derived substitutions has gone some way towards answering some of the outstanding questions about the functional role of alternative splicing (74) and we hope that this resource can inspire research into these conserved, clinically relevant splice events.
DATA AVAILABILITY
Data is available in supplementary material.
Supplementary Material
Contributor Information
Laura Martinez Gomez, Bioinformatics Unit, Spanish National Cancer Research Centre (CNIO), C. Melchor Fernandez Almagro, 3, 28029 Madrid, Spain.
Fernando Pozo, Bioinformatics Unit, Spanish National Cancer Research Centre (CNIO), C. Melchor Fernandez Almagro, 3, 28029 Madrid, Spain.
Thomas A Walsh, Bioinformatics Unit, Spanish National Cancer Research Centre (CNIO), C. Melchor Fernandez Almagro, 3, 28029 Madrid, Spain; Eukaryotic Annotation Team, EMBL-EBI, Wellcome Genome Campus, Hinxton, Cambridgeshire CB10 1SA. UK.
Federico Abascal, Somatic Evolution Group, Wellcome Sanger Institute, Wellcome Genome Campus, Hinxton, Cambridgeshire CB10 1SA, UK.
Michael L Tress, Bioinformatics Unit, Spanish National Cancer Research Centre (CNIO), C. Melchor Fernandez Almagro, 3, 28029 Madrid, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Human Genome Research Institute of the National Institutes of Health [2 U41 HG007234]. Funding for open access charge: NHGRI [3 U41 HG007234].
Conflict of interest statement. None declared.
REFERENCES
- 1.Wang E., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S., Schroth G., Burge C.. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008; 456:470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J.. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008; 40:1413–1415. [DOI] [PubMed] [Google Scholar]
- 3.Ezkurdia I., Juan D., Rodriguez J.M., Frankish A., Diekhans M., Harrow J., Vazquez J., Valencia A., Tress M.L.. Multiple evidence strands suggest that there may be as few as 19 000 human protein-coding genes. Hum. Mol. Genet. 2014; 23:5866–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J.et al.. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D.et al.. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016; 44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson J., Castle J., Garrett-Engele P., Kan Z., Loerch P., Armour C., Santos R., Schadt E., Stoughton R., Shoemaker D.. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003; 302:2141–2144. [DOI] [PubMed] [Google Scholar]
- 7.Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A.et al.. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 8.Weatheritt R.J., Sterne-Weiler T., Blencowe B.J.. The ribosome-engaged landscape of alternative splicing. Nat. Struct. Mol. Biol. 2016; 23:1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ezkurdia I., del Pozo A., Frankish A., Rodriguez J.M., Harrow J., Ashman K., Valencia A., Tress M.L.. Comparative proteomics reveals a significant bias toward alternative protein isoforms with conserved structure and function. Mol. Biol. Evol. 2012; 29:2265–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abascal F., Ezkurdia I., Rodriguez-Rivas J., Rodriguez J.M., del Pozo A., Vázquez J., Valencia A., Tress M.L.. Alternatively spliced homologous exons have ancient origins and are highly expressed at the protein level. PLoS Comput. Biol. 2015; 11:e1004325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tress M.L., Abascal F., Valencia A.. Most alternative isoforms are not functionally important. Trends Biochem. Sci. 2017; 42:408–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buljan M., Frankish A., Bateman A.. Quantifying the mechanisms of domain gain in animal proteins. Genome Biol. 2010; 11:R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmitz J., Brosius J.. Exonization of transposed elements: a challenge and opportunity for evolution. Biochimie. 2011; 93:1928–1934. [DOI] [PubMed] [Google Scholar]
- 14.Martinez-Gomez L., Abascal F., Jungreis I., Pozo F., Kellis M., Mudge J.M., Tress M.L.. Few SINEs of life: Alu elements have little evidence for biological relevance despite elevated translation. NAR Genom. Bioinform. 2020; 2:lqz023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Avgan N., Wang J.I., Fernandez-Chamorro J., Weatheritt R.J.. Multilayered control of exon acquisition permits the emergence of novel forms of regulatory control. Genome Biol. 2019; 20:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pohl M., Bortfeldt R.H., Grützmann K., Schuster S.. Alternative splicing of mutually exclusive exons–a review. Biosystems. 2013; 114:31–38. [DOI] [PubMed] [Google Scholar]
- 17.Hatje K., Kollmar M.. Expansion of the mutually exclusive spliced exome in Drosophila. Nat. Commun. 2013; 4:2460. [DOI] [PubMed] [Google Scholar]
- 18.Sawaya M.R., Wojtowicz W.M., Andre I., Qian B., Wu W., Baker D., Eisenberg D., Zipursky S.L.. A double S shape provides the structural basis for the extraordinary binding specificity of Dscam isoforms. Cell. 2008; 134:1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerstein M.B., Rozowsky J., Yan K-K., Wang D., Cheng C., Brown J.B., Davis C.A., Hillier L., Sisu C., Li J.J.et al.. Comparative analysis of the transcriptome across distant species. Nature. 2014; 512:445–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park J.W., Graveley B.R.. Complex alternative splicing. Adv. Exp. Med. Biol. 2007; 623:50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burley S.K., Berman H.M., Kleywegt G.J., Markley J.L., Nakamura H., Velankar S.. Protein Data Bank (PDB): the single global macromolecular structure archive. Methods Mol. Biol. 2017; 1607:627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li S.A., Cheng L., Yu Y., Wang J.H., Chen Q.. Structural basis of Dscam1 homodimerization: insights into context constraint for protein recognition. Sci. Adv. 2016; 2:e1501118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kondrashov F.A., Koonin E.V.. Evolution of alternative splicing: deletions, insertions and origin of functional parts of proteins from intron sequences. Trends Genet. 2003; 19:115–119. [DOI] [PubMed] [Google Scholar]
- 25.Copley R.R.Evolutionary convergence of alternative splicing in ion channels. Trends Genet. 2004; 20:171–176. [DOI] [PubMed] [Google Scholar]
- 26.Letunic I., Copley R.R., Bork P.. Common exon duplication in animals and its role in alternative splicing. Hum. Mol. Genet. 2002; 11:1561–1567. [DOI] [PubMed] [Google Scholar]
- 27.Hatje K., Rahman R.U., Vidal R.O., Simm D., Hammesfahr B., Bansal V., Rajput A., Mickael M.E., Sun T., Bonn S., Kollmar M.. The landscape of human mutually exclusive splicing. Mol. Syst. Biol. 2017; 13:959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodriguez J.M., Pozo F., di Domenico T., Vazquez J., Tress M.L.. An analysis of tissue-specific alternative splicing at the protein level. PLoS Comp. Biol. 2020; 16:e1008287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abascal F., Tress M.L., Valencia A.. The evolutionary fate of alternatively spliced homologous exons after gene duplication. Genome Biol. Evol. 2015; 7:1392–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lam S.D., Babu M.M., Lees J., Orengo C.A.. Biological impact of mutually exclusive exon switching. PLoS Comput. Biol. 2021; 17:e1008708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cunningham F., Achuthan P., Akanni W., Allen J., Amode M.R., Armean I.M., Bennett R., Bhai J., Billis K., Boddu S.et al.. Ensembl 2019. Nucleic Acids Res. 2019; 47:D745–D751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017; 45:D158–D159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thurmond J., Goodman J.L., Strelets V.B., Attrill H., Gramates L.S., Marygold S.J., Matthews B.B., Millburn G., Antonazzo G., Trovisco V.et al.. FlyBase 2.0: the next generation. Nucleic Acids Res. 2019; 47:D759–D765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodriguez J.M., Rodriguez-Rivas J., Di Domenico T., Vázquez J., Valencia A., Tress M.L.. APPRIS 2017: principal isoforms for multiple gene sets. Nucleic Acids Res. 2018; 46:D213–D217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 36.Abascal F., Juan D., Jungreis I., Martinez L., Rigau M., Rodriguez J.M., Vazquez J., Tress M.L.. Loose ends: almost one in five human genes still have unresolved coding status. Nucleic Acids Res. 2018; 46:7070–7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pozo F., Martinez-Gomez L., Walsh T.A., Rodriguez J.M., Di Domenico T., Abascal F., Vazquez J., Tress M.L.. Assessing the functional relevance of splice isoforms. NAR Genom. Bioinform. 2021; 3:lqab044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amemiya C.T., Alföldi J., Lee A.P., Fan S., Philippe H., Maccallum I., Braasch I., Manousaki T., Schneider I., Rohner N.et al.. The African coelacanth genome provides insights into tetrapod evolution. Nature. 2013; 496:311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amores A., Catchen J., Ferrara A., Fontenot Q., Postlethwait J.H.. Genome evolution and meiotic maps by massively parallel DNA sequencing: spotted gar, an outgroup for the teleost genome duplication. Genetics. 2011; 188:799–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aparicio S., Chapman J., Stupka E., Putnam N., Chia J.M., Dehal P., Christoffels A., Rash S., Hoon S., Smit A.et al.. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002; 297:1301–1310. [DOI] [PubMed] [Google Scholar]
- 41.Howe K., Clark M.D., Torroja C.F., Torrance J., Berthelot C., Muffato M., Collins J.E., Humphray S., McLaren K., Matthews L.et al.. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013; 496:498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith J.J., Kuraku S., Holt C., Sauka-Spengler T., Jiang N., Campbell M.S., Yandell M.D., Manousaki T., Meyer A., Bloom O.E.et al.. Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution. Nat. Genet. 2013; 45:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lassmann T.Kalign 3: multiple sequence alignment of large data sets. Bioinformatics. 2019; 26:btz795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edgar R.C.MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004; 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar S., Stecher G., Suleski M., Hedges S.B.. TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017; 34:1812–1819. [DOI] [PubMed] [Google Scholar]
- 46.Parfrey L.W., Lahr D.J., Knoll A.H., Katz L.A.. Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:13624–13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soria-Carrasco V., Castresana J.. Diversification rates and the latitudinal gradient of diversity in mammals. Proc. Biol. Sci. 2012; 279:4148–4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Betancur-R R., Broughton R.E., Wiley E.O., Carpenter K., López J.A., Li C., Holcroft N.I., Arcila D., Sanciangco M., Cureton Ii J.C.et al.. The tree of life and a new classification of bony fishes. PLoS Curr. 2013; 5:ecurrents.tol.53ba26640df0ccaee75bb165c8c26288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gold D.A., Runnegar B., Gehling J.G., Jacobs D.K.. Ancestral state reconstruction of ontogeny supports a bilaterian affinity for Dickinsonia. Evol. Dev. 2015; 17:315–324. [DOI] [PubMed] [Google Scholar]
- 50.dos Reis M., Thawornwattana Y., Angelis K., Telford M.J., Donoghue P.C., Yang Z.. Uncertainty in the timing of origin of animals and the limits of precision in molecular timescales. Curr. Biol. 2015; 25:2939–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Antonelli A., Hettling H., Condamine F.L., Vos K., Nilsson R.H., Sanderson M.J., Sauquet H., Scharn R., Silvestro D., Töpel M.et al.. Toward a self-updating platform for estimating rates of speciation and migration, ages, and relationships of taxa. Syst. Biol. 2017; 66:152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delsuc F., Philippe H., Tsagkogeorga G., Simion P., Tilak M.K., Turon X., López-Legentil S., Piette J., Lemaire P., Douzery E.J.P.. A phylogenomic framework and timescale for comparative studies of tunicates. BMC Biol. 2018; 16:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim M.S., Pinto S.M., Getnet D., Nirujogi R.S., Manda S.S., Chaerkady R., Madugundu A.K., Kelkar D.S., Isserlin R., Jain S.et al.. A draft map of the human proteome. Nature. 2014; 509:575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang D., Eraslan B., Wieland T., Hallström B., Hopf T., Zolg D.P., Zecha J., Asplund A., Li L.H., Meng C.et al.. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019; 15:e8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deutsch E.W., Csordas A., Sun Z., Jarnuczak A., Perez-Riverol Y., Ternent T., Campbell D.S., Bernal-Llinares M., Okuda S., Kawano S.et al.. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic. Acids. Res. 2017; 45:D1100–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eng J.K., Jahan T.A., Hoopmann M.R.. Comet: an open-source MS/MS sequence database search tool. Proteomics. 2013; 13:22–24. [DOI] [PubMed] [Google Scholar]
- 57.The M., MacCoss M.J., Noble W.S., Käll L.. Fast and accurate protein false discovery rates on large-scale proteomics data sets with percolator 3.0. J. Am. Soc. Mass. Spectrom. 2016; 27:1719–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ezkurdia I., Calvo E., Del Pozo A., Vázquez J., Valencia A., Tress M.L.. The potential clinical impact of the release of two drafts of the human proteome. Expert Rev. Proteomics. 2015; 12:579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ezkurdia I., Vázquez J., Valencia A., Tress M.L.. Analyzing the first drafts of the human proteome. J. Proteome Res. 2014; 13:3854–3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gabler F., Nam S.Z., Till S., Mirdita M., Steinegger M., Söding J., Lupas A.N., Alva V.. Protein sequence analysis using the MPI bioinformatics toolkit. Curr. Protoc. Bioinformatics. 2020; 72:e108. [DOI] [PubMed] [Google Scholar]
- 61.Tweedie S., Braschi B., Gray K., Jones T.E.M., Seal R.L., Yates B., Bruford E.A.. Genenames.org: the HGNC and VGNC resources in 2021. Nucleic Acids Res. 2021; 49:D939–D946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Resch A., Xing Y., Alekseyenko A., Modrek B., Lee C.. Evidence for a subpopulation of conserved alternative splicing events under selection pressure for protein reading frame preservation. Nucleic Acids Res. 2004; 32:1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Irimia M., Maeso I., Gunning P.W., Garcia-Fernàndez J., Roy S.W.. Internal and external paralogy in the evolution of tropomyosin genes in metazoans. Mol. Biol. Evol. 2010; 27:1504–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lek M., MacArthur D.G., Yang N., North K.N.. Phylogenetic analysis of gene structure and alternative splicing in alpha-actinins. Mol. Biol. Evol. 2010; 27:773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Santos M.E., Athanasiadis A., Leitão A.B., DuPasquier L., Sucena E.. Alternative splicing and gene duplication in the evolution of the FoxP gene subfamily. Mol. Biol. Evol. 2011; 28:237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R., Thormann A., Flicek P., Cunningham F.. The ensembl variant effect predictor. Genome Biol. 2016; 17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W.et al.. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018; 46:D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ezkurdia I., Rodriguez J.M., Carrillo-de Santa Pau E., Vázquez J., Valencia A., Tress M.L.. Most highly expressed protein-coding genes have a single dominant isoform. J. Proteome Res. 2015; 14:1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beenken A., Mohammadi M.. The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009; 8:235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zinkle A., Mohammadi M.. Structural biology of the FGF7 subfamily. Front. Genet. 2019; 10:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liao R.G., Jung J., Tchaicha J., Wilkerson M.D., Sivachenko A., Beauchamp E.M., Liu Q., Pugh T.J., Pedamallu C.S., Hayes D.N.et al.. Inhibitor-sensitive FGFR2 and FGFR3 mutations in lung squamous cell carcinoma. Cancer Res. 2013; 73:5195–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kohmura N., Senzaki K., Hamada S., Kai N., Yasuda R., Watanabe M., Ishii H., Yasuda M., Mishina M., Yagi T.. Diversity revealed by a novel family of cadherins expressed in neurons at a synaptic complex. Neuron. 1998; 20:1137–1151. [DOI] [PubMed] [Google Scholar]
- 73.Wu Q., Maniatis T.. A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell. 1999; 97:779–790. [DOI] [PubMed] [Google Scholar]
- 74.Tress M.L., Abascal F., Valencia A.. Alternative splicing may not be the key to proteome complexity. Trends Biochem. Sci. 2017; 42:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is available in supplementary material.