


Abstract
Newly synthesized mRNA is translated during its export through the nuclear pore complex, when its 5′-cap structure is still bound by the nuclear cap-binding complex (CBC), a heterodimer of cap-binding protein (CBP) 80 and CBP20. Despite its critical role in mRNA surveillance, the mechanism by which CBC-dependent translation (CT) is regulated remains unknown. Here, we demonstrate that the CT initiation factor (CTIF) is tethered in a translationally incompetent manner to the perinuclear region by the DEAD-box helicase 19B (DDX19B). DDX19B hands over CTIF to CBP80, which is associated with the 5′-cap of a newly exported mRNA. The resulting CBP80–CTIF complex then initiates CT in the perinuclear region. We also show that impeding the interaction between CTIF and DDX19B leads to uncontrolled CT throughout the cytosol, consequently dysregulating nonsense-mediated mRNA decay. Altogether, our data provide molecular evidence supporting the importance of tight control of local translation in the perinuclear region.
INTRODUCTION
In the nucleus of mammalian cells, newly synthesized pre-mRNAs are processed into mature mRNAs via multiple steps, including 5′ capping, splicing, and 3′ polyadenylation. The 5′-cap structure of mRNAs is recognized by the nuclear cap-binding complex (CBC), a heterodimer of cap-binding protein (CBP) 80 and CBP20 (1–4). After complete processing in the nucleus, mRNAs are exported to the cytoplasm via the nuclear pore complex (NPC) (5). During or immediately after the export, the mRNAs are subjected to the pioneer (or first) round of translation, which frequently occurs with the CBC bound to the 5′-cap of the mRNAs. CBC-dependent translation (CT) preferentially involves the CT initiation factor (CTIF), which links the CBC and eukaryotic translation initiation factor (eIF) 3, promoting the recruitment of a small ribosomal subunit to the 5′-end of the mRNA (6).
In the cytoplasm, the CBC associated with the 5′-cap structure of the mRNAs is eventually replaced by eIF4E, the major cytoplasmic cap-binding protein, in a translation-independent manner and mediated by importins α and β (7,8). A recent study further showed that the CBC-to-eIF4E replacement step is regulated by the coordinated actions of UPF1 and Staufen1, a negative and positive regulator, respectively (9). The resulting eIF4E-bound mRNAs are actively translated using eIF4G, which acts as a scaffold protein to link eIF4E and eIF3 to recruit the small ribosomal subunit to the mRNA (10).
CT plays an important role in nonsense-mediated mRNA decay (NMD), a well-characterized mRNA surveillance pathway that degrades a subset of normal mRNAs as well as faulty mRNAs containing a premature termination codon (PTC) (2,11–14). In addition, several recent studies have suggested that CT is associated with a protein surveillance mechanism called the aggresome–autophagy pathway, in which newly generated misfolded polypeptides are selectively transported to the perinuclear aggresome and eventually eliminated from the cell by autophagy (15–17).
The DEAD-box RNA helicase 19B (DDX19B, a human homolog of yeast Dbp5) consists of an N-terminal extension helix and two RecA-like domains, which are structurally similar to each other and participate in ATP binding and hydrolysis (18,19). DDX19B localizes in the cytoplasmic side of the NPC in the perinuclear region via its association with NUP214 (a cytoplasmic NPC component) and plays a critical role in mRNA export from the nucleus to the cytoplasm with the help of GLE1 and inositol 1,2,3,4,5,6-hexakisphosphate (20–22). Furthermore, DDX19B interacts with eukaryotic release factor 1 (eRF1) and is involved in translation termination (23–25). In addition, DDX19B stabilizes ribosome complexes with eukaryotic elongation factor (eEF) 1 and eEF2 (26).
In this study, we demonstrate that CT is restricted to the perinuclear region via an interaction between CTIF and DDX19B. Disruption of the CTIF–DDX19B interaction causes CTIF redistribution and uncontrolled CT throughout the cytoplasm, consequently leading to the dysregulation of NMD. Therefore, our data elucidate the importance of tight control of local translation in the perinuclear region.
MATERIALS AND METHODS
Cell culture and generation of stable cell lines
HeLa (female; ATCC), HEK293T (fetal; ATCC) and HEK293FT (fetal; ATCC) cells stably expressing Myc-eIF4E were cultured in Dulbecco's modified Eagle's medium (DMEM; Capricorn Scientific) supplemented with 10% (v/v) fetal bovine serum (Capricorn Scientific) and 1% (v/v) of a penicillin/streptomycin solution (Capricorn Scientific).
To create HEK293FT cells stably expressing Myc-eIF4E, HEK293FT cells were co-transfected with a plasmid expressing Myc-eIF4E and a plasmid encoding a puromycin resistance gene. Two days later, the cells were serially diluted and grown in DMEM supplemented with 1 μg/ml puromycin (MilliporeSigma). Single-cell colonies were isolated and analyzed by western blotting. The resulting cell lines were cultured in DMEM with 0.5 μg/ml puromycin.
Plasmid construction
The following plasmids described previously have been used: pcDNA3-FLAG and pcDNA3-FLAG-CBP80 (27); pcDNA3-FLAG-eIF4E, pcDNA3-FLAG-CTIF, pcDNA3-FLAG-CTIF (1–305), pcDNA3-FLAG-CTIF (306–598) (6), pmCMV-Gl-Norm and -Ter, pmCMV-GPx1-Norm and -Ter, and phCMV-MUP (6); pcDNA3.1-HA (28); pCMV-Myc-eIF4E (29); pcDNA3.1-CBP80-HA (9); pCI-neo (Promega); and pCMV-Myc (Clontech).
pcDNA3-FLAG-CTIF-F460A was generated by site-directed mutagenesis of pcDNA3-FLAG-CTIF as a template.
To construct pcDNA3-FLAG-CTIF-WT-KASH1 and pcDNA3-FLAG-CTIF-F460A-KASH1, an in vitro-synthesized DNA fragment encoding the KASH1 domain (including the transmembrane domain) of the nesprin-1 gene (NM_182961.4) was inserted into the 3′-end of CTIF cDNA in pcDNA3-FLAG-CTIF or pcDNA3-FLAG-CTIF-F460A, respectively.
To generate pCI-neo-DDX19B-WT-HA, pCI-neo-HA was first constructed by inserting a fragment encoding HA from pcDNA3.1-HA into pCI-neo. Next, the cDNA fragment encoding full-length DDX19B (NM_007242) was amplified by PCR from pOTB7-DDX19B (Korea Human Gene Bank; Daejeon, Korea). The PCR-amplified fragment was inserted into pCI-neo-HA. pCI-neo-DDX19B-W6A/V10A-HA was constructed by site-directed mutagenesis of pCI-neo-DDX19B-WT-HA as a template.
pCI-neo-DDX19BR-WT-HA and pCI-neo-DDX19BR-W6A/V10A-HA were constructed by replacing DDX19B siRNA target sequences from 5′-CCTGAACTGAAGCTAGCTT-3′ to 5′-CCAGAGTTAAAATTGGCAT-3′.
pCI-neo-DDX19B(1–300)-HA expressing the N-terminal half of DDX19B and pCI-neo-DDX19B(301–479)-HA expressing the C-terminal half of DDX19B were generated by replacing the DDX19B-WT sequences in pCI-neo-DDX19B-WT-HA with a PCR-amplified fragment encoding the N- and C-terminal halves of DDX19B, respectively.
To construct pBiFC-HA-CTIF-WT-VC155 and pBiFC-HA-CTIF-F460A-VC155, a cDNA fragment encoding full-length CTIF or CTIF-F460A, respectively, was inserted between HA and VC155 of pBiFC-HA-VC155 (30).
To construct pBiFC-FLAG-DDX19B-VN173, a PCR-amplified DDX19B fragment was inserted between FLAG and VN173 of pBiFC-FLAG-VN173 (30).
DNA or siRNA transfection
HeLa cells were transiently transfected with plasmid(s) using either Lipofectamine 2000 (Life Technologies) or jetPRIME® (Polyplus Transfection) as described previously (31–33). For immunoprecipitation (IP) or polysome fractionation analysis using HEK293T or HEK293FT cells, the calcium phosphate transfection method was employed.
To downregulate endogenous proteins, cells were transfected with 100 nM in vitro-synthesized siRNAs (GenePharma) using Lipofectamine 3000 (Invitrogen). The following siRNA sequences were used in this study: 5′-r(GGAAGAAGCUAAAGAGAAA)d(TT)-3′ (human CBP80 siRNA), 5′-r(AAAAACAAAUCAUUAGUAG)d(TT)-3′ (human DDX19A siRNA), 5′-r(CCUGAACUGAAGCUAGCUU)d(TT)-3′ (human DDX19B siRNA) and 5′-r(ACAAUCCUGAUCAGAAACC)d(TT)-3′ (nonspecific control siRNA).
Antibodies
Primary antibodies against the following proteins were utilized for western blotting, IPs, or immunostaining [listed in the format ‘protein name (catalog number, supplier)’]: DDX19A (ARP36455_T100, Aviva System Biology); DDX19B (ab70305, Abcam); lamin A/C (sc-376248, Santa Cruz Biotechnology); eIF4E (2067, Cell Signaling Technology); eIF3b (sc-16377, Santa Cruz Biotechnology), β-actin (A5441, Sigma); GAPDH (LF-PA0212, AbFrontier); FLAG (A8592 or F1804, Sigma); HA (11867431001, Roche); Myc (9E10; OP10L, Calbiochem); U1 snRNP 70 (sc-390899, Santa Cruz Biotechnology); CBP80, eIF4G1 and CTIF (6); and eIF4A3 (32).
Immunoprecipitations and RNA immunoprecipitation
These procedures were performed as previously described (15,31,33). Briefly, HEK293T cells were harvested and resuspended in NET-2 buffer [50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride (Sigma), 2 mM benzamidine hydrochloride (Sigma), 0.05% NP-40 (IGEPAL® CA-630; Sigma), 10 mM sodium fluoride (Sigma), and 0.25 mM sodium orthovanadate (Sigma)]. The resuspended cells were lysed by sonication and centrifuged for 10 min at 4°C and 13 800 × g. Cell lysates were pre-cleared using protein G or A agarose 4B (Incospharm) for 1 h at 4°C. The pre-cleared supernatant was incubated with antibody-conjugated beads for 3 h at 4°C. The protein-bound beads were washed five times with NET-2 buffer, and the proteins were eluted with 2× sample buffer [10% β-mercaptoethanol, 4% SDS, 100 mM Tris–HCl (pH 6.8), 15% glycerol and 0.008% bromophenol blue]. The coimmunoprecipitated proteins were analyzed by western blotting. For quantification, the signal intensities of western blot bands were quantified using ImageJ software (National Institutes of Health, Bethesda, MD).
The RNA-IP protocol was similar to the IP protocol, except that tRNA-saturated and antibody-conjugated beads were used. After RNA-IPs, coimmunoprecipitated RNA and proteins were eluted with 2× sample buffer. The eluted coimmunoprecipitated RNAs were extracted with phenol, chloroform and isoamyl alcohol and subjected to cDNA synthesis using RevertAid Reverse Transcriptase (Thermo Fisher Scientific).
Quantitative real-time RT-PCR
qRT-PCR analyses were performed with gene-specific oligonucleotides and the Light Cycler 480 SYBR Green I Master Mix (Roche) on a Light Cycler 480 II machine (Roche). The following gene-specific oligonucleotides were used in this study: 5′-ACCACCGTAGAACGCAGATCG-3′ (sense) and 5′-CTTCTCACCATTCACCTCGCACTT-3′ (antisense) for the amplification of GPx1-Norm or -Ter mRNAs; 5′-ACCACCGTAGAACGCAGATCG-3′ (sense) and 5′-GGGTTTAGTGGTACTTGTGAGC-3′ (antisense) for the amplification of Gl-Norm or -Ter mRNAs; 5′-CTGATGGGGCTCTATG-3′ (sense) and 5′-TCCTGGTGAGAAGTCTCC-3′ (antisense) for the amplification of MUP mRNAs; 5′-GTGAAAAGCCTCCTTCTGG-3′ (sense) and 5′-CTGTAGAACTGAGGCAAGG-3′ (antisense) for the amplification of COMMD7 mRNAs; 5′-CAACATGACGCTGGAAGAGC-3′ (sense) and 5′-TCAATGGCCAAGAGGCAGAG-3′ (antisense) for the amplification of GADD45B mRNAs; and 5′-TGGCAAATTCCATGGCACC-3′ (sense) and 5′-AGAGATGATGACCCTTTTG-3′ (antisense) for the amplification of GAPDH mRNAs.
Polysome fractionation analysis
Polysome fractionation of HEK293FT cells was carried out as described previously (6,32). Briefly, HEK293FT cells were cultured in 150 mm culture dishes and preincubated in a culture medium containing 100 μg/ml cycloheximide (Sigma) for 10 min before cell harvest. The cells were washed with PBS containing 100 μg/ml cycloheximide and collected by centrifugation at 2000 × g for 10 min. The pellet was resuspended and lysed in polysome cell extraction buffer [50 mM MOPS (3-(N-morpholino) propanesulfonic acid; Sigma), 15 mM MgCl2 (Sigma), 150 mM NaCl (Sigma), 100 μg/ml cycloheximide, 0.5% (v/v) Triton X-100 (Sigma), 1 mg/ml heparin sodium salt (Sigma), 0.2 U/μl RiboLock RNase inhibitor (Thermo Fisher Scientific), 2 mM phenylmethanesulfonyl fluoride (Sigma), and 1 mM benzamidine hydrochloride (Sigma)]. After centrifugation at 13 000 × g for 10 min, the supernatant was loaded onto a sucrose gradient bar (10–50%) and subjected to centrifugation at 36 000 rpm for 2 h (Optima XE-90 Ultracentrifuge, Beckman Coulter) with maximum speed acceleration and natural deceleration. For collecting fractions according to A254, a Foxy Jr Density Gradient System (Teledyne ISCO) was used. The polysome-fractionated samples were collected as 11 fractions for western blotting (Figures 5 and 6) or seven fractions for IP experiments (Figure 7). The intensities of the western blot images in each fraction were quantitated using Multi Gauge (Fujifilm), and the values of each fraction were presented as a percentage of the sum of the intensities.
Figure 5.

CTIF unable to interact with DDX19B co-fractionates to a higher extent with polysomes. (A) Polysome fractionation analysis of FLAG-CTIF-WT or of one of its variants. Cytoplasmic extracts of HEK293FT cells transiently expressing either FLAG-CTIF-WT or FLAG-CTIF-F460A were subjected to polysome fractionation, followed by analysis of the protein samples in each fraction by western blotting with the indicated antibodies. The peaks corresponding to the small ribosomal subunits, large ribosomal subunits, and monosomes are denoted as 40S, 60S, and 80S, respectively. (B) Quantitative representation of the relative distribution of FLAG-CTIF-WT or FLAG-CTIF-F460A across the fractions. The intensities of FLAG-CTIF-WT or FLAG-CTIF-F460A in each fraction of panel A were quantitated. To rule out any possibility or bias caused by differences in protein expression and exposure time during western blotting, relative levels of FLAG-CTIF-WT (red) and FLAG-CTIF-F460A (blue) are presented as a percentage of the sum of the intensities of the respective protein. Mean values and standard deviations from two biological replicates are shown. (C) Quantitative representation of the relative levels of endogenous CBP80, as determined in panel B. (D) Quantitative representation of relative levels of endogenous eIF4E, as determined in panel B.
Figure 6.

DDX19B inhibits polysome association of CTIF in the perinuclear region. (A) Schematic illustration of FLAG-CTIF-WT-KASH1 and FLAG-CTIF-F460A-KASH1. The FLAG tag, CBP80-interacting domain, MIF4G domain, and KASH1 domain are depicted. The position of the F460A mutation is indicated by a black arrow. (B) Immunostaining of FLAG-CTIF-WT and its variants. HeLa cells transiently expressing the indicated protein were subjected to immunostaining with α-FLAG antibody (green) and DAPI (blue); n = 2; Scale bar, 10 μm. (C) Polysome fractionation analysis of cytoplasmic extracts of HEK293FT cells transiently expressing FLAG-CTIF-WT-KASH1 or FLAG-CTIF-F460A-KASH1. The protein samples in each fraction after polysome fractionation experiments were analyzed by western blotting with α-FLAG antibody. (D) Quantitative representation of relative distributions of FLAG-CTIF-WT-KASH1 or FLAG-CTIF-F460A-KASH1. Mean values and standard deviations from three biological replicates are shown; n = 3; #, not significant; *P < 0.05, **P < 0.01.
Figure 7.
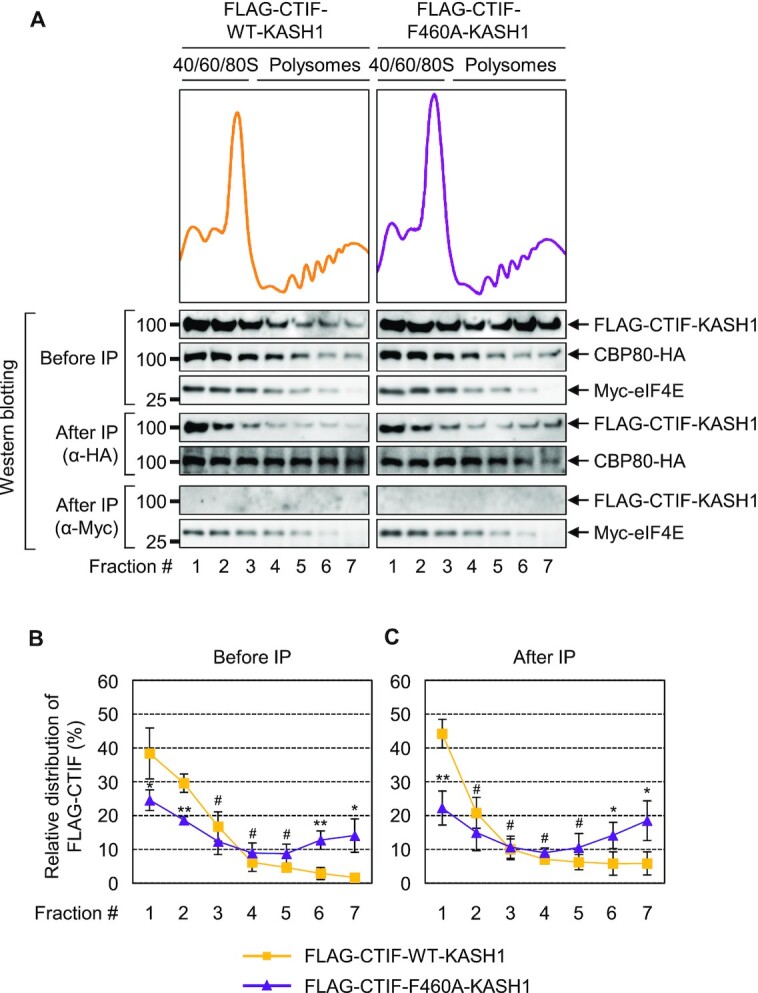
Inhibition of polysome association of CTIF by DDX19B mainly occurs on CBC-bound mRNA. HEK293FT cells stably expressing Myc-eIF4E were transiently co-transfected with a plasmid expressing CBP80-HA and a plasmid expressing either FLAG-CTIF-WT-KASH1 or FLAG-CTIF-F460A-KASH1. The cytoplasmic extracts were subjected to polysome fractionation. Next, total fractions were pooled into seven fractions and subjected to IP with either α-HA antibody or α-Myc antibody. (A) Relative distribution of the indicated proteins. Protein samples in each fraction before or after IP were analyzed by western blotting. (B and C) Quantitative representation of the relative distribution of FLAG-CTIF-WT-KASH1 and FLAG-CTIF-F460A-KASH1 across the gradient before (B) and after IP (C), as shown in Figure 5B–D; n = 3.
Nucleocytoplasmic fractionation
Nuclear and cytoplasmic fractions were obtained from HEK293T cells, as previously described (34). Briefly, cells were lysed with hypotonic buffer [10 mM Tris–HCl (pH 7.4), 10 mM NaCl, 0.1% Triton X-100 (Sigma), 10 mM EDTA (pH 8.0), 1 mM phenylmethylsulfonyl fluoride (Sigma) and 2 mM benzamidine hydrochloride (Sigma)] and incubated for 10 min on ice. After centrifugation at 13 800 × g for 10 min at 4°C, the supernatant was considered as the cytoplasmic fraction. The pellets were washed three times with a hypotonic buffer and centrifuged at 13 800 × g for 3 min at 4°C. The resuspended pellet was sonicated and centrifuged for 10 min at 4°C at 13 800 × g. The supernatant was considered as the nuclear fraction.
Yeast two-hybrid assay
Yeast two-hybrid (Y2H) screening assays were performed by Panbionet (Pohang, Korea).
Immunostaining
This procedure was performed on HeLa cells as described previously (15). The cells were treated with formaldehyde (Sigma, F8775) and Triton X-100 (0.5%; Sigma, T8787) for fixation and permeabilization, respectively. Next, the cells were blocked with bovine serum albumin (BovoStar) and incubated for 1 h with a primary antibody and for another 1 h with secondary antibodies conjugated with Alexa Fluor 488, Alexa Fluor 568 or rhodamine. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Biotium). The cells were visualized under an LSM 510 Meta, LSM700 or LSM 800 microscope (Carl Zeiss). Zeiss LSM Image Browser and Zen 2.1 (black; Carl Zeiss) were used for the analysis of immunostained cell images. Fluorescence intensities were measured across the indicated lengths and directions using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Bimolecular fluorescence complementation (BiFC) assay
This assay was conducted as described previously (30,35,36). To minimize the background signal of VFP, HeLa cells transiently transfected with BiFC plasmids were incubated for 8 h at 37°C and then fixed in 2% formaldehyde for 10 min at room temperature.
Statistics
Two-tailed, equal-sample variance Student's t-test was performed for statistical analysis, with the difference being considered statistically significant at *P < 0.05 and **P < 0.01. In most cases, data obtained from at least two independently performed biological replicates were analyzed, unless indicated otherwise in the figure legends. Data are presented as the mean ± standard deviation.
RESULTS
Identification of DDX19B as a CTIF-interacting protein
To gain more insights into the molecular mechanism underlying CT, we searched for CTIF-interacting proteins. To this end, we conducted yeast two-hybrid screening using (i) the GAL4 DNA-binding domain (BD)-fused CTIFΔN (lacking the N-terminal half of CTIF) as bait and (ii) the GAL4 transcription activation domain (AD)-fused human thymus cDNA library. In two independent yeast two-hybrid screening assays, 47 independent positive colonies were obtained. Among them, 19, 7 and 17 colonies contained a plasmid encoding DDX19B, DDX19A and stem–loop binding protein (SLBP), respectively (Supplementary Figure S1). The remaining colonies contained a plasmid encoding RPS20, MIF4GD, DDX50 or ZNF26. Notably, the rapid degradation of replication-dependent histone mRNAs via an interaction between CTIF and SLBP largely occurs on CBC-associated mRNAs (36).
Because DDX19B (and its paralogue, DDX19A) was screened out most frequently in our yeast two-hybrid screening assays, we next validated the interaction between CTIF and DDX19B (and DDX19A). First, a yeast two-hybrid assay involving BD-CTIFΔN and either AD-DDX19B or AD-DDX19A yielded positive signals in three different assays, indicating an interaction between these two proteins: (i) blue color on a medium containing X-gal (Figure 1A, the filter assay), (ii) growth on a medium lacking uracil (Figure 1A, SD-LWU) and (iii) growth on a medium lacking adenine (Figure 1A, SD-LWA). Second, endogenous DDX19B was found to be preferentially enriched in immunoprecipitations (IPs) of FLAG-CTIF (Figure 1B) in an RNase A-resistant manner, indicating that this interaction is not mediated by RNA. In addition, endogenous CTIF was preferentially copurified with endogenous DDX19B (Figure 1C). In agreement with previous studies (6,29,32,36), CBP80, eIF3b (a component of the eIF3 complex), and eIF4A3 (a component of the exon junction complex), but not eIF4E and eIF4G1, were also specifically detected in the IPs of FLAG-CTIF in an RNase A treatment-resistant manner (Figure 1B and C). Confirming the efficiency of RNase A treatment, semiquantitative reverse-transcription PCR (sqRT-PCR) showed almost complete depletion of endogenous GAPDH mRNA after RNase A treatment (Figure 1B, lower panel). Finally, DDX19B was found to be preferentially enriched in the IPs of CTIF wild-type (WT) or CTIF 306–598 (corresponding to CTIFΔN) but not of CTIF 1–305 (the N-terminal half of CTIF; Figure 1D). In addition, endogenous CTIF selectively coimmunoprecipitated with DDX19B-WT-HA or the N-terminal half of DDX19B (1–300) but not with the C-terminal half of DDX19B (301–479) (Figure 1E). Collectively, our data indicate that the C-terminal MIF4G domain of CTIF interacts with the N-terminal half of DDX19B.
Figure 1.

CTIF interacts with DDX19B. (A) Yeast two-hybrid analysis using BD-CTIFΔN and AD-DDX19. The yeast strain PBN204 was co-transformed with two plasmids expressing the indicated BD- and AD-fusion proteins. Yeast cells harboring the two plasmids were selected on a synthetic defined (SD) medium lacking leucine and tryptophan (Master plate; SD-LW). In our yeast two-hybrid system, a specific interaction between the BD- and AD-fusion proteins enables (i) the appearance of a blue color (LacZ expression; the filter assay), (ii) the growth on an SD medium lacking leucine, tryptophan, and uracil (SD-LWU), and (iii) the growth on an SD medium lacking leucine, tryptophan, and adenine (SD-LWA). The well-characterized dimerization of polypyrimidine tract–binding protein served as the positive control (+), and empty vectors expressing only BD and AD served as the negative control (–). (B) Immunoprecipitation (IP) of FLAG-CTIF before or after RNase A treatment. HEK293T cells were transiently transfected with a plasmid expressing either FLAG alone or FLAG-tagged CTIF-WT. Two days later, the cells were harvested, lysed, and the cell extracts were either treated or not treated with RNase A before IP with anti-(α-) FLAG antibody. The protein samples before or after IP were analyzed by western blotting with antibodies against the indicated proteins. Efficient RNA degradation by the treatment with RNase A was assessed by detecting endogenous GAPDH mRNA using semiquantitative reverse-transcription PCR (sqRT-PCR). (C) IP of endogenous DDX19B before or after RNase A treatment. As performed in panel B, except that IP experiments were carried out with α-DDX19B antibody or nonspecific rabbit IgG (rIgG). To show that western blots were semiquantitative, the cell extracts before IP were serially diluted by 3-fold and loaded in the four leftmost lanes. (D) IP of FLAG-CTIF-WT or its deletion variants. As performed in panel B, except that the cells were transiently transfected with a plasmid expressing FLAG, FLAG-CTIF-WT, or one of its deletion variants, and RNase A treatment was applied before IP. Band intensities in each western blot image were quantified. Intensities of coimmunoprecipitated DDX19B and CBP80 were normalized to those of immunoprecipitated FLAG-CTIF-WT or its variants. The normalized levels of coimmunoprecipitated DDX19B and CBP80 in the IP of FLAG-CTIF-WT were arbitrarily set to 1.0. (E) IP of DDX19B-WT-HA or one of its deletion variants. As performed in panel D, except that the cells were transfected with a plasmid expressing DDX19B-WT-HA or one of its deletion variants, and IPs were carried out with the α-HA antibody or nonspecific rat IgG. The cell extracts were treated with RNase A before IP. Representative western blot images from two biological replicates (n = 2) are shown.
CBP80 partially competes with DDX19B to bind to CTIF
Interestingly, an increased amount of endogenous DDX19B and CBP80 was enriched in the IPs of the C-terminal half and N-terminal half of CTIF, respectively, compared with that of the full-length CTIF-WT (Figure 1D), and CBP80 was not detectable in the IPs of endogenous DDX19B (Figure 1C). These observations prompted us to hypothesize that CBP80 might compete with DDX19B for binding to CTIF, even though the N- and C-terminal halves of CTIF are sufficient for interacting with CBP80 and DDX19B, respectively (Figure 1).
To test this hypothesis, we carried out IPs in the extracts of cells that transiently expressed FLAG-CTIF and were either depleted or not of endogenous DDX19B by means of a specific small interfering RNA (siRNA). Downregulation of DDX19B to ∼10% of the level in undepleted cells increased the amount of coimmunoprecipitated CBP80 by 2.6-fold in the IPs of FLAG-CTIF (Figure 2A). Conversely, ∼2.5-fold more DDX19B was found to be enriched in the IP of FLAG-CTIF when the cells were depleted of endogenous CBP80 (Figure 2B), indicating a competition between CBP80 and DDX19B for binding to CTIF. Notably, DDX19A also had the ability to compete with CBP80 for binding to CTIF, showing an additive effect with DDX19B (Supplementary Figure S2). However, the ability of DDX19A was relatively weak compared with that of DDX19B (Supplementary Figure S2), suggesting a predominant role of DDX19B in the competition.
Figure 2.

CBP80 competes with DDX19B for binding to CTIF. (A) IP of FLAG-CTIF in the extracts of cells depleted of DDX19B. HEK293T cells either depleted or not depleted of endogenous DDX19B were transiently transfected with a plasmid expressing either FLAG or FLAG-CTIF-WT. The relative levels of coimmunoprecipitated DDX19B and CBP80 were quantified and are presented at the bottom of each image. Data are presented as the mean ± standard deviation; n = 2. (B) IPs of FLAG-CTIF in the extracts of cells depleted of CBP80. As performed in panel A, except that HEK293T cells were either depleted or not depleted of endogenous CBP80; n = 2. (C) Schematic illustration of DDX19B, CTIF and CBP80. CTIF consists of a CBP80-interacting domain (yellow) and a middle domain of eIF4G (MIF4G) domain (orange). The point mutations characterized in this study are indicated by red arrowheads. (D) Structure of a Danio rerio SLIP1 (DrSLIP1) and DrDBP5 complex (PDB: 4JHJ). The hydrophobic property is presented in the molecular surface of DrSLIP1: the more intense the red color is, the stronger is the hydrophobicity. Ribbon diagrams of DrDBP5 are shown in blue. Two important residues (W6 and V10) of DrDBP5 for interacting with DrSLIP1 are specified. In addition, the amino acid residues involved in the hydrophobic interaction between DrDBP5 and DrSLIP1 are indicated. The predicted amino acid residues important for an interaction between Homo sapiens CTIF (HsCTIF; homologous to DrSLIP1) and HsDDX19B (homologous to DrDBP5) are indicated in brackets. (E) IP of DDX19B-WT-HA or its W6A/V10A variant. Extracts of HEK293T cells expressing either DDX19B-WT-HA or DDX19B-W6A/V10A-HA were subjected to IPs with either α-HA antibody or rat IgG; n = 2. (F) IP of FLAG-CTIF or its F460A variant. Extracts of HEK293T cells expressing FLAG, FLAG-CTIF-WT or FLAG-CTIF-F460A were subjected to IP with the α-FLAG antibody; n = 2.
The C-terminal MIF4G domain of Homo sapiens CTIF (HsCTIF) is similar to the MIF4G domain of SLBP-interacting protein 1 (SLIP1) in the amino acid sequence, and HsDDX19B is homologous to the DBP5 of zebrafish (Danio rerio, Dr). Based on available structural data on the interaction between DrSLIP1 and DrDBP5 (37) (Figure 2C and D), we determined the amino acid residues that were most likely critical for the CTIF–DDX19B interaction. Along with the sequence alignment analysis of HsDDX19B and DrDBP5, we predicted that the amino acids, W6 and V10, of DDX19B would be important for its binding to CTIF (Supplementary Figure S3A). In addition, after sequence alignment analysis of the MIF4G domain of HsCTIF, DrSLIP1, and HsSLIP1 (Supplementary Figure S3B), the amino acid F460 of CTIF was predicted to be important for its binding to DDX19B, since the corresponding residue of DrSLIP1 (F87) is required for binding to DrDBP5 (Figure 2D). As predicted, substitutions of tryptophan and valine with alanines at positions 6 and 10 of DDX19B (DDX19B-W6A/V10A) abrogated the interaction between DDX19B and CTIF (Figure 2E). Consistently, the substitution of phenylalanine with alanine at position 460 of CTIF (CTIF-F460A) also disrupted the interaction between CTIF and DDX19B (Figure 2F). Notably, approximately 2.8-fold more endogenous CBP80 was found to be enriched in the IP of CTIF-F460A compared to the IP of CTIF-WT, providing further evidence that CBP80 and DDX19B compete for binding to CTIF. It is likely that the observed competition may be due to steric hindrance between CBP80 and DDX19B, even though the minimal regions responsible for binding to CBP80 and DDX19B were assigned to the N- and C-terminal halves of CTIF, respectively.
DDX19B tethers CTIF to the perinuclear region
It has been previously shown that DDX19B localizes throughout the cytoplasm with pronounced enrichment in the perinuclear region owing to its interaction with NUP214 (38,39). In addition, CTIF is largely enriched in the cytoplasmic side of the nuclear envelope (6) as well as in the perinuclear aggresome (15,16) with some degree of cytoplasmic accumulation. Based on these observations, we tested whether the interaction between CTIF and DDX19B is responsible for the perinuclear localization of CTIF or DDX19B. Confocal microscopy revealed that the majority of CTIF-WT overlapped with endogenous lamin A/C, which is a component of the nuclear lamina and is located on the nucleoplasmic side of the inner nuclear membrane (6). In contrast, CTIF-F460A was evenly distributed throughout the cytoplasm (Figure 3A), indicating that the perinuclear localization of CTIF requires its interaction with DDX19B. In support of this notion, downregulation of endogenous DDX19B caused redistribution of endogenous CTIF from the perinuclear region into the entire cytoplasm (Figure 3B).
Figure 3.

DDX19B tethers CTIF to the perinuclear region. (A) Immunostaining of FLAG-CTIF-WT or FLAG-CTIF-F460A. HeLa cells transiently expressing FLAG, FLAG-CTIF-WT, or FLAG-CTIF-F460A were fixed with formaldehyde and stained with α-FLAG (red) and α-lamin A/C antibodies (green). Nuclei were visualized by DAPI (blue). Representative confocal images from three biological replicates (n = 3) are provided. (B) Immunostaining of endogenous CTIF in HeLa cells depleted of endogenous DDX19B. HeLa cells were either depleted or not depleted of endogenous DDX19B. The cells were stained with α-CTIF antibody (red) and α-lamin A/C antibodies (green); n = 2. (C) Immunostaining of DDX19B-WT-HA or DDX19B-W6A/V10A-HA; n = 3. (D) In vivo bimolecular fluorescence complementation (BiFC) assay. HeLa cells were transiently co-transfected with plasmids expressing DDX19B-VN and either CTIF-WT-VC or CTIF-F460A-VC. The Venus fluorescent signal (yellow) was examined by confocal microscopy; n = 2; scale bar, 10 μm.
Next, we monitored the relative distribution of DDX19B-WT-HA and DDX19B-W6A/V10A-HA. Both proteins showed similar intracellular distributions: localization throughout the cytoplasm with enrichment in the perinuclear region (Figure 3C). These data indicate that, unlike DDX19B-dependent perinuclear localization of CTIF, the perinuclear localization of DDX19B is independent of its interaction with CTIF.
We also carried out a BiFC assay (30,35), which allows us not only to determine a specific interaction between two proteins of interest within the cell but also to visualize where an interaction between two proteins takes place in the cell. To this end, in HeLa cells, we transiently expressed two fusion proteins: (i) a DDX19B-fused N-terminal fragment of Venus [an enhanced yellow fluorescent protein (VFP); VN] and (ii) either a CTIF-WT–fused or CTIF-F460A–fused C-terminal fragment of VFP (VC). In this assay, an interaction between the two studied fusion proteins brings VN and VC into close proximity, allowing their assembly into a functionally active VFP, which emits VFP signals. The BiFC results revealed that DDX19B-VN and CTIF-WT-VC yielded ample VFP signals in the perinuclear region, whereas DDX19B-VN and CTIF-F460A-VC emitted hardly any detectable VFP signals (Figure 3D), suggesting a specific interaction between CTIF and DDX19B in the perinuclear region. Collectively, our results demonstrate that CTIF interacts with DDX19B predominantly in the perinuclear region and that this interaction is crucial for the perinuclear localization of CTIF.
Failure of an interaction between DDX19B and CTIF causes accumulation of CBP80 in the cytoplasm
The failure of an interaction between DDX19B and CTIF releases a perinuclear CTIF to the cytoplasm (Figure 3A and B). Because CBP80 competes with DDX19B for binding to CTIF, the released cytoplasmic CTIF may be associated with CBP80 and is expected to drive the accumulation of CBP80 in the cytoplasm. As expected, we observed that a small portion of endogenous CBP80 was redistributed from the nucleus to the cytoplasm, either when the cells were overexpressed with CTIF-F460A, but not with CTIF-WT (Figure 4A), or when the cells were depleted of DDX19B. Remarkably, the expression of siRNA-resistant DDX19B (DDX19BR)-WT, but not DDX19BR-W6A/V10A, in DDX19B-depleted cells reversed the redistribution of endogenous CBP80 (Figure 4B). The observed partial redistribution of CBP80 was further validated by western blotting using nuclear and cytoplasmic fractions (Supplementary Figure S4). Collectively, these data indicate that the CTIF-DDX19B interaction helps the nuclear enrichment of CBP80, possibly by blocking the release of CTIF into the cytoplasm.
Figure 4.

CTIF-DDX19B interaction helps the nuclear enrichment of CBP80. (A) Immunostaining of endogenous CBP80 and FLAG, FLAG-CTIF-WT or FLAG-CTIF-F460A. HeLa cells transiently expressing FLAG, FLAG-CTIF-WT or FLAG-CTIF-F460A were stained with α-CBP80 antibody (red), α-FLAG antibody (green), and DAPI (blue). The intensities of endogenous CBP80 (red) and FLAG-CTIF (green) along the white arrows are quantitated and presented in the rightmost panel as a percentage of the sum of each intensity. Mean values and standard deviations of the relative intensities obtained from 30 individual cells are shown as solid lines and semi-transparent lines, respectively. Scale bar, 10 μm; n = 3; Scale bar, 10 μm. (B) Immunostaining of endogenous CBP80 in DDX19B-depleted cells. HeLa cells either depleted or not depleted of DDX19B were transiently transfected with a plasmid expressing HA, DDX19BR-WT-HA or DDX19BR-W6A/V10A-HA. After fixation, the cells were stained with α-CBP80 antibody (red) and DAPI (blue); n = 3; scale bar, 20 μm.
DDX19B inhibits polysome association of the CT complex
What is the molecular function of the CTIF–DDX19B interaction? Based on a previous study showing that CTIF associates with polysomes through its direct interaction with CBP80 and thus preferentially directs efficient CT (6) and the present study suggesting that disruption of the CTIF–DDX19B interaction causes the accumulation of CBP80 in the cytoplasm, we investigated the possible role of DDX19B in CT. For this purpose, we assessed the relative distributions of CTIF-WT and CTIF-F460A in polysome gradients (Figure 5). Polysome fractionation experiments with the cytoplasmic extracts of HEK293FT cells revealed that a greater amount of CTIF-F460A than CTIF-WT was detected in the polysomal fractions (Figure 5A and B). In addition, the expression of CTIF-F460A increased the association of CBP80 with polysomes (Figure 5A and C) without significantly affecting the relative distribution of eIF4E (Figure 5A and D). These data suggest that the interaction between CTIF and DDX19B prevents the polysome association of the CT complex.
DDX19B inhibits the polysome association of CTIF in the perinuclear region
Since the perinuclear localization of CTIF depends on DDX19B (Figure 3), we reasoned that the increased association of CTIF-F460A with polysomes might be caused either by its inability to interact with DDX19B or by the relocalization of CTIF from the perinuclear region to the cytoplasm. To clarify the underlying mechanism, we designed FLAG-CTIF constructs (either WT or F460A) fused to the Klarsicht, ANC-1, and syne homology 1 (KASH1) domain of nesprin-1 (Figure 6A). The KASH1 domain has been shown to target proteins on the cytoplasmic side of the outer nuclear membrane (40). As expected, although CTIF-WT and CTIF-F460A manifested perinuclear and cytoplasmic distributions, respectively, both CTIF-WT-KASH1 and CTIF-F460A-KASH1 predominantly localized to the perinuclear region (Figure 6B). Overall, the fusion of CTIF-WT or CTIF-F460A with KASH1 increased the association between CTIF and CBP80 and between CTIF and eIF3b compared with CTIF-WT or CTIF-F460A lacking KASH1 (Supplementary Figure S5), suggesting that artificial tethering of CTIF to the perinuclear region increases the chance that CTIF interacts with CBP80 and eIF3b. Remarkably, CTIF-F460A-KASH1 was found to be more strongly enriched in the polysome fractions than CTIF-WT-KASH1 (Figure 6C and D). Taken together, these data indicate that the association of the CT complex with polysomes is mostly influenced by both the ability of CTIF to interact with DDX19B and the perinuclear localization of CTIF.
Inhibition of the polysomal association of CTIF by DDX19B occurs on CBC-bound mRNAs
Because CTIF is a specific factor responsible for efficient CT (6), we next investigated whether DDX19B-mediated inhibition of CTIF association with polysomes occurs during CT. To this end, we conducted polysome fractionation experiments on cytoplasmic extracts of cells that stably expressed Myc-eIF4E and transiently expressed CBP80-HA and either FLAG-CTIF-WT-KASH1 or FLAG-CTIF-F460A-KASH1 (Figure 7). The polysome-fractionated samples were then subjected to IPs with either α-HA or α-Myc antibody to separate CBP80-bound messenger ribonucleoproteins (mRNPs) and eIF4E-bound mRNPs. Specific and efficient IPs of CBP80-HA or Myc-eIF4E and the relative distributions of coimmunoprecipitated FLAG-CTIF were analyzed by western blotting (Figure 7A). In agreement with our results from IPs (Figures 1 and 2), both CTIF-WT-KASH1 and CTIF-F460A-KASH1 selectively coimmunopurified with CBP80-HA but not with Myc-eIF4E (Figure 7A), supporting that CTIF selectively associates with the CBC-bound mRNP complex. Furthermore, consistent with the polysome fractionation experiments in Figure 6, CTIF-F460A-KASH1 was found to be more enriched in the polysome fractions before IP of CBP80-HA in comparison to CTIF-WT-KASH1 (Figure 7A and B). Notably, strong enrichment of CTIF-F460A-KASH1 in the polysome fractions was still observed even after the IP of CBP80-HA (Figure 7A and C). These data indicate that DDX19B-mediated inhibition of CTIF association with polysomes largely occurs on CBC-bound mRNPs rather than on eIF4E-bound mRNPs.
Interaction between CTIF and DDX19B inhibits NMD
Given that (i) CTIF is a specific factor involved in CT (6), (ii) CT plays an important role in NMD (1,2,11) and (iii) DDX19B plays an inhibitory role in CT (Figures 5 and 6), we hypothesized that an interaction between DDX19B and CTIF most likely reduces NMD efficiency. To test this hypothesis, we compared the effects of overexpressed CTIF-WT or CTIF-F460A on NMD efficiency of two NMD reporter mRNAs: globin mRNA (Figure 8A), which harbors either no PTC (Norm) or a PTC (Ter) at the 39th codon of the globin gene (Gl) (41) and glutathione peroxidase 1 (GPx1) mRNA (Figure 8B), which harbors either no PTC (Norm) or PTC (Ter) at the 46th codon of GPx1 (42). Results of qRT-PCR revealed that overexpression of CTIF-F460A significantly reduced the abundance of Gl-Ter mRNA (by ∼3.1-fold; Figure 8A) and GPx1-Ter mRNA (by ∼2.7-fold; Figure 8B) as compared to overexpression of CTIF-WT. Furthermore, overexpression of CTIF-F460A increased the efficiency of NMD of previously known endogenous NMD substrates (COMMD7 mRNA and GADD45B mRNA) (Figure 8C and D). These results indicate that the CTIF-DDX19B interaction plays an inhibitory role in NMD.
Figure 8.

Interaction between CTIF and DDX19B affects NMD. (A) The effect of overexpressed FLAG-CTIF-WT or FLAG-CTIF-F460A on NMD of Gl reporter mRNAs. HEK293T cells were co-transfected with a plasmid expressing the Gl NMD reporter (either Norm or Ter), a phCMV-MUP reference plasmid, and a plasmid expressing FLAG, FLAG-CTIF-WT, or FLAG-CTIF-F460A. The levels of Gl mRNA were normalized to those of MUP mRNA, and the normalized levels of Gl Norm mRNA were arbitrarily set to 100%; n = 3; **P < 0.01. (B) The effect of overexpressed FLAG-CTIF-WT or FLAG-CTIF-F460A on NMD of GPx1 reporter mRNAs. As performed in panel A, except that GPx1 NMD reporter mRNAs were analyzed; n = 3; **P < 0.01. (C and D) The influence of overexpressed FLAG-CTIF-WT or FLAG-CTIF-F460A on endogenous NMD substrates, COMMD7 mRNA, and GADD45B mRNA. The levels of COMMD7 mRNA (C) and GADD45B mRNA (D) were normalized to those of GAPDH mRNA; n = 3; **P < 0.01. (E–I) Complementation experiments using DDX19B siRNA and siRNA-resistant DDX19BR-HA. HEK293T cells either depleted or not depleted of DDX19B were transiently co-transfected with a plasmid expressing NMD reporter (either Norm or Ter), phCMV-MUP reference plasmid, and plasmid expressing HA, DDX19BR-WT-HA, or DDX19BR-W6A/V10A-HA. Specific downregulation of DDX19B and expression of DDX19BR-HA comparable to that of endogenous DDX19B are shown in panel E. Normalized levels of Gl mRNA (F), GPx1 mRNA (G), endogenous COMMD7 mRNA (H) and endogenous GADD45B mRNA (I) were analyzed; n = 3; *P < 0.05; **P < 0.01.
DDX19B can act on multiple steps of translation, either positively or negatively. For instance, DDX19B is known to interact with eRF1, consequently promoting translation termination (23–26). In contrast, we found that DDX19B targets the initiation step of CT, inhibiting the efficiency of CT. Because of the opposite functions of DDX19B at different translation stages, it would be difficult to interpret the data obtained after DDX19B downregulation. To pinpoint the effect of the CTIF-DDX19B interaction on NMD, we carried out complementation experiments using DDX19B siRNA and DDX19BR either WT or W6A/V10A variant (Figure 8E–I). Downregulation of DDX19B inhibited NMD of reporter mRNAs by ∼2.3-fold (Figure 8F and G) and endogenous NMD substrates by ∼1.7–1.9-fold (Figure 8H and I), suggesting that the inhibitory effect of DDX19B on CT is masked by its promoting effect on translation termination. Intriguingly, whereas the expression of DDX19BR-WT almost completely rescued NMD efficiency, DDX19B-depleted cells expressing DDX19BR-W6A/V10A exhibited more efficient NMD than those expressing DDX19BR-WT. Therefore, these data indicate that the CTIF-DDX19B interaction inhibits NMD.
NMD inhibition by CTIF-DDX19B interaction mostly occurs on CBC-bound mRNA
We next investigated whether the increase in NMD efficiency upon CTIF-F460A overexpression occurs on mRNA bound by either CBC or eIF4E. To this end, we carried out IPs in the extracts of cells expressing Gl reporter mRNA (either Norm or Ter) and CTIF (either WT or F460A) with either an α-CBP80 or α-eIF4E antibody. Endogenous CBP80, but not eIF4E, was specifically enriched in CBP80 IP, whereas endogenous eIF4E, but not CBP80, was specifically enriched in eIF4E IP, confirming the specificity of the IPs of endogenous CBP80 or eIF4E (Figure 9A). The results of qRT-PCR of coimmunoprecipitated mRNAs showed significant reductions in the abundance of both CBP80-bound and eIF4E-bound Gl-Ter reporter mRNAs upon overexpression of CTIF-F460A as compared to those upon overexpression of CTIF-WT (Figure 9B). Considering that CBC-bound mRNAs are precursors of eIF4E-bound mRNAs (11,43), our observations suggest that DDX19B largely inhibits the NMD of CBC-bound mRNAs.
Figure 9.
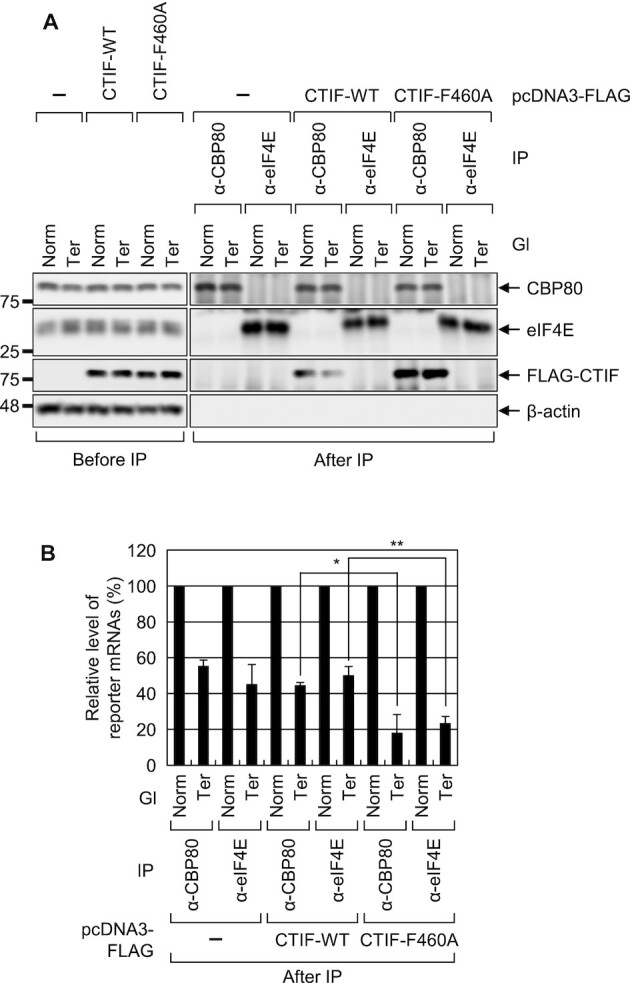
Overexpression of CTIF-F460A increases NMD of mRNAs bound by CBP80. As performed in Figure 8A, except that the extracts of the cells were subjected to IPs with either α-CBP80 or α-eIF4E antibody. Total cell protein and RNA were analyzed by western blotting (A) and qRT-PCR (B), respectively; n = 3; *P < 0.05, **P < 0.01.
DISCUSSION
Translation within a specialized intracellular compartment (e.g. the endoplasmic reticulum or axon of neurons) is subject to strict regulations. Otherwise, translation in inappropriate cellular compartments may cause cellular defects and proteotoxic stress (44,45). In this regard, during or immediately after mRNA export, the regulation of appropriate assembly of RNA-binding proteins into complexes with newly exported mRNAs in the perinuclear region might be critical for cell viability or functionality. In this study, we revealed that CTIF interacts with DDX19B through the C-terminal MIF4G domain of CTIF and the N-terminal extension region of DDX19B (Figures 1 and 2). The binding of CTIF to DDX19B tethers CTIF to the perinuclear region (Figure 3) and hinders the interaction between CBP80 and CTIF (Figure 2), thereby inhibiting the association of CTIF with polysomes in the perinuclear region (Figure 5–7). Consequently, uncontrolled CT throughout the cytoplasm is prevented, and CT-coupled NMD is brought under tight control (Figures 8 and 9).
A previous study (6) and the present study showed that CBP80 and DDX19B bind to the N- and C-terminal halves of CTIF, respectively. Nonetheless, our results uncovered a competition between CBP80 and DDX19B for binding to CTIF (Figures 1 and 2). The observed competition may result from a possible conformational change in CTIF after it interacts with either CBP80 or DDX19B. Alternatively, the mutually exclusive interactions may be due to steric hindrance between CBP80 and DDX19B. Given that (i) CBP80 binds to a newly synthesized mRNA in the nucleus, (ii) the mRNA is exported to the cytoplasm with CBP80 bound to the 5′-cap structure and (iii) both CTIF and DDX19B are mostly enriched in the perinuclear region, it is likely that the CTIF-DDX19B interaction in the perinuclear region is disrupted when CBP80-bound mRNA emerges on the cytoplasmic side of the nuclear envelope (Figure 10). The resulting CBP80-CTIF-associated mRNA may then recruit eIF3 and a small ribosomal subunit primed for translation initiation (6,29,32), thereby initiating CT in the perinuclear region. If the mRNA harbors a PTC, it may be rapidly degraded via NMD in the perinuclear region during CT. In the support of this idea, temporal and spatial characterization of NMD by single-RNA fluorescent in situ hybridization revealed that a PTC-containing β-globin reporter mRNA is rapidly degraded immediately after its export to the cytoplasm (46). After CT, CTIF may translocate back to the cytoplasmic side of the nuclear envelope through its interaction with DDX19B. In this way, DDX19B may keep CTIF tethered to the perinuclear region and allow for local CT of newly synthesized mRNAs being exported. Concomitantly, cytoplasmic CBP80 enters the nucleus and binds to newly synthesized mRNAs for the next round of CT (Figure 4). Although less effective than DDX19B, DDX19A also contributes to the above-mentioned regulation in an additive manner with DDX19B.
Figure 10.

Working model of DDX19B-mediated CT regulation in the perinuclear region. See text in the Discussion for more details.
Failure of CTIF to interact with DDX19B causes the release of CTIF from the perinuclear region and its distribution across the entire cytoplasm (Figure 3). The released CTIF may be more accessible to CBP80, thus leading to uncontrolled CT in inappropriate cellular compartments. Accordingly, NMD would then be less restricted to the perinuclear region and could instead proceed throughout the cytoplasm, resulting in an overall higher efficiency (Figures 8 and 9). Active NMD in inappropriate cellular compartments may be deleterious to the cell because it is believed that NMD acts as a double-edged sword, and its efficiency should be properly regulated depending on the cellular environment (14,47). For instance, proper regulation of NMD efficiency is crucial for animal development, self-renewal of embryonic or neural stem cells, and differentiation of stem cells (48–52). Therefore, inappropriate NMD regulation caused by the lack of a controlled hand-over of CTIF from DDX19B to CBP80 may be detrimental to normal cell functions. Alternatively, the NMD substrates that escape tight regulation through the CTIF-DDX19B interaction may be subject to another surveillance pathway, that is, NMD occurring on eIF4E-associated mRNAs (53,54). Future studies will address the molecular details of local translation in the perinuclear region.
DATA AVAILABILITY
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary Material
Contributor Information
Yeonkyoung Park, Creative Research Initiatives Center for Molecular Biology of Translation, Korea University, Seoul 02841, Republic of Korea; Division of Life Sciences, Korea University, Seoul 02841, Republic of Korea.
Joori Park, Creative Research Initiatives Center for Molecular Biology of Translation, Korea University, Seoul 02841, Republic of Korea; Division of Life Sciences, Korea University, Seoul 02841, Republic of Korea.
Hyun Jung Hwang, Creative Research Initiatives Center for Molecular Biology of Translation, Korea University, Seoul 02841, Republic of Korea; Division of Life Sciences, Korea University, Seoul 02841, Republic of Korea.
Leehyeon Kim, Division of Life Sciences, Korea University, Seoul 02841, Republic of Korea.
Kwon Jeong, Creative Research Initiatives Center for Molecular Biology of Translation, Korea University, Seoul 02841, Republic of Korea; Division of Life Sciences, Korea University, Seoul 02841, Republic of Korea.
Hyun Kyu Song, Division of Life Sciences, Korea University, Seoul 02841, Republic of Korea.
Simone C Rufener, Department of Chemistry and Biochemistry, University of Bern, 3012 Bern, Switzerland.
Oliver Mühlemann, Department of Chemistry and Biochemistry, University of Bern, 3012 Bern, Switzerland.
Yoon Ki Kim, Creative Research Initiatives Center for Molecular Biology of Translation, Korea University, Seoul 02841, Republic of Korea; Division of Life Sciences, Korea University, Seoul 02841, Republic of Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Research Foundation (NRF) of Korea grant funded by the Korean government (Ministry of Science, ICT and Future Planning) [NRF-2015R1A3A2033665, NRF-2018R1A5A1024261]; Korea UniversityGrant. Funding for open access charge: National Research Foundation (NRF) of Korea [NRF-2015R1A3A2033665].
Conflict of interest statement. None declared.
REFERENCES
- 1.Maquat L.E., Tarn W.Y., Isken O.. The pioneer round of translation: features and functions. Cell. 2010; 142:368–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryu I., Kim Y.K.. Translation initiation mediated by nuclear cap-binding protein complex. BMB Reports. 2017; 50:186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonatopoulos-Pournatzis T., Cowling V.H.. Cap-binding complex (CBC). Biochem. J. 2014; 457:231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muller-McNicoll M., Neugebauer K.M.. Good cap/bad cap: how the cap-binding complex determines RNA fate. Nat. Struct. Mol. Biol. 2014; 21:9–12. [DOI] [PubMed] [Google Scholar]
- 5.Katahira J.Nuclear export of messenger RNA. Genes. 2015; 6:163–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim K.M., Cho H., Choi K., Kim J., Kim B.W., Ko Y.G., Jang S.K., Kim Y.K.. A new MIF4G domain-containing protein, CTIF, directs nuclear cap-binding protein CBP80/20-dependent translation. Genes Dev. 2009; 23:2033–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato H., Maquat L.E.. Remodeling of the pioneer translation initiation complex involves translation and the karyopherin importin beta. Genes Dev. 2009; 23:2537–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dias S.M., Wilson K.F., Rojas K.S., Ambrosio A.L., Cerione R.A.. The molecular basis for the regulation of the cap-binding complex by the importins. Nat. Struct. Mol. Biol. 2009; 16:930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeong K., Ryu I., Park J., Hwang H.J., Ha H., Park Y., Oh S.T., Kim Y.K.. Staufen1 and UPF1 exert opposite actions on the replacement of the nuclear cap-binding complex by eIF4E at the 5′ end of mRNAs. Nucleic Acids Res. 2019; 47:9313–9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tahmasebi S., Khoutorsky A., Mathews M.B., Sonenberg N.. Translation deregulation in human disease. Nat. Rev. Mol. Cell Biol. 2018; 19:791–807. [DOI] [PubMed] [Google Scholar]
- 11.Ishigaki Y., Li X., Serin G., Maquat L.E.. Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell. 2001; 106:607–617. [DOI] [PubMed] [Google Scholar]
- 12.Maquat L.E., Hwang J., Sato H., Tang Y.. CBP80-promoted mRNP rearrangements during the pioneer round of translation, nonsense-mediated mRNA decay, and thereafter. Cold Spring Harb. Symp. Quant. Biol. 2010; 75:127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim Y.K., Maquat L.E.. UPFront and center in RNA decay: UPF1 in nonsense-mediated mRNA decay and beyond. RNA. 2019; 25:407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurosaki T., Popp M.W., Maquat L.E.. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019; 20:406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park J., Park Y., Ryu I., Choi M.H., Lee H.J., Oh N., Kim K., Kim K.M., Choe J., Lee C.et al.. Misfolded polypeptides are selectively recognized and transported toward aggresomes by a CED complex. Nat. Commun. 2017; 8:15730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park Y., Park J., Kim Y.K.. Crosstalk between translation and the aggresome-autophagy pathway. Autophagy. 2017; 14:1079–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park Y., Park J., Hwang H.J., Kim B., Jeong K., Chang J., Lee J.B., Kim Y.K.. Nonsense-mediated mRNA decay factor UPF1 promotes aggresome formation. Nat. Commun. 2020; 11:3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collins R., Karlberg T., Lehtio L., Schutz P., van den Berg S., Dahlgren L.G., Hammarstrom M., Weigelt J., Schuler H.. The DEXD/H-box RNA helicase DDX19 is regulated by an {alpha}-helical switch. J. Biol. Chem. 2009; 284:10296–10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tieg B., Krebber H.. Dbp5 - from nuclear export to translation. Biochim. Biophys. Acta. 2013; 1829:791–798. [DOI] [PubMed] [Google Scholar]
- 20.Napetschnig J., Kassube S.A., Debler E.W., Wong R.W., Blobel G., Hoelz A.. Structural and functional analysis of the interaction between the nucleoporin Nup214 and the DEAD-box helicase Ddx19. PNAS. 2009; 106:3089–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montpetit B., Thomsen N.D., Helmke K.J., Seeliger M.A., Berger J.M., Weis K.. A conserved mechanism of DEAD-box ATPase activation by nucleoporins and InsP6 in mRNA export. Nature. 2011; 472:238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin D.H., Correia A.R., Cai S.W., Huber F.M., Jette C.A., Hoelz A.. Structural and functional analysis of mRNA export regulation by the nuclear pore complex. Nat. Commun. 2018; 9:2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gross T., Siepmann A., Sturm D., Windgassen M., Scarcelli J.J., Seedorf M., Cole C.N., Krebber H.. The DEAD-box RNA helicase Dbp5 functions in translation termination. Science (New York, N.Y.). 2007; 315:646–649. [DOI] [PubMed] [Google Scholar]
- 24.Beissel C., Neumann B., Uhse S., Hampe I., Karki P., Krebber H.. Translation termination depends on the sequential ribosomal entry of eRF1 and eRF3. Nucleic Acids Res. 2019; 47:4798–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beissel C., Grosse S., Krebber H.. Dbp5/DDX19 between translational readthrough and nonsense mediated decay. Int. J. Mol. Sci. 2020; 21:1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mikhailova T., Shuvalova E., Ivanov A., Susorov D., Shuvalov A., Kolosov P.M., Alkalaeva E.. RNA helicase DDX19 stabilizes ribosomal elongation and termination complexes. Nucleic Acids Res. 2017; 45:1307–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oh N., Kim K.M., Cho H., Choe J., Kim Y.K.. Pioneer round of translation occurs during serum starvation. Biochem. Biophys. Res. Commun. 2007; 362:145–151. [DOI] [PubMed] [Google Scholar]
- 28.Cho H., Han S., Park O.H., Kim Y.K.. SMG1 regulates adipogenesis via targeting of staufen1-mediated mRNA decay. Biochim. Biophys. Acta. 2013; 1829:1276–1287. [DOI] [PubMed] [Google Scholar]
- 29.Choe J., Oh N., Park S., Lee Y.K., Song O.K., Locker N., Chi S.G., Kim Y.K.. Translation initiation on mRNAs bound by nuclear cap-binding protein complex CBP80/20 requires interaction between CBP80/20-dependent translation initiation factor and eukaryotic translation initiation factor 3g. J. Biol. Chem. 2012; 287:18500–18509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shyu Y.J., Liu H., Deng X., Hu C.D.. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. BioTechniques. 2006; 40:61–66. [DOI] [PubMed] [Google Scholar]
- 31.Cho H., Park O.H., Park J., Ryu I., Kim J., Ko J., Kim Y.K.. Glucocorticoid receptor interacts with PNRC2 in a ligand-dependent manner to recruit UPF1 for rapid mRNA degradation. PNAS. 2015; 112:E1540–E1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choe J., Ryu I., Park O.H., Park J., Cho H., Yoo J.S., Chi S.W., Kim M.K., Song H.K., Kim Y.K.. eIF4AIII enhances translation of nuclear cap-binding complex-bound mRNAs by promoting disruption of secondary structures in 5′UTR. PNAS. 2014; 111:E4577–E4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park O.H., Park J., Yu M., An H.T., Ko J., Kim Y.K.. Identification and molecular characterization of cellular factors required for glucocorticoid receptor-mediated mRNA decay. Genes Dev. 2016; 30:2093–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ryu I., Won Y.S., Ha H., Kim E., Park Y., Kim M.K., Kwon D.H., Choe J., Song H.K., Jung H.et al.. eIF4A3 phosphorylation by CDKs Affects NMD during the Cell Cycle. Cell Rep. 2019; 26:2126–2139. [DOI] [PubMed] [Google Scholar]
- 35.Kerppola T.K.Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 2006; 1:1278–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choe J., Kim K.M., Park S., Lee Y.K., Song O.K., Kim M.K., Lee B.G., Song H.K., Kim Y.K.. Rapid degradation of replication-dependent histone mRNAs largely occurs on mRNAs bound by nuclear cap-binding proteins 80 and 20. Nucleic Acids Res. 2013; 41:1307–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.von Moeller H., Lerner R., Ricciardi A., Basquin C., Marzluff W.F., Conti E.. Structural and biochemical studies of SLIP1-SLBP identify DBP5 and eIF3g as SLIP1-binding proteins. Nucleic Acids Res. 2013; 41:7960–7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmitt C., von Kobbe C., Bachi A., Pante N., Rodrigues J.P., Boscheron C., Rigaut G., Wilm M., Seraphin B., Carmo-Fonseca M.et al.. Dbp5, a DEAD-box protein required for mRNA export, is recruited to the cytoplasmic fibrils of nuclear pore complex via a conserved interaction with CAN/Nup159p. EMBO J. 1999; 18:4332–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.von Moeller H., Basquin C., Conti E.. The mRNA export protein DBP5 binds RNA and the cytoplasmic nucleoporin NUP214 in a mutually exclusive manner. Nat. Struct. Mol. Biol. 2009; 16:247–254. [DOI] [PubMed] [Google Scholar]
- 40.Wilhelmsen K., Ketema M., Truong H., Sonnenberg A.. KASH-domain proteins in nuclear migration, anchorage and other processes. J. Cell Sci. 2006; 119:5021–5029. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J., Sun X., Qian Y., Maquat L.E.. Intron function in the nonsense-mediated decay of beta-globin mRNA: indications that pre-mRNA splicing in the nucleus can influence mRNA translation in the cytoplasm. RNA. 1998; 4:801–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moriarty P.M., Reddy C.C., Maquat L.E.. Selenium deficiency reduces the abundance of mRNA for Se-dependent glutathione peroxidase 1 by a UGA-dependent mechanism likely to be nonsense codon-mediated decay of cytoplasmic mRNA. Mol. Cell. Biol. 1998; 18:2932–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lejeune F., Ishigaki Y., Li X., Maquat L.E.. The exon junction complex is detected on CBP80-bound but not eIF4E-bound mRNA in mammalian cells: dynamics of mRNP remodeling. EMBO J. 2002; 21:3536–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Costa C.J., Willis D.E.. To the end of the line: Axonal mRNA transport and local translation in health and neurodegenerative disease. Dev. Neurobiol. 2018; 78:209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Costa E.A., Subramanian K., Nunnari J., Weissman J.S.. Defining the physiological role of SRP in protein-targeting efficiency and specificity. Science (New York, N.Y.). 2018; 359:689–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trcek T., Sato H., Singer R.H., Maquat L.E.. Temporal and spatial characterization of nonsense-mediated mRNA decay. Genes Dev. 2013; 27:541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han X., Wei Y., Wang H., Wang F., Ju Z., Li T.. Nonsense-mediated mRNA decay: a ‘nonsense’ pathway makes sense in stem cell biology. Nucleic Acids Res. 2018; 46:1038–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jaffrey S.R., Wilkinson M.F.. Nonsense-mediated RNA decay in the brain: emerging modulator of neural development and disease. Nat. Rev. Neurosci. 2018; 19:715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li T., Shi Y., Wang P., Guachalla L.M., Sun B., Joerss T., Chen Y.S., Groth M., Krueger A., Platzer M.et al.. Smg6/Est1 licenses embryonic stem cell differentiation via nonsense-mediated mRNA decay. EMBO J. 2015; 34:1630–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lou C.H., Dumdie J., Goetz A., Shum E.Y., Brafman D., Liao X., Mora-Castilla S., Ramaiah M., Cook-Andersen H., Laurent L.et al.. Nonsense-mediated RNA decay influences human embryonic stem cell fate. Stem Cell Reports. 2016; 6:844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong J.J., Ritchie W., Ebner O.A., Selbach M., Wong J.W., Huang Y., Gao D., Pinello N., Gonzalez M., Baidya K.et al.. Orchestrated intron retention regulates normal granulocyte differentiation. Cell. 2013; 154:583–595. [DOI] [PubMed] [Google Scholar]
- 52.Nasif S., Contu L., Muhlemann O.. Beyond quality control: the role of nonsense-mediated mRNA decay (NMD) in regulating gene expression. Semin. Cell Dev. Biol. 2018; 75:78–87. [DOI] [PubMed] [Google Scholar]
- 53.Durand S., Lykke-Andersen J.. Nonsense-mediated mRNA decay occurs during eIF4F-dependent translation in human cells. Nat. Struct. Mol. Biol. 2013; 20:702–709. [DOI] [PubMed] [Google Scholar]
- 54.Rufener S.C., Muhlemann O.. eIF4E-bound mRNPs are substrates for nonsense-mediated mRNA decay in mammalian cells. Nat. Struct. Mol. Biol. 2013; 20:710–717. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.