

Amyloid beta oligomers cause glutamatergic synapse degeneration by targeting the planar cell polarity components in the synapses.
Abstract
The signaling pathway directly controlling the maintenance of adult glutamatergic synapses has not been well understood. Planar cell polarity (PCP) signaling components were recently shown to play essential roles in the formation of glutamatergic synapses. Here, we show that they are localized in the adult synapses and are essential for their maintenance. Synapse loss at early stages of Alzheimer’s disease is thought to be induced by β-amyloid (Aβ) pathology. We found that oligomeric Aβ binds to Celsr3 and assists Vangl2 in disassembling synapses. Moreover, a Wnt receptor and regulator of PCP signaling, Ryk, is also required for Aβ-induced synapse loss. In the 5XFAD mouse model of Alzheimer’s disease, Ryk conditional knockout or a function-blocking monoclonal Ryk antibody protected synapses and preserved cognitive function. We propose that tipping of the fine balance of Wnt/PCP signaling components in glutamatergic synapses may cause synapse degeneration in neurodegenerative disorders with Aβ pathology.
INTRODUCTION
Glutamatergic synapses are the main excitatory synapses in mammalian brain, and their overall numbers are largely stable in adulthood (1). The signaling pathway that directly regulates adult synapse maintenance within the synapses has not been well understood. Planar cell polarity (PCP) signaling establishes the global polarity of cellular and tissue morphology along the tissue plane (2, 3). Six conserved core components—Frizzled, Flamingo, Van Gogh (Vang), Prickle, Dishevelled, and Diego—form dynamic and asymmetric protein complexes at the Cadherin-mediated adherens junctions that connect neighboring cells (4, 5). Frizzled, Flamingo, and Vang are transmembrane proteins, whereas Prickle, Dishevelled, and Diego are cytosolic proteins that interact with these transmembrane proteins. The PCP components form protein complexes that are asymmetrically distributed in cells and have complex, sometimes, antagonistic functions (2, 3). PCP components were found localized in glutamatergic synapses in a similar fashion to the asymmetric epithelial cell-cell junctions during PCP signaling and were shown to play essential roles in glutamatergic synapse formation in development (6, 7). Frizzled3 is enriched in the synaptic vesicles and on the plasma membrane of the presynaptic boutons, and Vangl2 is enriched on the plasma membrane of the postsynaptic neurons and in the postsynaptic density (PSD), whereas Celsr3 is present on the plasma membranes of both pre- and postsynaptic neurons. Celsr3 promotes synapse formation, whereas Vangl2 inhibits synapse formation during development (6, 7). However, whether PCP signaling plays a role in mature neural circuits in adulthood is not known. Synapse degeneration occurs in most of the neurodegenerative disorders. Amyloid precursor protein and its metabolites regulate synaptic transmission, plasticity, and calcium homeostasis (8). While β-amyloid (Aβ) at picomolar concentrations is essential for long-term potentiation (LTP) and long-term depression (LTD), Aβ at nanomolar concentrations inhibits LTP induction (9). Aβ self-associates to form a range of soluble neurotoxic oligomers and insoluble deposited fibers (10). In a number of experimental systems, soluble Aβ oligomers induce loss of glutamatergic synapses, loss of LTP, and decrease of dendritic spine density (11–16).
Using super-resolution microscopy, we show here that the PCP components and a Wnt receptor, Ryk, are also present in the adult glutamatergic synapses. We found that PCP components regulate the maintenance of adult glutamatergic synapses. Knocking out Celsrs in the adult hippocampus led to the loss of approximately 50% of synapses 2 months later. In contrast, knocking out Vangl2 in the adult hippocampus led to a 27% increase of synapse numbers 2 months later. Furthermore, our results showed that Aβ oligomers were unable to cause synapse loss when Vangl2 was conditionally knocked out in vitro or in vivo in the adult hippocampus. PCP signaling is known to be mediated by a set of dynamic protein-protein interactions. One such essential interaction is an asymmetric intercellular complex composed of Frizzled and Celsr3 (17). We show that Vangl2 may disassemble glutamatergic synapses by disrupting the Celsr3/Frizzled3-Celsr3 intercellular complex. Ryk regulates PCP signaling by directly interacting with and promoting the function of Vangl2 (18, 19). We found that Ryk is also required for Aβ oligomer–induced synapse loss in vitro and in vivo as shown by Ryk conditional knockout (cKO) and a function-blocking monoclonal Ryk antibody. Ryk cKO or infusion of a function-blocking monoclonal Ryk antibody prevented the loss of synapses and preserved cognitive functions in 5XFAD, a mouse model of Alzheimer’s disease (AD). Collectively, these findings reveal a previously unknown function of PCP signaling in regulating the numbers of the adult synapses and provide valuable insights into the mechanisms of synapse degeneration in neurodegenerative disorders.
RESULTS
Wnt/PCP signaling components are essential for glutamatergic synapse maintenance and regulate synapse numbers in the adult nervous system
Recent studies showed that PCP signaling components are localized to glutamatergic synapses, interact with multiple key synaptic proteins, and function as essential regulators of synapse formation during early postnatal development (6). Biochemical fractionation showed that Celsr3 is localized on the plasma membrane of both pre- and postsynaptic sides in the synaptic cleft. Frizzled3 is localized on the presynaptic membrane, whereas Vangl2 is localized on the postsynaptic membrane (6). To test whether PCP signaling plays a role in the adult brain, we evaluated whether the PCP components are also present in the glutamatergic synapses of the adult hippocampus and localized in similar ways using confocal microscopy and super-resolution microscopy, the 3D STORM (fig. S1A and Fig. 1, A and B) (20, 21). We observed that the PCP components, Celsr3 and Vangl2, and the Wnt receptor, Ryk, are present in the adult glutamatergic synapses (at 2, 4, and 6 months of age). Super-resolution images showed that Celsr3 is colocalized with both the presynaptic marker (Bassoon) and the postsynaptic marker (PSD-95) (Fig. 1B and movie S1). Using the same strategies, Frizzled3 was shown colocalized with Bassoon, and Vangl2 was found colocalized with PSD-95 (Fig. 1B and movies S2 and S3). Ryk is a coreceptor of Frizzled and also interacts with Vangl2, regulating Vangl2 function. Using 3D STORM, we found that Ryk is present on both the pre- and postsynaptic sides (Fig. 1B and movie S4). Thus, the PCP components maintain their expression and display similar subcellular localization in both developing and adult glutamatergic synapses (Fig. 1C). The specificity of the antibodies used here has been confirmed using knockouts or cKOs in our previous studies (6, 22–24).
Fig. 1. Localization of Wnt/PCP signaling components in glutamatergic synapses in adult hippocampus and role of Celsrs and Vangl2 in synapse maintenance.

(A) Schematic diagram showing the hippocampal areas. (B) Representative 3D STORM images showing the localization of PCP proteins and Ryk (red) with synaptic markers. (C) Schematic of the distribution of Wnt/PCP components in glutamatergic synapses. mo, months. (D) Experimental design of viral injection. (E and F) Representative images and quantification of synaptic puncta in the stratum radiatum (SR) (circles indicate colocalization). n = 4 animals in each group. (G and H) Representative images and quantification of dendritic spine density. Thirteen dendrites from three control animals and 10 dendrites from four animals with Celsr2/3 sgRNAs. (I) Experimental design of viral injection. (J and K) Representative images and quantification of synaptic puncta in the SR. n = 5 control animals; n = 3 Vangl2 cKO animals. (L and M) Representative images and quantification of dendritic spine density. Twenty-one dendrites from three control animals and 17 dendrites from four Vangl2 cKO animals. Student’s t test. *P < 0.05, **P < 0.01, and ***P < 0.001. Scale bars, 100 nm (B) and 1 μm (E, G, J, and L). Error bars represent SEM.
To assess whether PCP signaling continues to regulate synapse maintenance, we first knocked out Celsrs2 and Celsr3, which are the two abundantly expressed homologs in the adult brain, in adult hippocampus using the CRISPR-Cas9 system. CRISPR-induced genome editing was first tested in Neuro2A cells (fig. S2, A and B), and knockout efficiency was evaluated by Western blots of protein extracts from cultured hippocampal neurons (fig. S2C). We then injected both adeno-associated virus (AAV)–sgCelsr2-Cre and AAV-sgCelsr3-Cre into the CA1 region of the adult hippocampus of the Cre-dependent Cas9 mice and analyzed the synapse numbers first by costaining with synaptic markers 2 months later. We used Image J plugin called “synapse counter” to measure the puncta that are pre, post, or colocalized. When the pre and post are overlapped by 33 to 100%, they are considered colocalized. We found that knocking out Celsr2 and Celsr3 significantly reduced the glutamatergic synapse numbers (colocalized) in the stratum radiatum (Fig. 1, D to F). To assess the change of synapse number using a different approach, we then sparsely labeled the dendrites with a lower titer of the AAV–human synapsin (hSyn)–mCherry to visualize the dendritic spines. Knocking out Celsr2 and Celsr3 significantly reduced the number of the dendritic spines of the CA1 pyramidal neurons (Fig. 1, G and H). We next conditionally knocked out Vangl2 by injecting AAV1-hSyn–enhanced green fluorescent protein (eGFP)–Cre (AAV that harbors the hSyn promotor driving the Cre recombinase) to the CA1 region of the adult hippocampus (at the age of 2 months) of the Vangl2fl/fl mice (fig. S2D) and then analyzed the synapse numbers 2 months later (Fig. 1I). We observed a 27% increase of synapse numbers in Vangl2 cKO by constaining with synaptic markers (Fig. 1, J and K). Dendrites were labeled sparsely by coinjecting AAV1-hSyn-Cre (at a lower titer) and AAV1-CAG-Flex-eGFP into the Vangl2 cKO. We found that conditionally knocking out Vangl2 led to an increase of dendritic spines of the CA1 pyramidal neurons 2 months later (Fig. 1, L and M), suggesting that similar to development, Vangl2 also negatively regulates synapse numbers in adult mice. Therefore, the functions of Celsrs and Vangl2 extend beyond synapse formation and they continue to regulate synapse numbers in adulthood.
Vangl2 is required for Aβ oligomer–induced synapse loss in vitro and in vivo
Because PCP signaling components control the maintenance of a large number of glutamatergic synapses in adulthood, we then hypothesized that PCP signaling components may be involved in synapse loss in neurodegeneration. Oligomeric amyloid-β (Aβ oligomer) is a well-established model for synapse degeneration. We first tested whether Vangl2 is required for Aβ oligomer–induced synapse loss in cultured hippocampal neurons isolated from embryonic day 18 (E18.5) of Vangl2fl/fl mice. AAV1-hSyn-eGFP-Cre was added into the culture on the seventh day after the start of culture [day in vitro 7 (DIV7)]. At DIV14, 400 nM monomer equivalent of Aβ oligomers were added, and the cultures were fixed 12 hours later for staining and analysis (Fig. 2A). Figure S3 shows that the effective concentration of the dimers added was 80 nM and that the effective concentration of the tetramers was ~152 nM. The calculation of concentration is described in Materials and Methods (25, 26). Neurons isolated from littermate control (Vangl2+/+, control) mice were also treated with AAV1-hSyn-eGFP-Cre. Vangl2 protein levels were successfully reduced in Vangl2fl/fl neurons treated with the AAV1-hSyn-eGFP-Cre (Fig. 2B). We found that, in control neurons, Aβ oligomers reduced the synapse numbers by 30% as shown by costaining with the Bassoon and PSD-95 antibodies (Fig. 2, C and D). However, the number of glutamatergic synapses in Vangl2 cKO neurons was unchanged when treated with Aβ oligomers (Fig. 2, C and D). Consistent with our previous finding that Vangl2 inhibits synapse formation, Vangl2 cKO itself led to 40% increase of synapse numbers during the 7.5 days of culture of the E18.5 embryonic neurons, suggesting that 1 week was long enough time to observe the increased synapse formation in these cultures from E18.5 neuronal cultures (Fig. 2, C and D) (6).
Fig. 2. Vangl2 is required for Aβ oligomer–induced synapse loss in vitro and in vivo.

(A) Schematic illustrating the experimental design. AAV1-hSyn-eGFP-Cre virus was added to hippocampal neuron cultures on DIV7 for 7 days and then oligomeric Aβ42 was added. Aβ-O, Aβ oligomer. Twelve hours later cultures were fixed for staining with synaptic markers. (B) Western blot showing the level of Vangl2 proteins in cultures infected with the AAV1-hSyn-eGFP-Cre virus. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (C) Immunostaining for presynaptic (green) and postsynaptic (red) puncta of glutamatergic synapses (arrowheads) in 14-DIV hippocampal cultures from littermate Vangl2+/+ or Vangl2fl/fl with or without oligomeric Aβ42. (D) Quantification of (C). n = 3 for Vangl2+/+ mice, n = 4 for Vangl2fl/fl from three independent experiments. (E) Schematic illustrating the experimental design. AAV1-hSyn-eGFP-Cre virus was injected into the CA1 region of the hippocampus bilaterally. Two weeks later, Aβ oligomeric was injected intracerebroventricularly (i.c.v.). Five days after Aβ oligomer injection, animals were fixed with perfusion and sectioning and stained with synaptic markers. (F) Vangl2 protein level in the total hippocampal extracts from animals injected with the AAV1-hSyn-eGFP-Cre virus. (G) Representative images of Bassoon (red)– and PSD-95 (green)–immunoreactive puncta (arrowheads) in the stratum radiatum of Vangl2+/+ and Vangl2fl/fl hippocampus (CA1) with or without Aβ oligomeric injection and quantification of synapse numbers. (H) Quantification of (G). One-way ANOVA. n = 8 of Vangl2+/+ mice, n = 3 of Vangl2+/+ mice with Aβ oligomeric injection, n = 6 of Vangl2fl/fl mice, and n = 5 of Vangl2fl/fl mice with Aβ oligomeric injection. *P < 0.05 and **P < 0.01. One-way ANOVA. Scale bars, 2 μm (C and G). Error bars represent SEM.
To test whether Vangl2 is required for Aβ oligomer–induced synapse loss in vivo in adulthood, we injected AAV1-hSyn-eGFP-Cre into the hippocampal CA1 region of Vangl2+/+ (control) and Vangl2fl/fl mice at the age of 2 months. Two weeks later, we bilaterally injected 5 ng of Aβ oligomers into the cerebral ventricles (Fig. 2E) (27). Five days after injection of Aβ oligomers, animals were perfused, and the synapse numbers were analyzed. Vangl2 protein levels were found significantly reduced in Vangl2 cKO (Fig. 2F). We observed a significant loss of synapses in control animals injected with Aβ oligomers but not in the Vangl2 cKO mice injected with Aβ oligomers (Fig. 2, G and H). In contrast to the experiment with cultured neurons from E18.5 embryos, Vangl2 cKO in adulthood showed no significant changes in synapse numbers 19 days after the AAV1-hSyn-eGFP-Cre injection (Fig. 2, G and H). It is probably because the synapse turnover is not as rapid in adulthood such that 19 days is not long enough to observe changes in synapse numbers.
Binding of Aβ oligomers to Celsr3 is required for Aβ oligomer–induced synapse loss
To understand how PCP signaling may mediate Aβ oligomer–induced synapse loss, we investigated a potential direct interaction between Aβ oligomers and the PCP components. PCP components are localized in glutamatergic synapses in a similar fashion to the asymmetric epithelial cell-cell junctions during PCP signaling (Fig. 1, B and C) (6). Among the six core PCP components, Celsrs, Frizzleds, and Vangls are present on the plasma membrane. To determine whether Aβ oligomers target any one of those proteins, we measured binding of biotin-Aβ42 oligomers to human embryonic kidney (HEK) 293T (HEK293T) cells expressing mouse Vangl2 (Vangl2-Flag), Frizzled3 (Frizzled3-HA), Celsr3 (Celsr3-Flag), or control vector (pCAGEN). We found that Aβ oligomers only bound to Celsr3 but not to Vangl2 or Frizzled3 (Fig. 3A), with an apparent dissociation constant (Kd) of ~40 nM equivalent of total Aβ peptide (Fig. 3B). Aβ monomers did not bind to Celsr3 (fig. S4).
Fig. 3. Celsr3 is a binding protein for Aβ oligomeric, and the binding of Aβ oligomeric to the EGF7 and EGF8 domains of Celsr3 is required for synapse loss.

(A) Staining of the HEK293T cells transfected with Vangl2-Flag, Frizzled3-HA, Celsr3-Flag, or control vector (pCAGEN) and incubated with oligomeric Aβ42 (200 nM total peptide, monomer equivalent). Bound oligomeric Aβ42 (green) was visualized using Alexa-488-conjugated streptavidin. (B) Binding curve of oligomeric Aβ42 to Celsr3-expressing HEK293T cells (concentration shown as total peptide, monomer equivalent). (C) Schematic of mouse Celsr3 extracellular domain with nine cadherin domains, eight EGF domains, and three laminin domains. (D) Staining of bound Aβ42 (200 nM total peptide, monomer equivalent) with Celsr3-Flag–transfected or truncated Celsr3-Flag–transfected HEK293T cells. Bound oligomeric Aβ42 (green) was visualized using Alexa-488-conjugated streptavidin. Scale bar, 10 μm. (E and F) Immunoprecipitated assays testing the interaction between Frizzled3 and Celsr3 or with truncated Celsr3 and quantification data of the expression level of co-IPed Frizzled3. A.U., arbitrary units. *P < 0.05. One-way ANOVA. Means ± SEM. (G) Pull-down assay for purified GST function proteins mixed with biotinylated Aβ42 oligomers by streptavidin. (H and I) Aβ oligomer–induced synapse loss in the presence of EGF7-GST or EGF8-GST fusion proteins. Arrowheads indicate the Bassoon/PSD95 colocalized puncta. *P < 0.05 and ** P < 0.01. Student’s t test. Scale bars, 10 μm (A and D) and 1 μm (H). Error bars represent SEM.
Celsr3 belongs to the family of adhesion G protein–coupled receptors with a large extracellular region, which contains nine cadherin domains, eight EGF repeats, and three laminin domains (Fig. 3C). Cadherin domains are required for homophilic binding of Celsr3. To determine the domain(s) of Celsr3 that bind to Aβ oligomers, we first made a deletion construct that lacks all the EGF repeats and laminin domains and tested binding in HEK293T cells. We found that Aβ oligomers did not bind to this truncated protein, suggesting that Aβ oligomers do not bind to the cadherin domains but rather bind to the EGF repeats and the laminin domains (Fig. 3D and fig. S5A). We then made a series of Celsr3 constructs that lack the individual EGF and laminin domains (fig. S5B) and tested for binding to Aβ oligomers in HEK293T cells. We found that two EGF domains, EGF7 and EGF8, and one laminin domain, laminin G1, are required for binding of Aβ oligomers (Fig. 3D and fig. S5C). The human homolog of murine Celsr3 also contains nine cadherin domains, eight EGF repeats, and three laminin domains. The laminin G1 and EGF7 domains of hCelsr3 align closely with that of mCelsr3 with a homology of 98.537 and 80%, respectively. The amino acid sequence of the EGF8 domain of hCelsr3 is 100% homologous with that of the EGF8 domain of mCelsr3 (fig. S6A). We found that Aβ oligomers also bound to hCelsr3 with an apparent Kd of ~70 nM equivalent of total Aβ peptide (fig. S6B). Similar to mCelsr3, EGF7 and EGF8 and laminin G1 of hCelsr3 are required for binding with Aβ oligomers (fig. S6B).
In PCP signaling, protein-protein interaction is essential for the establishment of cell and tissue polarity along the tissue plane (2). Celsr3 forms a complex with Frizzled3 on the plasma membrane of one cell, and Celsr3 forms a complex with Vangl2 on the plasma membrane of the neighboring cells. We then tested whether the Aβ oligomer–binding domains of Celsr3 are involved in the protein-protein interactions among PCP components. We expressed Frizzled3 or Vangl2 together with WT Celsr3 or mutant Celsr3 (with domain deletions) in HEK293T cells. We found that deletion of all eight EGF repeats and three laminin domains caused a 68% reduction of the interaction between Celsr3 and Frizzled3. Deletion of laminin G1 alone led to a 66% reduction of the interaction between Celsr3 and Frizzled3, whereas deleting EGF7 or EGF8 did not affect the interaction between Celsr3 and Frizzled3 (Fig. 3, E and F). The interaction between Vangl2 and Celsr3 did not require the EGF repeats or the laminin domains (fig. S7). To determine whether the binding of Aβ oligomers on these domains is important for synapse loss, we tested the role of EGF7 and EGF8 domains. We performed binding assays with purified EGF7–glutathione S-transferase (GST) or EGF8-GST fusion proteins and found that both domains can bind to Aβ oligomers, as pulling down biotinylated Aβ oligomers with streptavidin (NeutrAvidin agarose) can pull down the EGF7-GST and EGF8-GST fusion proteins (Fig. 3G). Last, we added EGF7-GST or EGF8-GST fusion proteins to neuronal hippocampal culture and found that they both blocked Aβ oligomer–induced synapse loss (Fig. 3, H and I). Therefore, the EGF7 and EGF8 domains of Celsr3 are likely direct targets of Aβ oligomers.
Aβ oligomers enhance the function of Vangl2 in disrupting the Celsr3/Frizzled3-Celsr3 intercellular complex essential for PCP signaling
PCP signaling is known to be mediated by a set of dynamic protein-protein interactions (2). One such essential interaction is an asymmetric intercellular complex made of Frizzled and Celsr (17). The Frizzled/Celsr complex on the plasma membrane on the distal side of a cell forms an intercellular complex with Celsr on the plasma membrane on the proximal side of the neighboring cell (distal to the first cell), bridging the two cells via the homophilic interaction of the cadherin repeats of the two Celsr proteins. Such an asymmetric intercellular bridge is sufficient to polarize both cells even in the absence of Vangl and, therefore, is thought to be essential for PCP signaling (17).
Because the Aβ oligomers bind to the laminin G1 domain of Celsr3, which is also required for Frizzled3 binding, we hypothesized that Aβ oligomers may weaken the interaction between Celsr3 and Frizzled3, allowing Vangl2 to more efficiently disrupt the asymmetric intercellular complex of Celsr3/Frizzled3-Celsr3 and thus disassemble synapses. We established an assay to test the intercellular PCP complex, similar to the “transcellular interaction assay” that we published previously (28). We cotransfected Frizzled3 (HA-tagged) and Celsr3 (untagged) in one dish of HEK293T cells and transfected Celsr3 (Flag-tagged) and Vangl2 in another. After culturing them separately for one day, we mixed them together and cultured for one more day and then performed coimmunoprecipitation to test the protein-protein interactions (Fig. 4A). To test whether Vangl2 disrupts the intercellular bridge, we pulled down Frizzled3 and measured how much Celsr3 from the neighboring cell was coimmunoprecipitated. We found that Vangl2 disrupted this intercellular complex as much less Flag-tagged Celsr3 was pulled down by HA-tagged Frizzled3 (fig. S8, A to C). We think that Vangl2 is likely disrupting this intercellular complex by weakening the interaction between Ceslr3 and Frizzled3, because the presence of Vangl2 alone from the neighboring cell caused the reduction of the interaction between Frizzled3 and Celsr3 by 30 to 40% (fig. S8, D to F). Celsr3 from the neighboring cell does not affect the complex between Frizzled3 and Celsr3 (fig. S8, G to I).
Fig. 4. Aβ oligomer enhances Vangl2’s function in disrupting the intercellular complex of Celsr3/Frizzled3-Celsr3.

(A) Schematic illustrating the experimental design of the “transcellular interaction” assay. (B) Testing the intercellular complex by immunoprecipitating Celsr3 in one cell and detecting Frizzled3 in the neighboring cell with or without Vangl2. (C) Quantification of (B). (D) Representative 3D STORM images showing the endogenous Celsr3 (red), Frizzled3 (green), and Bassoon (blue) on the secondary dendrite of the cultured hippocampal neuron. (d1 to d3) Representative 3D STORM images of synapses in the cultured hippocampal neuron that were labeled for Celsr3 (red), Frizzled3 (green), and Bassoon (blue). (E) The distribution of localization points along the trans-synaptic axis was measured and fit with Gaussian functions, and the distances between the centroid positions of the two Gaussians were defined as the distances between the secondary antibodies detecting Celsr3 and Frizzled3 antibodies, which reflect the distances between Celsr3 and Frizzled3. (F) The histogram of the distances between the secondary antibodies detecting Celsr3 and Frizzled3 antibodies, which reflect the distances between Celsr3 and Frizzled3. (G) The distribution of localization points along the trans-synaptic axis was measured and fit with Gaussian functions, and the distances between the centroid positions of the two Gaussians were defined as the distances between the secondary antibodies detecting Celsr3 and Frizzled3 antibodies with different concentration (monomer equivalent) of Aβ oligomer. (H) The histogram of the distances with different concentration (monomer equivalent) of Aβ oligomer between the secondary antibodies detecting Celsr3 and Frizzled3 antibodies, which reflect the distances between Celsr3 and Frizzled3. *P < 0.05 and ***P < 0.001. One-way ANOVA. Western blot results are representative of four biological replicates. Scale bars, 1 μm (D) and 100 nm (d1 to d3). Error bars represent SEM.
To determine how Aβ oligomers cause synapse loss, we tested whether Aβ oligomers facilitate the function of Vangl2 in disrupting the intercellular complex. First, we found that Aβ oligomers did not disrupt the interaction between Celsr3 and Frizzled3 that were transfected and expressed in the same cell, suggesting that Aβ oligomers themselves are not sufficient to disrupt the Ceslr3-Frizzled3 complex (fig. S9, A to C). As shown previously, Vangl2 expressed in the neighboring cell can decrease the interaction between Celsr3 and Frizzled3 (fig. S8, D to F). The interaction between Frizzled3 and Celsr3 was reduced to a greater extent when Aβ oligomers were added to the culture (fig. S9, D to F), indicating that Aβ oligomers may enhance the function of Vangl2 in disrupting the Celsr3-Frizzled3 complex across the cell-cell junction. Furthermore, we found that Aβ oligomers can also disrupt the intercellular complex, as the HA-tagged Frizzled3 in one cell pulled down significantly less Flag-tagged Celsr3 from a neighboring cell when Aβ oligomers were added to the mixed culture (fig. S9, G to I). This suggests that the intercellular interaction of Celsr3 between two neighboring cells may weaken the intracellular interaction between Celsr3 and Frizzled3 within the same cell, allowing Aβ oligomers to more efficiently disrupt the entire intracellular interaction. To test the role of Aβ oligomers in enhancing the function of Vangl2, we reduced the amount of transfected Vangl2 by half (at 0.5 μg per well) so that Vangl2 is at suboptimal concentration (Fig. 4, A to C). We found that adding Aβ oligomers to mixed cultures with Vangl2 led to the greater disruption of this intercellular complex (Fig. 4, A to C).
To further test our model that Aβ oligomers may promote synapse disassembly by interfering with the interactions between Celsr3 and Frizzled3, we performed super-resolution microscopy and characterized the distance between Celsr3 and Frizzled3 on cultured hippocampal neurons using 3D STORM. We labeled Celsr3, Frizzled3, and a presynaptic marker Bassoon using the corresponding primary antibodies and secondary antibodies conjugated with fluorophore and collected the STORM signals in the synapses on the secondary dendrite (Fig. 4D). We observed three types of colocalization: Celsr3 and Frizzeld3 colocalized with Bassoon (Fig. 4D, d1), Celsr3 colocalized with Bassoon (Fig. 4D, d2), and Frizzled3 colocalized with Bassoon (Fig. 4D, d3). We then rotated the three-dimensional (3D) images so that Celsr3 and Frizzled3 are axially distributed and measured the distance between the centroids of the two Gaussian areas (Fig. 4D, d1 to d3) (21). We found that the distance between the signals that represent Celsr3 and Frizzled3 was 181.2 ± 5.2 (±SEM) nm in the control group (192 pairs quantified in control group). Thirty minutes after the treatment of Aβ oligomers (400 nM), the distance became larger (236.5 ± 5.9 nm, 174 pairs). One hour after the treatment, the distance was 245.8 ± 5.4 nm (179 pairs). Two hours after the treatment, it became shorter (213.1 ± 5.3 nm, 177 pairs) (Fig. 4, E and F). These distances represent the two secondary antibodies (with a size of ~15 nm of each antibody) that correspond to Celsr3 and Frizzled3 and, therefore, are greater than the actual distances between Celsr3 and Frizzled3. Nonetheless, they do reflect the distances between Celsr3 and Frizzled3. In addition, we observed a dose response. The Celsr3-Frizzled3 distance was slightly increased when the neurons were treated with 100 nM Aβ oligomers for 1 hour (224.3 ± 3.4 nm, 50 pairs) and further increased when the neurons were treated with 400 nM Aβ oligomers for 1 hour (256.3 ± 5.3 nm, 29 pairs). The distance became shorter with 800 nM Aβ oligomer treatment (238.9 ± 4.3 nm, 38 pairs) (Fig. 4, G and H). The increased distance may represent the change of Celsr3 and Frizzled3 interaction with the synapses. It may also reflect the change of the histogram of distances because the disassembly of synapses increases the average distances. Future experiments will be needed to distinguish these possibilities. The fact that the average distance of Celsr3 and Frizzled3 is changed within 30 min after the addition of Aβ oligomers supports our model that Aβ oligomers directly target the PCP proteins in the synapses and disrupt the intercellular complex of Celsr3/Frizzled3-Celsr3 by interfering with the interactions between Celsr3 and Frizzled3.
The Wnt/Ryk/Vangl2 signaling axis mediates Aβ oligomer–induced synapse loss in vitro and in vivo
To further address how Vangl2 mediates Aβ oligomer–induced synapse loss and identifies potential therapeutic targets to protect synapses, we explored regulators of core PCP signaling components. As Ryk is a coreceptor for Wnt in PCP signaling by directly interacting with Vangl2 and promoting the function of Vangl2, we sought to test whether Wnt/Ryk signaling is involved in Vangl2 function in this context (18, 19). Wnt5a causes a reduction of synapse numbers probably by causing Frizzled3 endocytosis (6, 29). We first tested whether Ryk mediates Wnt5a function in regulating synapse numbers and whether Ryk does so in a Vangl2-dependent manner. Hippocampal neurons isolated from E18.5 WT mice were treated with Wnt5a on DIV14 for 12 hours or pretreated with a function blocking monoclonal Ryk antibody, which blocks the binding between Wnts and Ryk, for 2 hours (Fig. 5A) (24). Normal mouse immunoglobulin G (IgG) was used as control. Wnt5a caused a 30% reduction in the number of colocalized puncta. In contrast, Wnt5a did not produce a significant difference in synapse numbers when the cultures were pretreated with the function-blocking Ryk monoclonal antibody (Fig. 5, A and B), suggesting that Wnt5a inhibits synapse formation through binding to Ryk. During the 14 hours of culture, Ryk antibody itself did not cause any significant changes in synapse numbers (Fig. 5B). To test whether Vangl2 mediates the function of Wnts downstream of Ryk, we cultured embryonic hippocampal neurons from Vangl2+/+ (control) and Vangl2 cKO (transduced with AAV1-hSyn-eGFP-Cre) and treated neurons with Wnt5a on DIV14 for 12 hours. We found that Wnt5a addition to Vangl2+/+ caused a 30% reduction in the number of colocalized puncta, whereas Wnt5a addition to Vangl2 cKO neurons did not produce a significant difference compared with untreated Vangl2 cKO neurons (Fig. 5, C and D), suggesting that Vangl2 is required for the inhibitory function of Wnt5a in synapse formation. Consistent with our previous finding (Fig. 2, C and D), Vangl2 cKO itself led to more synapses in the cultured embryonic neurons 7.5 days after the introduction of Cre (Fig. 5D). Finally, we pretreated cultured hippocampal neurons with the function-blocking anti-Ryk monoclonal antibody for 2 hours before adding the Aβ oligomers (24). We found that Aβ oligomers failed to reduce synapse numbers in the presence of the Ryk antibody (Fig. 5, E and F). Again, during the 14 hours of time, Ryk antibody itself did not cause any change in synapse numbers in these cultures (Fig. 5F). Therefore, Ryk, together with Vangl2, is also required for Aβ oligomer–induced loss of glutamatergic synapses. As Aβ oligomers do not bind to Ryk (fig. S10), we propose that Wnt/Ryk and Vangl2 function as one signaling axis that is required for synapse disassembly and Aβ oligomers enhance their function in disrupting the synapses.
Fig. 5. The Wnt/Ryk/Vangl2 signaling axis mediates synapse loss induced by Aβ oligomeric.

(A and B) Representative images and quantification of synaptic puncta (arrowheads) testing the effect of the Ryk antibody on Wnt5a-induced synapse reduction in cultured hippocampal neurons. n = 3 experiments (n = 27 neurons in IgG control, n = 22 neurons in Ryk antibody, n = 24 in IgG + Wnt5a, and n = 20 neurons in Ryk antibody + Wnt5a). (C and D) Representative images and quantification of synaptic puncta (arrowheads) testing the role of Vangl2 in Wnt5a-induced synapse reduction in cultured hippocampal neurons. n = 3 WT mice and n = 4 Vangl2 cKO mice. (E and F) Representative images and quantification of synaptic puncta (arrowheads) testing the effect of the Ryk antibody in oligomeric Aβ–induced synapse reduction in cultured hippocampal neurons. n = 3 experiments (n = 26 neurons in IgG control, n = 33 neurons in Ryk antibody, n = 34 neurons in Aβ oligomeric, and n = 39 neurons in Ryk antibody + Aβ oligomeric). (G) Schematic illustrating the experimental design. AAV1-hSyn-eGFP-Cre virus was injected into the CA1 and CA3 regions of the hippocampus bilaterally. Two weeks later, Aβ oligomers were injected into the lateral ventricle for 5 days. Five days later, animals were fixed with perfusion and sectioning and stained with synaptic markers. (H and I) Representative images and quantification of synaptic puncta detected by costaining for Bassoon (red)– and PSD-95 (green)–immunoreactive puncta (arrowheads) in the stratum radiatum. n = 4 for Ryk+/+ mice, n = 3 for Ryk+/+ mice with Aβ oligomeric injection, n = 3 for Ryk cKO mice, and n = 3 for Ryk cKO mice with Aβ oligomeric injection. *P < 0.05, **P < 0.01, and ***P < 0.001. One-way ANOVA. Scale bars, 1 μm. Error bars represent SEM.
To further test the role of Ryk in vivo, we injected of Aβ oligomers into the Ryk cKO mice previously generated in our laboratory and analyzed synapse numbers (24). Because Ryk is likely involved on both the presynaptic and the postsynaptic side, AAV1-hSyn-eGFP-Cre was injected into the CA1 and CA3 regions of the hippocampus of 8-week-old mice to conditionally knock out Ryk in adulthood. In Ryk+/+ (control), Aβ oligomers led to a 60% reduction of synapse numbers. However, the synapse numbers were not affected in the Ryk cKO mice that received intracerebroventricular injection of Aβ oligomers (Fig. 5, G to I).
Ryk cKO prevents the loss of synapses and preserves cognitive function in the 5XFAD mice
To determine whether Wnt-Ryk signaling is required for Aβ oligomer–induced synapse loss in a mouse model of Aβ deposition, we crossed the Ryk cKO with the 5XFAD transgenic mouse. PCP components are present in the glutamatergic synapses of the adult 5XFAD animals (fig. S1B). AAV1-hSyn-eGFP-Cre was injected into the hippocampal CA1 and CA3 regions of the hippocampus of 8-week-old mice to conditionally knock out Ryk in adulthood. Glutamatergic synapse numbers were analyzed 2 months later (Fig. 6A). We found that Ryk cKO in hippocampal neurons prevented the synapse loss (Fig. 6, B and C). We stained for Celsr3 and found that the protected synapses are Celsr3-positive (Fig. 6D). Ryk cKO itself showed a statistically insignificant (P = 0.107) trend toward increases in synapse numbers 2 months after the cKO. Compared to the 5XFAD animals, Ryk cKO resulted in a 400% increase of synapse numbers in 5XFAD, preserving the synapse numbers to the control level (Fig. 6, B and C). To validate the protection of synapses, we analyzed spine density in vivo using sparse labeling of dendrites in 4-month-old mice with a virus mixture of AAV1-hSyn-Cre (at a lower titer) and AAV1-CAG-Flex-eGFP (Fig. 6E). The 5XFAD mice showed a decreased spine density compared with age-matched control animals. Ryk cKO itself led to no significant difference in spine density, whereas Ryk cKO preserved the dendritic spines in the 5XFAD animals (Fig. 6, F and G).
Fig. 6. Ryk cKO prevented synapse loss and preserved cognitive function in the 5XFAD mice.
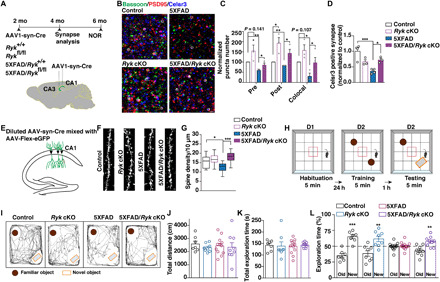
(A) Schematic of timeline and experimental design. AAV1-hSyn-eGFP-Cre virus was injected into the CA1 and CA3 region of the hippocampus bilaterally. Animals were fixed with perfusion and sectioning and stained with synaptic markers at 4 months of age. A separate set of animals were injected and subjected to NOR at 6 months of age. (B and C) Representative images and quantification of synapse numbers. (D) Quantification of Celsr3-positive glutamatergic synapse (Celsr3 colocalized with PSD-95 and bassoon). (E) Schematic illustrating the experimental design for sparse labeling of neurons to visualize dendritic spines. (F and G) Representative images and quantification of dendritic spine density. Nineteen dendrites from four animals in the control group, 15 dendrites from three animals in the Ryk cKO group, 19 dendrites from three animals in the 5XFAD group, and 17 dendrites from four animals in the 5XFAD/Ryk cKO group. (H) Schematic showing the design of novel object recognition (NOR). Mice were subjected to an open arena for three trails to evaluate the memory of objects. (I) Trajectories of mice in the NOR test session. (J) Quantification of locomotion. (K) Quantification of total time for exploration of the two objects. (L) Quantification of NOR. *P < 0.05, **P < 0.01, and ***P < 0.001. One-way ANOVA. Scale bars, 1 μm. Error bars represent SEM.
To test whether Ryk cKO can preserve cognitive function, we performed the novel object recognition (NOR) test with a new set of mice at 6 months of age (Fig. 6A). NOR tests were carried out in an open-field arena measuring 0.4 m by 0.4 m by 0.45 m (Fig. 6H). The total distance was recorded during the 5-min open-field session as locomotor activity; no differences were found among the groups (Fig. 6J). The total time spent exploring the familiar and novel objects was also measured; no differences were found among the groups (Fig. 6K). We found that the Ryk cKO itself did not cause any behavioral defects, but it rescued the impaired NOR behavior of the 5XFAD mice (Fig. 6, I and L). We asked whether the Ryk cKO in neurons affected the development of amyloid plaques in 5XFAD mice and found that the number and total areas of plaques were unchanged at the matching 4 months of age when we quantified synapse numbers (fig. S11).
Ryk monoclonal antibody infusion prevents the loss of synapses and preserves cognitive function in the 5XFAD mice
To investigate whether Wnt/Ryk/PCP signaling can be a potential therapeutic target, we tested the aforementioned function-blocking anti-Ryk monoclonal antibody by intracerebroventricular infusion into the 8-week-old 5XFAD mice (Fig. 7A) (24). Intracerebroventricular administration is a reliable way to deliver antibodies into the brain parenchyma (30). The purified Ryk monoclonal antibody (at 1 mg/ml in 100 μl of artificial cerebrospinal fluid) was loaded in a subcutaneously implanted osmotic minipump connected to a stainless steel cannula. The cannula was stereotaxically implanted into the lateral ventricle, and the infusion was performed at a flow rate of 0.25 μl/hour for 14 days. Therefore, each animal received a total of 84 μg of the purified Ryk monoclonal antibody. Synapse numbers were then analyzed at the age of 4 months, and behavioral tests were performed at the age of 6 months (with a different group of animals). We found that the Ryk monoclonal antibody infusion was able to block the loss of glutamatergic synapse numbers in the hippocampus of the adult 5XFAD transgenic mice (Fig. 7, B and C), as measured by immunostaining with synaptic markers and the density of the dendritic spines (Fig. 7, D and E). We infused a separate cohort of animals with the Ryk monoclonal antibodies, and LTP of the protected CA3-CA1 synapses was assessed at the age of 4 to 5 months. We found that Ryk monoclonal antibody infusion preserved LTP in the 5XFAD animals (Fig 7, F and G). In the NOR test with another cohort of animals (at 6 months of age), Ryk monoclonal antibody infusion preserved the cognitive function of the 5XFAD mice (Fig. 7, H to K). We asked whether the Ryk monoclonal antibody infusion affected the development of the amyloid plaques in 5XFAD mice and found that the number and total areas of plaques were unchanged at the matching 4 months of age (Fig. 7, L and M). These results are consistent with the observation that amyloid plaques were unchanged in 5XFAD when Ryk was conditionally knocked out (fig. S11). Astrogliosis and microglia activation are observed in the 5XFAD mice and other Aβ models (27, 31). We then investigate whether Ryk monoclonal antibody could affect astrogliosis or microgliosis in the 5XFAD mice. We observed a significant increase in glial fibrillary acidic protein (GFAP)+ and Iba-1+ (Ionized calcium binding adaptor molecule 1) area coverage in the mouse IgG-infused 5XFAD mice. Ryk monoclonal antibody reduced the increase of both in the 5XFAD mice (Fig. 7, N and O). We currently cannot distinguish whether the reduction of astrogliosis or microgliosis is a secondary effect due to the protection of synapses in neurons or a direct effect of the Ryk antibody on astrocytes or microglia. However, the reduced astrogliosis or microgliosis is consistent with the preservation of the cognitive function and supportive of the Ryk antibody as a potential treatment option. To confirm that the Ryk monoclonal antibody is effectively delivered using intracerebroventricular administration, we also stained these tissues using a secondary antibody against mouse IgG and found that the mouse IgG signals can be detected in the animals infused with Ryk monoclonal antibody but not in those animals infused with control mouse IgG (fig. S12). Some signals appear to be distributed on neurites, most likely dendrites, suggesting that the Ryk monoclonal antibody diffused into the hippocampal parenchyma and became enriched on the dendrites where Ryk protein and the synapses are located. In addition to the signals on neurites, the signals appear to have some colocalization/close apposition with Iba-1 immunoreactivity and GFAP immunoreactivity (potentially more with Iba-1 than with GFAP). However, with the current resolution, we are not able to be certain about the exact distribution of the Ryk monoclonal antibody. These observations motivated us to use higher resolution and a more complete set of markers in our future studies to explore whether Ryk monoclonal antibody is engaging the glutamatergic synapses or the processes of the microglia or astrocytes or both.
Fig. 7. Function blocking Ryk antibody can prevent synapse loss and preserve cognitive function in the 5XFAD mice.

(A) Timeline and schematic of intracerebroventricular infusion of the Ryk monoclonal antibody. Cannula and preinfused osmotic minipumps were implanted at ~2 months old. Minipumps were removed 2 weeks later. Synapse number analysis was done at the age of 4 months and NOR was done at the age of 6 months. (B and C) Staining and quantification of glutamatergic synapses in the 5XFAD mice infused with the Ryk monoclonal antibody. (D and E) Representative images and quantification of dendritic spine density. Twenty-one dendrites from four animals in the control_mIgG group, 22 dendrites from four animals in the 5XFAD_mIgG group, and 23 dendrites from four animals in the 5XFAD_Ryk mAb group. One-way ANOVA. (F) Average traces for field excitatory postsynaptic potentials (fEPSPs) in hippocampal slices obtained from four animals in the control_mIgG group, five animals in the 5XFAD_mIgG group, and six animals in the 5XFAD_Ryk mAb group. (G) Mean changes in the EPSP amplitude between 45 and 60 min after HFS (high-frequency stimulation) protocol. (H) Trajectories of mice in the NOR test session. (I) Quantification of locomotion. (J) Quantification of total time for exploration of the two objects. (K) Quantification of NOR. (L and M) Representative images of hippocampi stained for Aβ using antibody 6E10 and quantification of number of Aβ-immunopositive deposits and total Aβ-immunostained area. n = 4 animals in the 5XFAD_mIgG group and n = 6 animals in the 5XFAD_Ryk mAb group. Student’s t test. (N) Representative images of hippocampi stained for GFAP+ astrocytes and (O) Iba-1+ microglia, and quantification of GFAP+ and Iba-1+ area coverage. n = 4 to 6 animals per group. (P) Schematic diagram showing the balance of Wnt/PCP signaling in synapse maintenance and the binding site of Aβ oligomeric. *P < 0.05, **P < 0.01, and ***P < 0.001. Scale bars, 1 μm (B and D), 100 μm (L, N, and O), and 10 μm (N and O insets). Error bars represent SEM.
DISCUSSION
Glutamatergic synapse numbers in adulthood are largely stable with a small proportion of synapses undergoing dynamic changes, such as during learning and memory (1, 32). Dysregulation of synapse numbers and functions is implicated in many neurological and psychiatric disorders. Although an extensive body of literature has shown that transcription factors, neurotrophins, and other signaling cascades regulate the stability and plasticity of the synapses, the signaling pathways that operate directly in the synapse to maintain or disassemble synapses locally have not been well understood. We showed previously that the PCP components are localized in the pre- and postsynaptic compartments in glutamatergic synapses, interact with synaptic proteins, and are the key regulators of a large number of glutamatergic synapse formation in development (6). We show here that PCP proteins are also localized in adult synapses in the same fashion and continue to regulate the maintenance of a large number of synapses in the mature nervous system. We tested the role of the PCP components in experimental models of Aβ-associated synapse degeneration and found that Celsr3 and the Wnt/PCP signaling components are a direct target of Aβ oligomers. Vangl2 disrupts the intercellular complex of Celsr3/Frizzled3-Celsr3 between neighboring cells. Aβ oligomers bind to three of the Celsr3 domains, one of which mediates the formation of the Frizzled3/Celsr3 complex, and may disrupt this intercellular complex by weakening the interaction between Frizzled3 and Celsr3, promoting the function Vangl2 in synapse disassembly. This function of PCP components in synapse assembly and disassembly is also regulated by Wnt/Ryk signaling. Ryk cKO in hippocampal neurons and a function-blocking Ryk monoclonal antibody can protect synapses and preserve cognitive function in the 5XFAD mice, an Aβ model of Alzheimer’s disease. Our study suggests that the PCP signaling components are not only essential for normal behavior but also a key target in neurological and psychiatric disorders that involve misregulated synapse numbers or synapse degeneration, such as in Alzheimer’s disease. Our previous studies showed that the apical-basal polarity signaling components, atypical protein kinase C (aPKC) and Lgl1, also regulate glutamatergic synapse numbers in adulthood, with aPKC maintaining synapses and Lgl1 (lethal giant larvae 1) functioning in the opposite way (7, 33). We found that aPKC may affect PCP signaling via regulating Frizzled3 phosphorylation and membrane localization, whereas Lgl1 may affect PCP signaling by regulating the level of Vangl2 (23, 33). Therefore, the apical-basal and planar polarity signaling components may interact in a highly organized molecular network to mediate the intricate regulations of synapse numbers and functions, potentially providing inputs from diverse molecular or electrical signals. In addition to the core components of these polarity signaling pathways, regulators of these pathways, such as Ryk and LRRK2 (Leucine Rich Repeat Kinase 2), may also exert their function on synapses by regulating the core components. We showed previously that LRRK2 regulates Frizzled3 phosphorylation and trafficking and its interaction with Vangl2 across the intercellular complex bridged via Celsr3 (28). A growing body of literature suggests that the role of LRRK2 in synaptic functions may be relevant to Parkinson’s disease (34). We showed previously that aPKC and Ryk levels were altered in a mouse model of ALS (amyotrophic lateral sclerosis) (35). Therefore, the cell polarity signaling components and their regulators deserve more investigation in neurodegenerative disorders.
Amyloid precursor protein and its metabolites regulate synaptic transmission, plasticity, and calcium homeostasis (8, 9). In general, Aβ and their derivatives may be a negative feedback mechanism to reduce neural activity in neuronal networks. Soluble Aβ oligomers have been shown to induce loss of glutamatergic synapses, loss of LTP, and a decrease in dendritic spine density (11–15). Several Aβ receptors, including cellular prion protein (PrPC), EphB2 (Ephrin type-B receptor 2), and paired immunoglobulin-like receptor B or its human ortholog leukocyte immunoglobulin-like receptor B2, have been reported and regulate synaptic plasticity. These receptors mediate the function of Aβ oligomers in altering synaptic function and plasticity, but their role in synapse loss has not been documented (26, 36, 37). Our study is the first to identify the binding protein of Aβ oligomers that directly regulates synapse stability and maintenance. Our study also suggests that the Wnt/PCP pathway is a target of Aβ-mediated synapse loss, which may be a normal negative feedback of neuronal activity, but hijacked in pathological conditions, such as when Aβ is overproduced in patients with Alzheimer’s disease (38). A number of studies showed that a Ryk inhibitor, Wnt-inhibitory factor-1, is down-regulated in patients with Alzheimer’s disease (39, 40).
The extracellular Aβ plaques and intracellular neurofibrillary tangles, formed by hyperphosphorylated tau, are the primary neuropathologic hallmarks for Alzheimer’s disease (41, 42). They have both been proposed to cause Alzheimer’s disease (43–47). More recent findings suggest that a strong but complex involvement of the innate immune system may be a key downstream event to either respond to or enhance amyloid pathology or exacerbate tau-associated pathology. The majority of cases of Alzheimer’s disease are late-onset, with APOE4 as the strongest risk factor. Increased Aβ seeding and reduced Aβ clearance appear to be related to the risk, suggesting that APOE4 may affect Alzheimer’s disease risk, at least in part, by regulating amyloid pathology (48–50). The relationship between Aβ pathology and tauopathy, which corelates better with the progression of cognitive impairment, has not been sorted out as to which one is causal or whether they actually synergize (51). Our findings identified the Wnt/PCP pathway as a synaptic target of Aβ-associated synaptotoxicity, which starts long before the appearance of cognitive symptoms and still causes synapse degeneration in later stages of development. Therefore, targeting the Wnt/PCP pathway has great therapeutic potential for both early and late stages of disease progression. Immune responses and tauopathy are thought to be triggered by Aβ-associated toxicity and play important roles in driving disease progression in the later stage. Great efforts have gone into developing therapies targeting the immune responses and tauopathy. However, an amyloid-based therapy would still be necessary even if the therapies based on immune responses and tauopathy are successful. We still need to stop the synapse loss caused by Aβ-associated toxicity, which takes place in a large proportion of patients with Alzheimer’s disease. Based on our current data, it appears that Ryk is a better therapeutic target than Vangl2 as Vangl2 cKO itself leads to behavioral deficits and cannot improve cognitive function of the 5XFAD mice and Ryk cKO itself does not cause behavioral deficits and can significantly preserve cognitive function of the 5XFAD mice (Fig. 6L). Infusion of a function blocking monoclonal antibody against Ryk showed promising efficacy in protecting the glutamatergic synapses and cognitive functions (Fig. 7, C, D, and K). We found that Ryk monoclonal antibody treatment also reduced astrogliosis and microgliosis in a mouse model of Alzheimer’s disease, 5XFAD (Fig. 7, N and O). Although we currently cannot determine whether the reduction of astrogliosis and microgliosis is secondary, due to the protection of synapses, and thus preserving the function of the circuits, or due to a direct function of our Ryk monoclonal antibody on astrocytes and microglia, these results are consistent with the observed preservation of cognitive function, suggesting that the Ryk monoclonal antibodies may be effective therapeutic agents, which can address both synapse loss and gliosis.
MATERIALS AND METHODS
Study design
This study was designed to evaluate the role of PCP signaling pathway in synapse degeneration in mature neural circuits. Experiments were performed in biological replicates of three or more. The researchers were blinded to data collection.
Animals
All animal work in this research was approved by the University of California, San Diego Institutional Animal Care and Use Committee. The 5XFAD transgenic mice carrying the five mutations Swedish (K670N and M671L), Florida (I716V), and London (V717I) in human APP695 and human PS1 cDNA (M146L and L286V) under the transcriptional control of the neuron-specific Thy-1 promoter were purchased from the Jackson Laboratory (stock no. 34848) (31). CRISPR-Cas9 knockin mice were purchased from the Jackson Laboratory (stock no. 026175). Vangl2fl/fl (cKO) was provided by Y. Yang at Harvard Medical School (52). Rykfl/fl (cKO) was generated in the Zou laboratory (24). Roughly equal numbers of male (56%) and female (44%) animals were used in this study.
Hippocampal neuron culture
Hippocampi were dissected from E18.5 mice, and hippocampal neuron culture was performed as previously described (6). Briefly, cells were pelleted and resuspended in Neurobasal medium supplemented with 1% B27 (Invitrogen), penicillin/streptomycin (CellGro), and GlutaMAX (Invitrogen) and plated on poly-d-lysine (Millipore)–coated glass coverslips in a 24-well plate at a density of 2 × 104 cells/cm2 for immunostaining. Medium was changed every 4 days. Cultures were maintained for 14 DIV at 37°C with 5% carbon dioxide atmosphere.
Immunofluorescence staining and image analysis of cultured neurons
For synaptic puncta density analysis in cultured hippocampal neurons, neurons on DIV14 were fixed for 20 min in 4% PFA (paraformaldehyde). After fixation, cells were incubated in a blocking solution [1% bovine serum albumin and 5% goat serum in tris buffer saline (TBS) solution with 0.1% Triton X-100] for 1 hour and then stained overnight at 4°C with primary antibodies chicken anti-MAP2 (Microtubule Associated Protein 2) (neuronal marker; Abcam), guinea pig anti-Bassoon (presynaptic marker; Synaptic Systems), and goat anti–PSD-95 (postsynaptic marker; Millipore). Afterward, cells were incubated with fluorochrome-conjugated secondary antibodies Alexa 488 anti-chicken, Alexa 647 anti–guinea pig, and Alexa 568 anti-goat solution for 2 hours at room temperature (RT) and mounted in mounting media. Z-stacked images were obtained with a Carl Zeiss microscope using a 63× oil immersion objective. Three or more neurons with pyramidal morphology and at least two diameters’ distance from the neighboring neurons were selected per coverslip. Three coverslips were used for each group per experiment. Secondary dendrites were chosen for puncta analysis. The number of puncta was analyzed using the ImageJ Synapse Counter plug-in, and the length of the dendrite was analyzed by ImageJ [National Institutes of Health (NIH)]. When the pre and post are overlapped by 33 to 100%, they are considered colocalized. Data in Figs. 2D and 5 (B, D, and F) were quantified in single blind.
Aβ oligomer preparation
Human Aβ42 (AnaSpec) or human biotin-Aβ (1 to 42) (AnaSpec) was dissolved in dimethyl sulfoxide, sonicated, and diluted with F12 medium for Aβ monomerization to a concentration of 100 μM. For oligomerization, the solution was incubated for 24 to 26 hours at 4°C and centrifuged at 16,000g for 20 min, and the supernatant was collected as oligomerized Aβ. The oligomerized Aβ42 preparations were analyzed via SDS–polyacrylamide gel electrophoresis using 12% tris-glycine gels. Aβ42 peptides (50 μg) were loaded and electrophoretically separated at 25 mA. Gels were transferred onto a polyvinylidene difluoride membrane. Forms of Aβ42 were detected using antibody 6E10 (BioLegend); 26.94 ± 7.43% of Aβ42 existed as monomers [molecular weight (MW), 2.5 to 6.5 kDa] (n = 4); 2.06 ± 0.41% existed as dimers (MW, 6.5 to 11.5 kDa); 17.84 ± 0.97% existed as trimers (MW, 11.5 to 15.5 kDa); 38.15 ± 5.09% existed as tetramers (MW, 15.5 to 20.5 kDa); and 15.01 ± 7.95% existed as high-n oligomers (fig. S2).
Intracerebroventricular injection of Aβ oligomer
Adult (2 to 3 months old) mice were deeply anesthetized with an intraperitoneal injection of ketamine/xylazine cocktail until unresponsive to toe and tail pinch. Aβ oligomers (5 ng; volume, 250 nl) or phosphate-buffered saline (PBS) (volume, 250 nl) was stereotaxically injected into bilateral ventricles (−0.1 mm anteroposterior, 1 mm mediolateral, and −2.5 mm dorsoventral). Five days after intracerebroventricular injection, brains were harvested for immunohistochemistry.
Intrahippocampal injection of AAV1-hSyn-eGFP-Cre
Adult Vangl2 cKO, Ryk cKO, and WT littermate controls (2 to 3 months old) were deeply anesthetized with an intraperitoneal injection of ketamine/xylazine cocktail until unresponsive to toe and tail pinch. AAV1-hSyn-eGFP-Cre (Addgene) was stereotaxically injected into bilateral hippocampal CA1 (160 nl per site) (1.7 mm anteroposterior, 1 mm mediolateral, and −1.3 mm dorsoventral; and 2.4 mm anteroposterior, 2.25 mm mediolateral, and − 1.5 mm dorsoventral) in Vangl2 cKO, and CA1 and CA3 (2.06 mm anteroposterior, 2.5 mm mediolateral, and 2.25 mm dorsoventral) in Ryk cKO.
sgRNA design, expression analysis, and virus generation
The Brie database was used for the design of single-guide RNAs (sgRNAs). To validate the efficiency of sgRNA candidates, individual sgRNA was cloned into PX549-SpCas9 vector (Addgene, no. 62988). Neuro2A cells were cultured on a 12-well plate. Cells were then transfected with 1 μg of sgRNA plasmid by using polyethylenimine MAX (1 mg/ml; Polyscience). Puromycin was used to select the transfected cells. Genomic DNA of Neuro2A was purified by using the chloroform-ethanol method. The amplification of target DNA fragment and efficiency testing of individual sgRNAs were performed with the manufacturer’s protocol of Surveyor assay.
Selected sgRNA sequences are as follows: Celsr2 exon1, 5′-GTACACCGTTCGGCTCAACG-3′; and Ceslr3 exon1, 5′-CGTTCGGGTGTTATCAGCAC-3′. The sgRNAs cloned the AAV vector (Addgene, no. 60231) for virus package. The LacZ sgRNA was used as a negative control. AAV was produced by transfection of 293 cells with three plasmids: an AAV vector expressing target constructs, an AAV helper plasmid (pHELPER; Agilent), and an AAV rep-cap helper plasmid (pRC-DJ). Transfection of 293 cells was carried out using calcium phosphate. The cells were collected and lysed 72 hours after transfection. Viral particles were purified by an iodixanol step-gradient ultracentrifugation method. Quantitative reverse transcription polymerase chain reaction (PCR) was used for virus titer measurement. The virus titer was ~1012 GC (genome copies)/ml. AAV-sgRNA-Cre virus was added into the cultured hippocampal neurons derived from Cas9 embroys for 2 weeks (added on DIV7 and harvested on DIV21). Cells were then lysed for Western blotting test. For the in vivo experiment, AAV-sgRNA-Cre virus was stereotaxically injected into the hippocampal CA1 region of the Cas9 mice.
Immunofluorescence staining and image analysis of brain sections
For in vivo synaptic protein immunostaining, mice were deeply anesthetized with an intraperitoneal injection of ketamine/xylazine until unresponsive to toe and tail pinch and perfused with PBS followed by 4% PFA. Brains were removed and postfixed in 4% PFA overnight at 4°C. Afterward, brains were cryoprotected in 30% sucrose for 2 days and coronal free-floating sections were prepared at 30 μm in a vibratome. The sections obtained were treated with 1% SDS for 5 min at RT for antigen retrieval, incubated in a blocking solution (1% bovine serum albumin and 5% goat serum in TBS solution with 0.1% Triton X-100) for 1.5 hours, and then stained overnight at 4°C with primary antibodies guinea pig anti-Bassoon (presynaptic marker; Synaptic Systems) and goat anti–PSD-95 (postsynaptic marker; Millipore). Afterward, sections were incubated with fluorochrome-conjugated secondary antibodies Alexa 647 anti–guinea pig and Alexa 568 anti-goat solution for 2 hours at RT, counterstained with 4′,6-diamidino-2-phenylindole (DAPI), and mounted in mounting media. The synapses formed between the Schaffer collaterals and the hippocampal CA1 pyramidal neuron apical dendrites spanning the mouse stratum radiatum were imaged. Fluorescent z-stack images were acquired with an LSM510 Zeiss confocal microscope using a 63× oil immersion objective with 2× zoom-in. Twelve to 16 images from six to eight brain sections were analyzed per animal for staining quantification. A number of puncta from each image were analyzed using the ImageJ Synapse Counter plug-in. When the pre and post are overlapped by 33 to 100%, they are considered colocalized.
Rabbit anti-human Aβ (6E10; Novus Biologicals) was used to test Aβ load. Fluorescent z-stack images were acquired with an LSM510 Zeiss confocal microscope using a 10× objective. The number of Aβ plaques and the Aβ plaque area in the hippocampal area from three to four brain sections per animal were analyzed using ImageJ. Rabbit anti-Iba-1 (Wako) and rabbit anti-GFAP (Abcam) were used to test gliosis. Three to four brain sections per animal with three areas in the stratum radiatum per brain section were quantified for glial cell coverage using ImageJ. Both image acquisition and analysis (Figs. 1, F, H, I, and M; 2H; 5I, 6, C and D; and 7, C and D) were performed by two experienced researchers blinded to the experimental conditions.
Sparse labeling of neurons for analysis of dendritic spines
The mixture of AAV-Cre (100 nl;109 infective particles/ml) with AAV-Flex-eGFP (1013 infective particles/ml) was injected into each site. Secondary and tertiary dendritic branches (30- to 40-μm segments of CA1 oblique apical dendrites), located 100 to 200 μm from the CA1 pyramidal neuronal cell bodies in the stratum radiatum, were imaged using a ×63 magnification oil objective. Spine density analysis was performed using Bitplane Imaris software. Protrusions not clearly defined or with lengths >5 μm were excluded from analysis. Both image acquisition and analysis were double-blinded to the genotypes or experimental conditions.
Immunohistochemistry and sample preparation for 3D STORM
Cryostat tissue sections with 10 μm thickness were quenched with 20 mM glycine in DPBS (Dulbecco’s phosphate-buffered saline) and then permeabilized and blocked with 10% donkey serum and 0.3% Triton X-100 in PBS for 1 hour, followed by incubation with primary antibodies (overnight at 4°C) and secondary antibodies (1 hour at RT). Sections were lastly fixed for 5 min in 3% PFA with 0.05% glutaraldehyde after antibody labeling. Guinea pig anti-Bassoon (Synaptic Systems), goat anti–PSD-95 (Abcam) or mouse anti–PSD-95 (Millipore), rabbit anti-Celsr3 (generated in the Zou laboratory), mouse anti-Vangl2 (Santa Cruz Biotechnology), goat anti-Frizzled3 (R&D Systems), and mouse anti-Ryk (generated in the Zou laboratory) were used. For secondary antibodies (Jackson ImmunoResearch), Alexa Fluor 647, Alexa Fluor 568, and Alexa Fluor 488 were used for three-color staining. Imaging buffer (100 μl) was dropped onto glass slides holding the brain sections and covered with #1.5 rectangular glass coverslips (Thermo Fisher Scientific). Samples were sealed using nail polish. The imaging buffer was prepared according to the N-STORM protocol from Nikon.
Hippocampal neurons were cultured in glass-bottom dishes (no. 1.5, MatTek). On DIV14, neurons were fixed for 15 min in 4% PFA at 37°C. After fixation, cells were washed with PBS and then incubated in a blocking solution (3% bovine serum albumin, 3% goat serum, 0.1% Tween 20, and 0.02% sodium azide in PBS), followed by incubation with primary antibodies (overnight at 4°C) and secondary antibodies at RT for 1 hour. Cells were then fixed with 4% PFA for 5 min. Guinea pig anti-Bassoon (Synaptic Systems), rabbit anti-Celsr3 (generated in the Zou laboratory), and goat anti-Frizzled3 (R&D Systems) were used. For secondary antibodies (Jackson ImmunoResearch), Alexa Fluor 647, Alexa Fluor 568, and Alexa Fluor 488 were used for three-color staining. Imaging buffer (4 ml) was added into the dish before imaging.
3D STORM imaging and analysis
Imaging was performed on a Nikon A1R Confocal STORM super-resolution system with a 100×/1.49 TIRF (total internal reflection fluprescence) oil immersion objective. Images were collected on an ANDOR IXON3 Ultra DU897 EMCCD camera using the multicolor mode setting in the Nikon Elements software. Collection was stopped after 10,000 periods of each channel were collected, and the super-resolution images were reconstructed with the Nikon STORM software. The position of individual molecules has been localized with high accuracy by switching them on and off sequentially using the 488, 568, and 647 lasers at appropriate power settings. The substantial drift generated by the multiple switching cycles is considerably reduced by calculating and correcting for sample drift over the course of the experiment by an autocorrelation method used by the Nikon software. This is done by correlating STORM images reconstructed from subsets of localizations at different time segments to that from the beginning of the acquisition. Axial drift over the course of the acquisition is minimized by engaging the Nikon perfect focus system. Images obtained on the Nikon N-STORM (Nikon Inc.) system were imported into the Nikon NIS-Elements analyzer. Then, the individual Bassoon containing the Celsr3-Frizzled3 pair was rotated in 3D. The axial distance between Celsr3 and Frizzled3 was calculated using the NIS-Elements analyzer.
Plasmid, inhibitors, and antibodies
Celsr3-Flag, Fzd3-HA, Vangl2-Myc, and tdTomato expressing constructs were described previously (22, 23). Sulfo-NHS-LC-Biotin was purchased from Pierce. The antibodies used in this study include α-Vangl2 (Santa Cruz Biotechnology), α-Celsr3 (rabbit polyclonal antibodies were generated in the Zou laboratory), α-Flag (Sigma-Aldrich), α-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Chemicon), α-insulin Rβ (Santa Cruz Biotechnology), and α-HA (BioLegend).
To generate truncated Celsr3 constructs, the full-length Celsr3 extracellular domain is amplified by PCR, digested with Eco RV/Nhe I, and subcloned into pCAGEN vector using the following primers:
ΔEGF/Lam_Celsr3 forward primer 1: 5′-GATCGATATC TTCTCTGGAGAGCTCACAGC-3′
ΔEGF/Lam_Celsr3 reverse primer 1: 5′-GCAGGCATCGTA AAAGGGCAGCACGTCGAG-3′
ΔEGF/Lam_Celsr3 forward primer 2: 5′-GTGCTGCCCTTT TACGATGCCTGCCCCAAG-3′
ΔEGF/Lam_Celsr3 reverse primer 2: 5′-GATCGCTAGCAAGTAGGCCAGCAAG-3′
ΔEGF1_Celsr3 forward primer: 5′-TGCTGCCCTTTACAGAGCTCGACCTCTGTTAC-3′
ΔEGF1_Celsr3 reverse primer: 5′-CGAGCTCTGTAAAGGGCAGCACGTCGAG-3′
ΔEGF2_Celsr3 forward primer: 5′-TCTGTGAGACACTGGACACTGAAGCTGGACG-3′
ΔEGF2_Celsr3 reverse primer: 5′-TCAGTGTCCAGTGTCTCACAAGAAGTCTCCCG-3′
ΔEGF3_Celsr3 forward primer: 5′-GCTGGACACTGTGGCCGCACGCTCCTTTC-3′
ΔEGF3_Celsr3 reverse primer: 5′-GTGCGGCCACAGTGTCCAGCTCGCAGTC-3′
ΔLaminin G1_Celsr3 forward primer: 5′-ACGCTGTGAGCAGGCCAAGTCACACTTTTGTG-3′
ΔLaminin G1_Celsr3 reverse primer: 5′-ACTTGGCCTGCTCACAGCGTGGACCATC-3′
ΔEGF4_Celsr3 forward primer: 5′-AGGCTGCCAGCTCACAATGGCCCATCCCTAC-3′
ΔEGF4_Celsr3 reverse primer: 5′-CCATTGTGAGCTGGCAGCCTGCCATAGTG-3′
ΔLaminin G2_Celsr3 forward primer: 5′-CTGTCGACTCACTGTGACCAACCCCTGTG-3′
ΔLaminin G2_Celsr3 reverse primer: 5′-TGGTCACAGTGAGTCGACAGTCTTTGCCACC-3′
ΔEGF5_Celsr3 forward primer: 5′-TGGCTGTACTGATGCCTGCCTCCTGAACC-3′
ΔEGF5_Celsr3 reverse primer: 5′-GGCAGGCATCAGTACAGCCAGGCTCCACATTC-3′
ΔEGF6_Celsr3 forward primer: 5′-AGGCTGTGTGTATTTTGGTCAGCACTGTGAGCAC-3′
ΔEGF6_Celsr3 reverse primer: 5′-GCTGACCAAAATACACACAGCCTGGGCCATAG-3′
ΔEGF7_Celsr3 forward primer: 5′-TGTGAGTGGCAAGACGAATGGCCAGTGCC-3′
ΔEGF7_Celsr3 reverse primer: 5′-CCATTCGTCTTGCCACTCACAGTCACAAG-3′
ΔEGF8_Celsr3 forward primer: 5′-CAACTGCAACCCCCACAGCGGGCAGTG-3′
ΔEGF8_Celsr3 reverse primer: 5′-CTGTGGGGGTTGCAGTTGGGGTCAAAGC-3′
ΔLaminin EGF_Celsr3 forward primer: 5′-ATGCCACCCACTCTACGATGCCTGCCCCAAG-3′
ΔLaminin EGF_Celsr3 reverse primer: 5′-GCATCGTAGAGTGGGAGGCATGAGTCACTG-3′
HEK293T cell transfection
HEK293T cells were purchased from the American Type Culture Collection and maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum. Transfection of HEK293T cells was carried out using polyethylenimine MAX (1 mg/ml; Polyscience). Mycoplasma contamination was monitored by DAPI staining.
Aβ oligomer binding assay
HEK293 cells were transiently transfected (polyethylenimine) with expression vectors encoding TdTomato, Celsr3-Flag, or control empty vectors (pCAGEN). Two days after transfection, cells were treated with biotinylated Aβ oligomer for 2 hours at 37°C, washed twice, fixed with 4% PFA for 20 min, and blocked with 5% donkey serum in PBS with 0.1% Triton X-100. The bound Aβ peptides were visualized with streptavidin–Alexa fluorophore conjugates (Alexa 488). DAPI was used to counterstain cell nuclei; TdTomato was used to monitor construct transfection. Anti-flag antibody was used to stain Celsr3. Fluorescent images were captured with Zeiss LSM 880 fast Airyscan using a 63× oil immersion objective. For saturation binding assays and calculation of Kd, 48 hours after transfection, cells were incubated with serial dilutions of biotinylated Aβ oligomer for 2 hours at 37°C. Control experiments were performed using the maximum amount of biotinylated Aβ oligomers, but HEK293T cells were transiently transfected with the pCAGEN vector. Cells were washed and fixed with 4% PFA for 10 min, washed with 0.1% Triton X-100/PBS, and incubated for 30 min with extra-avidin peroxidase (Sigma-Aldrich). Cells were then extensively washed, and bound peroxidase was quantified using TMB (3,3′,5,5′,-tetramethylbenzidine) substrate (Thermo Fisher Scientific). The reaction was terminated by the addition of TMB stop solution (Thermo Fisher Scientific). Absorbance was read at 450 nm in an ultraviolet-visible microplate reader. Nonspecific binding was determined in the presence of 100 μm of nonbiotinylated Aβ oligomer, and specific binding was calculated by subtracting absorbance values for nonspecific binding from total binding values.
Surface biotinylation assay to characterize cell surface expression levels of Celsr3 and Celsr3 deletion constructs (fig. S5)
The surface biotinylation and NeutrAvidin pull-down methods have been described previously (22, 23). Briefly, 48 hours after transfection with indicated plasmids, HEK293T cells [seeded on a PDL (poly-d-lysine) (20 μg/ml)–coated six-well plate] were washed with ice-cold PBS (pH 8.0) three times and incubated with Sulfo-NHS-LC-Biotin (1 mg/ml; Thermo Fisher Scientific)/PBS for 2 min at RT to initiate the reaction, followed by incubation on ice for 1 hour. After quenching active biotin by washing with ice-cold 100 mM glycine/PBS twice followed by normal ice-cold PBS, the cell lysates were incubated with NeutrAvidin agarose for 2 hours and then precipitated. For quantification, three independent experiments were performed, and the band intensity was quantified with ImageJ (NIH).
Coimmunoprecipitation
Forty-eight hours after transfection with the indicated plasmids, HEK293T cells were lysed with IP (immunoprecipitation) buffer [20 mM tris HCl (pH 7.5), 150 mM NaCl, 1 mM EGTA, 5 mM NaF, 10 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM dithiothreitol and protease inhibitor cocktail (Sigma-Aldrich), and 0.1% Triton X-100]. Lysates were immunoprecipitated with anti-HA, anti-Myc, or anti-Flag antibodies and with protein A/G agarose (Santa Cruz Biotechnology). Experiments were repeated three times and showed similar results.
Ryk monoclonal antibody infusion
Osmotic minipumps (Model 1002, Alzet, Cupertino, CA) were prefilled with either mouse IgG or Ryk monoclonal IgG (clone 25.5.5), expressed, and produced by Antibody Solutions (Santa Clara, CA) in 100 μl of artificial cerebrospinal fluid. The osmotic pump was subcutaneously implanted and connected by polyvinylchloride tubing to a stainless steel cannula, which is stereotaxically implanted into the lateral ventricle (Brain Infusion Kit 1; Alzet). Mice were randomly selected for mouse IgG or Ryk monoclonal IgG treatment. Osmotic minipumps were removed after 14 days.
GST fusion protein generation and biotinylated Aβ pull-down assay
GST or GST fusion of the EGF7 domain and EGF8 domain of Celsr3 (GST-EGF7 and GST-EGF8) was generated using pGEX4T-1. All GST fusions were expressed in BL21 Escherichia coli and purified with glutathione–Sepharose 4B (glutathione beads; GE Healthcare). Biotinylated Aβ oligomer (100 μM) and 200 μg of GST, GST-EGF7, and GST-EGF8 were incubated with NeutrAvidin agarose slurry for 2.5 hours in a cold room and then precipitated. The protein before and after precipitation was analyzed by Western blotting.
Electrophysiology
Mice weighing 25 to 30 g (4 to 5 months old) were used for the experiments. The animals were anesthetized and the brain was rapidly removed and placed in an ice-cold cutting solution containing (in millimolar) 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 7 MgSO4, 0.5 CaCl2, 25 glucose, 110 Na-pyruvate, 1.3 ascorbic acid, and 110 choline chloride (pH 7.4, 95% O2 and 5% CO2). The brain was cut in 300-μm-thick sections on a vibratome (VT1000S; Leica Microsystems, Nussloch, Germany), and the obtained sections in the region of the hippocampus were incubated in aCSF (artificial cerebrospinal fluid) before transferring them to the recording chamber (in millimolar): 119 NaCl, 2.5 KCl, 1 NaH2PO4, 26.2 NaHCO3, 11 glucose, 1.3 MgSO4, and 2.5 CaCl2 bubbled with 95% O2 and 5% CO2 at 37°C for 45 min and then at RT (20° to 25°C) until use. Standard field excitatory postsynaptic potentials (fEPSPs) were recorded in the CA1 region on acute mouse hippocampal slices as previously described. A stimulating electrode was placed in Schaffer collaterals, and a glass capillary microelectrode recording electrode filled with aCSF (resistance, 4 to 6 megohms) was positioned 200 to 300 μm from the stimulating electrode in the stratum radiatum of CA1. Stimulation was given at 0.033 Hz to evoke fEPSP. After 30 min of stable baseline recording, LTP was induced by two consecutive (1 s) stimuli at 100 Hz separated by 20 s. The data were sampled at 10 kHz and filtered at 2 kHz (Axon Instrument 200B and Digitdata 1440A). Traces were obtained by pClamp 10.3 and analyzed using Clampfit 10.3 and MATLAB 2018b.
Novel objective recognition behavioral testing
Object recognition tests were carried out in double blind in an open-field arena measuring 0.4 m by 0.4 m by 0.45 m. The total distance was recorded during a 5-min open-field session as locomotor activity; no differences were found among the groups. For the NOR test, the animals were placed at the center of the arena in the presence of two identical (familiar) objects for a 5-min training session. Exploratory behavior (amount of time exploring each object) was recorded by experienced researchers. One hour after training, animals were replaced into the arena for the testing session, in which one of the objects had been replaced by an unfamiliar (novel) object. The time spent exploring familiar and novel objects was measured blinded using SMART video tracking software (PanLab Harvard Apparatus, Holliston, MA, USA). Animals that had a total exploration time below 8 s were excluded from the NOR tests. Results are expressed as the percentage of time exploring each object during the training or testing session and were analyzed using a one-sample Student’s t test comparing the mean exploration time for each object with a fixed value of 50% (chance level).
Statistical analysis
Comparisons between multiple experimental groups were performed by one-way analysis of variance (ANOVA) followed by Tukey-Kramer post hoc test, when appropriate. Comparisons between two experimental groups were performed by Student’s t test. All statistical analyses were performed using GraphPad Prism software (La Jolla, California, USA). A value of P < 0.05 was considered significant.
Acknowledgments
We would like to thank K. Wang, Z. He, and members of the Zou laboratory for critical reading and comments on the manuscript. Funding: This project was supported by RO1 MH116667 to Y.Z. Airyscan confocal microscopy imaging was performed at the University of California San Diego (UCSD) School of Medicine Light Microscopy Facility (grant P30 NS047101). Author contributions: Y.Z., B.F., and A.E.F. designed the experiments. B.F., A.E.F., L.G., R.T., J.W., A.S.G., and Y.R.L. performed all experiments under the supervision of Y.Z. B.F., R.T., A.E.F., and Y.Z. analyzed and interpreted data. B.F. and Y.Z. wrote the paper. All authors read and commented on the manuscript. Competing interests: Y.Z., B.F., R.T., and A.E.F. are inventors on a PCT (PCT/US20/39194), “Methods and compositions for treating Alzheimer’s disease,” which was filed by the UCSD on 23 June 2020. Y.Z. is the founder of VersaPeutics and has equity, compensation and interim managerial role. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict-of-interest policies. The other authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/34/eabh2307/DC1
REFERENCES AND NOTES
- 1.Yang G., Pan F., Gan W. B., Stably maintained dendritic spines are associated with lifelong memories. Nature 462, 920–924 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butler M. T., Wallingford J. B., Planar cell polarity in development and disease. Nat. Rev. Mol. Cell Biol. 18, 375–388 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Humphries A. C., Mlodzik M., From instruction to output: Wnt/PCP signaling in development and cancer. Curr. Opin. Cell Biol. 51, 110–116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y., Nathans J., Tissue/planar cell polarity in vertebrates: New insights and new questions. Development 134, 647–658 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Zallen J. A., Planar polarity and tissue morphogenesis. Cell 129, 1051–1063 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Thakar S., Wang L., Yu T., Ye M., Onishi K., Scott J., Qi J., Fernandes C., Han X., Yates J. R. III, Berg D. K., Zou Y., Evidence for opposing roles of Celsr3 and Vangl2 in glutamatergic synapse formation. Proc. Natl. Acad. Sci. U.S.A. 114, E610–E618 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zou Y., Breaking symmetry - cell polarity signaling pathways in growth cone guidance and synapse formation. Curr. Opin. Neurobiol. 63, 77–86 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hefter D., Ludewig S., Draguhn A., Korte M., Amyloid, APP, and electrical activity of the brain. Neuroscientist 26, 231–251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puzzo D., Privitera L., Leznik E., Fa M., Staniszewski A., Palmeri A., Arancio O., Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 28, 14537–14545 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walsh D. M., Klyubin I., Fadeeva J. V., Cullen W. K., Anwyl R., Wolfe M. S., Rowan M. J., Selkoe D. J., Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Kamenetz F., Tomita T., Hsieh H., Seabrook G., Borchelt D., Iwatsubo T., Sisodia S., Malinow R., APP processing and synaptic function. Neuron 37, 925–937 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Cleary J. P., Walsh D. M., Hofmeister J. J., Shankar G. M., Kuskowski M. A., Selkoe D. J., Ashe K. H., Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 8, 79–84 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Townsend M., Shankar G. M., Mehta T., Walsh D. M., Selkoe D. J., Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: A potent role for trimers. J. Physiol. 572, 477–492 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shankar G. M., Bloodgood B. L., Townsend M., Walsh D. M., Selkoe D. J., Sabatini B. L., Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shankar G. M., Li S., Mehta T. H., Garcia-Munoz A., Shepardson N. E., Smith I., Brett F. M., Farrell M. A., Rowan M. J., Lemere C. A., Regan C. M., Walsh D. M., Sabatini B. L., Selkoe D. J., Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei G., Jewett A. I., Shea J. E., Structural diversity of dimers of the Alzheimer amyloid-β(25-35) peptide and polymorphism of the resulting fibrils. Phys. Chem. Chem. Phys. 12, 3622–3629 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Struhl G., Casal J., Lawrence P. A., Dissecting the molecular bridges that mediate the function of Frizzled in planar cell polarity. Development 139, 3665–3674 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macheda M. L., Sun W. W., Kugathasan K., Hogan B. M., Bower N. I., Halford M. M., Zhang Y. F., Jacques B. E., Lieschke G. J., Dabdoub A., Stacker S. A., The Wnt receptor Ryk plays a role in mammalian planar cell polarity signaling. J. Biol. Chem. 287, 29312–29323 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andre P., Wang Q., Wang N., Gao B., Schilit A., Halford M. M., Stacker S. A., Zhang X., Yang Y., The Wnt coreceptor Ryk regulates Wnt/planar cell polarity by modulating the degradation of the core planar cell polarity component Vangl2. J. Biol. Chem. 287, 44518–44525 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang B., Wang W., Bates M., Zhuang X., Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 319, 810–813 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dani A., Huang B., Bergan J., Dulac C., Zhuang X., Superresolution imaging of chemical synapses in the brain. Neuron 68, 843–856 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shafer B., Onishi K., Lo C., Colakoglu G., Zou Y., Vangl2 promotes Wnt/planar cell polarity-like signaling by antagonizing Dvl1-mediated feedback inhibition in growth cone guidance. Dev. Cell 20, 177–191 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Onishi K., Shafer B., Lo C., Tissir F., Goffinet A. M., Zou Y., Antagonistic functions of Dishevelleds regulate Frizzled3 endocytosis via filopodia tips in Wnt-mediated growth cone guidance. J. Neurosci. 33, 19071–19085 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hollis E. R. II, Ishiko N., Yu T., Lu C. C., Haimovich A., Tolentino K., Richman A., Tury A., Wang S. H., Pessian M., Jo E., Kolodkin A., Zou Y., Ryk controls remapping of motor cortex during functional recovery after spinal cord injury. Nat. Neurosci. 19, 697–705 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lacor P. N., Buniel M. C., Furlow P. W., Clemente A. S., Velasco P. T., Wood M., Viola K. L., Klein W. L., Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 27, 796–807 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lauren J., Gimbel D. A., Nygaard H. B., Gilbert J. W., Strittmatter S. M., Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 457, 1128–1132 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hong S., Beja-Glasser V. F., Nfonoyim B. M., Frouin A., Li S., Ramakrishnan S., Merry K. M., Shi Q., Rosenthal A., Barres B. A., Lemere C. A., Selkoe D. J., Stevens B., Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Onishi K., Tian R., Feng B., Liu Y., Wang J., Li Y., Zou Y., LRRK2 mediates axon development by regulating Frizzled3 phosphorylation and growth cone-growth cone communication. Proc. Natl. Acad. Sci. U.S.A. 117, 18037–18048 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davis E. K., Zou Y., Ghosh A., Wnts acting through canonical and noncanonical signaling pathways exert opposite effects on hippocampal synapse formation. Neural Dev. 3, 32 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Funk K. E., Mirbaha H., Jiang H., Holtzman D. M., Diamond M. I., Distinct therapeutic mechanisms of Tau antibodies: Promoting microglial clearance versus blocking neuronal uptake. J. Biol. Chem. 290, 21652–21662 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oakley H., Cole S. L., Logan S., Maus E., Shao P., Craft J., Guillozet-Bongaarts A., Ohno M., Disterhoft J., Van Eldik L., Berry R., Vassar R., Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhatt D. H., Zhang S., Gan W. B., Dendritic spine dynamics. Annu. Rev. Physiol. 71, 261–282 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Scott J., Thakar S., Mao Y., Qin H., Hejran H., Lee S. Y., Yu T., Klezovitch O., Cheng H., Mu Y., Ghosh S., Vasioukhin V., Zou Y., Apical-basal polarity signaling components, Lgl1 and aPKCs, control glutamatergic synapse number and function. iScience 20, 25–41 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuhlmann N., Milnerwood A. J., A critical LRRK at the synapse? The neurobiological function and pathophysiological dysfunction of LRRK2. Front. Mol. Neurosci. 13, 153 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tury A., Tolentino K., Zou Y., Altered expression of atypical PKC and Ryk in the spinal cord of a mouse model of amyotrophic lateral sclerosis. Dev. Neurobiol. 74, 839–850 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cisse M., Halabisky B., Harris J., Devidze N., Dubal D. B., Sun B., Orr A., Lotz G., Kim D. H., Hamto P., Ho K., Yu G. Q., Mucke L., Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim T., Vidal G. S., Djurisic M., William C. M., Birnbaum M. E., Garcia K. C., Hyman B. T., Shatz C. J., Human LilrB2 is a β-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science 341, 1399–1404 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masliah E., Mechanisms of synaptic dysfunction in Alzheimer’s disease. Histol. Histopathol. 10, 509–519 (1995). [PubMed] [Google Scholar]
- 39.Humphries C. E., Kohli M. A., Nathanson L., Whitehead P., Beecham G., Martin E., Mash D. C., Pericak-Vance M. A., Gilbert J., Integrated whole transcriptome and DNA methylation analysis identifies gene networks specific to late-onset Alzheimer’s disease. J. Alzheimers Dis. 44, 977–987 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Liu H., Luo K., Luo D., Guanosine monophosphate reductase 1 is a potential therapeutic target for Alzheimer’s disease. Sci. Rep. 8, 2759 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hyman B. T., Phelps C. H., Beach T. G., Bigio E. H., Cairns N. J., Carrillo M. C., Dickson D. W., Duyckaerts C., Frosch M. P., Masliah E., Mirra S. S., Nelson P. T., Schneider J. A., Thal D. R., Thies B., Trojanowski J. Q., Vinters H. V., Montine T. J., National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 8, 1–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long J. M., Holtzman D. M., Alzheimer disease: An update on pathobiology and treatment strategies. Cell 179, 312–339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hardy J. A., Higgins G. A., Alzheimer’s disease: The amyloid cascade hypothesis. Science 256, 184–185 (1992). [DOI] [PubMed] [Google Scholar]
- 44.Hardy J., Selkoe D. J., The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297, 353–356 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Ballatore C., Lee V. M., Trojanowski J. Q., Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 8, 663–672 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Musiek E. S., Holtzman D. M., Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen’. Nat. Neurosci. 18, 800–806 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Selkoe D. J., Hardy J., The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bales K. R., Verina T., Dodel R. C., Du Y., Altstiel L., Bender M., Hyslop P., Johnstone E. M., Little S. P., Cummins D. J., Piccardo P., Ghetti B., Paul S. M., Lack of apolipoprotein E dramatically reduces amyloid β-peptide deposition. Nat. Genet. 17, 263–264 (1997). [DOI] [PubMed] [Google Scholar]
- 49.Castellano J. M., Kim J., Stewart F. R., Jiang H., DeMattos R. B., Patterson B. W., Fagan A. M., Morris J. C., Mawuenyega K. G., Cruchaga C., Goate A. M., Bales K. R., Paul S. M., Bateman R. J., Holtzman D. M., Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 3, 89ra57 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verghese P. B., Castellano J. M., Garai K., Wang Y., Jiang H., Shah A., Bu G., Frieden C., Holtzman D. M., ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. U.S.A. 110, E1807–E1816 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Busche M. A., Hyman B. T., Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 23, 1183–1193 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song H., Hu J., Chen W., Elliott G., Andre P., Gao B., Yang Y., Planar cell polarity breaks bilateral symmetry by controlling ciliary positioning. Nature 466, 378–382 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/34/eabh2307/DC1