

Formation of transplantable reproducible hiPSC organoids can be aided with real-time label-free intracellular spectroscopy.
Abstract
Organoids formed from human induced pluripotent stem cells (hiPSCs) could be a limitless source of functional tissue for transplantations in many organs. Unfortunately, fine-tuning differentiation protocols to form large quantities of hiPSC organoids in a controlled, scalable, and reproducible manner is quite difficult and often takes a very long time. Recently, we introduced a new approach of rapid organoid formation from dissociated hiPSCs and endothelial cells using microfabricated cell-repellent microwell arrays. This approach, when combined with real-time label-free Raman spectroscopy of biochemical composition changes and confocal light scattering spectroscopic microscopy of chromatin transition, allows for monitoring live differentiating organoids without the need to sacrifice a sample, substantially shortening the time of protocol fine-tuning. We used this approach to both culture and monitor homogeneous liver organoids that have the main functional features of the human liver and which could be used for cell transplantation liver therapy in humans.
INTRODUCTION
Chronic liver failure is a potentially lethal condition, with liver transplantation being the only treatment option. However, this treatment option suffers from a shortage of donors and has high perioperative risks, and patients are confined to lifelong immunosuppression (1). Liver organoids formed with human induced pluripotent stem cells (hiPSCs) could be an inexhaustible source of liver tissue with limited immune rejection (2) that could be used for liver transplantations. However, the current hepatic differentiation strategies involving hiPSCs unfortunately suffer from several common problems (3–5).
A critical challenge of the hiPSC organoid technology is the inability to rapidly form abundant quantities of uniform, homogeneous, and sufficiently large three-dimensional (3D) clusters, called embryoid bodies, from dissociated undifferentiated single stem cells (6). The size of the embryoid bodies is a critical factor in organoid formation, as embryoid bodies over 500 μm in size have a much greater ability to differentiate into endoderm, the germ layer from which most internal organs arise. Although three main existing approaches using forced aggregation, bioreactors, and micromolds can fulfill some subset of the requirements, none of them can satisfy all the requirements. It is possible to form large, uniform, and homogenous embryoid bodies using forced aggregation (7), but it is impossible to generate many embryoid bodies using this approach (8). The bioreactor-based approach allows for the formation of large amounts of embryoid bodies; however, they are neither uniform nor homogenous (9). Last, the existing micromold approaches allow for the formation of large, over 500 μm in size, embryoid bodies; however, because of poor oxygen permeability and nutrient diffusion, these approaches often result in the formation of a necrotic core in large embryoid bodies, making them nonviable (10).
Previously, we introduced a new approach of high-throughput organoid formation from dissociated hiPSCs with microfabricated cell-repellent microwell arrays (11). This approach allows for a scalable and reproducible derivation of multiple large hiPSC organoids cocultured with other cell types and is sufficiently rapid to ensure that a necrotic core is not formed. Here, we demonstrate that when this approach is used to form functional liver organoids, along with efficiently blocking the Sonic hedgehog and Notch signaling pathways during the initial stages of differentiation, it is possible to achieve substantial improvements in production and enzymatic activity of critical proteins, detoxification, and coagulation factors in the organoids. These levels in the formed liver organoids are comparable to and sometimes even exceed the levels observed in the functional human liver.
While organoid formation presents various challenges, another critical challenge of the hiPSC organoid technology is the difficulty in monitoring the differentiation process. Often, several markers at the gene, protein, and functional levels must be screened simultaneously, using gene expression analysis, immunofluorescence imaging, analytical biochemistry assays, Western blotting, and other molecular biology techniques. Unfortunately, a substantial drawback of all these conventional techniques, including gene expression analysis with quantitative real-time polymerase chain reaction (PCR), is the need to sacrifice the sample to collect the data (12, 13). In addition, because of the heterogeneity of the hiPSCs, these methods cannot guarantee that the remaining cells or organoids have the same features as the sacrificed ones, even if they are from the same batch. Several approaches have been recently tested to overcome this problem and allow in vivo gene expression monitoring (14, 15). Recent work on the insertion of fluorescent tags by homologous recombination could allow in vivo observation of gene expression with fluorescence microscopy (16). Unfortunately, these approaches are limited by the labor-intensive fluorescent-tagging development for each new set of genes. The resulting genetically modified cells or organoids could not be used for therapeutic purposes (17, 18).
The approach of organoid formation from dissociated hiPSCs described here is well suited for label-free spectroscopic-based downstream phenotypic analysis, which allows for monitoring the differentiation state of organoids in real time. This real-time monitoring can be potentially useful for manipulating organoids into the desirable cell lineages. The important indicators of differentiation are subtle changes in biochemical composition of differentiating hiPSCs, and some of these changes could be detected using label-free Raman spectroscopy (19); for example, it has recently been demonstrated that it is possible to detect changes in glycogen, cytochrome, protein, and lipid composition in fixed samples of differentiating hiPSCs (20).
In addition to changes in biochemical composition, chromatin architecture reorganization has recently been identified as an important marker of pluripotency and stem cell differentiation (21). Compared to differentiated cells, undifferentiated cells lack highly condensed transcriptionally inactive heterochromatin and are significantly richer in lightly packed euchromatin composed of the most active portion of the genome (22, 23). As differentiation progresses, the amount of heterochromatin markedly increases. This is demonstrated indirectly by histological analysis of the nucleus (24) or chromatin immunoprecipitation (ChIP) accompanied by microarray hybridization (ChIP-chip) and high-throughput sequencing analyses (25, 26). It is also demonstrated directly with electron microscopy–based techniques (27). Unfortunately, these techniques are destructive, involve extensive manipulations of the sample, and hence cannot be used in living systems for real-time differentiation monitoring. Because of their nondestructive nature, optical microscopic techniques, such as superresolution microscopic visualization of histone proteins combined with fluorescent tags (28, 29), could be used for sensing chromatin organization in live cells. Sensing chromatin using its intrinsic properties without exogenous labels could be even more challenging. Backman and colleagues (30, 31) recently demonstrated label-free nanosensing of chromatin packing and dynamics with partial wave spectroscopic microscopy of live cells. This suggests that confocal light absorption and scattering spectroscopic (CLASS) microscopy (32), another native contrast functional imaging technique, could serve as a useful tool for label-free monitoring of differentiation in live hiPSC organoids. CLASS microscopy extends the principles of light scattering spectroscopy (33–35) to subcellular imaging and can also sense the state of the chromatin organization.
Here, we show that a high-throughput organoid formation technique, when combined with efficient liver-specific signal pathway blocking, can produce liver organoids with properties similar to functional liver tissue. Furthermore, a combination of chromatin-sensitive CLASS microscopy and biochemical composition–sensitive Raman spectroscopy can be used to monitor the differentiation progress toward liver organoids in an efficient and nondisruptive manner.
RESULTS
Rapid formation and evaluation of liver organoids
Human embryoid bodies (hEBs) were created with our previously developed cell-repellent agarose microwell array–based method, which overcomes the challenge of rapidly forming homogeneous organoids (see Fig. 1A and Materials and Methods). Agarose, which has non–cell-adhesive properties, allows spontaneous and rapid aggregation of the initially dissociated hiPSCs into precisely one hEB in each microwell, in approximately 18 to 24 hours, considerably faster than most of the other methods, with neither Rho-associated protein kinase inhibitor nor spinning used throughout the process. The size and uniformity of hEBs are dependent on the number of hiPSCs placed in a single 700-μm-diameter round-bottom microwell (Fig. 1B). While placing less than 15,000 hiPSCs per microwell results in unstable aggregation of mostly inhomogeneous hEBs, depositing 15,000 to 35,000 hiPSCs results in a substantial improvement in aggregation stability and uniformity of hEBs. The optimal conditions were reached at 35,000 cells per microwell with hEB sizes being in the 450- to 500-μm range and the aggregation becoming the most stable. Further increase in the number of hiPSCs in a single microwell causes the formed hEBs to once again become small and unstable.
Fig. 1. Formation of liver organoids using microwell arrays in cell-repellent agarose.

(A) Workflow for the formation of large amounts of hEBs with HAMECs mixed with hiPSCs in a 1:3 ratio. (B) The hEB gross morphology when formed with different hiPSC seeding densities within the agarose microwell. Scale bars, 500 μm. (C) The hiPSC suspension aggregation immediately after seeding within the microwells, and after 6- and 24-hour formation. (D) Schematic representation of the differentiation protocol for forming functional liver organoids.
Endothelial cells, which are among the first cells present under natural conditions at the initial stages of embryogenesis (36) and eventually constitute approximately one-fifth of the total adult liver cell mass (37), were incorporated into the organoids. Endothelial cells perform an important role in organogenesis by forming vascular networks that secrete angiocrine factors, which are organ-specific growth factors (38, 39). Endothelial cells also maintain homeostasis and metabolism, along with providing passive channels for blood and nutrient distribution to several organs. During the formation of the liver organoids, we used human adipose microvascular endothelial cells (HAMECs) mixed with hiPSCs in a 1:3 ratio within the embryoid bodies. Both hiPSCs and endothelial cells self-organize within the organoids over time by forming endothelial cell channels at the end of the differentiation process (40).
After the formation of hEBs in 18 to 24 hours (Fig. 1C), we used the protocol that we developed for forming functional hepatic organoids consisting of mature hepatocytes and endothelial cells (see Fig. 1D and Materials and Methods). The four stages of the protocol are definitive endoderm, foregut endoderm, hepatoblasts, and mature hepatocytes, with each stage lasting for 4 days. To mimic processes taking place during in vivo embryogenesis, we used pathway inhibitors specific to each of the four stages of differentiation. For example, the MK-4101 inhibitor of the Sonic hedgehog pathway along with activin A, basic fibroblast growth factor (bFGF), and transforming growth factor–β1 (TGF-β1) was used to develop definitive endoderm, Notch signaling pathway inhibitor LY-41575 was used in combination with bone morphogenetic protein 4 (BMP-4) and FGF4 to form the foregut endoderm, while WNT inhibitory factor 1 (WIF-1) and dickkopf WNT signaling pathway inhibitor 1 (DKK-1) inhibitors of Wnt/β-catenin were used for inducing differentiation of hepatoblasts.
To investigate whether the protocol successfully produced functional hepatic organoids, we monitored stage-specific markers throughout the protocol. We observed that formed liver organoids demonstrated high expression levels of liver-specific genes associated with multiple functions typical of human primary hepatocytes (HPHs). One of the main functions of the liver is the production of albumin, which is responsible for most of the oncotic pressure in the plasma of blood vessels, preventing fluids in the bloodstream from leaking into the surrounding tissue. In addition, albumin also plays an important role in antioxidation, immunomodulatory activity, and endothelial regulation. As can be seen in Fig. 2A, albumin gene expression in the liver organoids showed levels comparable with that of the in vitro HPHs and with published RNA sequencing in vivo data (41) (see also table S1). Another important liver function is related to its detoxification ability. We therefore tested the liver organoids for the presence of gene expression of cytochromes cytochrome P450 1A2 (CYP1A2), CYP3A4, CYP2B6, (Fig. 2A), CYP1B1, and CYP2C9 (fig. S1A), which are most prominent in phase 1 of liver detoxification, and enzymes UDP glucuronosyltransferase family 1 member A1 (UGT1A1), UGT1A3 (fig. S1A), and UGT1A4 (Fig. 2A), which are prominent in phase 2 of liver detoxification. Once again, their expression levels were very similar to those observed in vitro in HPHs and in the in vivo data (table S1). We also observed comparable levels to the HPHs in other liver genetic markers, such as α-fetoprotein (AFP), ornithine transcarbamylase (OTC), cytokeratin 18 (CK-18), alpha-1 antitrypsin (A1AT), phenylalanine hydroxylase (PAH), hepatocyte nuclear factor–1β (HNF-1β), HNF-4α, HNF-6, glycogen synthase 2, and fatty acid synthase 1 (fig. S1A).
Fig. 2. Functional characteristics of liver organoids.
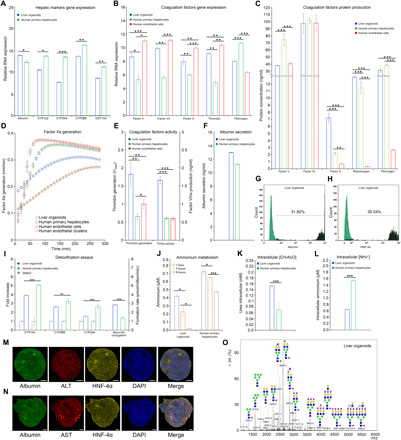
Real-time PCR measurements of (A) hepatic markers and (B) coagulation factor markers. (C) ELISA measurements of coagulation factor markers. (D) Factor Xa generation assay. HPH (green) and HAMEC (red) in 2D culture showed similar curve slopes, while 3D culture of HAMEC (brown) and liver organoids (blue) exhibited same slope pattern. The maximum rate of reaction Vmax of liver organoids (0.26) was lower than all positive controls (HPH, 0.40; HAMEC 2D, 0.41; HAMEC 3D, 0.33), indicating better enzymatic activity of organoids. (E) Thrombin generation assay and factor VIIIa activity displayed better results in organoids when compared with HPH and HAMEC. (F) ELISA measurement of albumin in liver organoids showed comparable results with HPH (13.05 ng/ml versus 11.30 ng/ml, respectively). (G) Fluorescence-activated cell sorting (FACS) analysis of albumin. (H) FACS analysis of HNF-4α. (I) CYP450 liver detoxification I analysis (induction of CYP450 showed doubled activity after drug administration in CYP1A2 and CYP2B6) and resorufin conjugation for liver detoxification II analysis (resorufin formation rate was statistically higher than HPH). (J) Ammonium metabolism assay. (K) Intracellular urea concentration analysis. (L) Intracellular ammonium concentration analysis. (M and N) Immunofluorescence of hepatic markers. Scale bars, 50 μm. (O) Glycan profile for mature liver organoids. Data are presented as means ± SD (n = 3). *P < 0.05, **P < 0.01, and ***P < 0.001.
Another important liver function is its role in hemostasis, which involves blood coagulation. The liver synthesizes almost all coagulation factors and their inhibitors, while liver damage is commonly associated with impairment of coagulation. We observed that the gene expression from major coagulation factors associated with intrinsic, extrinsic, and common pathways of the coagulation cascade, fibrinolysis, and platelet regulation was comparable with that observed in HPHs (Fig. 2B and fig. S1B). Some of the main factors responsible for the final and most important part of the coagulation cascade, namely, factors V, VII, X, and thrombin, were expressed at the levels even higher than that in HPHs (Fig. 2B and table S2). The substantial improvements in the functional characteristics of the liver organoids with endothelial cells, compared to the liver organoids formed with only hiPSCs, including both hepatic markers and coagulation factors, are presented in fig. S2.
In addition to quantifying gene expression, it is also important to evaluate the corresponding production of protein. Using the enzyme-linked immunosorbent assay (ELISA), we established that a number of coagulation factors, such as factors IX and X, plasminogen, ADAM metallopeptidase with thrombospondin type 1 motif 13 (ADAMTS13), antithrombin III, protein C, and protein S were expressed at levels comparable to that of HPH and HAMEC (Fig. 2C and fig. S1C). In addition, coagulation factors normally produced by the endothelial cells [tissue plasminogen activator and von Willebrand factor(VWF)] were also expressed in the liver organoids, demonstrating a successful integration of HAMEC within the organoids (fig. S1C). We also checked enzymatic activity of three main coagulation factors that are part of the common pathway, namely, factor Xa, thrombin generation, and factor VIIIa activity. Once again, we found that they were comparable to the enzymatic activity of HPH and HAMEC (Fig. 2, D and E).
We also used ELISA to analyze the production of albumin and established that it was comparable with protein production in HPH (Fig. 2F), while fluorescence-activated cell sorting (FACS) analysis of albumin and HNF-4α resulted in 31.82% (Fig. 2G) and 35% (Fig. 2H) of positive cells, respectively. Liver phase 1 detoxification was also tested at functional levels by inducing the major cytochromes P450 (CYP1A2, CYP2B6, and CYP3A4) with different drugs. Increased levels of cytochromes were observed after induction when compared with basal levels [dimethyl sulfoxide (DMSO)] similar to those observed in HPH (Fig. 2I). Resorufin production was also tested as a liver phase 2 marker, demonstrating a significantly higher formation rate than that in HPH (Fig. 2I). Ammonium metabolism assay of the liver organoids displayed a reduction of ammonium in the medium that was halving every 3 hours (Fig. 2J), confirmed also by the increased amount of intracellular urea (Fig. 2K) and reduction of intracellular ammonium (Fig. 2L) as a result of ammonium clearance efficiency of the liver organoids.
Organoid immunofluorescence imaging of albumin coupled with several liver markers [alanine aminotransferase (ALT), aspartate transaminase (AST), HNF-4α, HNF-1α HNF-3β, A1AT, tyrosine-protein kinase Met (C-MET), and CK18] was performed to test the ability of the organoid to simultaneously coexpress multiple liver proteins (Fig. 2, M and N, and fig. S1D). We also observed the formation of the CD31-positive tubular-like structures with circular cross sections within the differentiated liver organoids. As CD31 is a known marker for vascular development, this could be an early sign of vascular formation (fig. S1E). Microscopy images of the organoid morphology demonstrated a gradual increase in the size and elongation of the organoids and a change in the cellular morphology to a polygonal shape (fig. S1, F and G). An indocyanine green (ICG) test was carried out as an indicator of the ability to intake and secrete albumin from interstitial fluids (fig. S3, A and B). Lipid storage and uptake, as well as glycogen storage, were tested (fig. S3, C to E), proving that the organoids were capable of storing lipids and glycogen. The uptake of acetylated low-density lipoproteins (Ac-LDLs) was used as an indicator of complete functionality of the organoids.
Last, we performed glycomic analysis to compare the glycan profile of liver organoids with that of HPH (fig. S1H). While the developed liver organoids presented a fairly different N-glycan profile compared to cryopreserved HPH (42), they had a rather similar profile to that observed in fresh isolated HPH (41) and real liver tissues, with a lower occurrence of high mannoses, as opposed to the high amounts that are normally observed in cell cultures (Fig. 2O).
Raman spectroscopy of live organoids during differentiation
While conventional techniques were shown to be useful for monitoring biochemical changes, we also used Raman spectroscopy to monitor biochemical composition since it can be performed on live cells. Raman spectra were collected from live differentiating hiPSC organoids over a period of 16 days, with this 16-day period corresponding to the four stages of the protocol. While most Raman systems analyze by-products downstream, our system (see Materials and Methods) focuses a near-infrared excitation laser beam directly on the organoids. The organoids were initially taken from the incubator and placed into a quartz dish filled with the basal differentiation medium, without the addition of the growth factor. The measurements were performed every other day on multiple individual unstained organoids, with the duration of the measurements short enough to keep the organoids alive. Figure 3A shows the average spectra collected in hEBs and in organoids during the four stages. Within a single organoid, the Raman spectra had approximately 3.5% point-to-point variation, while the organoid-to-organoid variation of the Raman spectra was 5.8%. Because of cell cycle arrest during differentiation (43), the DNA content remains stable (20), and therefore, it makes sense to normalize the spectra by the DNA-related peak at 722 cm−1 (44). Although some of the prominent peaks in the Raman spectra of liver organoids can be mostly associated with the specific biochemicals important for hepatic function, such as albumin (1006 cm−1), AFP (1746 cm−1), glycogen (490 cm−1), or hepatic lipids (1452 cm−1), each of these biochemicals has multiple peaks (see Fig. 3A) overlapping with each other. In addition, biochemicals present in hEBs at the start of the differentiation process also have peaks overlapping with the aforementioned biomolecules. Therefore, to quantify the changes in individual biochemical composition of hiPSC organoids during the course of differentiation, we used the spectral unmixing algorithm (45, 46), with some improvements.
Fig. 3. Raman spectra of differentiating liver organoids.
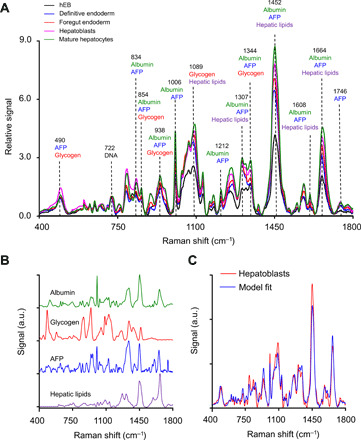
(A) Experimentally measured average Raman spectra of hEBs and liver organoids during the four stages of differentiation with prominent peaks specific to main Raman contributing biomolecules marked. (B) Four base Raman spectra: albumin, AFP, glycogen, and hepatic lipids. a.u., arbitrary units. (C) Experimentally measured Raman spectrum of the liver organoid in the hepatoblasts stage and model fit. a.u., arbitrary units.
The experimentally observed Raman spectrum of organoids during stage k of differentiation S(k)(λ), where k changes from 1 to 4 for the stages of definitive endoderm, foregut endoderm, hepatoblasts, and mature hepatocytes, respectively, and λ are Raman frequency shifts, can be written as
| (1) |
where N is the number of known main biochemicals contributing to the organoid spectrum, is the concentration of biochemical i during stage k, Si(λ) is the base spectrum of biochemical i, ShEB(λ) is the spectrum of the hEB at the start of the differentiation process, and ε(λ) is the error that includes experimental noise and contributions of the biochemicals either not present in the hEBs or not accounted for among the main biochemicals known to be present in the differentiating organoids. Experimental spectra can be fit to Eq. 1 using a linear least-square minimization approach. However, as it is known to be an ill-posed problem (47), one needs to use additional prior information about the spectrum. Therefore, to improve the problem conditioning, we used the cost function
| (2) |
with the regularization term P(λ) equal to the difference between the experimental and the modeled spectra at Raman frequency shifts where this difference is negative, which penalizes the potential solution when the modeled spectra exceed the experimental spectrum.
Before using the described linear least-square minimization approach, we first had to identify the main biochemicals contributing to the organoid spectra. In addition to the hEB Raman spectra, we identified four main Raman contributors to the differentiating liver organoid spectra. These contributors are albumin, AFP, glycogen, and hepatic lipids. The base spectra of albumin, AFP, and glycogen were measured using the Raman system. Hepatic cells feature a variety of fatty acids and triglycerides, and although most lipids have similar Raman spectra, we decided to use published spectra of hepatic lipid droplets (48) as the most representative spectrum of lipids in hepatic cells. All base spectra are presented in Fig. 3B.
Following the identification of the main biochemicals and collection of the related base spectra, we fitted the experimentally measured Raman spectra of liver organoids for each stage of differentiation. A typical comparison of the Raman spectrum for the stage of hepatoblasts and model fit is presented in Fig. 3C. The production of albumin, AFP, glycogen, and hepatic lipids detected in live organoids during differentiation with Raman spectroscopy is shown in Fig. 4 (A to D) and is consistent with the changes observed previously with conventional techniques. For example, relative concentration of albumin (Fig. 4A) steadily increases throughout each stage of differentiation with an accelerated rate during the hepatoblasts and mature hepatocytes stages, the two final stages of hepatic differentiation. Production of AFP (Fig. 4B), a typical marker of hepatoblasts (49), decreased somewhat during the foregut endoderm stage before significantly increasing and reaching its maximum during the stage of hepatoblasts and decreasing once again during the stage of mature hepatocytes, consistent with the published real-time PCR (50, 51) results. On the other hand, both glycogen (Fig. 4C) and hepatic lipids (Fig. 4D) steadily increase their relative concentration during all stages of differentiation, which, once again, is consistent with the published results (52).
Fig. 4. Reconstructed concentrations of four biomolecules during liver organoid differentiation.
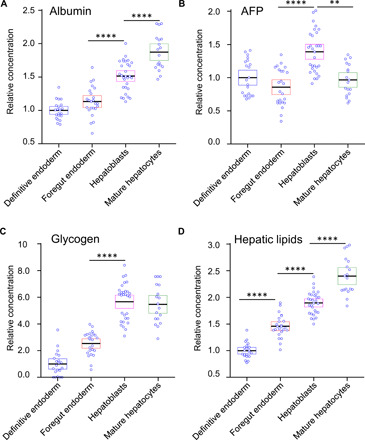
Comparisons of individual data points for (A) albumin, (B) AFP, (C) glycogen, and (D) hepatic lipids for each of the stages of differentiation relative to the hEB stage. Each data point represents a single-collection volume containing approximately 10,000 cells. A one-way analysis of variance (ANOVA) with post hoc Tukey’s test was performed. All experiments were performed with three biological replicates, from three technical replicates. The results represent means ± SEM. **P < 0.01 and ****P < 0.0001.
CLASS microscopy of chromatin in live differentiating organoids
In addition to monitoring changes in gene expression and biochemical composition with both conventional techniques and Raman spectroscopy, we also monitored changes in chromatin distribution within live differentiating cells. To pack a 2-m-long DNA into a several micrometer-sized nucleus, chromatin must be organized into distinct domains. Some of these domains display a more open form of chromatin, with high gene density and high gene expression, while others are more densely packed, exhibiting a closed chromatin state. These domains are called euchromatin and heterochromatin, respectively. Chromatin can consist of other types of domains referred to as topologically associating domains (TADs) (53). The exact nature of TADs has not been clarified, leading to inconsistent definitions of their boundaries in the genome (54).
Initially, it was postulated (55) that these domains form a highly knotted equilibrium state with the probability of contact being Pc(s) ~ s−3/2 for two loci, which are a genomic distance s apart. Later, it was experimentally established (56) that Pc(s) ~ s−1.08. The only conformation consistent with this almost linear behavior of the contact probability is a nonequilibrium state organized in a self-similar fashion (57), such as a fractal (58). Fractal organization results in contiguous spatial regions with transcriptionally active and inactive chromatin, facilitating easy unfolding and refolding during gene activation or repression. Both properties are observed in eukaryotic chromatin, providing further evidence that chromatin has fractal organization.
Self-similar properties of the chromatin fractal could be reflected in the chromatin light scattering spectra. Light scattering spectra are not limited by diffraction and can characterize subcellular and subnuclear structures significantly smaller than the wavelength of light (45). We chose to use CLASS microscopy (see Materials and Methods and Fig. 5A), which uses light scattering spectra as a native marker (32), allowing label-free sensing of chromatin at nanometer scales in live cells. Scattering by chromatin in a cell nucleus can be described as the scattering by the collection of N densely packed small scatterers with volumes V and close to unity relative refractive indices m. The scattering intensity detected by the CLASS microscope from a focal spot can be found (59) by integrating the squared electromagnetic field amplitude over the solid angle of the objective Ω related to its numerical aperture (NA), giving the following expression
| (3) |
where ri is the position vector of individual scatterers relative to the focus, λ is the wavelength of the incident and scattered light (since we only consider elastic scattering), and are unit vectors in the propagation direction of the incident and scattered light, respectively, θ is the scattering angle between and , R is the distance to the detector, E0 is the amplitude of the incident field, is the objective pupil function (the light delivery and collection are described by the same pupil function), and the sum is performed over all scatterers forming chromatin. Because of an angle averaging (60) for a large NA expression (Eq. 3), the scattering intensity can be related to the structure factor and approximated as , where g(r) is a pair correlation function of chromatin. Therefore, the explicit wavelength dependence of the scattering intensity of chromatin depends on the pair correlation function form, which, in turn, could be characterized by a statistical index of complexity associated with fractals called the Hausdorff dimension (58).
Fig. 5. CLASS microscopy measurements of chromatin packing in live cells.
(A) SolidWorks rendering of the CLASS microscopy system. Shown in the schematic is the light path in the customized confocal scanning unit and optical elements, which includes a supercontinuum broadband laser source, custom combiner of supercontinuum laser and conventional lasers, and complementary metal-oxide semiconductor spectrometer. (B) Rendering of the CLASS microscopy measurements of chromatin packing in a cell nucleus with various TADs highlighted using different colors and one of the TADs illuminated.
The confocal diffraction-limited interrogation volume of the CLASS microscope is comparable in size with the characteristic dimensions of the TADs (61) (Fig. 5B). Therefore, when associating CLASS spectra with the fractal organization of chromatin, it would make sense to consider both mass fractal organization of the chromatin within the domain and surface fractal organization of the domain boundary. This allows us to describe chromatin using the most general case of mass fractals bounded by surface fractals. While the explicit dependence of the pair correlation function and structure factor on mass and surface fractal dimensions dm and ds, respectively, are not well known (62), one can use the semiempirical form of the structure factor proposed by Sinha (63) and similar relationship obtained from the scaling considerations by Oh and Sorensen (64). It yields I ~ λ −4 + χ for the scattering intensity of the mass fractal bounded by a surface fractal with χ = 2dm − ds. We should note here that several important characteristics of chromatin could be described using the concept of mass fractal (30, 31), with mass fractal dimension dm being higher in the denser heterochromatin (65). On the boundary of the domain, the increase in the surface fractal dimension ds would be indicative of the presence of transcriptionally active euchromatin exposing a larger surface to nuclear proteins and thus facilitating gene transcription due to better transcription factor access to DNA. Therefore, on the boundary of the domain, the presence of heterochromatin will result in surface fractal dimension ds being lower. Both of these trends should result in a higher value of χ for heterochromatin. As χ can be directly evaluated from the CLASS spectra, we will therefore use χ to characterize fractal packing density of the chromatin within the confocal volume of the CLASS microscope.
We monitored chromatin changes in live hiPSC-only organoids undergoing differentiation by collecting CLASS microscopy images over a period of 16 days. Similar to our Raman spectroscopy measurements, this 16-day period corresponded to the four stages of the protocol (definitive endoderm, foregut endoderm, hepatoblasts, and mature hepatocytes). Measurements were carried out by first removing organoids from the incubator and placing them in a glass bottom 35-mm dish filled with the basal differentiation medium, without the addition of the growth factor (Fig. 6A). The duration of the measurements was short enough to keep the hiPSCs in the organoids alive, and no staining was used. Nuclei located approximately 15 to 25% deep inside the organoid were initially identified with white light reflectance imaging. Following nucleus identification, CLASS microscopy images and white light images were acquired simultaneously, allowing easy identification of the nucleus for data processing. With the progression of differentiation, the slope of the scattering spectra from the chromatin in live hiPSCs showed a highly significant change (Fig. 6B). This result is consistent with the steady increase in the packing parameter χ due to the expected increase in the heterochromatin fraction as differentiation progresses. The scattering spectra shown in Fig. 6B are the average spectra for the entire nucleus. In addition, the spectra can be analyzed locally, allowing the corresponding slope and packing parameter χ to also be obtained locally. This is illustrated in Fig. 6C, where the spatial maps of packing parameter χ are presented in pseudo-colors. Figure 6D shows the average values of χ for several organoids, calculated across the entire nucleus, at each stage of differentiation. As can be seen in Fig. 6D, hEBs are characterized by a low value of χ, reflecting the typical high percentage of decondensed euchromatin in undifferentiated stem cells. This high level of euchromatin is preserved throughout the definitive endoderm stage and can also be expected for the first germ layer developed after the initiation of the differentiation process. After that, when cells reach the foregut endoderm stage, which is a more specialized tissue from which most visceral organs arise, more and more genes are segregated as inactive, and only genes specific for the specialized tissue remain in the decondensed chromatin. This significant reduction in decondensed chromatin at the foregut endoderm stage is reflected in the increase of parameter χ. This continues through the hepatoblasts and mature hepatocytes stages, with the maximum heterochromatin presence observed at the final stage of differentiation. These results are in excellent agreement with the transmission electron microscopy (TEM) measurements we performed upon the same batches of differentiated hiPSC organoids on the same days as CLASS microscopy measurements (Fig. 6, E and F), where TEM images of cell nuclei were binarized to categorize areas of dense heterochromatin and loose euchromatin (see Materials and Methods). The CLASS results are also in good agreement with published studies (66), which demonstrate that stem cells having over 75% of chromatin in the transcriptionally active euchromatin state undergo reorganization and accumulate highly condensed and inactive heterochromatin upon differentiation, with a reduction in euchromatin regions by approximately 40%.
Fig. 6. Chromatin packing in live organoids at different stages of differentiation.
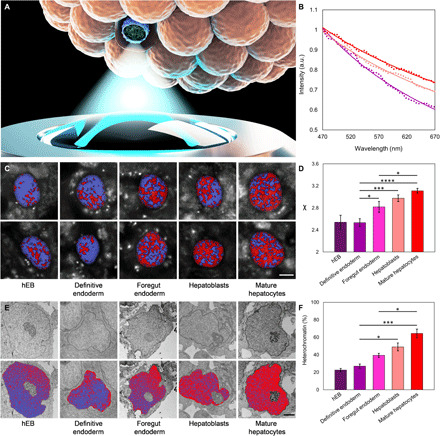
(A) Rendering of the measurements of chromatin packing within one of the nuclei in live differentiating hiPSC organoids using the CLASS microscope. (B) Average light scattering spectra collected with CLASS microscope for hiPSC organoids. Purple dots show spectra of chromatin at definitive endoderm stage (n = 19), pink at hepatoblasts stage (n = 21), and red at mature hepatocytes stage (n = 24). Model fits are presented with solid lines. (C) Spatial maps of fractal packing density of chromatin χ in live hiPSC organoids at different stages of differentiation. Two typical maps per stage overlaid over white light reflectance images of nuclei are shown. Values of χ in the range of 2.3 to 2.9 are shown as shades of purple and can be loosely associated with euchromatin, while values in the range of 2.9 to 3.5 are shown as shades of red and can be associated with heterochromatin. Scale bar, 5 μm. (D) Parameter χ reflecting changes in the heterochromatin to euchromatin ratio and SEs for hEB (n = 9), definitive endoderm (n = 19), foregut endoderm (n = 21), hepatoblasts (n = 21), and mature hepatocytes (n = 24) stages of differentiation. (E) Representative TEM images of nuclei in hiPSC organoids at different stages of differentiation. Top row: Original images. Bottom row: Same images overlaid with pseudo-color maps with heterochromatin highlighted in red and euchromatin in purple. Scale bars, 2 μm. (F) Percentage of heterochromatin at hEB (n = 4), definitive endoderm (n = 4), foregut endoderm (n = 3), hepatoblasts (n = 3), and mature hepatocytes (n = 3) stages of differentiation. Data are presented as means ± SEM. Statistical significance was evaluated with a Tukey’s multiple comparison test. *P < 0.05, ***P < 0.001, and ****P < 0.0001.
DISCUSSION
The emergence of hiPSCs was a substantial development in stem cell research. Initially obtained from human adult cells (67, 68), these pluripotent cells provide a similar long-term unlimited self-renewal and pluripotent differentiation capacity, when compared with human embryonic stem cells (69, 70). In terms of applications, hiPSCs can be used for disease modeling and drug screening. They can also be applied to regenerative medicine since they provide a potentially limitless source of autologous stem cells, which can be differentiated into numerous tissues. These tissues have the potential to limit immune rejection. However, their full potential is critically limited by the inability to direct the differentiation of large quantities of hiPSCs toward desirable cell lineages in a controlled, scalable, and reproducible way. Moreover, differentiation protocols developed and optimized for specific hiPSC lines are generally not applicable to different hiPSC lines, including patient-derived hiPSCs, due to their well-known epigenetic variability (71, 72).
Here, we report a method of rapid high-throughput formation of fully functional liver organoids derived from dissociated induced pluripotent stem cells and adipose microvascular endothelial cells using microfabricated cell-repellent microwell arrays. The organoids formed using this method are well organized, feature three distinct germ layers, are capable of multilineage differentiation, and demonstrate high viability. They can easily be made 500 μm in size or even larger, without any signs of core necrosis, and allow for straightforward incorporation of multiple cell types, making it possible to recreate the natural microenvironment of a specific organ. Several recently developed approaches (4, 73, 74) demonstrated the formation of liver organoids exhibiting a number of important functions characteristic of liver tissue. The method described here allows all of these liver-specific functions to be present in a single type of organoid. We were able to form large and uniform hEBs in only 18 to 24 hours from a single-cell suspension of hiPSCs and HAMEC mixture, with no core necrosis at any time throughout the differentiation process. Moreover, by mimicking the in vivo embryogenesis processes by efficiently blocking the Sonic hedgehog and Notch signaling pathways during the initial stages of differentiation, the production of albumin in the liver organoids improved more than 10 times, the production of the main coagulation factors increased up to five times, the production of detoxification factors doubled, enzymatic activity of factor VIIIa increased five times, and the rate of generation of factor Xa increased by approximately 50%. As a result, the production levels and rates of generation for all biomolecules critical for liver function either became comparable or sometimes even exceeded the corresponding levels and rates of the functional human liver.
The method we presented can also be easily combined with label-free optical spectroscopic methods. We showed the benefit of two complementary techniques, namely, Raman spectroscopy and CLASS microscopy, for monitoring organoid differentiation without the need to sacrifice or even modify the samples. While the Raman and CLASS system measure different properties, they also function at different scales. Our Raman system focuses light on the organoid, thereby probing several cells within the organoid. On the other hand, our CLASS system, which contains high NA optics, focuses and scans light across individual nuclei.
Label-free Raman spectroscopy is capable of detecting subtle changes in the biochemical intracellular composition of differentiating organoids, allowing for monitoring of their progression toward cell transplantation therapy compatible functional tissue. The most important difference between Raman spectroscopy and conventional techniques such as real-time PCR or ELISA is its ability to obtain this information in real time, in vivo, and without the need to sacrifice the organoids. It is worth noting that conventional approaches used in label-free Raman spectroscopy analysis rely either on monitoring changes in individual peaks or using various types of principal components analysis to characterize the spectra. Unfortunately, individual peaks often contain contributions from multiple biochemicals, while principal components analysis requires a training step. Since multiple biochemicals were present in the organoids and considering that we were following the organoids for 16 days, neither analysis approach was therefore satisfactory. With the Raman spectral unmixing approach that we used, we were able to quantitatively evaluate the concentrations of the biomolecules by reconstructing the Raman spectra of organoids from the base spectra of biochemicals whose concentrations are known to be changing during differentiation. Raman spectroscopy allowed us to monitor changes in the intracellular albumin production, which, as expected, steadily increased throughout the progression of the organoids from the initial differentiation stages toward the committed hepatocytes.
On the other hand, intracellular levels of AFP demonstrated a somewhat phasic behavior, increasing during the definitive endoderm and hepatoblasts stages (I and III) while decreasing during the foregut endoderm maturation and again at the stage of mature hepatocytes (II and IV). This is once again an expected behavior of this known marker of definitive endoderm and hepatoblasts stages.
Raman spectroscopy of two other important intracellular liver biomolecules, namely, glycogen and hepatic lipids, also demonstrated the behavior indicative of the progression toward the mature liver phenotype. Glycogen synthesis and storage are major aspects of glucose metabolisms in cells (75). In human pluripotent stem cells, an up-regulated glycogen synthesis can be observed by differentiation signaling molecules such as BMP-4 (76). This suggests that glycogen synthesis could be an efficient marker of lineage-related differentiation. However, direct observation of the intracellular glycogen content in live cells with Raman spectroscopy could be an even more accurate marker of hepatic differentiation. Hepatic lipids are another important class of biochemicals involved in stem cell differentiation to metabolically active hepatocytes, due to the fundamental role of lipids in energy metabolism. The continuing increase in the intracellular level of hepatic lipids observed with Raman spectroscopy during differentiation, with terminally differentiated hepatocytes displaying the highest lipid content compared to other differentiation stages, is in good agreement with published studies (75).
Our CLASS microscopy measurements showed that sensing of nanoscale changes in chromatin allows spatiotemporal monitoring of the dynamic chromatin distribution within live cells over the course of the differentiation process. This capability could be important not only to adjust the differentiation protocols but also to provide a better understanding of the differentiation process, as certain factors affecting regulation of chromatin structure might either inhibit normal cell differentiation pathways or even stimulate the uncontrolled cell proliferation toward oncogenesis (76). As an optical spectroscopic technique, CLASS microscopy is well suited to monitoring chromatin changes. While spectroscopic techniques are not diffraction limited, their interrogation volume is limited by diffraction and other optical effects. The confocal nature of CLASS microscopy significantly reduces the interrogation volume, allowing smaller length scales to be probed and spatial maps to be built up. In addition, CLASS microscopy provides a straightforward method for continuously monitoring chromatin changes throughout the differentiation process rather than static measurements of stem cells at the beginning and end of differentiation. The steady increase in the fractal packing parameter χ observed in our studies, which could be associated with the increase in mass fractal dimension, the decrease in surface fractal dimension, or both are consistent with a steady decrease in the euchromatin content as differentiation progresses. This result was in excellent agreement with the TEM measurements and with the published studies (66).
CLASS microscopy has numerous advantages when compared to TEM and other modalities. TEM imaging requires chemical fixation and can only analyze chromatin distribution at a single instance, while CLASS microscopy is label free and can be used on live cells. In addition, TEM analysis requires spatial maps to be generated to quantitively understand changes at the nucleus level. In the case of CLASS microscopy, nucleus-level changes can be obtained by simply averaging the spectra across the entire nucleus and finding the slope of the corresponding spectrum, providing an easy quantitative value for comaprison. TEM can obviously provide higher-resolution spatial maps, and this difference can clearly be seen in our results. However, both sets of spatial maps showed the same progression. Many existing methods that measure chromatin at the single-cell level, such as TEM, are not scalable, mostly due to time or cost constraints. Population-based methods can analyze many cells but do not provide information about the single-cell level. CLASS microscopy is both quick and extremely cost-effective, allowing population-based averages to be built up while also maintaining information about the single-cell level. The quick acquisition time has other important implications. While 2D images are presented here, a simple z-stack could be performed to build up a 3D distribution of chromatin within live cells.
We note that while chromatin organization is an important marker of stem cell differentiation, it also has a significance in many other areas. In addition to regulating many cellular processes, large-scale changes can be attributed to cancer, several neurological and autoimmune conditions, and other complex diseases (31). Therefore, CLASS microscopy, which allows straightforward chromatin monitoring in live cells, could be a valuable tool for future studies.
In summary, we presented a method for rapid high-throughput formation of fully functional liver organoids derived from dissociated induced pluripotent stem cells and adipose microvascular endothelial cells using microfabricated cell-repellent microwell arrays. Combining this method with the real-time label-free monitoring of intracellular biochemical composition changes using Raman spectroscopy and intranuclear chromatin changes with CLASS microscopy can significantly shorten the time of protocol fine-tuning, eliminating the need to sacrifice the sample to collect the data. Homogeneous liver organoids formed with this approach have the main functional features of human liver and could potentially be applied to cell transplantation liver therapy in humans.
MATERIALS AND METHODS
Culture conditions and sources of cells
We purchased iPS(foreskin)-3, foreskin fibroblast–derived hiPSCs, (WB0002, WiCell Research Institute) and cultured them using the mTeSR1 basal medium with the mTeSR1 supplement (85850, STEMCELL Technologies) on Matrigel hESC-Qualified Matrix lactose dehydrogenase elevating virus (LDEV)–free (354277, Corning)–coated plates. The passage of the hiPSC colonies was performed using Versene (EDTA) (BE17-711E, Lonza) at room temperature for 8 min. We purchased HAMEC (7200, ScienCell Research Laboratories) and cultured them in the endothelial cell medium (1001, ScienCell Research Laboratories) on flasks coated with fibronectin (2 mg/cm2; 8248, ScienCell Research Laboratories) following the manufacturer’s instructions. HPHs were purchased (HMCPMS, Gibco) and cultured following the manufacturer’s instructions.
hEB formation
We developed an agarose microwell array system that uses a custom Teflon stamp and a low melting point agarose. We dissolved agarose in phosphate-buffered saline (PBS), heated to 100°C, to reach a concentration of 40 g/liter and dispensed it into a 24-well culture plate. The Teflon stamps were pressed onto the agarose solution for about 5 min. When the agarose solidified in approximately 2 min, the stamps were withdrawn, and the microwell arrays were formed. We subsequently incubated the arrays with the basal differentiation medium overnight at 37°C and 5% CO2.
hEBs were formed by dissociating 1.2 × 106 hiPSCs and resuspending them into 50 μl of basal differentiation medium. The hEB suspension was then allowed to sediment in microwell arrays. After 24 hours of incubation at 37°C, hEBs were transferred to a 35-mm culture dish.
hEBs containing hiPSCs with HAMECs were formed by dissociating each cell type into a single cell suspension, counted using a cell counter, and mixed with a ratio of 3:1. A total number of cells per hEB was 1.2 × 106. The hiPSCs mixed with HAMEC were then placed into the agarose microwell arrays to allow hEBs formation. After the formation, both hiPSCs and hiPSCs with HAMEC were maintained in the suspension culture for the whole duration of the differentiation procedure.
Hepatic differentiation protocol
We used a basal differentiation medium consisting of Iscove’s modified Dulbecco’s medium with Ham’s F-12 Nutrient Mixture (Gibco), 5% fetal bovine serum (Gibco), 1% insulin transferrin selenium-A supplement (Gibco), 55 μM monothioglycerol (Sigma-Aldrich), penicillin (100 U/ml), and streptomycin (0.1 mg/ml; Sigma-Aldrich). The differentiation protocol consisted of four steps, through which we drove our hEBs from an undifferentiated stage to mature hepatocyte-like cells. To develop the definitive endoderm in 4 days, the Sonic hedgehog pathway was blocked with a MK-4101 potent inhibitor at 0.5 μM, used together with activin A (100 ng/ml), bFGF (10 ng/ml), and TGF-β1 (10 ng/ml; all from PeproTech). The definitive endoderm was confirmed with immunofluorescence by the presence of the markers forkhead box A2 (FOXA2) and SRY-box transcription factor 17 (SOX17) (fig. S4A). In the second stage of differentiation, which led to the formation of the foregut endoderm, we used the Notch inhibitor LY-41575 as inhibition of the Notch pathway is an important step for the formation of anterior foregut endoderm, from which all the visceral organs arise. We used LY-41575 at 0.5 μM in combination with BMP-4 (10 ng/ml; Invitrogen) and FGF-4 (10 ng/ml; PeproTech). The successful differentiation of the foregut endoderm was confirmed with immunofluorescence by the presence of the markers hematopoietically expressed homeobox (HHEX) and GATA Binding Protein 4 (GATA4) (fig. S4B). The third step used two Wnt/β-catenin inhibitors: WIF-1 (1 μg/ml; R&D System) and DKK-1 (0.1 μg/ml; PeproTech), which are responsible for the inhibition of both the canonical and noncanonical Wnt/β-catenin pathways. This step is important for the formation of hepatoblast cells, since if the Wnt/β-catenin pathway is active, it will lead to the formation of cholangioblast cells, the precursors of bile duct cells. Using these Wnt/β-catenin inhibitors, we have been able to significantly reduce the formation of unwanted nonhepatic cells. The formation of hepatoblasts was confirmed by the presence of hepatocyte progenitor cell markers, AFP and HNF-4α (fig. S4C). The last stage of the protocol is the final maturation step of the hepatoblast into mature hepatocyte-like cells. To achieve this, oncostatin M (30 ng/ml) and hepatocyte growth factor (HGF) (50 ng/ml; PeproTech) along with 10 μM dexamethasone were used. These growth factors are well known to induce maturation of hepatocyte progenitor cells into mature hepatocytes. The immunofluorescence of the albumin and CK18 markers indicated that the final maturation had occurred (fig. S4D). The differentiation factors for each stage were administrated every 2 days in fresh basal medium, and the hEBs were maintained in suspension culture during the whole process of differentiation. Undifferentiated hEBs from the same batches were used as negative controls. For morphology assessment, when terminal differentiation was reached, liver organoids were transferred onto a Matrigel-coated plate for imaging by light microscopy.
FACS analysis and cell sorting
To evaluate the viability of the organoids, we used the Live/Dead Zombie NIR Fixable Viability Kit (423105, BioLegend). The Fixation/Permeabilization Staining Buffer Set (00-5523-00, eBioscience) was used intracellularly for albumin staining. The BD FACSAria II cell sorter was used to collect the data, and 95% purity was routinely obtained. The human HNF-4α/NR2A1 Alexa Fluor 647–conjugated antibody (IC4605R, R&D Systems) and the anti-human serum albumin allophycocyanin (APC)–conjugated antibody (IC1455A, R&D Systems) were used. To setup the threshold, the control sample was stained with live/dead stain for viability, but not for albumin, with a gate for control sample set to zero. For the analysis with the FlowJo software, the events exceeding the threshold for the albumin-stained samples were considered to be positive. The geometric means of the appropriate channels were used to calculate the mean fluorescence intensities and the expansion indices in FlowJo software.
Gene expression analysis with real-time PCR
Gene expression was performed by real-time PCR. The RNA was extracted and treated using ribonuclease-free deoxyribonuclease (M6101, Promega). A reverse transcription was performed with iScript cDNA synthesis kit (1708891, Bio-Rad) following the manufacturer’s protocol. A 384-well plate from Bio-Rad was used to test in triplicate all genes of interest. The conditions for 40 cycles of PCR amplification with Bio-Rad CFX96 Touch consisted of a 2-min activation step at 95°C, followed by a 5-s naturation step at 95°C, and finished with a 30-s annealing step at 60°C, melt curve at 65° to 95°C with 0.5°C increments for 5 s per step. The data were processed with Bio-Rad CFX Manager 3.1. The results, relative to the glyceraldehyde-3-phosphate housekeeping gene, were normalized to undifferentiated hEBs, used as a negative control. Three independent experiments were represented by the SD error bars. List of the genetic markers used can be found in the Supplementary Materials.
Protein quantification
Protein quantification for both liver markers and coagulation factors was performed using conditioned medium collected after 48 hours of postterminal differentiation using ELISA and following the manufacturer’s protocol. See the Supplementary Materials for the list of proteins measured. To normalize the results, they were related to the number of cells, with 1 million cells used.
Immunofluorescence analysis
For the immunofluorescence analysis, hEBs were fixed for 90 min using 4% paraformaldehyde (28906, Pierce), permeabilized in PBS for 1 hour with 0.3% Triton X-100 (85111, Thermo Fisher Scientific), and blocked using 0.5% goat serum (G9023-5ML, Sigma-Aldrich). The primary antibody was incubated for 3 days at 4°C, and after several washes, the liver organoids were incubated with the secondary antibody for 2 hours at room temperature. The nuclei were stained using 4′,6-diamidino-2-phenylindole (DAPI) for 1 hour. Fluorescence images were obtained with the Olympus FV1000 confocal microscope.
Liver detoxification assays
Ammonia metabolism assay
Ammonium chloride was added to evaluate ammonia metabolism via the variation of the amount of ammonia in the collected supernatant over a 6-hour period. The culture medium with 100 liver organoids in suspension was supplemented with 1 mM NH4Cl. The ammonium concentration was measured 1, 3, and 6 hours after NH4Cl addition with a colorimetric ammonia assay kit (K470, BioVision).
Resorufin conjugation assay
Liver organoids were incubated for 3 days in a medium containing rifampicin. Subsequently, the organoids were moved to a black 96-well plate in groups of three and resuspended in a 40 μl of Hanks’ balanced salt solution (HBSS) with 40 μl of resorufin sodium salt (R3257, Sigma-Aldrich). The plate was incubated for 30 min at 37°C in the dark. After the incubation, all the samples were analyzed and read in triplicate by measuring the fluorescence excited at 535 nm and emitted at 581 nm with 35-nm resolution using a multimode microplate reader (SpectraMax i3x, Molecular Devices). The fluorescence from HBSS sample was used as background fluorescence and subtracted from the measurements. The results were normalized on the basis of the number of live cells using Quant-iT PicoGreen double-stranded DNA (P7589, Thermo Fisher Scientific).
CYP activity assay
The activity of cytochromes P450 was tested by incubating the liver organoids for 3 days using various inducers. For CYP1A2 activity assay, liver organoids and HPH were placed for 3 days in a basal medium with 50 μM omeprazole solution (O104, Sigma-Aldrich) or 0.1% DMSO. For the CYP3A4 activity, liver organoids and HPH were placed for 3 days in a basal medium with 20 μM rifampicin solution (R3501, Sigma-Aldrich) or 0.1% DMSO. For the CYP2B6 activity, liver organoids and HPH were placed for 3 days in basal medium with 1000 μM phenobarbital solution (P1636, Sigma-Aldrich) or 0.1% DMSO. A multimode microplate reader (SpectraMax i3x, Molecular Devices) was used to measure the enzyme activity by measuring the samples’ luminescence. See the Supplementary Materials for kit information.
Storage, uptake, and release assays
For all of the assays listed here, the liver organoids were plated in the Matrigel-coated chamber slides and allowed to form monolayers.
ICG uptake and release assay
Liver organoids were incubated with ICG (1340009, Sigma-Aldrich) in a basal medium at 37°C for 1 hour following the instructions provided by the manufacturer. To measure the ICG uptake, fluorescence signals were detected with a fluorescence microscope, and the ICG release was confirmed after 6 hours to guarantee that every positive cell released ICG.
Oil red staining
Lipid vesicle storage in liver organoids was assessed with Oil Red O staining in accordance with the manufacturer’s protocol (ab150678, Abcam).
Periodic acid–Schiff staining
Glycogen storage in liver organoids was assessed with Periodic acid–Schiff staining in accordance with the manufacturer’s protocol (395B, Sigma-Aldrich).
Uptake of LDL assay
Liver organoids were incubated overnight at 37°C with a preincubation medium consisting of serum-free medium with the addition of 0.1% of bovine serum albumin. The next day, the liver organoids were allowed to sit for 5 hours in preincubation medium also containing Dil-Ac-LDL (10 μg/ml; J65597, Alfa Aesar). After incubation, the cells were rinsed several times and fixed with 4% paraformaldehyde for 60 min and counterstained with DAPI for nuclei. All fluorescence images for storage, uptake, and release assays were acquired with the Olympus FV1000 confocal microscope.
Glycan preparation
Liver organoids were digested and homogenized using a buffer solution of NaCl (150 mM), EDTA (5 mM), tris (25 mM), and 0.5% CHAPS at pH 7.4. The sample was dialyzed overnight at 4°C using 50 mM NH4HCO3 buffer. The following day, the resulting cells were reduced by a 2-hour incubation at 50°C in a solution composed of 1,4-dithiothreitol (2 mg/ml) and 0.6 M tris at pH 8.5 and then alkylated with a 0.6 M tris at pH 8.5 solution with iodoacetamide (12 mg/ml) for 2 hours at room temperature without light. Following these steps, the cells were dialyzed once again overnight at 4°C using 50 mM NH4HCO3 before being lyophilized. The resulting sample was then resuspended into 1 ml of solution composed of 50 mM NH4HCO3 and tosyl phenylalanyl chloromethyl ketone (TPCK)–treated trypsin (500 μg/ml) and incubated overnight at 37°C. The sample was purified using a C18 Sep-Pak column (200 mg), preconditioned with one column volume (CV) of 5% acetic acid, one CV of methanol, and one CV of 1-propanol. After an initial wash with 5% acetic acid, the column was progressively eluted using 20, 40, and 100% of 1-propanol. The eluted samples were combined and then lyophilized. The lyophilized sample was resuspended into a solution composed of 50 mM NH4HCO3 with the addition of 3 μl of Peptide:N-glycosidase F (PNGase F) followed by 4-hour incubation at 37°C, and a further addition of another 3 μl of PNGase F that was incubated at 37°C overnight. The N-glycans were purified using a C18 Sep-Pak column (200 mg), preconditioned with one CV of 5% acetic acid, one CV of methanol, and one CV of 1-propanol. After an initial wash with 5% acetic acid, the N-glycan–released peptides were progressively eluted using 20, 40, and 100% of 1-popanol. The eluted samples were combined and then lyophilized.
Mass spectroscopy
Mass spectroscopy data were obtained using the Bruker UltraFlex II matrix-assisted laser desorption/ionization–time-of-flight mass spectrometer. For the N-glycans, the data were recorded between 500 and 6000 mass/charge ratio (m/z), and for the O-glycans, the data were recorded between 0 and 4000 m/z. In both cases, the reflective positive mode was used. At least 20,000 laser shots were accumulated for each N-glycan and O-glycan profile. For the data to be included, the signal-to-noise ratio of at least 2 was required. Analysis was performed only on signals matching N-glycan or O-glycan composition. The postdata acquisition analysis was performed using the open source mass spectrometry tool mMass.
Raman spectroscopy system
The Raman spectroscopy system used a low-magnification Raman microscopy setup with a 785-nm excitation wavelength 450-mW power diode laser (MDL-III-785-450 mW, CNI) delivering excitation light on the organoids with an 11-mm working distance focusing lens. The Raman signal was collected with the same lens and after passing through a notch filter and was detected with a Raman spectrometer (WP-785-R-SR-S-25, Wasatch Photonics) with 6 cm−1 of spectral resolution. The collection volume of the system was approximately 10 nl. During measurements, the organoids were placed in a customized quartz-bottom chamber with beam positioning achieved with a motorized XYZ stage (PT3-Z8, Thorlabs).
CLASS microscopy system
The CLASS microscopy system was described previously (29). Briefly, the system is based on an Olympus FluoView 1000 confocal microscope and uses a broadband supercontinuum laser source (WL-SC-400-4, Fianium) (see Fig. 3A). A high NA objective is used to focus the broadband source on the sample (Olympus UPlanSApo 60X; NA, 1.35), while a high-speed complementary metal-oxide semiconductor spectrometer (AvaSpec-ULS2048CL-EVO, Avantes) is used to detect the spectra. The spectrometer provides a 2.4-nm spectral resolution in the 400- to 800-nm wavelength range. The scanning head controller is controlled with a custom-written LabView (National Instrument, LabVIEW 8.5) code. White light imaging, which uses the microscope photomultiplier tubes and broadband supercontinuum laser source, is used to determine the nuclei boundary. The white light images are obtained simultaneously with the CLASS images, allowing exact coregistration. Data acquisition and analysis are performed with MATLAB. When analyzing the data, all spectra within the nucleus can be averaged to produce a nucleus average, which can be subsequently fit to a model. Alternatively, the CLASS images can be analyzed on the pixel level, allowing a pseudo-color map of the packing parameter to be generated.
TEM imaging of chromatin
TEM images of nuclei in hiPSC organoids at hEB, definitive endoderm, foregut endoderm, hepatoblasts, and mature hepatocytes stages of differentiation were collected at Harvard Medical School Electron Microscopy Facility using JEOL 1200EX, 80-kV microscope. Images of nuclei with nucleolus excluded were binarized to separate dark pixels representing heterochromatin and light pixels representing euchromatin (66). To prevent bias in image binarization, all pixels were normalized with the pixel values chosen between 0 and 1, and the global threshold was then selected (77) to minimize the intraclass variance of black and white pixels.
Statistical analysis
Statistical analysis was performed with GraphPad Prism v7.00. Normality of the data was tested using the Shapiro-Wilk test or the D’Agostino-Pearson test. When comparing two groups, we used the unpaired two-tailed Student’s t test when the data were normally distributed. When the data were not normally distributed, we performed the Mann-Whitney test. Data that contained more than two groups, and which showed normal distribution, was analyzed with a one-way analysis of variance (ANOVA) test and Tukey post hoc test. A Kruskal-Wallis nonparametric test with Dunn’s correction for multiple comparisons was used when data were not normally distributed. Asterisks were used to indicate significance: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Values of >0.05 were considered not significant.
Acknowledgments
Funding: This work was supported by U.S. NIH grants R01 CA228029, R01 EB003472, R01 CA205431, R01 EB025173, and R01 CA218382 and U.S. NSF grants EFRI-1830878, CBET-1605116, and CBET-1948722. Author contributions: G.P. and L.T.P. conceptualized and designed the study. G.P., L.Z., L.Q., and L.T.P. supervised the project. G.P., L.Z., L.C., M.G., A.B.D., and R.A.F. provided the samples. G.P. developed the organoid formation method. L.Q., M.F.C., X.Z., U.K., P.K.U., and Y.N.Z. constructed CLASS microscopy and Raman spectroscopy systems. M.F.C., C.J.S., X.Z., E.V., and L.Q. developed algorithms and software, M.F.C. performed the CLASS microscopy measurements and data analysis. X.Z. and L.C. performed the Raman spectroscopy measurements and analysis. M.F.C., X.Z., L.C., E.V., L.Q., and L.T.P. evaluated the data. G.P., M.F.C., L.C., L.Z., I.I., and L.Q. contributed to the writing of the manuscript. U.K. created the graphic art. L.T.P. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/34/eabj2800/DC1
REFERENCES AND NOTES
- 1.Zarrinpar A., Busuttil R. W., Liver transplantation: Past, present and future. Nat. Rev. Gastroenterol. Hepatol. 10, 434–440 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Bhatia S. N., Underhill G. H., Zaret K. S., Fox I. J., Cell and tissue engineering for liver disease. Sci. Transl. Med. 6, 245sr2 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thi V. L. D., Wu X., Belote R. L., Andreo U., Takacs C. N., Fernandez J. P., Vale-Silva L. A., Prallet S., Decker C. C., Fu R. M., Qu B., Uryu K., Molina H., Saeed M., Steinmann E., Urban S., Singaraja R. R., Schneider W. M., Simon S. M., Rice C. M., Stem cell-derived polarized hepatocytes. Nat. Commun. 11, 1677 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takebe T., Sekine K., Enomura M., Koike H., Kimura M., Ogaeri T., Zhang R. R., Ueno Y., Zheng Y. W., Koike N., Aoyama S., Adachi Y., Taniguchi H., Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 499, 481–484 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Schneemann S. A., Boers S. N., van Delden J. J. M., Nieuwenhuis E. E. S., Fuchs S. A., Bredenoord A. L., Ethical challenges for pediatric liver organoid transplantation. Sci. Transl. Med. 12, eaau8471 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Garreta E., Kamm R. D., Chuva de Sousa Lopes S. M., Lancaster M. A., Weiss R., Trepat X., Hyun I., Montserrat N., Rethinking organoid technology through bioengineering. Nat. Mater. 20, 145–155 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Ng E. S., Davis R., Stanley E. G., Elefanty A. G., A protocol describing the use of a recombinant protein-based, animal product-free medium (APEL) for human embryonic stem cell differentiation as spin embryoid bodies. Nat. Protoc. 3, 768–776 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Orlova V. V., Van Den Hil F. E., Petrus-Reurer S., Drabsch Y., Ten Dijke P., Mummery C. L., Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nat. Protoc. 9, 1514–1531 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Qian X., Jacob F., Song M. M., Nguyen H. N., Song H., Ming G. L., Generation of human brain region–specific organoids using a miniaturized spinning bioreactor. Nat. Protoc. 13, 565–580 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas D., O’Brien T., Pandit A., Toward customized extracellular niche engineering: Progress in cell-entrapment technologies. Adv. Mater. 30, 1703948 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Pettinato G., Wen X., Zhang N., Formation of well-defined embryoid bodies from dissociated human induced pluripotent stem cells using microfabricated cell-repellent microwell arrays. Sci. Rep. 4, 7402 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aoi T., Yae K., Nakagawa M., Ichisaka T., Okita K., Takahashi K., Chiba T., Yamanaka S., Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science 321, 699–702 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Bock C., Kiskinis E., Verstappen G., Gu H., Boulting G., Smith Z. D., Ziller M., Croft G. F., Amoroso M. W., Oakley D. H., Gnirke A., Eggan K., Meissner A., Reference maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 144, 439–452 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts B., Haupt A., Tucker A., Grancharova T., Arakaki J., Fuqua M. A., Nelson A., Hookway C., Ludmann S. A., Mueller I. A., Yang R., Horwitz R., Rafelski S. M., Gunawardane R. N., Systematic gene tagging using CRISPR/Cas9 in human stem cells to illuminate cell organization. Mol. Biol. Cell 28, 2854–2874 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lahm H., Doppler S., Dreßen M., Werner A., Adamczyk K., Schrambke D., Brade T., Laugwitz K. L., Deutsch M. A., Schiemann M., Lange R., Moretti A., Krane M., Live fluorescent RNA-based detection of pluripotency gene expression in embryonic and induced pluripotent stem cells of different species. Stem Cells 33, 392–402 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Tebo A. G., Pimenta F. M., Zhang Y., Gautier A., Improved chemical-genetic fluorescent markers for live cell microscopy. Biochemistry 57, 5648–5653 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Ang L. T., Tan A. K. Y., Autio M. I., Goh S. H., Choo S. H., Lee K. L., Tan J., Pan B., Lee J. J. H., Lum J. J., Lim C. Y. Y., Yeo I. K. X., Wong C. J. Y., Liu M., Oh J. L. L., Chia C. P. L., Loh C. H., Chen A., Chen Q., Weissman I. L., Loh K. M., Lim B., A roadmap for human liver differentiation from pluripotent stem cells. Cell Rep. 22, 2190–2205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bailey S. R., Maus M. V., Gene editing for immune cell therapies. Nat. Biotechnol. 37, 1425–1434 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Schulze H. G., Konorov S. O., Caron N. J., Piret J. M., Blades M. W., Turner R. F., Assessing differentiation status of human embryonic stem cells noninvasively using Raman microspectroscopy. Anal. Chem. 82, 5020–5027 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Hsu C.-C., Xu J., Brinkhof B., Wang H., Cui Z., Huang W. E., Ye H., A single-cell Raman-based platform to identify developmental stages of human pluripotent stem cell-derived neurons. Proc. Natl. Acad. Sci. U.S.A. 117, 18412–18423 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dixon J. R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J. E., Lee A. Y., Ye Z., Kim A., Rajagopal N., Xie W., Diao Y., Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaspar-Maia A., Alajem A., Meshorer E., Ramalho-Santos M., Open chromatin in pluripotency and reprogramming. Nat. Rev. Mol. Cell Biol. 12, 36–47 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meshorer E., Misteli T., Chromatin in pluripotent embryonic stem cells and differentiation. Nat. Rev. Mol. Cell Biol. 7, 540–546 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Spangrude G. J., Heimfeld S., Weissman I. L., Purification and characterization of mouse hematopoietic stem cells. Science 241, 58–62 (1988). [DOI] [PubMed] [Google Scholar]
- 25.Wen B., Wu H., Shinkai Y., Irizarry R. A., Feinberg A. P., Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat. Genet. 41, 246–250 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hawkins R. D., Hon G. C., Lee L. K., Ngo Q., Lister R., Pelizzola M., Edsall L. E., Kuan S., Luu Y., Klugman S., Antosiewicz-Bourget J., Ye Z., Espinoza C., Agarwahl S., Shen L., Ruotti V., Wang W., Stewart R., Thomson J. A., Ecker J. R., Ren B., Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 6, 479–491 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Efroni S., Duttagupta R., Cheng J., Dehghani H., Hoeppner D. J., Dash C., Bazett-Jones D. P., Le Grice S., McKay R. D. G., Buetow K. H., Gingeras T. R., Misteli T., Meshorer E., Global transcription in pluripotent embryonic stem cells. Cell Stem Cell 2, 437–447 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nozaki T., Imai R., Tanbo M., Nagashima R., Tamura S., Tani T., Joti Y., Tomita M., Hibino K., Kanemaki M. T., Wendt K. S., Okada Y., Nagai T., Maeshima K., Dynamic organization of chromatin domains revealed by super-resolution live-cell imaging. Mol. Cell 67, 282–293.e7 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Hihara S., Pack C. G., Kaizu K., Tani T., Hanafusa T., Nozaki T., Takemoto S., Yoshimi T., Yokota H., Imamoto N., Sako Y., Kinjo M., Takahashi K., Nagai T., Maeshima K., Local nucleosome dynamics facilitate chromatin accessibility in living mammalian cells. Cell Rep. 2, 1645–1656 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Virk R. K. A., Wu W., Almassalha L. M., Bauer G. M., Li Y., VanDerway D., Frederick J., Zhang D., Eshein A., Roy H. K., Szleifer I., Backman V., Disordered chromatin packing regulates phenotypic plasticity. Sci. Adv. 6, eaax6232 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y., Eshein A., Virk R. K. A., Eid A., Wu W., Frederick J., VanDerway D., Gladstein S., Huang K., Shim A. R., Anthony N. M., Bauer G. M., Zhou X., Agrawal V., Pujadas E. M., Jain S., Esteve G., Chandler J. E., Nguyen T.-Q., Bleher R., de Pablo J. J., Szleifer I., Dravid V. P., Almassalha L. M., Backman V., Nanoscale chromatin imaging and analysis platform bridges 4D chromatin organization with molecular function. Sci. Adv. 7, eabe4310 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itzkan I., Qiu L., Fang H., Zaman M. M., Vitkin E., Ghiran I. C., Salahuddin S., Modell M., Andersson C., Kimerer L. M., Cipolloni P. B., Lim K.-H., Freedman S. D., Bigio I., Sachs B. P., Hanlon E. B., Perelman L. T., Confocal light absorption and scattering spectroscopic microscopy monitors organelles in live cells with no exogenous labels. Proc. Natl. Acad. Sci. U.S.A. 104, 17255–17260 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perelman L. T., Backman V., Wallace M., Zonios G., Manoharan R., Nusrat A., Shields S., Seiler M., Lima C., Hamano T., Itzkan I., Van Dam J., Crawford J. M., Feld M. S., Observation of periodic fine structure in reflectance from biological tissue: A new technique for measuring nuclear size distribution. Phys. Rev. Lett. 80, 627–630 (1998). [Google Scholar]
- 34.Qiu L., Pleskow D. K., Chuttani R., Vitkin E., Leyden J., Ozden N., Itani S., Guo L., Sacks A., Goldsmith J. D., Modell M. D., Hanlon E. B., Itzkan I., Perelman L. T., Multispectral scanning during endoscopy guides biopsy of dysplasia in Barrett’s esophagus. Nat. Med. 16, 603–606 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang L., Pleskow D. K., Turzhitsky V., Yee E. U., Berzin T. M., Sawhney M., Shinagare S., Vitkin E., Zakharov Y., Khan U., Wang F., Goldsmith J. D., Goldberg S., Chuttani R., Itzkan I., Qiu L., Perelman L. T., Light scattering spectroscopy identifies the malignant potential of pancreatic cysts during endoscopy. Nat. Biomed. Eng. 1, 0040 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsumoto K., Yoshitomi H., Rossant J., Zaret K. S., Liver organogenesis promoted by endothelial cells prior to vascular function. Science 294, 559–563 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Poisson J., Lemoinne S., Boulanger C., Durand F., Moreau R., Valla D., Rautou P.-E., Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 66, 212–227 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Rafii S., Butler J. M., Ding B. S., Angiocrine functions of organ-specific endothelial cells. Nature 529, 316–325 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nolan D. J., Ginsberg M., Israely E., Palikuqi B., Poulos M. G., James D., Ding B. S., Schachterle W., Liu Y., Rosenwaks Z., Butler J. M., Xiang J., Rafii A., Shido K., Rabbany S. Y., Elemento O., Rafii S., Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev. Cell 26, 204–219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pettinato G., Lehoux S., Ramanathan R., Salem M. M., He L. X., Muse O., Flaumenhaft R., Thompson M. T., Rouse E. A., Cummings R. D., Wen X., Fisher R. A., Generation of fully functional hepatocyte-like organoids from human induced pluripotent stem cells mixed with endothelial cells. Sci. Rep. 9, 8920 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I. M., Edlund K., Lundberg E., Navani S., Szigyarto C. A.-K., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P.-H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J. M., Hamsten M., von Feilitzen K., Forsberg M., Persson L., Johansson F., Zwahlen M., von Heijne G., Nielsen J., Pontén F., Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Montacir H., Freyer N., Knöspel F., Urbaniak T., Dedova T., Berger M., Damm G., Tauber R., Zeilinger K., Blanchard V., The cell-surface N-glycome of human embryonic stem cells and differentiated hepatic cells thereof. Chembiochem 18, 1234–1241 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Myster D. L., Duronio R. J., Cell cycle: To differentiate or not to differentiate? Curr. Biol. 10, R302–R304 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Niu J. J., Schrlau M. G., Friedman G., Gogotsi Y., Carbon nanotube-tipped endoscope for in situ intracellular surface-enhanced Raman spectroscopy. Small 7, 540–545 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Fang H., Ollero M., Vitkin E., Kimerer L. M., Cipolloni P. B., Zaman M. M., Freedman S. D., Bigio I. J., Itzkan I., Hanlon E. B., Perelman L. T., Noninvasive sizing of subcellular organelles with light scattering spectroscopy. IEEE J. Sel. Top. Quant. Elect. 9, 267–276 (2003). [Google Scholar]
- 46.Turzhitsky V., Zhang L., Horowitz G. L., Vitkin E., Khan U., Zakharov Y., Qiu L., Itzkan I., Perelman L. T., Picoanalysis of drugs in biofluids with quantitative label-free surface enhanced Raman spectroscopy. Small 14, 1802392 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.C. Lawson, R. Hanson, Solving Least Squares Problems (Prentice Hall, 1974). [Google Scholar]
- 48.Szafraniec E., Kus E., Wislocka A., Kukla B., Sierka E., Untereiner V., Sockalingum G. D., Chlopicki S., Baranska M., Raman spectroscopy–based insight into lipid droplets presence and contents in liver sinusoidal endothelial cells and hepatocytes. J. Biophotonics 12, e201800290 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Kubota H., Reid L. M., Clonogenic hepatoblasts, common precursors for hepatocytic and biliary lineages, are lacking classical major histocompatibility complex class I antigen. Proc. Natl. Acad. Sci. U.S.A. 97, 12132–12137 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elmaouhoub A., Dudas J., Ramadori G., Kinetics of albumin- and alpha-fetoprotein-production during rat liver development. Histochem. Cell Biol. 128, 431–443 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Baharvand H., Hashemi S. M., Shahsavani M., Differentiation of human embryonic stem cells into functional hepatocyte-like cells in a serum-free adherent culture condition. Differentiation 76, 465–477 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Behbahan I. S., Duan Y., Lam A., Khoobyari S., Ma X., Ahuja T. P., Zern M. A., New approaches in the differentiation of human embryonic stem cells and induced pluripotent stem cells toward hepatocytes. Stem Cell Rev. Rep. 7, 748–759 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nora E. P., Lajoie B. R., Schulz E. G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N. L., Meisig J., Sedat J., Gribnau J., Barillot E., Blüthgen N., Dekker J., Heard E., Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dixon J. R., Gorkin D. U., Ren B., Chromatin domains: The unit of chromosome organization. Mol. Cell 62, 668–680 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lua R., Borovinskiy A. L., Grosberg A. Y., Fractal and statistical properties of large compact polymers: A computational study. Polymer 45, 717–731 (2004). [Google Scholar]
- 56.Lieberman-Aiden E., van Berkum N. L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B. R., Sabo P. J., Dorschner M. O., Sandstrom R., Bernstein B., Bender M. A., Groudine M., Gnirke A., Stamatoyannopoulos J., Mirny L. A., Lander E. S., Dekker J., Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mirny L. A., The fractal globule as a model of chromatin architecture in the cell. Chromosome Res. 19, 37–51 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.B. Mandelbrot, Fractals and Chaos: The Mandelbrot Set and Beyond (Springer, 2013). [Google Scholar]
- 59.Fang H., Qiu L., Vitkin E., Zaman M. M., Andersson C., Salahuddin S., Kimerer L. M., Cipolloni P. B., Modell M. D., Turner B. S., Keates S. E., Bigio I., Itzkan I., Freedman S. D., Bansil R., Hanlon E. B., Perelman L. T., Confocal light absorption and scattering spectroscopic microscopy. Appl. Optics 46, 1760–1769 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Hasmy A., Foret M., Pelous J., Jullien R., Small-angle neutron-scattering investigation of short-range correlations in fractal aerogels: Simulations and experiments. Phys. Rev. B Condens. Matter 48, 9345–9353 (1993). [DOI] [PubMed] [Google Scholar]
- 61.Sigal Y. M., Zhou R., Zhuang X., Visualizing and discovering cellular structures with super-resolution microscopy. Science 361, 880–887 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wong P., Cao Q., Correlation function and structure factor for a mass fractal bounded by a surface fractal. Phys. Rev. B 45, 7627–7632 (1992). [DOI] [PubMed] [Google Scholar]
- 63.Sinha S. K., Scattering from fractal structures. Physica D 38, 310–314 (1989). [Google Scholar]
- 64.Oh C., Sorensen C. M., Scaling approach for the structure factor of a generalized system of scatterers. J. Nanopart. Res. 1, 369–377 (1999). [Google Scholar]
- 65.Yi J., Stypula-Cyrus Y., Blaha C. S., Roy H. K., Backman V., Fractal characterization of chromatin decompaction in live cells. Biophys. J. 109, 2218–2226 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ugarte F., Sousae R., Cinquin B., Martin E. W., Krietsch J., Sanchez G., Inman M., Tsang H., Warr M., Passegué E., Larabell C. A., Forsberg E. C., Progressive chromatin condensation and H3K9 methylation regulate the differentiation of embryonic and hematopoietic stem cells. Stem Cell Rep. 5, 728–740 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S., Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Yu J., Vodyanik M. A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J. L., Tian S., Nie J., Jonsdottir G. A., Ruotti V., Stewart R., Slukvin I. I., Thomson J. A., Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Park I. H., Zhao R., West J. A., Yabuuchi A., Huo H., Ince T. A., Lerou P. H., Lensch M. W., Daley G. Q., Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451, 141–146 (2008). [DOI] [PubMed] [Google Scholar]
- 70.Wernig M., Meissner A., Foreman R., Brambrink T., Ku M., Hochedlinger K., Bernstein B. E., Jaenisch R., In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448, 318–324 (2007). [DOI] [PubMed] [Google Scholar]
- 71.Osafune K., Caron L., Borowiak M., Martinez R. J., Fitz-Gerald C. S., Sato Y., Cowan C. A., Chien K. R., Melton D. A., Marked differences in differentiation propensity among human embryonic stem cell lines. Nat. Biotechnol. 26, 313–315 (2008). [DOI] [PubMed] [Google Scholar]
- 72.Cahan P., Daley G. Q., Origins and implications of pluripotent stem cell variability and heterogeneity. Nat. Rev. Mol. Cell Biol. 14, 357–368 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Basma H., Soto-Gutiérrez A., Yannam G. R., Liu L., Ito R., Yamamoto T., Ellis E., Carson S. D., Sato S., Chen Y., Muirhead D., Navarro-Alvarez N., Wong R. J., Roy-Chowdhury J., Platt J. L., Mercer D. F., Miller J. D., Strom S. C., Kobayashi N., Fox I. J., Differentiation and transplantation of human embryonic stem cell-derived hepatocytes. Gastroenterology 136, 990–999.e4 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marongiu F., Gramignoli R., Dorko K., Miki T., Ranade A. R., Paola Serra M., Doratiotto S., Sini M., Sharma S., Mitamura K., Sellaro T. L., Tahan V., Skvorak K. J., Ellis E. C., Badylak S. F., Davila J. C., Hines R., Laconi E., Strom S. C., Hepatic differentiation of amniotic epithelial cells. Hepatology 53, 1719–1729 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Petersen M. C., Vatner D. F., Shulman G. I., Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 13, 572–587 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lara-Astiaso D., Weiner A., Lorenzo-Vivas E., Zaretsky I., Jaitin D. A., David E., Keren-Shaul H., Mildner A., Winter D., Jung S., Friedman N., Amit I., Chromatin state dynamics during blood formation. Science 345, 943–949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Otsu N., A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybern. 9, 62–66 (1979). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/34/eabj2800/DC1