


Abstract
DNMT3A/3L heterotetramers contain two active centers binding CpG sites at 12 bp distance, however their interaction with DNA not containing this feature is unclear. Using randomized substrates, we observed preferential co-methylation of CpG sites with 6, 9 and 12 bp spacing by DNMT3A and DNMT3A/3L. Co-methylation was favored by AT bases between the 12 bp spaced CpG sites consistent with their increased bending flexibility. SFM analyses of DNMT3A/3L complexes bound to CpG sites with 12 bp spacing revealed either single heterotetramers inducing 40° DNA bending as observed in the X-ray structure, or two heterotetramers bound side-by-side to the DNA yielding 80° bending. SFM data of DNMT3A/3L bound to CpG sites spaced by 6 and 9 bp revealed binding of two heterotetramers and 100° DNA bending. Modeling showed that for 6 bp distance between CpG sites, two DNMT3A/3L heterotetramers could bind side-by-side on the DNA similarly as for 12 bp distance, but with each CpG bound by a different heterotetramer. For 9 bp spacing our model invokes a tetramer swap of the bound DNA. These additional DNA interaction modes explain how DNMT3A and DNMT3A/3L overcome their structural preference for CpG sites with 12 bp spacing during the methylation of natural DNA.
Graphical Abstract
Graphical Abstract.
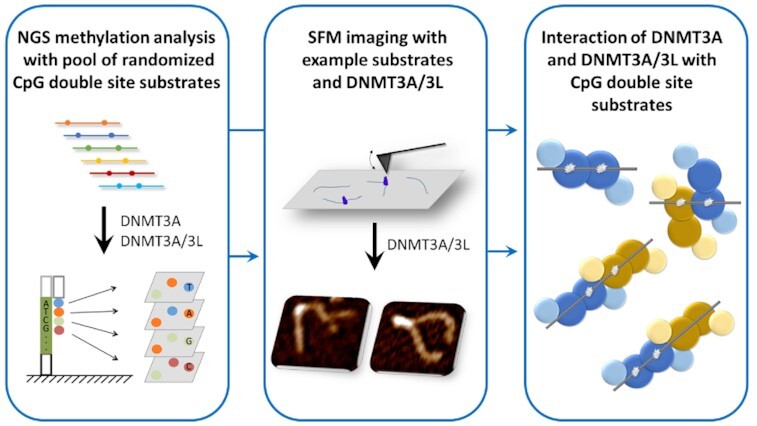
NGS methylation analysis with pool of randomized CpG double site substrates and SFM imaging of enzyme-DNA complexes with example substrates revealed novel interaction modes of DNMT3A and DNMT3A/3L with CpG double site substrates.
INTRODUCTION
DNA methylation plays essential roles in gene regulation and chromatin biology (reviews: 1–3). The DNA methyltransferase paralogs DNMT3A and DNMT3B were discovered in 1999 (4). Both enzymes catalyze the methylation of cytosine residues in DNA with a preference for CpG sites (reviews: 5,6) and set up DNA methylation patterns during gametogenesis and post-implantation development (reviews: 7,8). DNMT3A is essential for the development of mammals (review: 7,8), but it also has important roles in carcinogenesis (reviews: 9,10). The catalytically inactive DNMT3-like protein (DNMT3L) has an important regulatory role in this process by acting as a stimulator of DNMT3A (review: 6). One important role of both of these proteins is the setting of imprints in the mammalian germline (11–13). The catalytically active C-terminal domain of DNMT3A (14) in complex with the C-terminal domain of DNMT3L forms a linear heterotetrameric complex with the two DNMT3A subunits in the center and the DNMT3L at the edges (3L-3A-3A-3L) (15). The central DNMT3A subunit interface in this heterotetramer is based on the symmetric interaction of two arginine and aspartate residues and therefore designated RD interface (15). This region forms the DNA binding site of the complex such that each DNMT3A subunit can methylate with its active site one CpG site if these sites are placed at an appropriate distance. Based on the structural constraints these methylation events occur on opposite DNA strands. The DNMT3L subunits at the edges of the heterotetrameric complex can be replaced by two additional subunits of DNMT3A (16) yielding a DNMT3A homotetramer as the smallest catalytically active form of DNMT3A that contains two central DNMT3A subunits in the same arrangement as the DNMT3A/3L complex. Homotetramer formation of DNMT3A2 and DNMT3A catalytic domain and heterotetramer formation of the DNMT3A catalytic domain/DNMT3L C-terminal domain complex has been observed by analytical ultracentrifugation and size exclusion chromatography as well (16–18). However, larger multimers of DNMT3A catalytic domain and DNMT3A2 can also form in the absence of DNMT3L (17–19). DNMT3L does not contain an RD interface and thus prevents the formation of larger protein multimers than the heterotetramer (17,19). DNMT3A and DNMT3A/3L bind to DNA in a cooperative manner (16,20) and previous scanning force microscopy (SFM) imaging studies showed that both protein complexes multimerize on DNA at higher protein concentrations forming large protein-DNA fibers (17,20). SFM studies also showed different DNA interaction properties of DNMT3A and DNMT3A/3L. While larger complexes containing only DNMT3A were able to interact with two separate DNA molecules forming different irregular structures, this was not observed for DNMT3A/3L (17).
The catalytic cycle of DNMTs is initiated by the rotation of the target cytosine out of the DNA helix, followed by the attack of an active site cysteine residue on the C6-position of the target base leading to the transient formation of a covalent enzyme-DNA complex, which is resolved during later steps of the reaction (review: 6). Replacement of the target cytosine in DNA by Zebularine (Z) still allows covalent complex formation and methyl group transfer, but blocks further the steps of the reaction leading to a stable enzyme-DNA complex (21). Systematic analyses revealed that one DNMT3A/3L tetramer forms a tight complex with a 25mer DNA substrate containing two ZpG sites on different DNA strands at a distance of 12 bp between the CpG sites (corresponding to 14 bp between the Z-residues). This complex could be crystallized and the resulting structure revealed both target Z-residues flipped out of the DNA helix and placed in the active center of the two DNMT3A subunits (22). In addition, a structure was solved for DNMT3A/3L tetramer with two short 11mer DNA substrates each containing one ZpG site bound to one of the two DNMT3A subunits (22). The DNMT3A/3L tetramer in both structures did not undergo larger conformational changes when compared with a DNA-free structure solved earlier (15). While the short DNA molecules bound to one DNMT3A subunit were unbent, the long DNA interacting with both DNMT3A subunits showed an approximately 40° bending centered in the middle between the two ZpG sites. Subsequent kinetic studies showed that the interaction of the DNMT3A/3L complex with both Z-residues is strongly preferred for a spacing of 12 bp between these sites (23).
These structural and binding studies have significantly contributed to the understanding of DNMT3A-DNA interactions. However, previous SFM studies were carried out with DNA that was lacking specific target sites of DNMT3A, and the two-site binding experiments and crystallization studies mentioned above involved trapping of covalent complexes with ZpG sites, which could lead to a shift of conformational equilibria. Moreover, recent work has shown that the activity of DNMTs is strongly dependent on the sequences flanking the CpG sites (24–27) suggesting that sequence preferences could influence the bending necessary for the interaction of DNMT3A with two CpG sites. Here, we focused on the catalytic domain of DNMT3A and the complex formed by the catalytic domain of DNMT3A together with the C-terminal domain of DNMT3L and study their interaction with DNA containing pairs of CpG sites at different distances.
Using a next generation sequencing (NGS) coupled Deep-Enzymology approach, we investigated the preferences for DNMT3A and DNMT3A/3L catalyzed co-methylation of CpG sites at different distances under catalytic conditions and determined sequence effects on the efficiency of co-methylation. Unexpectedly, our data indicated several distinct modes of co-methylation of CpG sites by DNMT3A and DNMT3A/3L, which were associated with characteristic distances between the CpG sites of mainly 6, 9 and 12 bp. We then applied SFM imaging to characterize DNMT3A/3L complexes with DNA containing two CpGs at these distances. As a single molecule technique, SFM provides a powerful approach for conformational analyses of protein-DNA complexes formed at different DNA target sites and at unspecific sites (reviews: 28,29). The high, molecular resolution of SFM allows the distinction between (and separate analysis of) protein complexes bound at specific sites (here paired CpG sites with 6, 9 and 12 bp spacing), which are incorporated in the DNA substrates comprising about 400 bp at defined positions (here at 50% of the DNA length), and complexes bound elsewhere on the DNA. To further enhance the power of SFM for studies of protein-DNA interactions, we have previously developed an automated analysis that provides DNA bend angles at protein peak positions on the DNA in an experimenter independent and high throughput fashion (30). Here, we have further expanded this tool to detect the protein binding positions on the DNA correlated with the individual DNA bending angles at each of the peaks. Our studies revealed interesting differences between DNMT3A/3L complexes for the different DNA substrates. SFM imaging of DNMT3A/3L bound to substrates with two CpG sites at 12 bp spacing indeed revealed DNA bending consistent with the crystal structure (22). SFM data with substrates containing pairs of CpG sites at distances of 6 or 9 bp revealed distinct novel higher order complex arrangements for DNMT3A/3L tetramers. For 6 bp spacing between CpG sites, two DNMT3A/3L heterotetramers bound to the DNA side-by-side, while in case of pairs of DNMT3A/3L heterotetramers bound to CpG sites with 9 bp spacing, only one DNMT3A subunit of each heterotetramers contacts the DNA leading to a tetramer swap of the bound DNA. Importantly, these novel DNA interaction modes provide an explanation for why DNMT3A and DNMT3A/3L can function as global de novo DNA methylation devices in vivo without generating an obvious pattern of preferentially co-methylated CpG sites at a distance of 12 bp in cellular DNA.
MATERIALS AND METHODS
Protein expression and purification
The catalytic domain of murine DNMT3A (residues 608-908 of O88508) and C-terminal domain of DNMT3L (residues 207–421 of AAH83147) were overexpressed in BL21 Codon plus (DE3)-RIL Escherichia coli cells (Stratagene) and purified as described (17,31). The catalytic domains of human and murine DNMT3A are identical in amino acid sequence, the amino acid sequence identity between the C-terminal domains of the murine and human orthologs of DNMT3L is 74%. The purity of the protein preparations was estimated to be >95% based on Coomassie stained SDS gels (Supplemental Figure S1A). The concentrations of the proteins were determined by UV spectrophotometry and confirmed by densitometric analysis of Coomassie stained SDS–polyacrylamide gels. Complexes of DNMT3A catalytic domain with the C-terminal domain of DNMT3L were formed by equimolar mixing of both proteins and incubation for at least 30 min. Complex formation was confirmed by radioactive methylation kinetics using a 30mer oligonucleoide substrate with one CpG sites as described (32), where the activity of DNMT3A is stimulated about 5–10-fold in the presence of DNMT3L (32).
Methylation of substrate libraries
Single-stranded DNA oligonucleotides used for generation of double stranded substrates with two CpG sites at different distances were obtained from IDT (Supplemental table S1). Sixteen single-stranded oligonucleotides were pooled in equimolar amounts and the second strand synthesis was conducted by a primer extension reaction using one universal primer. The obtained mix of double-stranded DNA oligonucleotides (107 nM) (Supplemental Figure S1B) was methylated by DNMT3A catalytic domain and DNMT3A catalytic domain mixed with DNMT3L C-terminal domain, and incubated for 60 min at 37°C in the presence of 0.8 mM S-adenosyl-l-methionine (Sigma) in reaction buffer (20 mM HEPES pH 7.5, 1 mM EDTA, 50 mM KCl, 0.05 mg/ml bovine serum albumin). For DNMT3A, concentrations of 0.25, 0.5, 1 and 2 μM were used, for DNMT3A/3L 0.125 and 0.25 μM. In addition, a no-enzyme control was processed identically as all other samples. The methylation reactions were stopped by shock freezing in liquid nitrogen, then treated with proteinase K (NEB) for 2 h at 42°C, and purified by PCR cleanup kit (Macherey-Nagel). Afterwards, the DNA was digested with BsaI-HFv2 (NEB) and a hairpin (pGAGAAGGGATGTGGATACACATCCCT) was ligated onto the cut end using T4 DNA ligase (NEB). DNA was bisulfite-converted using EZ DNA Methylation-Lightning kit (ZYMO RESEARCH) according to the manufacturer protocol and eluted with 10 μl ddH2O.
NGS library generation
Libraries for Illumina Next Generation Sequencing (NGS) were produced with a two-step PCR approach (Supplemental Figure S1C). In the first PCR, 2 μl of bisulfite-converted DNA were amplified with the HotStarTaq DNA Polymerase (QIAGEN) and primers containing internal barcodes using following conditions: 15 min at 95°C, 10 cycles of 30 s at 94°C, 30 s at 50°C, 1 min and 30 s at 72°C, and final 5 min at 72°C; using a mixture containing 1× PCR Buffer, 1× Q-Solution (QIAGEN), 0.2 mM dNTPs, 0.05 U/μl HotStarTaq DNA Polymerase, 0.4 μM forward and 0.4 μM reverse primers in a total volume of 20 μl. In the second PCR, 1 μl of obtained products were amplified by Phusion HF Polymerase (Thermo) with another set of primers to introduce adapters and indices needed for NGS (30 s at 98°C, 10 cycles of 10 s at 98°C, 40 s at 72°C, and finally 5 min at 72°C). PCRII was carried out in 1× Phusion HF Buffer, 0.2 mM dNTPs, 0.02 U/μl Phusion HF DNA Polymerase, 0.4 μM forward and 0.4 μM reverse primers in a total volume of 20 μl. Obtained libraries were pooled in equimolar amounts, purified by PCR cleanup kit (Macherey-Nagel) and sequenced in the Max Planck Genome Centre Cologne.
Bioinformatic analysis
Bioinformatic analysis of obtained NGS data was conducted with a local Galaxy server (33) and with home written scripts. Briefly, fastq files were analyzed by FastQC (Galaxy Version 0.72+galaxy1, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), the 3′ ends of the reads with a quality lower than 20 were trimmed using Trim Galore! (Galaxy Version 0.4.3.1, https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), and reads containing both full-length sense and antisense strands were selected. Next, the samples were split with respect to the different experimental conditions using the internal barcodes. Afterwards, the insert DNA sequence was extracted and using the information of both strands of the bisulfite-converted substrate, the original DNA sequence and methylation state of both CpG sites was reconstituted. The sequences were sorted according to the distances between the CpG sites and DNA methylation was analyzed on both sense and anti-sense strands. Finally, the co-occurrence of methylation on sense and anti-sense strands as well as the average base composition in the middle of co-methylated substrates was extracted.
Structural modelling
Structural modelling was done in Chimera 1.4 (34) using the DNMT3A/3L complexes 6F57 and 6BRR (22). Higher order complexes binding one molecule of DNA at different distances were generated by placing two tetrameric complexes in the desired geometry and manually overlapping an appropriate number of base pairs of two complexes to place the adjacent CpG sites in the desired distance.
Design and generation of the SFM substrate
Three different substrates containing CpG sites at distances of 6 (substrate D6), 9 (substrate D9) and 12 bp (substrate D12) within a central CpG free sequence were designed for SFM experiments (Supplemental Figure S2A and S2B). The D6, D9 and D12 substrates had lengths of 406, 409 and 412 bp, respectively, with the dual CpG sites located at 50% of their overall lengths. Outside of the central CpG free region that surrounds the designed CpG double sites (total length 102 bp), the substrates contained 20 CpG sites with variable spacing. Combined analyses of binding properties to these parts of the DNA substrates (between DNA ends and 40% of DNA length), which includes binding to non-specific sites (non-CpG sites), single CpG sites, and two CpG sites with various spacing will be referred to here as binding to ‘average DNA’. The substrates were produced by primer extension to generate the double stranded oligos harboring CpG site pairs in a CpG free regions. In the second step, this inner unit was TOPO TA cloned into the pCR2.1 TOPO vector (Agilent). Using this vector as template, the final SFM substrates were amplified by PCR using primer sets, which are complementary to the vector backbone. The substrates were cleaned up according to the protocol of the PCR cleanup kit (Macherey-Nagel) (Supplemental Figures S2C).
SFM experiments
Preformed DNMT3A/3L complexes (100 nM) were incubated in SFM buffer containing 25 mM HEPES pH 7.5, 25 mM sodium acetate and 10 mM magnesium acetate at ambient temperature for 20 min with DNA substrates (4 nM). A volume of 20 μl of the incubations was deposited onto freshly cleaved mica (Grade V5, SPI Supplies). For control experiments, samples with DNA substrates without DNMT3A/3L (Supplemental Figure S3) were also imaged. The samples were allowed to spread evenly on the mica surface for 1 min and were then rinsed gently with ultrapure water and dried under a stream of nitrogen. Imaging was conducted in air using a Molecular Force Probe (MFP) 3D-Bio SFM (Asylum Research, Oxford Instruments) and AC240TSA silicon probes (Olympus, nominal end diameter < 10 nm) with spring constants of ∼2 N/m and resonance frequencies of ∼70 kHz in tapping mode. Image sizes of 2 μm × 2 μm, 4 μm × 4 μm and 8 μm × 8 μm were collected with a pixel resolution of ∼1.95 nm at a scan speed of 2.5 μm/s. All experiments were performed in triplicate.
Data analysis of the SFM experiments
SFM micrographs were plane-fitted and flattened to third order using MFP software on Igor Pro interactive software environment and were exported in Tiff format. DNA bend angles and protein binding positions on DNA were obtained using an extended version of our automated, high-throughput open source MatLab tool (available at Open Science Framework at https://osf.io/76e9s/). The detailed procedure involving image pre-processing, DNA skeletonization, protein localization, and DNA bend angle measurement has been previously described (30). In addition, we extended the MatLab script for SFM data analysis to allow the automated localization of protein positions on the DNA based on DNA skeletonization, a height cut-off to determine protein peak positions, and distances measured from protein peak centers to DNA ends (see also Supplemental text S1, detailed instructions can also be found at https://osf.io/76e9s/). Only DNA substrates of the correct length were included in the analyses to vouchsafe correct positioning of CpG target sites at 50% DNA length (see Supplemental text S1). The resulting position distributions of DNMT3A/3L on the DNA fragments were plotted using Origin Pro (OriginLab). Because we cannot distinguish the two (unlabeled) DNA ends in our substrates, position distributions are displayed between DNA ends (0% of DNA length) and the center of the DNA (50% of the DNA length). To estimate binding specificities for dual CpG target sites, the DNA was subdivided in 20 segments each comprising 5% of DNA length (2.5% on either side from the center position). The number of binding events observed in each segment was compared with the number expected for no binding preferences (equal probability of binding to all DNA sites, Nexp = Ntotal/20).
The volumes of the protein peaks on the DNA were measured using the density slice option in ImageSXM software (S. Barrett, University of Liverpool). Volume distributions were plotted and Gaussian fitted using Origin Pro (OriginLab Corporation, Northampton, USA). The volumes of the complexes were derived from the centers of the Gaussians. SFM volumes (V) can be translated into approximate molecular weights (MW) of protein complexes using our previously reported SFM volume calibration: MW = (V + 5.9)/1.2 (35), which can serve as a crude size estimate for DNA bound complexes as well (36). Lengths and heights of protein peaks at the 50% positions on the DNA substrate with volumes consistent with two DNMT3A/3L heterotetramers were measured manually with Image J (https://imagej.nih.gov/ij/) and Image SXM software, respectively.
RESULTS
Analysis of co-methylation frequencies of CpGs at different distances
We used a Deep-Enzymology approach to investigate the activity of DNMT3A and DNMT3A/3L complexes on different DNA substrates. These analyses revealed detailed mechanistic information regarding the concerted activity of both active DNMT3A subunits forming the RD interface in these complexes. In Deep-Enzymology approaches, single molecule DNA methylation kinetics are conducted on substrates containing partially randomized sequences to obtain novel insights into the influence of the substrate sequence on enzyme activity (25–27,37). In the current study, a library of DNA molecules containing pairs of CpG sites with different spacings was prepared and used for kinetic experiments with DNMT3A or DNMT3A/3L (Figure 1A). To provide a neutral sequence context and be able to later investigate the influence of the DNA sequence on co-methylation of CpG sites at different distances, we embedded the two CpG sites in a context of 23 randomized bases (Supplemental table S1). Using partially randomized oligonucleotides with the corresponding sequences, a mixed library pool of double stranded DNA was prepared, which was then methylated by DNMT3A or DNMT3A/3L. Afterwards, the methylation of both DNA strands was simultaneously detected by hairpin bisulfite conversion followed by deep NGS sequencing. Illumina sequencing yielded high NGS read coverages for each individual substrate (Supplemental table S2). Methylation reactions were conducted at different enzyme concentrations using DNMT3A (0.25, 0.5, 1 and 2 μM) or DNMT3A/3L (0.125 and 0.25 μM) and overall methylation levels were observed to increase accordingly from 1% to 11.6% and they reflected the known enhancement of the activity of DNMT3A by addition of DNMT3L (32) (Figure 1B). Control reactions were conducted without addition of enzyme and analyzed through the same pipeline. The obtained results revealed very low apparent methylation levels of 0.16%, which represent background from incomplete bisulfite conversion. These results document high conversion rates in the bisulfite analysis and correct data analysis.
Figure 1.

DNMT3A and DNMT3A/3L catalyzed co-methylation of CpG sites in different distances. (A) Schematic view of the Deep-Enzymology workflow applied here. A library of synthetic oligonucleotides with CpG sites in variable distances embedded in random flanking sequence context is synthesized. After second strand synthesis, the pool of substrates with different sequences is methylated by DNMT3A or DNMT3A/3L, followed by hairpin ligation and bisulfite conversion. Barcodes and indices are added by PCR and the library is subjected to Illumina NGS sequencing. After quality control of the obtained reads, the original sequence and methylation state is extracted followed by analysis with respect to co-methylation events and sequence enrichment. M represents 5mdC. Methylation in the upper DNA strand is indicated by ‘M’, methylation in the lower DNA strand by ‘W’. (B) Overall methylation levels in the DNA methylation experiments with different enzyme concentrations and in the no-enzyme control. The data show average methylation levels at all 4 target cytosines in the two CpG sites averaged for all distances. (C) Schematic illustration of the different types of co-methylation of pairs of CpG sites considered in this study. Note that mechanistically MM and WW methylation are equivalent.
We then determined the fraction of co-methylation events of CpG sites at spacings of 2 to 15 bp in these data (Supplemental table S3). For this, three types of co-methylation were distinguished (Figure 1C): (i) co-methylation of site 1 in the upper (‘M’) and site 2 in the lower (‘W’) DNA strand (MW co-methylation), (ii) co-methylation of site 1 in the lower and site 2 in the upper DNA strand (WM co-methylation), and iii) co-methylation of site 1 and 2 in the same DNA strand (MM co-methylation). Of note, co-methylation in the MW and WM mode is structurally and mechanistically distinct. In the conformation seen in the crystal structure, the tetrameric DNMT3A/3L complex would be able to introduce MW co-methylation at CpG sites in about 12 bp distance (22,23), but it could not generate WM or MM co-methylation. Co-methylation profiles were determined at different enzyme concentrations and after normalizing to the average values of the individual experiments, the relative co-methylation levels observed in the different experiments could be averaged with small error bars. MM and WW co-methylation are symmetrically related and refer to the same process once occurring on the upper and once on the lower DNA strand. Therefore, MM and WW co-methylation data were mixed and they are presented here as ‘MM’ co-methylation. For all types of co-methylation, a peak of occurrence was observed at distances of 2 bp (Figures 2, 3 and Supplemental Figure S4). For MW co-methylation (Figure 2), our data revealed two additional peaks for CpG distances of 9 bp and 12 bp in case of DNMT3A. For DNMT3A/3L, the peak at 12 bp was further enhanced while the peaks at 2 and 9 bp were less pronounced. Regarding WM co-methylation (Figure 3), a second maximum at 5-6 bp was evident for DNMT3A/3L. For MM co-methylation, apart from the enrichment at 2 bp, no strong preferences for CpG spacings were observed with DNMT3A or DNMT3A/3L (Supplemental Figure S4). All these peaks were reproducibly detected in all underlying independent methylation experiments, which were conducted at different enzyme concentrations (Figures 2, 3 and Supplemental Figure S4).
Figure 2.

Frequencies of MW co-methylation of CpG sites in variable distances observed with DNMT3A and DNMT3A/3L. (A) Scheme of MW co-methylation (B) Frequencies of MW co-methylation of CpG sites in variable distances observed with DNMT3A. The left panel shows the four independent data sets, the right panel shows averages and SEM obtained after normalization of the data sets to their average values. (C) Frequencies of MW co-methylation of CpG sites in variable distances observed with DNMT3A/3L. The left panel shows the two independent data sets, the right panel shows averages and SEM obtained after normalization of the data sets to their average values.
Figure 3.

Frequencies of WM co-methylation of CpG sites in variable distances observed with DNMT3A and DNMT3A/3L. (A) Scheme of WM co-methylation (B) Frequencies of WM co-methylation of CpG sites in variable distances observed with DNMT3A. The left panel shows the four independent data sets, the right panel shows averages and SEM obtained after normalization of the data sets to their average values. (C) Frequencies of WM co-methylation of CpG sites in variable distances observed with DNMT3A/3L. The left panel shows the two independent data sets, the right panel shows averages and SEM obtained after normalization of the data sets to their average values.
Base enrichment in the center of 12 base pair MW co-methylation
While abundant co-methylation at short distances, which is declining with increasing distance, can be explained by short-range movements of DNMT3A or DNMT3A/3L on the DNA (as described in the discussion section), enhanced co-methylation peaks at distances of 6, 9 or 12 bp require a different mechanism. To investigate the underlying mechanism for preferential MW co-methylation by DNMT3A or DNMT3A/3L, we first analyzed the effects of the sequence between two CpG sites separated by 12 bp (as in the crystal structure) on the efficiency of this type of co-methylation. The crystal structure indicates that DNA bending is necessary for the MW co-methylation of CpG sites at 12 bp distances, which is known to depend on the nucleotide composition of the DNA sequence (review: 38). Therefore, we were interested to find out if the DNA sequences of substrates with co-methylation of CpG sites in 12 bp distance differ. We focused on the region in the midpoint between the two CpG sites (position 5–8), which is at the center of the bending. As shown in Figure 4, clear sequence preferences were detected with a strong and highly significant enrichment of T in the upper DNA strand both for DNMT3A and DNMT3A/3L indicating a stimulation of co-methylation by the presence of an A-tract in the lower strand at the center of bending. At the same time, a strong depletion of G in the upper DNA strand was observed. As a control, we also extracted the NNNCGNNN flanking sequences of the subset of methylated CpGs, and compared them with published data revealing very high similarity between these two independent studies as expected (Supplemental Figure S5).
Figure 4.

Observed/expected frequencies of bases at the center of bending in MW co-methylated substrates with CpG sites in 12 bp distance. To analyze the DNA sequence at the center of bending positions 5–8 were considered. (A) Schematic view. (B) Averages of the enrichment and depletion of bases in substrates co-methylated by DNMT3A and DNMT3A/3L. Error bars display the SEM. (C) P-values of enrichment or depletion. P-values for a significant deviation from 1.0 were calculated using the averages and SD values with Z-statistic.
SFM analysis of DNMT3A/3L DNA complexes
Next, we characterized the interaction of DNMT3A/3L with pairs of CpG sites at different distances using single molecule imaging by scanning force microscopy (SFM). These studies were carried out with DNMT3A/3L, because the known complex structure provides a reliable starting point for modelling of higher order complexes and interpretation of the data. For these experiments, DNA substrates of ∼400 bp length were generated that contained one pair of CpG sites with spacings of 6, 9 or 12 bp (substrates D6, D9 and D12, respectively) in their center (at 50% of the DNA length). This allowed us to identify complexes bound specifically at the double-CpG target site based on their position on the DNA (Supplemental Figure S2). The intervening sequence was designed based on the base preferences observed in the methylated sequences (see above), but avoiding repetitive and palindromic sequences and strong biases. The 12 bp distance corresponds to the available structural data (22) and the preferences of MW co-methylation of DNMT3A and DNMT3A/3L at this distance. The distance of 9 bp corresponds to the strong preference of DNMT3A (and weaker preference of DNMT3A/3L) for MW co-methylation at this distance. Finally, the 6 bp distance corresponds to the WM co-methylation peak observed with DNMT3A/3L. Protein concentrations were kept low, in order to avoid the formation of fully occupied protein-DNA fibers as observed previously (16,17,20). For SFM data analysis, we developed an automated MatLab tool that provides the lengths of the DNA in the images. DNA length distributions of protein-DNA complexes as well as unbound DNAs are shown for the three substrates D6, D9 and D12 in Supplemental Figure S6. The software then locates the binding positions of protein complexes on the DNA and measures DNA bending in the individual complexes (see Methods and Supplemental text S1). Labeling of the protein peaks on the DNA by the procedure also enables us to specifically measure the size (volume, length and height) for each of the DNA bound complexes. DNMT3A/3L complexes were thus investigated with respect to their distribution on the DNA, complex volumes and dimensions, and their DNA bend angle.
Our SFM data revealed strong preferences of DNMT3A/3L for binding to double CpG sites with spacings of 9 and 12 bp (D9 and D12 substrates, respectively), indicating high affinities for these CpG pairs (Figure 5). For the D12 substrate, 43% of all binding events occurred at the central CpG sites, translating into a high preference (ratio of observed over expected number of events, obs/exp = 8.6). Although the enhancement of binding to the central CpG sites was lower for the D9 substrate (maximum obs/exp ratio of 3.5 at the 50% position), the peak in binding events was considerably broader for this substrate resulting in overall 52% of all binding events located in the region from 40% to 50% DNA length. In both cases (D9 and D12), a second local maximum was observed at 25-30% of the DNA length at the place of the transition of CpG free to average DNA. Importantly, this position is well separated from the specific sites at 50% of DNA length in our DNA substrates. For the D6 substrate, the peak at 25–30% DNA length was comparable in size to the binding peak at the central region (with the two CpG sites separated by 6 bp), indicative of only moderate affinity for the dual CpG sites with 6 bp spacing (maximum obs/exp = 3.1 at 50% DNA length, 16% of all binding events).
Figure 5.

SFM imaging of DNMT3A/3L binding preferences for pairs of CpG sites separated by 6, 9 and 12 bp. Position distributions of protein complexes bound to DNA substrates containing two CpGs with different spacings at 50% of DNA length: 6 bp (D6 substrate), 9 bp (D9 substrate), 12 bp (D12 substrate). Since the two DNA ends are not distinguished in our substrates, position distributions are plotted to 50% of DNA length (with distances measured for each protein peak to the closer end of the DNA). Protein positions were fitted with two normal distributions to highlight the bimodal distribution. Nexp (horizontal dashed lines) indicates the expected number of binding events in each binning interval assuming equal binding probability to all positions on the DNA substrates, i.e. complete lack of specificity. The triangles indicate exemplary complexes bound at the center of the DNA fragments (corresponding to the positions of CpG pairs with 6–12 bp spacing).
To investigate the stoichiometry of protein complexes bound at pairs of CpG sites with spacings of 6, 9 or 12 bp, we determined their volumes in the SFM images (Figure 6A). Interestingly, volumes at CpG sites with 6 and 9 bp distance indicated the predominant presence of two DNMT3A/3L heterotetramers (volumes of ∼200 nm3, corresponding to ∼200 kDa), while DNMT3A/3L complexes at 12 bp spacings showed a bimodal distribution of volumes corresponding to a single heterotetramer (slightly larger than 100 nm3, corresponding to ∼100 kDa) and volumes corresponding to two heterotetramers bound at the central double CpG sites. As a reference, we also measured the volumes of DNMT3A/3L on DNA between DNA ends and 40% of the DNA length (average DNA, Figure 6A, left panel). These volumes indicated that mostly single heterotetramers of DNMT3A/3L are bound to these DNA regions.
Figure 6.

Characterization of DNMT3A/3L complexes with CpG containing DNA substrates by SFM imaging. (A) Protein complex volumes and (B) DNA bending angles in the complexes situated at 50% of DNA length on substrates containing a pair of CpG sites separated by 6 bp (D6 substrate), 9 bp (D9 substrate), and 12 bp (D12 substrate). Average DNA refers to complexes formed at other sites (between DNA ends and 40% DNA length). (C) Separate volume distributions for species with moderate (∼40°, left) and strong DNA bending (∼80°, right) for the D12 substrate.
To further characterize the different types of complexes, we analyzed the bending introduced into the DNA by DNMT3A/3L bound at the center of the D6, D9 and D12 substrates and at average DNA (Figure 6B). For the 12 bp CpG spacing, we observed two species of complexes characterized by DNA bending angles of ∼40° and ∼80°. Separate volume distributions for these two different species for CpGs with 12 bp spacing (Figure 6C) revealed that the 40° bent species corresponds to a single heterotetramer of DNMT3A/3L (∼100 nm3), while the more strongly bent species corresponds to two DNMT3A/3L tetramers bound (∼200 nm3). DNA bend angles of 40° in the complexes are consistent with bending in the crystal structure of a single heterotetramer of DNMT3A/3L bound to two ZpG sites at 12 bp distance (22), while we attribute the larger complexes with 80° bending to two heterotetramers binding to the DNA side-by-side, each causing a bend of approximately 40° (see also below). At average DNA sites, DNMT3A/3L introduced a broad DNA bend angle distribution with maximum also at ∼40°. Importantly, the free D6, D9 and D12 substrates did not display bending at the central position (Supplemental Figure S3B). The SFM data thus show that DNMT3A/3L complexes actively introduce bending into the DNA.
Analysis of the bending angle distribution of DNMT3A/3L complexes bound at the central position of the D6 and D9 substrates revealed that the 6 and 9 bp spacing of CpG sites resulted in complexes with stronger DNA bending by ∼100°. Further analysis focused on the most abundant large volume complexes (∼200 nm3, corresponding to two DNMT3A/3L heterotetramers) at CpGs with 6 and 9 bp spacing revealed heights of ∼0.5 and ∼1.1 nm, respectively (Figure 7). Measurements of the length of the complexes on the DNA showed ∼20 and ∼10 nm for complexes bound to CpG sites with 6 and 9 bp spacing, respectively (Figure 7). These results indicate that complexes bound to the D6 substrate have a relatively flat and extended shape, while the complexes bound to CpGs at 9 bp distance appear to be more ‘knotty’ and compacted. As a control, we also measured lengths and heights of DNA complexes of DNMT3A/3L with volumes of ∼100 nm3 corresponding to a single heterotetramer (Supplemental Figure S7). Consistently, these all showed lengths of ∼8 nm (only slightly shorter than the D9 dimer) and heights of ∼0.5 nm (comparable to the D6 dimer).
Figure 7.

Characterization of large volume DNMT3A/3L complexes with CpG containing DNA substrates by SFM imaging. (A) SFM images of representative large volume DNMT3A/3L complexes bound to the D6 and D9 substrates in 3D (top) and top view (bottom). Image areas are ∼130 × 130 nm2. The scale bar at the right indicates the heights in the 3D images in units of nm. Complex volumes are 206 nm3 (D6) and 217 nm3 (D9), both consistent with two heterotetramers. The heights of these complexes are 0.69 nm and 1.18 nm and their lengths on the DNA are 18.9 and 9.4 nm for D6 and D9, respectively. (B) Heights (left) and lengths (right) of large volume DNMT3A/3L complexes bound at CpG sites separated by 6 bp (D6 substrate) and 9 bp (D9 substrate). The Gaussian fit centers indicate average heights of complexes on CpG pairs of ∼0.6 nm for D6 and ∼1.1 nm for D9. Average lengths of complexes on D6 and D9 are ∼20 and 10 nm, respectively. The distributions of heights and lengths of single heterotetramers (based on volumes) bound to the different DNA substrates are shown in Supplemental Figure S7.
Structural interpretation of the different co-methylation modes
The preference for MW co-methylation at a distance of 12 bp (Figure 8A) as observed in our methylation assays and for binding of a single DNMT3A/3L heterotetramer to the D12 substrate in our SFM data is in perfect agreement with the co-crystal structure of DNMT3A/3L bound to ZpG-DNA (22) as well as with additional biochemical data (23). Methylation at slightly larger or smaller distances (e.g. 11 or 13 bp) may be achieved by minor conformational changes. However, on the basis of the structural analyses and very low conformational flexibility observed in the various different DNMT3A/3L structures (15,22,39), it appears highly unlikely, that a single tetramer could prefer co-methylation of CpG sites at a distance of 9 bp, as observed for DNMT3A and to a lesser degree also for DNMT3A/3L. To understand our results regarding MW and WM co-methylation preferences in the kinetic experiments and the structural and stoichiometric properties of DNMT3A/3L bound to MW CpG sites at different spacings determined by SFM, we modelled different types of orientations for two heterotetrameric DNMT3A/3L complexes bound to CpG sites with different spacings (Figure 8).
Figure 8.

Schematic pictures, structural snapshots and models explaining MW and WM co-methylation at difference distances. (A) MW co-methylation at 12 bp distance is in agreement with the crystal structure of DNMT3A/3L (6BRR) (22). The two DNMT3A subunits in the heterotetramer are colored blue, DNMT3L is colored cyan, DNA orange and AdoHcy is shown in yellow. (B) MW co-methylation at shorter distances can be explained, if two adjacent tetramers interact with the DNA with only one DNMT3A subunit leading to a tetramer swap of the DNA. The model was generated by using two structures of DNMT3A/3L complexes with two short DNA substrates each bound to one DNMT3A subunit (6F57) (22). The DNA molecules of both tetramers were fused leaving a distance of 9 bp between the CpG sites. (C) WM co-methylation can be explained by two adjacent DNMT3A tetramers interacting with the DNA side-by-side. The model was generated by using two DNMT3A/3L structures with long DNA substrates bound to both DNMT3A subunits (6BRR (22)). The DNA molecules were aligned leaving a distance of 7 bp between the CpG sites.
To rationalize MW co-methylation at shorter distances, we considered the DNMT3A/3L structure with two short CpG containing DNA substrates, each bound separately to one DNMT3A subunit in a heterotetrameric complex (6F57) (22). These DNA molecules can be fused generating one DNA with two CpG sites each of which interact with one DNMT3A subunit of the two adjacent DNMT3A/3L tetramers leading to a tetramer swap of the DNA at the RD interfaces (Figure 8B). Modelling showed that the tetramers can approach each other to as close as 8–9 bp between the CpG sites without major steric clashes leading to relatively compact structures. This model was strongly supported by the SFM experiments showing that complexes at CpGs with 9 bp spacing had volumes corresponding to two tetramers, and that they have a condensed shape with short complex length and large height. However, in this model the centrally bound DNA does not show bending, while strong bending is clearly observed in our SFM images. However, both of the DNMT3A/3L tetramers in the model still contain one additional DNMT3A subunit, which is not involved in the central interaction with the two CpG sites. These free DNMT3A subunits could bind to the flanks of the DNA molecule, potentially leading to DNA bending and further compaction of the complex as described in more detail in the discussion section.
WM co-methylation can be explained by two adjacent DNMT3A tetramers each interacting with one CpG site in the DNA. Modelling was based on the DNMT3A/3L structures bound to a long DNA containing two CpG sites at 12 bp distance (6BRR) (22). By connecting the bound DNAs of two DNMT3A/3L tetramers, both complexes can be arranged side-by-side (Figure 8C). Modelling shows that adjacent tetramers can approach each other up to distances of 7 bp between the CpG sites without steric clashes, suggesting that slightly shorter distances will be possible with some conformational adjustments. In fact, a closer approach up to 5 or 6 bp would allow the two complexes to form an extensive interface, in agreement with the previous observation that DNMT3A binds cooperatively to DNA (16,17,20). In further agreement with previous data, one part of the putative interface between the tetramers is formed by the loop (R831-K855) which was previously shown to be essential for the multimerization of DNMT3A on DNA (20) (Supplemental Figure S8). The resulting dimers of DNMT3A/3L heterotetrameric complexes generate an elongated shape following the DNA in a moderately extended conformation. This interpretation is supported by the SFM results indicating large volumes of these complexes corresponding to two DNMT3A/3L tetramers, as well as long complex lengths, low heights, and strong DNA bending.
DISCUSSION
The smallest catalytically active unit of DNMT3A is a homotetramer of DNMT3A or a heterotetramer of DNMT3A and DNMT3L, in which the two centrally placed DNMT3A subunits can interact with two CpG sites spaced 12 bp apart and methylate them on opposite DNA strands (22). This observation immediately leads to the question of why this enzyme with an in-built preference for interaction with CpG sites 12 bp apart can more or less equally methylate natural DNA, which contains isolated CpG sites and pairs of CpG sites randomly spaced from one another. To approach this challenging question, in this study we investigated co-methylation by DNMT3A and DNMT3A/3L of CpG sites at different distances from each other. As it has been shown that flanking sequences have a strong impact on the activity of DNMTs (25–27,37), we embedded the two CpG sites with variable spacing in a random flanking sequence context to avoid potential bias of results caused by fixed sequence substrates. Co-methylation of CpG sites can be classified into three different types, either co-methylation of two CpG sites within the same DNA strand (MM co-methylation) or in different DNA strands (MW and WM co-methylation). MW co-methylation targets site 1 in the upper and site 2 in the lower DNA strand, while WM co-methylates site 1 in the lower and site 2 in the upper DNA strand (see schematic in Figure 1C). Of note, the latter two modes of co-methylation in opposite DNA strands are not symmetry related (they can be compared to diastereomers in chemical structures). Hence, distinct DNMT3A complex arrangements are required for their generation.
For all three types of co-methylation, distinct and highly reproducible patterns of over- and underrepresentation in defined distances were observed. We first investigated, if a simple model assuming independent methylation at both CpG sites could explain these data. In this scenario, the probability of co-methylation is identical at all distances, because the two methylation events are not connected to one another. However, statistical analysis of the observed levels of co-methylation in different distances is not in agreement with this model (Supplemental Figures S9 and S10).
Next, we considered if diffusion of the DNMT3A complexes along the DNA can explain our findings. For MM co-methylation, we observed a distinct preference for CpGs separated by very short distances, which decreased with larger distances. This result can be explained by sliding of the enzyme complex along the DNA after one turnover followed by a second turnover by the same complex or even subunit. Interestingly, preferences of MW and WM co-methylation at short distances were also detected, which cannot be explained by DNA sliding, because in these cases the second methylation event occurs on the opposite DNA strand. This observation is most readily explained by rapid dissociation/re-association cycles of DNMT3A on DNA, a process also called ‘hopping’. To explain co-methylation in opposite DNA strands, it is plausible to assume that during the hopping process the CpG site changes from one of the central DNMT3A subunits to the other, which would automatically lead to a switch of the DNA strand that is methylated. One-dimensional diffusion on DNA by sliding and hopping processes is well-established and regularly involved in target site location of DNA binding proteins after initial unspecific binding (reviews: 40,41). Being diffusional processes, both are most efficient at short distances and expected to decline continuously roughly with the square of the distance. Hence, peaks of co-methylation appearing at larger distances cannot be explained by these mechanisms.
For the MW and WM types of co-methylation, we observed peaks of occurrences at distances of 6, 9 and 12 bp. To more closely understand the mechanisms behind these distinct preferences in methylation pattern, we investigated the distribution of DNMT3A/3L complexes bound to DNA substrates with pairs of CpG sites in these distances by single molecule SFM imaging. Our co-methylation activity and SFM studies clearly showed a high preference of DNMT3A and DNMT3A/3L for binding to and MW co-methylation of CpG sites separated by 12 bp (Figures 2 and 5), which is fully consistent with the crystal structure and previous biochemical data (22,23). Comparison of the distance preference of DNMT3A with DNMT3A/3L shows that the preference for MW co-methylation is more stringent in the case of DNMT3A/3L. Such enhanced adaptability of DNMT3A versus DNMT3A/3L to target site distances may be related to the fact that in DNMT3A tetramers the DNMT3L subunits, which do not interact with DNA, are replaced by additional DNMT3A subunits that provide additional options for DNA interaction, in particular in the context of larger multimers (17). In previous hairpin bisulfite studies with a repetitive (CGA)9 sequence, three major peaks of DNMT3A catalyzed methylation were observed in the upper and lower DNA strands revealing several MW co-methylation events at distances of 7 bp (16,20). However, due to the repetitive structure of this substrate, the same data also correspond to MW co-methylation at distances of 13 bp, which coincides with the second most preferred distance in the current data for DNMT3A/3L. In this context, it needs to be considered, that in the previous studies only distance steps by multiples of 3 bp could be analyzed (7, 10 or 13 bp) such that the preferred distance of 12 bp was not available.
In addition to the preferential MW co-methylation of CpGs with 12 bp distance, our data unexpectedly revealed preferences of 8-9 bp for MW co-methylation and 5-6 bp for WM co-methylation. Consistent with these findings, our SFM data showed strong peaks of protein occupancy at central positions of D12 as well as D9 substrates (where the paired CpG sites are located), indicating a strong preference for binding to two CpG sites with 12 bp and 9 bp distances. With the D6 substrate (containing a central pair of CpG sites with 6 bp spacing), the peak at 50% DNA length was also present, though reduced, consistent with a lower preference for binding to CpG pairs with 6 bp spacing. Strikingly, in all three substrates an additional peak of protein occupancy was observed at about 25-30% of the DNA length. This region corresponds to the transition from the central (102 bp) CpG free region of all SFM substrates that surrounds the central pair of CpG sites to the rest of the DNA sequence, which contains CpG sites in average density and distances. This observation is consistent with DNMT3A/3L sliding and hopping along the DNA in search of CpG target sites after initial unspecific binding. If a DNMT3A/3L complex binds to the CpG free region of the substrate it has two options: Either it reaches the double CpG site at the center of the DNA fragment or it moves outwards. When it does not find the central CpG sites, the first option to bind a CpG will be at the transition of the CpG free region and normal DNA at ∼25–30% of the DNA length, explaining why protein complexes would be enriched in this region. In addition, two pairs of CpG sites in a distance of 13 bp at 23% and 28% of the length of the substrate could contribute to this peak.
Structural characterization of DNMT3A/3L complexes at the central CpG sites in DNA substrates by single molecule SFM further revealed interesting differences for CpG spacings of 6, 9 and 12 bp (Figure 6). SFM volume analyses showed interaction of a single DNMT3A/3L tetramer with the D12 substrate and bend angle measurements revealed DNA bending by approximately 40° in these complexes, as also seen in the crystal structure (Figure 8A) (22). Notably, DNMT3A/3L complexes with single CpG sites and non-specific DNA (average DNA) also showed similar bending by ∼40°. This suggests that the protein complex dictates bending in the DNA through the protein-DNA interface. DNA bending as an energetic test for the presence of their target sites based on altered DNA flexibility is a commonly applied strategy by proteins (reviews: 42,43). It has been described in particular for DNA repair enzymes, whose target sites typically introduce distortion or destabilization of DNA (30,36,44,45). The importance of DNA bending in the context of recognition of CpG sites has also been demonstrated (46). To analyze the connection of DNA bending, co-methylation of CpG sites, and DNA sequence, we determined the distribution of nucleotides at the center of bending between substrates with 12 bp distance between the CpG sites and observed that a high AT/GC ratio and presence of A-tracts increase MW co-methylation by DNMT3A and DNMT3A/3L (Figure 4). This result can be rationalized, because DNA bending is supported by a high AT/GC ratio and A-tracts are known to induce intrinsic bending (review: 38). Moreover, the DNMT3A-DNA structure shows a minor groove compression in this region, which can be supported by the absence of guanosine, because of its N2-amino group pointing into the minor groove.
The SFM data also revealed a second class of complexes bound to the D12 substrate, which showed volumes about twice the volume of a single tetramer and DNA bending of approximately 80° (Figure 6C). Previous work has shown that DNMT3A/3L binds cooperatively to DNA (16,17,20). Hence, the probability of a second DNMT3A/3L complex binding next to the first one is high (Supplemental Figure S11). Therefore, the large volume/large bending complexes presumably correspond to two tetramers bound to the DNA in a side-by-side arrangement. In this model, the individual DNA bending angles would be expected to be additive after spotting of the DNA on the mica surface, exactly as observed here.
Our SFM data of DNMT3A/3L complexes bound at CpG pairs with 6 and 9 bp spacings revealed large volumes indicating preferential binding of pairs of two DNMT3A/3L heterotetramers at these double CpG sites. Modelling showed that the WM co-methylation in a distance of 6 bp can be explained by two adjacent DNMT3A tetramers, in which each DNMT3A subunit interacts with the DNA. The resulting side-by-side dimer of DNMT3A/3L tetramers has an elongated shape following the DNA in a moderately extended conformation. This interpretation is also consistent with the SFM data that indicate long and low complex geometries on the DNA and strong DNA bending by ∼100° in these complexes. Note that this side-by-side model is similar to the model proposed for the large volume complexes formed on the D12 substrate, which also show strong (additive) bending. However, the pattern of CpG site interaction is different in each case (Figure 8C and Supplemental Figure S11). In the case of two tetramers bound to the D12 substrate, one tetramer interacts with both CpG sites, which are optimally spaced within the tetramer. The second tetramer interacts non-specifically with the DNA. In the case of the two tetramers bound on the D6 substrate, modelling suggests that each tetramer binds to one CpG site with one of its DNMT3A subunits, and to non-specific DNA with the second one. In this model, a loop (R831-K855) that was previously identified to be involved in the multimerization of DNMT3A on DNA (20) is placed at the interface of DNMT3A subunits of adjacent heterotetramers. This suggests direct interactions between the two tetramers. The concomitant conformational rearrangements may further lead to the enhanced DNA bending of ∼100° (slightly stronger bending than the sum from the two individual tetramers) and further supports the differences between D12 and D6 dimers of DNMT3A/3L tetramers.
To understand the mechanism of co-methylation of two CpG sites in 9 bp distances by dimers of DNMT3A/3L tetramers, we developed a structural model, in which two DNMT3A/3L tetramers bind to the DNA with only one DNMT3A subunit of each tetramer (Figure 8B). By this unique way, two CpG sites could approach either other up to 9 bp distance in a geometry consistent with MW co-methylation. This results in a tetramer swap of the bound DNA and very compact complexes in agreement with SFM analyses, which consistently showed very compact complex features with large height and short length for this type of complexes. In addition, these complexes induced strong DNA bending by ∼100° although the two DNA molecules used for the modeling of the central DNA with two CpG sites in 9 bp distance could, in principle, be connected without bending (Figure 8B). However, both DNMT3A/3L tetramers contain one additional DNMT3A subunit that is not involved in the interaction with the central DNA containing the pair of CpG sites. These orphan subunits could bind to the free DNA flanks, potentially forming loops. These additional DNA interactions likely occur asymmetrically in the sense that either the left or the right DNA flank will fold back and form additional interactions with the unbound DNMT3A subunits (dotted lines in Figure 8B), with the concomitant structural adjustments preventing subsequent interaction with the second flank. This process could explain the strong DNA bending of these complexes observed in the SFM images, as well as the high level of compaction of these structures. Of note, the secondary DNA interaction proposed here would lead to a shift of the center of the complex (by several tens of base pairs), which could explain the significantly broader positional distribution of complexes formed at the CpG double site in the D9 substrate (∼10% SD instead of ∼2% for D12 and D6). The observation in our kinetic studies that the 9 bp MW co-methylation peak was much more pronounced with DNMT3A than DNMT3A/3L is also supported in the framework of secondary DNA interactions of the complex, because in DNMT3A complexes additional DNMT3A subunits are replacing DNMT3L at the edges and these are available for DNA interactions (which is not the case for DNMT3L). These additional options for DNA interaction may further stabilize this arrangement and lead to stronger preferences for MW co-methylation of CpG sites with this spacing. However, although the secondary DNA interaction model of the D9 complexes is plausible and in agreement with all biochemical and structural data and it has high explanatory power, further details of these structures need to be experimentally established in future work.
In conclusion, our combined kinetic and SFM study revealed novel dimeric arrangements of DNMT3A or DNMT3A/3L tetramers on the DNA in a side-by-side and tetramer-swap mode. This variability in complex quaternary structures leads to a high adaptability in the interactions of DNMT3A and DNMT3A/3L with DNA, with preferential co-methylation not only of CpG sites at distances of ∼12–13 bp, but also at ∼2-3, ∼5-6 or ∼8-9 bp. These additional modes of DNA interactions explain how DNMT3A and DNMT3A/3L can overcome their in-built structural preference for interaction with CpG sites with 12 bp spacing. Thereby, DNMT3 enzymes can introduce DNA methylation into natural DNA without obvious preferences for CpG sites in a 12 bp spacing and without leaving a 12 bp co-methylation footprint in cellular DNA methylation patterns. The resulting flexibility in target site selection is highly important in vivo for global de novo DNA methylation and essential roles of DNMT3A and DNMT3A/3L in the generation of imprints and in development (4,12,13).
DATA AVAILABILITY
NGS kinetic raw data have been deposited at DaRUS, the Data Repository of the University of Stuttgart (https://doi.org/10.18419/darus-1781). Source files of the MatLab tool for SFM data analysis were uploaded at Open Science Framework at https://osf.io/76e9s/.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge support by the Max Planck-Genome-Centre Cologne in NGS sequencing. The funder had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author Contributions: M.E. conducted all biochemical experiments with DNMT3A and purified all proteins. S.K. conducted the DNMT3A/3L methylation reactions. S.A. prepared the substrate library and the sequencing libraries. S.A. and P.B. conducted the bioinformatic work. M.E. provided DNA and protein samples for the SFM experiments. D.B. conducted all SFM experiments. H.S.H. and K.H. contributed the script for automated SFM protein position analysis. A.J. and I.T. conceived and devised the study. A.J., I.T. and P.B. supervised the work. I.T. and A.J. prepared the manuscript draft and figures. All authors contributed to data interpretation and editing of the manuscript. The final manuscript was approved by all authors.
Contributor Information
Max Emperle, Institute of Biochemistry and Technical Biochemistry, Department of Biochemistry, University of Stuttgart, Stuttgart, Germany.
Disha M Bangalore, Rudolf Virchow Center for Integrative and Translational Bioimaging, University of Würzburg, Würzburg, Germany.
Sabrina Adam, Institute of Biochemistry and Technical Biochemistry, Department of Biochemistry, University of Stuttgart, Stuttgart, Germany.
Stefan Kunert, Institute of Biochemistry and Technical Biochemistry, Department of Biochemistry, University of Stuttgart, Stuttgart, Germany.
Hannah S Heil, Rudolf Virchow Center for Integrative and Translational Bioimaging, University of Würzburg, Würzburg, Germany.
Katrin G Heinze, Rudolf Virchow Center for Integrative and Translational Bioimaging, University of Würzburg, Würzburg, Germany.
Pavel Bashtrykov, Institute of Biochemistry and Technical Biochemistry, Department of Biochemistry, University of Stuttgart, Stuttgart, Germany.
Ingrid Tessmer, Rudolf Virchow Center for Integrative and Translational Bioimaging, University of Würzburg, Würzburg, Germany.
Albert Jeltsch, Institute of Biochemistry and Technical Biochemistry, Department of Biochemistry, University of Stuttgart, Stuttgart, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft [JE 252/6, JE 252/15, TE671/4-2]. The open access publication charge for this paper has been waived by Oxford University Press - NAR Editorial Board members are entitled to one free paper per year in recognition of their work on behalf of the journal.
Conflict of interest statement. None declared.
REFERENCES
- 1.Jones P.A.Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012; 13:484–492. [DOI] [PubMed] [Google Scholar]
- 2.Bergman Y., Cedar H.. DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol. 2013; 20:274–281. [DOI] [PubMed] [Google Scholar]
- 3.Schubeler D.Function and information content of DNA methylation. Nature. 2015; 517:321–326. [DOI] [PubMed] [Google Scholar]
- 4.Okano M., Bell D.W., Haber D.A., Li E.. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999; 99:247–257. [DOI] [PubMed] [Google Scholar]
- 5.Jeltsch A., Jurkowska R.Z.. Allosteric control of mammalian DNA methyltransferases - a new regulatory paradigm. Nucleic Acids Res. 2016; 44:8556–8575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gowher H., Jeltsch A.. Mammalian DNA methyltransferases: new discoveries and open questions. Biochem. Soc. Trans. 2018; 46:1191–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeng Y., Chen T.. DNA methylation reprogramming during mammalian development. Genes. 2019; 10:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Z., Zhang Y.. Role of mammalian DNA methyltransferases in development. Annu. Rev. Biochem. 2020; 89:135–158. [DOI] [PubMed] [Google Scholar]
- 9.Yang L., Rau R., Goodell M.A.. DNMT3A in haematological malignancies. Nat. Rev. Cancer. 2015; 15:152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamidi T., Singh A.K., Chen T.. Genetic alterations of DNA methylation machinery in human diseases. Epigenomics. 2015; 7:247–265. [DOI] [PubMed] [Google Scholar]
- 11.Bourc’his D., Xu G.L., Lin C.S., Bollman B., Bestor T.H.. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001; 294:2536–2539. [DOI] [PubMed] [Google Scholar]
- 12.Hata K., Okano M., Lei H., Li E.. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development. 2002; 129:1983–1993. [DOI] [PubMed] [Google Scholar]
- 13.Kaneda M., Okano M., Hata K., Sado T., Tsujimoto N., Li E., Sasaki H.. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004; 429:900–903. [DOI] [PubMed] [Google Scholar]
- 14.Gowher H., Jeltsch A.. Molecular enzymology of the catalytic domains of the Dnmt3a and Dnmt3b DNA methyltransferases. J. Biol. Chem. 2002; 277:20409–20414. [DOI] [PubMed] [Google Scholar]
- 15.Jia D., Jurkowska R.Z., Zhang X., Jeltsch A., Cheng X.. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007; 449:248–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jurkowska R.Z., Anspach N., Urbanke C., Jia D., Reinhardt R., Nellen W., Cheng X., Jeltsch A.. Formation of nucleoprotein filaments by mammalian DNA methyltransferase Dnmt3a in complex with regulator Dnmt3L. Nucleic Acids Res. 2008; 36:6656–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jurkowska R.Z., Rajavelu A., Anspach N., Urbanke C., Jankevicius G., Ragozin S., Nellen W., Jeltsch A.. Oligomerization and binding of the Dnmt3a DNA methyltransferase to parallel DNA molecules: heterochromatic localization and role of Dnmt3L. J. Biol. Chem. 2011; 286:24200–24207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen T.V., Yao S., Wang Y., Rolfe A., Selvaraj A., Darman R., Ke J., Warmuth M., Smith P.G., Larsen N.A.et al.. The R882H DNMT3A hot spot mutation stabilizes the formation of large DNMT3A oligomers with low DNA methyltransferase activity. J. Biol. Chem. 2019; 294:16966–16977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kareta M.S., Botello Z.M., Ennis J.J., Chou C., Chedin F.. Reconstitution and mechanism of the stimulation of de novo methylation by human DNMT3L. J. Biol. Chem. 2006; 281:25893–25902. [DOI] [PubMed] [Google Scholar]
- 20.Rajavelu A., Jurkowska R.Z., Fritz J., Jeltsch A.. Function and disruption of DNA methyltransferase 3a cooperative DNA binding and nucleoprotein filament formation. Nucleic Acids Res. 2012; 40:569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou L., Cheng X., Connolly B.A., Dickman M.J., Hurd P.J., Hornby D.P.. Zebularine: a novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J. Mol. Biol. 2002; 321:591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z.M., Lu R., Wang P., Yu Y., Chen D., Gao L., Liu S., Ji D., Rothbart S.B., Wang Y.et al.. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature. 2018; 554:387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao L., Anteneh H., Song J.. Dissect the DNMT3A- and DNMT3B-mediated DNA Co-methylation through a covalent complex approach. J. Mol. Biol. 2020; 432:569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emperle M., Rajavelu A., Kunert S., Arimondo P.B., Reinhardt R., Jurkowska R.Z., Jeltsch A.. The DNMT3A R882H mutant displays altered flanking sequence preferences. Nucleic Acids Res. 2018; 46:3130–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao L., Emperle M., Guo Y., Grimm S.A., Ren W., Adam S., Uryu H., Zhang Z.M., Chen D., Yin J.et al.. Comprehensive structure-function characterization of DNMT3B and DNMT3A reveals distinctive de novo DNA methylation mechanisms. Nat. Commun. 2020; 11:3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dukatz M., Adam S., Biswal M., Song J., Bashtrykov P., Jeltsch A.. Complex DNA sequence readout mechanisms of the DNMT3B DNA methyltransferase. Nucleic Acids Res. 2020; 48:11495–11509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adam S., Anteneh H., Hornisch M., Wagner V., Lu J., Radde N.E., Bashtrykov P., Song J., Jeltsch A.. DNA sequence-dependent activity and base flipping mechanisms of DNMT1 regulate genome-wide DNA methylation. Nat. Commun. 2020; 11:3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buechner C.N., Tessmer I.. DNA substrate preparation for atomic force microscopy studies of protein-DNA interactions. J. Mol. Recognit. 2013; 26:605–617. [DOI] [PubMed] [Google Scholar]
- 29.Bangalore D.M., Tessmer I.. Unique insight into protein-DNA interactions from single molecule atomic force microscopy. Aims Biophys. 2018; 5:194–216. [Google Scholar]
- 30.Bangalore D.M., Heil H.S., Mehringer C.F., Hirsch L., Hemmen K., Heinze K.G., Tessmer I.. Automated AFM analysis of DNA bending reveals initial lesion sensing strategies of DNA glycosylases. Sci. Rep. 2020; 10:15484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emperle M., Rajavelu A., Reinhardt R., Jurkowska R.Z., Jeltsch A.. Cooperative DNA binding and protein/DNA fiber formation increases the activity of the Dnmt3a DNA methyltransferase. J. Biol. Chem. 2014; 289:29602–29613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gowher H., Liebert K., Hermann A., Xu G., Jeltsch A.. Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J. Biol. Chem. 2005; 280:13341–13348. [DOI] [PubMed] [Google Scholar]
- 33.Afgan E., Baker D., van den Beek M., Blankenberg D., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Eberhard C.et al.. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016; 44:W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang C.C., Meng E.C., Morris J.H., Pettersen E.F., Ferrin T.E.. Enhancing UCSF chimera through web services. Nucleic Acids Res. 2014; 42:W478–W484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rill N., Mukhortava A., Lorenz S., Tessmer I.. Alkyltransferase-like protein clusters scan DNA rapidly over long distances and recruit NER to alkyl-DNA lesions. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:9318–9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beckwitt E.C., Jang S., Carnaval Detweiler I., Sauer F., Simon N., Bretzler J., Watkins S.C., Carell T., Kisker C.et al.. Single molecule analysis reveals monomeric XPA bends DNA and undergoes episodic linear diffusion during damage search. Nat. Commun. 2020; 11:1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emperle M., Adam S., Kunert S., Dukatz M., Baude A., Plass C., Rathert P., Bashtrykov P., Jeltsch A.. Mutations of R882 change flanking sequence preferences of the DNA methyltransferase DNMT3A and cellular methylation patterns. Nucleic Acids Res. 2019; 47:11355–11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allemann R.K., Egli M.. DNA recognition and bending. Chem. Biol. 1997; 4:643–650. [DOI] [PubMed] [Google Scholar]
- 39.Anteneh H., Fang J., Song J.. Structural basis for impairment of DNA methylation by the DNMT3A R882H mutation. Nat. Commun. 2020; 11:2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Halford S.E., Szczelkun M.D.. How to get from A to B: strategies for analysing protein motion on DNA. Eur. Biophys. J. 2002; 31:257–267. [DOI] [PubMed] [Google Scholar]
- 41.Halford S.E., Marko J.F.. How do site-specific DNA-binding proteins find their targets. Nucleic Acids Res. 2004; 32:3040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garvie C.W., Wolberger C.. Recognition of specific DNA sequences. Mol. Cell. 2001; 8:937–946. [DOI] [PubMed] [Google Scholar]
- 43.Rohs R., Jin X., West S.M., Joshi R., Honig B., Mann R.S.. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010; 79:233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang W.Structure and mechanism for DNA lesion recognition. Cell Res. 2008; 18:184–197. [DOI] [PubMed] [Google Scholar]
- 45.LeBlanc S.J., Gauer J.W., Hao P., Case B.C., Hingorani M.M., Weninger K.R., Erie D.A.. Coordinated protein and DNA conformational changes govern mismatch repair initiation by MutS. Nucleic Acids Res. 2018; 46:10782–10795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pan H., Bilinovich S.M., Kaur P., Riehn R., Wang H., Williams D.C. Jr. CpG and methylation-dependent DNA binding and dynamics of the methylcytosine binding domain 2 protein at the single-molecule level. Nucleic Acids Res. 2017; 45:9164–9177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NGS kinetic raw data have been deposited at DaRUS, the Data Repository of the University of Stuttgart (https://doi.org/10.18419/darus-1781). Source files of the MatLab tool for SFM data analysis were uploaded at Open Science Framework at https://osf.io/76e9s/.