

NMR and MD simulations provide insights into the mechanisms governing activation and substrate selectivity of Alkbh5.
Abstract
Alkbh5 catalyzes demethylation of the N6-methyladenosine (m6A), an epigenetic mark that controls several physiological processes including carcinogenesis and stem cell differentiation. The activity of Alkbh5 comprises two coupled reactions. The first reaction involves decarboxylation of α-ketoglutarate (αKG) and formation of a Fe4+═O species. This oxyferryl intermediate oxidizes the m6A to reestablish the canonical base. Despite coupling between the two reactions being required for the correct Alkbh5 functioning, the mechanisms linking dioxygen activation to m6A binding are not fully understood. Here, we use solution NMR to investigate the structure and dynamics of apo and holo Alkbh5. We show that binding of m6A to Alkbh5 induces a metal-centered rearrangement of αKG that increases the exposed area of the metal, making it available for binding O2. Our study reveals the molecular mechanisms underlying activation of Alkbh5, therefore opening new perspectives for the design of novel strategies to control gene expression and cancer progression.
INTRODUCTION
AlkB homolog 5 (Alkbh5) is an essential regulator of RNA epigenetics that controls several metabolic pathways by modulating transcription, translation, and cellular localization of RNA (1–5). Overexpression of Alkbh5 links to development of various types of cancer, including leukemia, breast, and brain cancer (6–8). Deletion of the Alkbh5 gene or inactivation of the enzyme via mutations or small-molecule inhibitors increases the sensitivity of tumors to immunotherapy (9). Therefore, Alkbh5 is actively researched to understand the fundamental mechanisms regulating gene expression and as a promising therapeutic target for anticancer treatments.
Alkbh5 belongs to the nonheme Fe2+ and α-ketoglutarate (αKG)–dependent AlkB dioxygenases and catalyzes oxidative demethylation of N6-methyladenosine (m6A) residues in single-stranded nucleic acids (10) with little to no activity against other modifications (11, 12). Biochemical and structural studies have led to a consensus mechanism for this family of dioxygenases that involves ordered binding of cofactor and substrate molecules and two coupled reactions (13–15). In particular, binding of the metal cofactor (Fe2+) and of the secondary substrate (αKG) precedes binding of the primary substrate (the m6A-containing oligonucleotide for Alkbh5) to the enzyme. Once the catalytic complex is fully assembled, binding of O2 to Fe2+ triggers rapid decarboxylation of αKG to generate carbon dioxide, succinate, and a reactive oxyferryl (Fe4+═O) intermediate that oxidizes the primary substrate to generate the unmodified nucleotide (Fig. 1A). While it is established that the highly reactive and potentially cytotoxic oxyferryl intermediate cannot form in the absence of the primary substrate (16, 17), the mechanisms coupling the binding of the primary substrate to dioxygen activation are not fully understood, and it is not clear whether all αKG dioxygenases share a common activation pathway.
Fig. 1. Postulated Alkbh5 activation pathway.
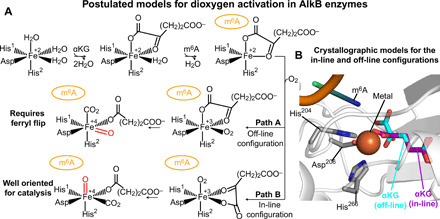
(A) Postulated paths for formation of the catalytically competent oxyferryl species. (B) Close-up view of the active site from the crystal structure of Alkbh5 in complex with Mn2+ (orange sphere) and off-line αKG (cyan and red sticks) [Protein Data Bank (PDB) code: 4NRO]. The side chains forming the 2-His-1-carboxylate facial-triad are shown as sticks (carbon, nitrogen, and oxygen atoms are colored gray, blue, and red, respectively). The in-line αKG configuration (purple and red sticks) was modeled in the active site based on the x-ray structure of Alkbh2 in complex with the primary and secondary substrate (PDB code: 3RZJ). An m6A-containing oligonucleotide was modeled in the active site (orange, blue, and green cartoon) based on the x-ray structure of AlkB in complex with the primary and secondary substrate (PDB code: 4NID).
In the complex formed by AlkB dioxygenases with Fe2+ and αKG, the metal binding site is provided by a 2-His-1-carboxylate facial-triad motif (18) formed by the side chains of His204, His266, and Asp206 in Alkbh5 (19). αKG is directly coordinated to the Fe2+ center in an off-line geometry, in which the C2 carbonyl oxygen is positioned trans with respect to the carboxylate group of the facial-triad motif (Asp206 in Alkbh5) (Fig. 1, A and B) (19). Binding of the primary substrate displaces the water molecule occupying the sixth coordination site, enabling the metal center to bind and activate dioxygen for catalysis (20–22). If the O2 molecule binds at the position previously occupied by the water molecule (path A in Fig. 1A), the subsequently formed Fe4+═O species points away from the primary substrate (Fig. 1), and a structural rearrangement (commonly referred to as “ferryl flip”) is required for efficient catalysis. Alternatively, αKG can transition from the off-line to the in-line geometry before O2 binding to the metal site (path B in Fig. 1A). Such metal-centered rearrangement allows formation of the reactive Fe4+═O adjacent to the primary substrate and ready for catalysis. Although computational studies indicate the latter mechanism (path B in Fig. 1A) to be energetically favored (23, 24), analysis of the crystal structures reported for AlkB dioxygenases in complex with their primary and secondary substrates reveals contrasting results. While the structure of Alkbh2 in complex with an m6A-containing nucleotide shows αKG in the in-line configuration compatible with path B (25), αKG adopts the off-line configuration in the complex of AlkB and FTO with their primary substrates (note that an αKG analog is used for FTO) (Fig. 1B) (16, 17, 26–28), suggesting that dioxygen activation might follow path A in these enzymes.
Here, we investigate the structure and dynamics of the apo and holo human Alkbh5 by solution nuclear magnetic resonance (NMR) and molecular dynamics (MD) simulations. We show that addition of the secondary and primary substrates introduces a progressive quenching of picosecond-nanosecond (ps-ns) and microsecond-millisecond (μs-ms) conformational dynamics, confirming that modulation of intramolecular flexibility plays an important role in regulating the sequential binding of substrate and cofactor molecules to AlkB enzymes (29, 30). We show that binding of the primary substrate introduces a rearrangement within the αKG binding site that is consistent with the conformational transition from the off-line to the in-line configuration. Such conformational switch, which is not triggered by binding of oligonucleotides containing the unmodified A or other methyl modifications, effectively exposes the metal center, making it available for binding O2. Our study reveals the molecular mechanism of Alkbh5 activation and highlights the ability of solution NMR spectroscopy to detect and characterize structural heterogeneity at protein active sites, which is critical for understanding functional regulation of complex enzymes (31–33).
RESULTS
Identification of an NMR-friendly divalent cation
As all other members of the AlkB dioxygenase family, Alkbh5 requires Fe2+ to bind the primary and secondary substrate (13–15). However, other divalent cations such as Mn2+ and Co2+ are commonly used in structural and thermodynamic studies on AlkB enzymes (16, 17, 19, 26–28). These metals preserve the octahedral geometry typical of Fe2+, are more stable in solution, and do not activate dioxygen for catalysis, therefore facilitating structural investigation of the holo enzyme. Nonetheless, Fe2+, Mn2+, and Co2+ are not ideal metals for NMR studies on AlkB dioxygenases, as their paramagnetism results in extensive line broadening and complete loss of the NMR signals from the protein active site. To overcome this limitation, we have evaluated the ability of diamagnetic cations such as Zn2+, Cd2+, and Mg2+ to stabilize the binding of αKG and an m6A-containing oligonucleotide to Alkbh5 by isothermal titration calorimetry (ITC) and pull-down experiments, respectively. As a reference, the same experiments were also acquired in the presence of Mn2+, which was shown to bind to AlkB enzymes with the same geometry as the physiological Fe2+ (28).
Analysis of the ITC data returned equilibrium dissociation constants (Kd) for the Alkbh5-αKG complex of 5.9 ± 0.4, 4.0 ± 0.4, or 2.4 ± 0.3 μM when the metal cofactor is Mn2+, Zn2+, or Cd2+, respectively (Fig. 2A and Table 1). No binding was detected in the presence of Mg2+ (Fig. 2A). Results of the pull-down assay (Fig. 2B) indicate that Alkbh5 binds the primary substrate in the presence of either Mn2+ or Zn2+ (lanes 2 and 4 in Fig. 2B) but fails to recognize the m6A-containing nucleotide when Cd2+ or Mg2+ are used as the metal cofactors (lanes 7 and 9 in Fig. 2B), indicating that the latter two cations are not suitable diamagnetic analogs of Fe2+ for NMR studies on Alkbh5. The enzyme is not able to recognize the primary substrate in the absence of αKG, as evidenced by the lack of a protein band in the pull-down assay run in the presence of Mn2+ and Zn2+ without αKG (lanes 1 and 3 in Fig. 2B). This observation is consistent with the sequential binding model in which binding of the metal cofactor and αKG precedes binding of the primary substrate to the enzyme (13–15).
Fig. 2. Identification of an NMR-friendly divalent cation.
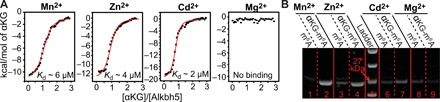
(A) ITC of the interaction between αKG and Alkbh5 in the presence of Mn2+ (panel 1), Zn2+ (panel 2), Cd2+ (panel 3), and Mg2+ (panel 4) obtained at pH 6.0 and 25°C. Integrated heats of injection (black squares) and least-squares best-fit curves (red lines) derived from a simple one-site binding model are shown. The equilibrium dissociation constants and thermodynamic parameters are given in Table 1. (B) Pull-down assay on the interaction between Alkbh5 and a DNA oligonucleotide containing the m6A modification. Lanes 1, 3, 6, and 8 show results for the pull-downs acquired in the presence of Mn2+, Zn2+, Cd2+, and Mg2+, respectively, and in the absence of αKG. Lanes 2, 4, 7, and 9 show results for the pull-downs acquired in the presence of Mn2+, Zn2+, Cd2+, and Mg2+, respectively, and in the presence of αKG. Lane 5 contains a molecular weight marker.
Table 1. Equilibrium dissociation constants and thermodynamic parameters for the Alkbh5-αKG complex.
| Divalent cation | Kd (μM) | ΔH (kcal mol−1) | ΔS (cal mol−1 K−1) |
| Mn2+ | 5.9 ± 0.4 | −10.7 ± 0.2 | −11.9 ± 0.8 |
| Zn2+ | 4.0 ± 0.4 | −13.1 ± 0.3 | −19.4 ± 1.1 |
| Cd2+ | 2.4 ± 0.3 | −20.4 ± 0.5 | −42.0 ± 1.8 |
In summary, the data reported here indicate that replacing Mn2+ with Zn2+ does not alter substantially the ability of Alkbh5 to bind its primary and secondary substrate and that the sequential binding of αKG and m6A, which is a hallmark of AlkB enzymes, is fully retained by Alkbh5 in the presence of Zn2+. In addition, in the presence of Zn2+, Alkbh5 is able to recognize m6A-containing oligonucleotides over oligonucleotides incorporating the canonical A or other methylated bases (see below). Therefore, Zn2+ is a suitable diamagnetic cation for investigating the holo form of Alkbh5 by solution NMR spectroscopy. Notably, Zn2+ has been previously used as a diamagnetic analog of Fe2+ for structural and thermodynamics studies on the bacterial enzyme AlkB and its complexes (29).
Effect of the primary and secondary substrates on the NMR spectra of Alkbh5
We have recently reported the assignment of the 1H-15N and 1H-13Cmethyl transverse relaxation–optimized spectroscopy (TROSY) spectra of the apo form of human Alkbh5 at pH 7.4 (34, 35). In the present work, solution NMR data are measured at pH 6.0 to increase the solubility of the metal cofactor. Backbone resonance assignments for apo Alkbh5 and for its complexes with αKG and αKG/m6A were obtained at pH 6.0 by using conventional triple resonance NMR experiments. In particular, the 5-mer DNA 5′-GG(m6A)CT-3′ was used as the primary substrate in this work because previous studies have shown that Alkbh5 demethylates this oligonucleotide with the same turnover (kcat ~ 0.15 min−1) and Michaelis constant (Km ~ 2.5 μM) of its RNA counterpart (36, 37). In total, 93, 92, and 79% of the expected protein backbone resonances were assigned for Alkbh5, Alkbh5-αKG, and Alkbh5-αKG/m6A, respectively. The assigned chemical shifts are consistent with the secondary structure of Alkbh5 revealed by x-ray crystallography (fig. S1) and were deposited on the Biological Magnetic Resonance Data Bank (accession numbers 50820, 50821, and 50831 for Alkbh5, Alkbh5-αKG, and Alkbh5-αKG/m6A, respectively).
As expected, analysis of the 1H-15N TROSY spectra indicates that addition of a saturating amount of Zn2+ and αKG to ~0.5 mM Alkbh5 (see Materials and Methods) induces chemical shift perturbations (ΔH/N) within the jelly-roll motif containing the biding site for αKG and the metal cofactor (Fig. 3, A and C). Addition of the primary substrate results in large ΔH/N values in flip 1 (residues 117 to 129), flip 2 (residues 136 to 165), and flip 3 (residues 229 to 242) and, to a minor extent, in the interior of the jelly-roll motif (Fig. 3, B and D). Although an experimental structure of Alkbh5 bound to its primary substrate is not available, our chemical shift perturbations are in good agreement with mutagenesis studies, suggesting that several residues from the flip regions and residues 209 and 210 from the jelly-roll motif are directly involved in recognition of the methylated oligonucleotide (19, 38).
Fig. 3. Solution NMR of human Alkbh5.
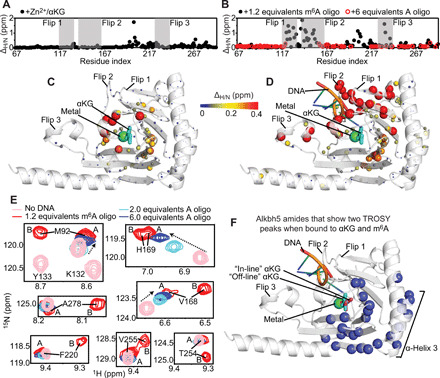
(A) ΔH/N’s induced by a saturating concentration of Zn2+ and αKG on the 1H-15N TROSY spectrum of Alkbh5 are plotted versus the residue index. (B) The ΔH/N’s measured upon addition of 1.2 equivalents of 5′-GG(m6A)CT-3′ (black circles) or 6 equivalents of 5′-GGACT-3′ (red circles) to Alkbh5-αKG are plotted versus the residue index. Flips 1, 2, and 3 are highlighted by gray boxes. (C) ΔH/N’s induced by addition of Zn2+ and αKG to Alkbh5 are plotted on the structure of Alkbh5. (D) ΔH/N’s induced by addition of 5′-GG(m6A)CT-3′ to Alkbh5-αKG are plotted on the structure of Alkbh5. The relationship between ΔH/N and sphere color and size is depicted by the color bar. (E) Selected region of the 1H-15N TROSY spectrum showing the splitting of some NMR resonances into two peaks upon addition of 5′-GG(m6A)CT-3′. The spectrum acquired in the absence of DNA (but in the presence of Zn2+ and αKG) is colored pink. The spectrum measured at a saturating concentration of 5′-GG(m6A)CT-3′ is colored red. Spectra acquired at 1 mM and 3 mM 5′-GGACT-3′ are colored cyan and blue, respectively. The NMR peaks assigned to states A and B are labeled on the NMR spectra of the Alkbh5-αKG/m6A complex. Populations of 51 ± 10 and 49 ± 10% were calculated for states A and B, respectively, from integration of the NMR cross peaks. (F) The 28 residues for which two separate NMR correlations are detected in the 1H-15N TROSY spectrum of the Alkbh5-αKG/m6A complex are shown as blue spheres on the structure of Alkbh5.
Notably, while performing resonance assignment on the Alkbh5-αKG/m6A complex, we noticed that 28 protein residues (I89, Q91, M92, R93, L94, E109, E166, W167, V168, H169, Q170, Q174, L176, E178, D194, Y195, V218, F220, F221, S224, L226, V248, T254, V255, L256, S257, Y259, and A278) could be assigned to two different peaks in the 1H-15N TROSY spectrum (i.e., for each residue, we have two possible assignments that can satisfy the experimental NMR data) (Fig. 3E), as each pair of 1H-15N TROSY peaks in this group shares similar 13C connectivity in the set of triple resonance experiments acquired for the resonance assignment (i.e., the two NH peaks correlate to the same Cα, Cβ, Cα′, Cβ′, and CO′, where ′ indicates the preceding residue in the amino acid sequence). These 28 residues cluster in the vicinity of the αKG molecule and on the α-helix 3 (residues 166 to 181) that is packed against the jelly-roll motif (Fig. 3F), suggesting that the αKG binding site in the Alkbh5-αKG/m6A complex exists in two different states with different NMR chemical shifts. As two sets of peaks of similar intensity are observed in the 1H-15N TROSY spectrum, the two states have similar abundance in the NMR sample and exchange on the slow time scale regime.
The interaction that the enzyme establishes with nucleic acids has been further characterized by investigating the effect of the unmodified 5′-GGACT-3′ oligonucleotide on the 1H-15N TROSY spectrum of the Alkbh5-αKG complex. Addition of the unmodified DNA to the NMR sample causes line broadening beyond detection level for all the NMR signals that experienced large ΔH/N values upon addition of the m6A-containing oligonucleotide (Fig. 3B). This observation implies that the exchange rate between the free and DNA-bound states of Alkbh5 is similar to the value of ΔH/N measured (in rad s−1) for these peaks. However, NMR signals whose chemical shift is affected to a lower extent by the DNA are still visible in the presence of the unmodified oligonucleotide (Fig. 3E). For such signals, the binding kinetics are on the fast time scale regime, and their chemical shift can be monitored to report on saturation of the enzyme-DNA complex during the NMR titration experiment (Fig. 3E) (39). As expected, the unmodified DNA has a lower affinity for Alkbh5 than its methylated analog, and a 6:1 DNA-to-protein ratio is required to nearly saturate binding (Fig. 3E). Only one set of NMR peaks is observed for the jelly-roll motif and α-helix 3, indicating that the αKG binding site is present in one single configuration when Alkbh5 is bound to the unmodified DNA. The latter conformation will be referred to as state A. The alternative configuration, accessible by the αKG binding site when Alkbh5 is bound to the methylated oligonucleotide, will be referred to as state B. Assignment of the 1H-15N TROSY peaks of states A and B was performed by comparison of the NMR spectra measured for the Alkbh5 complexes with methylated and unmethylated DNA, as shown in Fig. 3E. It is also important to notice that the presence of the unmodified DNA causes line broadening beyond detection level at residues Phe134, Phe135, Gly136, Glu137, Arg149, Gly149, Ile208, His209, Ile210, and Arg213 for which ΔH/N ~ 0 ppm (parts per million) was measured upon addition of m6A (Fig. 3B). Because these residues are localized within flip 2 or are directly facing flip 2, these data suggest that the interaction with the unmodified DNA perturbs the conformational dynamics of this long loop, further contributing to the observed line broadening.
We have also investigated the ability of other methylated nucleotides to generate a double conformation in the αKG binding site by measuring the perturbations produced by the 5-mer DNAs 5′-GG(m1A)CT-3′, 5′-GG(m3T)CT-3′, and 5′-GG(m5C)CT-3′ (where m1A, m3T, and m5C are N1-methyladenosine, N3-methylthymine, and 5-methylcytosine, respectively) on the 1H-15N TROSY spectrum of Alkbh5. Although none of these oligonucleotides bind to Alkbh5 with sufficient affinity to saturate binding at a DNA-to-protein ratio of 6:1 (fig. S2), several conclusions on the Alkbh5-DNA interaction can be drawn from these experiments. The fact that many NMR signals from the flip regions disappear upon addition of the three oligonucleotides (fig. S2) indicates that flips 1, 2, and 3 are capable of establishing weak interactions with the DNA irrespective of the presence of an m6A residue in the sequence. In addition, while binding of Alkbh5 to a 5-mer DNA containing the unmodified A or the m6A nucleotide generates chemical shift perturbations that extend all the way to α-helix 3 (Fig. 3E and fig. S2), oligonucleotides containing m1A, m3T, and m5C only perturb the NMR signals originating from the flip regions and, to a minor extent, the jelly-roll motif (fig. S2), suggesting that the latter three DNAs produce only minor and localized perturbations on the Alkbh5 structure.
As a final note, it is important to highlight that the 1H-15N TROSY spectrum acquired for the Alkbh5-αKG/m6A complex is of lower quality (in terms of both signal overlap and signal-to-noise ratio) than the ones measured for the apo protein and the Alkbh5-αKG complex (fig. S3). This reduced spectral quality diminishes the number of NMR peaks that can be unambiguously assigned and used to measure NMR relaxation and residual dipolar coupling (RDC) parameters for the DNA-bound state (see below).
An m6A-induced metal-centered transition
Analysis of the solution NMR data acquired on the Alkbh5-αKG/m6A complex indicates that the αKG binding site exists in two equally populated states (referred to as states A and B) with different 1H-15N chemical shifts. The two states share identical Cα and Cβ chemical shifts, suggesting that they do not originate from a transition in the backbone structure but are rather the result of a conformational change involving αKG and the surrounding side chains. Here, we hypothesize that states A and B correspond to the off-line and in-line configurations of αKG observed in the crystal structures of AlkB and Alkbh2, respectively, bound to their primary and secondary substrates.
To investigate the stability of these configurations in Alkbh5, 1.2-μs MD simulations of the Alkbh5-αKG and Alkbh5-αKG/m6A complexes were run with Fe2+ as the metal center and αKG either in the off-line or in the in-line conformation. The starting structure for Alkbh5-αKG/m6A in the off-line geometry was prepared by modeling the 5-mer oligonucleotide used for solution NMR (see above) into the crystal structure of the Alkbh5-αKG complex [Protein Data Bank (PDB) code: 4NRO]. The in-line geometry of αKG was modeled on the basis of the crystal structure of Alkbh2 in complex with αKG and the primary substrate (PDB code: 3RZJ) (more details on the modeling and equilibration of the starting structures are provided in Materials and Methods). Analysis of the MD simulations on the Alkbh5-αKG/m6A complex indicates that the metal coordination geometry of αKG is stable in both trajectories, with the dihedral angle formed by Nε1 of H204 and O1, C1, and C2 of αKG fluctuating around 44 ± 8° and −50 ± 9° for the off-line and in-line geometry, respectively (Fig. 4, A and B). On the other hand, only the off-line configuration provides a stable metal coordination geometry in the MD simulations of the Alkbh5-αKG complex (Fig. 4, A and B), suggesting that the methylated oligonucleotide is absolutely required to provide structural stabilization to the in-line form of αKG.
Fig. 4. Structural stability of off-line and in-line αKG.
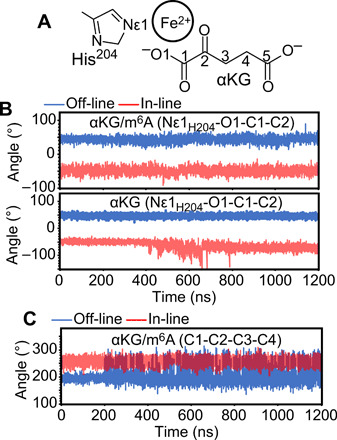
(A) Schematic representation of αKG, the metal center, and His204 from the Alkbh5 active site with specified atom numbering and nomenclature. (B) Evolution of the dihedral angle formed by Nε1 of H204 and O1, C1, and C2 of αKG during the 1.2-μs MD simulations run on the Alkbh5-αKG/m6A (top) and Alkbh5-αKG (bottom) complexes. (C) Evolution of the dihedral angle formed by C1, C2, C3, and C4 of αKG during the 1.2-μs MD simulations run on the Alkbh5-αKG/m6A complex. Results from the simulations started with αKG in the off-line or in-line configurations are shown in blue and red, respectively.
Analysis of the MD simulations on the Alkbh5-αKG/m6A complex also reveals that in-line αKG is more conformationally restrained than off-line αKG. The C1-C2-C3-C4 dihedral angle of αKG experiences larger fluctuations when off-line (Fig. 4, A and C), indicating that the in-line configuration results in a more efficient packing of the jelly-roll motif around the αKG molecule compared to the off-line configuration. The observation that the jelly-roll motif has different structural properties when αKG is off-line or in-line agrees well with our NMR data, indicating that the residues displaying different 1H-15N TROSY signals for states A and B are spread throughout the jelly-roll motif (Fig. 3F).
Overall, the results of the MD study support our hypothesis that the two states identified by NMR correspond to the off-line and in-line geometries of αKG. In particular, state A, which is the state adopted by αKG in the presence of unmodified DNA, corresponds to the off-line configuration observed in the crystal structure of Alkbh5-αKG. State B, which is only accessible in the presence of the m6A-containing nucleotide, corresponds to the in-line configuration.
Effect of the primary and secondary substrates on the conformational dynamics of Alkbh5
We have recently shown that the active site and the N-terminal region of apo Alkbh5 experience conformational dynamics on both the ps-ns and μs-ms time scale (34). Here, the effect of the primary and secondary ligands on the Alkbh5 conformational dynamics is investigated by means of 15N NMR relaxation measurements.
In particular, residue-specific 15N longitudinal (R1) and transverse (R2) relaxation rates were obtained at 800 MHz and 25°C by acquisition of TROSY-detected R1 and R1ρ experiments (40) on U-[2H-15N]–labeled Alkbh5. Analysis of the 15N-R2/R1 ratios is a convenient method to investigate ps-ns dynamics in proteins. For a rigid protein, where global tumbling is the only significant contribution to the ps-ns dynamics, the R2/R1 values are expected to be constant throughout the amino acid sequence and proportional to the rotational correlation time (τc) (41). On the other hand, the presence of flexible structural elements within the protein (such as long and flexible loops) that increase locally the ps-ns dynamics experienced by the backbone amide groups is revealed by a local shift of the R2/R1 ratios toward lower than average values (41). The measured R2/R1 ratios are graphed as a function of the residue index in Fig. 5A and plotted as a gradient on the structure of Alkbh5 in Fig. 5B. The data show a comparable trend for the apo enzyme and for the Alkbh5-αKG and Alkbh5-αKG/m6A complexes, with mean ratios of 40 ± 12, 36 ± 10, and 37 ± 12, respectively. These values translate to τc ~ 15 ns that is consistent with the τc (~16 ns) predicted at 25°C for a globular protein of the size of Alkbh5 using the Stokes’ law. For all three Alkbh5 states, the N-terminal segment (residues 67 to 74) and flip 2 show lower than average R2/R1 values, indicative of enhanced local motion in addition to overall molecular tumbling (Fig. 5, A and B). Noteworthy, the flip 2 region produces average R2/R1 of 16 ± 4, 15 ± 5, and 26 ± 8 for the apo, αKG-bound, and αKG/m6A-bound states, respectively. Such local increase in R2/R1 observed for flip 2 upon addition on nucleic acid suggests that DNA binding reduces the flexibility of this long loop, which is consistent with the direct participation of flip 2 in DNA binding revealed by NMR chemical shift perturbation (Fig. 3A) and mutagenesis experiments (19, 38). In summary, analysis of the R2/R1 data presented here indicates that binding of the metal and αKG has a negligible influence on the ps-ns dynamics of the Alkbh5 backbone, while DNA binding results in rigidification of flip 2. It should also be observed that, in apo and αKG-bound Alkbh5, a lower than average R2/R1 value is measured for Gly198, which is located on a marginal turn of the jelly-roll motif (Fig. 5, A and B). However, this NMR correlation was not assigned in the spectrum of the Alkbh5-αKG/m6A complex, and the effects of nucleic acid biding on the ps-ns dynamics of this residue are unknown.
Fig. 5. Dynamics of Alkbh5 and its complexes.
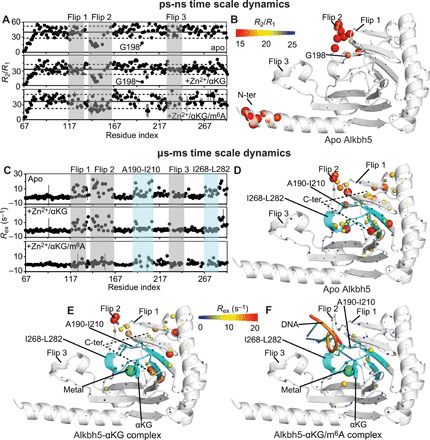
(A) The 15N-R2/R1 ratios measured for apo Alkbh5 (top), Alkbh5-αKG (middle), and Alkbh5-αKG/m6A (bottom) are plotted versus the residue index. The localization of flips 1, 2, and 3 is shown by transparent gray boxes. The average R2/R1 value in each panel is indicated by a horizontal black line. The horizontal dashed lines indicate a displacement of one SD from the average R2/R1 value. (B) The R2/R1 values measured for the apo enzyme are plotted on the crystal structure of Alkbh5. The relationship between experimental values and the color and size of the spheres is provided by the color bar. (C) The 15N-Rex values measured for apo Alkbh5 (top), Alkbh5-αKG (middle), and Alkbh5-αKG/m6A (bottom) are plotted versus the residue index. The localization of fragments 190 to 210 and 268 to 282 from the jelly-roll motif is shown by transparent light blue boxes. The localization of flips 1, 2, and 3 is shown by transparent gray boxes. The Rex values measured for (D) apo Alkbh5, (E) Alkbh5-αKG, and (F) Alkbh5-αKG/m6A are plotted on the crystal structure of the enzyme. The relationship between experimental values and the color and size of the spheres is provided by the color bar. The ribbon of fragments 190 to 210 and 268 to 282 is shown in cyan. Mn2+ and αKG are shown as green spheres and cyan sticks, respectively. The modeled 5′-GG(m6A)CT-3′ DNA is shown as orange, blue, and green cartoon. Fragment 145 to 149 from flip 2 (which is missing in the x-ray structure) is modeled as described in Materials and Methods. Quantitative analysis of the relaxation dispersion data is reported in figs. S4 and S5.
The μs-ms time scale dynamics in Alkbh5 were investigated by measuring the exchange contribution to the 15N-R2 (Rex) by Carr-Purcell-Meiboom-Gill relaxation dispersion NMR experiments (42) acquired at 800 MHz and 25°C. Backbone amides sensing conformational dynamics on the μs-ms time scale produce Rex values significantly larger than 0 s−1 and can be easily identified by plotting the experimental Rex’s versus the residue index (Fig. 5C). In the apo enzyme, large (>10 s−1) Rex values are measured for several amino acids located within flip 1, flip 2, the αKG biding site, and the C-terminal fragment (residues 284 to 292) (Fig. 5, C and D). Addition of Zn2+ and αKG results in a generalized reduction of Rex across the protein (Fig. 5, C and F), indicating that binding of the metal cofactor and of the secondary substrate induces a rigidification of the enzyme structure. Complete quenching of Rex is observed for several amino acids located within the Ala190-Ile210 and Ile268-Leu282 fragments that form part of the αKG binding site and provide the platform for DNA binding to Alkbh5 (Fig. 5, C, F, and G). Last, binding of the methylated nucleic acid results in further stabilization of the Alkbh5 structure, as evidenced by the near-complete quenching of the Rex contributions throughout the entire protein (Fig. 5, C and G). In summary, analysis of the 15N relaxation dispersion experiments indicates that binding of the secondary and primary substrate to Alkbh5 results in a progressive stabilization of the enzyme structure into a well-defined fold, which is competent for catalysis.
Effect of the primary and secondary substrates on the structure of Alkbh5
Analysis of the NMR data indicates that addition of αKG does not alter the 13C chemical shifts of the Alkbh5 backbone and that binding of m6A is associated with slight changes in the Cα secondary chemical shifts for residues located within flips 2 and 3 and for residue 203 from the m6A binding site (Fig. 6A). These data suggest that the interactions with the primary and secondary substrate generate minor and localized changes in the backbone of Alkbh5. To further characterize structural changes associated with substrate binding, we collected backbone amide 1DNH RDC data for the apo Alkbh5 and for the Alkbh5-αKG and Alkbh5-αKG/m6A complexes. Singular value decomposition (SVD) fitting of the full set of 1DNH RDCs to the x-ray coordinates of the Alkbh5-αKG complex (PDB: 2NRO) yielded R-factors of 48% for all the investigated samples (Fig. 6B and Table 2). As previously shown for apo Alkbh5 at pH 7.4 (34), the poor correlation between experimental and back-calculated RDC data originates from the high degree of conformational disorder within the active site that is not properly captured by the static crystallographic structure. Repeating the SVD fits on a subset of experimental RDCs from which all data coming from the active site residues are removed substantially improves the agreement between solution NMR data and crystal structure, returning R-factors of 20, 20, and 21% for apo Alkbh5, Alkbh5-αKG, and Alkbh5-αKG/m6A, respectively (Fig. 6B and Table 2). The fact that the RDCs originating from the jelly-roll motif and the three α helices of apo Alkbh5, Alkbh5-αKG, and Alkbh5-αKG/m6A can be fit using the same crystallographic model confirms that binding of the primary and secondary substrate does not alter the backbone fold and relative orientation of these rigid structural elements.
Fig. 6. Solution structure of apo and holo Alkbh5.
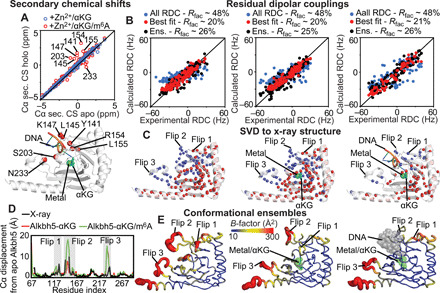
(A) The secondary Cα chemical shifts (CS) measured for Alkbh5-αKG (blue circles) and Alkbh5-αKG/m6A (red circles) are plotted versus the Cα secondary chemical shifts measured for apo Alkbh5 (top). Cα atoms that show a significant change in secondary chemical shift upon addition of m6A are plotted as red spheres on Alkbh5 (bottom). (B) The RDCs back-calculated from the Alkbh5 structure are plotted versus the experimental data. SVD fitting of the full set of RDCs against the x-ray structure is shown as blue circles. SVD fitting performed discarding all data from the active site is shown as red circles. RDCs back-calculated from the conformational ensemble are shown as black circles. Data are shown for apo Alkbh5 (left), Alkbh5-αKG (middle), and Alkbh5-αKG/m6A (right). (C) Structures of Alkbh5 (left), Alkbh5-αKG (middle), and Alkbh5-αKG/m6A (right) showing the amide groups whose experimental RDCs are in good agreement with back-calculated values from the x-ray structure (red spheres). Blue spheres indicate the RDCs removed from the SVD analysis. The lower number of analyzed RDCs for Alkbh5-αKG/m6A reflects the lower quality of the NMR spectrum measured in the presence of methylated oligonucleotide (fig. S3). (D) The average structures of the conformational ensembles are used to calculate the Cα displacement of Alkbh5-αKG (red line) and Alkbh5-αKG/m6A (green line) from the structure of the apo Alkbh5. The Cα displacement of the apo Alkbh5 average structure from the x-ray structure is shown as black line. (E) Conformational ensembles generated for apo Alkbh5 (left), Alkbh5-αKG (middle), and Alkbh5-αKG/m6A (right). Cartoons are colored according to the B-factor.
Table 2. Backbone amide (1DNH) RDC analysis of apo and holo Alkbh5.
| Used RDCs | No. of RDCs | (Hz)* | η* | Rfac (%)† |
| Back-calculation of RDC data from x-ray structure | ||||
| Apo—All RDC | 184‡ | 8.5 | 0.30 | 47.9 |
| Apo—Best fit | 121 | 15.6 | 0.50 | 20.1 |
| Zn2+/αKG—All RDC | 185‡ | 10.4 | 0.32 | 47.7 |
| Zn2+/αKG—Best fit | 121 | 15.5 | 0.51 | 19.8 |
| Zn2+/αKG/m6A—All RDC | 110‡ | 14.0 | 0.63 | 48.0 |
| Zn2+/αKG/m6A—Best fit | 76 | 21.2 | 0.39 | 21.4 |
| Ensemble refinement | ||||
| Apo—All RDC l | 192‡ | – | – | 25.8 |
| Zn2+/αKG—All RDC | 193‡ | – | – | 25.1 |
| Zn2+/αKG/m6A—All RDC | 116‡ | – | – | 25.6 |
* and η are the magnitude and rhombicity of the alignment tensor, respectively.
†The RDC Rfac is given by , where Dobs and Dcalc are the observed and calculated RDCs, respectively (57, 58).
‡More RDCs are used in the ensemble refinement than in SVD, as the data from residues that are missing from the x-ray structure could not be used for the SVD fitting.
To visualize the structural changes in the nucleotide recognition loops associated with substrate binding, we have calculated structural ensembles for the apo Alkbh5 and for the Alkbh5-αKG and Alkbh5-αKG/m6A complexes by coupling the experimental 1DNH RDC data with accelerated MD (aMD) simulations. This protocol has been proven successful in generating MD-derived structural ensembles of dynamical proteins that satisfy solution NMR data (34, 43). Analysis of the changes in R-factor as a function of the ensemble sizes indicates that 13-, 18-, and 13-member ensembles are required to fully satisfy the experimental RDC data measured for apo Alkbh5 (R-factor ~ 26%), Alkbh5-αKG (R-factor ~ 25%), and Alkbh5-αKG/m6A (R-factor ~ 26%), respectively (fig. S4 and Table 2). The modeled structural ensembles are visualized in Fig. 6E and confirm that addition of αKG and m6A causes a progressive quenching of protein conformational dynamics. Analysis of the average structures calculated from the conformational ensembles indicates that structural transitions associated with αKG and m6A binding are localized within the nucleotide recognition loops. While the average structure calculated for apo Alkbh5 closely resembles the crystallographic model of the enzyme (Fig. 6D), the average structure obtained for Alkbh5-αKG and Alkbh5-αKG/m6A show structural rearrangements associated with flip 3 and flips 2 and 3, respectively (Fig. 6D). Note that similar conclusions can be drawn by comparing structures of Alkbh5, Alkbh5-αKG, and Alkbh5-αKG/m6A obtained by direct refinement of the crystallographic structure of apo Alkbh5 against the experimental RDC data (fig. S7).
Overall, the data presented here indicate that addition of the primary and secondary substrate is coupled with small changes in the fold of the Alkbh5 backbone, mostly localized within flips 2 and 3. These conclusions support the hypothesis that the changes in μs-ms dynamics (Fig. 4) and the appearance of two sets of 1H-15N TROSY signals (Fig. 3) caused within the jelly-roll motif by addition of αKG and m6A originate from a conformational change that involves αKG and the surrounding side chains.
DISCUSSION
Structural heterogeneity at the active site is emerging as a ubiquitous phenomenon governing enzymatic activity. Enzymes are dynamic nanomachines that can adopt many conformational states with different catalytic abilities (31–33). For several enzymes, modulation of structural disorder at the active site upon cofactor/substrate binding was shown to control the thermodynamic balance between active and inactive states and to provide an important source of functional regulation (31, 44, 45). In this work, we investigate the molecular mechanisms governing activation of the human RNA demethylase Alkbh5, an oncoprotein that is a promising target for the development of anticancer drugs, by solution NMR spectroscopy and MD simulations.
By using NMR relaxation experiments, we have shown that the active site of apo Alkbh5 is highly disordered and undergoes conformational dynamics on both the ps-ns and μs-ms time scale (Fig. 5). We found that αKG binding to the enzyme does not affect the ps-ns dynamics of the protein backbone (Fig. 5A) but results in quenching of the μs-ms motions of Alkbh5 (Fig. 5C). In particular, the μs-ms dynamics in the Ile268-Leu282 fragment are completely suppressed by αKG (Fig. 5E). As this segment establishes several contacts with the oligonucleotide in the Alkbh5-αKG/m6A complex (Fig. 5F), our data suggest that its structural stabilization in the αKG-bound form promotes optimal docking of m6A onto Alkbh5 and is responsible for orchestrating sequential binding of the secondary and primary substrate to the enzyme. Suppression μs-ms dynamics upon ligand binding was also shown to promote formation of interdomain interactions in bacterial Enzyme I (46), suggesting that ligand-induced suppression of conformational dynamics is a common mechanism that mediates signal transduction in biological systems.
Binding of the primary substrate to Alkbh5 completely abolishes μs-ms time scale motions within the active site of the enzyme (Fig. 5, C and F). However, NMR analysis of the Alkbh5-αKG/m6A complex reveals that the αKG binding site exists as two equally populated species that are in slow exchange on the NMR chemical shift time scale (≥ s) and result in two sets of 1H-15N TROSY peaks (Fig. 3, E and F). Analysis of 1.2-μs MD simulations acquired on the Alkbh5-αKG and Alkbh5-αKG/m6A complexes (Fig. 4) indicates that the two states detected by NMR correspond to the off-line and in-line geometries of αKG (Fig. 1B). Oligonucleotides that do not contain an m6A modification are not able to generate the in-line configuration of αKG (Fig. 3E and fig. S2) and are not demethylated by Alkbh5 (Fig. 7A), suggesting that the off-line to in-line structural transition might be required for activation of Alkbh5. Notably, the transition from the off-line to the in-line configuration of αKG effectively increases the solvent-accessible surface area (SASA) of the metal center from 0.0 to 2.5 Å2 in the Alkbh5-αKG complex and from 0.0 to 1.3 Å2 in the Alkbh5-αKG/m6A complex (Fig. 7B). From these data, one can conclude that the off-line configuration is required to keep the metal center in the inactive +2 oxidation state when the primary substrate is not present in the active site. On the contrary, binding of m6A to Alkbh5 promotes transition of αKG to the in-line geometry, which exposes the metal, making it available to coordinate and activate O2 for catalysis (Fig. 1A, path B).
Fig. 7. The off-line to in-line transition activates Alkbh5.
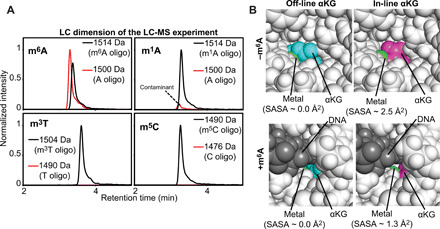
(A) Demethylation activity of Alkbh5 against 5′-GG(m6A)CT-3′ (top left), 5′-GG(m1A)CT-3′ (top right), 5′-GG(m3T)CT-3′ (bottom left), and 5′-GG(m5C)CT-3′ (bottom right). Each panel shows the liquid chromatography (LC) dimension of an LC–mass spectroscopy (MS) experiment run after 60-min incubation of 10 μM substrate with 5 μM Alkbh5, 300 μM αKG, and 150 μM Fe2+ at 25°C (see Materials and Methods for further details). The red and black lines correspond to the elution profiles of the species with molecular masses corresponding to the demethylated and methylated oligonucleotides, respectively. The minor peak at 1500 Da present in the LC-MS data acquired for the 5′-GG(m1A)CT-3′ does not grow with time (i.e., it is present with the same intensity even before addition of Alkbh5 to the reaction mixture) and it was ascribed to the presence of a small contamination of nonmethylated oligonucleotide in the starting sample. (B) Solvent accessibility of the metal cofactor. Close-up view of the active site in (top left) the crystal structure of Alkbh5 bound to off-line αKG, (top right) the modeled structure of Alkbh5 bound to in-line αKG, (bottom left) the modeled structure of Alkbh5 bound to off-line αKG and 5′-GG(m6A)CT-3′, and (bottom right) the modeled structure of Alkbh5 bound to in-line αKG and 5′-GG(m6A)CT-3′. The protein is shown as white spheres. The metal is shown as a green sphere. Off-line and in-line αKG are shown as cyan and purple spheres, respectively. The 5-mer DNA is shown as gray spheres. The SASA of the metal center was calculated using the PyMOL command get_area with dot_solvent = 1 and dot_density = 3. Structural models were generated and equilibrated as described in Materials and Methods.
The observation of two active site configurations with different catalytic abilities calls to mind previous works on the DNA polymerase in which transition to a protein conformation with altered essential motions was observed to trigger catalysis (47, 48). However, this does not seem to be the case for Alkbh5 as (i) principal components analysis of the MD simulations on the Alkbh5-αKG/m6A complex (fig. S8) does not highlight obvious changes in confrontational dynamics at the active site upon transition of αKG to the in-line conformation, and (ii) the NMR signals assigned to Alkbh5 bound to in-line or off-line αKG display similar relaxation parameters.
Last, it is worth noting that our NMR study on the interaction between Alkbh5 and oligonucleotides revealed that while nucleic acids that do not contain the m6A modification are not able to stabilize the in-line αKG configuration and activate the enzyme for catalysis, they are still capable of establishing weak interactions with the nucleotide recognition loops (flips 1, 2, and 3) (Fig. 3E and fig. S2). These weak binding events might facilitate the search for the m6A modification site on long nucleic acids, in a way similar to the target search process, whereby transcription factors locate their specific DNA binding site (49, 50).
In conclusion, our experiments highlight the ability of solution NMR to detect and characterize subtle changes in structure and dynamics at enzyme active sites and suggest that binding of the primary substrate to Alkbh5 is required to promote transition of αKG to the in-line configuration, which, in turn, allows the metal cofactor to bind dioxygen and activate the enzyme for catalysis.
MATERIALS AND METHODS
Protein expression and purification
All experiments were performed using a truncated version of the human Alkbh5 comprising residues 66 to 292. This selected construct has been used in crystallographic studies and was shown to retain the enzymatic activity of the full-length protein (19, 38). Alkbh5 was expressed and purified as previously described (36). The M9 medium for the U-[2H-15N]–labeled sample was prepared in 99.9% 2H2O using 15NH4Cl as the sole nitrogen source. For U-[2H-15N-13C]–Alkbh5, the M9 medium was prepared in 99.9% 2H2O using 15NH4Cl and 2H,13C-glucose as the sole nitrogen and carbon sources, respectively.
Isothermal titration calorimetry
ITC experiments on the Alkbh5-αKG interactions were acquired using a previously described protocol for AlkB dioxygenases (29). Briefly, 1 mM divalent cation (either Mn2+, Zn2+, Cd2+, or Mg2+) and 1 mM αKG were titrated into the sample cell containing 70 μM Alkbh5. Samples were prepared in 20 mM tris-HCl (pH 7.4) and 150 mM NaCl. All experiments were performed at 25°C and 500 rpm using MicroCal iTC200 (GE Healthcare). Analysis of the data was performed using the Origin software provided with the instrument.
Pull-down assays
Pull-down assays were performed in a total volume of 100 μl consisting of 20 μl of the NeutrAvidin Plus UltraLink Resin (Thermo Fisher Scientific), 5 μM biotinylated DNA (5′-biotin-TEG-TTTTTTGG(m6A)CT-3′ (GenScript), 25 μM Alkbh5, 500 μM αKG, and 500 μM X2+ (where X = Mn2+, Zn2+, Cd2+, or Mg2+). The solution was rocked at 4°C for 30 min and spun at 13,000g for 30 s, and the supernatant was discarded. The beads were washed three times with 100 μl of H2O to remove any residual unbound protein and then resuspended in 20 μl of SDS loading buffer, boiled for 10 min, spun at 13,000g for 30 s, and run (10 μl of the supernatant) on a Bolt 4 to 12% Bis-Tris Plus polyacrylamide gel (Invitrogen).
Enzymatic assays
The demethylation activity of AlkBH5 toward methylated substrates was tested as previously described (36). Briefly, a reaction mixture containing 5 μM Alkbh5 and 10 μM substrate was prepared in 2 mM l-ascorbic acid, 150 μM Fe2+, 300 μM αKG, and 50 mM tris-HCl (pH 7.4). The reaction mixtures were incubated at 37°C. Aliquots (50 μl) were taken at 0 and 60 min of reaction time and quenched with a 1:1 ratio of 20% (v/v) formic acid. The nucleic acids were separated from the protein by passing the quenched reaction through a 0.2-μm, 0.4-ml Ultrafree-MC Centrifugal Filter column (Millipore) by centrifugation (16,000g for 5 min). The pH of the flow through, containing a mixture of the methylated and demethylated substrate, was neutralized by addition of high-performance liquid chromatography (LC)–grade NH4OH and analyzed by LC–mass spectrometry (MS).
NMR spectroscopy
NMR samples (~0.5 mM) of the apo Alkbh5 were prepared in 20 mM MES (pH 6.0), 150 mM NaCl, 1 mM EDTA, 2 mM dithiothreitol (DTT), 1× protease inhibitor (EDTA-free), 50 mM arginine + glutamic acid, 0.02% (w/v) NaN3, and 90% H2O/10% D2O (v/v). To study Alkbh5 in the αKG-bound state, 500 μl of ~0.5 mM Alkbh5 was diluted into a 10-ml 20 mM MES (pH 6.0), 150 mM NaCl, 100 μM ZnCl2, 1 mM αKG, 1× protease inhibitor (EDTA-free), 50 mM arginine + glutamic acid, 0.02% (w/v) NaN3, and 90% H2O/10% D2O (v/v) and concentrated appropriately to reach a final protein concentration of ~0.5 mM. EDTA and DTT were removed from the buffer to eliminate any potential chelation of Zn2+. Saturation of the enzyme with Zn2+ and αKG is confirmed by 1H-15N TROSY. To study the DNA-bound state, the appropriate oligonucleotide was titrated directly into the NMR tube containing Alkbh5, Zn2+, and αKG.
NMR spectra were acquired on Bruker 600-, 700-, and 800-MHz spectrometers equipped with Z-shielded gradient triple resonance cryoprobes. Spectra were processed using NMRPipe and analyzed using NMRFAM-SPARKY. The sequential 1H/15N/13C backbone assignments of Alkbh5 in the apo, αKG-bound, and αKG/m6A-bound states were achieved using TROSY versions of conventional three-dimensional triple resonance correlation experiments [HNCO, HNCA, HNCACB, HN(CO)CA, and HN(CO)CACB] (51).
For chemical shift perturbation experiments, the weighted combined 1H/15N shift (∆H/N) was measured from the 1H-15N TROSY spectra of the free and bound states using the equation
| (1) |
where WH (=1) and WN (=0.154) are the weighting factors for the 1H and 15N amide chemical shifts, and ∆δH and ∆δN are the 1H and 15N chemical shift differences in ppm when comparing the NMR spectra of the free and bound states.
Backbone amide 1DNH RDCs were measured at 25°C by taking the difference in 1JNH scalar couplings in isotropic and alignment media. Phage pf1 (8 mg/ml; ASLA Biotech) was the used alignment media, and the 1JNH couplings were measured using the RDCs by TROSY pulse scheme (52). Xplor-NIH was used to compute SVD analysis of the RDC values.
15N-R1 and 15N-R1ρ experiments were acquired at 25°C and a 1H frequency of 800 MHz, using heat-compensated pulse schemes with a TROSY readout (40). A recycle delay of 1.5 s was used for R1 and R1ρ experiments, with the spin-lock field (ω1) for the R1ρ experiments set to 1 kHz. Relaxation delay durations were 0, 80, 200, 320, 440, 560, 720, and 840 ms for R1 and 0.2, 1.4, 2.4, 5.0, 7.8, 10.8, 14.9, 17.4, and 20.0 ms for R1ρ, respectively. 15N-R1 and 15N-R1ρ values were calculated by fitting the time-dependent exponential restoration of 1H-15N cross-peak intensities at increasing relaxation delays using a monoexponential function. Transverse relaxation rates (R2) were extracted from the measured R1 and R1ρ values.
15N relaxation dispersion experiments were performed at 25°C and 800 and 600 MHz using a pulse sequence that measures the exchange contribution for the TROSY component of the 15N magnetization (53). Data were modeled using the Carver-Richards equation.
MD simulations and structure modeling
The crystal structure of Alkbh5 in complex with Mn2+ and αKG (PDB code: 4NRO) was used as the starting structure for all simulations on the apo enzyme and on the Alkbh5-αKG complex with the substrate in the off-line configuration. Missing residues from the x-ray structure (fragments 145 to 149) were modeled using the software Modeler. The starting structures incorporating the in-line configuration were created by replacing the off-line αKG molecule in the crystal structure of the Alkbh5-αKG complex with the in-line αKG found in the structure of Alkbh2 bound to the primary and secondary substrate (PDB code: 3RZJ). The starting structures for simulations on the αKG/m6A-bound state were created by modeling the 5-mer DNA 5′-GG(m6A)CT-3′ into the starting structures created for the Alkbh5-αKG with the secondary substrate in the off-line or in-line configuration. Modeling was performed by enforcing the oligonucleotide backbone and the m6A base to adopt the structure and orientation within the active site observed in the crystallographic model of the AlkB-αKG/m6A complex (PDB code: 4NID). Each modeled structure was equilibrated by running 1-μs simulations with weak position restraints applied to retain the starting geometry of the active site (see below).
MD simulations were run using the Amber 16 package and the Amber ff14SB force field (54). αKG was parameterized with the AM1-BCC charge model (55) and the GAFF force field (56). The initial structure was centered in a truncated octahedron, filled with TIP3P water model and neutralizing ions, and the distance between the protein atoms and the boundaries was set to 10 Å. Energy minimization of the initial structures, including 1000 steps of steepest descent and 1000 steps of conjugate gradient, was performed in three stages. First, ions and water positions were relaxed. Then, the protein (or protein complex) was allowed to relax. Last, the full system was energy-minimized. The system was equilibrated with a 1-ns run in which the temperature was gradually raised from 0 to 310 K, followed by a 5-ns run in which the temperature was held constant at 310 K. For simulations on the Alkbh5 complexes with αKG and αKG/m6A, Fe2+ was used as the divalent cation and an additional 1-μs equilibration step at 310 K was performed during which the geometry of the active site (defined by a bubble of 5 Å radius centered on the αKG center of mass) was restrained to its starting coordinates by a restraining weight of 2 kcal mol−1 Å−2. The restraining potential was kept during the first half and gradually relaxed during the second half of the 1-μs equilibration protocol. The equilibrated system was simulated for 1.2 μs by keeping the temperature (310 K) and pressure (1 atm) constant. Periodic boundary conditions were applied, and bonds were restrained with the SHAKE algorithm. An integration step of 2 fs was used. Weak coupling to an external pressure and temperature bath was used. Particle mesh Ewald summation with a cutoff of 10 Å for long-range interactions was used to treat electrostatic interactions.
The 6-ns restrained MD simulations were run in Amber 16 using similar parameters as the unrestrained simulations. The force constant for the RDC restraints was set to 0.1 kcal mol−1 Hz−2.
Calculation of conformational ensembles
Conformational ensembles for Alkbh5, Alkbh5-αKG, and Alkbh5-αKG/m6A were calculated by combining Gaussian aMD (GaMD) simulations and the NMR-derived 1DNH RDC data (see the “NMR spectroscopy” section), as recently described (34).
The GaMD module implemented in the graphics processing unit (GPU) version of AMBER 16 was applied to each system, which included a 10-ns-short MD simulation that was used to collect the potential statistics for calculating GaMD acceleration parameters, a 50-ns equilibration after adding the boost potential, and, finally, a 500-ns GaMD production simulation with randomized initial atomic velocities. All GaMD simulations were run at the “dual-boost” level, in which the total potential energies and the dihedral energies were boosted.
To generate structural ensemble representations, each trajectory was clustered to produce representative structures of the aMD with a high degree of structural diversity. Each representative structure was energy-minimized, and the ensemble of representative structures was used to fit the experimental RDC data (34). Briefly, back-calculation of RDCs from conformational ensembles was done using the following equation
| (2) |
where θ is the angle formed between the internuclear bond vector of the amide group of residue i and the z axis of the alignment tensor, ϕ is the angle between the xy plane projection of the internuclear bond vector and the x axis, and Dk is the magnitude of the alignment tensor for ensemble member k multiplied by its fractional population in the ensemble. Dk, θ, and ϕ were optimized to reduce the discrepancy between experimental and back-calculated RDCs using the MATLAB script downloadable at http://group.chem.iastate.edu/Venditti/downloads.html.
The consistency between experimental and back-calculated RDC data was evaluated in terms of R-factor
| (3) |
where and are the experimental and back-calculated RDC for residue i, respectively. The protocol was iterated by increasing the number of clusters (and therefore the representative structures in the pool) until a stable R-factor was obtained.
Acknowledgments
We thank B. Chen and D. Kramer for the help in setting up the pull-down assays and J. Roche for critical reading of the manuscript. Funding: This work was supported by funds from NIGMS R35GM133488 (to V.V.). Author contributions: J.A.P. and V.V. designed the study. J.A.P., T.T.N., B.K., A.S., and V.V. acquired and analyzed the data. J.A.P. and V.V. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/34/eabi8215/DC1
REFERENCES AND NOTES
- 1.Meyer K. D., Saletore Y., Zumbo P., Elemento O., Mason C. E., Jaffrey S. R., Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149, 1635–1646 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X., Lu Z., Gomez A., Hon G. C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G., Ren B., Pan T., He C., N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y., Li Y., Toth J. I., Petroski M. D., Zhang Z., Zhao J. C., N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 16, 191–198 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu R., Jiang D., Wang Y., Wang X., N6-methyladenosine (m6A) methylation in mRNA with a dynamic and reversible epigenetic modification. Mol. Biotechnol. 58, 450–459 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Yue Y., Liu J., He C., RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 29, 1343–1355 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng X., Su R., Feng X., Wei M., Chen J., Role of N6-methyladenosine modification in cancer. Curr. Opin. Genet. Dev. 48, 1–7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng X., Su R., Weng H., Huang H., Li Z., Chen J., RNA N6-methyladenosine modification in cancers: Current status and perspectives. Cell Res. 28, 507–517 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esteller M., Pandolfi P. P., The epitranscriptome of noncoding RNAs in cancer. Cancer Discov. 7, 359–368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li N., Kang Y., Wang L., Huff S., Tang R., Hui H., Agrawal K., Gonzalez G. M., Wang Y., Patel S. P., Rana T. M., ALKBH5 regulates anti–PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc. Natl. Acad. Sci. U.S.A. 117, 20159–20170 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng G., Dahl J. A., Niu Y., Fedorcsak P., Huang C.-M., Li C. J., Vågbø C. B., Shi Y., Wang W.-L., Song S.-H., Lu Z., Bosmans R. P. G., Dai Q., Hao Y.-J., Yang X., Zhao W.-M., Tong W.-M., Wang X.-J., Bogdan F., Furu K., Fu Y., Jia G., Zhao X., Liu J., Krokan H. E., Klungland A., Yang Y.-G., He C., ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toh J. D. W., Sun L., Lau L. Z. M., Tan J., Low J. J. A., Tang C. W. Q., Cheong E. J. Y., Tan M. J. H., Chen Y., Hong W., Gao Y.-G., Woon E. C. Y., A strategy based on nucleotide specificity leads to a subfamily-selective and cell-active inhibitor of N6-methyladenosine demethylase FTO. Chem. Sci. 6, 112–122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang T., Cheong A., Mai X., Zou S., Woon E. C. Y., A methylation-switchable conformational probe for the sensitive and selective detection of RNA demethylase activity. Chem. Commun. 52, 6181–6184 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Zheng G., Fu Y., He C., Nucleic acid oxidation in DNA damage repair and epigenetics. Chem. Rev. 114, 4602–4620 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fedeles B. I., Singh V., Delaney J. C., Li D., Essigmann J. M., The AlkB family of Fe(II)/α-ketoglutarate-dependent dioxygenases: Repairing nucleic acid alkylation damage and beyond. J. Biol. Chem. 290, 20734–20742 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinez S., Hausinger R. P., Catalytic mechanisms of Fe(II)- and 2-oxoglutarate-dependent oxygenases. J. Biol. Chem. 290, 20702–20711 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu B., Edstrom W. C., Benach J., Hamuro Y., Weber P. C., Gibney B. R., Hunt J. F., Crystal structures of catalytic complexes of the oxidative DNA/RNA repair enzyme AlkB. Nature 439, 879–884 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Yu B., Hunt J. F., Enzymological and structural studies of the mechanism of promiscuous substrate recognition by the oxidative DNA repair enzyme AlkB. Proc. Natl. Acad. Sci. U.S.A. 106, 14315–14320 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Que L. Jr., One motif—Many different reactions. Nat. Struct. Biol. 7, 182–184 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Feng C., Liu Y., Wang G., Deng Z., Zhang Q., Wu W., Tong Y., Cheng C., Chen Z., Crystal structures of the human RNA demethylase Alkbh5 reveal basis for substrate recognition. J. Biol. Chem. 289, 11571–11583 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z., Ren J., Stammers D. K., Baldwin J. E., Harlos K., Schofield C. J., Structural origins of the selectivity of the trifunctional oxygenase clavaminic acid synthase. Nat. Struct. Biol. 7, 127–133 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Pavel E. G., Zhou J., Busby R. W., Gunsior M., Townsend C. A., Solomon E. I., Circular dichroism and magnetic circular dichroism spectroscopic studies of the non-heme ferrous active site in clavaminate synthase and its interaction with α-ketoglutarate cosubstrate. J. Am. Chem. Soc. 120, 743–753 (1998). [Google Scholar]
- 22.Zhou J., Kelly W. L., Bachmann B. O., Gunsior M., Townsend C. A., Solomon E. I., Spectroscopic studies of substrate interactions with clavaminate synthase 2, a multifunctional α-KG-dependent non-heme iron enzyme: Correlation with mechanisms and reactivities. J. Am. Chem. Soc. 123, 7388–7398 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Chaturvedi S. S., Ramanan R., Lehnert N., Schofield C. J., Karabencheva-Christova T. G., Christov C. Z., Catalysis by the non-heme iron(II) histone demethylase PHF8 involves iron center rearrangement and conformational modulation of substrate orientation. ACS Catal. 10, 1195–1209 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waheed S. O., Ramanan R., Chaturvedi S. S., Lehnert N., Schofield C. J., Christov C. Z., Karabencheva-Christova T. G., Role of structural dynamics in selectivity and mechanism of non-heme Fe(II) and 2-oxoglutarate-dependent oxygenases involved in DNA repair. ACS Cent. Sci. 6, 795–814 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yi C., Chen B., Qi B., Zhang W., Jia G., Zhang L., Li C. J., Dinner A. R., Yang C.-G., He C., Duplex interrogation by a direct DNA repair protein in search of base damage. Nat. Struct. Mol. Biol. 19, 671–676 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holland P. J., Hollis T., Structural and mutational analysis of Escherichia coli AlkB provides insight into substrate specificity and DNA damage searching. PLOS ONE 5, e8680 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu C., Yi C., Switching demethylation activities between AlkB family RNA/DNA demethylases through exchange of active-site residues. Angew. Chem. Int. Ed. 53, 3659–3662 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Zhang X., Wei L.-H., Wang Y., Xiao Y., Liu J., Zhang W., Yan N., Amu G., Tang X., Zhang L., Jia G., Structural insights into FTO’s catalytic mechanism for the demethylation of multiple RNA substrates. Proc. Natl. Acad. Sci. U.S.A. 116, 2919–2924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ergel B., Gill M. L., Brown L., Yu B., Palmer A. G. III, Hunt J. F., Protein dynamics control the progression and efficiency of the catalytic reaction cycle of the Escherichia coli DNA-repair enzyme AlkB. J. Biol. Chem. 289, 29584–29601 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bleijlevens B., Shivarattan T., van den Boom K. S., de Haan A., van der Zwan G., Simpson P. J., Matthews S. J., Changes in protein dynamics of the DNA repair dioxygenase AlkB upon binding of Fe2+ and 2-oxoglutarate. Biochemistry 51, 3334–3341 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Dotas R. R., Nguyen T. T., Stewart C. E. Jr., Ghirlando R., Potoyan D. A., Venditti V., Hybrid thermophilic/mesophilic enzymes reveal a role for conformational disorder in regulation of bacterial Enzyme I. J. Mol. Biol. 432, 4481–4498 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghose R., Nature of the pre-chemistry ensemble in mitogen-activated protein kinases. J. Mol. Biol. 431, 145–157 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Elias M., Wieczorek G., Rosenne S., Tawfik D. S., The universality of enzymatic rate-temperature dependency. Trends Biochem. Sci. 39, 1–7 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Purslow J. A., Nguyen T. T., Egner T. K., Dotas R. R., Khatiwada B., Venditti V., Active site breathing of human Alkbh5 revealed by solution NMR and accelerated molecular dynamics. Biophys. J. 115, 1895–1905 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Purslow J. A., Venditti V., 1H, 15N, 13C backbone resonance assignment of human Alkbh5. Biomol. NMR Assign. 12, 297–301 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Khatiwada B., Purslow J. A., Underbakke E. S., Venditti V., N-terminal fusion of the N-terminal domain of bacterial enzyme I facilitates recombinant expression and purification of the human RNA demethylases FTO and Alkbh5. Protein Expr. Purif. 167, 105540 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zou S., Toh J. D. W., Wong K. H. Q., Gao Y.-G., Hong W., Woon E. C. Y., N6-methyladenosine: A conformational marker that regulates the substrate specificity of human demethylases FTO and ALKBH5. Sci. Rep. 6, 25677 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aik W., Scotti J. S., Choi H., Gong L., Demetriades M., Schofield C. J., McDonough M. A., Structure of human RNA N6-methyladenine demethylase ALKBH5 provides insights into its mechanisms of nucleic acid recognition and demethylation. Nucleic Acids Res. 42, 4741–4754 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Purslow J. A., Khatiwada B., Bayro M. J., Venditti V., NMR methods for structural characterization of protein-protein complexes. Front. Mol. Biosci. 7, 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lakomek N.-A., Ying J., Bax A., Measurement of 15N relaxation rates in perdeuterated proteins by TROSY-based methods. J. Biomol. NMR 53, 209–221 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kay L. E., Torchia D. A., Bax A., Backbone dynamics of proteins as studied by nitrogen-15 inverse detected heteronuclear NMR spectroscopy: Application to staphylococcal nuclease. Biochemistry 28, 8972–8979 (1989). [DOI] [PubMed] [Google Scholar]
- 42.Mittermaier A., Kay L. E., New tools provide new insights in NMR studies of protein dynamics. Science 312, 224–228 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Nguyen T. T., Ghirlando R., Roche J., Venditti V., Structure elucidation of the elusive Enzyme I monomer reveals the molecular mechanisms linking oligomerization and enzymatic activity. Proc. Natl. Acad. Sci. U.S.A. 118, e2100298118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonk B. M., Weis J. W., Tidor B., Machine learning identifies chemical characteristics that promote enzyme catalysis. J. Am. Chem. Soc. 141, 4108–4118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liao Q., Kulkarni Y., Sengupta U., Petrović D., Mulholland A. J., van der Kamp M. W., Strodel B., Kamerlin S. C. L., Loop motion in triosephosphate isomerase is not a simple open and shut case. J. Am. Chem. Soc. 140, 15889–15903 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Venditti V., Tugarinov V., Schwieters C. D., Grishaev A., Clore G. M., Large interdomain rearrangement triggered by suppression of micro- to millisecond dynamics in bacterial Enzyme I. Nat. Commun. 6, 5960 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liptak C., Mahmoud M. M., Eckenroth B. E., Moreno M. V., East K., Alnajjar K. S., Huang J., Towle-Weicksel J. B., Doublié S., Loria J. P., Sweasy J. B., I260Q DNA polymerase β highlights precatalytic conformational rearrangements critical for fidelity. Nucleic Acids Res. 46, 10740–10756 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Selvaratnam R., VanSchouwen B., Fogolari F., Mazhab-Jafari M. T., Das R., Melacini G., The projection analysis of NMR chemical shifts reveals extended EPAC autoinhibition determinants. Biophys. J. 102, 630–639 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Halford S. E., Marko J. F., How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 32, 3040–3052 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takayama Y., Clore G. M., Intra- and intermolecular translocation of the bi-domain transcription factor Oct1 characterized by liquid crystal and paramagnetic NMR. Proc. Natl. Acad. Sci. U.S.A. 108, E169–E176 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clore G. M., Gronenborn A. M., Determining the structures of large proteins and protein complexes by NMR. Trends Biotechnol. 16, 22–34 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Fitzkee N. C., Bax A., Facile measurement of 1H–15N residual dipolar couplings in larger perdeuterated proteins. J. Biomol. NMR 48, 65–70 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Loria J. P., Rance M., Palmer A. G. III, A TROSY CPMG sequence for characterizing chemical exchange in large proteins. J. Biomol. NMR 15, 151–155 (1999). [DOI] [PubMed] [Google Scholar]
- 54.Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., Simmerling C., ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jakalian A., Jack D. B., Bayly C. I., Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 23, 1623–1641 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Huang L., Roux B., Automated force field parameterization for nonpolarizable and polarizable atomic models based on ab initio target data. J. Chem. Theory Comput. 9, 3543–3556 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clore G. M., Garrett D. S., R-factor, free R, and complete cross-validation for dipolar coupling refinement of NMR structures. J. Am. Chem. Soc. 121, 9008–9012 (1999). [Google Scholar]
- 58.Venditti V., Egner T. K., Clore G. M., Hybrid approaches to structural characterization of conformational ensembles of complex macromolecular systems combining NMR residual dipolar couplings and solution X-ray scattering. Chem. Rev. 116, 6305–6322 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nguyen T. T., Venditti V., An allosteric pocket for inhibition of bacterial Enzyme I identified by NMR-based fragment screening. J. Struct. Biol. X 4, 100034 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Aalten D. M. F., Amadei A., Linssen A. B. M., Eijsink V. G. H., Vriend G., Berendsen H. J. C., The essential dynamics of thermolysin: Confirmation of the hinge-bending motion and comparison of simulations in vacuum and water. Proteins 22, 45–54 (1995). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/34/eabi8215/DC1