

A perfusable vascularized 3D-bioprinted cancer model with stromal cells can be used for personalized therapy and target discovery.
Abstract
Many drugs show promising results in laboratory research but eventually fail clinical trials. We hypothesize that one main reason for this translational gap is that current cancer models are inadequate. Most models lack the tumor-stroma interactions, which are essential for proper representation of cancer complexed biology. Therefore, we recapitulated the tumor heterogenic microenvironment by creating fibrin glioblastoma bioink consisting of patient-derived glioblastoma cells, astrocytes, and microglia. In addition, perfusable blood vessels were created using a sacrificial bioink coated with brain pericytes and endothelial cells. We observed similar growth curves, drug response, and genetic signature of glioblastoma cells grown in our 3D-bioink platform and in orthotopic cancer mouse models as opposed to 2D culture on rigid plastic plates. Our 3D-bioprinted model could be the basis for potentially replacing cell cultures and animal models as a powerful platform for rapid, reproducible, and robust target discovery; personalized therapy screening; and drug development.
INTRODUCTION
Cancer is the second leading cause of death globally (1). It is estimated that around 30 to 40% of patients with cancer are being treated with ineffective drugs (2); therefore, preclinical drug screening platforms attempt to overcome this challenge. Several approaches, such as whole-exome or RNA sequencing (RNA-seq), aim to identify druggable, known mutations or overexpressed genes that may be exploited as a therapeutic target for personalized therapy. More advanced approaches offer to assess the efficacy of a drug or combinations of drugs in patient-derived tumor xenograft models or in vitro three-dimensional (3D) organoids. Unfortunately, most of the existing methods face unmet challenges, which limit their efficacy. For instance, cells can become quiescent or acquire somatic mutations while growing many generations on plastic (3) under the influence of static mechanical forces and in the absence of functional vasculature. In addition, predictions of drug sensitivity derived from microarray and sequencing methods cannot always be validated, likewise the lack of information about the tumor microenvironment (TME), and hence do not offer reliable evaluations of tumor response. Currently, the development of drugs, until reaching approval and commercialization, may take more than a decade and costs approximately 2.6 billion U.S. dollars (4). Moreover, most of the anticancer compounds fail in clinical trials due to lack of efficacy, and thus, the overall likelihood of approval from phase 1 for all oncology approaches is 5.3% (5). Therefore, there is a need for a reliable, clinically relevant platform for high-throughput drug screening.
The conventional preclinical drug development process relies on in vitro evaluation of the drug toxicity/efficacy usually in 2D cell culture followed by animal studies. 2D culture methods have a tremendous value in biomedical research and in drug screening because of their low costs and ease of use. However, these methods do not support tissue-specific architecture and do not maintain the diverse functions of the multiple cell types that play important roles in the progression of the disease. Furthermore, 2D cultures struggle to predict the diverse effects of a given treatment in vivo (6). For in vivo studies of human source, traditionally, a small chunk of tumor tissue obtained from patients during surgery is implanted into immunodeficient mice [i.e., patient-derived xenografts (PDXs)]. These models include tumor cells and their stroma; while these tumors are implanted with their stroma, the human stroma is replaced by murine equivalents with time (7). Furthermore, drug screening using PDX models is an expensive and time-consuming process that can take up to 6 to 9 months to receive an answer regarding the suitable drug to a patient. This prolonged time phrase until a treatment can be offered makes, in many cases, the outcome irrelevant, either because the tumor cells have mutated and changed substantially from those cells that were resected originally or because the patient already succumbed to the disease. In addition, PDX models require the use of immunocompromised animals that lack a fully functional immune system and therefore cannot be used to test immunotherapies.
Numerous 3D cancer models have been developed over the years in the attempt to overcome the limitations of current cancer models and reduce the costs of preclinical drug evaluation. Tumor spheroids and organoids are the most commonly used 3D cancer models in preclinical research. These models better mimic the origin tissue architecture in terms of structural organization, cell-cell interactions, and cell–extracellular matrix (ECM) interactions. The heterogeneity of the TME can be partially recapitulated by coculturing cancer cells with stromal cells. This is important as, in many cases, drugs do not target directly the cancer cells but rather the interactions between the cancer cells and the TME cells. Moreover, tumor spheroids and organoids can be embedded in naturally derived hydrogel such as Matrigel or in other synthetic hydrogels that enable cells to polarize and interact with other cells. Although these 3D models have shown great promises, they often do not reflect the complexity of tumors in vivo, as they lack the whole plethora of stromal cells and functional blood vessels (8–14), which are critical for the development and progression of the disease and evaluation of the response to the therapy (15).
A novel promising technology in the field of tissue regeneration is 3D-bioprinting, a technology for precise 3D construction of complex tissues and organs. 3D-bioprinting technology allows the position of cells and biocompatible materials layer by layer, with spatial control of each component location. 3D-bioprinting allows the use of a wide range of biomaterials at different viscosities and cell densities. Considering that the 3D-bioprinting process can control the organization of multiple cell types in 3D architecture, 3D-bioprinted models have the potential to better imitate the TME. Nonetheless, many of the 3D-bioprinted tumor models established thus far were designed without the complete physiological characteristics of the tumor, such as multiscale architecture (8, 9) and blood vessels (8, 9, 16, 17), and if included, the blood vessels are not frequently perfused (14). The establishment of physiologically relevant ex vivo vascularized 3D tumor model is of high importance, as without perfusable vasculature, the model lacks one of the main cancer hallmarks, angiogenesis (18). However, achieving hierarchical vascularization networks inside the desired 3D tumor model is a great challenge in 3D-bioprinting (19). Therefore, some of the existing models rely on artificial microfluidic channel chips that lack the tissue-tissue interfaces (6, 10–13, 20), while other models rely on casted hydrogels on top of a needle or fiber vessels (21). A recent study had demonstrated the importance of accurate tomographic analysis of cancer 3D-bioprinted model with perfused vasculature (22).
We present here a 3D-bioprinted engineered tumor model as a clinically relevant platform for rapid drug screening and prediction of treatment outcome. Our 3D engineered tissue constructs are based on two bioinks, a tumor bioink and a vascular bioink. The tumor bioink is composed of the natural polymers fibrinogen and gelatin, while the vascular bioink is composed of the thermo-reversible biocompatible synthetic polymer, Pluronic F127. Since the gelation process of Pluronic F127 is reversible, it was used as a sacrificial material to fabricate embedded vasculature inside the 3D model and liquefied to create a 3D lumen vascular structure.
As a proof of concept, we focused on 3D-bioprinting of glioblastoma (GB), the most common and aggressive primary brain cancer (23). With the current standard of care that includes surgery, chemotherapy, and radiotherapy, the 5-year survival rates of patients with GB are less than 10% (24). Poor patient outcome is attributed to the highly invasive and aggressive nature of GB tumors and to the development of resistance to chemotherapy. The complex intratumoral heterogeneity and the TME are the major contributors of GB cell resistance to therapy. The brain TME is known to be a dynamic and complex population that plays a major role in tumor progression, involving various signaling pathways that affect GB cell growth and invasion (24). Therefore, models that better mimic the intricate microenvironment of GB have the potential to facilitate the development of effective alternative treatment options.
In this study, we investigated the mechanical properties and biological functionality of our biocompatible fibrin 3D-bioink, exploiting an array of murine, human, and patient-derived GB (PD-GB) models. By incorporating into our 3D-bioink a penta-culture of GB tumor cells and stromal cells as well as a perfusable vascular network, our models are able to mimic GB cellular heterogeneity, cell-cell interaction, and spatial tomography. We evaluated the ability of these 3D-bioprinted GB models to recapitulate the various in vivo characteristics of GB tumors such as their growth kinetics, invasion capabilities, response to therapies, and their genetic signatures. We show that our model provides a more accurate evaluation of the native molecular and cellular characteristics of GB establishment and progression and the response to therapies compared to 2D plasticware models. Our 3D-bioprinted models can be exploited to better emulate the clinical scenario of various cancer types and can potentially serve as a powerful clinically accurate platform for preclinical research and drug discovery.
MATERIALS AND METHODS
Materials
Dulbecco’s modified Eagle’s medium (DMEM; Gibco), fetal bovine serum (FBS; Gibco), l-glutamine, penicillin, streptomycin, mycoplasma detection kit, EZ-RNA II total RNA isolation kit, and fibronectin (1 mg/ml; 1:100 dilution) were purchased from Biological Industries Ltd. (Kibbutz Beit HaEmek, Israel). Percoll medium (catalog no. p4937), gelatin (type A, 300 bloom from porcine skin), Pluronic F127, 70-kDa dextran-FITC (fluorescein isothiocyanate), Latex beads for phagocytosis assays (catalog no. L4655), and all other chemical reagents, including salts and solvents, were purchased from Sigma-Aldrich (Rehovot, Israel). Milli-Q water was prepared using a Millipore water purification system. Fibrinogen, thrombin (Th), poly-l-lysine (PLL; 0.1 mg/ml; catalog no. A-005-C), Endo-Gro medium, and bovine serum albumin were purchased from Merck Millipore (Burlington, MA, USA). Transglutaminase (TG) was purchased from Moo Glue (Modernist Pantry, Eliot, Maine, USA). Collagenase II, collagenase IV, dispase II (neutral protease), and deoxyribonuclease I were purchased from Worthington Biochemical Corporation (NJ, USA). Red blood cell lysis solution (catalog no. 420301) was purchased from BioLegend (San Diego, CA, USA). Magnetic-activated cell sorting (MACS) MS columns for cell separation (catalog no. 130-042-201), CD11b MicroBeads for cell isolation (catalog no. 130-093-634), CD31 MicroBeads for cell isolation (catalog no. 130-097-418), anti-AN2 MicroBeads for cell isolation (catalog no. 130-097-170), and CD144 MicroBeads for cell isolation (catalog no. 130-097-857) were purchased from Miltenyi Biotec (Bergisch Gladbach, Germany). P-selectin inhibitor (SELPi) KF38789 (catalog no. 2748) was purchased from Tocris BioScience (Bristol, UK). ProLong Gold mounting with DAPI (4′,6-diamidino-2-phenylindole; catalog no. p36935) and Hoechst 33342 (catalog no. H3570) were purchased from Invitrogen (Carlsbad, California, USA). Mayer’s hematoxylin solution (catalog no. 05-06002) and eosin Y solution (catalog no. 05-10002) were purchased from Bio-Optica (Milano, Italy). PresoBlue Cell Viability Reagent was purchased from Thermo Fisher Scientific (MA, USA).
Plasmids
mCherry and iRFP (near-infrared fluorescent protein) were subcloned by our group into the pQCXIP vector (Clontech, USA) as previously described (25).
Primary immunostaining antibodies
Rabbit anti-mouse/human glial fibrillary acidic protein (GFAP) (1:500 dilution; Dako, catalog no. 20025480; lot. no. 41556) and rabbit anti-mouse/human Iba1 (1:200 dilution; catalog no. NBP2-19019; lot no. 41556) were purchased from Novus (CO, USA). Mouse anti-human P-selectin (1:20 dilution; catalog no. BBA1; lot nos. APB081704 and APB0818111; clone BBIG-E) was purchased from R&D Systems (Minneapolis, MN, USA).
Secondary immunostaining antibodies
Goat anti-rabbit Alexa Fluor 488 (1:300 dilution; catalog no. ab150077; lot no. GR315933-2) was purchased from Abcam (Cambridge, UK), and goat anti-mouse Alexa-568 (1:400 dilution; catalog no. ab175473; lot no. GR3246243-3) was purchased from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA, USA).
Flow cytometry antibodies
FITC rat anti-mouse CD62P P-selectin clone RB 40.34 (1:20 dilution; catalog no. 561923; lot no. 8340691) and FITC rat immunoglobulin G1 (IgG1) isotype control (1:20 dilution; catalog no. 553995; lot no. 5100888) were purchased from BD Biosciences (NJ, USA). Mouse anti-human P-selectin (1:20 dilution; catalog no. BBA1; lot. no. APB0818111; clone BBIG-E) and mouse IgG1 isotype control (1:20 dilution; catalog no. mab002; lot. no. IX2417011) were purchased from R&D Systems (Minneapolis, MN, USA). Mouse-IgG ƙ BP-CFL 488 secondary antibody (1:20 dilution; catalog no. sc-516176; lot nos. K1119 and HO118) was purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA).
Fibrin 3D-bioink
Fibrinogen is a glycoprotein that has an important role in the formation of blood clots in vertebrates; it forms a fibrin gel when mixed with Th. We combined fibrinogen with the hydrolyzed form of collagen, gelatin, which is a low-cost, abundant, and biocompatible material and the major component of the ECM in most tissues. We allowed slow cross-linking to the fibrin gel by TG, a natural, nontoxic enzyme. TG can catalyze intramolecular and intermolecular covalent bonds between the glutamine and lysine residues of gelatin. Gelatin solutions at 7.5% (w/v), 15% (w/v), and 30% (w/v) were dissolved in phosphate-buffered saline (PBS) without calcium and magnesium at 70°C for 12 hours under vigorous stirring. Then, the pH was adjusted dropwise to 7.5 using 1 M NaOH. The solution was filtered through a 0.2-μm filter and stored at 4°C. Fibrinogen solution (50 mg/ml) was produced by dissolving lyophilized human blood plasma protein at 37°C in sterile PBS without calcium and magnesium for 45 min. The pH was adjusted dropwise to 7.5 using 0.5 M NaOH, and the solution was stored at −20°C. TG solution (100 mg/ml) was dissolved in PBS without calcium and magnesium, gently mixed for 20 min at 37°C, and sterile-filtered. Stock solution of 250 mM CaCl2 was prepared by dissolving CaCl2 powder in PBS without calcium and magnesium. To prepare stock solution of Th, lyophilized Th was reconstituted at 2000 U/ml using sterile PBS without calcium and magnesium and stored at −20°C. The bioink solutions were mixed together at 37°C.
Rheological characterization
The bioinks must flow through the deposition nozzle without clogging yet quickly solidify. To that end, the ink must be both shear thinning and viscoelastic, i.e., with a shear elastic modulus (G′) that exceeds the loss modulus (G″). The ink rheology, mechanical properties, and cross-linking kinetics were measured using a controlled stress rheometer (AR-G2, TA Instruments). Time-sweep oscillatory tests in a 20-mm cone plate with 1° geometry were performed using 350 μl of fresh fibrinogen solutions (resulting in a gap size of 600 μm), 1 min after its preparation at 37°C, 0.01% strain, and frequency of 1 Hz. The gelation time was determined by the first point at which G′ raises. Each bioink formulation was tested three independent times.
Mechanical characterization
Young’s modulus (E) was evaluated using LS1 tensile testing instrument (Lloyd Instruments Ltd.) equipped with a ultrasensitive 5-N load cell as previously described (26). Young’s modulus (E) is defined as
where σ is the stress applied on the sample and ε is the strain in the linear region of the stress-strain curve.
Briefly, disk-shaped samples of fibrin 3D-bioink were prepared as described above, applied into silicone molds (8 mm in diameter and 2 mm in thickness; Sigma-Aldrich), and incubated at 37°C and 5% CO2 for 1.5 hours. Young’s modulus was then evaluated by measuring the slope of the compressive stress-strain curve in the linear region. The samples were compressed at a rate of 1 mm/min up to 80% strain. The strain was calculated by determining the initial thickness of the sample at the point at which the stress values changed from negative to positive and started to increase during compressive test. The Young’s modulus was calculated from the linear region of the stress-strain curve, as the slope between 1 and 10% strain. Device control, data acquisition, and processing were performed with NEXYGEN plus 3.0 software (Lloyd Instruments Ltd.). Each bioink formulation has six to eight samples per group, and the experiments were repeated in three independent times.
Cell culture
Human GB cells (U-87MG, T98G, and U373), human embryonic kidney 293T cells, human osteosarcoma (Saos-2) cells, and MDA-MB-231 human breast cancer cells were obtained from the American Type Culture Collection (Manassas, VA, USA). GL261 murine GB cell line was obtained from the National Cancer Institute (Frederick, MD, USA). Cancer cells were grown in DMEM supplemented with 10% FBS (Gibco), streptomycin (100 μg/ml), penicillin (100 U/ml), nystatin (12.5 U/ml), and 2 mM l-glutamine. Human astrocytes (hAstro), human microglia (hMG), and primary human microvascular brain pericytes (hPericytes) were purchased from ScienCell (CA, USA). hAstro and freshly isolated murine astrocytes (mAstro) cells were grown in the supplied astrocyte medium supplemented with 2% FBS, penicillin (100 IU/ml), streptomycin (100 μg/ml), nystatin (12.5 U/ml), and 1% astrocyte growth supplements (ScienCell). Primary hMG and freshly isolated murine microglia (mMG) cells were grown on PLL (Sigma-Aldrich)–coated plates in the supplied microglia medium supplemented with 5% FBS, penicillin (100 IU/ml), streptomycin (100 μg/ml), nystatin (12.5 U/ml), and 1% microglia growth supplements (ScienCell). hPericytes were grown on PLL-coated plates in the supplied pericyte medium supplemented with 2% FBS, penicillin (100 IU/ml), streptomycin (100 μg/ml), nystatin (12.5 U/ml), and 1% pericyte growth supplements (ScienCell). Human umbilical vein endothelial cells (HUVECs) were purchased from Lonza and cultured in EGM-2 medium (Lonza, Switzerland) or Endo-Gro medium on fibronectin-coated plates. All cells were grown at 37°C and 5% CO2. All primary cell lines were grown up to passage 7. Mycoplasma tests were done routinely with a mycoplasma detection kit.
Ethical statement
All animals were housed in Tel Aviv University animal facility. The experiments were approved by the Animal Care and Use Committee of Tel Aviv University (approval nos. 01-19-015 and 01-19-097) and conducted in accordance with the National Institutes of Health guidelines. Experiments involving human tissues were performed with the approval of the Institutional Review Board (IRB) and in compliance with all legal and ethical considerations for human subject research (approval no. 0735-13-TLV). All studies were approved by the local ethics committee, and written informed consent was obtained from all patients.
Human primary GB cells
PD-GB tissues were obtained during surgical procedures from Tel Aviv Sourasky Medical Center (Tel Aviv, Israel) under an approved IRB (0735-12-TLV and 920130237). Tumor tissues were kept in cold PBS and processed within 40 min. Tumor specimens were dissected to 0.5-mm pieces and incubated in rotation with collagenase IV/dispase solution for a minimum of 3 hours at 37°C. Cells were passed through a 70-μm strainer, and red blood cell lysis was carried out. Isolated cells containing mainly tumor cells were plated and grown in DMEM growing media. Following continuous media replacement, viable cancer cells remained attached to culture plates and kept growing in culture, while stroma and cell debris were washed.
Isolation of murine brain stromal cells
Brains from 6- to 10-week-old male C57BL/6 mice (Envigo CRS, Israel) were harvested, chopped, and incubated in rotation with collagenase IV/dispase II solution for 50 min at 37°C. Red blood cell lysis was carried out followed by Percoll gradient for myelin separation. Cell suspension was then incubated with CD11b or CD31 MicroBeads for microglia or endothelial cell separation, respectively, and placed in MACS MS columns. Murine pericytes were isolated using anti-AN2 MicroBeads combined with anti-CD144 MicroBeads. The remaining cells collected following bead separation were plated and grown in complete astrocyte medium for a week. Culture enrichment of astrocytes was evaluated for cell type and purity by fluorescence-activated cell sorting (FACS; GLAST-APC antibody, Miltenyi Biotec; 5 μl/1 × 106 cells).
Swelling
To determine the swelling ability and the time required for swelling equilibrium, kinetic experiments were performed with fibrin 3D-bioink as previously described (27). Briefly, fibrin 3D-bioink samples were prepared in the absence or presence of GL261 or PD-GB4 cells (1 × 106 cells/ml). The samples were placed on a stainless-steel grid submerged in a six-well plate containing 15 ml of DMEM to simulate sink conditions and were incubated at 37°C and 5% CO2. The swelling kinetics was determined gravimetrically; each fibrin 3D-bioink sample was weighed at scheduled time intervals after gently removing excess water from the grid using a Kimwipe and returned immediately to the well plate. The swelling percentage at each time interval was calculated as follows
where wo is the initial weight of the fibrin 3D-bioink sample, right after cross-linking is ended and before swelling, and w is the weight of the sample at any other time.The wo refers to the original dimension at the time of fibrinogen polymerization and not to the dimension of the dry state of the bioink since it mimics better the experimental setup that the bioink has to tackle. Swelling at equilibrium of polymeric fibrin 3D-bioink was estimated from the swelling at the last time point (day 28). All swelling experiments were performed using six to eight samples per group, and the experiments were repeated in three independent times.
3D metabolic activity assays
GB cells, hAstro, mAstro, hMG, and osteosarcoma cells were cultured in fibrin 3D-bioink at different cell densities and gelatin concentrations. Cell viability and growth rates up to 6 to 7 weeks in fibrin 3D-bioink were evaluated using PresoBlue Cell Viability Reagent. Cells were counted using Countess automated cell counter (Invitrogen). To evaluate the growth curve of GB cells (GL261, T98G, U-87MG, U373, PD-GB1, and PD-GB4), the initial concentration was 1 × 106 cells/ml in DMEM. To evaluate the growth curve of human primary cells, the initial cell concentrations were 1 × 105 cells/ml up to 2 × 106 cells/ml in astrocytes and microglia medium, respectively. To evaluate the growth curve of cells grown in coculture, GB cells (PD-GB4, GL261, PD-GB1, and U-87MG-D) and astrocytes (hAstro or mAstro) were seeded at the final concentration of 2 × 106 cells/ml at a ratio of 1:1 and 1:2 and grown in mixed medium (1:1 ratio) of DMEM and astrocytes medium. To evaluate the growth curve of fast-growing and dormant GB cell types (T98G and U-87MG) in fibrin 3D-bioink, cells were seeded at the concentration of 1 × 106 cells/ml, while osteosarcoma cells (Saos-2) were seeded at the concentration of 1 × 107 cells/ml in DMEM.
The 3D-bioink samples were placed in 500 μl of PrestoBlue in a 24-well plate and incubated at 37°C for 30 min or up to 4 hours (depending on the cell type) for growth curve experiments. We then took three samples of 100 μl from each fibrin 3D-bioink sample to a 96-well plate and measured the fluorescence by excitation of 545 nm, auto emission cutoff (590 nm), and emission of 600 nm. Measurements were averaged to each sample, a control measurement of a clear fibrin 3D-bioink was subscribed, and the final result was normalized to the values obtained at the first day of incubation. The fibrin 3D-bioink samples were washed with PBS for 15 min at 37°C and then incubated in fresh cell medium. Each group contained four to six samples, and the experiments were repeated in three independent times.
Generation of fluorescently labeled cell lines
Cells were labeled with pQC-mCherry retroviral particles as previously described (25). Briefly, GL261, U-87MG, T98G, PD-GB3, and HUVEC were labeled with mCherry. Cells were selected for stable expression 48 hours following infection by puromycin (2 to 3 μg/ml; TOKU-E, Singapore). hAstro and hMG cells, patient-derived cells (PD-GB1), and Saos-2 were labeled with green fluorescent protein (GFP). GFP-positive cells were then selected using G418 (0.5 mg/ml for Saos-2 and 2500 μg/ml for hAstro and hMG; TOKU-E, Singapore) for 2 weeks. Human pericytes and patient-derived cells (PD-GB4) were labeled with iRFP, and positive cells were then selected using hygromycin (2.5 mg/ml for PD-GB4 and 250 μg/ml for human pericytes; Sigma-Aldrich) for 2 weeks. PD-GB4 were also labeled with Azurite, and positive cells were selected using hygromycin (250 μg/ml) for 2 weeks. All retroviral infected cells showed more than 90% positive fluorescent labeling.
Imaging and image analysis
To evaluate the viability of the labeled cells in the cell-laden bioink, fibrin 3D-bioink samples were prepared on a 35-mm petri dish with a bottom grid glass (ibidi, Gräfelfing, Germany). Medium was replaced every other day. Fluorescent images were obtained using SP8 confocal microscope (Leica, Wetzlar, Germany) or with EVOS FL Auto cell imaging system (Thermo Fisher Scientific). 3D projections and Z-stacks were generated using manual and automated processes in Leica and Imaris (Imaris 8.4.1, Bitplane Scientific Software). Each group contained three samples, and the experiments were repeated in three independent times.
Frozen OCT fibrin 3D-bioink samples fixation
Fibrin 3D-bioink samples (day 7 or 14 after cell seeding) were incubated with 4% paraformaldehyde for 4 hours, followed by 0.5 M sucrose (BioLab) for 1 hour, and 1 M sucrose overnight. The samples were then embedded in optimal cutting temperature (OCT) compound (Scigen) on dry ice and stored at −80°C.
Immunostaining
Fibrin 3D-bioink samples embedded in OCT were cryosectioned into 5-μm-thick sections by a cryostat (Leica). Sections were stained by hematoxylin and eosin (H&E) and for the relevant markers using antibodies. Immunostaining was performed using BOND RX autostainer (Leica). Briefly, slides were incubated with goat serum (10% goat serum in PBS + 0.02% Tween 20 + 0.02% gelatin) for 30 min to block nonspecific binding sites. Slides were then added with rabbit anti-mouse/human GFAP, rabbit anti-mouse IBA1, and mouse anti-human P-selectin (1:20 dilution; R&D Systems). Following 1 hour of incubation, slides were incubated with the following secondary antibodies for an additional 1 hour: goat anti-rabbit Alexa Fluor 488 for GFAP and IBA1 and goat anti-mouse Alexa-594 for human P-selectin and muse P-selectin. They were then washed and treated with ProLong Gold mounting with DAPI or Hoechst 33342 before being covered with coverslips. Images were recorded using the EVOS FL Auto cell imaging system.
Invasion in 3D-bioink
We established a modified invasion assay in which a fibrin 3D GB tumor core is surrounded by brain-mimicking fibrin 3D-bioink tissue, allowing monitoring of tumor cell invasion into the outer core (fig. S2C). PD-GB4 iRFP-labeled were casted into 96-well glass-bottom plates (Cellvis, CA, USA) in a core tumor at a density of 1 × 107 cells/ml and surrounded with unlabeled hAstro (1 × 106 cells/ml). Experiments were done in a medium mixture at 1:1:1 ratio of DMEM, astrocyte medium, and microglia medium. For invasion estimation of dormant versus fast-growing cell types, mCherry-labeled U-87MG and T98-G human GB cells (2 × 106 cells/ml) were plated in glass-bottom 96-well plates in the core of fibrin 3D-bioink. The surrounding fibrin 3D-bioink was an empty, clear fibrin 3D-bioink. Experiments were done in a medium mixture at 1:1:1:1:1 ratio of DMEM, astrocyte medium, microglia medium, pericyte medium, and Endo-Gro medium. Cell invasion was monitored for 2 to 5 weeks using EVOS FL Auto cell imaging system and quantified by ImageJ software, presented here as relative light units (RLUs). Cell invasion was normalized by comparing the size of the cell core at day 1 (polygon drawing shape) up to day 14. Each group contained 6 to 12 samples, and the experiments were repeated in three independent times. For the evaluation of proliferation in the presence of P-selectin inhibitor (SELPi), mCherry-labeled U-87MG-F and T98G-F cells generating fast-growing tumors or iRFP-labeled PD-GB4 cells (4 × 106 cells/ml) were seeded in a glass-bottom 96-well plate in the core of fibrin 3D-bioink. The surrounding fibrin 3D-bioink was an empty, clear fibrin 3D-bioink. Cells were then treated with 0.5 μM KF38789, which is a selective inhibitor of P-selectin (SELPi) that we chose to serve as a small-molecule drug. It selectively inhibits P-selectin binding in lymphocytes at the half-maximal inhibitory concentrations (IC50) of 2 μM. P-selectin functions as a cell adhesion molecule on the surfaces of cells, which binds carbohydrates (18). It is also involved in tumor cell binding and promotes their invasion by supporting a permissive metastatic microenvironment, and P-selectin is highly expressed in GB (28). Control groups were treated with dimethyl sulfoxide (DMSO) vehicle at the corresponding volume percentage [0.2% (v/v)], prepared in a medium mixture at 1:1:1:1:1 ratio of DMEM, astrocyte medium, microglia medium, pericyte medium, and Endo-Gro medium. Proliferation was quantified during time with ImageJ software by RLU. Each group contained 6 to 12 samples, and the experiments were repeated in three independent times.
Scanning electron microscope (SEM)
For SEM analysis, acellular and cell-laden fibrin 3D-bioink sample were prepared and incubated for 14 days at 37°C and 5% CO2. Cell-laden samples were prepared using PD-GB4 cells (1 × 107 cell/ml) alone or in coculture with hAstro (1 × 107 cell/ml) and hMG (1 × 106 cell/ml). Fibrin 3D-bioink samples were then removed from the culture medium after 14 days, washed in PBS, and fixed with 2.5% (v/v) glutaraldehyde (Sigma-Aldrich) in PBS overnight at 4°C. The samples were snap-frozen with liquid nitrogen and lyophilized. The same procedure was done for both healthy and tumorous (GL261 intracranially injected cells, 15 to 20 days after implantation) brain hemispheres of C57BL/6 mice. Samples were stored under vacuum in a desiccator before sputter-coating with gold (~ 20 nm). SEM images were taken using a Quanta 200 FEG Environmental SEM (FEI, OR, USA) at high vacuum and 10.0 kV. Lengths and pore size were measured using ImageJ software.
Preparation of the vascular bioink
Pluronic F127 is a biologically inert triblock copolymer based on polyethylene oxide and polypropylene oxide, which exhibits reversible thermal gelation and a lower critical solution temperature behavior. At lower temperatures, Pluronic F127 solution is liquid, and upon heating to room temperature, it transforms into a gel. Pluronic F127 [40% (w/v)] was dissolved in double-distilled water using an overhead mechanical stirrer at 4°C. The solution was stored at 4°C. Before use, Th solution (2000 U/ml) was added to create the fugitive vascular bioink of final 38% (w/v) Pluronic F127 and Th (100 U/ml). The vascular bioink was loaded into a 30-ml syringe at 4°C and centrifuged to remove air bubbles.
Fabrication of 3D-bioprinted vascularized tumor model
Printing formulation of fibrin 3D-bioink [composed of 1% (w/v) fibrinogen and 6% (w/v) gelatin] was mixed with PD-GB4 cells labeled with Azurite (1 × 106 cells/ml), hAstro labeled with GFP (1 × 106 cells/ml), and hMG cells (1 × 105 cells/ml). The mixture forming the GB-bioink was loaded in a syringe (Nordson, CA, USA) and tapered with a needle tip (Nordson) of varying size attached via a luer-lock. The GB-bioink was cooled to 4°C for 15 min and then loaded into the 3D printer’s cartridge. Temperature was set to 24°C and held for 15 min before 3D-bioprinting initiation. 3D structures were printed using a 3D-Bioplotter (Manufacturer Series, EnvisionTEC, Gladbeck, Germany) equipped with five independent ink cartridges. Before each 3D-bioprinting, the nozzles were calibrated to determine their respective X, Y, and Z offsets. Following calibration, six to eight layers were printed on a thin coverslip, framed by a polydimethylsiloxane (PDMS) (Sylgard 184, Dow, MI, USA) gasket, creating the bottom platform onto which vasculature structure could be printed. Cell-laden bioinks were printed for a time period up to 2 hours to prevent cell death. The printed layers were left to dry for up to 1 hour. Casting formulation of fibrin 3D-bioink [composed of 1% (w/v) fibrinogen and 6% (w/v) or 12% (w/v) gelatin] was mixed with Azurite-labeled PD-GB4 cells (1 × 106 cells/ml), GFP-labeled hAstro (1 × 106 cells/ml), and hMG (1 × 105 cells/ml). The mixture forming the GB-bioink was casted on a thin coverslip, framed by a PDMS gasket, creating the bottom platform onto which vasculature structure could be printed. Cell-laden fibrin 3D-bioink constructs were left to fully cross-link for up to 2 hours. Then, the vascular bioink composed of Pluronic F127 38% (w/v) and Th (100 U/ml) was loaded onto a printer cartridge (Nordson) at 4°C, warmed to 29°C, and printed using a 0.25-mm needle tip attached via a luer-lock. 3D structures were printed with the vascular bioink according to a bioengineered design of vasculature, created with Rhino 6 3D modeling software (Rhinoceros). Immediately after the vascular bioink 3D-bioprinting was completed, connectors (Darwin Microfluidics, Paris, France) were inserted to the inlet and outlet positions of the PDMS gasket to allow future flow by peristaltic pump, and a fresh formulation of the fibrin 3D-bioink with PD-GB4 and microenvironment cells was casted, covering the printed vasculature and filling the PDMS frame completely. The sample was then covered with an additional coverslip glass and sealed in a metal frame (creating the perfusable chip), self-designed for optimal noninvasive monitoring of fluorescent signals (fig. S3A), and incubated at 37°C for a minimum of 3 hours until complete cross-linking is achieved, reaching the physiological stiffness of the brain. To liquefy the fugitive Pluronic F127 bioink, the sample was cooled to 4°C. Cold PBS was then injected into the mold’s inlet by applying positive pressure while applying negative pressure through the outlet, leaving the model with the desired 3D-bioprinted lumens. Following Pluronic F127 wash, fibronectin (100 μg/ml) was injected into the lumens’ inlet and incubated in rotation for 3 hours at 37°C to prime the vasculature wall and create an adherable interface. Next, a mixture of mCherry-labeled HUVEC (8 × 106 cells/ml) and iRFP-labeled human microvascular brain pericytes (2 × 106 cells/ml) (named vascular bioink) at 4:1 ratio was injected into the vessel and incubated for 2 hours. Cell seeding steps were repeated four times, and each time sample incubation positioning was rotated by 90° to maximize cell attachment area. The model was then incubated in rotation overnight at 37°C to allow cell attachment to the lumens’ walls with a full coverage of the lumen. The next day, the sample was connected to a peristaltic pump (EBERS, Zaragoza, Spain), incubated at 37°C, and perfused with medium mixture of all the cells in the samples at a 1:1:1:1:1 ratio (DMEM, astrocyte medium, microglia medium, pericyte medium, and EGM-2) for 5 days. After confluent cover was validated by confocal imaging, 70-kDa dextran-FITC (1 and 0.1 mg/ml) was perfused through the vasculature using a syringe pump (Braintree Scientific, Braintree, MA, USA) at a flow rate of 25 μl/min. Dextran-FITC flow-through was imaged by EVOS FL Auto cell imaging system at 20-s intervals. 3D-printed GB models used for dextran-FITC perfusion were created with unlabeled cancer and stromal cells (hAstro and hMG).
Creating the metastatic model
We have created a simplified vascularized 3D model of breast cancer brain metastasis using a 23-gauge needle inside our perfusion chip. Brain metastatic human breast cancer cells MDA-MB-231, astrocytes, and microglia were mixed with the fibrin 3D-bioink under similar conditions as the GB microenvironment according to our calibrations (cell type ratio was 1:1:0.1, respectively). Moreover, functional blood vessels were prepared using human endothelial cells and human microvascular brain pericytes in the same ratio of 4:1 following the same cross-linking and seeding protocol.
2D proliferation assays
For growth curve evaluation, U-87MG and T98G human GB cells generating dormant or fast-growing tumors (25,000 cells per well) (29) were seeded onto 24-well plates in complete DMEM and incubated for 72 hours (37°C; 5% CO2). For the comparison of Saos-2 cell type growth curves in 2D culture, 10,000 cells per well were cultured in DMEM following a daily incubation with PrestoBlue reagent for 4 hours. For proliferation evaluation in the presence of SELPi, U-87MG and T98G human GB cells generating dormant and fast-growing tumors or PD-GB4 cells (25,000 cells per well) were plated onto a 24-well plate in complete DMEM and incubated for 24 hours (37°C; 5% CO2) to allow cell attachment. Cells were then treated with either 0.5 μM SELPi or with DMSO vehicle at corresponding percent (v/v) of 0.2% (v/v), prepared in medium mixture at 1:1:1:1:1 ratio of DMEM, astrocyte medium, microglia medium, pericyte medium, and Endo-Gro medium, for 30 min. All wells were washed twice with PBS and incubated further with fresh medium mixture. Cell proliferation was monitored by IncuCyte Zoom Live cell analysis system (Essen BioScience), while the first images were taken right before the treatment. Each group was assayed in triplicates, and the experiment was repeated in three independent times. Proliferation of cells was normalized to time 0 and presented as percent (%) of cell confluence.
Wound healing assay
U-87MG and T98G human GB cells (7.5 × 104 cells per well) were plated onto a 96-well plate (ImageLock, Essen BioScience, Hertfordshire, UK) in DMEM medium. A scratch was performed using a WoundMaker, followed by two washes with PBS. Cells were then treated with either 0.5 μM SELPi or with DMSO vehicle at corresponding percent (v/v). Wound density percent was monitored and quantified by Incucyte ZOOM (Essen BioScience). Each treatment was assayed in triplicates, and the experiment was repeated in three independent times.
Animal models
For gene expression analysis, mCherry-labeled murine GL261 GB cells (5 × 104) were stereotactically implanted into the striatum of 7-week-old male C57BL/6 immunocompetent mice (n = 5, Envigo CRS), with prior anesthesia using ketamine (150 mg/kg) and xylazine (12 mg/kg) injected intraperitoneally. Mice were euthanized on day 14 after injection, and the brains were resected for further evaluation of gene expression. For the establishment of human GB dormancy model, T98-G cells (1 × 106) were injected subcutaneously into the flank of 6- to 8-week-old male severe combined immunodeficient (SCID) mice (Envigo CRS). Tumor volume was monitored by caliper measurement (width2 × length × 0.52). Body weight was monitored q.o.d.
Flow cytometry for P-selectin expression
For 2D culture samples, GB cells (1 × 106 cells per 10-cm2 petri) were grown in DMEM medium for 24 hours, then scraped with 1 ml of PBS, and centrifuged for 5 min at 4°C and 2000 rpm. The pellet was resuspended in FACS buffer (PBS supplemented with 0.5% bovine serum albumin and 0.5 mM EDTA; Sigma-Aldrich), and cells were counted using Countess automated cell counter (Invitrogen). For 3D-bioink samples, GB cells (1 × 106 cells/ml) were grown in fibrin 3D-bioink for 10 days in DMEM medium. Then, the fibrin 3D-bioink was digested with collagenase II (600 U/ml, 1.5 hours) and filtered through a 70-μm mesh to avoid undigested polymeric clots or particles. The suspension was centrifuged for 5 min at 4°C and 2000 rpm, and supernatant was discarded. Murine GL261 cells were incubated with FITC rat anti-mouse anti–P-selectin or FITC rat IgG1 isotype control for 1 hour on ice at a concentration of 3 μl per 1 × 106 cells. After incubation, FACS buffer was added to the pellet, and cells were washed twice by centrifugation for 5 min at 4°C and 2000 rpm. Human cells (PG-GB4, T98G, and U-87MG) were first incubated with nonlabeled primary antibody (mouse anti-human E-selectin/P-selectin or mouse IgG1 I.C.) for 1 hour on ice. Then, FACS buffer was added to the pellet, and cells were washed by centrifugation for 5 min at 4°C and 2000 rpm. Cells were then incubated with mouse-IgG ƙ BP-CFL 488 secondary for 1 hour on ice, followed by two washes as described above. Unstained sample and the corresponding isotype control were used as internal control for unspecific antibodies epitope binding. Fluorescent intensity was evaluated using Attune NxT Acoustic Focusing flow cytometer (Thermo Fisher Scientific) and analyzed by the Kaluza 1.3 software (Beckman Coulter). Each group contained four to eight bioink samples that were pooled after the bioink digestion, and the experiments were repeated in three independent times. For 3D-bioink samples containing PD-GB(1-4) cells, the same procedure was used with 1 × 107 cells/ml (fig. S5A).
RNA sequencing
To evaluate changes in gene expression in our fibrin 3D-bioink compared to 2D culture and in vivo models, we performed RNA-seq analysis. For 2D culture and 3D-bioink models, a murine penta-culture composed of GL261 mCherry-labeled and freshly isolated murine primary astrocytes, microglia, pericytes, and endothelial cells was grown on a 2D petri dish, or in fibrin 3D-bioink, GL261 cells were seeded at the concentration of 1 × 106 cells/ml (in fibrin 3D-bioink or in 2D culture) at the ratio of 1:1:2:0.1:0.1, creating the penta-culture. For the in vivo GB model, mCherry-labeled GL261 tumor cells were sorted from tumor-bearing mice at day 14 after stereotactic implantation. 3D-bioink was cut into 1- to 2-mm fragments, digested with collagenase II (600 U/ml) for 3 hours, and filtered with a 70-μm mesh before RNA extraction. Total RNA was then isolated from all samples using the EZ-RNA II Total RNA Isolation Kit (Biological Industries, CA, USA), following the manufacturer’s protocol. RNA quality was evaluated by TapeStation RNA Assay (Agilent Technologies). Libraries were constructed with the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina [#E7760, New England Biolabs (NEB)] using the manufacturer’s instructions. Twenty-five to 75 ng of RNA were amplified using 14 cycles of polymerase chain reaction. Final quality was evaluated by TapeStation DNA HS Assay (Agilent Technologies). Equimolar pooling of libraries was performed on the basis of Qubit values and loaded (in triplicates) onto an Illumina NextSeq 500 platform (Illumina, CA, USA) to evaluate changes in gene expression between the tested groups.
RNA-seq analysis
Data analysis was aligned to mice genome. RNA-seq reads were aligned using Kallisto (30) to mouse genome version mm10, and expression levels were calculated using RSEM (31), followed by further processing using the Bioconductor package DESeq2 1.24.0 in R (32). The data were normalized using Trimmed mean of M values (TMM) normalization, and differentially expressed genes were defined using the differential expression pipeline on the raw counts with a single call to the function DESeq [false discovery rate (FDR)–adjusted P < 0.05]. For global transcriptome quantification and comparison between the samples (2D culture, fibrin 3D-bioink, and in vivo), we performed principal components analysis (PCA) and calculated the Euclidean distance between each pair of transcriptional profiles.
Temozolomide resistance evaluation in 2D versus 3D
The resistance of patient-derived cells to temozolomide (TMZ; LIXIN, China) was assessed in fibrin 3D-bioink and in 2D culture. Patient-derived cells were seeded in fibrin 3D-bioink at a density of 1 × 107 cells/ml to match resemble a very dense GB tissue in different TMZ concentrations (2000 to −0.01 μM) for 7 days. For the 2D culture TMZ resistance evaluation, we seeded 20,000 to 40,000 cells per well in a 24-well plate in fresh DMEM. We replaced the media with different TMZ concentrations (10 to −0.01 μM) for 3 days. Then, the 3D-bioink samples were placed in 500 μl of PrestoBlue in a 24-well plate and incubated at 37°C for 3 hours.
Statistical methods
Data are expressed as means ± standard error of the mean. Statistical significance was determined using an unpaired two-sided Student’s t test when comparing between two groups and analysis of multiple comparisons [analysis of variance (ANOVA) test] when comparing more than two groups. P < 0.05 was considered statistically significant. All experiments were performed in three biological replicates with cells from different batches to ensure reliable and repetitive results. Statistical analysis was performed using GraphPad Prism 8.
RESULTS
Fibrin 3D-bioink formulation and characterization
We created a polymeric bioink based on fibrinogen and gelatin, which can provide a supporting 3D structure for GB cells and their microenvironment. First, we evaluated the effect of the addition of the cross-linker, Th, on the gelation time of a mixture of gelatin, fibrinogen, and TG [6% (w/v), 1% (w/v), and 3% (w/v), respectively] at different Th concentrations (0, 0.5, and 1 U/ml) via time sweep rheological test. Th induced rapid cross-linking and reached higher elastic modulus (G′) in a dose-dependent manner until complete cross-linking (Fig. 1A). On the contrary, a mixture of fibrinogen with TG alone and serial gelatin concentrations, without Th, was not sufficient to create a matrix with the appropriate tissue-like stiffness (fig. S1A). Furthermore, we have used a lower Th concentration (0.5 U/ml) to ensure homogeneous mixing of the 3D-bioink before casting. Moreover, Young’s modulus (E) of the created fibrin 3D-bioink was evaluated by calculating and comparing the slope of a stress-strain curve, obtained by performing a compression test of several 3D-bioink formulations at constant fibrinogen [1% (w/v)], Th (0.5 U/ml), and TG [3% (w/v)] and gelatin at serial concentrations [3% (w/v), 6% (w/v), and 12% (w/v)]. We found that the stiffness can be controlled by varying the final concentration of the gelatin to generate the desired mechanical property of GB tumor that is compatible with cell growth. No significant differences were measured at the same gelatin concentration; however, significant differences were evaluated at different gelatin concentrations [3% (w/v) versus 6% (w/v), P = 0.0037; 6% (w/v) versus 12% (w/v), P = 0.0003; 3% (w/v) versus 12% (w/v), P < 0.0001 by two-way ANOVA Tukey’s multiple comparisons test]. Young’s modulus of 24.5 ± 2.6 kPa was achieved when the final gelatin concentration was 6% (w/v) (Fig. 1B). Our results are in agreement with a previous study conducted on U-87MG xenografts, reporting a tumor Young’s modulus of 26.6 kPa (33). The Young’s modulus of the cell containing formulation (PD-GB cells PD-GB4 or murine GB cell type GL261) remained constant for 28 days. Another important physicochemical parameter of a bioink is its swelling behavior, which determines the accuracy of the 3D-bioprinted final structure. Furthermore, we performed a kinetic swelling study in the absence or presence of murine GB cells at different gelatin concentrations. No significant differences were measured between 3% (w/v) and 6% (w/v) gelatin concentration; a significant difference was evaluated between 3% (w/v) and 6% (w/v) gelatin concentration to 12% (w/v) (P = 0.0014 and P = 0.0011, respectively, by one-way ANOVA Tukey’s multiple comparisons test). Fibrin 3D-bioink samples containing 3% and 6% (w/v) gelatin reached an equilibrium state at around 5% swelling after 28 days, while samples containing 12% (w/v) gelatin reached an equilibrium state at around 20% swelling, indicating the ability of our bioink to retain its deposited dimensions (Fig. 1C). These findings are in agreement with previous reports (34). Next, we set to calibrate the optimal seeding density of different GB models (GL261, human T98G, U373, and U-87MG, as well as PD-GB4) in our fibrin 3D-bioink at 6% (w/v) gelatin. We found that the initial optimal cell seeding density is 1 × 106 cells/ml to achieve rapid growth rates. Cells from different origins (murine, human, and patient-derived) showed different growth rates in our fibrin 3D-bioink (fig. S1B). Next, we set to evaluate the proliferation rate of GL261 and PD-GB4 at different gelatin concentrations at a seeding density of 1 × 106 cells/ml. Cells at 6% (w/v) gelatin showed the highest proliferation rates in our fibrin 3D-bioink (Fig. 1D). Therefore, we decided to continue with further investigations using the fibrin 3D-bioink at the final concentrations of 1% (w/v) fibrinogen, 6% (w/v) gelatin, 3% (w/v) TG, and Th (0.5 U/ml) (unless stated otherwise). Diverse cell morphology, as demonstrated by fluorescence imaging of live and fixed fibrin 3D-bioink samples as well as by H&E staining (Fig. 1E and fig. S1C), indicated that GB cells in our 3D-bioink may interact differently with the bioink, creating cell-matrix interactions that are unique to each cell type.
Fig. 1. Bio-mechanical characterization determined the bioink composition that best mimics the elasticity and composition of the brain tissue.
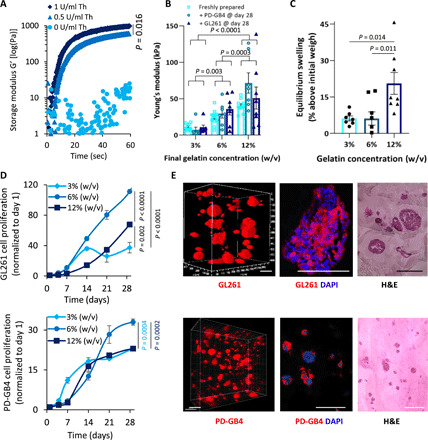
(A) G′ of fibrin 3D-bioink formation at different Th concentrations with 3% (w/v) TG at 37°C (average shown of n = 3 per group). (B) Young’s modulus of fibrin 3D-bioink at different concentrations of gelatin [3%, 6%, and 12% (w/v)] with 3% (w/v) TG and Th (0. 5 U/ml) as a clear bioink and as a cell-laden bioink composed of PD-GB4 or GL261 at 1 × 106 cells/ml (n = 8 to 13 per group). (C) Swelling at equilibrium of fibrin 3D-bioink at different concentrations of gelatin [3%, 6%, and 12% (w/v)] with 3% (w/v) TG and Th (0.5 U/ml) (n = 8 per group). (D) Growth curves at different concentrations of gelatin [3%, 6%, and 12% (w/v)] with 3% (w/v) TG and 0. 5 U/ml of Th of GL261 [top: 3% (w/v) versus 6% (w/v), P < 0.00001; 6% (w/v) versus 12% (w/v), P < 0.00001; 3% (w/v) versus 12% (w/v), P = 0.002 by t test] and PD-GB4 [bottom: 3% (w/v) versus 6% (w/v), P = 0.0004; 6% (w/v) versus 12% (w/v), P = 0.0004, by t test] in fibrin 3D-bioink (n = 4 per group). (E) Representative images demonstrating the morphology of mCherry-labeled GL261 (top) and iRFP-labeled PD-GB4 cells (bottom) following 14 days in fibrin 3D-bioink [6% (w/v) gelatin, 3% (w/v) TG, and Th (0.5 U/ml)]. Cells were analyzed by live confocal Z-stack imaging of the whole bioink and by fluorescence imaging and H&E staining of bioink sections (n = 3 to 4 per group). Scale bars, 100 μm.
Fibrin 3D-bioink is suitable for in vitro and ex vivo assays
The TME is critical for a proper evaluation of cancer cell growth and response to drugs. Therefore, we set to recapitulate the complexity of the brain microenvironment by the addition of stromal cells in our fibrin GB 3D-bioink. We evaluated the growth of two of the main cellular components of the brain TME, astrocytes and microglia, at different cell densities according to their abundance in human brain samples; 20 to 60% of the total number of glial cells in the human brain are astrocytes (35). The growth rate of hAstro positively correlated with the number of cells seeded in the 3D-bioink. On the contrary, microglia cell viability was enhanced when primary hMG cells were seeded in low amount (fig. S2A). These results showed that our fibrin 3D-bioink can support the growth of the brain microenvironment cells over time. Next, we cocultured PD-GB4 cells together with hAstro and hMG using the fibrin 3D-bioink. We observed the expression of hAstro and hMG activation markers (GFAP and IBA1, respectively) in sections of our fibrin 3D-bioink 7 days after cell seeding (Fig. 2A and fig. S2B). Activation of astrocytes and microglia was similarly detected in ex vivo analysis comparing the stromal activation of murine and human GB tumors versus healthy tissue (36, 37). This suggests that cells grown in our 3D system have similar biological properties as observed in human GB samples and in mouse models. The addition of hAstro enhanced the growth rate of patient-derived PD-GB4 cells, suggesting that hAstro secrete factors that enhance cancer cell proliferation (Fig. 2B). Similar increase in GB cell growth was observed using three additional GB models: GL261, PD-GB1, and U-87MG (fig. S2D). In addition, we found that the invasion ability of PD-GB4 was enhanced by 50% toward the outer 3D-bioink containing hAstro compared to naïve surrounding bioink (Fig. 2C). We then evaluated the structure of our fibrin 3D-bioink using scanning electron microscopy in comparison to healthy and tumor-bearing mice brain tissues from C57BL/6 mice. Acellular fibrin 3D-bioink and healthy hemisphere show smaller pores compared to the larger pores observed in the cell-laden fibrin 3D-bioink and tumor-containing hemisphere. Nonhomogeneous structure and an average pore size of 4.7 μm in diameter were measured in acellular fibrin 3D-bioink; therefore, small molecules such as oxygen, nutrients, and drug molecules can diffuse freely through the model. The pore size of the GB-bioink was not significantly increased when PD-GB4 cells were seeded alone or cocultured with brain stromal cells (hAstro and hMG) in the 3D-bioink (7.2 to 7.1 μm in diameter, respectively). The addition of activated and proliferating tumor and stromal cells alters the appearance of synthetic and natural ECM and significantly increases their pore size (P = 2 × 10−13, t test). Similarly, the pore size of the tumor-containing hemisphere was higher than the pore size in a healthy hemisphere (P = 0.0002, t test). In addition, the cell-laden fibrin 3D-bioink shows cellular filopodia toward the surrounding matrix, suggesting the presence of cell-cell and cell-ECM interactions (Fig. 2D and fig. S2E).
Fig. 2. 3D brain-mimicking bioink is biocompatible and promotes long-term cell viability of GB cells and brain stromal cells.
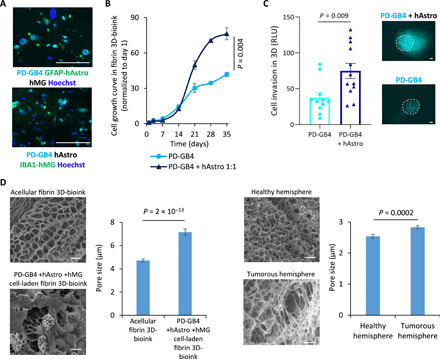
(A) Representative immunostaining images of GFAP (top, in green) or IBA1 (bottom, in green) in fibrin 3D-bioink seeded with GB and stromal cells cocultured for 7 days. Images depict Hoechst-stained nucleus (in blue), iRFP-labeled PD-GB cells (PD-GB4; in cyan). Scale bars, 100 μm. (B) Growth curves of PD-GB cells (PD-GB4), alone or cocultured with hAstro (1:1 ratio) in fibrin 3D-bioink (P = 0.004, t test; n = 4 per group). (C) The invasion of iRFP-labeled PD-GB4 cells from the inner core to the surrounding area in the absence or presence of hAstro (1 × 106 cells/ml) was evaluated by fluorescent microscopy imaging. The invasion was calculated as the total area density in outer bioink and quantified using ImageJ by RLU (P = 0.004, t test; n = 12 per group). Representative images of cell invasion are shown; dashed lines delineate the edge between the core and the surrounding tissue according to the images on day 1. Scale bars, 100 μm. (D) SEM images of acellular fibrin 3D-bioink (top left); cell-laden fibrin 3D-bioink composed of patient-derived PD-GB4 cells, hAstro, and hMG cells (bottom left); healthy hemisphere of a C57BL/6 mouse (top right); and GL261 tumor-containing hemisphere of the same mouse (bottom right). Scale bars, 10 μm. The pore size diameter of each group was quantified using ImageJ (n = 2 to 20 photos per group, n = 13 to 170 measurements in each photo) and showed that enlarged and diverse pore sizes characterized the cell-laden bioink and brain tissue bearing the GB tumor.
3D-printed vascularization
Functional vasculature is essential for the growth and function of the tumor cells and their surrounding TME, as it supplies oxygen and nutrients, and essential for the clearance of metabolic excrements (23). In the absence of perfusable vasculature, cells are unable to remain viable if they are far from a blood vessel beyond the oxygen diffusion distance of approximately 200 μm, leading to the development of necrotic regions. Therefore, we have engineered a functional vascular included in our GB-bioink as illustrated in Fig. 3A. We designed a pattern resembling vascular structure—including curves, branching, and anastomosis—using Rhino 3D modeling software. A GB-bioink was created by mixing the fibrin 3D-bioink with PD-GB4 cells, hAstro, and hMG. The vascular network was created using a fugitive bioink, composed of Pluronic F127 and Th, which was printed on top of the 3D printed/casted fibrin 3D-bioink according to the bioengineered design (Fig. 3B). After casting another layer of GB-bioink on top of the printed vasculature, the vascularized 3D-bioprinted model was sealed in our self-designed metal perfusion chip (Fig. 3C and fig. S3A). Following the evacuation of the vascular ink, a mixture of HUVEC and human pericytes was injected directly into the hollow channel and incubated in rotation to allow cell attachment homogeneously to the lumens’ walls. The resulting penta-culture, vascularized 3D-bioprinted GB model, was connected to a peristaltic pump to facilitate the formation of the vascular lumen layer under flow for 5 days (Fig. 3D and schematic illustration in fig. S3B). We emphasize the perfusion ability of our 3D-bioprinted vascular tumor model in movie S1 and fig. S3C. We monitored cell arrangement by fluorescence and confocal microscopy to evaluate the coverage of the hollow channels with endothelial cells and pericytes. We indeed confirmed the formation of the desired 3D-bioprinted lumens (Fig. 3E and movie S2). Similar 3D-bioprinting protocol was performed with a penta-culture mixture of cells using a bioink prepared at a final gelatin concentration of 6% with PD-GB4 cells (fig. S3D) or PD-GB3 cells (fig. S3E). To evaluate the functionality of the vascular network in our bioengineered GB model, we created the vascularized 3D-bioprinted model with the same composition of unlabeled cells in the GB-bioink to avoid overlapping of fluorescent signals. We perfused the vasculature with 70-kDa dextran-FITC solution using a syringe pump (fig. S3F) and monitored the flow of dextran-FITC by fluorescent microscopy. The use of the dextran FITC shows the potential to perfuse the sample through the vessels that are used as channels to transport nutrients and waste into and out of the system, respectively. These perfusable blood vessels offer the possibility to mimic the blood-brain barrier (BBB), allowing the evaluation of the integrity and functionality of the endothelial barrier in the presence or absence of cancer cells (Fig. 3F and movie S3).
Fig. 3. Fibrin brain-mimicking 3D-bioink integrated with 3D engineered printed perfusable vascular network.
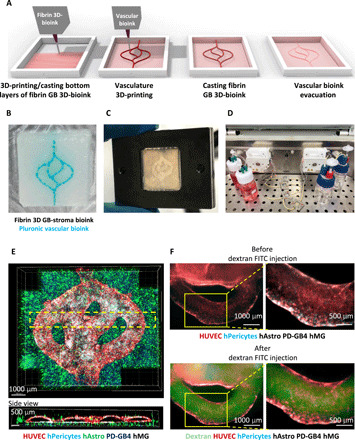
(A) Schematic illustration of the 3D-bioprinting model multistage process. (B) 3D-printed Pluronic-based vascular bioink (in cyan) on top of 3D-printed layers of fibrin 3D GB-stroma bioink (in white). (C) 3D-bioprinted vascularized GB model sealed in a metal frame showing the complete perfusion chip. (D) The vascularized 3D-bioprinted GB model is connected to a peristaltic pump through a tubing system, placed in a designated incubator. (E) Tiled Z-stack confocal microscopy images of the 3D-printed penta-culture vascularized GB model. Blood vessels are lined with iRFP-labeled hPericytes (in cyan) together with mCherry-labeled HUVEC (in red) (107 cells/ml; 4:1 ratio) and surrounded by azurite-labeled PD-GB4 (in blue), GFP-labeled hAstro (in green), and nonlabeled hMG (2.1 × 106 cells/ml; 1:1:0.1 ratio). The dashed box represents a coronal cross-sectional plane of the vessel. (F) Fluorescence microscopy images of the 3D-bioprinted vascularized GB model before (top) and after (bottom) perfusion of 70-kDa dextran-FITC. The 3D-bioprinted model is composed of a fluorescently labeled vascular network (mCherry-labeled HUVEC and iRFP-labeled hPericytes) surrounded by nonlabeled GB-bioink (hAstro, PD-GB4, and hMG).
3D GB model resembles the in vivo tumor settings better than 2D models
We have previously established and comprehensively characterized two pairs of GB cell types by generating dormant (“D”) and fast-growing (“F”) GB models (29, 38). The first pair was established from the human T98G GB cell line (T98G-D), which generates small, undetectable tumors following inoculation into mice that remain dormant for prolonged periods of time until they spontaneously escape and grow. From the “escaped” tumors, we isolated their “fast-growing” clone (T98G-F), which generates tumors rapidly (39). The second pair was established using the aggressive human U-87MG GB cell line (U-87MG-F), from which we isolated a dormant tumor-generating clone (U-87MG-D) out of many single-cell clones that we isolated. Despite the differences in their in vivo growth patterns, this pair of cells displays similar characteristics in vitro, showing similar growth rate kinetics when grown in vitro in 2D culture (Fig. 4, A and B) (29, 38). We therefore evaluated whether our fibrin 3D-bioink was able to mimic in vivo tumor dormancy or fast growing using our two models of dormant and fast-growing GB models. In accordance with our previous publications, inoculation of T98G-F cells into SCID mice resulted in tumor growth within 2 months, while T98G-D remained dormant for up to 5.5 months. Similar tumor growth patterns were observed when U-87MG-F and D were inoculated in SCID mice (Fig. 4B). However, when cultured in 2D monolayer, both pairs of dormant and fast-growing cells displayed similar growth rates and invasion capabilities, losing their growth characteristics and differences observed in vivo (Fig. 4, C and D). Cells grown in our fibrin 3D-bioink showed similar tumorigenic characteristics as in the in vivo settings. In particular, cell generating fast-growing tumors displayed a significantly higher (P = 0.01 for T98G and P = 0.00008 for U-87MG cell types, t test) proliferation rate compared to cells generating dormant tumors (Fig. 4E). Invasion (experimental setup in fig. S2C) of T98G-F cells was 100-fold higher than that of T98G-D in fibrin 3D-bioink on day 14 (P = 4 × 10−8, t test), whereas invasion of U-87MG-F cells was twofold higher than U-87MG-D in fibrin 3D-bioink at day 14 (P = 0.002, t test) (Fig. 4F) in accordance to the in vivo observations (29).
Fig. 4. Fibrin 3D-bioink reproduced the dormancy phenomenon of two GB human cell types, which thus far could only be observed in SCID mice and not in 2D culture.
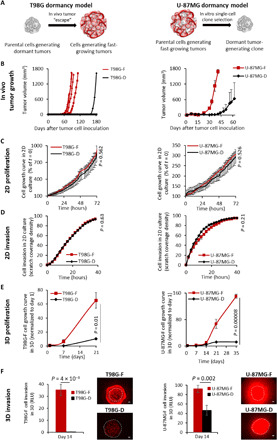
(A) Schematic illustration of T98-G and U-87MG human dormancy models. (B) In vivo growth kinetics of dormant (T98G-D and U-87MG-D) and fast-growing (T98G-F and U-87MG-F) cell types. n = 4 in the T98G-F group and n = 3 in the T98G-D group. Values for U-87MG growth in mice were averaged from data previously presented (29). (C) Cell growth evaluation of both GB pairs in 2D culture. n = 3 per group. (D) Cell invasion evaluation of both GB pairs in 2D culture. n = 3 per group. (E) Growth kinetics of both dormant and fast-growing cell types were evaluated in fibrin 3D-bioink. n = 4 per group. (F) Cell invasion ability in fibrin 3D-bioink was quantified using ImageJ. n = 12 per group. Representative fluorescent images of the invasion from the core tumor model to the surrounding area are presented. Scale bars, 100 μm. Dashed lines delineate the edge between the core and the surrounding tissue according to the images on day 1.
In addition, we have previously established and comprehensively characterized pairs of dormant (Saos-2-D) and fast-growing (Saos-2-E) human osteosarcoma models in mice. Similarly to our GB dormancy models, Saos-2-E fast-growing cells generate tumors within a month following inoculation into SCID mice, while Saos-2-D dormant cells remain dormant for up to 7 months (40). However, when cultured in 2D, both dormant and fast-growing tumor-generating cells display similar growth rates. We have evaluated the growth rate of these additional cell types in our fibrin 3D-bioink and found that their growth patterns resemble the in vivo setting (P = 0.0002, t test; fig. S4A). These results suggest that our fibrin 3D-bioink could serve as a reliable ex vivo model, preserving the in vivo phenotype of a given tumor.
Therapeutic effect of SELPi in fibrin 3D-bioink tumor model
We set to evaluate the differences in response to therapeutics in cells grown in 2D culture, in our fibrin 3D-bioink and in vivo. For this purpose, we used a commercially available P-selectin inhibitor. SELPi did not affect the proliferation of PD-GB4 seeded in 2D cultures. However, a significant inhibition of the PD-GB4 cell proliferation (P = 0.0001, t test) was observed when cells were grown in fibrin 3D-bioink. We have recently observed a similar inhibitory effect in vivo administering SELPi (16 mg/kg, i.v., q.o.d.) to GB-bearing mice (36). We speculated that these differences in response to treatment with SELPi are corresponding to the different expression levels of P-selectin in 2D culture versus 3D. P-selectin is an adhesion molecule that was previously demonstrated by our group to have a role in GB progression. We previously found elevated expression of P-selectin in GB 3D-spheroid models compared to 2D culture (28). Therefore, we evaluated P-selectin expression in GB cells grown in our GB fibrin 3D-bioink. Flow cytometry analysis demonstrated high P-selectin expression levels on PD-GB4 cells grown in fibrin 3D-bioink compared to 2D culture (Fig. 5A). Similar high expression of P-selectin in fibrin 3D-bioink compared to 2D culture was observed in additional four PD-GB cell types (fig. S5A). Immunostaining for P-selectin showed positive expression in fibrin 3D-bioink composed of PD-GB4 cells in the presence or absence of hAstro and hMG (fig. S5B) or in murine GB cells after 14 days in culture (fig. S5C). SELPi did not affect two additional human GB cell types in 2D culture. Significant inhibition of T98G cells (P = 0.00003, t test) and U-87MG (P = 4 × 10−6, t test) fast-growing cell types was obtained when cells were treated with SELPi in our fibrin 3D-bioink, which correlates to the higher expression levels of P-selectin in 3D-bionk versus 2D culture (Fig. 5, B and C).
Fig. 5. Treatment with SELPi resulted in a substantial reduction in GB cell proliferation in fibrin 3D-bioink compared to 2D culture.
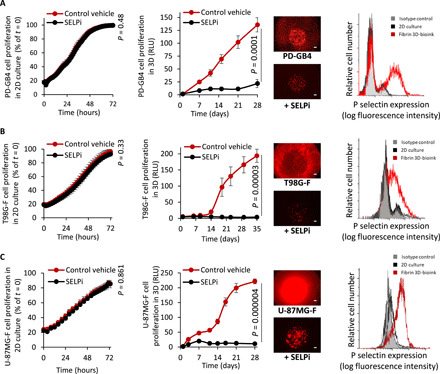
(A to C) Response of PD-GB4 (A), T98G-F (B), and U-87MG-F (C) to treatment with SELPi in 2D culture (left, n = 3 per group) and in 3D-bioink (center, n = 8/12 per group), representative images of labeled cells at the end of evaluation. Scale bars, 100 μm. Flow cytometry analysis of P-selectin expression of cells grown in 2D culture and in fibrin 3D-bioink (right, n = 3 per group).
Higher similarities in the transcriptional profile between in vivo and 3D compared to 2D models
To further understand the changes attributed to the differences between 2D and 3D cell cultures, we evaluated the transcriptome of GB cells grown in 2D, in our 3D-bioink or isolated from GB tumors in mice. We performed RNA-seq of GB tumors grown in mice and compared it to the penta-culture composed of GL261 murine GB cells, primary astrocytes, microglia, pericytes, and endothelial cells grown in 2D or in 3D-bioink (fig. S6A and Materials and Methods). To specifically compare between tumor cells in all conditions, we focused on genes that have been previously shown to be intrinsically expressed in gliomas (9511 genes of 17,810) (41). The PCA (Fig. 6A) shows that the transcriptional signature of cells from the three conditions differs. PC1, which contains the most variance (56%), indicates that cells grown in fibrin 3D-bioink are more similar to the signature of GB tumor cells isolated from GB tumor-bearing mice, as opposed to cells grown in 2D culture. To further investigate these differences, we calculated the Euclidian distance between the samples (Fig. 6B). This analysis quantitively shows that the cells grown in the fibrin 3D-bioink are indeed more similar to the cells isolated from mice brain than the 2D culture samples (P = 2.8 × 107; Fig. 6C). Analysis of differentially expressed genes between the 3D and the 2D culture of the 9511 glioma-associated genes identified 6936 differentially expressed genes. Gene set enrichment analysis shows several enriched pathways (according to their FDR q < 0.05) including proliferation, cell-cell interaction, adhesion, inflammatory response, angiogenesis, and several oncogenic markers (fig. S6B). Among all enriched pathways, we focused on GB-related pathways [angiogenesis hallmark, Janus kinase–signal transducer and activator of transcription (JAK-STAT) signaling pathway, interferon-γ (IFN-γ)–related response, vascular endothelial growth factor (VEGF) pathway up-regulation, epidermal growth factor receptor pathway up-regulation, and transforming growth factor–β signaling pathway] and evaluated the up-regulated genes in each examined group (Fig. 6D and fig. S6C). We observed similar gene expression signature in cells grown in fibrin 3D-bioink as observed in cells isolated from intracranial GB tumors, suggesting that our system recapitulates various genetic programs that are also activated in vivo. For example, RNA-seq analysis revealed activation of the JAK/STAT pathway, suggesting that our system preserved GB-stroma interactions (42) better than the 2D model. We have also found enhanced expression of the following genes both in fibrin 3D-bioink and iv vivo: JAK3 and SOCS7, which are known GB oncogenes (43), while Pik3cd was found to regulate GB migration (44), and IFNGR2 was found to be an immune modulator. Among the gene signature analyzed, we found a significant up-regulation in the gene expression of markers involved in macrophage recruitment (the chemoattractant CCL2 and ARG-1), angiogenesis (VEGFB and SPP1), matrix remodeling enzymes (MMP9), and genes related to endothelial cell junction molecules (PECAM1). We found that RNA isolated from cells grown in our 3D-bioink expressed high levels of GB markers (GFAP, CHIL1, and NTN1) and of MPZL3, CD79b, PTPRN, RQS14, TIMP4, and NDRG1, which were recently identified as potentially poor prognostic biomarkers of GB (45–47). These genes were also found to be highly expressed in the RNA isolated from the in vivo GB model.
Fig. 6. RNA-seq analysis shows higher similarities between the gene expression of GL261 grown in fibrin 3D-bioink and GL261 cells grown in mice compared to those grown in 2D culture.
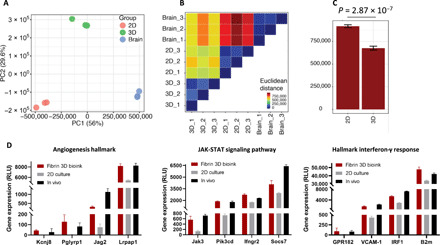
(A) PCA analysis showing gene expression profile derived from 2D culture, 3D-bioink, and GB tumors in mice in vivo (n = 3 per group). (B) Euclidian distance matrix between samples showing a closer distance between the in vivo samples and the fibrin 3D-bioink compared to increased distance between the in vivo samples and the 2D culture. (C) Summary comparison between the Euclidian distance of the 3D-bioink and the 2D culture to the in vivo samples (P = 2.8 × 107, t test). (D) A comparison of gene expression levels of enriched pathways, displaying similarly high levels of expression both in fibrin 3D-bioink and in vivo.
Clinical relevance
TMZ is an alkylating agent used as a standard-of-care chemotherapy for patients with GB according to the Stupp protocol (48). Therefore, we have evaluated the response of three patient-derived cells to TMZ when grown in 2D culture and in fibrin 3D-bioink (fig. S7). The IC50 values observed in 2D cultures were almost identical between the different PD-GB cells; however, each cell type exhibited a different IC50 value when grown in our 3D model. This correlates with the fact that each patient responds differently to the same therapy and emphasizes the need for this personalized approach. These patients showed a different response to TMZ, and each of them survived for a different period of time.
As another evidence for the robustness and the broad possible uses of our system, we have established a metastatic breast cancer model (fig. S8). We successfully created a vascularized 3D model of breast cancer brain metastasis, consisting of MDA-MB-231 human breast cancer cells, astrocytes, and microglia, as well as functional blood vessels prepared using human endothelial cells and pericytes. This is a proof of concept for the use of our 3D models for the study of brain metastasis in addition to GB primary models.
DISCUSSION
Nowadays, the discovery, development, and evaluation of therapies are performed using human cancer cells grown in 2D culture dish, which are frequently subjected to mechanical stresses and bounded to the rigid plastic, followed by in vivo testing in immunodeficient mice. These experimental settings constitute a fundamental hurdle in the translation of preclinical discoveries into clinical practice, with only 1 of 10,000 potential drugs successfully reaching the market (49). Therefore, there is an unmet need for alternative drug discovery and screening platforms that can accurately predict clinical response to therapies.
We report here a perfusable 3D-bioprinted tumor model based on biocompatible polymers containing tumor and stromal cells. Exploiting GB as a proof-of-concept model, we set to demonstrate the feasibility of our 3D-bioprinted model to recapitulate the TME. Cells within tissues are exposed to physical forces such as hydrostatic pressure, shear stress and tension, and compression forces. These forces, generated by cell-cell or cell-ECM interactions, play major roles in regulating cell and tissue behavior (50). Furthermore, desmoplastic stroma, which is present in many solid tumors, is usually substantially stiffer than normal tissues (51). Another vital parameter is the swelling equilibrium, which describes the water content within the bioink sample at equilibrium. It relies on the cross-linking density, hydrophilicity, the polymer’s structure at equilibrium, and degree of ionization of the functional groups in the polymeric network (52). To that end, we first formulated and characterized a brain-mimicking fibrin 3D-bioink. We optimized the fibrin 3D-bioink’s gelation time as a function of the concentrations and ratios of the gelatin and the cross-linking enzymes, Th and TG. Rheological and mechanical characterization confirmed that our fibrin 3D-bioink shares similar mechanical properties of GB brain tissue (33) and can be tailored to resemble other tissue types by altering the bioink composition. Furthermore, our fibrin 3D-bioink exhibited a moderate swelling degree of approximately 5 to 20%, a desirable aspect of the bioink characteristics, since low swelling degree (less than 40%) is important to accurately maintain the printed model dimensions and the desired optimal mechanical properties (53). The swelling at equilibrium of our fibrin 3D-bioink suggests that it has the potential for high water content, high porosity, and efficient nutrient diffusion, same as other hydrogels with similar mechanical properties (54). Moreover, our fibrin 3D-bioink demonstrated a long-term culture capability, as it supported GB cell viability for up to 5 weeks. Similar cell morphology was exhibited by live confocal Z-stack images and in histological H&E staining of GB slides from mice (55). Murine GL261 GB and PD-GB4 cells demonstrated round morphology when grown in our fibrin 3D-bioink, while human PD-GB1 and U-87MG cells exhibited elongated cell morphology. Wang et al. (56) attributed different cell morphologies to the properties of the matrix. Cell morphology in stiff matrix will often be round, while cells in soft matrix will be elongated. Since both cell types were seeded in the same fibrin 3D-bioink formulation, we hypothesize that the changes in cell morphology are attributed to the difference in cell origin. As GB consists of several molecular subtypes exhibiting different morphology and behavior (24), preserving the intrinsic properties of the different GB models is important when constructing drug screening systems As the brain TME plays a vital role in tumor progression (24), we incorporated into our bioink both astrocytes and microglia, which both have been shown to have a role in GB progression and constitute most of the glial cells in the brain (35). We found that fibrin 3D-bioink supports the viability of GB TME cells for up to 8 weeks after seeding. PD-GB cells proliferated and invaded more rapidly when cocultured with hAstro in fibrin 3D-bioink. This is in coordination with other studies, which showed that astrocytes facilitate GB aggressiveness in vivo (57). Furthermore, immunostaining showed that hAstro and hMG in the 3D-GB-bioink were in their activated state, expressing GFAP and IBA1, respectively. This is supported by other reports demonstrating that hAstro and hMG are activated in GB tumors but not in normal brain tissues (37).
To further recapitulate tumor architecture and cellular heterogeneity, we 3D-bioprinted GB-stroma bioink composed of GB cells, astrocytes, and microglia together with a 3D-printing vascular bioink, composed of the thermo-reversible Pluronic F127, to generate a vascular lumen coated with endothelial cells and pericytes (58), generating a 3D-bioprinted penta-culture. This 3D-bioprinted tumor model, sealed in our self-designed perfusion chip, was perfused through its hollow channels by a peristaltic pump through a tubing system over 5 days. Metabolized cell media was pumped through the outlet vessel. Moreover, our 3D perfusion chip allows long-term flow, imaging, and drug response assessment. To the best of our knowledge, this is the first report of a perfusable 3D-bioprinted engineered GB penta-culture. Some of the existing 3D-bioprinted tumor models lack endothelial cells or blood vessels (8, 9, 16, 17), or in case there are endothelial cells, they lack lumen and therefore are not perfusable (59). A recent study had demonstrated 3D-printed model with perfused vasculature (22); however, only cancer and HUVECs were used.
Our system allows incorporation of peripheral blood mononuclear cells, circulating tumor cells, and/or a variety of anticancer drugs that can be redirected either into a waste container after they perfuse through the 3D-bioprinted model or back toward the inlet vessel, generating a closed circulatory system. Hence, our perfusable 3D-printed model can be manufactured on demand to serve as a drug screening array for the evaluation of drug response, customized to each patient individually by including patient-derived tumor, stromal, and immune cells in the 3D-bioink. The 3D tumor model can be printed rapidly and robustly, allow testing of several drugs or their combinations simultaneously with such perfusion system.
We demonstrate here that our 3D-bioink can serve as an alternative to mouse models, as it is able to mimic key features of tumors grown in vivo. Using our previously established GB dormancy models, we performed a set of experiments evaluating tumor growth and invasion in our 3D-bioink compared to traditional 2D culture and in vivo models. We show that the dormancy state of human cell line, thus far only observed in SCID mice and not in 2D culture (29, 38), can be recapitulated in our 3D-bioink. Furthermore, we demonstrate that our 3D-bioink can be used for a more accurate evaluation of response to therapies compared to 2D culture methods. Inhibition of P-selectin, overexpressed in GB tumors (28), by SELPi did not have any effect on cells grown in 2D culture but resulted in a remarkable reduction in GB cell proliferation in our fibrin 3D-bioink. These differences can be attributed to the elevated expression levels of P-selectin by GB cells grown in fibrin 3D-bioink and in vivo (36), compared to cells grown in 2D cultures. These results correspond to the effect of SELPi in mice bearing intracranial GB tumors, recently reported by our group (36). While GB-bearing mice treated with SELPi exhibited smaller tumor volumes compared to those of the control group 1 month following treatment initiation, tumor growth inhibition in our fibrin 3D-bioink was observed in a shorter timeframe already after 1 to 2 weeks (depending on the cell source). Yi et al. (59) evaluated the resistance of their 3D-bioprinted human GB on a chip to chemotherapies such as cisplatin and TMZ in combination with radiotherapy. They observed higher IC50 values when treating their 3D model, which was based on decellularized porcine brain, than those of the 2D culture; however, they only cocultured patient GB cells with HUVECs (59). Similarly, Tang et al. (45) observed higher IC50 values when treating their hyaluronic acid–based 3D model, compared to 3D spheres; however, they have also used only a coculture of GB cells and HUVECs. Moreover, 3D models based on gelatin methacrylate and glycidyl methacrylate-hyaluronic acid, previously created by Tang et al. (46), displayed enhanced resistance to erlotinib, gefitinib, and TMZ treatment, in contrast to 3D spheres. Moreover, neither of those models can be perfused. Therefore, our insights on the fibrin 3D-bioink corroborate its ability to serve as a rapid predictive tool for in vivo drug response. A quick reply is crucial in GB because of its lethal nature; therefore, a rapid, robust, and reproducible 3D-printed ex vivo array of personalized GB models, as can be achieved using our platform, is extremely valuable. In addition, a common challenge with testing and targeting adhesion molecules is that they detach the cells in 2D culture (60). Using our platform, we can screen traditional chemotherapeutics, biological treatments, and immunotherapies as well as target adhesion molecules. These evaluations provide additional evidence that conventional 2D culture strategies relying on monolayers of cancer cells may not be sufficient to capture the complex TME.
Gene expression profiling showed that the transcriptional signature of murine GB cells grown in fibrin 3D-bioink is more similar to the cells grown orthotopically in mice than to cells grown in 2D culture. The analysis highlighted several genes that were similarly up-regulated in cells grown in 3D-bioink and in vivo compared to cells grown in 2D culture, among them several oncogenes and prognosis biomarkers for GB patient survival. For example, MMP9 is a key contributor to tumor growth and progression by degrading the basement membrane that further allows angiogenesis (61). Moreover, different genes involved in stroma-tumor cell crosstalk (IRF-1 and SEMA7A) were up-regulated both in fibrin 3D-bioink and in vivo but not in the 2D samples. Furthermore, higher expression of IRF-1 in RNA levels induces the cross-talk between astrocytes and GB cells (62). Moreover, IRF1 may be an important mediator of GB resistant to bevacizumab (63), while SEMA7A was described as a promigratory cue under physiological and pathological conditions. As SEMA7A expression was indeed found in invasive U-87MG GB cells (64), it may be one of the contributors to tumor cell invasive phenotype (65). In addition, vascular cell adhesion protein 1 (VCAM-1) expression promotes macrophage–tumor cell interaction and tumor cell invasion and indicates that VCAM-1 may be a potential molecular target for improving cancer therapy (66). Specifically, GPR182 is a marker for endothelial differentiation (67), and a recent reported silencing of GPR182 reduced the growth and metastasis of pancreatic cancer, without excessive harmful effects, which indicated that GPR182 might be a valuable therapeutic target (68). These genes can be the basis for novel therapies since many of them are associated with proliferation, survival, and invasion. We also show that expressed genes in our fibrin 3D-bioink provides essential cues to enhance tumor growth and survival; however, they are inefficiently expressed in standard 2D culture. Up-regulated gene expression levels in our system were also seen in similar GB 3D models (17, 45, 46); however, these models lack the whole complexity of GB TME (astrocytes, microglia, endothelial cells, and pericytes) together with the GB cells, as presented in this manuscript. This further emphasized that our 3D-bioink can serve not only as a drug development tool but also as an important basic, preclinical research tool.
Regarding the standard of care for GB, we have evaluated the response of three patient-derived cells to treatment with TMZ. The results showed different responses to TMZ between the different patient-derived cells when grown in our 3D models, while there were no differences observed in the response to TMZ when the cells were grown as 2D models. This demonstrates that our model reflects the broad spectrum of drug response seen in patients better than the traditional 2D models that resulted in similar responses for TMZ on different origins of GB cells from several patients. In addition, patient-derived cells grown in our fibrin 3D-bioink tumor models were more resistant to TMZ treatment with diverse IC50 values in the micromolar scale (1000, 1400, and 280 μM), compared to cells grown in 2D culture with an IC50 value in the nanomolar scale (all around 0.004 μM). These findings are in agreement with previous reports (8, 9), stating that 3D models are more resistant to chemotherapy due to lower diffusion rates, enzymatic cleavage, and other morphological and kinetic aspects reflecting better the clinical scenario. These evaluations provide additional evidence that conventional 2D culture strategies relying on 2D monolayer of cancer cells may not be sufficient to determine drug response and resistance.
In summary, using human and murine GB models, we show here that our perfusable 3D-bioprinted platform can serve as a reliable alternative preclinical tool. Our 3D-GB bioink was able to recapitulate fundamental aspects of in vivo GB models, including cell proliferation, invasion, response to therapies, and gene profiling. As we can control the mechanical properties of the 3D-bioink to emulate several other tissues, this platform can be easily modified to other cancer types beyond GB. As a proof of concept, we show here that our 3D-bioink also successfully recapitulated growth patterns of a pair of dormant and fast-growing osteosarcoma cell lines originating from the same parental tumor sample (40). In addition, this platform could be easily converted to a brain metastasis model, by replacing GB cells with other cancer cell types such as melanoma or breast cancer cells, which frequently metastasize to the brain (69). We performed comprehensive calibrations and optimizations for protocol standardization regarding the mechanical properties of the bioink, cell density, cell viability, and cell type–reflective 3D models of multiple cell types and origins (murine cell lines, human cell lines, and patient-derived samples). We also calibrated the ratio between the cancer cells and the brain microenvironment cells (astrocytes and microglia) to be 1:1:0.1. Moreover, the ratio between the pericytes and the endothelial cells was calibrated to 1:4 to form a confluent coverage of the 3D-bioprinted blood vessels. We believe that our thorough standardization protocol will serve as the starting parameters for a given patient sample. However, as each patient is unique, the parameters may require mild adjustments in a personalized manner (Fig. 7).
Fig. 7. Bridging the translational gap from bedside to bench and back.
Schematic illustration of the methodological approach using a perfusable microengineered vascular 3D-bioprinted tumor model for drug screening and target discovery. MRI, magnetic resonance imaging; μ-CT, micro–computed tomography.
Moreover, there is a need to develop reliable 3D multicellular BBB models for the assessment of drug delivery to the central nervous system. Recently developed BBB models have shown higher similarity to the in vivo scenario compared to traditional TransWell models. Some disadvantages of current BBB microfluidic systems include limited scalability and complex fabrication. Here, we show the ability of our 3D-bioprinted cancer model to include functional, intact blood vessels preventing 70-kDa dextran to extravasate into the outer gel. This shows that our model allows for proper arrangement and communication of pericytes and endothelial cells forming tight tumor vasculature. Thus, our model may be adjusted and used in the future for high-throughput drug screening with a special focus on transport through the 3D-bioprinted blood vessels consisting of using brain microvascular endothelial cells. These optimized models will provide invaluable insights to brain disorders. There is still a need for standardized quantification of parameters such as viability, expression of tight junction proteins, various molecular transporters, and efflux pumps to enable correlation studies (70). Today, most preclinical cancer drug screening studies use animal models. In GB, these models are particularly difficult to establish and monitor, as there is no “one model fits all” since each patient is unique and requires a complex cranial surgery followed by evaluation of drug efficacy by an endpoint histological analyses or via time-consuming noninvasive imaging techniques. Moreover, a high number of animals are required to achieve low-variance results, since this procedure can cause animal loss. Therefore, we postulate that our bioengineered, 3D-printed GB model could help in narrowing the “valley of death” in drug development by advancing the preclinical studies and will revolutionize the way we treat patients with cancer by adequately mimicking the brain tissue and predicting the suitable clinical treatment to each person, which has special importance in tumors with short-term survival like GB.
Acknowledgments
We thank the Tel Aviv University Sackler Cellular and Molecular Imaging Center (SCMIC) for the imaging services, and special thanks to A. Lichtenstein for professional assistance with confocal microscopy and to A. Barbul for professional assistance with confocal microscopy at the core facility at the Life Science faculty, Tel Aviv University. We also express our thankfulness to O. Yaron and S. Karako-Lampert from the NGS and Molecular Biology Unit at Bar-Ilan University. L.N. and N.R. were TEVA BioInnovation fellows in advanced research. L.N. was also a fellow of Dan David Prize. Funding: The Satchi-Fainaro laboratory’s research leading to these results has received funding from the Morris Kahn Foundation, a partial funding from the European Research Council (ERC) Consolidator Grant (617445; PolyDorm), ERC Advanced Grant (835227; 3DBrainStrom), and ERC Proof of Concept (PoC) Grant (862580; 3DCanPredict); from the Israel Science Foundation (1969/18), from Nancy and Peter Brown friends of The Israel Cancer Association (ICA) USA, in memory of Kenny and Michael Adler (20150909), from the Israel Cancer Research Fund (ICRF) Professorship award (PROF-18-682); and from the Check Point Software Technologies Ltd. Author contributions: L.N. and R.S.-F. conceived and designed the research. E.Y., L.N., N.R., Y.S., D.B.-S., S.P., A.E.-B., and S.F. performed the research. L.N., A.M., G.T., D.B.-S., and R.S.-F. analyzed the data. R.G. and Z.R. contributed materials or tissues. L.N., G.T., D.B.-S., and R.S.-F. wrote the manuscript. All authors reviewed the manuscript and provided comments. R.S.-F. was in charge of the overall direction and planning of this study. Competing interests: R.S.-F. is a Board Director at Teva Pharmaceutical Industries Ltd. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The sequence data generated in this study have been deposited in the Gene Expression Omnibus (GEO) and are accessible through the GEO Series accession number GSE178371.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/34/eabi9119/DC1
REFERENCES AND NOTES
- 1.World Health Organization. Cancer. March 3, 2021. www.who.int/news-room/fact-sheets/detail/cancer (2018).
- 2.Jakka S., Rossbach M., An economic perspective on personalized medicine. HUGO J. 7, 1 (2013). [Google Scholar]
- 3.Ben-David U., Siranosian B., Ha G., Tang H., Oren Y., Hinohara K., Strathdee C. A., Dempster J., Lyons N. J., Burns R., Nag A., Kugener G., Cimini B., Tsvetkov P., Maruvka Y. E., O’Rourke R., Garrity A., Tubelli A. A., Bandopadhayay P., Tsherniak A., Vazquez F., Wong B., Birger C., Ghandi M., Thorner A. R., Bittker J. A., Meyerson M., Getz G., Beroukhim R., Golub T. R., Genetic and transcriptional evolution alters cancer cell line drug response. Nature 560, 325–330 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DiMasi J. A., Grabowski H. G., Hansen R. W., Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 47, 20–33 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Clinical Development Success Rates and Contributing Factors 2011–2020 (BIO, QLS Advisors, Informa UK Ltd, 2021).
- 6.Bhatia S. N., Ingber D. E., Microfluidic organs-on-chips. Nat. Biotechnol. 32, 760–772 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Yoshida G. J., Applications of patient-derived tumor xenograft models and tumor organoids. J. Hematol. Oncol. 13, 4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dai X., Ma C., Lan Q., Xu T., 3D bioprinted glioma stem cells for brain tumor model and applications of drug susceptibility. Biofabrication 8, 045005 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Zhao Y., Yao R., Ouyang L., Ding H., Zhang T., Zhang K., Cheng S., Sun W., Three-dimensional printing of Hela cells for cervical tumor model in vitro. Biofabrication 6, 035001 (2014). [DOI] [PubMed] [Google Scholar]
- 10.DelNero P., Song Y. H., Fischbach C., Microengineered tumor models: Insights & opportunities from a physical sciences-oncology perspective. Biomed. Microdevices 15, 583–593 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trujillo-de Santiago G., Flores-Garza B. G., Tavares-Negrete J. A., Lara-Mayorga I. M., González-Gamboa I., Zhang Y. S., Rojas-Martínez A., Ortiz-López R., Álvarez M. M., The tumor-on-chip: Recent advances in the development of microfluidic systems to recapitulate the physiology of solid tumors. Materials 12, 2945 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sobrino A., Phan D. T. T., Datta R., Wang X., Hachey S. J., Romero-López M., Gratton E., Lee A. P., George S. C., Hughes C. C. W., 3D microtumors in vitro supported by perfused vascular networks. Sci. Rep. 6, 31589 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim S., Lee H., Chung M., Jeon N. L., Engineering of functional, perfusable 3D microvascular networks on a chip. Lab Chip 13, 1489–1500 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Huang T. Q., Qu X., Liu J., Chen S., 3D printing of biomimetic microstructures for cancer cell migration. Biomed. Microdevices 16, 127–132 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.C. Box, S. J. Rogers, M. Mendiola, S. A. Eccles, Seminars in Cancer Biology (Elsevier, 2010), vol. 20, pp. 128–138. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y. S., Duchamp M., Oklu R., Ellisen L. W., Langer R., Khademhosseini A., Bioprinting the cancer microenvironment. ACS Biomater. Sci. Eng. 2, 1710–1721 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heinrich M. A., Bansal R., Lammers T., Zhang Y. S., Michel Schiffelers R., Prakash J., 3D-bioprinted mini-brain: A glioblastoma model to study cellular interactions and therapeutics. Adv. Mater. 31, 1806590 (2019). [DOI] [PubMed] [Google Scholar]
- 18.H. Lodish, A. Berk, S. Lawrence Zipursky, P. Matsudaira, D. Baltimore, J. Darnell, Molecular Cell Biology (Macmillan, 2008). [Google Scholar]
- 19.Skylar-Scott M. A., Uzel S. G. M., Nam L. L., Ahrens J. H., Truby R. L., Damaraju S., Lewis J. A., Biomanufacturing of organ-specific tissues with high cellular density and embedded vascular channels. Sci. Adv. 5, eaaw2459 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma L., Barker J., Zhou C., Li W., Zhang J., Lin B., Foltz G., Küblbeck J., Honkakoski P., Towards personalized medicine with a three-dimensional micro-scale perfusion-based two-chamber tissue model system. Biomaterials 33, 4353–4361 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng F., Meyer C. M., Joung D., Vallera D. A., McAlpine M. C., Panoskaltsis-Mortari A., 3D bioprinted in vitro metastatic models via reconstruction of tumor microenvironments. Adv. Mater. 31, 1806899 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ozturk M. S., Lee V. K., Zou H., Friedel R. H., Intes X., Dai P. G., High-resolution tomographic analysis of in vitro 3D glioblastoma tumor model under long-term drug treatment. Sci. Adv. 6, eaay7513 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El Demerdash N., Kedda J., Ram N., Brem H., Tyler B., Novel therapeutics for brain tumors: Current practice and future prospects. Expert Opin. Drug Deliv. 17, 9–21 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Yeini E., Ofek P., Albeck N., Ajamil D. R., Neufeld L., Eldar-Boock A., Kleiner R., Vaskovich D., Koshrovski-Michael S., Dangoor S. I., Krivitsky A., Luna C. B., Shenbach-Koltin G., Goldenfeld M., Hadad O., Tiram G., Satchi-Fainaro R., Targeting glioblastoma: Advances in drug delivery and novel therapeutic approaches. Adv. Therapeut. 4, 2000124 (2021). [Google Scholar]
- 25.Segal E., Pan H., Ofek P., Udagawa T., Kopecková P., Kopecek J., Satchi-Fainaro R., Targeting angiogenesis-dependent calcified neoplasms using combined polymer therapeutics. PLOS ONE 4, e5233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yom-Tov O., Neufeld L., Seliktar D., Bianco-Peled H., A novel design of injectable porous hydrogels with in situ pore formation. Acta Biomater. 10, 4236–4246 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Neufeld L., Bianco-Peled H., Designing a biocompatible hydrogel for the delivery of mesalamine. Int. J. Pharm. 491, 170–179 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Ferber S., Tiram G., Sousa-Herves A., Eldar-Boock A., Krivitsky A., Scomparin A., Yeini E., Ofek P., Ben-Shushan D., Vossen L. I., Licha K., Grossman R., Ram Z., Henkin J., Ruppin E., Auslander N., Haag R., Calderón M., Satchi-Fainaro R., Co-targeting the tumor endothelium and P-selectin-expressing glioblastoma cells leads to a remarkable therapeutic outcome. eLife 6, e25281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Satchi-Fainaro R., Ferber S., Segal E., Ma L., Dixit N., Ijaz A., Hlatky L., Abdollahi A., Almog N., Prospective identification of glioblastoma cells generating dormant tumors. PLOS ONE 7, e44395 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.N. L. Bray, H. Pimentel, P. Melsted, L. Pachter, Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 34, 525–527, (2016). Epub 2016 Apr 4. Erratum in: Nat Biotechnol. 2016 Aug 9;34(8):888. doi:10.1038/nbt.3519. PMID: 27043002. [DOI] [PubMed]
- 31.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anders S., Huber W., Differential expression analysis for sequence count data. Nat. Precedings , 1 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Netti P. A., Berk D. A., Swartz M. A., Grodzinsky A. J., Jain R. K., Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 60, 2497–2503 (2000). [PubMed] [Google Scholar]
- 34.Rahimi N., Molin D. G., Cleij T. J., van Zandvoort M. A., Post M. J., Electrosensitive polyacrylic acid/fibrin hydrogel facilitates cell seeding and alignment. Biomacromolecules 13, 1448–1457 (2012). [DOI] [PubMed] [Google Scholar]
- 35.von Bartheld C. S., Bahney J., Herculano-Houzel S., The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J Comp Neurol 524, 3865–3895 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeini E., Ofek P., Pozzi S., Albeck N., Ben-Shushan D., Tiram G., Golan S., Kleiner R., Sheinin R., Dangoor S. I., Reich-Zeliger S., Grossman R., Ram Z., Brem H., Hyde T. M., Magod P., Friedmann-Morvinski D., Madi A., Satchi-Fainaro R., P-Selectin axis plays a key role in microglia immunophenotype and glioblastoma progression. Nat Commun. 12, 1912 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matias D., Balça-Silva J., da Graça G. C., Wanjiru C. M., Macharia L. W., Nascimento C. P., Roque N. R., Coelho-Aguiar J. M., Pereira C. M., Dos Santos M. F., Pessoa L. S., Lima F. R. S., Schanaider A., Ferrer V. P., Leite de Sampaio e Spohr T. C., Moura-Neto V., Microglia/astrocytes–glioblastoma crosstalk: Crucial molecular mechanisms and microenvironmental factors. Front. Cell. Neurosci. 12, 235 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tiram G., Ferber S., Ofek P., Eldar-Boock A., Ben-Shushan D., Yeini E., Krivitsky A., Blatt R., Almog N., Henkin J., Amsalem O., Yavin E., Cohen G., Lazarovici P., Lee J. S., Ruppin E., Milyavsky M., Grossman R., Ram Z., Calderón M., Haag R., Satchi-Fainaro R., Reverting the molecular fingerprint of tumor dormancy as a therapeutic strategy for glioblastoma. FASEB J. 32, 5835–5850 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Almog N., Ma L., Raychowdhury R., Schwager C., Erber R., Short S., Hlatky L., Vajkoczy P., Huber P. E., Folkman J., Abdollahi A., Transcriptional switch of dormant tumors to fast-growing angiogenic phenotype. Cancer Res. 69, 836–844 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Tiram G., Segal E., Krivitsky A., Shreberk-Hassidim R., Ferber S., Ofek P., Udagawa T., Edry L., Shomron N., Roniger M., Kerem B., Shaked Y., Aviel-Ronen S., Barshack I., Calderón M., Haag R., Satchi-Fainaro R., Identification of dormancy-associated microRNAs for the design of osteosarcoma-targeted dendritic polyglycerol nanopolyplexes. ACS Nano 10, 2028–2045 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Wang Q., Hu B., Hu X., Kim H., Squatrito M., Scarpace L., deCarvalho A. C., Lyu S., Li P., Li Y., Barthel F., Cho H. J., Lin Y.-H., Satani N., Martinez-Ledesma E., Zheng S., Chang E., Sauvé C.-E. G., Olar A., Lan Z. D., Finocchiaro G., Phillips J. J., Berger M. S., Gabrusiewicz K. R., Wang G., Eskilsson E., Hu J., Mikkelsen T., De Pinho R. A., Muller F., Heimberger A. B., Sulman E. P., Nam D.-H., Verhaak R. G. W., Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42–56. e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heiland D. H., Ravi V. M., Behringer S. P., Frenking J. H., Wurm J., Joseph K., Garrelfs N. W. C., Strähle J., Heynckes S., Grauvogel J., Franco P., Mader I., Schneider M., Potthoff A.-L., Delev D., Hofmann U. G., Fung C., Beck J., Sankowski R., Prinz M., Schnell O., Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 10, 2541 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brantley E. C., Benveniste E. N., Signal transducer and activator of transcription-3: a molecular hub for signaling pathways in gliomas. Mol. Cancer Res. 6, 675–684 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Azam Z., Shao W., Ng H.-k., Wang J., Chen Z.-p., To S.-s. T., Comprehensive RNAseq analysis reveals PIK3CD promotes glioblastoma tumorigenesis by mediating PI3K-Akt signaling pathway. Glioma 3, 135–142 (2020). [Google Scholar]
- 45.Tang M., Tiwari S. K., Agrawal K., Tan M., Dang J., Tam T., Tian J., Wan X., Schimelman J., You S., Xia Q., Rana T. M., Chen S., Rapid 3D bioprinting of glioblastoma model mimicking native biophysical heterogeneity. Small 17, 2006050 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang M., Xie Q., Gimple R. C., Zhong Z., Tam T., Tian J., Kidwell R. L., Wu Q., Prager B. C., Qiu Z., Yu A., Zhu Z., Mesci P., Jing H., Schimelman J., Wang P., Lee D., Lorenzini M. H., Dixit D., Zhao L., Bhargava S., Miller T. E., Wan X., Tang J., Sun B., Cravatt B. F., Muotri A. R., Chen S., Rich J. N., Three-dimensional bioprinted glioblastoma microenvironments model cellular dependencies and immune interactions. Cell Res. 30, 833–853 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yin W., Tang G., Zhou Q., Cao Y., Li H., Fu X., Wu Z., Jiang X., Expression profile analysis identifies a novel five-gene signature to improve prognosis prediction of glioblastoma. Front. Genet. 10, 419 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stupp R., Mason W. P., van den Bent M. J., Weller M., Fisher B., Taphoorn M. J. B., Belanger K., Brandes A. A., Marosi C., Bogdahn U., Curschmann J., Janzer R. C., Ludwin S. K., Gorlia T., Allgeier A., Lacombe D., Cairncross J. G., Eisenhauer E., Mirimanoff R. O., Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Unger C., Kramer N., Walzl A., Scherzer M., Hengstschläger M., Dolznig H., Modeling human carcinomas: Physiologically relevant 3D models to improve anti-cancer drug development. Adv. Drug Deliv. Rev. 79–80, 50–67 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Butcher D. T., Alliston T., Weaver V. M., A tense situation: Forcing tumour progression. Nat. Rev. Cancer 9, 108–122 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paszek M. J., Zahir N., Johnson K. R., Lakins J. N., Rozenberg G. I., Gefen A., Reinhart-King C. A., Margulies S. S., Dembo M., Boettiger D., Hammer D. A., Weaver V. M., Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241–254 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Krishnamoorthy S., Noorani B., Xu C., Effects of encapsulated cells on the physical–mechanical properties and microstructure of gelatin methacrylate hydrogels. Int. J. Mol. Sci. 20, 5061 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen K., Lin Q., Wang L., Zhuang Z., Zhang Y., Huang D., Wang H., An all-in-one tannic acid-containing hydrogel adhesive with high toughness, notch insensitivity, self-healability, tailorable topography, and strong, instant, and on-demand underwater adhesion. ACS Appl. Mater. Interfaces 13, 9748–9761 (2021). [DOI] [PubMed] [Google Scholar]
- 54.Naghieh S., Sarker M. D., Sharma N. K., Barhoumi Z., Chen X., Printability of 3D printed hydrogel scaffolds: Influence of hydrogel composition and printing parameters. Appl. Sci. 10, 292 (2020). [Google Scholar]
- 55.Schultz C., Lemke N., Ge S., Golembieski W. A., Rempel S. A., Secreted protein acidic and rich in cysteine promotes glioma invasion and delays tumor growth in vivo. Cancer Res. 62, 6270–6277 (2002). [PubMed] [Google Scholar]
- 56.Wang C., Tong X., Yang F., Bioengineered 3D brain tumor model to elucidate the effects of matrix stiffness on glioblastoma cell behavior using PEG-based hydrogels. Mol. Pharm. 11, 2115–2125 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Schwartz H., Blacher E., Amer M., Livneh N., Abramovitz L., Klein A., Ben-Shushan D., Soffer S., Blazquez R., Barrantes-Freer A., Müller M., Müller-Decker K., Stein R., Tsarfaty G., Satchi-Fainaro R., Umansky V., Pukrop T., Erez N., Incipient melanoma brain metastases instigate astrogliosis and neuroinflammation. Cancer Res. 76, 4359–4371 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Kolesky D. B., Homan K. A., Skylar-Scott M. A., Lewis J. A., Three-dimensional bioprinting of thick vascularized tissues. Proc. Natl. Acad. Sci. U.S.A. 113, 3179–3184 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yi H.-G., Jeong Y. H., Kim Y., Choi Y.-J., Moon H. E., Park S. H., Kang K. S., Bae M., Jang J., Youn H., Paek S. H., Cho D.-W., A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nat. Biomed. Eng. 3, 509–519 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Eldar-Boock A., Miller K., Sanchis J., Lupu R., Vicent M. J., Satchi-Fainaro R., Integrin-assisted drug delivery of nano-scaled polymer therapeutics bearing paclitaxel. Biomaterials 32, 3862–3874 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Palma M., Biziato D., Petrova T. V., Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 17, 457–474 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Yoshino A., Katayama Y., Yokoyama T., Watanabe T., Ogino A., Ota T., Komine C., Fukushima T., Kusama K., Therapeutic implications of interferon regulatory factor (IRF)-1 and IRF-2 in diffusely infiltrating astrocytomas (DIA): Response to interferon (IFN)-β in glioblastoma cells and prognostic value for DIA. J. Neurooncol 74, 249–260 (2005). [DOI] [PubMed] [Google Scholar]
- 63.Liang J., Piao Y., Henry V., Tiao N., de Groot J. F., Interferon-regulatory factor-1 (IRF1) regulates bevacizumab induced autophagy. Oncotarget 6, 31479–31492 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Manini I., Ruaro M. E., Sgarra R., Bartolini A., Caponnetto F., Ius T., Skrap M., di Loreto C., Beltrami A. P., Manfioletti G., Cesselli D., Semaphorin-7A on exosomes: A promigratory signal in the glioma microenvironment. Cancer 11, 758 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Angelucci C., Lama G., Sica G., Multifaceted functional role of semaphorins in glioblastoma. Int. J. Mol. Sci. 20, 2144 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu Y.-S., Lin H.-Y., Lai S.-W., Huang C.-Y., Huang B.-R., Chen P.-Y., Wei K.-C., Lu D.-Y., MiR-181b modulates EGFR-dependent VCAM-1 expression and monocyte adhesion in glioblastoma. Oncogene 36, 5006–5022 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Schmid C. D., Schledzewski K., Mogler C., Waldburger N., Kalna V., Marx A., Randi A. M., Géraud C., Goerdt S., Koch P.-S., GPR182 is a novel marker for sinusoidal endothelial differentiation with distinct GPCR signaling activity in vitro. Biochem. Biophys. Res. Commun. 497, 32–38 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Ramachandran V., Arumugam T., Hwang R. F., Greenson J. K., Simeone D. M., Logsdon C. D., Adrenomedullin is expressed in pancreatic cancer and stimulates cell proliferation and invasion in an autocrine manner via the adrenomedullin receptor, ADMR. Cancer Res. 67, 2666–2675 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Doron H., Pukrop T., Erez N., A blazing landscape: Neuroinflammation shapes brain metastasis. Cancer Res. 79, 423–436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sivandzade F., Cucullo L., In-vitro blood–brain barrier modeling: A review of modern and fast-advancing technologies. J. Cereb. Blood Flow Metab. 38, 1667–1681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/34/eabi9119/DC1