

ABSTRACT
Lung-localized CD4 T cells play a critical role in the control of influenza virus infection and can provide broadly protective immunity. However, current influenza vaccination strategies primarily target influenza hemagglutinin (HA) and are administered peripherally to induce neutralizing antibodies. We have used an intranasal vaccination strategy targeting the highly conserved influenza nucleoprotein (NP) to elicit broadly protective lung-localized CD4 T cell responses. The vaccine platform consists of a self-assembling nanolipoprotein particle (NLP) linked to NP with an adjuvant. We have evaluated the functionality, in vivo localization, and persistence of the T cells elicited. Our study revealed that intranasal vaccination elicits a polyfunctional subset of lung-localized CD4 T cells that persist long term. A subset of these lung CD4 T cells localize to the airway, where they can act as early responders following encounter with cognate antigen. Polyfunctional CD4 T cells isolated from airway and lung tissue produce significantly more effector cytokines IFN-γ and TNF-α, as well as cytotoxic functionality. When adoptively transferred to naive recipients, CD4 T cells from NLP:NP-immunized lung were sufficient to mediate 100% survival from lethal challenge with H1N1 influenza virus.
IMPORTANCE Exploiting new, more efficacious strategies to potentiate influenza virus-specific immune responses is important, particularly for at-risk populations. We have demonstrated the promise of direct intranasal protein vaccination to establish long-lived immunity in the lung with CD4 T cells that possess features and positioning in the lung that are associated with both immediate and long-term immunity, as well as demonstrating direct protective potential.
KEYWORDS: CD4 T cells, Trm, airway T cells, influenza A, influenza virus challenge, lung parenchyma vasculature, mucosal immunology, polyfunctional T cells, rational vaccine design, tissue resident memory
INTRODUCTION
Disease outbreaks caused by influenza A viruses result in considerable human morbidity, mortality, and economic burden each year (1–4). Currently, intramuscular vaccination with recombinant or split influenza vaccines that are solely composed of, or highly enriched for, HA is the primary clinical strategy employed to combat influenza infection, by seeking to elicit neutralizing HA-specific antibodies (5–7). This strategy can fail to elicit sufficiently protective responses (8–11), sometimes due to antigenic mismatch between circulating and vaccine viruses, emergence of novel viruses that arise from antigenic shift, or mutations that occur during vaccine production (12–14). Further, anti-HA antibody titers in vaccinated subjects can wane over time (15). To overcome the limitations of current subunit influenza vaccination strategies, development of universal influenza vaccines that elicit improved durability and breadth of immune protection has become a high priority in the field (16–20).
To achieve the goal of universal influenza vaccines that induce broadly protective immunity, one particularly advantageous strategy is to more fully induce and harness lung-localized and T cell-mediated immunity, with a focus on the immune response to influenza antigens that are broadly conserved among virus strains. Live-attenuated influenza vaccines (LAIV) allow priming of both peripheral and lung-localized T cells; however, efficacy is greatly reduced in adults, presumably because preexisting antibodies to HA in the human host blunts replication of LAIV in the respiratory tract (21, 22). A number of groups have explored the potential of intranasal protein-based vaccines to induce protective immunity to influenza, but these studies have been primarily limited to assessment of induced antibody responses and empirical virus challenge designs (23–25). Influenza virus-specific CD4 and CD8 T cells are known to play critical roles in mediating protection from influenza virus infection (14, 26–28). CD8 T cells primarily display cytotoxic activity against infected antigen-bearing cells in the respiratory tract, while CD4 T cells are more complex in their effector functions. Influenza virus-specific CD4 T cells can promote CD8 T cell expansion and development of CD8 memory. Influenza virus-specific CD4 T follicular helper cells can provide help to B cells for production of high-affinity, HA-specific B cells that can produce antibodies conveying sterilizing immunity (29–32). Within the respiratory tract, CD4 T cells recruited to the lung can induce antiviral cytokines and deliver direct cytotoxicity. In addition, memory CD4 T cells in the lung can facilitate innate immune responses and early activation of antigen-presenting cells through production of non-Th1 cytokines and through recruitment of innate cells into the respiratory tract that can suppress viral replication (33–36).
Here, we have explored a rational protein vaccination strategy that targets immune responses in the respiratory tract. By fostering influenza virus-specific CD4 T cell responses in the lung, we can exploit the multiplicity of functions delivered by CD4 T cells and the ability of these cells to form tissue-resident populations in the lung (37–41). The strategy employs codelivery of the genetically conserved influenza virus NP and a low-dose adjuvant, associated with a novel self-assembling nanolipoprotein complex. NLPs are nanoscale mimetics of nascent high-density lipoproteins (∼15 nm in diameter) and feature a lipid bilayer stabilized by apolipoproteins (42). The NLP platform presents the advantages of being self-assembling, nontoxic, not intrinsically immunogenic, and can bind to a broad array of antigens while remaining stable under biological conditions (42–45). We have assessed the seeding of antigen-specific CD4 T cells in the lung and have evaluated the functional potential, homing, and persistence of the elicited cells in both the periphery and the respiratory tract. Our studies demonstrate the promise of this approach to localize multifunctional, influenza virus-specific CD4 T cells to multiple compartments in the respiratory tract, as well as in the periphery, and their potential to protect from infection.
RESULTS
Potentiating lung-localized CD4 T cell immunity via intranasal immunization.
In order to better utilize animals for preclinical models of human vaccine studies, we considered the well-known feature of human immunity to influenza, characterized by immune memory that develops from intermittent exposure through vaccination and infection (46–48). Accordingly, we evaluated the immune response of intranasal vaccination in the context of influenza virus-specific memory established by priming mice intranasally with live-attenuated influenza virus (LAIV), which seed memory cells with diverse antigen specificity (49, 50) and across many tissues, as is likewise observed in human subjects (51–54). After intranasal vaccination with LAIV, mice were rested for 30 days to allow the immune response to contract and then boosted with influenza NP (Fig. 1A), a highly expressed internal virion protein that is conserved among diverse influenza virus strains (55), or boosted with LAIV. A nanolipoprotein nickel-chelating construct was employed for vaccination that allows coassembly of His-tagged antigen and cholesterol-tagged CpG, as previously reported (44) and described in the Materials and Methods. The features and characterization of the assembled vaccine are illustrated and described in Fig. 1B. This approach generates stable complexes of NP-antigen and cholesterol-tagged CpG conjugated to the NLP platform, and will be referred to as NLP:NP (Fig. 1C and D).
FIG 1.

Potentiating lung localized CD4 T cell immunity via intranasal immunization. (A) Schematic representation of immunization regimen. Naive C57BL/6 mice were immunized with monovalent live attenuated influenza virus (LAIV), a 6:2 reassortant of caA/Ann Arbor/6/60 and A/New Caledonia/99. Responses were allowed to contract for 30 days prior to intranasal boost with NLP. NLP immunizations were composed of 5 μg recombinant influenza NP and 5 μg CpG conjugated to the NLP at a molar ratio of 1:2.4:18.4, NLP to NP to CpG, respectively. Responses were analyzed at 10 days post boost. (B) Schematic demonstrating the NLP self-assembly process. Purified components (including lipids DGS-NTA-Ni and DOPC, with apolipoprotein) are solubilized with surfactant and mixed in aqueous solution. Removal of the surfactant initiates self-assembly of the functionalized NLPs. Conjugation of adjuvants and antigens is achieved by reacting with cognate NLP surface functionalities (e.g., His tag with chelated nickel) or through anchoring of lipidic moieties featured on amphiphilic cargo molecules (e.g., cholesterol-tagged CpG). (C) His-tagged NP antigen can be conjugated to nickel-chelating NLPs at ratios from 2 to 16 antigens per NLP. Size exclusion chromatography (SEC) is used to monitor the conjugation, evidenced by a dose-dependent decrease in retention time and an increase in NLP absorbance intensity, indicating successful conjugation of antigen to NLP. (D) SEC demonstrates the successful incorporation of both NP protein and cholesterol-modified CpG in the final vaccine assembly, evidenced by a shift in retention time and increase in absorbance intensity.
Intranasal NLP:NP boost elicits a higher frequency of NP-specific CD4 T cells in the lung than LAIV boost.
To quantify the CD4 T cell response to LAIV priming and NLP:NP vaccination, gamma interferon (IFN-γ) and interleukin 2 (IL-2) cytokine ELISpot assays were performed at several time points, including 7 days postvaccination with LAIV (Fig. 2A to C), 30 days postvaccination with LAIV (Fig. 3, open bars), and again following boost with LAIV (Fig. 3, hatched bars) or NLP:NP (Fig. 3, solid bars). Cells isolated from lung (Fig. 3A and D), the lung-draining mediastinal (mLN) lymph node (Fig. 3B and E), and spleen (Fig. 3C and F) were used to quantify and evaluate the distribution of antigen-specific cells extracted by these tissues. The peptides tested by highly sensitive and specific ELISpot assays included immunodominant I-Ab restricted peptides (Table 1) from HA, nonstructural protein 1 (NS1), NP, and neuraminidase (NA), all of which were previously defined (56). These studies revealed that after the intranasal NLP:NP vaccination, the frequency of IFN-γ-producing NP-specific CD4 T cells increased 117-fold in lung, 39-fold in mLN, and 89-fold in spleen, relative to LAIV D30 mice (Fig. 3A to C). By comparison, the frequency of IFN-γ-producing NP-specific CD4 T cells increased 3.3-fold in lung, 4.3-fold in mLN, and 0.4-fold in spleen of LAIV-boosted mice relative to LAIV D30 mice (Fig. 3A to C). Relative to LAIV D30, the frequency of IL-2-producing cells in NLP:NP-boosted mice increased 16-fold, 12-fold, and 45-fold in lung, mLN, and spleen, respectively (Fig. 3D to F). The frequency of IL-2-producing cells in LAIV-boosted mice increased 4.4-fold in lung, 4.5-fold in mLN, and 0.4-fold in spleen relative to LAIV D30 mice. Of note, boosting following NLP:NP vaccination was limited to NP-specific CD4 T cells and did not nonspecifically recruit or activate HA, NS1, or NA-specific CD4 T cells. This result demonstrates that the recruitment and boost are not induced by CpG administration alone, a conclusion supported by the failure to boost NP-specific cells with CpG-NLP vehicle alone (not shown). These results highlight the increased immunogenicity of the NLP:NP platform in boosting antigen-specific CD4 T cells relative to secondary exposure to LAIV. The advantages of NLP:NP boosting over LAIV boosting held across all tissues and mediators assessed.
FIG 2.
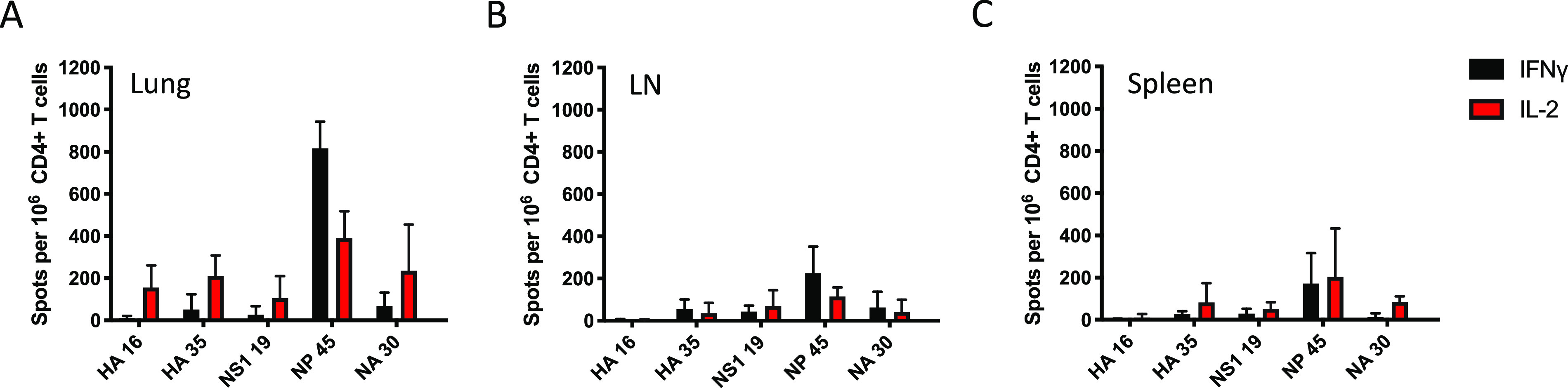
Live-attenuated virus establishes memory. (A to C) Naive C57BL/6 mice were primed with live-attenuated influenza virus to establish influenza virus-specific memory. The CD4 T cell response was assayed in lung (A), mLN (B), and spleen (C) at 7 days postinfection via IL-2 and IFN-γ cytokine ELISpot to identify immunodominant CD4 T cell epitopes across hemagglutinin (HA), nonstructural protein 1 (NS1), nucleoprotein (NP), and neuraminidase (NA) viral proteins. Results are presented as the mean number of cytokine-producing spots per million CD4 T cells with the standard deviation shown. The mean is representative of three independent experiments of five pooled mice each.
FIG 3.

Intranasal NLP:NP boost elicits a higher frequency of NP-specific CD4 T cells in the lung than does an LAIV boost. (A to F) Thirty days postimmunization with LAIV, CD4 T cell responses were assayed in lung, mLN, and spleen via IFN-γ (A to C) and IL-2 (D to F) cytokine ELISpot to characterize the immune response prior to intranasal boost with NLP:NP. At 10 days post intranasal immunization with NLP:NP, the extent of CD4 T cell boosting in lung (A and D), mLN (B and E), and spleen (C and F) was quantified by IL-2 and IFN-γ cytokine ELISpot. Results are presented as the mean number of cytokine-producing spots per million CD4 T cells with the standard deviation shown. The mean is representative of three to five independent experiments of five pooled mice each.
TABLE 1.
MHC Class II-restricted epitopes in H-2b mice
| I-Ab-restricted epitopes | |
|---|---|
| Epitope | Amino acid position and sequence |
| HA 16 | 90 KESWSYIVETPNPENGT 106 |
| HA 35 | 203 NQRALYHTENAYVSVVS 219 |
| NS1 19 | 108 KQKVAGPLCVRMDQAIM 124 |
| NP 45 | 261 RSALILRGSVAHKSCLP 277 |
| NA 30 | 171 NSKFESVAWSASACHDG 187 |
Intranasal NLP:NP vaccination establishes a large population of tissue-resident CD4 T cell populations in the lung.
The localization of CD4 T cells has a profound effect on their protective capacity, and their ability to rapidly respond to infection. The microenvironment, including cytokines, chemokines, and antigen-dependent interactions, can affect the recruitment, expansion, phenotype, and persistence of the antigen-specific cells (12, 34, 57–60). To determine the localization and phenotype of CD4 T cells in the lung following intranasal NLP:NP or LAIV immunization, intravascular (i.v.) labeling was employed in conjunction with multiparameter flow cytometry (59). Given the importance of localization within the lung, we were particularly interested in determining whether NLP:NP or LAIV boosting differed in their potential to elicit tissue-resident CD4 T cell populations in the lung. Using the gating scheme depicted in Fig. S1 in the supplemental material, live CD4 T cells were partitioned between vasculature (CD45 IV+) and tissue (CD45 IV−) compartments prior to additional analyses. This enabled assessment of these discrete populations on the basis of antigen experience, tissue homing potential, and memory precursor potential (61).
While LAIV boost increased the percentage of CD4 T cells localized to lung tissue relative to LAIV D30, only intranasal NLP:NP vaccination significantly increased both the percentage and total abundance of CD4 T cells localized to the lung tissue relative to LAIV D30 and LAIV-boosted mice (Fig. 4A to C). LAIV boost increased the percentage of antigen-experienced CD44+ CD62L− in the lung tissue relative to LAIV D30 controls. NLP:NP-boosted mice had a higher percentage of antigen-experienced CD44+ CD62L− cells in both tissue and vasculature, relative to LAIV boost and LAIV D30 controls (Fig. 4D and E). Further analyses revealed the lung tissue of NLP:NP-boosted mice had an increased total abundance of antigen-experienced CD44+ CD62L− cells relative to LAIV D30 and LAIV-boosted mice, and that there were more lung vasculature-localized antigen-experienced cells in NLP:NP-boosted mice than in LAIV-boosted mice (Fig. 4F).
FIG 4.

Intranasal NLP:NP vaccination establishes larger tissue-resident CD4 T cell populations in the lung than does an LAIV boost. (A) Analysis of lung localization among CD4 T cells, depicting representative flow plots. (B) Frequency of lung tissue-localized CD4 T cells. (C) Absolute abundance of CD4 T cells in the intravascular labeled CD45+ lung vasculature and unlabeled CD45− lung tissue. (D) CD4 T cells were assessed for their expression of CD44 and CD62L. (E and F) The frequency (E) and absolute abundance (F) of effector CD44+ CD62L− CD4 T cells was assessed in the lung tissue and vasculature. (G) CD44+ CD62L− effector CD4 T cells were subsequently gated on expression of CD11a and CD69. (H and I) The frequency (H) and abundance (I) of CD11a+ CD69+/− was calculated among effector CD4 T cells in the lung tissue. (J) Expression of memory-precursor marker Ly6C was assessed among CD44+ CD62L− effector CD4 T cells. (K and L) The frequency (K) and abundance (L) of Ly6C+ and Ly6C− CD4 T cells was assessed among lung tissue localized effector CD4 T cells. Results are presented as the mean number of the respective population with the standard deviation and individual mice shown. The mean is representative of three independent experiments of five individual mice each. With the exception of (B), significance (***, P < 0.001; ****, P < 0.0001) for comparisons of cellular frequency and abundance between LAIV and LAIV+NLP boost cohorts was determined by two-way ANOVA with Tukey’s correction for multiple comparisons.
Within the subset of antigen-experienced cells localized to lung tissue, we evaluated expression of lung residency markers CD11a and CD69. CD11a has been implicated as a critical marker of T cell homing to inflamed tissues. CD69 plays a role in retention of the recruited lymphocytes at the site of inflammation, especially within nonlymphoid tissues (62–69). Within each immunization group, we found increased percentages of CD11a+ CD69+ CD4 T cells in the lung tissue relative to their CD11a+ CD69− counterparts, but no significant differences existed between groups (Fig. 4G and H). NLP:NP vaccination was found to significantly increase the total abundance of CD11a+ CD69− and CD11a+ CD69+ CD4 T cells in the lung tissue relative to LAIV D30 or LAIV-boosted controls (Fig. 4I). Whereas CD4 T cells uniformly expressed high levels of CD11a, a substantial heterogeneity in the abundance of cells expressing CD69 was noted (Fig. 4G to I), as has been reported in studies of T cells in infection models of influenza (69, 70). Finally, we examined the relative proportion and overall abundance of short-lived effector Ly6C+ CD4 T cells, a marker associated with short term effector cells (61). When expression of Ly6C was examined on tissue-resident CD4 T cells, the relative proportion of Ly6C+ and Ly6C− negative cells was similar between the three groups; however, the abundance of Ly6C− cells was significantly increased in the NLP:NP-immunized group relative to LAIV D30 or LAIV-boosted controls (Fig. 4J to L). These data indicate that intranasal NLP:NP immunization increases the abundance of tissue-resident effector CD4 T cells relative to LAIV controls, including those with the potential to persist long term in the lung.
Use of pMHC-II tetramer to track NP-specific CD4 T cells reveals CD4 T cells localized primarily to the lung tissue, relative to lung vasculature.
The multiparameter flow cytometry data shown in Fig. 4 demonstrate boosting of lung-localized CD4 T cell responses, but do not define the antigen specificity of the cells. To explicitly study positioning of NP-specific CD4 T cells within the lung, an NP45 pMHC-II tetramer was generated and used in combination with intravascular labeling. In the gating scheme shown in Fig. S2, a double lineage-specific gating scheme was employed to ensure high specificity of pMHC-II tetramer staining. Compared to LAIV D30 mice, NLP:NP-vaccinated mice displayed an increased percentage of tetramer-positive cells (Fig. 5A). Importantly, most of the tetramer-positive CD4 T cells were localized to the lung tissue, with very few localized to the vasculature (Fig. 5B). However, in terms of absolute abundance of tetramer-positive CD4 T cells, NLP:NP-boosted mice had 94-fold more NP45-specific CD4 T cells than LAIV D30 controls (Fig. 5C). When assessing CD4 T cell localization using IV labeling, mean numbers of tetramer-positive CD4 T cells recovered from NLP:NP-immunized lung were 2.48 × 105 in lung tissue and 5.53 × 103 in lung vasculature. By comparison, mice that were only primed with LAIV had a mean of 2.61 × 103 tetramer-positive cells in lung tissue and 8.3 × 101 tetramer-positive cells in lung vasculature, representing 95-fold and 66-fold increases, respectively (Fig. 5D).
FIG 5.

Intranasal NLP:NP immunization boosts antigen-specific CD4 T cells which localize to the lung tissue. (A) Representative plot of NP-specific CD4 T cells detected by I-Ab pMHC-II NP45 tetramer staining in LAIV and LAIV+NLP immunized murine lung. (B) Frequency of vasculature-localized CD45+ and tissue-localized CD45− pMHC-II NP45 tetramer+ cells. (C and D) Quantification of the total number of NP45-specific CD4 T cells (C) and those localized to lung tissue and vasculature (D) of LAIV and LAIV+NLP immunized mice. Results are presented as the mean number of pMHC-II NP45 tetramer+ cells per lung with standard deviation and individual replicates shown. The mean is representative of 10 individual mice analyzed as pools of two lungs each. Cell numbers from pooled lungs are divided by two to reflect the cellularity of a single lung. Significance (***, P < 0.001) for comparisons of cellular abundance between LAIV and LAIV+NLP boost cohorts was determined by two-way ANOVA with Tukey’s correction for multiple comparisons.
Codelivery of antigen and adjuvant on NLP:NPs enhances CD4 T cell responses to intranasal vaccination.
Given the extent of CD4 T cell boosting observed following intranasal vaccination with NLP:NP, we sought to determine whether the responses are a consequence of codelivery of antigen and adjuvant on NLP. Responses to NLP:NP were analyzed in parallel with delivery of vaccine components separately but simultaneously at an identical dose and in soluble form, in mice that had been previously primed with LAIV (Fig. 6A). Immunization with NLP:NP complexes elicited 4.4-fold more IFN-γ-producing cells and 3-fold more IL-2-producing cells in the lung than did immunization with a matched dose of soluble NP and CpG (Fig. 6B and C). In the lung-draining mLN, NLP:NP immunization elicited 3.7-fold more IFN-γ-producing cells and 1.2-fold more IL-2-producing CD4 T cells than soluble NP and CpG (Fig. 6D and E). NLP:NP immunization also increased the frequency of IFN-γ- and IL-2-producing CD4 T cells in the spleen relative to soluble NP and CpG (Fig. 6F and G). In all tissues, CD4 T cell boosting was only observed among NP-specific cells. Overall, these experiments revealed that the conjugation of the vaccine components together elicits a higher frequency of NP-specific CD4 T cells than does soluble antigen and adjuvant at the same dose and does so in a highly antigen-specific manner.
FIG 6.
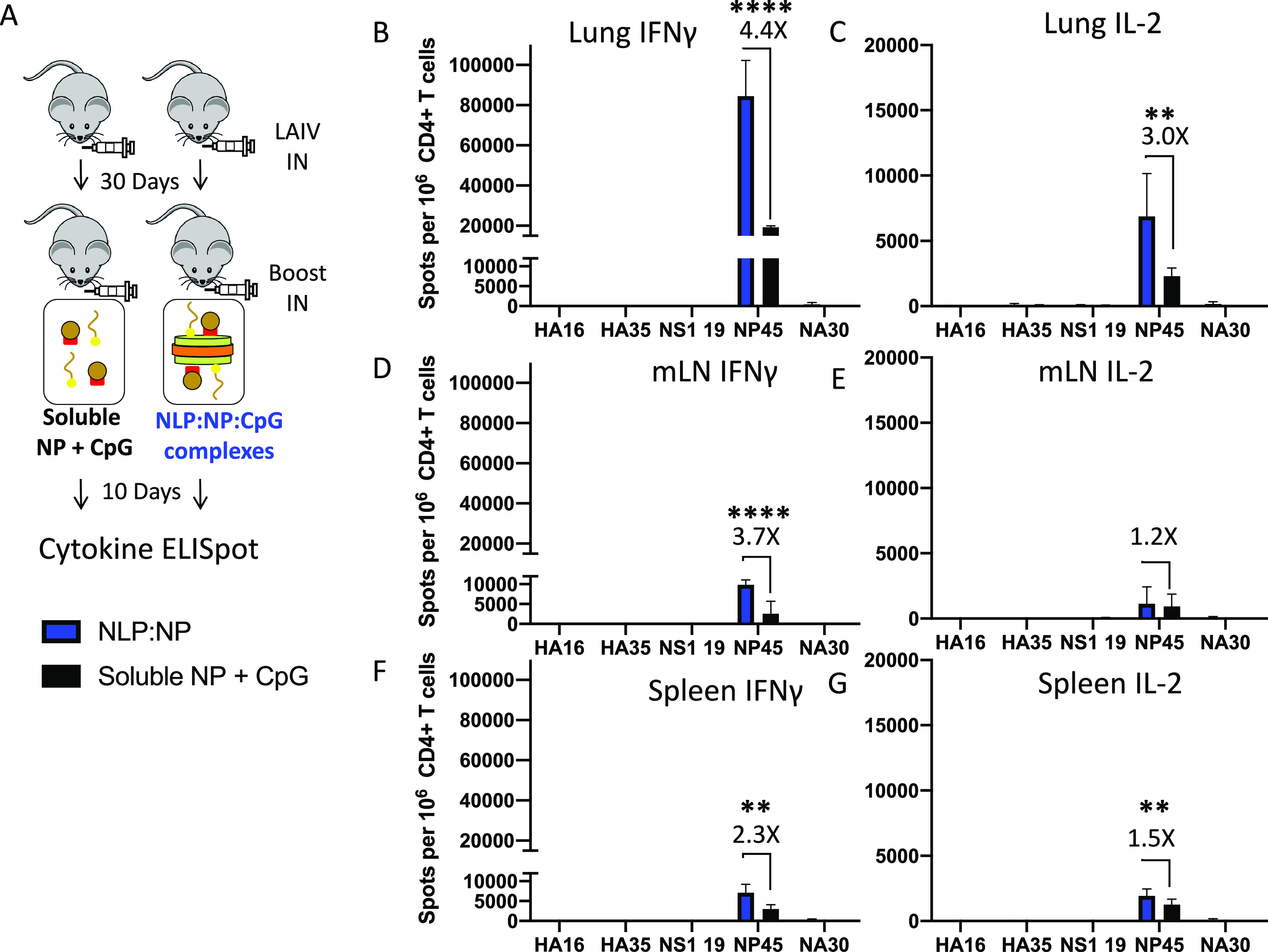
Codelivery of antigen and adjuvant on NLP:NP enhances CD4 T cell responses to intranasal vaccination. (A) Schematic representation of the immunization regimen, where mice were primed with LAIV and subsequently boosted with NLP:NP or soluble NP+CpG, and responses were assayed at 10 days post intranasal boost. The CD4 T cell response to NLP:NP and soluble NP+CpG immunization was compared via IFN-γ and IL-2 cytokine ELISpot assay in lung (B and C), mLN (D and E), and spleen (F and G). Results are presented as the mean with standard deviation of two independent experiments of five pooled mice each. Statistical significance was determined by unpaired, two-tailed t test with Welch’s correction.
Intranasal vaccination with NLP:NP elicits a higher frequency of antigen-specific CD4 T cells than peripheral vaccination.
We next formally addressed the impact that route of immunization has on frequency and localization of antigen-specific cells in the lung. Mice were primed with LAIV and vaccinated with NLP:NP via either an intranasal or subcutaneous (S.Q.) route (Fig. 7A). Use of cytokine ELISpot and flow cytometry showed that intranasal NLP:NP immunization elicited 14.7-fold more NP-specific IFN-γ-producing cells in the lung than S.Q. vaccination (Fig. 7B). Further, intranasal immunization seeded 5.4-fold and 1.9-fold more IFN-γ-producing cells to mLN and spleen, respectively (Fig. 7C and D). The frequency of NP-specific CD4 T cells was not significantly different between intranasal and S.Q. vaccination in the popliteal lymph node, which drains the site of subcutaneous immunization (Fig. 7E). These results indicate the critical role that direct delivery to the respiratory track plays in localizing influenza-specific CD4 T cells in the respiratory tract, in agreement with studies of attenuated and licensed influenza vaccine (71).
FIG 7.
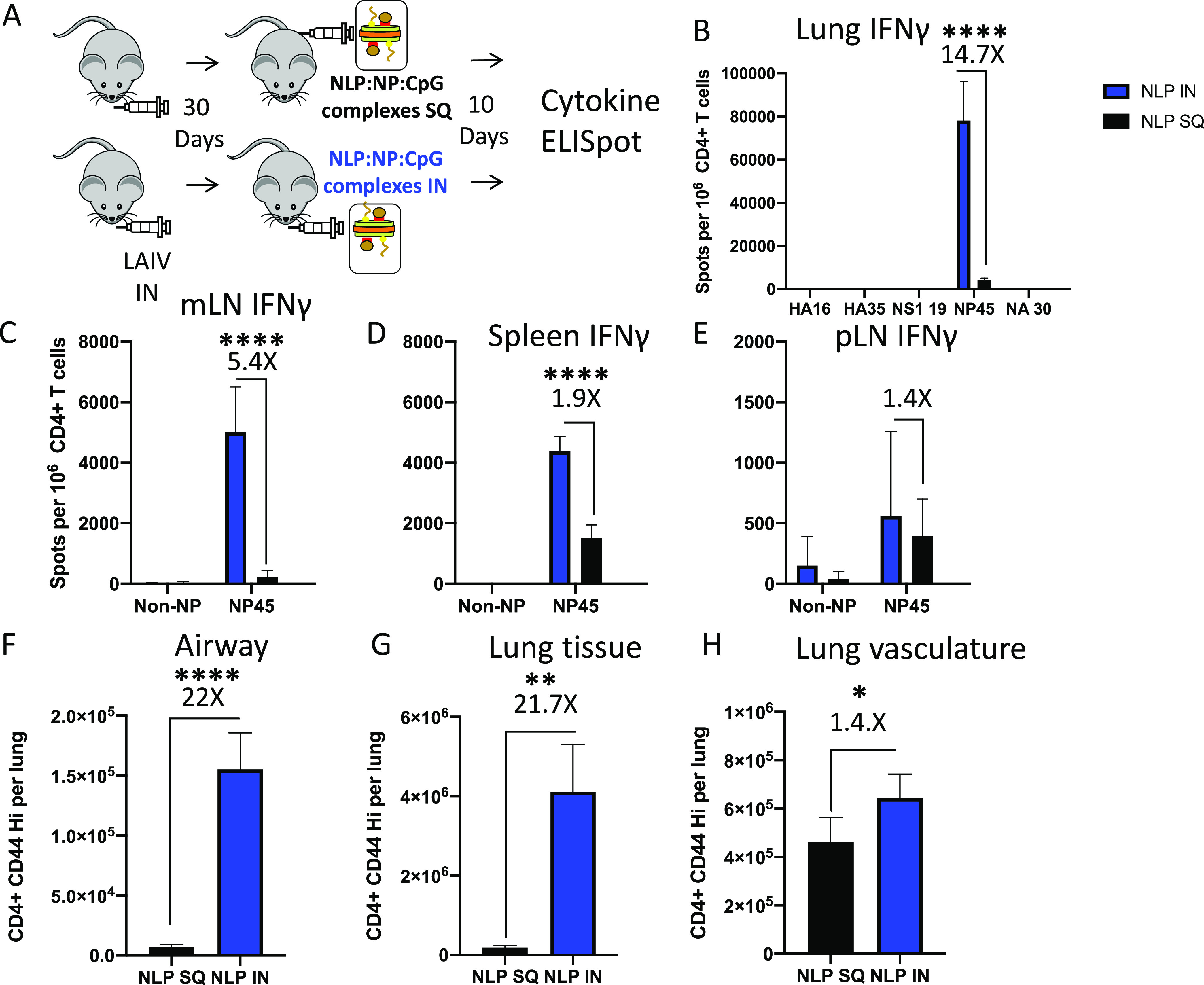
Intranasal vaccination with NLP:NP elicits a higher frequency of antigen-specific CD4 T cells than peripheral vaccination. (A) Schematic representation of the immunization regimen used to compare the effect of intranasal (i.n.) versus subcutaneous (S.Q.) immunization with NLP:NP. (B to D) Comparison of the frequency of lung- (B), mLN- (C), and spleen-localized (D) CD4 T cell responses following i.n. or S.Q. immunization as determined by cytokine ELISpot. (F to G) Assessment of the abundance of antigen-experienced CD4 T cells within the airway (F), lung tissue (G), and lung vasculature (H) following i.n. or S.Q. NLP:NP immunization. Results are presented as the mean with standard deviation of four individual mice per cohort. One cohort was assessed by cytokine ELISpot and a second cohort was assessed by flow cytometry. Statistical significance was determined by unpaired, two-tailed t test with Welch’s correction.
The preceding studies also revealed the enhanced overall immunogenicity of intranasal vaccine responses, in both lung and peripheral secondary lymphoid tissues. When the total abundance of antigen-experienced CD44 high CD4 T cells was compared between intranasal and subcutaneous vaccination, intranasal immunization was found to elicit more than 20-fold more CD4 T cells to the lung airway and lung tissue than subcutaneous immunization, as well as leading to a substantial increase in CD4 T cells localized to the lung vasculature (Fig. 7F to H). Representation of cells in the spleen increased almost 2-fold (Fig. 7D). Thus, lung delivery is critical in seeding tissue-resident CD4 T cells that home to the site of subsequent influenza virus infection but also enhance representation of viral antigen-specific cells in secondary lymphoid tissue.
Intranasal NLP:NP immunization elicits distinct populations of NP-specific CD4 T cells localized to the airway, lung tissue, and lung vasculature.
After finding that NLP:NP immunization elicits a population of NP-specific lung tissue-resident CD4 T cells, we sought to additionally evaluate whether the vaccine strategy elicits lung tissue-resident T cells that can localize directly to the airway. Previous studies of primary and secondary infections have shown that T cells localized to the airway have unique potential to serve as front-line defenders against viral infections, and may have altered functional potential relative to CD4 T cells isolated from other sites within lung or secondary lymphoid organs (72–76). We employed an intranasal antibody labeling technique in conjunction with intravascular antibody labeling to definitively identify CD4 T cells induced by the intranasal vaccination that had localized to airway, lung tissue, and lung vasculature (75). CD4 T cells isolated from mLN and spleen were examined in parallel. Additionally, NLP:NP-boosted mice were compared to LAIV D30 mice in terms of their ability to produce cytokines and degranulate in response to antigen. The strategy used to partition CD4 T cell populations from each of the tissues described above is shown in the representative gating schemes (Fig. S3).
In order to determine the cell surface phenotype and functional potential of CD4 T cells localized to the three distinct compartments of the lung, cells were restimulated with NP45 peptide and stained intracellularly for IFN-γ, tumor necrosis factor alpha (TNF-α), IL-2, and the putative cytotoxic degranulation marker CD107a (77). Employing a Boolean gating strategy to assess polyfunctionality in the antigen-experienced, cytokine-producing CD4 T cells, LAIV-primed mice immunized 30 days prior contained few cytokine-producing cells in the lung, with some IFN-γ single producers detectable in the BAL fluid and mLN, while IL-2 single producers predominated in the spleen (Fig. 8A). In contrast, following NLP:NP boost, NP-specific CD4 T cells from lung and peripheral sites exhibited multifunctionality, producing both IFN-γ and TNF-α upon restimulation with antigen (Fig. 8B). A smaller proportion of NP-specific CD4 T cells simultaneously produced IFN-γ, TNF-α, and IL-2, or else produced either IFN-γ or TNF-α alone (Fig. 8B). A significantly higher frequency of multifunctional CD4 T cells producing IFN-γ and TNF-α was isolated from the airway, relative to lung or peripheral lymphoid tissue (Fig. 8B). CD4 T cells were also examined for markers of cytolytic potential (60, 78–80). Vaccination with the NLP:NP conjugate increased the frequency of CD4 T cells in the lung airway and tissue that expressed the cytotoxic degranulation marker CD107a and the surface marker NKG2, relative to LAIV-alone controls (Fig. 8C and D). These results indicate that intranasal delivery of the NLP:NP conjugate poises a population of influenza virus-specific CD4 T cells that localize to the lung airway and tissue to produce IFN-γ, TNF-α, and degranulate upon restimulation with cognate antigen. Airway-localized CD4 T cells display enhanced production of antiviral cytokines and cytotoxic mediators relative to CD4 T cells localized to the lung tissue or vasculature. CD4 T cells localized to the lung possess enhanced effector functions relative to cells isolated from mLN or spleen. Production of IL-4 and IL-17 was also assessed by intracellular cytokine staining (ICS) to determine if vaccination elicits Th2 and Th17 polarized cells, respectively. NP-specific CD4 T cells restimulated with NP45 peptide (Fig. 9A) produced IL-4 or IL-17 at a frequency of less than 0.5% of CD44 high CD4 T cells.
FIG 8.

Intranasal NLP:NP immunization elicits distinct populations of NP-specific CD4 T cells localized to the airway, lung tissue, and lung vasculature. CD4 T cells isolated from the indicated tissue were restimulated with cognate antigen and stained intracellularly with IFN-γ, TNF-α, and IL-2 antibodies. Cells producing one or more cytokines were determined using Boolean gating. (A and B) The frequency of cells producing one or more cytokines was quantified among CD4 T cells from LAIV alone (A) or from LAIV+NLP-boosted (B) mice. (C and D) Quantification of antigen-experienced CD4 T cells that underwent degranulation (C), as evidenced by expression of CD107a+ in response to antigen stimulation or those expressing putative cytolytic marker NKG2 (D). Data are presented as the mean of two independent experiments of 15 pooled mice per treatment group (LAIV alone or LAIV+NLP boost) with standard deviation shown. Statistical significance (*, P < 0.05; **, P < 0.01 ***, P < 0.001; and ****, P < 0.0001) for comparisons of cellular frequency between tissues was determined by two-way ANOVA with Tukey’s correction for multiple comparisons.
FIG 9.

NP45 peptide-stimulated CD4 T cells produce prototype Th1 cytokines but not IL-4 or IL-17. CD4 T cells isolated from the indicated tissue were restimulated with cognate antigen and stained intracellularly with IFN-γ, TNF-α, IL-2, IL-4, and IL-17 antibodies as described in Fig. 8. (A) The frequency of cells singly producing each cytokine was quantified among CD4 T cells from NLP-boosted mice in the indicated tissues. Data are presented as the mean of five single mice.
We next applied the high-dimensional data visualization tool viSNE to our flow cytometry data. Based on the t-distributed stochastic neighbor embedding (t-SNE) algorithm, viSNE is an unsupervised dimensionality reduction algorithm that allows visualization of the relationships between single cells in two-dimensional plots, while faithfully capturing relationships between diverse populations in the data set (81). Within the viSNE data set (Fig. 10), clusters of interest have been labeled and outlined (black circle). The viSNE analyses identified populations of CD4 T cells based on their antibody labeling status that is associated with the two antibody-accessible compartments in the lung. Within the airway, a clear subset of intranasally labeled cells emerges (“IN Ab”), only detectable in the BAL fluid (Fig. 10A), whereas in the lung, a subset of intravenous antibody labeled cells (“IV Ab”) is detectable (Fig. 10B). Cytokine-positive cells are identifiable in the CD44 high population in each tissue and are circled (Fig. 10C). Also, a cluster of IFN-γ, TNF-α, and IL-2 positive CD4 T cells falls within a region of CD44 high CD4 T cells. These regions of IFN-γ, TNF-α, and IL-2 cells overlap with one another, demonstrating the markers associated with polyfunctional cytokine production (Fig. 10D). A subset of these cytokine-producing cells also overlaps with cells enriched for expression of the cytotoxic markers NKG2 and CD107a, suggesting that some of the antigen-specific CD4 T cell repertoire is capable of both classical Th1 cytokine production and cytotoxicity (Fig. 10D and E). An island of CD107a-positive cells overlaps with the region of CD44 high CD4 T cells but not cytokine production (Fig. 10E). This observation suggests there are different subsets of vaccine-elicited CD4 T cells that provide protective functions by distinct effector mechanisms. Higher intensity of cytokine expression and cytotoxic markers within the airway and lung tissue are in agreement with manually gated analyses. Overall, these results indicate the heterogeneity within the NP-specific CD4 T cell response to vaccination and highlight the polyfunctionality of these cells.
FIG 10.

Unsupervised clustering highlights polyfunctionality of CD4 T cells elicited by NLP:NP immunization. (A to E) viSNE pseudocolored maps depicting the relative expression of the indicated markers (columns) across CD4 T cells isolated from airway, lung, mLN, and spleen (rows). Features of interested are highlighted in the viSNE pseudocolored maps using circles and letters (A to E) corresponding to descriptions in the results. The scale bar reflects the relative expression difference of each marker indicated at the top of each column from low (blue) to high (red). Cells were stimulated as described in Fig. 7. Manually gated live CD4+ and CD8+ cells were input into the viSNE clustering algorithm, with 50,000 cells per population input into the clustering algorithm for a total of 200,000 cells. viSNE plots were generated with an iteration number of 3,000 and perplexity of 50 (final KL Divergence of run 3.87).
NLP:NP immunization elicits a population of antigen-specific CD4 T cells that persist in the lung long term.
Durability of memory T cells following intranasal vaccination is a high priority for future influenza vaccine platforms (16). Several studies have noted a steady decline of CD4 tissue-resident memory within the lung microenvironment after influenza virus infection (70, 82). To assess the durability of the response to intranasal immunization, the abundance of NP-specific CD4 T cells was examined late, at day 270 postvaccination. Despite attrition of CD4 T cells over time, NP-specific CD4 T cells were still readily detectable in lung, mLN, and spleen at 9 months postvaccination (Fig. 11A to C). The highest frequency of NP-specific CD4 T cells was detected in the lung, suggestive of either a stable population that persists long term in the lung or steady recruitment from the periphery to maintain a population of lung-localized cells (Fig. 11A). These findings demonstrate that intranasal NLP:NP immunization elicits influenza virus-specific CD4 T cells that can persist long term in the lung, a critical aspect of any intranasal vaccine candidate.
FIG 11.

NLP:NP immunization elicits a population of antigen-specific CD4 T cells that persist in the lung long term. Persistence of antigen-specific CD4 T cells was assessed at 9 months postimmunization to assess durability of responses without confounding effects of immunosenescence. (A to C) The frequency of IL-2- and IFN-γ-producing cells was assessed in lung (A), mLN (B), and spleen (C) by ELISpot assay. Data, presented as cytokine-producing spots per million CD4 T cells, are the mean of three independent experiments of five pooled mice each.
Adoptive transfer of CD4 T cells from NLP:NP-boosted, but not LAIV-boosted, mice is sufficient for protection of naive mice from pathogenic pH1N1 virus challenge.
Finally, we addressed whether the CD4 T cells elicited by our immunization regimen of LAIV prime and NLP:NP boost had the potential to mediate protection from lethal influenza virus challenge, and whether an NLP:NP boost provided enhanced protection relative to boost with LAIV. Five million purified CD4 T cells were adoptively transferred into naive CD45.1 congenic recipient mice 1 day prior to challenge with a lethal dose of A/California/09 pH1N1 virus. Challenge of mice that received CD4 T cells isolated from NLP:NP-immunized lung resulted in some transient weight loss, with a mean maximum weight loss of 8.1% on day 8 postinfection and 100% survival (Fig. 12A and B). All mock-immunized mice succumbed to infection by day 10 postinfection (Fig. 12B). In striking contrast, CD4 T cells isolated from lung of LAIV-boosted mice were not sufficient to mediate protection from a pH1N1 challenge (Fig. 12C and D). Mice that received lung CD4 T cells from LAIV-boosted donors both displayed severe weight loss and succumbed (100%) to infection by day 9 postinfection (Fig. 12C and D). Crucially, these results demonstrate that adoptive transfer of CD4 T cells boosted by NLP:NP immunization, but not by LAIV, is sufficient to mediate protection from lethal influenza virus challenge.
FIG 12.

Adoptive transfer of CD4 T cells from NLP:NP-boosted, but not LAIV-boosted, mice is sufficient for protection of naive mice from pathogenic pH1N1 virus challenge. The protective potential of CD4 T cells from NLP:NP-boosted and LAIV-boosted mice was assessed in the context of a lethal influenza virus challenge experiment. Five million purified CD4 T cells were adoptively transferred into naive CD45.1 congenic recipient mice at 1 day prior to challenge with a lethal dose of A/California/09 pH1N1 virus. Mice that lost >20% of their starting body weight were scored deceased and humanely euthanized in accordance with institutional guidelines. (A to D) Weight loss was tracked daily over the course of 15 days. Weight loss (A) and overall survival (B) are shown for mice that received CD4 T cells from NLP:NP-boosted mice. Matched adoptive transfer and viral challenge data showing weight loss (C) and survival (D) of mice that received CD4 T cells from LAIV-boosted mice. Data are presented as the mean of two independent experiments with the standard error of the mean (SEM) shown, where n = 5 to 8 for the lung-derived T cells and n = 18 for the mock control. Statistical significance was determined by one-way ANOVA and log rank (Mantel-Cox) test.
Adoptive transfer of CD4 T cells isolated from NLP:NP-immunized lung to naive recipients was sufficient to prevent weight loss-induced lethality in 100% of recipient mice. Transfer of lung-isolated CD4 T cells from LAIV-boosted mice to naive recipients was insufficient to mediate protection from lethal challenge. In summary, adoptive transfer of CD4 T cells from the lung of NLP:NP-boosted mice, which primarily target the highly conserved influenza nucleoprotein, are able to completely protect naive recipient mice from lethal influenza virus challenge.
DISCUSSION
In this study, we evaluated the capability of an intranasal nanoparticle vaccine bearing the highly conserved influenza antigen NP to potentiate lung-localized CD4 T cell immunity with the potential to protect against influenza virus infection. These studies revealed that intranasal vaccination with NLP:NP elicited NP-specific CD4 T cells localized to three distinct compartments within the lung, i.e., the vasculature, tissue, and airway. NP-specific CD4 T cells were also seeded to secondary lymphoid organs. Phenotypic analyses suggested that NLP:NP immunization elicited lung-resident effector CD4 T cell populations, including those with memory precursor potential. Substantiating this memory-forming potential, durable lung-localized CD4 T cell responses were found to persist at least 9 months postvaccination. Importantly, NLP:NP vaccination potentiated a population of airway-localized NP-specific CD4 T cells that responded robustly to restimulation with cognate antigen via production of antiviral cytokines and degranulation, and did so at a frequency higher than lung tissue, lung vasculature, or peripheral CD4 T cells. Lung tissue- and vasculature-localized cells also had enhanced effector functions relative to NP-specific CD4 T cells isolated from mLN or spleen. These lung-localized CD4 T cell responses were found to be dependent on codelivery of NP and CpG on the NLP platform, and immunization by the intranasal route. Results from intracellular cytokine staining experiments (Fig. 8) lead us to the conservative conclusion that at least 51.6% of the 4.6 × 106 tissue-resident effector CD4 T cells (Fig. 4) in an average NLP:NP-immunized lung produce at least a single cytokine in response to peptide restimulation. This estimate is conservative because it is known that not all cells of a given antigen specificity produce cytokines, potentially undercounting the total antigen-specific repertoire (83–85).
There were a number of notable features of the immunity generated by intranasal NLP:NP vaccination. A critical subset of CD4 T cells elicited by NLP:NP vaccination were airway-homing CD4 T cells that produced IFN-γ, as airway-localized CD4 and CD8 T cells have been implicated in protection from diverse viral infections such as SARS, RSV, Sendai virus, and influenza virus (72–76). Further, airway-localized CD4 T cells have been shown to mediate protection from virus infection after direct intratracheal transfer into naive mice, and have the potential to do so in a CD8-independent manner (75). Another defining feature of the multifunctional CD4 T cells elicited by NLP:NP immunization is production of TNF-α, which has been implicated as a critical mediator for limiting influenza virus replication in lung epithelial cells (86). Finally, airway-localized CD4 T cells elicited by NLP:NP vaccination display increased cytotoxic potential relative to lung tissue, vasculature, or periphery, as measured by degranulation in response to antigen and expression of the surface marker NKG2. Cytotoxic CD4 T cells expressing NKG2 have previously been implicated in protection from influenza virus infection through MHC-II-restricted killing via Granzyme B and CD107a (60). This result is in contrast to airway-homing CD8 T cells, which have inferior cytolytic capacity relative to lung tissue- or vasculature-localized CD8 T cells (87).
CD4 T cells elicited by the intranasal vaccine described here express markers associated with tissue residency in the lung, a subset of cells shown to be critical for early control of viral infection. Observed heterogeneity in expression of the marker CD69, often associated with tissue residency in the lung and noted in studies of lung T cells after infection, may be a consequence of local antigen reencounter, local cytokine milieu, or reflect positioning within a lung niche that enables CD69-independent retention of CD4 T cells (63, 65, 69). Heterogeneity in CD69 expression has also been noted after influenza virus infection (69, 70). The progressive loss of lung tissue-resident memory populations after infection (74, 88) has emphasized the importance of durable responses in vaccine design efforts (16). NP-specific CD4 T cells in the lung at the peak of the T cell response were found to lack expression of Ly6C. Previous studies have shown that Ly6C low CD4 T cells are longer lived and have greater proliferative potential in response to reinfection than Ly6C high CD4 T cells, a critical parameter for cells elicited by vaccination that can respond to subsequent infections (61). Our data from 9 months postvaccination demonstrated the durability of CD4 T cell responses to NLP vaccination, whereas other reports have found that antigen-specific CD4 T cell responses were found to be undetectable 2 months after the peak of the response (74). Durability of responses may be related to a recently described phenomenon termed retrograde migration, where lung-localized T cells emigrate from the lung to draining mLN and maintain a program of regionalized immune surveillance (89).
We found that CD4 T cells elicited by our immunization regimen of LAIV prime and NLP:NP boost had the potential to mediate protection from lethal influenza virus challenge, and that the NLP:NP boost had a protective advantage compared to LAIV boost alone. The transient weight loss and 100% survival observed in recipient mice that received CD4 T cells isolated from NLP:NP-immunized lung stands in stark contrast to LAIV-boosted recipients that succumbed to infection by day 9 postinfection. CD4 T cells isolated from lung of LAIV-boosted mice were sufficient to mediate protection from pH1N1 challenge. The enhanced magnitude of the CD4 T cell response to NLP:NP immunization relative to LAIV boost (Fig. 3 and 4) may underlie the lack of protection seen in the LAIV boost. As previously reported, preexisting immunity (most likely antibodies) to LAIV may blunt replication, reducing antigen abundance and resulting in decreased efficacy (21, 22), whereas a vaccine that does not require replication bypasses this negative impact. The mechanisms of protection induced by the NLP-NP boost are likely multifactorial. Others have shown that CD4 T cell memory cells can potentiate early recruitment of innate cells that have antiviral activity (33–36). Our results show that many of the CD4 T cells boosted by the NLP-NP vaccine that reside in the lung tissue and airways produced IFN-γ, a known antiviral mediator that contributes to control of viral infection by airway-localized CD8 T cells (87). Finally, our multiparameter flow cytometry data also suggest that cytolytic CD4 T cells are induced by the NLP-NP boost. Each of these activities may act synergistically to contribute to the protection conveyed by the transferred cells.
These studies collectively emphasize the key feature of CD4 T cells specific for the conserved vaccine antigen NP that can be induced by intranasal protein vaccination with a novel nanoparticle platform. The localization, polyfunctionality, and persistence in vivo, coupled with the multiplicity of function that memory CD4 T cells are known to convey (29–32), suggest the validity of pursuing intranasal vaccine approaches for induction of broadly protective immunity that can rapidly respond to influenza virus challenge.
MATERIALS AND METHODS
Animals.
Female C57BL/6 mice were obtained from the National Cancer Institute and maintained at a specific-pathogen-free facility at the University of Rochester Medical Center according to institutional guidelines. Mice were used at 8 to 10 weeks of age.
Ethics statement.
All mice were maintained under specific-pathogen-free conditions at the University of Rochester Medical Center according to institutional guidelines. All animal protocols adhere to AAALAC International, the Animal Welfare Act, the PHS Guide, and were approved by the University of Rochester Committee on Animal Resources, Animal Welfare Assurance Number A3291-01. The protocol under which the studies were conducted was first approved 4 March 2006 (protocol 2006-030) and has been reviewed and reapproved every 36 months, with the most recent reapproval on 23 January 2018.
NLP production and purification.
The 22 kDa N-terminal domain of apolipoprotein E4 was prepared as previously described (42), with the exception that the murine sequence was used to avoid any immune response against the NLP. NLPs were essentially prepared as described (44). Briefly, a thin lipid film of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-(N-[5-amino-1-carboxypentyl]iminodiacetic acid)succinyl (nickel salt) (DGS-NTA-Ni) (Avanti Polar Lipids, Alabaster, AL) was solubilized in phosphate-buffered saline (PBS) with 60 mM sodium cholate (Sigma-Aldrich). The molar lipid ratio was 35:65 of DGS-NTA-Ni and DOPC, respectively. Upon addition of the apolipoprotein, the reaction was dialyzed overnight against PBS to initiate self-assembly of the NLP. The apolipoprotein was prepared as previously described, with the exception of using the murine sequence, and particles were purified by size exclusion chromatography (SEC) on a Superdex 200 Increase 10/300 GL column (Cytiva) in PBS at a 1 ml/min flow rate. Elution fractions corresponding to the NLP peak were pooled, concentrated, filter sterilized (Corning Costar Spin-X filters, 0.22 μm), lyophilized in 0.1 M trehalose, and stored at −20°C until use. Recombinant influenza virus NP from A/New Caledonia/20/99 was produced as described (90). Antigen and adjuvant conjugations were assessed by SEC using a Superdex 200 Increase 5/150 GL column (Cytiva) in PBS at a 0.5 ml/min flow rate. To prepare vaccine formulations, NLPs were rehydrated with water and diluted in PBS. Cholesterol-modified CpG (5′-Chol-TCCATGACGTTCCTGACGTT-3′, custom order from BioSearch Technologies, Novato, CA) and NP protein were subsequently added to the NLP and used without further purification.
Intranasal priming and vaccination.
Intranasal immunization was performed under anesthesia with 250 mg/kg tribromoethanol delivered by an intraperitoneal route. All intranasal immunizations were performed at a final volume of 30 μl. Naive mice were primed with 1 × 107 50% egg infective dose (EID50) monovalent live attenuated influenza virus (LAIV), a 6:2 reassortant of caA/Ann Arbor/6/60 and A/New Caledonia/99. NLP immunizations comprised 5 μg recombinant influenza NP and 5 μg CpG conjugated to the NLP at a molar ratio of 1:2.4:18.4, NLP to NP to CpG, respectively. For subcutaneous immunizations, the same formulation as described above was delivered to the rear footpads.
Intravascular and intranasal antibody labeling.
Mice were anaesthetized via inhalation of isoflurane and retro-orbitally injected with 3 μg anti-mouse CD45 (30-F11, Tonbo) in a total volume of 100 μl (59). At 3 min following i.v. injection, mice were sacrificed by overdose with tribromoethanol and tissues were harvested. Intranasal antibody labeling (75) was performed by intranasal delivery of 0.2 μg anti-CD90 (30-H12, Biolegend).
Tissue processing and cell isolation.
Lung and lymphoid tissues were excised from euthanized mice. Lung tissues were minced using surgical scissors in medium containing collagenase type II or IV (103 units/ml) and DNase I (30 μg/ml) in RPMI supplemented with 2.5% fetal bovine serum (FBS) and 10 mM HEPES. Individual lungs were added to GentleMACS tubes (Miltenyi 130-093-237) and processed on GENTLEMACS setting Lung 01, incubated for 1 h at 37°C with constant shaking, then processed on setting Heart 01. Lung tissues were subsequently passed through 40-μM sterile nylon mesh and rinsed with Dulbecco’s modified Eagle medium (DMEM, Gibco) supplemented with 1% gentamicin and 10% heat-inactivated FBS. The lymph node was disrupted using 40-μM sterile nylon mesh and a 5 ml syringe plunger. Resulting single-cell suspensions were treated with ACK lysis buffer (0.15 M NH4Cl, 1.0 mM KHCO3, 0.1 mM NaEDTA, pH 7.2) to deplete red blood cells.
Flow cytometry.
For surface staining experiments, 2 × 106 cells were added to a U-bottom plate. Cells were washed twice with PBS, then incubated with fixable live/dead aqua (Life Technologies) for 30 min at 4°C according to the manufacturer’s instructions. Cells were subsequently washed twice with FC stain buffer (PBS plus 2% heat-inactivated FBS and 0.01% sodium azide) and resuspended in anti-mouse CD16/CD32 (BD FC block, 2.4G2) for 20 min at 4°C. Without washing, cells were stained for 30 min at 4°C with the following antibodies: CD4 (RM4-5, BD Biosciences), CD8 (5H10, Biolegend), CD11a (2D7, BD Biosciences), CD44 (IM7, Tonbo), CD62L (MEL-14, Biolegend), Ly6C (AL-21, BD Biosciences), NKG2A/C/E (20d5, BD Biosciences), and CD69 (H1.2F3, Biolegend). Data were acquired using a BD LSR-II, configured with 488 nm, 633 nm, 407 nm, and 532 nm lasers. Data were analyzed using FlowJo software version 10.7.1 (Becton, Dickinson and Company, Ashland, OR).
Intracellular cytokine staining.
Cells from LAIV-primed and NLP:NP-vaccinated mice were isolated and cocultured in U-bottom 96-well plates (3 × 105 cells/well) with splenocytes from naive syngeneic donors (5 × 105 cells/well) with or without NP-derived peptide. Brefeldin A, monensin, and anti-CD107a antibody (1D4B, eBioscience) were added to cultures 2 h after stimulation, then incubated for an additional 6 h for a total of 8 h. Plates were stored overnight at 4°C. Cells were washed and surface stained as described above. Cells were fixed and permeabilized using the eBioscience FoxP3 transcription factor staining kit according to the manufacturer’s instructions. Cells were stained for 1 h at 4°C with the following antibodies: IFN-γ (XMG1.2, BD Biosciences), IL-2 (JES6-5H4, BD Biosciences), TNF-α (MP6-XT22, Biolegend), IL-4 (11B11, Biolegend), and IL-17 (TC11-18H10, BD Biosciences).
MHC-II tetramer staining and enrichment.
Lung single cell suspensions were generated as described above. Cells were then stained with 10 nM NP261:I-Ab-streptavidin-phycoerythrin tetramer for 1 h at room temperature in diluted in FC block (2.4G2 from BD) as described (91). Cells were then washed with FC stain buffer. Tetramer-stained cells were resuspended in a volume of 200 μl and 50 μl of anti-PE microbeads (MACS 130-048-801) for 30 min at 4°C. Cells were washed with FC stain buffer and resuspended in 3 ml FC stain buffer before being passed over LS columns (MACS 130-042-401). The column was washed three times with 3 ml of FC stain buffer. The column was removed from the magnetic field and the bound fraction was eluted in 5 ml by plunging the column. The resulting column-bound and unbound fractions underwent viability staining and surface staining as described above. Cells were stained with a cocktail containing antibodies specific for B220 (RA3-6B2), CD11b (M1/70.15), CD11c (N418), F4/80 (BM8), CD3 (145-2C11), CD4 (RM4-5), CD8 (5H10), and CD44 (IM-7). The entire column-bound fraction was collected and surface stained as described above.
ELISpot Assay.
The 96-well filter plates (Millipore, Billerica, MA, USA) were coated with 2 μg/ml purified rat anti-mouse IL-2 (JES6-1A12, BD) or IFN-γ (AN-18, BD) in PBS overnight at 4°C. Prior to plating, wells were washed with medium to remove unbound antibody and incubated with medium for 1 h at room temperature to block nonspecific binding. CD4 T cells (200,000 mLN/spleen cells or 50,000 lung cells) were purified by negative selection (MACS 130-104-454) and cocultured with 500,000 syngeneic splenocytes and 5 μM peptide in a total volume of 200 μl for 16 to 18 h at 37°C with 5% CO2. Cells were subsequently removed from the filter plates and washed with ELISpot wash buffer (1× PBS with 0.1% Tween 20). Biotinylated rat anti-mouse IL-2 (JES6-1A12) or IFN-γ (XMG1.2) was diluted to 2 μg/ml in ELISpot wash buffer supplemented with 10% FBS in a volume of 50 μl for 30 min at room temperature. Plates were washed with ELISpot wash buffer, and streptavidin-conjugated alkaline phosphatase (Jackson Immuno Research, West Grove, PA, USA) was added at a 1:1,000 dilution in ELISpot wash buffer supplemented with 10% FBS and incubated for 30 min at room temperature. Plates were washed with ELISpot wash buffer and incubated with Vector Blue substrate kit III (Vector Laboratories, CA, USA) in 100 mM Tris (pH 8.2) for 5 min at room temperature. Following development, plates were washed with water and dried. Quantification of spots was performed using an Immunospot reader series 5.2 with Immunospot software version 5.1.
Adoptive transfer and virus challenge.
Cohorts of 15 mice were primed with LAIV and boosted with NLP:NP or LAIV as described above. At day 10 postboost, lungs were harvested and pooled prior to CD4 T cell purification by MACS CD4 T cell negative selection, as described above. Naive CD45.1 congenic recipients received mock adoptive transfer of 100 μl PBS or 5 million purified CD4 T cells isolated from lung by retro-orbital injection. CD4 T cells were transferred 1 day prior to challenge with 20 PFU A/California/4_NYICE_E3/2009 in 30 μl PBS. This dose resulted in 100% lethality in naive CD45.1 congenic recipient mice based on humane weight loss criteria. Mice that lost >20% of their starting body weight were scored deceased and humanely euthanized in accordance with institutional guidelines. Weight loss was tracked daily over the course of 15 days.
Statistical analyses.
Statistical analyses were performed using GraphPad Prism software version 8.4.3 (GraphPad Software, San Diego, CA). Significance was assigned as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Data were analyzed by two-tailed t test, one-way ANOVA, two-way ANOVA, or log rank tests. The specific test performed is indicated in the figure legend.
Synthetic peptides.
Seventeen-mer peptides overlapping by 11 amino acids encompassing the entire sequence of viral proteins were obtained from the NIH Biodefense and Emerging Infectious Disease Research Repository, NIAID, NIH. Individual peptides were reconstituted and used at a final concentration of 5 μM. Sequences of peptides used in this study are listed in Table 1.
ACKNOWLEDGMENTS
This work was supported by research grants from the NIAID Centers of Excellence for Influenza Research and Surveillance and the National Institutes of Health, including grants HHSN272201400005C, HHSN272201400005C-17A, and 5T32HL066988-19. A graphical summary of findings from this manuscript was generated using BioRender.
We declare no conflicts of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Andrea J. Sant, Email: Andrea_Sant@URMC.Rochester.edu.
Stacey Schultz-Cherry, St. Jude Children's Research Hospital.
REFERENCES
- 1.Molinari NA, Ortega-Sanchez IR, Messonnier ML, Thompson WW, Wortley PM, Weintraub E, Bridges CB. 2007. The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine 25:5086–5096. 10.1016/j.vaccine.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 2.Rolfes MA, Foppa IM, Garg S, Flannery B, Brammer L, Singleton JA, Burns E, Jernigan D, Olsen SJ, Bresee J, Reed C. 2018. Annual estimates of the burden of seasonal influenza in the United States: a tool for strengthening influenza surveillance and preparedness. Influenza Other Respir Viruses 12:132–137. 10.1111/irv.12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reed C, Chaves SS, Daily Kirley P, Emerson R, Aragon D, Hancock EB, Butler L, Baumbach J, Hollick G, Bennett NM, Laidler MR, Thomas A, Meltzer MI, Finelli L. 2015. Estimating influenza disease burden from population-based surveillance data in the United States. PLoS One 10:e0118369. 10.1371/journal.pone.0118369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iuliano AD, Roguski KM, Chang HH, Muscatello DJ, Palekar R, Tempia S, Cohen C, Gran JM, Schanzer D, Cowling BJ, Wu P, Kyncl J, Ang LW, Park M, Redlberger-Fritz M, Yu H, Espenhain L, Krishnan A, Emukule G, van Asten L, Pereira da Silva S, Aungkulanon S, Buchholz U, Widdowson M-A, Bresee JS, Azziz-Baumgartner E, Cheng P-Y, Dawood F, Foppa I, Olsen S, Haber M, Jeffers C, MacIntyre CR, Newall AT, Wood JG, Kundi M, Popow-Kraupp T, Ahmed M, Rahman M, Marinho F, Sotomayor Proschle CV, Vergara Mallegas N, Luzhao F, Sa L, Barbosa-Ramírez J, Sanchez DM, Gomez LA, Vargas XB, Acosta Herrera a, Llanés MJ, et al. 2018. Estimates of global seasonal influenza-associated respiratory mortality: a modelling study. Lancet 391:1285–1300. 10.1016/S0140-6736(17)33293-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richards KA, Moritzky S, Shannon I, Fitzgerald T, Yang H, Branche A, Topham DJ, Treanor JJ, Nayak J, Sant AJ. 2020. Recombinant HA-based vaccine outperforms split and subunit vaccines in elicitation of influenza-specific CD4 T cells and CD4 T cell-dependent antibody responses in humans. NPJ Vaccines 5:77. 10.1038/s41541-020-00227-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouvier NM. 2018. The future of influenza vaccines: a historical and clinical perspective. Vaccines (Basel) 6:58. 10.3390/vaccines6030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Del Giudice G, Rappuoli R. 2015. Inactivated and adjuvanted influenza vaccines. Curr Top Microbiol Immunol 386:151–180. 10.1007/82_2014_406. [DOI] [PubMed] [Google Scholar]
- 8.Belongia EA, Skowronski DM, McLean HQ, Chambers C, Sundaram ME, De Serres G. 2017. Repeated annual influenza vaccination and vaccine effectiveness: review of evidence. Expert Rev Vaccines 16:1–14. 10.1080/14760584.2017.1334554. [DOI] [PubMed] [Google Scholar]
- 9.Krammer F, Garcia-Sastre A, Palese P. 2018. Is it possible to develop a “universal” influenza virus vaccine? Potential target antigens and critical aspects for a universal influenza vaccine. Cold Spring Harb Perspect Biol 10:a028845. 10.1101/cshperspect.a028845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poland GA. 2018. Influenza vaccine failure: failure to protect or failure to understand? Expert Rev Vaccines 17:495–502. 10.1080/14760584.2018.1484284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang TT, Bournazos S, Ravetch JV. 2018. Immunological responses to influenza vaccination: lessons for improving vaccine efficacy. Curr Opin Immunol 53:124–129. 10.1016/j.coi.2018.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clemens EB, van de Sandt C, Wong SS, Wakim LM, Valkenburg SA. 2018. Harnessing the power of T cells: the promising hope for a universal influenza vaccine. Vaccines (Basel) 6:18. 10.3390/vaccines6020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim TS, Sun J, Braciale TJ. 2011. T cell responses during influenza infection: getting and keeping control. Trends Immunol 32:225–231. 10.1016/j.it.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schotsaert M, Garcia-Sastre A. 2014. Influenza vaccines: a moving interdisciplinary field. Viruses 6:3809–3826. 10.3390/v6103809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards KA, Shannon I, Treanor JJ, Yang H, Nayak JL, Sant AJ. 2020. Evidence that blunted CD4 T-cell responses underlie deficient protective antibody responses to influenza vaccines in repeatedly vaccinated human subjects. J Infect Dis 222:273–277. 10.1093/infdis/jiz433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erbelding EJ, Post DJ, Stemmy EJ, Roberts PC, Augustine AD, Ferguson S, Paules CI, Graham BS, Fauci AS. 2018. A universal influenza vaccine: the strategic plan for the National Institute of Allergy and Infectious Diseases. J Infect Dis 218:347–354. 10.1093/infdis/jiy103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei CJ, Crank MC, Shiver J, Graham BS, Mascola JR, Nabel GJ. 2020. Next-generation influenza vaccines: opportunities and challenges. Nat Rev Drug Discov 19:239–252. 10.1038/s41573-019-0056-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krammer F, Palese P. 2015. Advances in the development of influenza virus vaccines. Nat Rev Drug Discov 14:167–182. 10.1038/nrd4529. [DOI] [PubMed] [Google Scholar]
- 19.Estrada LD, Schultz-Cherry S. 2019. Development of a universal influenza vaccine. J Immunol 202:392–398. 10.4049/jimmunol.1801054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knight M, Changrob S, Li L, Wilson PC. 2020. Imprinting, immunodominance, and other impediments to generating broad influenza immunity. Immunol Rev 296:191–204. 10.1111/imr.12900. [DOI] [PubMed] [Google Scholar]
- 21.Mohn KG, Smith I, Sjursen H, Cox RJ. 2018. Immune responses after live attenuated influenza vaccination. Hum Vaccin Immunother 14:571–578. 10.1080/21645515.2017.1377376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mok DZL, Chan KR. 2020. The effects of pre-existing antibodies on live-attenuated viral vaccines. Viruses 12:520. 10.3390/v12050520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coulter A, Harris R, Davis R, Drane D, Cox J, Ryan D, Sutton P, Rockman S, Pearse M. 2003. Intranasal vaccination with ISCOMATRIX adjuvanted influenza vaccine. Vaccine 21:946–949. 10.1016/S0264-410X(02)00545-5. [DOI] [PubMed] [Google Scholar]
- 24.Deng L, Ibanez LI, Van den Bossche V, Roose K, Youssef SA, de Bruin A, Fiers W, Saelens X. 2015. Protection against influenza A virus challenge with M2e-displaying filamentous Escherichia coli phages. PLoS One 10:e0126650. 10.1371/journal.pone.0126650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herve PL, Raliou M, Bourdieu C, Dubuquoy C, Petit-Camurdan A, Bertho N, Eleouet JF, Chevalier C, Riffault S. 2014. A novel subnucleocapsid nanoplatform for mucosal vaccination against influenza virus that targets the ectodomain of matrix protein 2. J Virol 88:325–338. 10.1128/JVI.01141-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jansen JM, Gerlach T, Elbahesh H, Rimmelzwaan GF, Saletti G. 2019. Influenza virus-specific CD4+ and CD8+ T cell-mediated immunity induced by infection and vaccination. J Clin Virol 119:44–52. 10.1016/j.jcv.2019.08.009. [DOI] [PubMed] [Google Scholar]
- 27.Nussing S, Sant S, Koutsakos M, Subbarao K, Nguyen THO, Kedzierska K. 2018. Innate and adaptive T cells in influenza disease. Front Med 12:34–47. 10.1007/s11684-017-0606-8. [DOI] [PubMed] [Google Scholar]
- 28.Thomas PG, Keating R, Hulse-Post DJ, Doherty PC. 2006. Cell-mediated protection in influenza infection. Emerg Infect Dis 12:48–54. 10.3201/eid1201.051237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sant AJ, Richards KA, Nayak J. 2018. Distinct and complementary roles of CD4 T cells in protective immunity to influenza virus. Curr Opin Immunol 53:13–21. 10.1016/j.coi.2018.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DiPiazza A, Richards KA, Knowlden ZA, Nayak JL, Sant AJ. 2016. The role of CD4 T cell memory in generating protective immunity to novel and potentially pandemic strains of influenza. Front Immunol 7:10. 10.3389/fimmu.2016.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKinstry KK, Strutt TM, Swain SL. 2011. Hallmarks of CD4 T cell immunity against influenza. J Intern Med 269:507–518. 10.1111/j.1365-2796.2011.02367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strutt TM, McKinstry KK, Marshall NB, Vong AM, Dutton RW, Swain SL. 2013. Multipronged CD4(+) T-cell effector and memory responses cooperate to provide potent immunity against respiratory virus. Immunol Rev 255:149–164. 10.1111/imr.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strutt TM, McKinstry KK, Swain SL. 2011. Control of innate immunity by memory CD4 T cells. Adv Exp Med Biol 780:57–68. 10.1007/978-1-4419-5632-3_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zens KD, Farber DL. 2015. Memory CD4 T cells in influenza. Curr Top Microbiol Immunol 386:399–421. 10.1007/82_2014_401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strutt TM, McKinstry KK, Dibble JP, Winchell C, Kuang Y, Curtis JD, Huston G, Dutton RW, Swain SL. 2010. Memory CD4+ T cells induce innate responses independently of pathogen. Nat Med 16:558–564. 10.1038/nm.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swain SL, McKinstry KK, Strutt TM. 2012. Expanding roles for CD4(+) T cells in immunity to viruses. Nat Rev Immunol 12:136–148. 10.1038/nri3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beura LK, Fares-Frederickson NJ, Steinert EM, Scott MC, Thompson EA, Fraser KA, Schenkel JM, Vezys V, Masopust D. 2019. CD4(+) resident memory T cells dominate immunosurveillance and orchestrate local recall responses. J Exp Med 216:1214–1229. 10.1084/jem.20181365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brummelman J, Pilipow K, Lugli E. 2018. The single-cell phenotypic identity of human CD8(+) and CD4(+) T cells. Int Rev Cell Mol Biol 341:63–124. 10.1016/bs.ircmb.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 39.Masopust D, Soerens AG. 2019. Tissue-resident T cells and other resident leukocytes. Annu Rev Immunol 37:521–546. 10.1146/annurev-immunol-042617-053214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen QP, Deng TZ, Witherden DA, Goldrath AW. 2019. Origins of CD4(+) circulating and tissue-resident memory T-cells. Immunology 157:3–12. 10.1111/imm.13059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilk MM, Mills KHG. 2018. CD4 TRM cells following infection and immunization: implications for more effective vaccine design. Front Immunol 9:1860. 10.3389/fimmu.2018.01860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fischer NO, Blanchette CD, Chromy BA, Kuhn EA, Segelke BW, Corzett M, Bench G, Mason PW, Hoeprich PD. 2009. Immobilization of His-tagged proteins on nickel-chelating nanolipoprotein particles. Bioconjug Chem 20:460–465. 10.1021/bc8003155. [DOI] [PubMed] [Google Scholar]
- 43.Blanchette CD, Fischer NO, Corzett M, Bench G, Hoeprich PD. 2010. Kinetic analysis of his-tagged protein binding to nickel-chelating nanolipoprotein particles. Bioconjug Chem 21:1321–1330. 10.1021/bc100129s. [DOI] [PubMed] [Google Scholar]
- 44.Fischer NO, Rasley A, Corzett M, Hwang MH, Hoeprich PD, Blanchette CD. 2013. Colocalized delivery of adjuvant and antigen using nanolipoprotein particles enhances the immune response to recombinant antigens. J Am Chem Soc 135:2044–2047. 10.1021/ja3063293. [DOI] [PubMed] [Google Scholar]
- 45.Fischer NO, Weilhammer DR, Dunkle A, Thomas C, Hwang M, Corzett M, Lychak C, Mayer W, Urbin S, Collette N, Chiun Chang J, Loots GG, Rasley A, Blanchette CD. 2014. Evaluation of nanolipoprotein particles (NLPs) as an in vivo delivery platform. PLoS One 9:e93342. 10.1371/journal.pone.0093342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nelson SA, Sant AJ. 2019. Imprinting and editing of the human CD4 T cell response to influenza virus. Front Immunol 10:932. 10.3389/fimmu.2019.00932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Somes MP, Turner RM, Dwyer LJ, Newall AT. 2018. Estimating the annual attack rate of seasonal influenza among unvaccinated individuals: a systematic review and meta-analysis. Vaccine 36:3199–3207. 10.1016/j.vaccine.2018.04.063. [DOI] [PubMed] [Google Scholar]
- 48.Krammer F. 2019. The human antibody response to influenza A virus infection and vaccination. Nat Rev Immunol 19:383–397. 10.1038/s41577-019-0143-6. [DOI] [PubMed] [Google Scholar]
- 49.DiPiazza AT, Fan S, Rattan A, DeDiego ML, Chaves F, Neumann G, Kawaoka Y, Sant AJ. 2019. A novel vaccine strategy to overcome poor immunogenicity of avian influenza vaccines through mobilization of memory CD4 T cells established by seasonal influenza. J Immunol 203:1502–1508. 10.4049/jimmunol.1900819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richards KA, Chaves FA, Sant AJ. 2011. The memory phase of the CD4 T-cell response to influenza virus infection maintains its diverse antigen specificity. Immunology 133:246–256. 10.1111/j.1365-2567.2011.03435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pizzolla A, Nguyen TH, Sant S, Jaffar J, Loudovaris T, Mannering SI, Thomas PG, Westall GP, Kedzierska K, Wakim LM. 2018. Influenza-specific lung-resident memory T cells are proliferative and polyfunctional and maintain diverse TCR profiles. J Clin Invest 128:721–733. 10.1172/JCI96957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Purwar R, Campbell J, Murphy G, Richards WG, Clark RA, Kupper TS. 2011. Resident memory T cells (TRM) are abundant in human lung: diversity, function, and antigen specificity. PLoS One 6:e16245. 10.1371/journal.pone.0016245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shin H, Iwasaki A. 2013. Tissue-resident memory T cells. Immunol Rev 255:165–181. 10.1111/imr.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Szabo PA, Miron M, Farber DL. 2019. Location, location, location: tissue resident memory T cells in mice and humans. Sci Immunol 4:eaas9673. 10.1126/sciimmunol.aas9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kukol A, Hughes DJ. 2014. Large-scale analysis of influenza A virus nucleoprotein sequence conservation reveals potential drug-target sites. Virology 454-455:40–47. 10.1016/j.virol.2014.01.023. [DOI] [PubMed] [Google Scholar]
- 56.Nayak JL, Richards KA, Chaves FA, Sant AJ. 2010. Analyses of the specificity of CD4 T cells during the primary immune response to influenza virus reveals dramatic MHC-linked asymmetries in reactivity to individual viral proteins. Viral Immunol 23:169–180. 10.1089/vim.2009.0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrancois L, Farber DL. 2011. Cutting edge: tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol 187:5510–5514. 10.4049/jimmunol.1102243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McKinstry KK, Strutt TM, Kuang Y, Brown DM, Sell S, Dutton RW, Swain SL. 2012. Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J Clin Invest 122:2847–2856. 10.1172/JCI63689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, Masopust D. 2014. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc 9:209–222. 10.1038/nprot.2014.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marshall NB, Vong AM, Devarajan P, Brauner MD, Kuang Y, Nayar R, Schutten EA, Castonguay CH, Berg LJ, Nutt SL, Swain SL. 2017. NKG2C/E marks the unique cytotoxic CD4 T cell subset, ThCTL, generated by influenza infection. J Immunol 198:1142–1155. 10.4049/jimmunol.1601297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, Rutishauser R, Cui W, Kleinstein SH, Craft J, Kaech SM. 2011. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity 35:633–646. 10.1016/j.immuni.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schenkel JM, Masopust D. 2014. Tissue-resident memory T cells. Immunity 41:886–897. 10.1016/j.immuni.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takamura S, Yagi H, Hakata Y, Motozono C, McMaster SR, Masumoto T, Fujisawa M, Chikaishi T, Komeda J, Itoh J, Umemura M, Kyusai A, Tomura M, Nakayama T, Woodland DL, Kohlmeier JE, Miyazawa M. 2016. Specific niches for lung-resident memory CD8+ T cells at the site of tissue regeneration enable CD69-independent maintenance. J Exp Med 213:3057–3073. 10.1084/jem.20160938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takamura S. 2018. Niches for the long-term maintenance of tissue-resident memory T cells. Front Immunol 9:1214. 10.3389/fimmu.2018.01214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walsh DA, Borges da Silva H, Beura LK, Peng C, Hamilton SE, Masopust D, Jameson SC. 2019. The functional requirement for CD69 in establishment of resident memory CD8(+) T cells varies with tissue location. J Immunol 203:946–955. 10.4049/jimmunol.1900052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyarto BZ, Southern PJ, Masopust D. 2015. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell 161:737–749. 10.1016/j.cell.2015.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gebhardt T, Whitney PG, Zaid A, Mackay LK, Brooks AG, Heath WR, Carbone FR, Mueller SN. 2011. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature 477:216–219. 10.1038/nature10339. [DOI] [PubMed] [Google Scholar]
- 68.Iijima N, Iwasaki A. 2014. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science 346:93–98. 10.1126/science.1257530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Turner DL, Bickham KL, Thome JJ, Kim CY, D'Ovidio F, Wherry EJ, Farber DL. 2014. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol 7:501–510. 10.1038/mi.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.DiPiazza A, Laniewski N, Rattan A, Topham DJ, Miller J, Sant AJ. 2018. CD4 T cell epitope specificity and cytokine potential are preserved as cells transition from the lung vasculature to lung tissue following influenza virus infection. J Virol 92:e00377-18. 10.1128/JVI.00377-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zens KD, Chen JK, Farber DL. 2016. Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight 1:e85832. 10.1172/jci.insight.85832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Richter M, Ray SJ, Chapman TJ, Austin SJ, Rebhahn J, Mosmann TR, Gardner H, Kotelianski V, deFougerolles AR, Topham DJ. 2007. Collagen distribution and expression of collagen-binding alpha1beta1 (VLA-1) and alpha2beta1 (VLA-2) integrins on CD4 and CD8 T cells during influenza infection. J Immunol 178:4506–4516. 10.4049/jimmunol.178.7.4506. [DOI] [PubMed] [Google Scholar]
- 73.Hogan RJ, Zhong W, Usherwood EJ, Cookenham T, Roberts AD, Woodland DL. 2001. Protection from respiratory virus infections can be mediated by antigen-specific CD4(+) T cells that persist in the lungs. J Exp Med 193:981–986. 10.1084/jem.193.8.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Woodland DL, Scott I. 2005. T cell memory in the lung airways. Proc Am Thorac Soc 2:126–131. 10.1513/pats.200501-003AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao J, Zhao J, Mangalam AK, Channappanavar R, Fett C, Meyerholz DK, Agnihothram S, Baric RS, David CS, Perlman S. 2016. Airway memory CD4(+) T cells mediate protective immunity against emerging respiratory coronaviruses. Immunity 44:1379–1391. 10.1016/j.immuni.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guvenel A, Jozwik A, Ascough S, Ung SK, Paterson S, Kalyan M, Gardener Z, Bergstrom E, Kar S, Habibi MS, Paras A, Zhu J, Park M, Dhariwal J, Almond M, Wong EHC, Sykes A, Del Rosario J, Trujillo-Torralbo M-B, Mallia P, Sidney J, Peters B, Kon OM, Sette A, Johnston SL, Openshaw PJ, Chiu C. 2019. Epitope-specific airway-resident CD4+ T cell dynamics during experimental human RSV infection. J Clinical Invest 130:523–538. 10.1172/JCI131696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wolint P, Betts MR, Koup RA, Oxenius A. 2004. Immediate cytotoxicity but not degranulation distinguishes effector and memory subsets of CD8+ T cells. J Exp Med 199:925–936. 10.1084/jem.20031799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Juno JA, van Bockel D, Kent SJ, Kelleher AD, Zaunders JJ, Munier CM. 2017. Cytotoxic CD4 T cells—friend or foe during viral infection? Front Immunol 8:19. 10.3389/fimmu.2017.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takeuchi A, Badr Mel S, Miyauchi K, Ishihara C, Onishi R, Guo Z, Sasaki Y, Ike H, Takumi A, Tsuji NM, Murakami Y, Katakai T, Kubo M, Saito T. 2016. CRTAM determines the CD4+ cytotoxic T lymphocyte lineage. J Exp Med 213:123–138. 10.1084/jem.20150519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Takeuchi A, Saito T. 2017. CD4 CTL, a cytotoxic subset of CD4(+) T cells, their differentiation and function. Front Immunol 8:194. 10.3389/fimmu.2017.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe'er D. 2013. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 31:545–552. 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hayward SL, Scharer CD, Cartwright EK, Takamura S, Li ZT, Boss JM, Kohlmeier JE. 2020. Environmental cues regulate epigenetic reprogramming of airway-resident memory CD8(+) T cells. Nat Immunol 21:309–320. 10.1038/s41590-019-0584-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. 2006. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441:235–238. 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 84.Whitmire JK, Benning N, Whitton JL. 2006. Precursor frequency, nonlinear proliferation, and functional maturation of virus-specific CD4+ T cells. J Immunol 176:3028–3036. 10.4049/jimmunol.176.5.3028. [DOI] [PubMed] [Google Scholar]
- 85.Williams MA, Ravkov EV, Bevan MJ. 2008. Rapid culling of the CD4+ T cell repertoire in the transition from effector to memory. Immunity 28:533–545. 10.1016/j.immuni.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Seo SH, Webster RG. 2002. Tumor necrosis factor alpha exerts powerful anti-influenza virus effects in lung epithelial cells. J Virol 76:1071–1076. 10.1128/jvi.76.3.1071-1076.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McMaster SR, Wilson JJ, Wang H, Kohlmeier JE. 2015. Airway-resident memory CD8 T cells provide antigen-specific protection against respiratory virus challenge through rapid IFN-gamma production. J Immunol 195:203–209. 10.4049/jimmunol.1402975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Slutter B, Van Braeckel-Budimir N, Abboud G, Varga SM, Salek-Ardakani S, Harty JT. 2017. Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci Immunol 2:eaag2031. 10.1126/sciimmunol.aag2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stolley JM, Johnston TS, Soerens AG, Beura LK, Rosato PC, Joag V, Wijeyesinghe SP, Langlois RA, Osum KC, Mitchell JS, Masopust D. 2020. Retrograde migration supplies resident memory T cells to lung-draining LN after influenza infection. J Exp Med 217:e20192197. 10.1084/jem.20192197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Alam S, Sant AJ. 2011. Infection with seasonal influenza virus elicits CD4 T cells specific for genetically conserved epitopes that can be rapidly mobilized for protective immunity to pandemic H1N1 influenza virus. J Virol 85:13310–13321. 10.1128/JVI.05728-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moon JJ, Chu HH, Hataye J, Pagan AJ, Pepper M, McLachlan JB, Zell T, Jenkins MK. 2009. Tracking epitope-specific T cells. Nat Protoc 4:565–581. 10.1038/nprot.2009.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S3. Download JVI.00841-21-s0001.pdf, PDF file, 1.6 MB (1.6MB, pdf)