

ABSTRACT
Brucella abortus is a facultatively extracellular-intracellular pathogen that encounters a diversity of environments within the host cell. We report that bacteria extracted from infected cells at late stages (48 h postinfection) of the intracellular life cycle significantly increase their ability to multiply in new target cells. This increase depends on early interaction with the cell surface, since the bacteria become more adherent and penetrate more efficiently than in vitro-grown bacteria. At this late stage of infection, the bacterium locates within an autophagosome-like compartment, facing starvation and acidic conditions. At this point, the BvrR/BvrS two-component system becomes activated, and the expression of the transcriptional regulator VjbR and the type IV secretion system component VirB increases. Using bafilomycin to inhibit BvrR/BvrS activation and using specific inhibitors for VjbR and VirB, we showed that the BvrR/BvrS and VjbR systems correlate with increased interaction with new host cells, while the VirB system does not. Bacteria released from infected cells under natural conditions displayed the same phenotype as intracellular bacteria. We propose a model in which the B. abortus BvrR/BvrS system senses the transition from its replicative niche at the endoplasmic reticulum to the autophagosome-like exit compartment. This activation leads to the expression of VirB, which participates in the release of the bacterium from the cells, and an increase in VjbR expression that results in a more efficient interaction with new host cells.
KEYWORDS: brucellosis, two-component system, type IV secretion system, adherent, host cells, host-pathogen interactions, infectiousness, intracellular life cycle, transcriptional regulator VjbR, virulence circuit
INTRODUCTION
Brucella abortus is a pathogen of bovines and a zoonotic agent. Bovines acquire the infection through contacts with other infected animals or with aborted fetuses, whereas humans commonly become infected through ingestion of unpasteurized dairy products (1). After invasion, Brucella organisms come in contact with professional phagocytes (2). Although B. abortus does not replicate within neutrophils, these infected cells become efficient vehicles for transmitting the bacterium to other phagocytic cells (3). The preferred Brucella host cells, in which the bacterium replicates extensively, are macrophages, monocytes, dendritic cells, and epithelial cells (4, 5).
The ability of B. abortus to enter and multiply within host cells is essential for brucellosis pathogenesis and persistence. Once bacteria are internalized by host cells, they transit within a Brucella-containing vacuole (BCV), which interacts with early endosomes. At these early stages of the intracellular life cycle, the BvrR/BvrS two-component regulatory system senses the acidic and low-nutrient environment found during the endosomal route (6). The subsequent activation of this system allows B. abortus to trigger the transcriptional response required to redirect its trafficking to the endoplasmic reticulum (ER). This includes the positive regulation of the quorum-sensing transcriptional regulator VjbR and the VirB system (6–9). The delivery of effector proteins through VirB modulates host secretory functions and remodels BCVs into replication-permissive organelles (rBCV) derived from the host ER (10). After the replicative stage, bacteria reach an acidic, double-membrane, LAMP-1-positive, autophagy-related compartment (aBCV). To develop, the aBCV requires the autophagic initiation proteins ULK1, Beclin 1, and ATG14L, as well as phosphoinositide 3-kinase (PI3K) activity. The aBCV promotes cell-to-cell spreading, which completes the intracellular B. abortus life cycle. Finally, the exit of the bacterium seems to be governed by a specific process involving unknown mechanisms (9). The inactivation of VirB at late stages showed a decrease in aBCV formation and a lower level of bacterial release, revealing that at postreplication stages, VirB is required again (10). However, the molecular determinants involved in the late stages of infection and the fate of the released bacteria remain unclear.
In this work, we demonstrate that once B. abortus reaches the aBCV after successful replication in the ER, the bacterium senses intracellular environmental conditions, such as low pH, through BvrR/BvrS, triggering the expression of vjbR and the virB operon and thus promoting the assembly of the VirB secretion system. The induction of this virulence circuit is associated with more-competent bacteria with an increased ability to interact with their host cells in subsequent infection cycles.
RESULTS
Intracellular B. abortus increases its ability to interact with new host cells.
We sought to determine the ability of intracellular bacteria to initiate a new cycle of cellular infection. To do so, we purified bacteria derived from epithelial cells infected for 48 h (11). Purified intracellularly derived brucellae (referred to here as intracellular bacteria) were then used as an inoculum to infect epithelial host cells in a new round of infection. Bacteria grown in vitro in tryptic soy broth (TSB) to the exponential phase (the stage at which they are most infective) were used as a control (referred to here as extracellular bacteria). The concentration of intracellular bacteria could not be determined by spectrophotometric analysis, as is routinely done for extracellular bacteria. We therefore determined the multiplicity of infection (MOI) used for each experiment a posteriori by plating serial dilutions of the inoculum in tryptic soy agar (TSA) and counting colonies 72 h postinfection. The MOIs of the extracellular bacterial inoculum were adjusted in each experiment to match the MOIs of the intracellular bacterial inoculum. In agreement with previous reports (12, 13), infection of epithelial cells with extracellular bacteria was inefficient; it was necessary to explore several optical fields to find infected cells (Fig. 1A). In contrast, many infected cells per field were observed when epithelial cells were inoculated with intracellular bacteria with MOIs equivalent to, or even lower than, those used with extracellular bacteria (Fig. 1A). Quantitation of infected cells revealed similar infection efficiencies (∼2%) with an inoculum of intracellular bacteria at a MOI of 1 and an inoculum of extracellular bacteria at a MOI of 100. Moreover, the rate of infected cells reached 6% when intracellular bacteria were used at a MOI of 10 (Fig. 1B). Then, to compare the early infection dynamics of intracellular bacteria with those of extracellular bacteria, we performed a competition experiment. Epithelial cells were simultaneously infected with intracellular bacteria expressing green fluorescent protein (GFP) and with GFP-negative extracellular bacteria, and bacterial growth was monitored at different times by plate counting of serial dilutions. At the initial time of infection (t0), we observed a 2-log difference in the CFU count between intracellular and extracellular bacteria (Fig. 1C). This difference, observed throughout the experiment (Fig. 1C), suggested that the higher efficiency of intracellular bacteria was due not to improved ability to replicate intracellularly but to a higher efficiency of initial interaction with the host cell. Following this, we infected epithelial cells with either intracellular or extracellular bacteria and evaluated their initial interactions with host cells by using differential immunofluorescence labeling. Intracellular bacteria interacted with host cells more efficiently than extracellular bacteria, as indicated by the proportion of cells with associated bacteria and the mean number of bacteria per infected cell (Fig. 2A and B). Besides, intracellular bacteria were significantly more efficient in entering cells than extracellular bacteria (Fig. 2C). Using these three parameters, we calculated the number of intracellular bacteria per 100 cells. Again, the intracellular bacteria reached significantly higher numbers than the extracellular bacteria (Fig. 2D).
FIG 1.
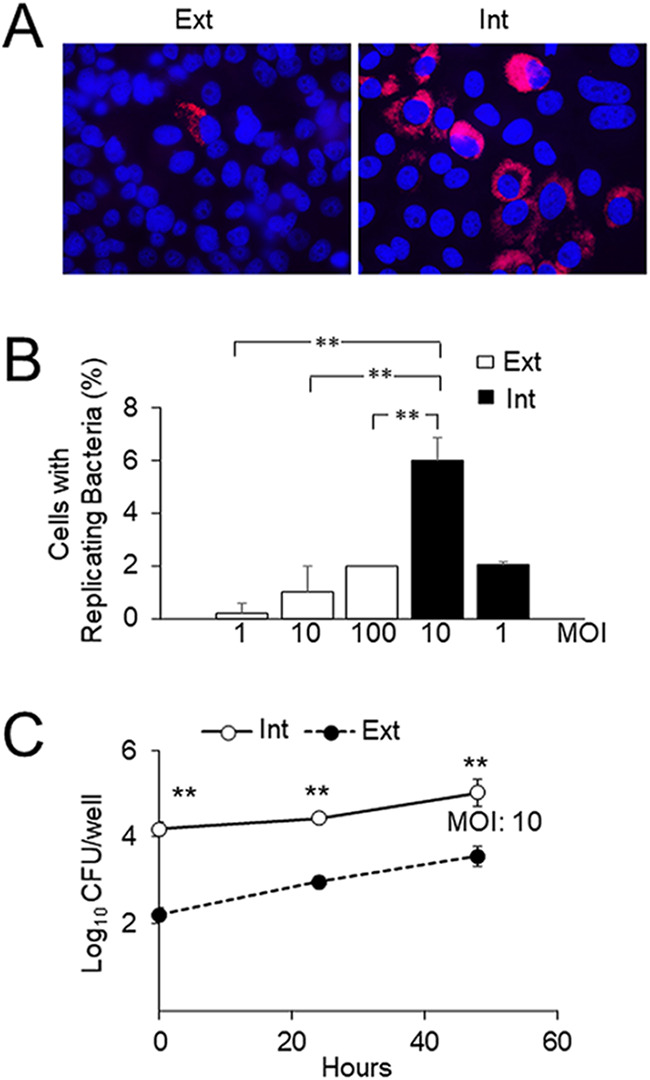
Intracellular B. abortus becomes highly infective for new host cells. (A) HeLa cells were infected with B. abortus-RFP, and at 48 h postinfection, intracellular bacteria were extracted and purified (Int). Extracted intracellular bacteria were used as the inoculum for a new round of HeLa cell infection using a gentamicin protection assay with a multiplicity of infection (MOI) of 10. As a control, HeLa cells were infected with B. abortus-RFP grown in TSB in vitro (Ext) at a MOI of 10. At 48 h postinfection, cells were processed by fluorescence microscopy. (B) HeLa cells were infected with Int or Ext bacteria processed as for panel A at the indicated MOIs. At 48 h postinfection, the percentage of cells with replicating bacteria (>20 bacteria per cell) was determined. Each value is the average of three independent determinations. Statistical significance was calculated by analysis of variance and Tukey's multiple-comparison test (**, P < 0.005). (C) At 48 h postinfection, Int-GFP bacteria prepared as for panel A were used in a coinfection experiment of HeLa cells with GFP-negative extracellular bacteria (Ext). At the indicated times, cells were lysed, and the CFU count of intracellular bacteria was determined by plate counting using UV light to distinguish GFP-positive from GFP-negative bacteria. Each value is the average of three independent determinations. Statistical significance was calculated by Student’s t test. Asterisks indicate significant differences (**, P < 0.005) from counts of extracellular bacteria.
FIG 2.
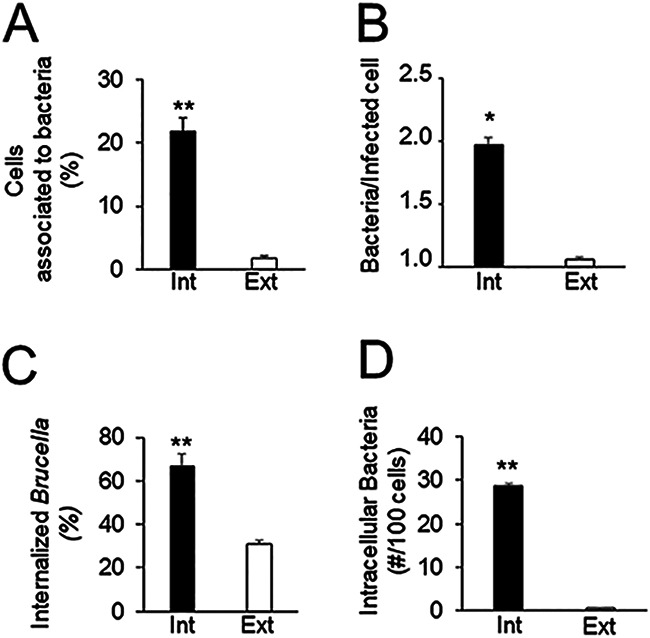
Intracellular B. abortus displays increased adhesion to, and internalization in, new host cells. HeLa cells grown on coverslips were infected with extracted intracellular B. abortus (Int) or in vitro-grown B. abortus (Ext) as indicated in Fig. 1 at a MOI of 10. At 2 h postinfection, cells were extensively washed, and living nonpermeabilized cells were incubated with an FITC-conjugated cow antibody against B. abortus for 30 min at 4°C to label extracellular bacteria (green). Cells were then fixed and permeabilized, and intracellular bacteria were further detected with a rabbit anti-Brucella antibody and Alexa Fluor Plus 594-conjugated goat anti-rabbit IgG (red). (A) Percentage of cells with associated bacteria; (B) mean number of bacteria per infected cell; (C) percentage of internalized Brucella; (D) number of intracellular bacteria in 100 cells. Each value is the average of at least three independent determinations. The statistical significance of differences from results for extracellular bacteria was calculated by Student’s t test (*, P < 0.05; **, P < 0.005).
B. abortus naturally released from infected cells interacts more efficiently with host cells than extracellular bacteria.
To determine if the findings described above for extracted intracellular B. abortus are observed for bacteria naturally released from infected epithelial cells, we removed gentamicin from the medium at the end of the infection (48 h postinfection) and collected the released bacteria present in the supernatant 4 h later. The interaction of these released bacteria with epithelial cells was assessed by observing that the infection yields were similar to those obtained with intracellular bacteria. Released B. abortus at a MOI of 10 infected epithelial cells twice as efficiently as extracellular bacteria at a MOI of 100 (Fig. 3A). Moreover, bacterial growth in infected cells revealed that naturally released B. abortus was better able than extracellular bacteria to interact with epithelial cells (Fig. 3B).
FIG 3.
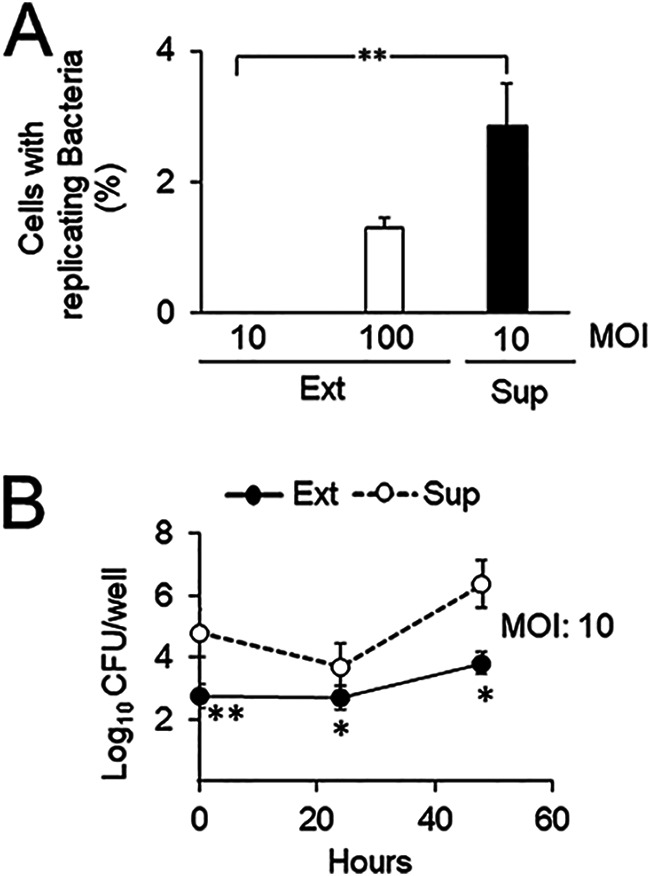
B. abortus released from cells interacts more efficiently than extracellular bacteria with new host cells. (A) HeLa cells were infected with B. abortus-RFP, and at 48 h postinfection, infected cells were washed three times with PBS and incubated for 4 h in DMEM without antibiotics. Bacteria from the supernatant (Sup) were collected by centrifugation and were used as the inoculum for a new round of HeLa cell infection using a gentamicin protection assay at the indicated MOI. As a control, HeLa cells were infected with B. abortus-RFP grown in TSB in vitro (Ext) at the indicated MOI. At 48 h postinfection, the percentage of cells with replicating bacteria (>20 bacteria per cell) was determined by immunofluorescence. Each value is the average of at least three independent determinations. Statistical significance was calculated by the Kruskal-Wallis test and Dunn's multiple-comparison test (**, P < 0.005). (B) GFP-bacteria collected from the supernatant as for panel A (Sup) were used in a coinfection experiment of HeLa cells with GFP-negative bacteria grown in TSB in vitro (Ext). At the indicated times, cells were lysed, and the CFU counts of intracellular bacteria were determined by plate counting using UV light to distinguish GFP-positive from GFP-negative bacteria. Each point is the average of at least three independent determinations. Statistical significance was calculated by Student’s t test. Asterisks indicate statistical significance (*, P < 0.05; **, P < 0.005) for differences from Sup bacteria.
To test if the increased interaction with host cells was dependent on the cell types of the primary and secondary infected cells, we then used different combinations of HeLa epithelial cells and RAW 264.7 murine macrophages. All combinations tested resulted in a dramatic increase in the proportion of infection over that with control extracellular bacteria when the secondary cell type was infected with either intracellular or naturally released bacteria (Fig. 4).
FIG 4.
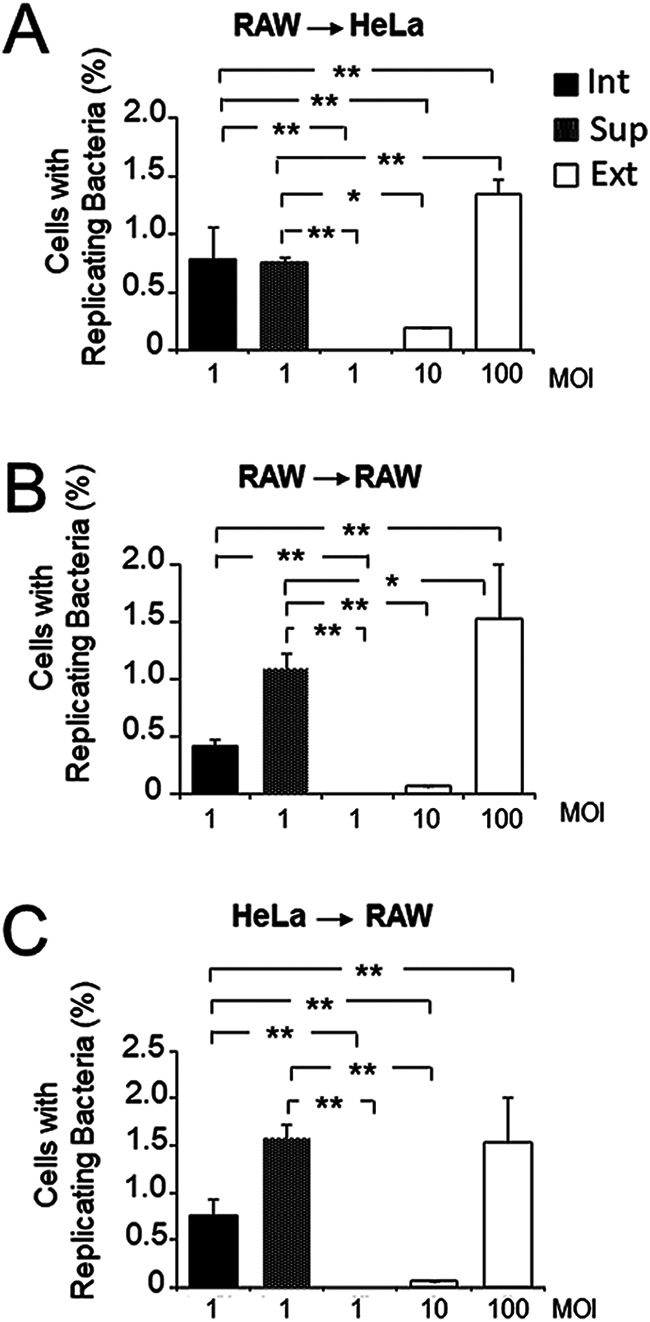
Intracellular B. abortus extracted from macrophages and epithelial cells becomes highly infective for new cells. RAW 264.7 macrophages (A, B) or HeLa cells (C) were infected with B. abortus-RFP, and at 48 h postinfection, infected cells were washed three times with PBS and incubated for 4 h in DMEM without antibiotics. Bacteria from the supernatant (Sup) were collected by centrifugation. In parallel, intracellular bacteria were extracted and purified. Extracted intracellular bacteria (Int) and Sup bacteria were used as inocula for a new round of infection of HeLa cells (A) or RAW 264.7 macrophages (B, C) using a gentamicin protection assay with the indicated multiplicity of infection (MOI). As a control, HeLa cells or RAW 264.7 macrophages were infected at the indicated MOI with B. abortus-RFP grown in TSB in vitro (Ext). At 48 h postinfection, cells were processed by fluorescence microscopy, and the percentage of cells with >20 replicating bacteria was calculated. Each value is the average of at least three independent determinations. Statistical significance was calculated by analysis of variance and Tukey’s multiple-comparison test (*, P < 0.05; **, P < 0.005).
Activation of BvrR/BvrS and increased VjbR expression correlate with the augmented efficiency of intracellular bacteria.
At the late stages of its intracellular cycle, B. abortus moves to an acidic aBCV (9). An acidic pH was previously shown to activate BvrR/BvrS and to induce vjbR and virB operon expression (6). To explore if the same molecular mechanism occurred in the aBCV, we infected macrophages with intracellular bacteria, and B. abortus was then purified at the late stages of infection. The isolated bacteria were then analyzed by immunochemical methods. For these experiments, we used macrophages instead of epithelial cells, because only from the former was it possible to render a sufficient yield of intracellular bacteria for the biochemical analysis. Through Phos-tag SDS-PAGE, we observed robust phosphorylation of BvrR 48 h postinfection, coinciding with the transit of B. abortus from the ER to the aBCV (Fig. 5A and B) (9). Furthermore, and as expected from the positive transcriptional regulation exerted by BvrR/BvrS, the protein levels of both VjbR and VirB increased at late times of infection (Fig. 5C). In agreement with the positive role of VjbR in virB operon expression, the increase in VirB protein levels was delayed relative to that of VjbR (Fig. 5C).
FIG 5.
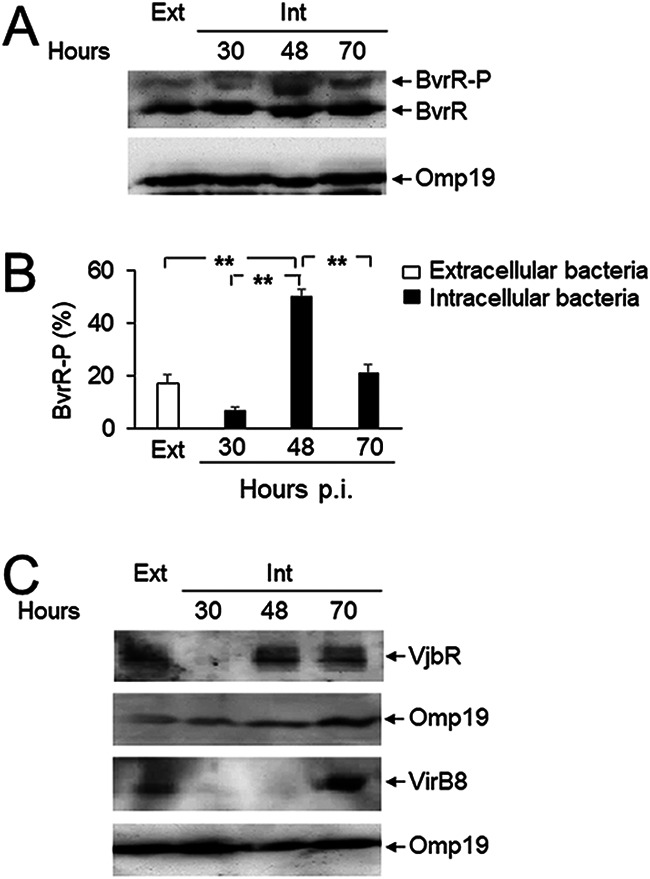
Increased activation of BvrR/BvrS and expression of vjbR and virB at late stages of the intracellular infection cycle. (A) RAW 264.7 macrophages were infected with B. abortus, and at the indicated times, intracellular bacteria were extracted and purified (Int). Bacterial lysates were prepared and separated by 10% Phos-tag SDS-PAGE, transferred to PVDF membranes, and probed with anti-BvrR or anti-Omp19 (loading control) antibodies. Bacteria grown in TSB in vitro were used as a control (Ext). (B) The percentage of total BvrR that was phosphorylated (BvrR-P) was calculated for each indicated condition by densitometry from at least three independent experiments. Statistical significance was calculated by analysis of variance and Tukey’s multiple-comparison test (**, P < 0.005). (C) Lysates from intracellular bacteria obtained as described for panel A were used for the detection of VjbR and VirB8 by Western blotting. These results are representative of three independent experiments.
It has been shown that VirB plays a role during the late stages of the intracellular life cycle (10). We therefore envisioned that the activation of this virulence circuit could also be involved in the increased ability of intracellular bacteria to interact with host cells. To test this hypothesis, cells were first infected with extracellular B. abortus for 48 h and then treated, before extraction of the intracellular bacteria, with either (i) bafilomycin, an antibiotic that hampers the acidic vacuolar environment required to promote BvrR phosphorylation, (ii) homoserine lactone, which specifically blocks the DNA-binding activity of VjbR, or (iii) B8I-2, which prevents VirB8 dimerization (14–16). Inhibition of VjbR had the greatest impact on bacterial interaction with cells, affecting the percentage of cells associated with bacteria, the mean number of bacteria per cell, and the efficiency of internalization (Fig. 6A to C); inhibition of BvrR phosphorylation reduced the proportion of cells associated with bacteria and the efficiency of internalization (Fig. 6A to C). In contrast, inhibition of VirB function did not alter the parameters for early bacterium-cell interactions (Fig. 6A to C). These results were consistent with the general decrease in the number of intracellular bacteria per 100 cells (Fig. 6D). Likewise, they agreed with a decrease in the number of bacteria interacting with cells, as determined by bacterial counting, after inhibition of VjbR DNA-binding activity or BvrR phosphorylation, but not after VirB inhibition (Fig. 6E). Since exposure of infected cells to B8I-2 was the only treatment that did not reduce the binding of intracellular bacteria, we tested that this compound inhibited VirB function as expected (15). Both extracellular and intracellular bacteria exposed to B8I-2 attached properly to cells but failed to replicate intracellularly (Fig. 6F).
FIG 6.

BvrR/BvrS and VjbR, but not VirB, participate in the increased interaction of extracted intracellular B. abortus with cells. HeLa cells were first infected with B. abortus 2308 at a MOI of 500 for 48 h and then either left untreated (–) or treated with either 50 nM bafilomycin (Bm), 50 μM homoserine lactone (HSL), or 10 μM VirB8 dimerization inhibitor B8I-2 for 4 h. Intracellular bacteria were then extracted and purified (Int) and were used in a reinfection experiment with HeLa cells. As a control, HeLa cells were infected with B. abortus 2308 grown in TSB in vitro at a MOI of 100 (Ext). After 2 h, cells were extensively washed and processed by differential intracellular/extracellular immunofluorescence as described for Fig. 2. (A) Percentage of cells with associated bacteria; (B) mean number of bacteria per infected cell; (C) percentage of internalized Brucella; (D) number of intracellular bacteria in 100 cells. (E) HeLa cells were infected and treated as described for panel A. At 2 h postinfection, cells were lysed, and intracellular bacteria were quantitated by plate counting. Each value is the average of at least three independent determinations. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.005) from intracellular bacteria that were left untreated. Statistical significance was calculated by the Kruskal-Wallis test and Dunn’s multiple-comparison test. (F) B. abortus 2308 grown in TSB (Ext) or intracellular bacteria extracted 48 h postinfection (Int) were either treated for 4 h with 10 μM VirB8 dimerization inhibitor B8I-2 (open symbols) or left untreated (filled symbols). HeLa cells were then infected with the indicated bacteria, and intracellular growth was monitored for 48 h by a gentamicin protection assay. Each point is the average of at least three independent determinations. Statistical significance was calculated by Student’s t test. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.005) from intracellular or extracellular bacteria that were left untreated.
Transit of B. abortus through autophagosomes triggers an adherent phenotype.
Our results indicate that exposure to the environmental conditions found by B. abortus at late times of its infection cycle in the aBCV that the bacterium reaches before exit is crucial for increased bacterial adherence in a subsequent infection cycle. To explore if the aBCV environment is required to trigger bacterial adherence, we used the autophagosome inhibitor 3-methyladenine (3MA), which hampers the function of class III PI3Ks (9). HeLa cells infected with B. abortus for 34 h were treated with 3MA for 14 additional hours. After treatment, bacteria were extracted and used in a new round of infection. This treatment dramatically decreased the ability of extracted intracellular bacteria to establish early interactions with host cells, as determined by both immunofluorescence and plate counting (Fig. 7). Since the autophagosome is acidic, and since bafilomycin inhibits the increased bacterial adherence acquired by intracellular passage, we explored if a low pH could mimic this stage. Bacteria were first exposed to a low-pH medium for 6 h and then tested for their ability to interact with host cells. This treatment did not increase their ability to establish initial interactions over the ability of control bacteria incubated in the rich medium at a neutral pH. This indicates that other factors besides pH are required in the aBCV for the development of the adherent phenotype.
FIG 7.
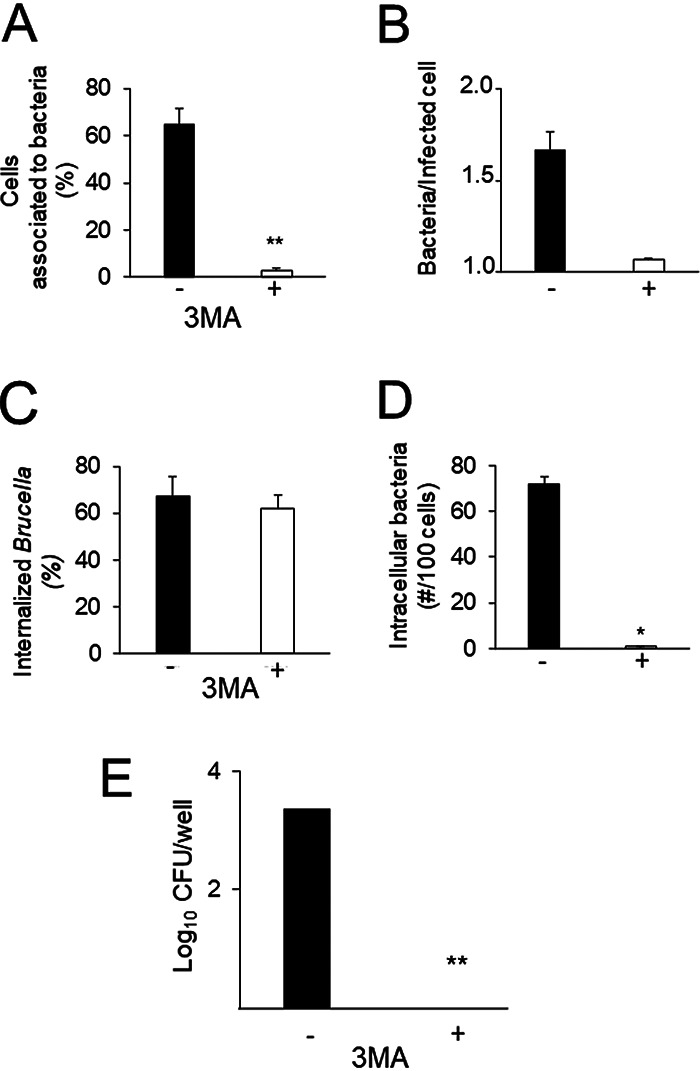
Transit through autophagosome-related compartments increases the interaction of B. abortus with new host cells. HeLa epithelial cells were infected with B. abortus 2308 at a MOI of 500 for 34 h and were then either treated with 10 mM 3-methyladenine (3-MA), an autophagosome inhibitor, or left untreated (–) for 14 h. Intracellular bacteria were then extracted, purified (Int), and used in a new round of infection on HeLa epithelial cells at a MOI of 10. After 2 h of incubation, cells were extensively washed and processed by differential intracellular/extracellular immunofluorescence as described for Fig. 2. (A) Percentage of cells with associated bacteria; (B) mean number of bacteria per infected cell; (C) percentage of internalized Brucella bacteria; (D) number of intracellular bacteria in 100 cells. (E) HeLa epithelial cells were infected and treated as described above. At 2 h postinfection, cells were lysed, and intracellular bacteria were quantitated by plate counting. Each value is the average of at least three independent determinations. Statistical significance was calculated by Student’s t test. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.005) from intracellular bacteria that were left untreated.
DISCUSSION
As a facultatively extracellular-intracellular pathogen, B. abortus needs to adapt its metabolism and structural composition to the variety of niches the bacterium encounters during the infection. At early times of the cell infection, B. abortus arrests its cell cycle after confronting the endosomal route (17, 18). Through its BvrR/BvrS two-component system, B. abortus detects the entrance to this potentially degradative pathway, triggering a transcriptional response that allows it to escape the lysosome as the final destination, instead redirecting its trafficking to the ER (6). Once in this compartment, B. abortus overcomes cell cycle arrest and assumes a replicative stage that resembles the proteomic phenotype of bacteria growing in vitro at an exponential rate (19). Finally, in the late stages of infection, the bacterium transits to an aBCV to prepare its exit from the infected cell and the subsequent infection events.
In this work, we report that B. abortus, precisely at these late stages of intracellular growth, reactivates its virulence circuit composed of the BvrR/BvrS two-component system, the transcriptional regulator VjbR, and VirB to allow egress from the host cell and preparation for a subsequent infection cycle. These molecular events result in bacteria with increased competitiveness for interaction with new host cells (Fig. 8). Better interaction with host cells and augmented competitiveness occurred in purified intracellular bacteria as well as in naturally released bacteria, indicating that our conclusions apply under physiological conditions. Using protocols that allow the extraction and purification of intracellular bacteria (11), we were able to demonstrate that BvrR is highly phosphorylated at late stages of the cell cycle and that, in turn, there was a sequential increase in the expression of VjbR and VirB. The activation of this virulence circuit correlated with an increase in the adherence and entrance of these bacteria into new host cells, and consistently, these molecular and phenotypic events were reversed by specific inactivation of VjbR or blockade of BvrR phosphorylation. While the acidic pH of the BCVs is required to trigger this molecular response, it is not sufficient, since in vitro exposure to this condition did not mimic the increased competitiveness reported here. Thus, additional signals detected by the bacterium within this environment are required.
FIG 8.

Model showing the activation of the BvrR/VjbR/VirB virulence circuit at late stages of B. abortus infection. After replication at the ER, B. abortus reaches an aBCV. There, BvrS senses environmental cues such as an acidic pH, triggering its autophosphorylation, and activates, in turn, the regulator protein BvrR. This transcriptional regulator then binds to promoters such as PvjbR and PvirB, enhancing their expression. The type IV secretion system component VirB promotes the exit of the bacterium from its host cell, whereas VjbR induces changes at the membrane that increase the interaction of the bacteria with the host cell in subsequent infection cycles. Plasma membrane (PM).
Our results are in agreement with the role of the VirB secretion machinery in the exit of bacteria from cells, observed previously (10), since we detected an increase in the expression of VjbR, a crucial transcriptional regulator of VirB expression, and the subsequent increase in the expression of this injection machinery. The use of specific inhibitors indicated that VjbR, but not VirB, was required for the increased competitiveness to establish a new infection cycle. However, since the B8I-2 compound was added to cells infected with bacteria for 48 h, a time at which the VirB injection complex might already be assembled, and considering that this inhibitor has not been demonstrated to induce disassembly of this machinery, we cannot completely rule out a role of VirB in the phenomena we are reporting. Integrating all our data, we postulate a model in which the BvrR/BvrS two-component system detects the environment of the aBCV and triggers the expression of VjbR (Fig. 8). As reported previously, this regulator has a deep impact on the transcriptome of B. abortus (16). Among the genes regulated by VjbR are several transcripts encoding outer membrane proteins; thus, it is reasonable to expect a remodeling of the outer membrane of B. abortus upon VjbR activation (Fig. 8). Some of the VjbR-regulated genes encode adhesins such as AidA, AidA-1, and BMEI1873, an ortholog of Brucella melitensis BtaE, as well as modulators of exopolysaccharide production (16), which can impact the ability of the bacterium to interact with the surfaces of eukaryotic cells. The expression of adhesin-encoding genes btaE and bmaC is weak in vitro but seems to increase within the host environment before bacterial internalization (20–23). It would then be relevant to study the expression of these adhesins in bacteria extracted from cells at the late stages of their intracellular cycle.
Taken together, our data indicate that VjbR functions as a master regulator to prepare the intracellular brucellae at later times of infection to infect new cells with higher efficiency. Additionally, and from its role in eliciting VirB expression, VjbR activation will promote the expression and assembly of this injection machinery, facilitating the exit of the bacterium from its host cell (10). It is tempting to speculate that the outer membrane remodeling induced by VjbR activation also enables the bacteria to deal with different components of the immune system encountered during the extracellular phase, as proposed previously (24, 25). Egress to the extracellular space exposes the pathogen to antibodies, complement, and other antimicrobial factors that can inhibit their growth or block their entry into new cells. It has been demonstrated that intracellular Brucella dissociation into a rough phenotype is involved in egress and dissemination (26), inducing chemokine and cytokine release and promoting macrophage recruitment to the site of infection. In agreement with our findings, Brucella dissociation is enhanced in acidic environments (27). In this vein, physiological Brucella dissociation might be related to the molecular events we described in this report.
Systems orthologous to BvrR/BvrS, such as the Bartonella henselae BatR/BatS, Agrobacterium tumefaciens ChvG/ChvI, and Sinorhizobium meliloti ExoS/ChvI systems (28–32), have been implicated in early interactions of alphaproteobacteria with their corresponding hosts. Furthermore, in Yersinia sp., the sensing of extracytoplasmic stress by the two-component system CpxAR contributes to productive interactions with animal cells (33).
In intracellular pathogens, egress from infected host cells is essential for dissemination and transmission to other hosts. This process can proceed by lysis of the host cell, protrusion, and engulfment by neighboring cells, extrusion into membrane-bound compartments, exocytic fusion, and induction of cell death (34). Specific virulence factors and molecular adaptations are involved in this step. In Shigella spp., virulence factors such as IpaB, IpaC, and MixE are essential for efficient cell-to-cell dissemination (35). Salmonella develops a hyperreplicating population in the cytosol, primed for an invasion, that is transcriptionally distinct from intravacuolar counterparts. This population expresses an invasion-associated type III secretion system and flagella (36).
In the present work, we described a role for a two-component system in late events of the intracellular life cycle dealing with bacterial egress from host cells. Following this, our results may work as a general model for understanding the infection cycles of other facultatively intracellular alphaproteobacterial pathogens of animals and plants.
MATERIALS AND METHODS
Bacterial strains.
B. abortus 2308 NaIr is a virulent smooth-lipopolysaccharide (LPS) strain (37). B. abortus 2308 expressing a constitutive red fluorescent protein (RFP) from Discosoma coral (B. abortus-RFP) was provided by Jean Jacques Letesson (Unité de Recherche en Biologie Moléculaire, Facultés Universitaires Notre-Dame de la Paix, Namur, Belgium). B. abortus 2308 expressing a constitutive green fluorescent protein (B. abortus 2308-GFP) was constructed using a mini-Tn7 transposon as described previously (10).
Bacterial growth conditions.
Brucella strains were grown in vitro at 37°C in tryptic soy broth (TSB) for 24 h to stationary phase, and aliquots were frozen at −70°C in TSB–20% glycerol. To prepare the bacterial preinoculum, a frozen stock of brucellae was thawed 48 h before the assays. Then the bacteria were grown in 20 ml of TSB in glass flasks at 200 rpm and 37°C for 18 h as described previously (11). To prepare the bacterial inoculum, the optical density of the bacterial preinoculum at 420 nm (OD420) was measured. The bacterial population was estimated by plotting the OD420 on a standard curve, and 5 × 109 bacteria were inoculated in 20 ml of TSB and were incubated with agitation at 200 rpm and 37°C for 16 h to obtain bacteria in the exponential-growth phase (OD420, 0.3 to 0.5).
Cell culture.
Murine RAW 264.7 macrophages (ATCC TIB-71) or HeLa epithelial cells (ATCC clone CCL-2) were cultivated and infected with B. abortus as described previously (11). For the intracellular replication experiments, cells were seeded in 24-well tissue culture plates 2 days before infection to obtain a final concentration of 5 × 105 cells per well. For the purification experiments with extracted intracellular bacteria and naturally released bacteria, cells were seeded in 6-well tissue culture plates 2 days before infection to obtain a final concentration of 3 × 106 cells per well. For immunofluorescence analysis, HeLa cells were seeded on 13-mm glass slides in 24-well tissue culture plates 2 days before infection to obtain a final concentration of 5 × 105 cells per well.
Gentamicin protection assay.
Cells were infected with B. abortus grown in TSB in vitro to the exponential-growth phase, in which they have the highest infectivity (6). The multiplicity of infection (MOI), indicated for each assay in the figures, was adjusted by estimating the bacterial population by optical density as described above and then diluting the bacteria in Dulbecco’s modified Eagle’s medium (DMEM). Infected cells were centrifuged for 5 min at 330 × g and 4°C, incubated for 45 min at 37°C under 5% CO2, and washed with phosphate-buffered saline (PBS). Extracellular bacteria were eliminated by treatment with 100 μg/ml gentamicin for 1 h, and cells were incubated in the presence of 5 μg/ml gentamicin for the times indicated in the figures.
Purification of intracellular bacteria.
Intracellular B. abortus bacteria were purified at various postinfection times using a standardized protocol based on sucrose gradients and ultracentrifugation (11). Briefly, after infection and incubation, cell monolayers were detached using a scraper. The cell suspension was then transferred to a Dounce homogenizer and disrupted. After benzonase treatment for 30 min at 37°C, the cell homogenate was layered on top of a 1-ml cushion of a 0.8 M sucrose solution in a 15-ml tube and was centrifuged at 500 × g for 5 min at 4°C. The homogenate at the surface of the 0.8 M sucrose cushion was diluted with 4 volumes of 3.0 mM imidazole (pH 7.4), layered onto a sucrose gradient (containing, from the bottom to the top, 1.5 M sucrose, 1 M sucrose, 0.1% SDS, and 0.8 M sucrose), and centrifuged in a swinging bucket rotor at 30,000 × g for 25 min at 4°C. The pellet containing the extracted and purified intracellular bacteria was then collected, diluted in DMEM, and used to infect a new batch of HeLa epithelial cells or murine RAW 264.7 macrophages. The MOI was determined a posteriori by plating serial dilutions of the inoculum in TSA and counting colonies 72 h postinfection. Alternatively, the pellet containing the purified intracellular bacteria was used for electrophoretic and immunochemical analyses.
Purification of naturally released bacteria.
Infected cells were washed three times with PBS at 44 h postinfection and were incubated with DMEM without antibiotics for 4 h. After incubation, the supernatant was collected, centrifuged, diluted in DMEM, and used to infect cells as described above. The MOI was calculated as described for extracted and purified intracellular bacteria.
Addition of inhibitory molecules to infected cells.
Bafilomycin (100 nM; Sigma-Aldrich), an inhibitor of phagosome acidification (14), homoserine lactone (50 μM; Sigma-Aldrich), an inhibitor of VjbR DNA-binding activity (16), and B8I-2 compound (10 μM; ChemBridge), an inhibitor of VirB8 dimerization (15), were added to infected HeLa cells 48 h postinfection and were incubated for 4 h before the purification of intracellular bacteria. Additionally, to demonstrate that the B8I-2 compound was working as expected (15), bacteria grown in vitro to the exponential-growth phase or extracted intracellular bacteria were incubated for 4 h with 10 μM B8I-2 molecule and used to infect HeLa cells in the presence of 10 μM B8I-2. In parallel, as a negative control, HeLa cells were infected with bacteria without treatment. Finally, to inhibit class III PI3K activity and block autophagocytosis, infected HeLa cells were incubated at 34 h postinfection with 10 mM 3-methyladenine (3-MA) (Sigma) for 14 h before the purification of intracellular bacteria (9).
Intracellular replication quantitation.
The number of viable intracellular B. abortus was determined at different hours postinfection. Briefly, after infection of RAW 264.7 macrophages or HeLa epithelial cells as described above, cells were washed twice with PBS and treated for 10 min with 0.1% Triton X-100. Lysates were serially diluted, plated on tryptic soy agar dishes, and incubated at 37°C under 5% CO2 for 3 days for the quantitation of CFU.
Immunofluorescence microscopy.
To determine the percentage of cells with associated bacteria, the number of bacteria per infected cell, and the proportion of internalized bacteria, immunofluorescence was performed 2 h postinfection, and each parameter was determined as described previously (38). To label extracellular bacteria, infected cells were washed three times with PBS and incubated with a homemade cow anti-B. abortus antibody conjugated with fluorescein isothiocyanate (FITC) for 30 min at 4°C. Cells were then fixed with ice-cold 3% paraformaldehyde for 10 min. Samples were washed once, incubated for 10 min with PBS containing 50 mM NH4Cl, and permeabilized for 10 min with 0.1% Triton X-100. Then all bacteria were stained with a homemade rabbit anti-Brucella antibody, washed three times with 0.1% Triton X-100, and incubated for 30 min with an Alexa Fluor Plus 594 (Invitrogen)-conjugated, highly cross-adsorbed goat anti-rabbit IgG (H+L) secondary antibody. After incubation, slides were washed three times with 0.1% Triton X-100, and the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific). Finally, slides were mounted using ProLong gold antifade mountant (Invitrogen) before analysis by fluorescence microscopy. Intracellularly located bacteria were exclusively labeled in red, whereas extracellular bacteria were stained red and green. Intracellular and extracellular bacteria, as well as the number of bacteria per cell, were counted from at least 100 infected cells. The percentage of cells with associated bacteria is expressed as the mean percentage of cells with bound bacteria in 10 different 40× fields, and error bars in the figures represent standard deviations (SD).
Exposure of B. abortus to minimal and low-pH medium.
B. abortus was grown in TSB in vitro to the exponential-growth phase. A volume of the culture that corresponds to a MOI of 100 was calculated as described above. Then bacteria were centrifuged at 10,000 × g for 3 min and resuspended in a minimal medium (33 mM KH2PO4, 60.3 mM K2HPO4, and 0.1% yeast extract) at pH 5.0 (adjusted with citric acid) or in a nutrient-rich medium (TSB) at pH 7.0 for 6 h at 37°C and 200 rpm. After incubation, bacteria were concentrated by centrifugation at 10,000 × g for 3 min, diluted in DMEM, and used to infect HeLa epithelial cells as described above.
Detection of BvrR phosphorylation and VirB8 and VjbR protein levels.
Intracellular bacteria were resuspended in Laemmli sample buffer and heated at 100°C for 20 min. Protein concentrations were determined by the Bio-Rad DC method according to the manufacturer’s instructions. Equal amounts of protein (20 μg) were loaded onto a 10% gel for SDS-PAGE. Separated proteins were transferred to a polyvinylidene difluoride (PVDF) membrane and were probed with antibodies. To analyze the phosphorylated status of BvrR, samples were solubilized in Laemmli sample buffer without heating, and equal amounts of protein (20 μg) were loaded onto a 10% gel for SDS-PAGE containing 100 mM Phos-tag and 0.2 mM MnCl2 (39).
Statistical analysis.
Statistical analyses were performed using GraphPad Prism software. Data were processed in Microsoft Office Excel 2016 and GraphPad Prism software.
ACKNOWLEDGMENTS
This work was funded by Fondos de Recursos del Sistema FEES/CONARE (C0456), Espacio Universitario de Estudios Avanzados, UCREA (B8762), from the presidency of the University of Costa Rica and by the Vice Presidency for Research, University of Costa Rica.
The funders had no role in the study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Contributor Information
Esteban Chaves-Olarte, Email: esteban.chaves@ucr.ac.cr.
Craig R. Roy, Yale University School of Medicine
REFERENCES
- 1.Moreno E, Moriyon I. 2006. The genus Brucella, p 315–456. In Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (ed), The prokaryotes. Springer, New York, NY. [Google Scholar]
- 2.Ackermann MR, Cheville NF, Deyoe BL. 1988. Bovine ileal dome lymphoepithelial cells: endocytosis and transport of Brucella abortus strain 19. Vet Pathol 25:28–35. 10.1177/030098588802500104. [DOI] [PubMed] [Google Scholar]
- 3.Gutiérrez-Jiménez C, Mora-Cartín R, Altamirano-Silva P, Chacón-Díaz C, Chaves-Olarte E, Moreno E, Barquero-Calvo E. 2019. Neutrophils as Trojan horse vehicles for Brucella abortus macrophage infection. Front Immunol 10:1012. 10.3389/fimmu.2019.01012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ko J, Splitter GA. 2003. Molecular host-pathogen interaction in brucellosis: current understanding and future approaches to vaccine development for mice and humans. Clin Microbiol Rev 16:65–78. 10.1128/cmr.16.1.65-78.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liautard JP, Gross A, Dornand J, Köhler S. 1996. Interactions between professional phagocytes and Brucella spp. Microbiologia 12:197–206. [PubMed] [Google Scholar]
- 6.Altamirano-Silva P, Meza-Torres J, Castillo-Zeledón A, Ruiz-Villalobos N, Zuñiga-Pereira AM, Chacón-Díaz C, Moreno E, Guzmán-Verri C, Chaves-Olarte E. 2018. Brucella abortus senses the intracellular environment through the BvrR/BvrS two-component system, which allows B. abortus to adapt to its replicative niche. Infect Immun 86:e00713-17. 10.1128/IAI.00713-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Celli J, Salcedo SP, Gorvel JP. 2005. Brucella coopts the small GTPase Sar1 for intracellular replication. Proc Natl Acad Sci U S A 102:1673–1678. 10.1073/pnas.0406873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gorvel JP, Moreno E. 2002. Brucella intracellular life: from invasion to intracellular replication. Vet Microbiol 90:281–297. 10.1016/s0378-1135(02)00214-6. [DOI] [PubMed] [Google Scholar]
- 9.Starr T, Child R, Wehrly TD, Hansen B, Hwang S, López-Otin C, Virgin HW, Celli J. 2012. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe 11:33–45. 10.1016/j.chom.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith EP, Miller CN, Child R, Cundiff JA, Celli J. 2016. Postreplication roles of the Brucella VirB type IV secretion system uncovered via conditional expression of the VirB11 ATPase. mBio 7:e01730-16. 10.1128/mBio.01730-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaves-Olarte E, Altamirano-Silva P, Guzmán-Verri C, Moreno E. 2014. Purification of intracellular bacteria: isolation of viable Brucella abortus from host cells. Methods Mol Biol 1197:245–260. 10.1007/978-1-4939-1261-2_14. [DOI] [PubMed] [Google Scholar]
- 12.Detilleux PG, Deyoe BL, Cheville NF. 1990. Penetration and intracellular growth of Brucella abortus in nonphagocytic cells in vitro. Infect Immun 58:2320–2328. 10.1128/IAI.58.7.2320-2328.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaves-Olarte E, Guzmán-Verri C, Méresse S, Desjardins M, Pizarro-Cerdá J, Badilla J, Gorvel JP, Moreno E. 2002. Activation of Rho and Rab GTPases dissociates Brucella abortus internalization from intracellular trafficking. Cell Microbiol 4:663–676. 10.1046/j.1462-5822.2002.00221.x. [DOI] [PubMed] [Google Scholar]
- 14.Porte F, Liautard JP, Köhler S. 1999. Early acidification of phagosomes containing Brucella suis is essential for intracellular survival in murine macrophages. Infect Immun 67:4041–4047. 10.1128/IAI.67.8.4041-4047.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paschos A, den Hartigh A, Smith MA, Atluri VL, Sivanesan D, Tsolis RM, Baron C. 2011. An in vivo high-throughput screening approach targeting the type IV secretion system component VirB8 identified inhibitors of Brucella abortus 2308 proliferation. Infect Immun 79:1033–1043. 10.1128/IAI.00993-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weeks JN, Galindo CL, Drake KL, Adams GL, Garner HR, Ficht TA. 2010. Brucella melitensis VjbR and C12-HSL regulons: contributions of the N-dodecanoyl homoserine lactone signaling molecule and LuxR homologue VjbR to gene expression. BMC Microbiol 10:167. 10.1186/1471-2180-10-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pizarro-Cerdá J, Méresse S, Parton RG, van der Goot G, Sola-Landa A, Lopez-Goñi I, Moreno E, Gorvel JP. 1998. Brucella abortus transits through the autophagic pathway and replicates in the endoplasmic reticulum of nonprofessional phagocytes. Infect Immun 66:5711–5724. 10.1128/IAI.66.12.5711-5724.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deghelt M, Mullier C, Sternon JF, Francis N, Laloux G, Dotreppe D, Van der Henst C, Jacobs-Wagner C, Letesson JJ, De Bolle X. 2014. G1-arrested newborn cells are the predominant infectious form of the pathogen Brucella abortus. Nat Commun 5:4366. 10.1038/ncomms5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamontagne J, Forest A, Marazzo E, Denis F, Butler H, Michaud JF, Boucher L, Pedro I, Villeneuve A, Sitnikov D, Trudel K, Nassif N, Boudjelti D, Tomaki F, Chaves-Olarte E, Guzmán-Verri C, Brunet S, Côté-Martin A, Hunter J, Moreno E, Paramithiotis E. 2009. Intracellular adaptation of Brucella abortus. J Proteome Res 8:1594–1609. 10.1021/pr800978p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Czibener C, Merwaiss F, Guaimas F, Del Giudice MG, Serantes DA, Spera JM, Ugalde JE. 2016. BigA is a novel adhesin of Brucella that mediates adhesion to epithelial cells. Cell Microbiol 18:500–513. 10.1111/cmi.12526. [DOI] [PubMed] [Google Scholar]
- 21.Posadas DM, Ruiz-Ranwez V, Bonomi HR, Martín FA, Zorreguieta A. 2012. BmaC, a novel autotransporter of Brucella suis, is involved in bacterial adhesion to host cells. Cell Microbiol 14:965–982. 10.1111/j.1462-5822.2012.01771.x. [DOI] [PubMed] [Google Scholar]
- 22.Ruiz-Ranwez V, Posadas DM, Estein SM, Abdian PL, Martin FA, Zorreguieta A. 2013. The BtaF trimeric autotransporter of Brucella suis is involved in attachment to various surfaces, resistance to serum and virulence. PLoS One 8:e79770. 10.1371/journal.pone.0079770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruiz-Ranwez V, Posadas DM, Van der Henst C, Estein SM, Arocena GM, Abdian PL, Martín FA, Sieira R, De Bolle X, Zorreguieta A. 2013. BtaE, an adhesin that belongs to the trimeric autotransporter family, is required for full virulence and defines a specific adherence pole of Brucella suis. Infect Immun 81:996–1007. 10.1128/IAI.01241-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barquero-Calvo E, Chaves-Olarte E, Weiss DS, Guzmán-Verri C, Chacón-Díaz C, Rucavado A, Moriyón I, Moreno E. 2007. Brucella abortus uses a stealthy strategy to avoid activation of the innate immune system during the onset of infection. PLoS One 2:e631. 10.1371/journal.pone.0000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martirosyan A, Moreno E, Gorvel JP. 2011. An evolutionary strategy for a stealthy intracellular Brucella pathogen. Immunol Rev 240:211–234. 10.1111/j.1600-065X.2010.00982.x. [DOI] [PubMed] [Google Scholar]
- 26.Pei J, Kahl-McDonagh M, Ficht TA. 2014. Brucella dissociation is essential for macrophage egress and bacterial dissemination. Front Cell Infect Microbiol 4:23. 10.3389/fcimb.2014.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braun W. 1946. Dissociation in Brucella abortus: a demonstration of the rôle of inherent and environmental factors in bacterial variation. J Bacteriol 51:327–349. 10.1128/JB.51.3.327-349.1946. [DOI] [PubMed] [Google Scholar]
- 28.Charles TC, Nester EW. 1993. A chromosomally encoded two-component sensory transduction system is required for virulence of Agrobacterium tumefaciens. J Bacteriol 175:6614–6625. 10.1128/jb.175.20.6614-6625.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng HP, Walker GC. 1998. Succinoglycan production by Rhizobium meliloti is regulated through the ExoS-ChvI two-component regulatory system. J Bacteriol 180:20–26. 10.1128/JB.180.1.20-26.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L, Jia Y, Hou Q, Charles TC, Nester EW, Pan SQ. 2002. A global pH sensor:Agrobacterium sensor protein ChvG regulates acid-inducible genes on its two chromosomes and Ti plasmid. Proc Natl Acad Sci U S A 99:12369–12374. 10.1073/pnas.192439499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quebatte M, Dehio M, Tropel D, Basler A, Toller I, Raddatz G, Engel P, Huser S, Schein H, Lindroos HL, Andersson SG, Dehio C. 2010. The BatR/BatS two-component regulatory system controls the adaptive response of Bartonella henselae during human endothelial cell infection. J Bacteriol 192:3352–3367. 10.1128/JB.01676-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao SY, Luo L, Har KJ, Becker A, Rüberg S, Yu GQ, Zhu JB, Cheng HP. 2004. Sinorhizobium meliloti ExoR and ExoS proteins regulate both succinoglycan and flagellum production. J Bacteriol 186:6042–6049. 10.1128/JB.186.18.6042-6049.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carlsson KE, Liu J, Edqvist PJ, Francis MS. 2007. Influence of the Cpx extracytoplasmic-stress-responsive pathway on Yersinia sp.-eukaryotic cell contact. Infect Immun 75:4386–4399. 10.1128/IAI.01450-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hybiske K, Stephens RS. 2008. Exit strategies of intracellular pathogens. Nat Rev Microbiol 6:99–110. 10.1038/nrmicro1821. [DOI] [PubMed] [Google Scholar]
- 35.Kane CD, Schuch R, Day WA, Maurelli AT. 2002. MxiE regulates the intracellular expression of factors secreted by the Shigella flexneri 2a type III secretion system. J Bacteriol 184:4409–4419. 10.1128/jb.184.16.4409-4419.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knodler LA, Vallance BA, Celli J, Winfree S, Hansen B, Montero M, Steele-Mortimer O. 2010. Dissemination of invasive Salmonella via bacterial-induced extrusion of mucosal epithelia. Proc Natl Acad Sci US A 107:17733–17738. 10.1073/pnas.1006098107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sangari F, Agüero J. 1991. Mutagenesis of Brucella abortus: comparative efficiency of three transposon delivery systems. Microb Pathog 11:443–446. 10.1016/0882-4010(91)90040-h. [DOI] [PubMed] [Google Scholar]
- 38.Guzmán-Verri C, Chaves-Olarte E, von Eichel-Streiber C, López-Goñi I, Thelestam M, Arvidson S, Gorvel JP, Moreno E. 2001. GTPases of the Rhosubfamily are required for Brucella abortus internalization in nonprofessional phagocytes: direct activation of Cdc42. J Biol Chem 276:44435–44443. 10.1074/jbc.M105606200. [DOI] [PubMed] [Google Scholar]
- 39.Lynch AS, Lin EC. 1996. Transcriptional control mediated by the ArcA two-component response regulator protein of Escherichia coli: characterization of DNA binding at target promoters. J Bacteriol 178:6238–6249. 10.1128/jb.178.21.6238-6249.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]