

ABSTRACT
The SOS response to DNA damage is a conserved stress response in Gram-negative and Gram-positive bacteria. Although this pathway has been studied for years, its relevance is still not familiar to many working in the fields of clinical antibiotic resistance and stewardship. Under some conditions, the SOS response favors DNA repair and preserves the genetic integrity of the organism. On the other hand, the SOS response also includes induction of error-prone DNA polymerases, which can increase the rate of mutation, called the mutator phenotype or “hypermutation.” As a result, mutations can occur in genes conferring antibiotic resistance, increasing the acquisition of resistance to antibiotics. Almost all of the work on the SOS response has been on bacteria exposed to stressors in vitro. In this study, we sought to quantitate the effects of SOS-inducing drugs in vivo, in comparison with the same drugs in vitro. We used a rabbit model of intestinal infection with enteropathogenic Escherichia coli strain E22. SOS-inducing drugs triggered the mutator phenotype response in vivo as well as in vitro. Exposure of E. coli strain E22 to ciprofloxacin or zidovudine, both of which induce the SOS response in vitro, resulted in increased antibiotic resistance to 3 antibiotics: rifampin, minocycline, and fosfomycin. Zinc was able to inhibit the SOS-induced emergence of antibiotic resistance in vivo, as previously observed in vitro. Our findings may have relevance in reducing the emergence of resistance to new antimicrobial drugs.
KEYWORDS: RecA, hypermutation, zinc, rabbit model, enteropathogenic E. coli, heteroresistance, minocycline, rifampin, fosfomycin, zidovudine
INTRODUCTION
The SOS response is a conserved bacterial stress response triggered primarily by DNA damage. The SOS response triggers and coordinates DNA repair pathways, arrest of bacterial cell division, and induction of latent bacteriophage. If the DNA damage is prolonged and severe, the SOS response also includes induction of DNA polymerases IV and V, capable of synthesizing across areas of damaged DNA, called translesion synthesis. DNA polymerases IV and V, encoded by the dinB and umuDC genes, respectively, are error prone, and thus, their expression results in an increased mutation rate (1–4). The increased rate of mutation observed during induction of the SOS response has been termed the mutator phenotype, the mutator response, or “hypermutation” (2). The molecular microbiology literature is replete with studies on the mechanisms of the SOS response because of the intriguing mechanisms involved and their relevance to the regulation of gene expression (5). Because of the heavy mechanistic emphasis, however, an understanding of the SOS response has been slow to percolate into fields such as clinical antibiotic resistance and antibiotic stewardship. In recent years, researchers have recognized that SOS-induced hypermutation may be a significant pathway for the generation of resistance to antibiotics. Almost all of the studies on the SOS response and antibiotic resistance were conducted with bacteria grown in vitro. Previous studies implicated the mutator phenotype in the ability of pathogens such as Haemophilus influenzae and Pseudomonas aeruginosa to cause chronic infections in cystic fibrosis patients, for example (6, 7). Activation of the SOS response was inferred to occur in vivo, for example, from studies of the genetic changes in strains recovered sequentially (8) and other less direct evidence (9, 10). We sought to extend the understanding of the impact of the SOS response in vivo using a rabbit model of gastrointestinal infection, which is relevant because this is frequently the anatomical site from which resistant bacteria emerge (11). We had previously used the ligated ileal loop model to study enteropathogenic Escherichia coli (EPEC) and Shiga-toxigenic E. coli (STEC) in vivo (12, 13).
Using the ligated intestinal segment model, we tested nonantibiotic inducers of the SOS response, such as the antiretroviral drug zidovudine (14), as well as the antibiotic ciprofloxacin with rabbit enteropathogenic E. coli strain E22 for their impact on the mutation frequency of E. coli strain E22. We compared the antibiotic resistance frequency in the intestinal segments treated with an inducer of the SOS response with that in untreated control loops, testing for resistance to three different antibiotics: minocycline, rifampin, and fosfomycin. Since we had previously shown that zinc could inhibit the SOS response in vitro (15, 16), we also tested whether this divalent metal could block SOS-induced hypermutation in vivo by measuring the frequencies of antibiotic resistance of E. coli E22 to three different antibiotics: minocycline, rifampin, and fosfomycin. In order to provide a basis for comparison, we also tested for SOS-induced emergence of resistance to these same three antibiotics in vitro. We hypothesized that the longer duration of exposure to the SOS-inducing drug in vivo would lead to a higher magnitude of hypermutation response than in vitro. Our results indicate that the E22 mutation frequency is increased following its exposure to zidovudine or ciprofloxacin in vivo and that treatment with zinc, but not iron or manganese, partially blocks this increase.
RESULTS
Induction of hypermutation using SOS-inducing antimicrobial drugs in vitro.
Before initiating experiments with rabbits, we conducted in vitro experiments to attempt to optimize the conditions for observing SOS-induced hypermutation capable of generating antibiotic resistance in two E. coli strains. We previously showed that SOS-inducing drugs triggered hypermutation in several other E. coli strains as well as in Klebsiella pneumoniae and Enterobacter cloacae (15, 16).
We began with the classic human EPEC strain B171-8, with which we had had previous laboratory experience (15, 17), and measured SOS-induced hypermutation in vitro. As shown in Fig. 1A, low concentrations of ciprofloxacin were able to induce an increase in the rifampin resistance frequency in this strain. As previously observed, zinc reversed the ciprofloxacin-induced increase in rifampin resistance (15). When we initiated animal experiments with this strain, however, this human EPEC strain failed to induce a fluid secretory response when injected into the rabbit intestinal segments. As a result, we could not obtain sufficient intestinal fluid for analysis, such as for dilutions and plate counts, inflammatory markers, and other biochemical testing. We therefore abandoned in vivo experiments with strain B171-8 and initiated work with rabbit EPEC strain E22.
FIG 1.

Effect of SOS inducers and inhibitors on hypermutation in vitro. Cultures of E. coli strains B171-8 and E22 grown overnight were subcultured into DMEM–F-12 broth medium and grown at 37°C with shaking at 300 rpm for 1.5 h, and the SOS-inducing drugs or metals were then added. The subcultures were collected at 4.5 h, and dilutions and plate counts were performed to measure total as well as antibiotic-resistant CFU per milliliter. Results are expressed as antibiotic resistance frequencies per 107 total CFU. Abbreviations: zido, zidovudine; cipro, ciprofloxacin; zinc, zinc acetate. The definitions of symbols used are indicated in each panel.
Using rabbit EPEC strain E22 and with ciprofloxacin as the SOS inducer, we were able to induce hypermutation, resulting in rifampin resistance (Fig. 1B), while zinc blocked this increase in the mutation frequency (Fig. 1C). When we used rifampin as the selecting antibiotic, we noticed that rifampin-resistant colonies appeared within 24 h, and few to no additional colonies appeared on the rifampin plates when the incubation was prolonged to 48 h. Low concentrations of zidovudine were also able to induce the emergence of rifampin resistance (Fig. 1D). As shown in Fig. 1D, zinc acetate once again was able to prevent SOS-induced hypermutation, while an equal concentration of FeSO4 did not block the hypermutation response.
Figure 1E shows that zidovudine was also able to induce an increase in resistance to minocycline, a tetracycline antibiotic, and that this induction was reversed by zinc acetate. When testing for minocycline resistance, however, at 24 h of incubation, only tiny pinpoint colonies were observed. Therefore, we had to prolong the incubation of our antibiotic-containing plates to 48 h for minocycline. Figure 1F shows that, unlike zinc, iron sulfate did not inhibit zidovudine-induced hypermutation to minocycline resistance. Manganese chloride also failed to block zidovudine-induced minocycline resistance, and in some experiments, MnCl2 actually increased zidovudine-induced minocycline resistance (Fig. 1G).
In summary, Fig. 1 shows that we were able to induce a hypermutation response in rabbit EPEC strain E22 using zidovudine and ciprofloxacin as inducers. Zinc acetate, but not iron sulfate or manganese chloride, inhibited the SOS-induced increase in the mutation frequency. An increase in the frequency of antibiotic resistance was observed toward two different antibiotics, rifampin and minocycline, which have different mechanisms of action in the bacterial cell. Zinc acetate, but not iron sulfate or manganese chloride, inhibited the SOS-induced increase in mutation frequency.
Optimization of concentrations of SOS-inducing drugs used in vivo in the rabbit intestinal loop model of infection.
We next used the results that we obtained from in vitro SOS experiments to determine if similar results would be observed in vivo, also using rabbit EPEC strain E22. In preliminary experiments using the concentrations of zidovudine used in Fig. 1D, the hypermutation response was not observed. Therefore, we increased the concentration of zidovudine used in vivo. Figure 2A shows a photograph of the rifampin-resistant colonies that emerged when we plated intestinal fluid from a loop infected with E22 alone (control) (left plate) and the colonies from an intestinal loop infected with E22 plus 0.6 μg/ml zidovudine (Fig. 2A, right plate). The quantitative data from this experiment are shown in Fig. 2B. The normal rabbit ileum has a microbiota, and coliform bacteria are present at ∼1,000 CFU/ml. Since the spontaneous antibiotic resistance frequencies for rifampin and minocycline are usually ∼1 colony per 107 bacteria, it is not surprising that we usually did not observe antibiotic-resistant colonies in the uninfected, control loops. Therefore, the resistance frequencies for the uninfected loops are omitted from this figure. Figure 2C shows data from another rabbit experiment testing zidovudine-induced hypermutation, illustrating that the magnitude of the hypermutation response can vary from experiment to experiment and from animal to animal. Figure 2C shows the data on a logarithmic scale, with a 1-log increase in the rifampin resistance frequency in the presence of 0.4 μg/ml zidovudine, compared to E22 alone.
FIG 2.

Effect of SOS inducers and inhibitors on hypermutation in vivo. Enteropathogenic E. coli (EPEC) strain E22, a rabbit-adapted strain, was used for the in vivo experiments. According to the procedures described in Materials and Methods, rabbits were subjected to a laparotomy, segments of the ileum were ligated, and two segments or “loops” were then injected with a narrow-gauge needle with 2 ml of an E22 bacterial suspension at 4 × 108 CFU/ml with or without inducers or inducers plus inhibitors. The surgical incisions were closed, and the rabbits were allowed to recover. After 20 h, the rabbits were euthanized, and loop fluid was collected for analysis. In panels C, E, and F, the results are shown using a logarithmic scale. In panel E, each line represents one rabbit, with duplicate loops subjected to each experimental condition. The paired t test was used to compare resistance frequencies in loops receiving E22 alone with those in loops receiving E22 plus the inducer (zidovudine) in the same rabbit. In panel G, the infected loop fluids were diluted 1:10, spread on MacConkey agar plates, and then overlaid with fosfomycin MIC strips to determine the MIC. White arrows indicate inlier colonies within the ellipses of inhibition. Panel H shows a comparison of the numbers of fosfomycin inlier colonies without and with zidovudine treatment in vivo.
We next tested the ability of zidovudine to induce a mutational response resulting in minocycline resistance in vivo. When minocycline at 6 μg/ml was used, as in Fig. 1D, uncountable smears of E22 bacteria were observed. Therefore, the minocycline concentration used was increased to 10 or 12 μg/ml in subsequent experiments. Figure 2D shows that zidovudine increased the minocycline resistance frequency observed in vivo, and this increase was significantly decreased by 0.3 mM zinc acetate. Figure 2E shows the combined results of 5 in vivo experiments with minocycline, each with 8 experimental loops (1 of which is the experiment shown in Fig. 2D, plus 4 other separate experiments). As shown in Fig. 2E, the zidovudine-induced increase in minocycline resistance was significant. In 5 experiments, the mean increase in the minocycline resistance frequency was 0.82 log units, or a 6.6-fold increase. Zinc’s inhibitory effect on minocycline resistance was also statistically significant. On average, zinc acetate reduced zidovudine-induced resistance to about one-half of that observed with zidovudine alone (Fig. 2E) (a 0.35-log decrease with zinc, or a 2.2-fold decrease). We tested the effect of FeSO4 on zidovudine-induced minocycline resistance in vivo and again observed that iron failed to block the hypermutation response (Fig. 2F), consistent with our in vitro results (Fig. 1D and F).
We next sought to determine if antibiotic MIC strips could be used as a faster way to screen for the emergence of antibiotic resistance, without having to do full sets of dilutions and plate counts for every infection condition. In Fig. 2G and H, we compared the growth of E22 bacteria from control and zidovudine-treated intestinal loops in the presence of fosfomycin MIC strips. As shown in the photograph in Fig. 2G, performed on MacConkey agar, zidovudine treatment did not result in an increase in the fosfomycin MIC. In fact, the fosfomycin MIC actually decreased a tiny amount from 0.25 mg/liter (control) to 0.19 mg/liter in the zidovudine-treated bacteria. What was notable, however, was that the number of colonies that emerged within the ellipse of inhibition (“inlier colonies”) was substantially higher in the E22 bacteria that had been treated with zidovudine (Fig. 2G, white arrows, and Fig. 2H, graph). Figure 2G and H suggest that the use of MIC strips may be a faster way to screen for SOS-induced hypermutation to various antibiotics, as we previously observed with doxycycline (15).
In summary, the results in Fig. 2 show that exposure to low concentrations of zidovudine triggered an increase in antibiotic resistance to rifampin and minocycline in vivo, and our semiquantitative results with the MIC strips suggest that this is the case for fosfomycin as well. We also performed hypermutation experiments in vivo using ciprofloxacin as the SOS-inducing drug. The induction of hypermutation by ciprofloxacin in vivo is shown in Table 2. In six separate rabbit experiments, ciprofloxacin increased the rifampin resistance frequency by 3.5- ± 1.2-fold compared to untreated control loops in the same rabbit. We failed, however, to observe a consistent increase in the magnitude of the hypermutation response with increasing concentrations of ciprofloxacin in vivo. This may be attributable to the fact that the basal (untreated) antibiotic resistance frequency varies so much from animal to animal, as is also observed in Fig. 2.
TABLE 2.
Ciprofloxacin as a trigger of SOS-induced hypermutation in E. coli E22 in vivo
| Rabbit expt | Ciprofloxacin concn (ng/ml) | Fold increase in rifampin resistance frequencya compared to E22 alone |
|---|---|---|
| 1 | 40 | 5.8 |
| 2 | 60 | 3.7 |
| 3 | 70 | 3.0 |
| 4 | 80 | 2.7 |
| 5 | 80 | 3.4 |
| 6 | 80 | 2.5 |
| Mean ± SD | 3.5 ± 1.2b | |
The rifampin concentration in the selection plates was 16 μg/ml.
Significant by a one-sided paired t test for nonparametric (skewed) data (P = 0.03).
We next tested whether we could find evidence of activation of the SOS response in our in vitro or in vivo hypermutation experiments. Induction of recA mRNA and, a bit later, RecA protein is an early and reliable marker of the onset of the SOS response (15, 18, 19). Recent reports have challenged the traditional idea that RecA remains in an intracellular location in bacteria interacting with the host. Instead, the release of RecA protein into the extracellular environment occurs in response to antimicrobial peptides, the induction of apoptotic-like bacterial cell death, or other stressors (20–22). This unusual finding suggests that RecA released into the extracellular space might continue to be biologically and catalytically active. Based on these reports, we tested whether we could detect RecA protein by Western immunoblotting in the supernatants of our E22 cultures and whether the abundance of RecA seen would change during SOS induction. As shown in Fig. 3A, RecA was present in sufficient quantities in the supernatant medium of E22 bacteria to be detected by Western immunoblotting. RecA from the experimental cultures migrated as a single band and at the same position as did the purified RecA protein used as the positive control. When treated with ciprofloxacin in vitro, the abundance of the RecA protein appeared to increase (Fig. 3A, middle 3 lanes). At a higher concentration of ciprofloxacin, however, the inhibition of E22 growth appeared to predominate, and the abundance of the RecA protein appeared to decrease (Fig. 3A, right 3 lanes). As shown in Fig. 3B, the increase in the abundance of RecA in the immunoblot was statistically significant when the raw data were scanned and quantitated. We next adjusted for the inhibitory effect of ciprofloxacin on bacterial growth. In Fig. 3C, the band density was adjusted for the culture turbidity of the initial bacterial culture, and the RecA band density again showed a significant increase, in the presence of 30 ng/ml ciprofloxacin, when adjusted for the optical density at 600 nm (OD600). We also adjusted for the effects on growth by normalization to viable E22 bacterial counts under these growth conditions. In Fig. 3D, both 30 ng/ml ciprofloxacin and 40 ng/ml ciprofloxacin showed statistically significant increases in RecA expression when the band densities were normalized to the values for total viable bacteria. In other words, the RecA band increased in expression whether corrected for the effects on growth (Fig. 3C and D) or left unadjusted (raw), as shown in Fig. 3B.
FIG 3.
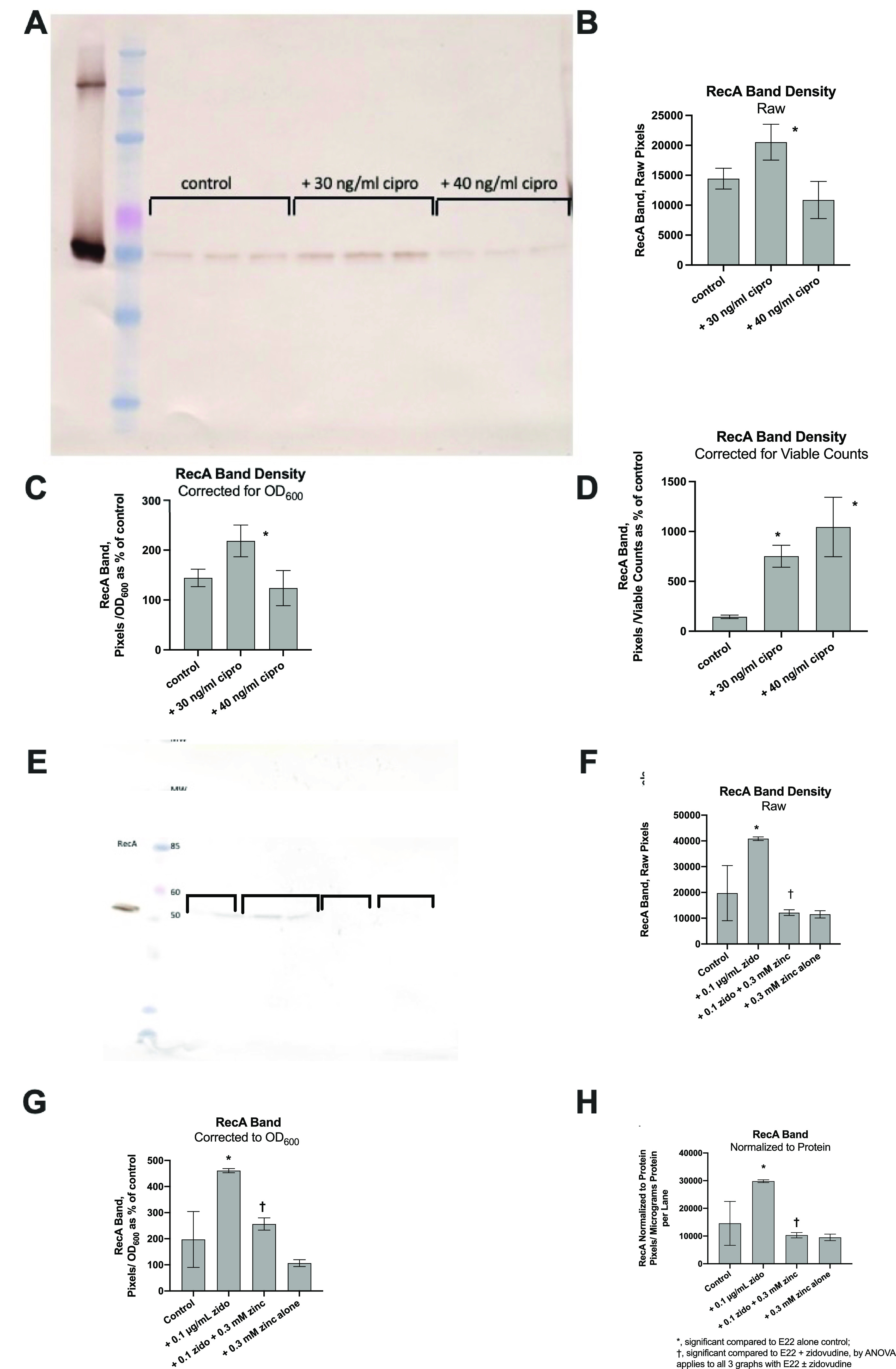
Detection of E. coli RecA protein by Western immunoblot analysis in the supernatants of E22 bacteria in vitro. (A) The supernatant media from in vitro hypermutation experiments such as those shown in Fig. 1 were stored frozen and then subjected to SDS-gel electrophoresis, transferred to nitrocellulose, and blotted for RecA using a rabbit polyclonal Ab against E. coli RecA (Abcam). For detection, a 2nd antibody of goat anti-rabbit IgG conjugated to horseradish peroxidase (HRP) was used. The blot was developed with diaminobenzidine (DAB) as the substrate in the presence of hydrogen peroxide, which generates a brown color. Lanes from left to right are as follows: lane 1, purified E. coli RecA, ∼25 ng of protein; lane 2, Invitrogen BenchMark prestained molecular weight markers (the pink marker is 60 kDa, and the blue marker below it is 50 kDa, the apparent molecular weight of the RecA protein); lanes 3 to 5, supernatants from control E22 cultures; lanes 6 to 8, supernatants from E22 cells treated with 30 ng/ml ciprofloxacin; lanes 9 to 11, supernatants from E22 cells treated with 40 ng/ml ciprofloxacin. (B to D) Analysis of the RecA band density subjected to various mathematical corrections. (B) Raw RecA density, without any corrections; (C) RecA band corrected by the culture turbidity (OD600) of the original cultures; (D) RecA band corrected based on viable E22 bacterial counts. (E) Immunoblot analysis of RecA on E22 supernatants treated with zidovudine with or without zinc in vivo. Lane 1 again shows purified RecA protein, and lane 2 shows molecular weight (MW) markers. Equal volumes of experimental samples were run in duplicate and were, from left to right, E22 alone; E22 plus 0.1 μg/ml zidovudine; E22, zidovudine, and 0.3 mM zinc; and 0.3 mM zinc alone, as shown by the brackets. The concentration of bacterial protein loaded per lane was 1.4 μg for the untreated, control lanes. (F) RecA band density, in pixels, without any mathematical correction. (G and H) Amounts of RecA adjusted for the OD600 (G) and for the bacterial supernatant protein (H). The * and † symbols apply similarly in panels F to H.
A similar Western immunoblot analysis was conducted using zidovudine as the SOS-inducing drug, and the results are shown in Fig. 3E to H. As shown in the figures, the addition of 0.1 μg/ml zidovudine triggered an increase in the amount of RecA protein on the immunoblots. Once again, the zidovudine-induced increase in RecA expression was observed when the band density was left unadjusted for effects on growth, or raw, as shown in Fig. 3F. When the band density was corrected for changes in the OD600, the stimulatory effect of zidovudine was preserved (Fig. 3G). Finally, we corrected RecA expression by normalization based on micrograms of the bacterial supernatant protein, and zidovudine-induced RecA expression remained statistically significant (Fig. 3H). Zinc acetate at 0.3 mM inhibited the stimulatory effect of zidovudine, and the zinc effect was significant based on the raw pixels as well as the results adjusted for the OD600 and total bacterial protein (Fig. 3F to H).
The RecA immunoblot results shown in Fig. 3 provide evidence that the SOS response was induced in vitro in strain E22 and that RecA protein was released from the E. coli bacterial cells. We also attempted to perform immunoblot analyses for RecA on the supernatant of loop fluids in vivo, but the interpretation of the blots was more difficult because fragments of immunoglobulin run at a position close to that of RecA and cross-react with the 2nd antibody (Ab) used for the blots. We are attempting to remove the interference by the addition of protein A/protein G agarose beads to deplete immunoglobulin from the lumen fluid.
The low concentrations of zidovudine and ciprofloxacin used in the in vivo experiments did not have a large inhibitory effect on the growth of E22, with most experiments showing very modest reductions in E22 CFU per milliliter of 10 to 20%. We wondered if this modest reduction in E22 bacteria affected the host responses to infection. Figure 4 shows the effect of zidovudine treatment on two host responses that we measured in vivo, fluid secretion and interleukin-8 (IL-8) production. EPEC, unlike commensal E. coli strains and extraintestinal pathogenic E. coli (ExPEC), triggers an outpouring of fluid into the intestinal lumen (23, 24). Figure 4A shows that zidovudine did not block the fluid secretory response induced by strain E22 in the rabbit intestine, as measured by the volume-to-length ratio. In a similar manner, 30 to 80 ng/ml ciprofloxacin did not block E22-induced fluid secretion in the rabbit ileal segments (Fig. 4B; higher concentrations of ciprofloxacin are not shown). EPEC infection is also accompanied by an influx of neutrophils into the gut lumen (these cells are called heterophils in rabbits). IL-8 is a neutrophil chemoattractant that is secreted by intestinal epithelial cells into the gut lumen as well as the bloodstream (25, 26). We measured interleukin-8 levels in the fluids collected from the rabbit intestinal loops. Figure 4C shows that zidovudine inhibited the amount of IL-8 released into the intestinal lumen in a single, typical experiment. In experiments similar to the one shown in Fig. 3B, the inhibitory effects of zidovudine (0.3 to 0.5 μg/ml) on IL-8 reached statistical significance in 5 of 7 experiments in vivo (including the experiment shown in Fig. 3B). In contrast, ciprofloxacin did not appear to reduce IL-8 concentrations in the rabbit ileum (Fig. 4D). The difference between the effects of zidovudine and ciprofloxacin on IL-8 levels may be due to the effects of zidovudine on host cells since this antiviral is known to have some inhibitory activity against phagocytic cells (27). The fact that IL-8 levels decreased in response to zidovudine, however, means that the hypermutation phenomenon observed in Fig. 2 cannot be attributed to an enhanced host inflammatory response triggered by the antiretroviral drug.
FIG 4.

Host responses to E22 infection in vivo. (A and B) Volume-to-length ratios, an indicator of the amount of fluid secreted into the intestinal lumen, using zidovudine (A) and ciprofloxacin (B) as SOS inducers. (C and D) Interleukin-8 (IL-8) levels in the loop fluids were measured using an EIA for rabbit IL-8, again using zidovudine (C) and ciprofloxacin (D) as inducers of the SOS response. HBS, HEPES-buffered saline.
Antibiotic resistance phenotypes of E22 clones that emerged after SOS induction in vitro and in vivo.
We next investigated the antibiotic resistance phenotypes of the bacteria that emerged after treatment with the SOS inducer, followed by selection for antibiotic resistance. We compared the resistant colonies that emerged after SOS induction in vitro with those that emerged in vivo. For these experiments, we turned to the antibiotic-resistant colonies that emerged from the hypermutation experiments shown in Fig. 1 and 2, which had been collected and archived at −70°C. The resistant bacteria were passaged in antibiotic-free Luria-Bertani (LB) broth medium and then retested for resistance using rifampin and minocycline MIC strips. Figure 5A shows the distribution of rifampin MICs from 16 unique rifampin-resistant E22 clones that had emerged as resistant in vitro. For comparison, the rifampin MIC of the E22 parental strain is 3 μg/ml (Table 1). Figure 5A shows that although we selected for the ability to grow on 16 μg/ml rifampin, the actual MICs observed from this collection were generally much higher. Indeed, 13 of 16 strains tested were resistant to rifampin at >256 μg/ml (i.e., no zone of inhibition), and all of the strains had MIC values well above the selecting concentration of 16 μg/ml.
FIG 5.
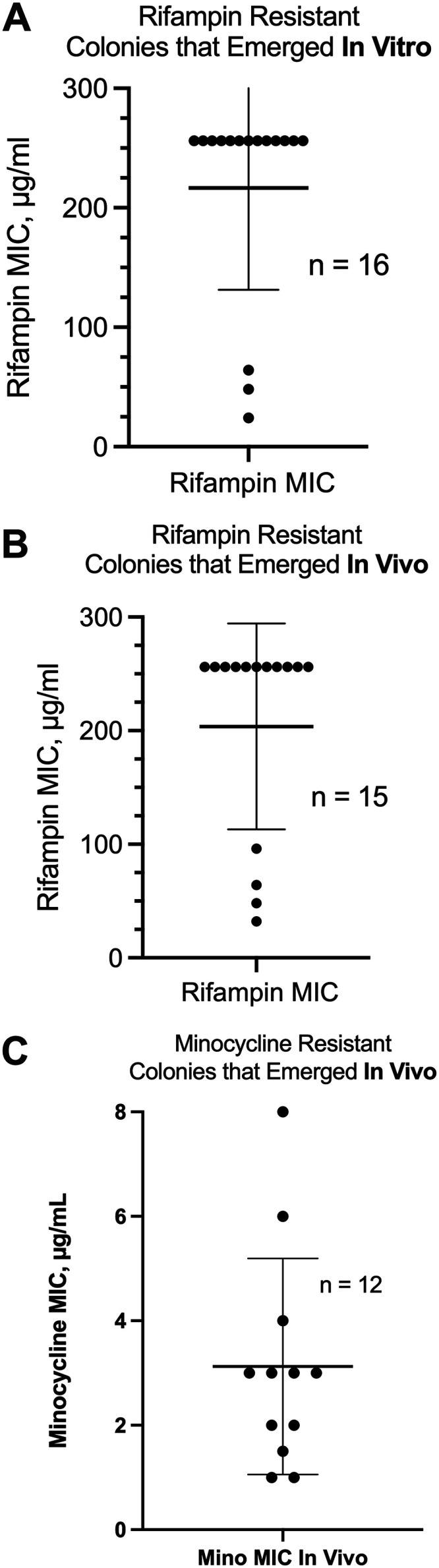
MICs of the antibiotic-resistant colonies that emerged from treatment with SOS inducers in vitro and in vivo. We collected 2 antibiotic-resistant colonies from the rifampin plates or minocycline plates from E22 cells exposed to zidovudine in vitro or in vivo and archived them at −70°C. After passaging in antibiotic-free LB broth, they were retested for rifampin or minocycline resistance using Liofilchem MIC strips. (A) Rifampin MIC scattergram of 16 colonies collected from 8 separate in vitro experiments. (B) Rifampin MIC scattergram of 15 colonies obtained from 7 separate rabbit experiments. The rifampin MIC of the E22 parental strain is 2.5 μg/ml (Table 1). (C) Minocycline MIC scattergram from minocycline-resistant colonies that emerged on LB medium with 12 μg/ml minocycline in vivo. (The minocycline MIC scattergram from in vitro experiments showed results similar to those in panel C and is not shown.)
TABLE 1.
Bacterial strains useda
| Strain | Serotype | Description | MIC (mg/liter) in LB broth |
Reference(s) or source (reference) | ||
|---|---|---|---|---|---|---|
| Ciprofloxacin | Minocycline | Rifampin | ||||
| Pathogenic E. coli strains | ||||||
| B171-8 | O111:NM | Classic human EPEC | 0.012 | 2.0 (in MM) | 2-3 | 15, 60 |
| E22 | O103:H2 | Rabbit EPEC | 0.064 | 0.19–0.38 | 3 | 61 |
| Laboratory E. coli strain | DH5α-(pGFP) | Created by electroporation of DH5α with plasmid pGFP-uv and selection for ampicillin resistance | pGFP-uv plasmid purchased from Clontech, now TaKaRa Bio (62) | |||
LB, Luria-Bertani; MM, minimal medium. Rifampin is also known as rifampicin.
We next tested E22-derived mutants that had emerged as resistant in vivo. As shown in Fig. 5B, these mutants also demonstrated rifampin MIC values well above the rifampin concentration in the selection plates (16 μg/ml). Eleven of 15 rifampin-resistant mutants that emerged in vivo had MIC values of >256 mg/liter. In summary, the rifampin-resistant mutants that emerged in vitro and in vivo were robustly resistant to rifampin, and the phenotype remained stable over time and with serial passages.
In contrast, the results that we observed with minocycline-resistant mutants that emerged in vivo were quite different. When minocycline-resistant colonies were grown in antibiotic-free broth and then replated on minocycline-containing agar, some of the minocycline-resistant mutants grew poorly, with small, slow-growing colonies (photographs not shown). For those strains that could be grown, the MIC results are shown in Fig. 5C. Even though these colonies were collected from plates containing 12 μg/ml minocycline (Fig. 2F), the MICs observed upon retesting often fell below this value, with the mean MIC value being 3 μg/ml. The MIC for the E22 parental strain is 0.38 μg/ml, so the minocycline MICs increased as a result of the in vivo treatment, but the resistance was not as robust or as genetically stable as that of the rifampin-resistant colonies shown in Fig. 5A and B. The explanation for the differing results with minocycline may be the genetic instability of minocycline resistance in the absence of continuous antibiotic selection (see Discussion, below) (28, 29).
The experiments shown in Fig. 1 to 5 all dealt with SOS-induced emergence of antibiotic resistance within a single strain of bacteria. But the transfer of antibiotic resistance genes between different strains or different species, or horizontal resistance, is also an important route by which antibiotic resistance emerges and spreads. In the rabbit ileal loop model, EPEC and STEC infection triggers a strong recruitment of acute inflammatory cells (heterophils) into the gut lumen, as suggested by the IL-8 experiments shown in Fig. 4C and D and as previously reported by our laboratory (13, 17, 30). These activated heterophils release neutrophilic extracellular traps (NETs) consisting of extracellular DNA decorated by additional proteins such as myeloperoxidase, cationic peptides, and histones (31, 32). We wondered whether the NETs triggered by EPEC infection could also entrap other bacteria. Figure 6A shows the formation of NETs in vivo in loop fluid obtained from rabbits infected with strain E22 and stained for DNA with propidium iodide. DNA strands often appear to emanate from degenerating heterophils (Fig. 6A, red arrow) and may take the form of elongated, stretched DNA strands (black arrow). E22 bacteria are visible as small pink coccobacillary forms in this fluorescence microscope photograph. Figure 6B shows what happened when we added a fluorescently labeled, laboratory strain of E. coli, DH5α-(pGFP), ex vivo. DH5α-(pGFP) is a nonpathogenic strain and does not trigger an acute inflammatory response or the generation of NETs. When added to the infected loop fluid, however, some green-fluorescing DH5α-(pGFP) bacteria became enmeshed in the NETs triggered by the E22 bacteria (Fig. 6B, red bacteria, black arrow). The inset photographs in Fig. 6B show red E22 bacteria (red arrows) in close contact with green-fluorescing DH5α bacterial cells (green arrows). The findings in Fig. 6 suggest that there may be pathways for the emergence of drug resistance in vivo that may not be operative in vitro. One pathway for horizontal gene transfer is by conjugation, which requires close contact of bacterial cells (33), and this close contact could be facilitated by coentrapment in NETs. The SOS response also increases the transfer of antibiotic resistance genes via transformation (34). We showed that the SOS-induced transfer of antibiotic resistance genes is inhibited by zinc (16), and Buberg et al. also confirmed that zinc and copper block the conjugative transfer of antibiotic resistance (35). In addition to promoting close contact between bacteria, DNA biofilms also reduce the susceptibility of bacteria to antibiotics, an effect that can be reversed by the addition of DNase (36).
FIG 6.

Fluorescence micrographs showing the formation of neutrophilic extracellular traps (NETs) in vivo in loop fluid from rabbits infected with E22. (A) Loop fluid from rabbit ileum infected in vivo for 20 h with EPEC strain E22 was subjected to low-speed centrifugation (21 × g for 5 min) onto glass microscope slides in a cytological “cytospin” centrifuge using Shandon centrifuge funnels. After fixation in alcohol-acetone, the glass slides were dried on a warmer plate and then stained for DNA with 10 μg/ml propidium iodide. An example of a DNA NET is indicated by the black arrow. E22 bacteria grow as short coccobacilli when in contact with host tissues and stain red or pink, often adhering to the DNA NETs. Host cells, mostly heterophils, the rabbit equivalent of neutrophils, also stain with propidium iodide (red arrow). The size bar is at the bottom right. The photograph was taken at a ×600 magnification under oil immersion. (B) Loop fluid from rabbit ileum was mixed with a fluorescently labeled, green fluorescent protein (GFP)-expressing laboratory strain of E. coli, DH5α-(pGFP), and allowed to incubate for 20 min at 37°C. The mixture was again applied to the funnels of a cytological centrifuge and spun onto glass slides as described above for panel A. After fixation and drying, the slides were again stained with propidium iodide. Panel B shows that the laboratory strain DH5α-(pGFP) becomes enmeshed in the DNA NETs along with the pathogenic strain E22. E22 bacteria, stained red by propidium iodide, are short, about 2 μm in length, and are indicated by the black arrow. The DH5α-(pGFP) bacteria fluoresce bright green and are about 4 μm in length. Diffuse pink background staining represents extracellular DNA. The main photograph in panel B is again at a ×600 magnification under oil immersion. The inset photographs, taken at a ×1,000 magnification, show red E22 bacteria (red arrows) in close contact with green-fluorescing DH5α bacteria (green arrows), and the size bar in the top left inset, 8 μm, applies to the top right inset photograph as well. The proximity of the two different bacterial strains, trapped together in DNA NETs, could facilitate the horizontal transfer of antibiotic resistance genes via conjugation or transformation.
DISCUSSION
Although the SOS response has been studied for years, its relevance to the emergence of clinically important antibiotic resistance has been slow to permeate the field. One of the first mentions of the “SOS repair” pathway appeared in the literature in 1975 (37), the same year that the ship Edmund Fitzgerald sank without sending out an SOS signal. Despite the long pedigree of research, the bacterial SOS response continues to be studied because new aspects continue to be discovered (38) and because the bacterial pathway remains a model for studies of DNA repair and mutagenesis in eukaryotic organisms (39), including in human cancers.
Recently, articles have emphasized the relevance of the SOS response to the emergence of antibiotic resistance (34, 40–43). The importance of the SOS response in antibiotic resistance is now recognized in many types of bacteria other than E. coli, including Gram-positive organisms and Mycobacterium tuberculosis (44–46). In addition, researchers have appreciated that the SOS response can be strongly induced by drugs that are commonly used clinically, including the quinolones, antivirals, and anticancer drugs such as 5-fluorouracil (14, 47). The vast majority of these studies, however, have been conducted solely in vitro. As stated above, we wished to test if the emergence of antibiotic resistance would occur in response to SOS-inducing drugs in vivo.
One hypothesis of this project was that the long duration of exposure to the SOS-inducing agent in vivo (20 h), compared to a 3- to 4-h exposure in vitro, would result in a higher magnitude of hypermutation. This hypothesis was not borne out by the actual data, in which the magnitude of the zidovudine-induced hypermutation response was about the same (∼6-fold increase for minocycline) (Fig. 2E) in vivo as that in vitro. Similarly, the 3.5-fold increase in the rifampin resistance frequency with ciprofloxacin as the SOS inducer in vivo (Table 2) was similar to the magnitude with ciprofloxacin in vitro (Fig. 1A to C). One reason for this may be that zidovudine and ciprofloxacin are likely absorbed systemically during the course of the infection, lowering the amount of SOS-inducing drug remaining in the intestinal lumen. In addition, EPEC E22 infection triggers an influx of watery fluid into the gut, thereby reducing the concentration of the SOS inducer by dilution (Fig. 4A and B).
A second goal of the current project was to test whether zinc would also inhibit SOS-induced antibiotic resistance in vivo. In vivo, the inhibitory effects of zinc were again observed, and zinc’s effects achieved statistical significance most of the time (Fig. 2B to E).
Nevertheless, the inhibitory effects of zinc were clearly different from those of iron and manganese, which showed either no inhibition of hypermutation or, sometimes, an increase (Fig. 1). Because the inhibitory effects of zinc were modest in vivo, we are planning to test whether zinc’s inhibitory effects can be enhanced by using “combination therapy,” such as zinc plus a nitric oxide donor (48) or zinc plus a peptide inhibitor of RecA, 4E1 (49), or other RecA inhibitors (50).
As summarized in Fig. 5, the colonies that emerged as resistant after treatment with an SOS inducer behaved differently depending on which antibiotic was used for selection. When resistance to 16 μg/ml rifampin was selected for, the resistant colonies were genetically stable, and, upon retesting, their MIC values were much higher than the concentration of rifampin used for selection (Fig. 5A and B). In contrast, when resistance to 12 μg/ml minocycline was selected for, some of the colonies appeared unstable and partially lost their resistance to minocycline when passaged in antibiotic-free medium (Fig. 5C). In other words, the resistant colonies regressed or reverted to the antibiotic phenotype of the wild-type, parental E22 strain. Resistance to minocycline and other tetracyclines is often due to the overexpression of efflux pumps (51, 52), and this overexpression can occur via duplication or amplification of the efflux pump genes, also called copy number variation (28, 29), or by inactivation of negative regulators of expression. Amplifications that can result in tetracycline resistance can be in genes such as tetA and ramA and in the acrAB and oqxAB loci (29, 51, 53, 54). These changes in copy number are reversible, and loss of gene copies can occur in the absence of ongoing antibiotic selection pressure. This variability in the level of resistance is a hallmark of heteroresistance, which can result in confusion in the clinical microbiology laboratory (55) but may also lead to treatment failures (56). Heteroresistance is clearly, to quote El-Halfawy and Valvano, “a field in need of clarity” (55).
Finally, we found that NETosis, or the formation of DNA NETs, can bring different strains of bacteria into close contact with one another ex vivo (Fig. 6), possibly favoring the SOS-induced transfer of antibiotic resistance genes within a DNA-containing biofilm. We believe that researchers interested in antibiotic resistance should include pathogenic strains in their strain collections under study and that the incorporation of in vivo models of infection may reveal new pathways for the emergence of antibiotic resistance that would not be revealed by in vitro experiments alone.
In summary, SOS-inducing drugs can trigger hypermutation in vivo, resulting in the emergence of antibiotic resistance. We showed that zidovudine and ciprofloxacin can trigger the emergence of resistance to rifampin, minocycline, and fosfomycin in vivo. Drugs that are not considered antibiotics can be strong SOS inducers, such as zidovudine used in this study as well as many others, including azathioprine, 5-fluorouracil, azacytidine, bleomycin, mitomycin C, and the herbicide paraquat (15, 47, 57). Treatment with an SOS-inducing drug followed by a short exposure to an antibiotic may be an efficient route for generating new antibiotic resistance. In the future, or perhaps even now, people involved in antibiotic stewardship may need to pay attention to the nonantibiotic, SOS-inducing drugs being administered to humans and animals as well as to the antibiotics themselves (58). In addition, the ability of zinc and other SOS inhibitors to block the emergence of antibiotic resistance deserves further study.
MATERIALS AND METHODS
Materials.
The following reagents were obtained from Sigma-Aldrich (now Millipore Sigma): ciprofloxacin, zidovudine, vancomycin, rifampin, minocycline, Luria-Bertani (LB) agar, diaminobenzidine (DAB) tablets, and urea-peroxide tablets. Zidovudine is also known as azidothymidine or “AZT.” Zinc acetate, MnCl2, and FeSO4 were also obtained from Millipore Sigma. Purified RecA protein and antibody against E. coli RecA were obtained from Abcam (Cambridge, MA). The antibody was rabbit polyclonal. As the 2nd antibody, we used goat anti-rabbit IgG conjugated to horseradish peroxidase (HRP), also from Abcam.
Antibiotic gradient strips, or MIC strips, were obtained from Liofilchem, Waltham, MA, and were used to determine the antibiotic MICs for various antibiotics in vitro.
Growth of bacterial strains for in vitro hypermutation studies.
Bacterial strains used are shown in Table 1.
E. coli strains were grown overnight in LB broth at 37°C with shaking at 300 rpm and then subcultured at a dilution of 1:100 into antibiotic-free Dulbecco’s modified Eagle’s medium (DMEM)–F-12 medium. Growth was continued by incubation at 37°C with shaking. Because SOS-inducing stimuli are more effective if applied when bacteria are growing rapidly, the addition of the SOS inducer was delayed until 1.5 h after the subculture was started, and growth was then continued for an additional 3 h (4.5 h total). Culture turbidity was measured by the OD600.
Growth of bacterial strains for in vivo hypermutation studies.
To prepare the inoculum for in vivo infections in the rabbit intestinal segments, E. coli bacteria were subcultured into minimal medium and grown at 37°C with shaking at 300 rpm for 6 h. The OD600 was measured, and sterile glycerol was added to yield a final concentration of 30% glycerol. The suspensions were divided into multiple aliquots and frozen at −70°C. A frozen aliquot was thawed, and the titer was then determined to obtain bacterial cell counts, usually >109 CFU/ml. When grown and stored in this way, bacterial viability declined only about 1% per month, meaning that the frozen aliquots can be used for many animal experiments. On the day of a rabbit experiment, the frozen aliquots were thawed, diluted in DMEM–F-12 medium to achieve a bacterial density of 4 × 108 CFU/ml, and used to infect intestinal segments. Dilutions and plate counts were performed for each experiment to verify the inoculum. Ligated rabbit loops were infected by injecting the loops with 2 ml of the diluted E22 suspension using a narrow, 26-gauge needle.
Preparation of antibiotic-containing plates for selection of resistant colonies.
LB agar was used to prepare rifampin plates as described previously (15), and they were kept protected from light. Vancomycin was added at 20 μg/ml to the plates used for the detection of antibiotic resistance in vivo because the rabbit intestine is colonized by large quantities of Bacillus species. Minocycline is less stable than other tetracyclines, and the active concentration diminishes with time in microbiological media (59). Therefore, we always prepared minocycline plates 48 h in advance of their use, and unused plates were discarded. Minocycline plates were also protected from light. Unless otherwise noted, rifampin was used at 16 μg/ml in LB medium for hypermutation experiments, while minocycline was used at 6 μg/ml for in vitro experiments and at 12 μg/ml in our in vivo experiments.
Determination of antibiotic resistance frequencies.
The resistance frequencies for the emergence of resistance to antibiotics were calculated as the ratio of the number of bacteria resistant to that antibiotic (in CFU per milliliter) to the total number of bacteria (in CFU per milliliter), as is standard in the methods described by other laboratories (40, 41). These ratios usually yielded antibiotic resistance frequencies in the range of 10−7. Therefore, on the graphs, the data are often plotted as antibiotic resistance frequencies per 107 bacteria.
Rabbit experiments.
Rabbit experiments were reviewed and approved by the IACUC of the University at Buffalo. Rabbits were New Zealand White (NZW) rabbits of both sexes, 2 to 2.5 kg, from Charles River Laboratories. Rabbits were housed in large cages with environmental enrichment, and the diet of rabbit chow was enhanced with timothy hay, a source of fiber. Animal experiments were reviewed and approved by the IACUC of the University at Buffalo. Details of the surgical procedures involved in the creation of the ligated intestinal segments, or “loops,” are described in the supplemental material. In this procedure, segments of the ileum are ligated and infected or not with bacteria, and the infection is allowed to proceed for 20 h. Uninfected loops received 2 ml of HEPES-buffered saline as a mock infection. Analgesia was provided using 12.5 μg fentanyl transdermal patches, placed on the back of the rabbit 24 h before surgery. At the end of the experiment, euthanasia was performed using intravenous (i.v.) pentobarbital in the form of Fatal Plus, a method recognized by the American Veterinary Medical Association (AVMA). After euthanasia, the fluid from the rabbit intestinal segments was collected and kept on ice.
Interleukin-8 assays.
Interleukin-8 (IL-8) was measured in intestinal loop fluids using the IL-8 enzyme immunoassay (EIA) development kit for rabbit from Kingfisher Biotech (St. Paul, MN).
Western immunoblot analysis of E. coli RecA.
Precast SDS-polyacrylamide gels were 10% acrylamide, Bis-Tris gels from Invitrogen (now part of Thermo Fisher). Electrophoresis running buffer (20×) and blot transfer buffer (20×) were also obtained from Invitrogen/Thermo Fisher. For Western immunoblotting, nitrocellulose was obtained from Pall Scientific, Pensacola, FL. Western blot analyses were carried out by using standard techniques as previously described for the detection of LexA (15). We used both the 1st antibody (anti-RecA) and the 2nd antibody (goat IgG-HRP) at final dilutions of 1:3,000 in 5% nonfat dry milk in phosphate-buffered saline. Due to the coronavirus disease 2019 (COVID-19) pandemic, core laboratory facilities for chemiluminescence imaging of the blots were unavailable. As a result, we used DAB as the substrate to develop the immunoblots, which generates a brown stain in the presence of hydrogen peroxide. Quantitation of the band intensity was done by scanning the blots on a visible-light scanner and then using the program Un-Scan-It Gel, version 6.3, for Macintosh (Silk Scientific, Orem, UT).
Statistical analysis.
t tests and analysis of variance (ANOVA) were performed using GraphPad Prism for Macintosh, versions 8 and 9. Error bars shown in the figures are standard deviations.
ACKNOWLEDGMENTS
This research was funded by the National Institutes of Health, NIAID, via grant R21 AI 145836-01.
None of the authors have any conflicts of interest to declare.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Sutton MD, Walker GC. 2001. umuDC-mediated cold sensitivity is a manifestation of functions of the UmuD2C complex involved in a DNA damage checkpoint control. J Bacteriol 183:1215–1224. 10.1128/JB.183.4.1215-1224.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goodman MF. 2016. Better living with hyper‐mutation. Environ Mol Mutagen 57:421–434. 10.1002/em.22023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pérez-Capilla T, Baquero M-R, Gómez-Gómez J-M, Ionel A, Martín S, Blázquez J. 2005. SOS-independent induction of dinB transcription by β-lactam-mediated inhibition of cell wall synthesis in Escherichia coli. J Bacteriol 187:1515–1518. 10.1128/JB.187.4.1515-1518.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanders LH, Rockel A, Lu H, Wozniak DJ, Sutton MD. 2006. Role of Pseudomonas aeruginosa dinB-encoded DNA polymerase IV in mutagenesis. J Bacteriol 188:8573–8585. 10.1128/JB.01481-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maslowska KH, Makiela-Dzbenska K, Fijalkowska IJ. 2019. The SOS system: a complex and tightly regulated response to DNA damage. Environ Mol Mutagen 60:368–384. 10.1002/em.22267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watson ME, Burns JL, Smith AL. 2004. Hypermutable Haemophilus influenzae with mutations in mutS are found in cystic fibrosis sputum. Microbiology (Reading) 150:2947–2958. 10.1099/mic.0.27230-0. [DOI] [PubMed] [Google Scholar]
- 7.Oliver A, Cantón R, Campo P, Baquero F, Blázquez J. 2000. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288:1251–1254. 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- 8.Hocquet D, Llanes C, Thouverez M, Kulasekara HD, Bertrand X, Plésiat P, Mazel D, Miller SI. 2012. Evidence for induction of integron-based antibiotic resistance by the SOS response in a clinical setting. PLoS Pathog 8:e1002778. 10.1371/journal.ppat.1002778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breidenstein EB, Bains M, Hancock RE. 2012. Involvement of the lon protease in the SOS response triggered by ciprofloxacin in Pseudomonas aeruginosa PAO1. Antimicrob Agents Chemother 56:2879–2887. 10.1128/AAC.06014-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rossi E, La Rosa R, Bartell JA, Marvig RL, Haagensen JAJ, Sommer LM, Molin S, Johansen HK. 2021. Pseudomonas aeruginosa adaptation and evolution in patients with cystic fibrosis. Nat Rev Microbiol 19:331–342. 10.1038/s41579-020-00477-5. [DOI] [PubMed] [Google Scholar]
- 11.Snitkin ES, Zelazny AM, Thomas PJ, Stock F, NISC Comparative Sequencing Program Group, Henderson DK, Palmore TN, Segre JA. 2012. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med 4:148ra116. 10.1126/scitranslmed.3004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crane J, Naeher T, Shulgina I, Zhu C, Boedeker E. 2007. Effect of zinc in enteropathogenic Escherichia coli infection. Infect Immun 75:5974–5984. 10.1128/IAI.00750-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crane JK, Broome JE, Reddinger RM, Werth BB. 2014. Zinc protects against Shiga-toxigenic Escherichia coli by acting on host tissues as well as on bacteria. BMC Microbiol 14:145. 10.1186/1471-2180-14-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mamber SW, Brookshire KW, Forenza S. 1990. Induction of the SOS response in Escherichia coli by azidothymidine and dideoxynucleosides. Antimicrob Agents Chemother 34:1237–1243. 10.1128/aac.34.6.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bunnell BE, Escobar JF, Bair KL, Sutton M, Crane J. 2017. Zinc blocks SOS-induced hypermutation via inhibition of RecA in Escherichia coli. PLoS One 12:e0178303. 10.1371/journal.pone.0178303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crane J, Cheema M, Olyer M, Sutton M. 2018. Zinc blockade of SOS response inhibits horizontal transfer of antibiotic resistance genes in enteric bacteria. Front Cell Infect Microbiol 8:410. 10.3389/fcimb.2018.00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crane J, Naeher TM, Broome J, Boedeker E. 2013. Role of xanthine oxidase in infection due to enteropathogenic and Shiga-toxigenic Escherichia coli. Infect Immun 81:1129–1139. 10.1128/IAI.01124-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Little JW, Edmiston SH, Pacelli LZ, Mount DW. 1980. Cleavage of the Escherichia coli LexA protein by the RecA protease. Proc Natl Acad Sci U S A 77:3225–3229. 10.1073/pnas.77.6.3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roca AI, Cox MM, Brenner SL. 1990. The RecA protein: structure and function. Crit Rev Biochem Mol Biol 25:415–456. 10.3109/10409239009090617. [DOI] [PubMed] [Google Scholar]
- 20.Ibáñez de Aldecoa AL, Zafra O, González-Pastor JE. 2017. Mechanisms and regulation of extracellular DNA release and its biological roles in microbial communities. Front Microbiol 8:1390. 10.3389/fmicb.2017.01390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee H, Lee DG. 2019. SOS genes contribute to Bac8c induced apoptosis-like death in Escherichia coli. Biochimie 157:195–203. 10.1016/j.biochi.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Lee JY, Lee JS, Materne EC, Rajala R, Ismail AM, Seto D, Dyer DW, Rajaiya J, Chodosh J. 2018. Bacterial RecA protein promotes adenoviral recombination during in vitro infection. mSphere 3:e00105-18. 10.1128/mSphere.00105-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neter E. 1959. Enteritis due to enteropathogenic Escherichia coli. J Pediatr 55:223–239. 10.1016/S0022-3476(59)80091-3. [DOI] [PubMed] [Google Scholar]
- 24.Levine M, Nataro J, Karch H, Baldini M, Kaper J, Black R, Clements M, O’Brien A. 1985. The diarrheal response of humans to some classic serotypes of enteropathogenic Escherichia coli is dependent on a plasmid encoding an enteroadhesiveness factor. J Infect Dis 152:550–559. 10.1093/infdis/152.3.550. [DOI] [PubMed] [Google Scholar]
- 25.Daig R, Rogler G, Aschenbrenner E, Vogl D, Falk W, Gross V, Schölmerich J, Andus T. 2000. Human intestinal epithelial cells secrete interleukin-1 receptor antagonist and interleukin-8 but not interleukin-1 or interleukin-6. Gut 46:350–358. 10.1136/gut.46.3.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evgenikos N, Bartolo D, Hamer-Hodges D, Ghosh S. 2002. Assessment of ileoanal pouch inflammation by interleukin 1β and interleukin 8 concentrations in the gut lumen. Dis Colon Rectum 45:249–255. 10.1007/s10350-004-6156-6. [DOI] [PubMed] [Google Scholar]
- 27.De Simone C, Maffione AB, Calvello R, Nacci C, Sciannameo G, Greco B, Caradonna L, Pece S, Antonaci S, Jirillo E. 1996. In vitro effects of 3′-azido-3′-deoxythymidine (AZT) on normal human polymorphonuclear cell and monocyte-macrophage functional capacities. Immunopharmacol Immunotoxicol 18:161–178. 10.3109/08923979609052730. [DOI] [PubMed] [Google Scholar]
- 28.Nicoloff H, Hjort K, Levin BR, Andersson DI. 2019. The high prevalence of antibiotic heteroresistance in pathogenic bacteria is mainly caused by gene amplification. Nat Microbiol 4:504–514. 10.1038/s41564-018-0342-0. [DOI] [PubMed] [Google Scholar]
- 29.Andersson DI, Nicoloff H, Hjort K. 2019. Mechanisms and clinical relevance of bacterial heteroresistance. Nat Rev Microbiol 17:479–496. 10.1038/s41579-019-0218-1. [DOI] [PubMed] [Google Scholar]
- 30.Crane JK, Broome JE, Lis A. 2016. Biological activities of uric acid in infection due to enteropathogenic and Shiga-toxigenic Escherichia coli. Infect Immun 84:976–988. 10.1128/IAI.01389-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303:1532–1535. 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 32.Kaplan MJ, Radic M. 2012. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol 189:2689–2695. 10.4049/jimmunol.1201719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christie PJ. 2016. Classic spotlight: the awesome power of conjugation. J Bacteriol 198:372. 10.1128/JB.00955-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beaber JW, Hochhut B, Waldor MK. 2004. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 427:72–74. 10.1038/nature02241. [DOI] [PubMed] [Google Scholar]
- 35.Buberg ML, Witsø IL, L’Abée-Lund TM, Wasteson Y. 2020. Zinc and copper reduce conjugative transfer of resistance plasmids from extended-spectrum beta-lactamase-producing Escherichia coli. Microb Drug Resist 26:842–849. 10.1089/mdr.2019.0388. [DOI] [PubMed] [Google Scholar]
- 36.Tetz GV, Artemenko NK, Tetz VV. 2009. Effect of DNase and antibiotics on biofilm characteristics. Antimicrob Agents Chemother 53:1204–1209. 10.1128/AAC.00471-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Radman M. 1975. SOS repair hypothesis: phenomenology of an inducible DNA repair which is accompanied by mutagenesis. Basic Life Sci 5A:355–367. 10.1007/978-1-4684-2895-7_48. [DOI] [PubMed] [Google Scholar]
- 38.Michel B. 2005. After 30 years of study, the bacterial SOS response still surprises us. PLoS Biol 3:e255. 10.1371/journal.pbio.0030255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klein HL, Symington LS. 2019. Recognition for discoveries in DNA repair. N Engl J Med 381:677–679. 10.1056/NEJMcibr1907358. [DOI] [PubMed] [Google Scholar]
- 40.Song LY, Goff M, Davidian C, Mao Z, London M, Lam K, Yung M, Miller JH. 2016. Mutational consequences of ciprofloxacin in Escherichia coli. Antimicrob Agents Chemother 60:6165–6172. 10.1128/AAC.01415-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kohanski MA, DePristo MA, Collins JJ. 2010. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol Cell 37:311–320. 10.1016/j.molcel.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Händel N, Hoeksema M, Mata MF, Brul S, ter Kuile BH. 2016. Effects of stress, reactive oxygen species, and the SOS response on de novo acquisition of antibiotic resistance in Escherichia coli. Antimicrob Agents Chemother 60:677–679. 10.1128/AAC.02684-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blázquez J, Rodríguez-Beltrán J, Matic I. 2018. Antibiotic-induced genetic variation: how it arises and how it can be prevented. Annu Rev Microbiol 72:209–230. 10.1146/annurev-micro-090817-062139. [DOI] [PubMed] [Google Scholar]
- 44.Maiques E, Úbeda C, Campoy S, Salvador N, Lasa Í, Novick RP, Barbé J, Penadés JR. 2006. β-Lactam antibiotics induce the SOS response and horizontal transfer of virulence factors in Staphylococcus aureus. J Bacteriol 188:2726–2729. 10.1128/JB.188.7.2726-2729.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van der Veen S, van Schalkwijk S, Molenaar D, de Vos WM, Abee T, Wells-Bennik MH. 2010. The SOS response of Listeria monocytogenes is involved in stress resistance and mutagenesis. Microbiology (Reading) 156:374–384. 10.1099/mic.0.035196-0. [DOI] [PubMed] [Google Scholar]
- 46.Nautiyal A, Patil KN, Muniyappa K. 2014. Suramin is a potent and selective inhibitor of Mycobacterium tuberculosis RecA protein and the SOS response: RecA as a potential target for antibacterial drug discovery. J Antimicrob Chemother 69:1834–1843. 10.1093/jac/dku080. [DOI] [PubMed] [Google Scholar]
- 47.Oda Y. 1987. Induction of SOS responses in Escherichia coli by 5-fluorouracil. Mutat Res 183:103–108. 10.1016/0167-8817(87)90051-4. [DOI] [PubMed] [Google Scholar]
- 48.Vareille M, de Sablet T, Hindré T, Martin C, Gobert A. 2007. Nitric oxide inhibits Shiga-toxin synthesis by enterohemorrhagic Escherichia coli. Proc Natl Acad Sci U S A 104:10199–10204. 10.1073/pnas.0702589104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yakimov A, Pobegalov G, Bakhlanova I, Khodorkovskii M, Petukhov M, Baitin D. 2017. Blocking the RecA activity and SOS-response in bacteria with a short alpha-helical peptide. Nucleic Acids Res 45:9788–9796. 10.1093/nar/gkx687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alam MK, Alhhazmi A, DeCoteau JF, Luo Y, Geyer CR. 2016. RecA inhibitors potentiate antibiotic activity and block evolution of antibiotic resistance. Cell Chem Biol 23:381–391. 10.1016/j.chembiol.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 51.Chen Y, Hu D, Zhang Q, Liao X-P, Liu Y-H, Sun J. 2017. Efflux pump overexpression contributes to tigecycline heteroresistance in Salmonella enterica serovar Typhimurium. Front Cell Infect Microbiol 7:37. 10.3389/fcimb.2017.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang P, McElheny CL, Mettus RT, Shanks RMQ, Doi Y. 2017. Contribution of the TetB efflux pump to minocycline susceptibility among carbapenem-resistant Acinetobacter baumannii strains. Antimicrob Agents Chemother 61:e01176-17. 10.1128/AAC.01176-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosenblum R, Khan E, Gonzalez G, Hasan R, Schneiders T. 2011. Genetic regulation of the ramA locus and its expression in clinical isolates of Klebsiella pneumoniae. Int J Antimicrob Agents 38:39–45. 10.1016/j.ijantimicag.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nicoloff H, Perreten V, McMurry LM, Levy SB. 2006. Role for tandem duplication and lon protease in AcrAB-TolC-dependent multiple antibiotic resistance (Mar) in an Escherichia coli mutant without mutations in marRAB or acrRAB. J Bacteriol 188:4413–4423. 10.1128/JB.01502-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.El-Halfawy OM, Valvano MA. 2015. Antimicrobial heteroresistance: an emerging field in need of clarity. Clin Microbiol Rev 28:191–207. 10.1128/CMR.00058-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Band VI, Weiss DS. 2019. Heteroresistance: a cause of unexplained antibiotic treatment failure? PLoS Pathog 15:e1007726. 10.1371/journal.ppat.1007726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Y, Pan C, Ye L, Si Y, Bi C, Hua X, Yu Y, Zhu L, Wang H. 2020. Nonclassical biofilms induced by DNA breaks in Klebsiella pneumoniae. mSphere 5:e00336-20. 10.1128/mSphere.00336-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ng SMS, Sioson JSP, Yap JM, Ng FM, Ching HSV, Teo JWP, Jureen R, Hill J, Chia CSB. 2018. Repurposing zidovudine in combination with tigecycline for treating carbapenem-resistant Enterobacteriaceae infections. Eur J Clin Microbiol Infect Dis 37:141–148. 10.1007/s10096-017-3114-5. [DOI] [PubMed] [Google Scholar]
- 59.Barry A, Badal R. 1978. Stability of minocycline, doxycycline, and tetracycline stored in agar plates and microdilution trays. Curr Microbiol 1:33–36. 10.1007/BF02601704. [DOI] [Google Scholar]
- 60.Bieber D, Ramer S, Wu C-Y, Murray W, Tobe T, Fernandez R, Schoolnik G. 1998. Type IV pili, transient bacterial aggregates, and virulence of enteropathogenic Escherichia coli. Science 280:2114–2118. 10.1126/science.280.5372.2114. [DOI] [PubMed] [Google Scholar]
- 61.Milon A, Oswald E, De Rycke J. 1999. Rabbit EPEC: a model for the study of enteropathogenic Escherichia coli. Vet Res 30:203–219. [PubMed] [Google Scholar]
- 62.Crameri A, Whitehorn EA, Tate E, Stemmer WP. 1996. Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat Biotechnol 14:315–319. 10.1038/nbt0396-315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download AAC.00013-21-s0001.pdf, PDF file, 0.05 MB (50.6KB, pdf)