

PURPOSE
Homologous recombination DNA repair deficiency (HRD) is associated with sensitivity to platinum and poly (ADP-ribose) polymerase inhibitors in certain cancer types, including breast, ovarian, pancreatic, and prostate. In these cancers, BRCA1/2 alterations and genomic scar signatures are useful indicators for assessing HRD. However, alterations in other homologous recombination repair (HRR)-related genes and their clinical significance in other cancer types have not been adequately and systematically investigated.
METHODS
We obtained data sets of all solid tumors in The Cancer Genome Atlas and Cancer Cell Line Encyclopedia, and comprehensively analyzed HRR pathway gene alterations, their loss-of-heterozygosity status, and per-sample genomic scar scores, that is, the HRD score and mutational signature 3 ratio, DNA methylation profiles, gene expression profiles, somatic TP53 mutations, sex, and clinical or in vitro response to chemical exposure.
RESULTS
Biallelic alterations in HRR genes other than BRCA1/2 were also associated with elevated genomic scar scores. The association between HRR-related gene alterations and genomic scar scores differed significantly by sex and the presence of somatic TP53 mutations. HRD tumors determined by a combination of indices also showed HRD features in gene expression analysis and exhibited significantly higher sensitivity to DNA-damaging agents than non-HRD cases in both clinical samples and cell lines.
CONCLUSION
This study provides evidence for the usefulness of HRD analysis in all cancer types, improves chemotherapy decision making and its efficacy in clinical settings, and represents a substantial advancement in precision oncology.

INTRODUCTION
Homologous recombination repair (HRR) is one of the most accurate DNA repair mechanisms for DNA double-strand breaks. Disruption of this mechanism (homologous recombination deficiency [HRD]) leads to a high degree of genetic instability and accumulation of genetic mutations, thus playing an important role in the development and progression of cancer. Thus far, mutations in the BRCA1 and BRCA2 genes are considered principal drivers of HRD1; germline BRCA1/2 mutation carriers more frequently develop BRCA-associated cancers, that is, those of the ovaries, breasts, prostate, and pancreas.2 Both germline and somatic BRCA1/2 mutations have demonstrated high sensitivity to DNA-damaging drugs such as platinum, doxorubicin, and topoisomerase inhibitors.1 The discovery and reproducibility of synthetic lethality in BRCA1/2-mutated cancers by poly (ADP-ribose) polymerase (PARP) inhibitors in 20053,4 has led to recent successive US Food and Drug Administration approvals of PARP inhibitors for BRCA-associated cancers.5,6
CONTEXT
Key Objective
Genomic alterations in BRCA1/2 and genomic scar signatures are associated with homologous recombination DNA repair deficiency (HRD) and serve as therapeutic biomarkers for platinum and poly (ADP-ribose) polymerase inhibitors in breast and ovarian cancers. However, pan-cancer significance of scar scores and mutations in other homologous recombination repair–related genes is not fully understood.
Knowledge Generated
Analysis of solid cancers in The Cancer Genome Atlas and Cancer Cell Line Encyclopedia demonstrated that association between biallelic alterations in homologous recombination pathway genes and genomic scar signatures differed depending on sex and the presence of somatic TP53 mutations. HRD cases identified by these indicators showed higher sensitivity to DNA-damaging drugs both in clinical samples and cell lines.
Relevance
This work provides evidence of the utility of HRD analysis for all cancer types and will improve tumor molecular diagnostics as well as chemotherapy selection in clinical oncology.
In experimental studies, the suppression of HRR pathway signaling confers HRD properties in various cancers,7 and thus, gene mutations in the HRR pathway have been considered useful in predicting drug sensitivity associated with HRD. However, in a clinical setting, HRR pathway mutations other than BRCA1/2 have not been sufficiently proven to be useful.8 Moreover, the efficacy of PARP inhibitors in non–BRCA-associated cancers remains low, even in patients with BRCA1/2 mutations.9-11
Alternative methods for assessing HRD status have been developed only in the past decade, following the large-scale accumulation and analysis of sequencing data.12,13 The HRD score,14 which quantifies chromosomal structural abnormalities by combining telomere allelic imbalances,15 large state transitions,16 and loss of heterozygosity (LOH),17 and the mutational signature 3 (Sig3) ratio, which quantifies the characteristic pattern of somatic mutations in HRD tumors, are referred to as genomic scar signatures.18,19 In ovarian and breast cancers, tumors with high scores for these indicators exhibit high sensitivity to platinum and PARP inhibitors, even in the absence of BRCA1/2 mutations.20
Tumor suppressor genes typically require not only a loss-of-function mutation in a single allele but rather a complete loss of the suppressive wild-type allele21 to cause tumorigenesis. Recently, integrated analysis of allele-specific copy-number changes and genomic scar signatures revealed that BRCA1/2 mutations require a loss of the nonmutated allele, termed locus-specific LOH, to acquire functional significance.22,23 However, these studies mainly focused on BRCA1/2 mutations in BRCA-associated cancers; other cancers have been insufficiently analyzed. Furthermore, the association between zygosity status and genomic scar signatures in HRR pathway gene mutations other than BRCA1/2 has yet to be investigated in detail.
With the increasingly widespread use of gene panel and sequencing-based testing for personalized medicine, it is important to comprehensively evaluate the significance of HRR pathway gene alterations and genomic scar signatures in a pan-cancer context. Here, we systematically evaluate biallelic HRR pathway gene alterations and genomic scar signatures in all solid cancers from The Cancer Genome Atlas (TCGA) and the Cancer Cell Line Encyclopedia via an ensemble of analytical techniques. We further assess the efficacy of DNA-damaging drugs in HRD cases, both in in vitro and clinical contexts.
METHODS
See the Data Supplement.
RESULTS
Correlations Between HRR Pathway Gene Alterations and Genomic Scar Signatures
The HRD score and the Sig3 ratio were used as the measures of genomic scarring associated with HRD. Combining results from the ASCAT24 and FACETS25 algorithms, which classify input into LOH-positive, LOH-negative, or undetermined statuses, allele-specific copy numbers were factored in at each locus in 29 selected HRR pathway genes to determine whether genes were classified as having locus-specific LOH (see Methods). Until stated otherwise, analyses focus on the TCGA data set.
We examined the relationship between the genomic scar scores and the status of mutations with locus-specific LOH in HRR pathway genes; specifically, we considered the following six stratifications: germline(g)BRCA1, gBRCA2, gHRR (all HRR pathway genes excluding BRCA1/2, n = 27), somatic(s)BRCA1, sBRCA2, and sHRR (Figs 1A and 1B). When all samples were arranged in order of resultant HRD score, high-scoring cases had demonstrably more frequent mutations with LOH, whereas mutations without LOH had systemically lower HRD scores; this trend was consistent in all six stratifications (Fig 1A). The same trend was observed for the Sig3 ratio (Fig 1B). Both the HRD score and the Sig3 ratio were statistically significantly higher in cases with LOH mutations than in cases with non-LOH mutations (Figs 1A and 1B box plots). For individual HRR pathway genes other than BRCA1/2, cases with LOH-positive mutations in gATM, gBRIP1, gFANCM, gPALB2, gRAD51C, sATM, sCDK12, and sFANCD2 were enriched with high HRD score or Sig3 ratio (statistically significant), whereas mutations at these loci not classified as LOH were not enriched (Data Supplement). Mutations in these LOH-detected genes have been previously reported to be found in tumors with molecular features similar to BRCA-mutant tumors, known as BRCAness.1
FIG 1.
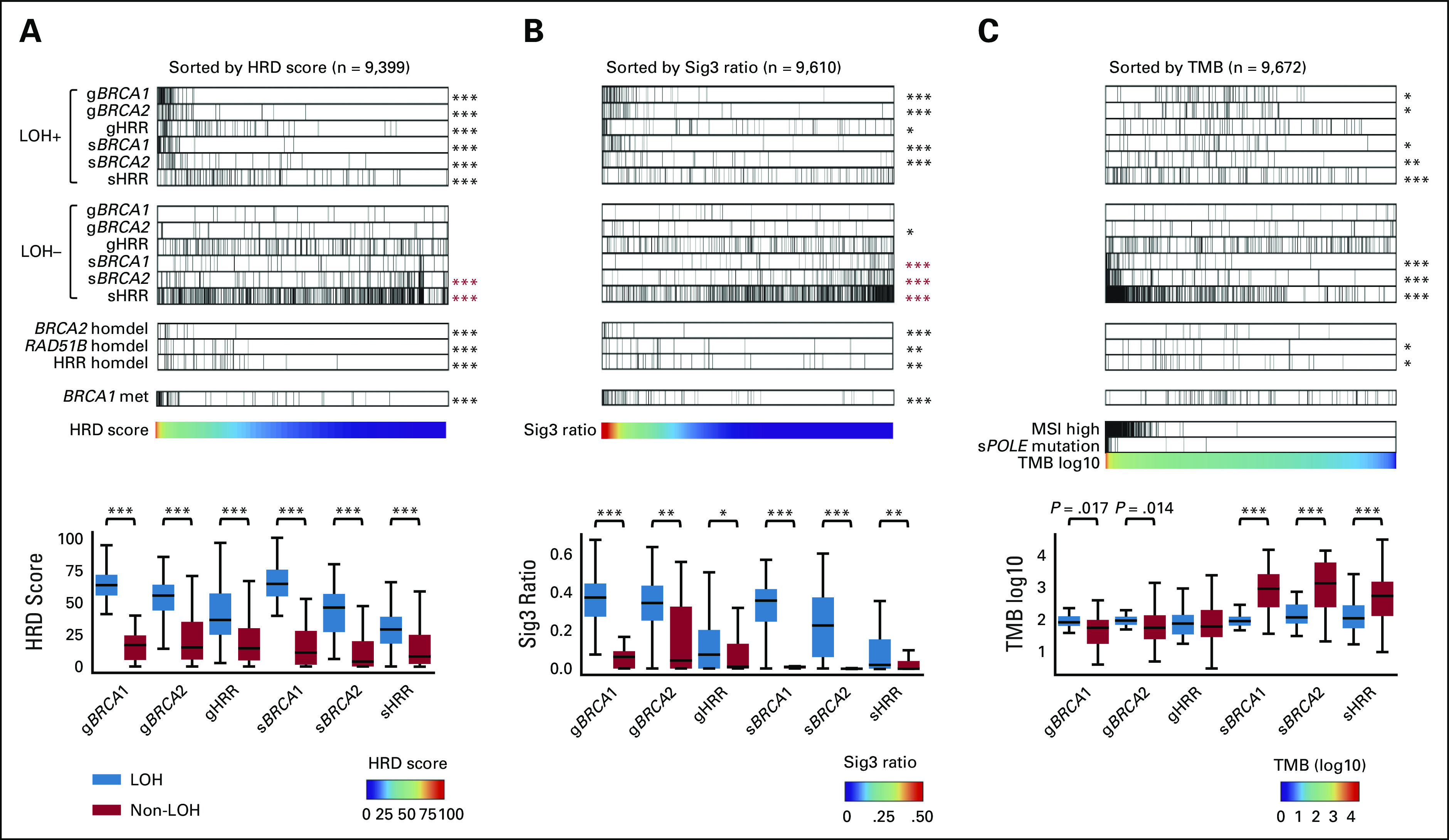
Association between HRR pathway gene alterations and genomic scar scores or TMB. Solid cancer samples from The Cancer Genome Atlas were arranged in order of (A) HRD score, (B) Sig3 ratio, and (C) TMB, respectively. The top and second panels show the distribution of mutations, respectively, with and without locus-specific LOH, stratified by germline (prefix g) or somatic (prefix s), and BRCA1, BRCA2, or non-BRCA HRR pathway genes. The third group of panels show homozygous deletions in BRCA2, RAD51B, and HRR pathway genes (excluding BRCA1/2 but including RAD51B), and a fourth panel shows BRCA1 methylation. The fifth panels contain the scar scores or TMB values used for ordering; for TMB-based analysis, samples are also annotated with MSI-high and somatic POLE mutation statuses. Boxplots in bottom panels assess the distribution of values for each stratification. *P < .01, **P < 1 × 10–4, and ***P < 1 × 10–6 in the Mann-Whitney U test. Asterisks in red color indicate a negative correlation with the corresponding signature value. HRD, homologous recombination DNA repair deficiency; HRR, homologous recombination repair; LOH, loss of heterozygosity; MSI, microsatellite instability; Sig3, signature 3; TMB, tumor mutation burden.
Alternatively, when all of the cases were arranged in order of tumor mutational burden, cases with LOH-negative sBRCA1/2 and sHRR mutations had systemically larger tumor mutation burden, including microsatellite instability-high and POLE-mutated cases (Fig 1C). The result suggests that these mutations are neutral, passenger mutations (Fig 1C, box plot). In gBRCA1/2, cases with LOH mutations had higher tumor mutational burden on average than cases with non-LOH mutations (gBRCA1 P = .017, gBRCA2 P = .014, Fig 1C box plot), which was consistent with previous reports that HRD tumors had moderately increased numbers of gene mutations.26,27
The relationship between homozygous deletions, HRD scores, and Sig3 ratios was investigated by using the ASCAT24 and ABSOLUTE28 methods independently (see Methods). Cases with BRCA2 homozygous deletion had higher genomic scar scores and lower gene expression (Figs 1A and 1B, and Data Supplement). RAD51B homozygous deletion was found in 20 patients with high genomic scar scores and reduced gene expression (Figs 1A and 1B, Data Supplement). Although RAD51B mutations have been reported in a few cases in breast cancer, their pathogenic significance remains controversial.29 To the best of our knowledge, there have been no reports on homozygous deletion in RAD51B.
Regarding the association between the genomic scar scores and promoter methylation of HRR pathway genes, only BRCA1 was significantly enriched in cases with a higher HRD score and Sig3 ratio (Figs 1A and 1B, Data Supplement, see Methods). Thus, subsequent analyses considered methylation status only for BRCA1.
Per-Cancer Differences in HRD Status, LOH, and Alteration Type
We next examined the mutation rate of HRR pathway genes and the frequency of locus-specific LOH for each cancer type. Consistent with previous reports,22,23,30 BRCA1/2 mutations tended to show higher LOH ratios in BRCA-associated cancers, that is, ovarian, breast, pancreatic, and prostate cancers. However, the mean HRD score and the Sig3 ratio were not particularly higher in breast, pancreatic, or prostate cancers compared with other cancers (Fig 2A). Similarly, the frequency of HRR pathway gene alterations was not much higher in pancreatic and prostate cancers (Fig 2B). After ovarian cancer (OV), the next highest frequency of HRR alterations was observed in testicular germ cell tumors, in which most of the alterations were derived from BRCA1 methylation. Biallelic alterations in ATM were observed the second most frequently after BRCA1/2 (Fig 2B).
FIG 2.
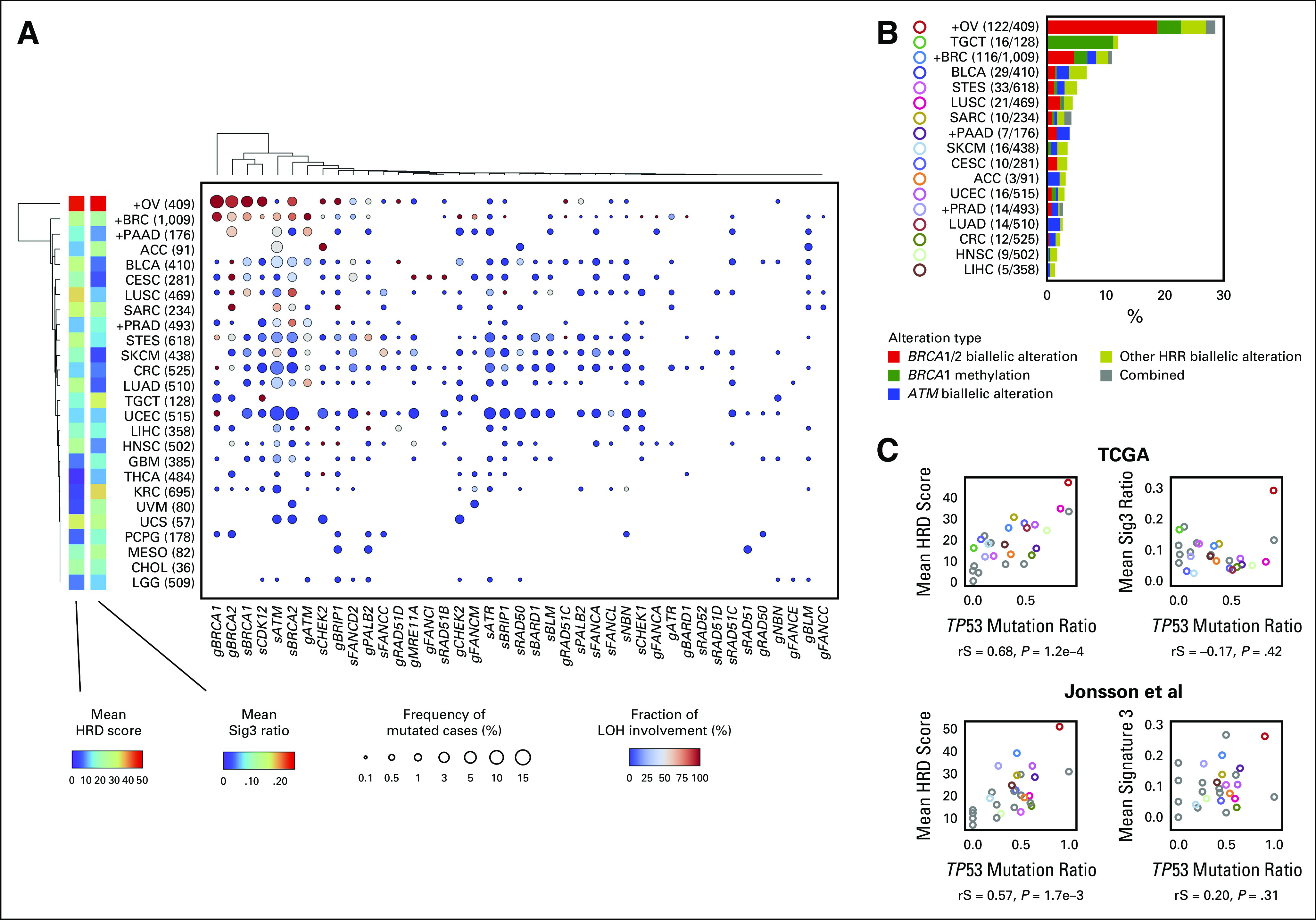
Detailed HRR pathway gene alterations per cancer type. (A) For each cancer type and HRR pathway gene, the percentage of samples with gene mutation detected and with LOH detected are annotated using size and color. The product of these two values is input to hierarchical clustering to generate the resulting type-gene ordering. Cancer types annotated with a + mark are BRCA-associated cancers, and values indicate number of cases in TCGA. (B) Types and proportions of biallelic HRR pathway gene alterations per cancer type. (C) Correlation between TP53 mutation ratio with mean HRD score (left) and mean Sig3 ratio (right) by cancer type. Upper and lower panels are from the TCGA and Jonsson et al data sets, respectively. Colors are identical to B, or otherwise gray. rS and P represent the Spearman correlation coefficient and its P value, respectively. HRD, homologous recombination DNA repair deficiency; HRR, homologous recombination repair; LOH, loss of heterozygosity; Sig3, signature 3; TCGA, The Cancer Genome Atlas.
We found a strong positive correlation between the mean HRD score per cancer type and the type's TP53 mutation ratio (rS = 0.68, P = 1.2 × 10–4, Fig 2C), whereas there was no correlation between the mean Sig3 ratio and TP53 mutation ratio. These results were similarly observed when applied to an external multi-cancer data set reported by Jonsson et al.23
Identification of Pan-Cancer HRD Cases Based on Genomic Scar Signatures
The number of carriers of germline BRCA1/2 variants is equal in women and men,31 although the lifetime cancer risk is higher in women.32 Therefore, we hypothesized that the effect of HRD on cancer development could also be sex-dependent. We evaluated the association between genomic scar scores and HRR pathway alterations, with patients split by sex and TP53 mutation status. Cases with BRCA1/2 alterations, meaning germline and somatic BRCA1/2 biallelic alteration plus BRCA1 methylation, had the highest average HRD and Sig3 values in females with TP53 mutations (Fig 3A, left). The results were also similar in cases with HRR pathway gene alterations (HA cases), meaning germline and somatic biallelic alteration in HRR pathway genes and BRCA1 methylation (Fig 3A, middle). Trends were mirrored in the Jonsson et al data set23 (Fig 3A, right). Even when excluding BRCA-associated cancers, similar trends were observed (Data Supplement). By contrast, even in the group without HRR alterations, the HRD score was elevated in the presence of TP53 mutation (Fig 3A middle). These results indicate that stratification by sex and the presence of TP53 mutation enriches the dimensionality of information applicable to the identification of HRD cases based on genomic scar scores. A systematic analysis for each TCGA cancer type is provided in the Data Supplement.
FIG 3.
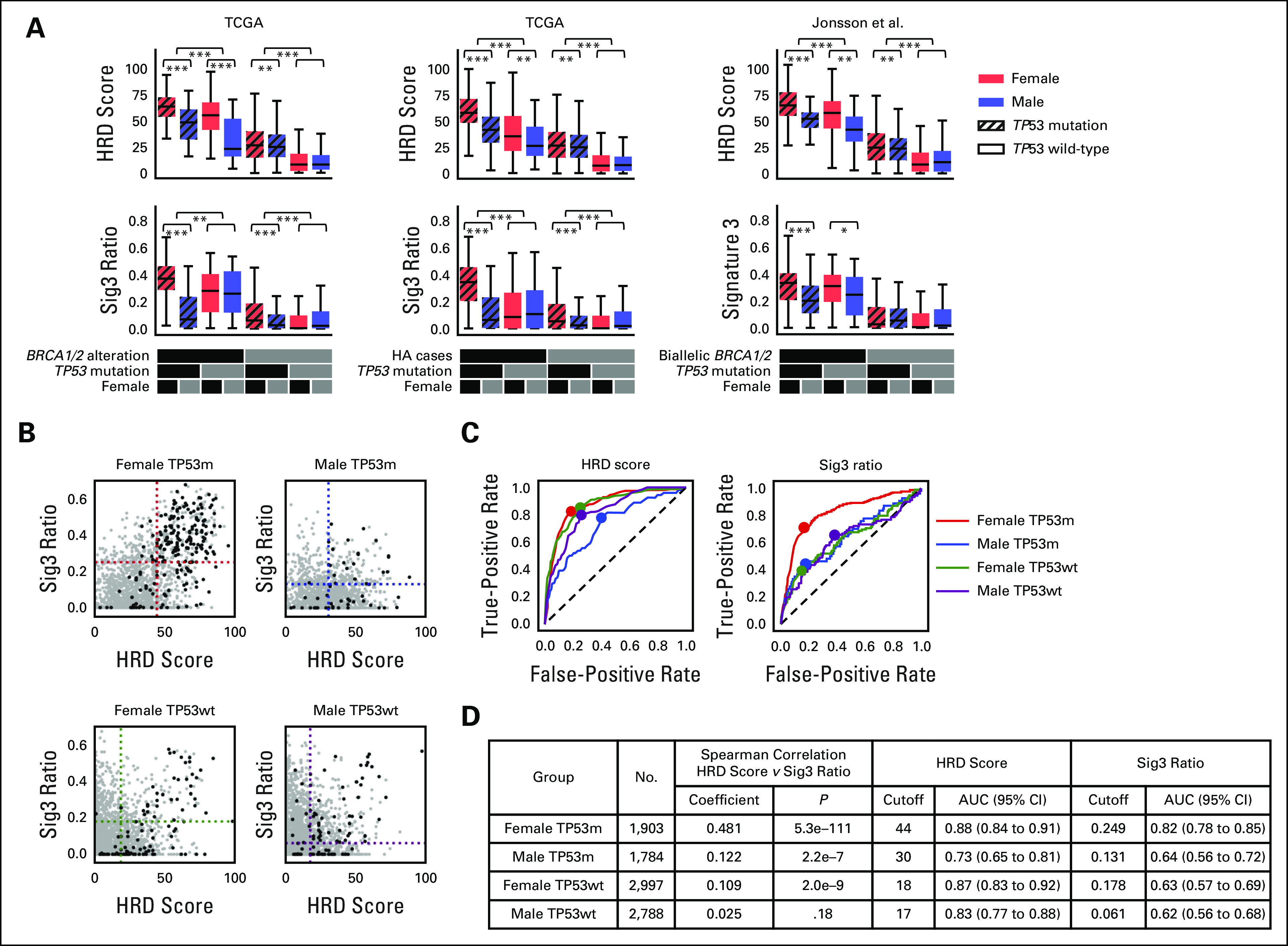
Systematization and results for genomic scar score-based classification. (A) Comparison of HRD score and Sig3 ratio stratified by TP53 mutation and sex. Further stratification is applied using biallelic BRCA1/2 alteration (including BRCA1 methylation, left), biallelic HRR pathway gene alteration (middle), and biallelic BRCA1/2 alteration in Jonsson's data set (right). *P < .05, **P < .01, and ***P < .001 in the Mann-Whitney U test. (B) HRD score and Sig3 ratio pairwise distribution when patients are divided by sex and TP53 mutation status. Black dots represent HRR pathway gene-altered cases and gray dots indicate nonaltered cases. (C) Dotted lines are placed at the optimal cutoff values determined by ROC curve analysis. (C) Differences in ROC curves and optimal cutoff values for HRR pathway gene alterations in HRD score and Sig3 ratio among the four groups. (D) Per stratification, Spearman correlation coefficients between HRD score and Sig3 ratio, optimized scar score cutoff values, and resulting AUCs are shown. AUC, area under the curve; HRD, homologous recombination DNA repair deficiency; HA, HRR-altered; HRR, homologous recombination repair; ROC, receiver operating characteristic; Sig3, signature 3; TCGA, The Cancer Genome Atlas.
The positive correlation between HRD score and Sig3 ratio was strongest in females with TP53 mutations, weaker in males with TP53 mutations and females without TP53 mutations, and not statistically significant in males without TP53 mutations (Fig 3B, rS = 0.481, 0.122, and 0.109, 0.025, respectively). Analogous results were observed even when excluding BRCA-associated cancers (Data Supplement). For each of the sex-mutation groups, we analyzed receiver operating characteristic curves to systematically extract optimal HRD and Sig3 cutoff values for determining HRR-altered (HA) cases (Fig 3C). As expected from the stratified score distributions, the optimal cutoffs and the areas under the curve differed among the four groups (Fig 3D). For each group, the cases that exceeded both cutoff values were defined as genomic scar high (GS) cases and analyzed below.
Gene Mutation and Expression Analyses in GS and HA Cases
The number of GS and/or HA cases was between 187 and 357 in each of the four patient groups (Fig 4A, details of cancer type stratification are in the Data Supplement). The biallelic ATM alteration ratio was lower in the TP53-mutated group than in the TP53 wild-type group; ATM and TP53 mutation were significantly mutually exclusive (Fig 4A, 0.19% and 1.2%, respectively, chi-square test P = 1.4 × 10–7).
FIG 4.
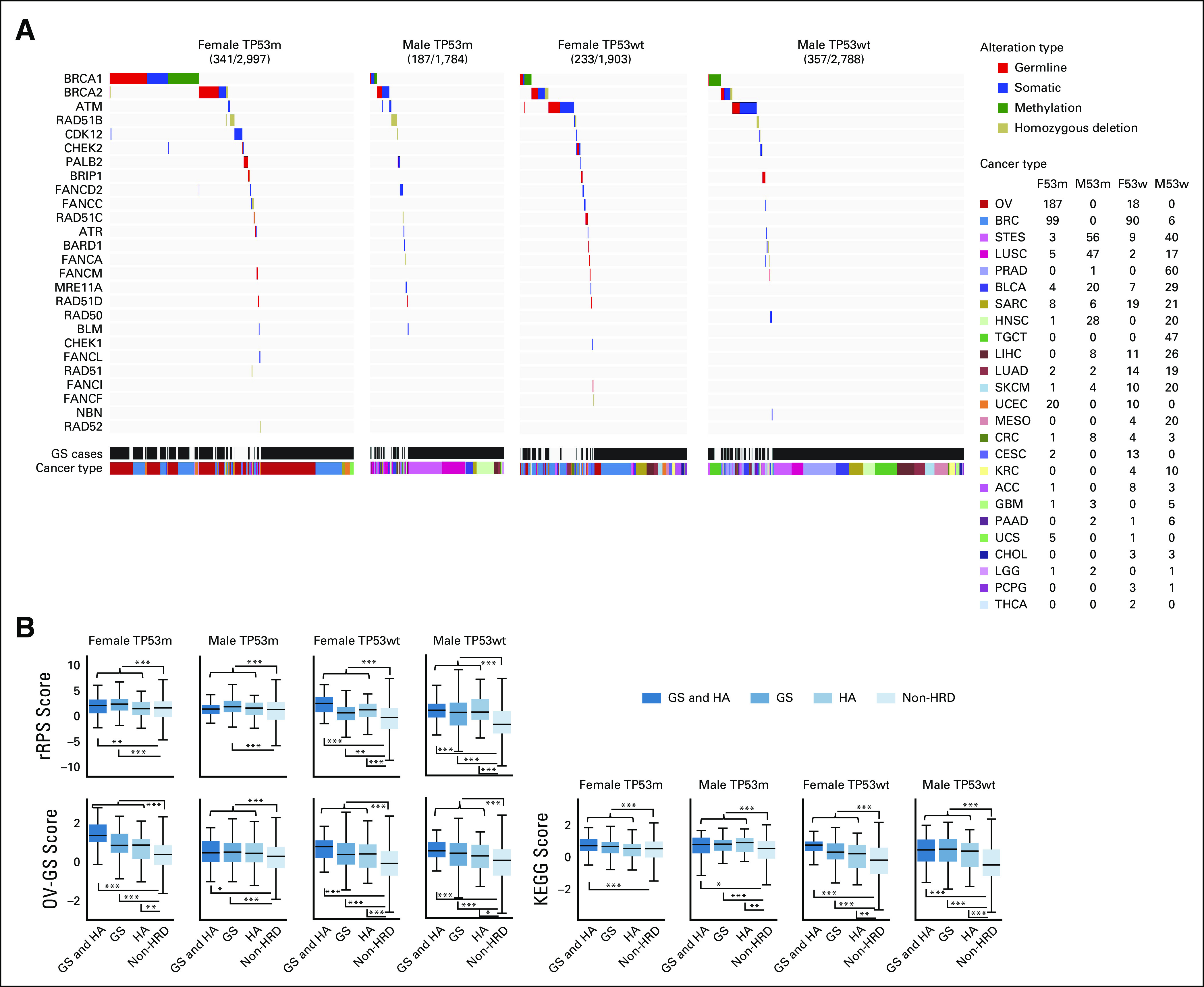
Characteristics of HA cases and GS score high cases. (A) Per-patient HRR alteration type analysis in HA and/or GS cases, following stratification by sex and TP53 mutation status. Numbers in parenthesis next to cancer types represent the number of cases in the four groups, respectively. (B) Comparisons of three gene expression–based HRD assessment scores after stratification by HA and GS. These scores were calculated from modification of the RPS, comparison between The Cancer Genome Atlas OV samples, and KEGG pathway database, respectively (see Methods). *P < .05, **P < .01, and ***P < .001 in the Mann-Whitney U test. GS, genomic scar; HA, HRR-altered; HRD, homologous recombination DNA repair deficiency; HRR, homologous recombination repair; rRPS, reversed Recombination Proficiency Score; OV, ovarian cancer.
GS and/or HA cases had significantly higher gene expression–based HRD-related scores, where we tested the reversed Recombination Proficiency Score,33 OV-GS, and KEGG scores (Fig 4B, see Methods), indicating that they have HRD features in terms of gene expression profiles. GS and/or HA cases are hereafter referred to as HRD cases.
To investigate sex differences in HRD status, we examined the association between X-chromosome inactivation and HRD. Female HRD tumors had significantly lower XIST expression, lower global promoter methylation of the X chromosome, and higher expression scores of all genes on the X chromosome, whereas scores for a subset of genes reported to escape X chromosome inactivation34 were not significantly elevated (Data Supplement, see Methods). These differences were consistent even when BRCA-associated cancers were excluded (Data Supplement), suggesting that X-chromosome inactivation may underlie differences in HRD by sex.
Association Between Chemotherapeutic Agents and Survival Outcomes in HRD Cases
To investigate the drug sensitivity of HRD cases, all TCGA cases were divided into one of two groups either with (n = 2,979) or without (n = 6,460) a treatment history of DNA-damaging agents, including alkylating agents, antibiotics, antimetabolites, platinum, and topoisomerases. While HRD cases had a significantly better overall survival than non-HRD cases in the group with DNA-damaging agent use, HRD cases in the group without DNA-damaging treatment had a significantly worse outcome than non-HRD cases (Fig 5A, log-rank test P = 5.1 × 10–4 and 1.1 × 10–10, respectively).
FIG 5.
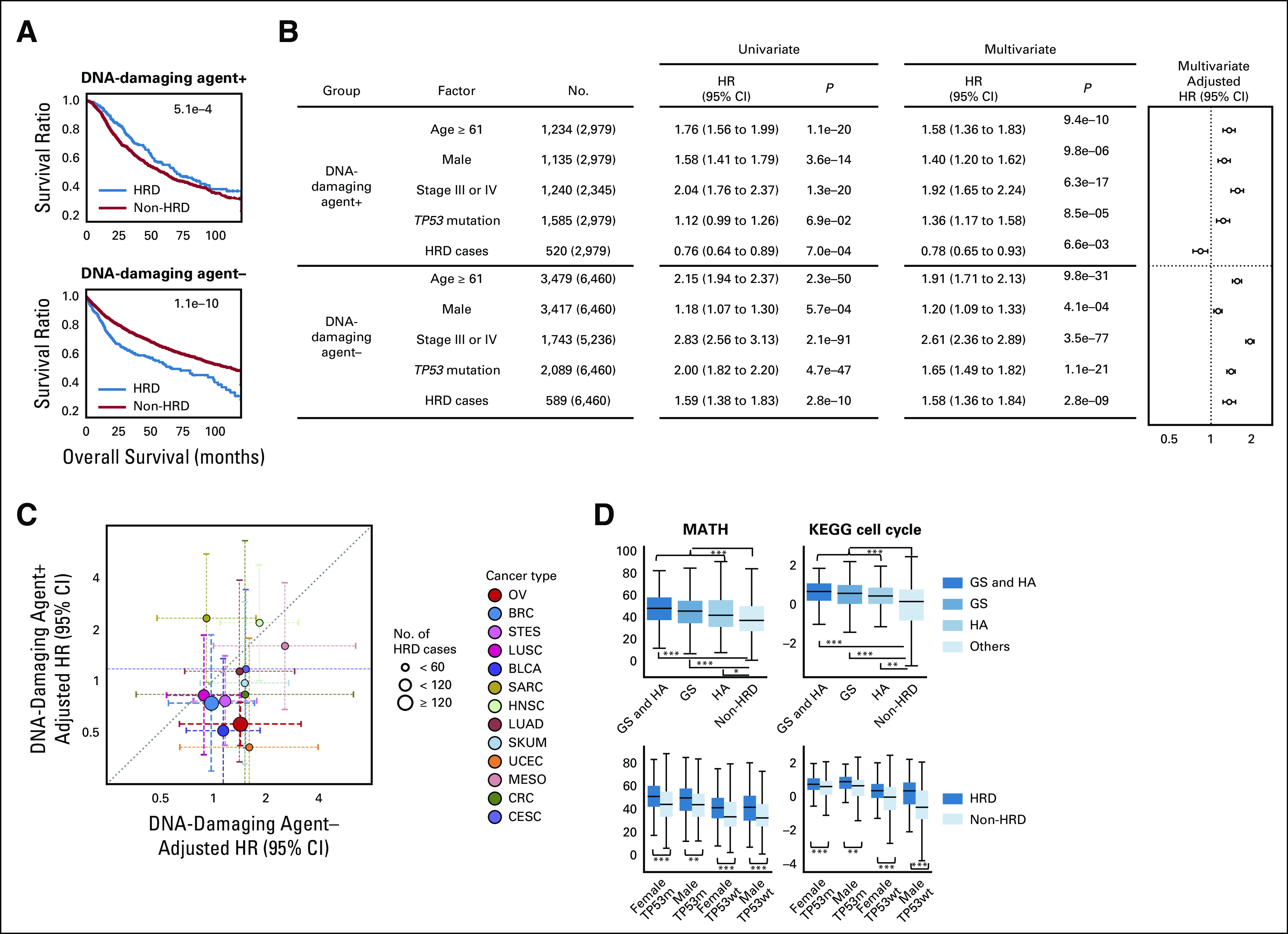
Survival analysis stratified by HRD status and DNA-damaging chemotherapy. (A) Comparison of overall survival between HRD and non-HRD cases, dividing patient groups by the presence or absence of DNA-damaging chemotherapy. The P value of the log-rank test is given. (B) Left: univariate and multivariate Cox proportional hazard model analyses with covariates of age, sex, TP53 mutation, clinical stage, and HRD assignment. Right: adjusted HRs from the multivariate Cox analysis. (C) For HRD-present cases, comparison of the two adjusted HRs in each of 13 cancer types that have five or more comparable cases. Identical to B's pan-cancer analysis, per-type samples were divided based on DNA-damaging chemotherapy presence or absence. (D) Comparisons of MATH index and KEGG cell cycle scores with stratifications similar to Figures 3 and 4. *P < .01, **P < 1 × 10–5, and ***P < 1 × 10–7 in the Mann-Whitney U test. GS, genomic scar; HA, HRR-altered; HR, hazard ratio; HRD, homologous recombination DNA repair deficiency.
Cox proportional hazard multivariate analysis with covariates of age, sex, stage, and TP53 mutation showed that HRD was a good independent prognostic factor in the DNA-damaging treatment group (adjusted hazard ratio [HR] 0.78, P = 6.6 × 10–3), whereas it was a poor factor in the nontreatment group (adjusted HR 1.58, P = 2.8 × 10–9; Fig 5B). Similar analyses were performed for each of the 13 cancer types that contained five or more comparable cases in both therapy groups. For 11 of 13 types (85%), the adjusted HRs of HRD were lower in the DNA-damaging agent group than in the nonagent group (Fig 5C, Data Supplement). These results suggest that DNA-damaging agents would improve the prognosis of HRD cases, regardless of the cancer type.
The MATH index,35 an indicator of intratumor heterogeneity, and KEGG cell cycle score calculated by single sample Gene Set Enrichment analysis (ssGSEA)36 were higher in HRD cases than in non-HRD cases (Fig 5D, see Methods). These results suggest that increased intratumor heterogeneity and proliferation capacity associated with HRD may lead to poor survival outcomes in the absence of DNA-damaging drugs.
We repeated analysis without applying the stratification by sex and TP53 mutation status, obtaining agnostic cutoff values for the HRD score and the Sig3 ratio (Data Supplement). Although the associations between gene expression profiles, drug administration, and survival outcomes of HRD cases showed trends similar to those with the stratification, the characteristics of the determined HRD cases, such as differences in HRs and P values in the survival analysis, were weaker (eg, adjusted HRs 1.58-v-0.78 stratified, 1.37-v-0.83 unstratified, Data Supplement).
High Sensitivity of DNA-Damaging Agents to HRD Tumor Cell Lines
The analyses described above were applied verbatim to data sets of human cancer cell lines from the Cancer Cell Line Encyclopedia, demonstrating similar trends in the associations between HRR pathway gene mutations and locus-specific LOH, TP53 mutations, sex, and genomic scar signatures (Data Supplement). The IC50 values of 198 compounds for these cell lines were analyzed using data from the Genomics of Drug Sensitivity in Cancer, where DNA-damaging drugs showed higher sensitivity to HRD tumors (Data Supplement, Fig 6A). Furthermore, DNA-damaging drugs showed higher sensitivity to HRD tumors compared with drugs targeting other mechanisms of action (eg, kinase inhibitors, Fig 6B).
FIG 6.
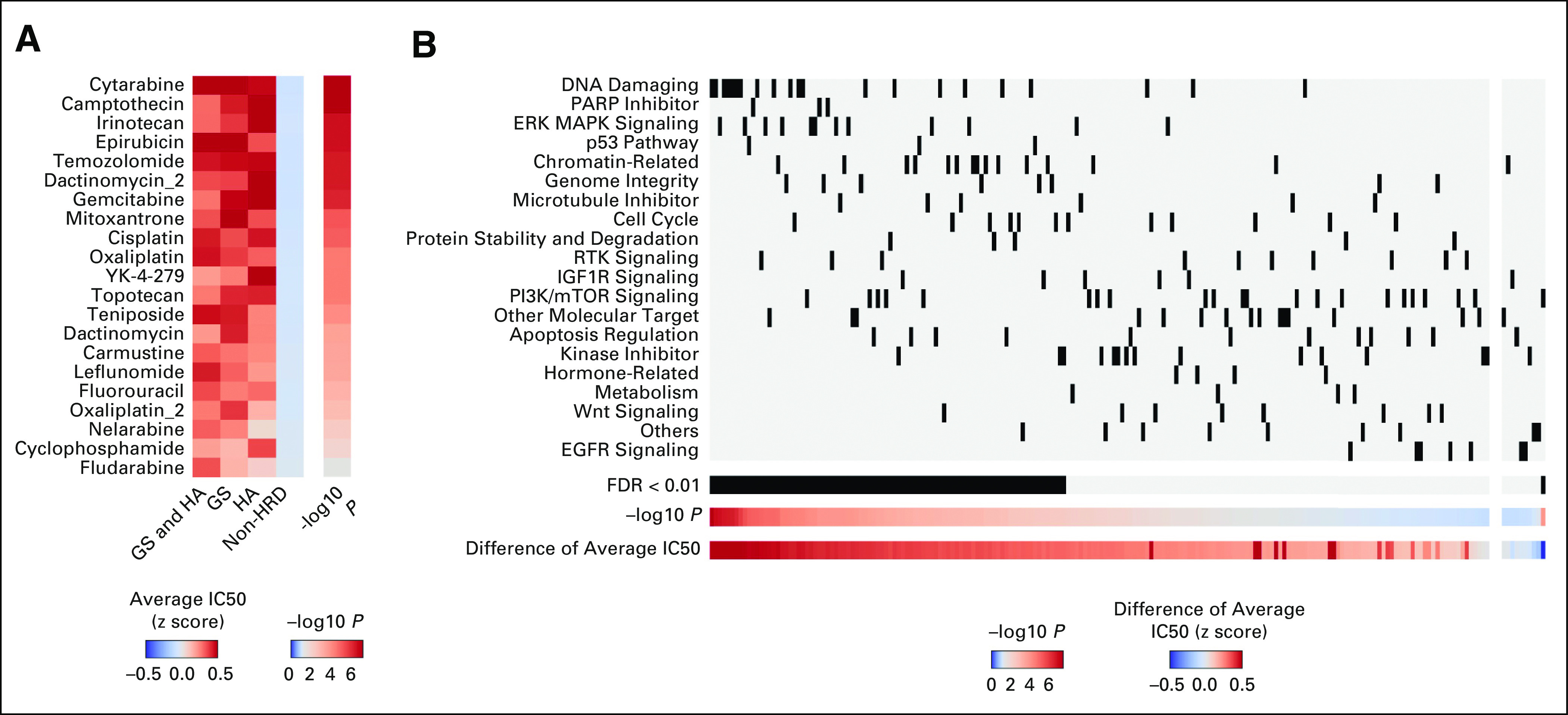
Association between HRD status and chemotherapy sensitivity in Cancer Cell Line Encyclopedia cell lines (n = 741). (A) HRD cell lines exhibit increased inhibition in response to DNA-damaging chemotherapy than non-HRD samples. The left heatmap indicates the average z-scaled IC50 value for each group, with accompanying Mann-Whitney test P values in the right heatmap, which is color-centered at P = .05. (B) Association between drug sensitivity to HRD cell lines and drug mechanism of action. IC50 values of each of 198 compounds from the Genomics of Drug Sensitivity in Cancer database were compared between HRD and non-HRD groups. In the left panels, compounds with higher sensitivity to HRD samples are arranged in decreasing order of Mann-Whitney test P value (log-transformed) on the horizontal axis, whereas in the right panels, those with higher sensitivity to non-HRD samples are in order of increasing P value. Drug categories were vertically ordered top-to-bottom by decreasing average P value (also log-transformed). Among the drugs that showed significantly higher sensitivity to HRD samples (FDR < 0.01, using Benjamini-Hochberg method), DNA-damaging agents were the most prevalent. EGFR, epidermal growth factor receptor; ERK MAPK, extracellular regulated kinase mitogen-activated protein kinase; FDR, false discovery rate; GS, genomic scar; HA, HRR-altered; HRD, homologous recombination DNA repair deficiency; mTOR, mammalian target of rapamycin; PARP, poly (ADP-ribose) polymerase; RTK, receptor tyrosine kinase.
DISCUSSION
Recently, several clinical trials of PARP inhibitors have been conducted in which HRR pathway gene mutations have been examined as biomarkers (eg, niraparib plus carboplatin in patients with homologous recombination-deficient advanced solid tumor malignancies, NCT03209401, talazoparib in treating patients with HRRD-positive recurrent stage IV squamous cell lung cancer, NCT03377556, and study of olaparib (MK-7339) in combination with pembrolizumab (MK-3475) in the treatment of homologous recombination-repair mutation (HRRm) and/or homologous recombination deficiency (HRD)-positive advanced cancer (MK-7339-007/KEYLYNK-007), NCT04123366). However, few have assessed whether these mutations are biallelic or not. Our findings here indicate that the zygosity of HRR variants is a very important consideration in future clinical trials and that robust molecular-scale technologies for determining the zygosity of these variants need to be further developed.
While one study reported a benefit of PARP inhibitors in BRCA-associated cancers even with monoallelic BRCA1/2 alterations,23 another study showed that tumors with germline BRCA1/2 mutations but without locus-specific LOH have a low HRD score and low drug sensitivity.22 Considering that these studies each used only one algorithm, the differences in the conclusions of those reports may be because of differences in methods. In this study, LOH and homozygous deletion were each determined using pairs of methods. These integrated analyses provided a clearer association between biallelic changes and genomic scar scores than when only a single method was used (Data Supplement), showing that using ensembles of algorithms can improve robustness.
The HRD score was higher in tumors with TP53 mutations (Fig 2C). Some have reported that TP53 mutations activate the HRR pathway under certain circumstances,37 whereas others suggest that they did not directly associate with HRD.38,39 In our data, TP53 mutation did not correlate with the Sig3 ratio (Fig 2C), whereas TP53 mutated cases, even without HRR alteration, had higher HRD scores than TP53 wild-type cases (Fig 3A). Recently, in prostate cancer, TP53 mutations were reported to be associated with increased HRD scores independently of HRR pathway gene alterations.40 This suggests that high HRD score in TP53-mutated cases could alternatively be because of chromosomal instability caused by mechanisms other than HRD (eg, loss of cell-cycle checkpoint mechanisms).
We observed that not only the two genomic scar scores themselves, but also their correlations and area under the curve cutoffs for classification differed greatly depending on sex and TP53 mutation status (Fig 3). Based on these observations, we would propose that differences in sex and TP53 should be taken into account when using these genomic scores to determine HRD cases. However, since the HRD score and the Sig3 ratio were originally developed in a rather limited analysis to identify BRCA1/2 mutant tumors in breast and ovarian cancers,15-19 it may be difficult to evaluate tumors in groups other than women with TP53 mutations. Given that large-scale multiomics databases are being constructed worldwide and that new methods for evaluating HRD using whole-genome sequencing are emerging,41,42 it is expected that more robust methods for identifying HRD cases in pan-cancer contexts will be developed in the future.
While the data of our pan-cancer analysis suggest that HRD involvement in tumors may differ by sex, the very small number of samples with HRR alterations in non–BRCA-associated cancers made it difficult to compare by cancer type at high resolution (Data Supplement). Given the relative prevalence of HRD in female tumors such as ovarian and breast cancers, it is also possible that differences in tumor type, not related to sex, may have an effect. By contrast, one of the major differences between the cells of men and women is the presence of X-chromosome inactivation. The loss of X-chromosome inactivation in some female cancers has long been known as the loss of the Barr body.43 It is reported that some molecules belonging to the DNA double-strand break repair pathway are involved in X-chromosome inactivation.44-46 In the present analysis, we found that the loss of X-chromosome inactivation was associated with female HRD tumor cells (Data Supplement). This suggests that the relatively high frequency of HRD in female tumors may be related to inactivation of the X chromosome. However, there have been no reports on the association between the genomic scar-based HRD status and the X chromosome; thus, more detailed studies are needed in the future.
This comprehensive pan-solid cancer HRD analysis revealed that stratified analysis by sex and TP53 mutation status can more accurately assess HRD status, suggesting that HRD is useful as a cancer type-agnostic therapeutic biomarker, and that DNA-damaging drugs could be beneficial for HRD cases across cancer types. Based on the results, it is rational to expect improvements in the implementation of personalized cancer medicine based on HRD. In parallel, further diagnostic methods can be developed for the pinpoint identification of clinically significant HRD cases.
J.B. Brown
Employment: Boehringer Ingelheim
Honoraria: Kyowa Kirin International, Daiichi Sankyo RD Novare
Research Funding: Daiichi Sankyo RD Novare
Travel, Accommodations, Expenses: Boehringer Ingelheim, Kyowa Kirin International
Junzo Hamanishi
Research Funding: MSD, Ono Pharmaceutical, Dainippon Sumitomo Pharma
Seiichi Mori
Consulting or Advisory Role: Konica Minolta Precision Medicine
Noriomi Matsumura
Leadership: Takara Bio
Speakers' Bureau: AstraZeneca
Research Funding: AstraZeneca
No other potential conflicts of interest were reported.
SUPPORT
Supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant No. 18H02945 and 18H02947.
DATA SHARING STATEMENT
Controlled access data including germline variant annotations, raw sequence data, and raw SNP array data are available through the dbGaP authorized access system (study accession phs000178). The processed data and codes to reproduce the main results of this work are available on the GitHub page (https://github.com/shirotak/pancancer_hrd_analysis). Other codes for preprocessing public or restricted-access data are available from the corresponding author upon reasonable request.
AUTHOR CONTRIBUTIONS
Conception and design: J.B. Brown, Masaki Mandai, Noriomi Matsumura
Administrative support: J.B. Brown, Koji Yamanoi
Provision of study materials or patients: Junzo Hamanishi
Collection and assembly of data: Shiro Takamatsu, Ken Yamaguchi, Hisamitsu Takaya, Masaki Mandai
Data analysis and interpretation: Shiro Takamatsu, J.B. Brown, Junzo Hamanishi, Koji Yamanoi, Tomoko Kaneyasu, Seiichi Mori
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by the authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
J.B. Brown
Employment: Boehringer Ingelheim
Honoraria: Kyowa Kirin International, Daiichi Sankyo RD Novare
Research Funding: Daiichi Sankyo RD Novare
Travel, Accommodations, Expenses: Boehringer Ingelheim, Kyowa Kirin International
Junzo Hamanishi
Research Funding: MSD, Ono Pharmaceutical, Dainippon Sumitomo Pharma
Seiichi Mori
Consulting or Advisory Role: Konica Minolta Precision Medicine
Noriomi Matsumura
Leadership: Takara Bio
Speakers' Bureau: AstraZeneca
Research Funding: AstraZeneca
No other potential conflicts of interest were reported.
REFERENCES
- 1.Lord CJ, Ashworth A: BRCAness revisited. Nat Rev Cancer 16:110-120, 2016 [DOI] [PubMed] [Google Scholar]
- 2.Dullens B, de Putter R, Lambertini M, et al. : Cancer surveillance in healthy carriers of germline pathogenic variants in BRCA1/2: A review of secondary prevention guidelines. J Oncol 2020;9873954, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farmer H, McCabe N, Lord CJ, et al. : Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434:917-921, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Bryant HE, Schultz N, Thomas HD, et al. : Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434:913-917, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Patel M, Nowsheen S, Maraboyina S, et al. : The role of poly(ADP-ribose) polymerase inhibitors in the treatment of cancer and methods to overcome resistance: A review. Cell Biosci 10:35, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nizialek E, Antonarakis ES: PARP inhibitors in metastatic prostate cancer: Evidence to date. Cancer Manag Res 12:8105-8114, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Póti Á, Gyergyák H, Németh E, et al. : Correlation of homologous recombination deficiency induced mutational signatures with sensitivity to PARP inhibitors and cytotoxic agents. Genome Biol 20:240, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stover EH, Fuh K, Konstantinopoulos PA, et al. : Clinical assays for assessment of homologous recombination DNA repair deficiency. Gynecol Oncol 159:887-898, 2020 [DOI] [PubMed] [Google Scholar]
- 9.Sandhu SK, Schelman WR, Wilding G, et al. : The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: A phase 1 dose-escalation trial. Lancet Oncol 14:882-892, 2013 [DOI] [PubMed] [Google Scholar]
- 10.de Bono J, Ramanathan RK, Mina L, et al. : Phase I, dose-escalation, two-Part Trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov 7:620-629, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Owonikoko TK, Redman MW, Byers LA, et al. : A phase II study of talazoparib (BMN 673) in patients with homologous recombination repair deficiency (HRRD) positive stage IV squamous cell lung cancer (lung-MAP sub-study, S1400G). J Clin Oncol 37, 2019. (abstr 9022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hutter C, Zenklusen JC: The cancer genome atlas: Creating lasting value beyond its data. Cell 173:283-285, 2018 [DOI] [PubMed] [Google Scholar]
- 13.International Cancer Genome Consortium, Hudson TJ, Anderson W, et al. : International network of cancer genome projects. Nature 464:993-998, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Telli ML, Timms KM, Reid J, et al. : Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res 22:3764-3773, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birkbak NJ, Wang ZC, Kim JY, et al. : Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov 2:366-375, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Popova T, Manié E, Rieunier G, et al. : Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res 72:5454-5462, 2012 [DOI] [PubMed] [Google Scholar]
- 17.Abkevich V, Timms KM, Hennessy BT, et al. : Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer 107:1776-1782, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. : Signatures of mutational processes in human cancer. Nature 500:415-421, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polak P, Kim J, Braunstein LZ, et al. : A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet 49:1476-1486, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gou R, Dong H, Lin B: Application and reflection of genomic scar assays in evaluating the efficacy of platinum salts and PARP inhibitors in cancer therapy. Life Sci 261:118434, 2020 [DOI] [PubMed] [Google Scholar]
- 21.Knudson AG: Two genetic hits (more or less) to cancer. Nat Rev Cancer 1:157-162, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Maxwell KN, Wubbenhorst B, Wenz BM, et al. : BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat Commun 8:319, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jonsson P, Bandlamudi C, Cheng ML, et al. : Tumour lineage shapes BRCA-mediated phenotypes. Nature 571:576-579, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Loo P, Nordgard SH, Lingjærde OC, et al. : Allele-specific copy number analysis of tumors. Proc Natl Acad Sci USA 107:16910-16915, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen R, Seshan VE: FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 44:e131, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Birkbak NJ, Kochupurakkal B, Izarzugaza JM, et al. : Tumor mutation burden forecasts outcome in ovarian cancer with BRCA1 or BRCA2 mutations. PLoS One 8:e80023, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pellegrino B, Musolino A, Llop-Guevara A, et al. : Homologous recombination repair deficiency and the immune response in breast cancer: A literature review. Transl Oncol 13:410-422, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carter SL, Cibulskis K, Helman E, et al. : Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30:413-421, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grundy MK, Buckanovich RJ, Bernstein KA: Regulation and pharmacological targeting of RAD51 in cancer. NAR Cancer 2:zcaa024, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sokol ES, Pavlick D, Khiabanian H, et al. : Pan-cancer analysis of BRCA1 and BRCA2 genomic alterations and their association with genomic instability as measured by genome-wide loss of heterozygosity. JCO Precis Oncol 4:442-465, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agnese DM: Battle of the BRCA1/BRCA2 (offspring) sex ratios: Truth or consequences. J Med Genet 43:201-202, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thompson D, et al. : Breast cancer linkage consortium. Cancer incidence in BRCA1 mutation carriers. J Natl Cancer Inst 94:1358-1365, 2002 [DOI] [PubMed] [Google Scholar]
- 33.Pitroda SP, Pashtan IM, Logan HL, et al. : DNA repair pathway gene expression score correlates with repair proficiency and tumor sensitivity to chemotherapy. Sci Transl Med 6:229ra42, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mroz EA, Rocco JW: MATH, a novel measure of intratumor genetic heterogeneity, is high in poor-outcome classes of head and neck squamous cell carcinoma. Oral Oncol 49:211-215, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wainer Katsir K, Linial M: Human genes escaping X-inactivation revealed by single cell expression data. BMC Genomics 20:201, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barbie DA, Tamayo P, Boehm JS, et al. : Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462:108-112, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moureau S, Luessing J, Harte EC, et al. : A role for the p53 tumour suppressor in regulating the balance between homologous recombination and non-homologous end joining. Open Biol 6:160225, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Willers H, McCarthy EE, Wu B, et al. : Dissociation of p53-mediated suppression of homologous recombination from G1/S cell cycle checkpoint control. Oncogene 19:632-639, 2000 [DOI] [PubMed] [Google Scholar]
- 39.Boehden GS, Akyüz N, Roemer K, et al. : p53 mutated in the transactivation domain retains regulatory functions in homology-directed double-strand break repair. Oncogene 22:4111-4117, 2003 [DOI] [PubMed] [Google Scholar]
- 40.Lotan TL, Kaur HB, Salles DC, et al. : Homologous recombination deficiency (HRD) score in germline BRCA2- versus ATM-altered prostate cancer. Mod Pathol 34:1185-1193, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davies H, Glodzik D, Morganella S, et al. : HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med 23:517-525, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen L, Martens JWM, Van Hoeck A, et al. : Pan-cancer landscape of homologous recombination deficiency. Nat Commun 11:5584, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barr ML, Moore KL: Chromosomes, sex chromatin, and cancer. Proc Can Cancer Conf 2:3-16, 1957 [PubMed] [Google Scholar]
- 44.Ouyang Y, Salstrom J, Diaz-Perez S, et al. : Inhibition of Atm and/or Atr disrupts gene silencing on the inactive X chromosome. Biochem Biophys Res Commun 337:875-880, 2005 [DOI] [PubMed] [Google Scholar]
- 45.ElInati E, Russell HR, Ojarikre OA, et al. : DNA damage response protein TOPBP1 regulates X chromosome silencing in the mammalian germ line. Proc Natl Acad Sci USA 114:12536-12541, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vancevska A, Ahmed W, Pfeiffer V, et al. : SMCHD1 promotes ATM-dependent DNA damage signaling and repair of uncapped telomeres. EMBO J 39:e102668, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Controlled access data including germline variant annotations, raw sequence data, and raw SNP array data are available through the dbGaP authorized access system (study accession phs000178). The processed data and codes to reproduce the main results of this work are available on the GitHub page (https://github.com/shirotak/pancancer_hrd_analysis). Other codes for preprocessing public or restricted-access data are available from the corresponding author upon reasonable request.