

Graphical abstract
Abbreviations: ACE2, angiotensin-converting enzyme 2; 3CLpro, 3C-Like protease; COVID-19, coronavirus disease 2019; cryo-EM, cryo-electron microscopy; DyKAT, dynamic kinetic asymmetric transformation; EBOV, Ebola virus; EC50, half maximal effective concentration; EMD, Electron Microscopy Data; FDA, U.S. Food and Drug Administration; HCoV-229E, human coronavirus 229E; HPLC, high-performance liquid chromatography; IC50, half maximal inhibitory concentration; MERS-CoV, Middle East respiratory syndrome coronavirus; MD, molecular dynamics; MMPBSA, molecular mechanics Poisson-Boltzmann surface area; Mpro, main protease; MTase, methyltransferase; Nsp, nonstructural protein; PDB, Protein Data Bank; PLpro, papain-like protease; RdRp, RNA-dependent RNA polymerase; RTP, ribonucleoside triphosphate; SAM, S-adenosylmethionine; SARS-CoV, severe acute respiratory syndrome coronavirus; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; SI, selectivity index; Ugi-4CR, Ugi four-component reaction
Keywords: SARS-CoV-2, Natural products, FDA-approved drugs, Candidate drugs, Co-crystal structures
Abstract
Safer and more-effective drugs are urgently needed to counter infections with the highly pathogenic SARS-CoV-2, cause of the COVID-19 pandemic. Identification of efficient inhibitors to treat and prevent SARS-CoV-2 infection is a predominant focus. Encouragingly, using X-ray crystal structures of therapeutically relevant drug targets (PLpro, Mpro, RdRp, and S glycoprotein) offers a valuable direction for anti–SARS-CoV-2 drug discovery and lead optimization through direct visualization of interactions. Computational analyses based primarily on MMPBSA calculations have also been proposed for assessing the binding stability of biomolecular structures involving the ligand and receptor. In this study, we focused on state-of-the-art X-ray co-crystal structures of the abovementioned targets complexed with newly identified small-molecule inhibitors (natural products, FDA-approved drugs, candidate drugs, and their analogues) with the assistance of computational analyses to support the precision design and screening of anti–SARS-CoV-2 drugs.
1. Introduction
The unprecedented coronavirus disease 2019 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spread quickly across continents, with 4,180,161 deaths currently recorded on a global basis [1]. In view of the high virulence and mortality rates, the World Health Organization declared SARS-CoV-2 as the sixth public health emergency of international concern [2]. The pandemic is ongoing, and the effects of this highly contagious virus are widespread. Efforts are continuing to combat the SARS-CoV-2 outbreak with a variety of drugs, including remdesivir [3], dalbavancin [4], clofazimine [5], mycophenolic acid [6], plitidepsin [7], and Chinese herbal medicines such as Lianhuaqingwen capsules [8] and Qingfei Paidu decoction [9]. However, despite considerable effort, no effective countermeasures for controlling or eventually eradicating this dangerous virus have emerged [10], [11]. Rapid discovery and development of novel, efficacious, safe, and stable agents to treat COVID-19 is currently the focus of intense worldwide research.
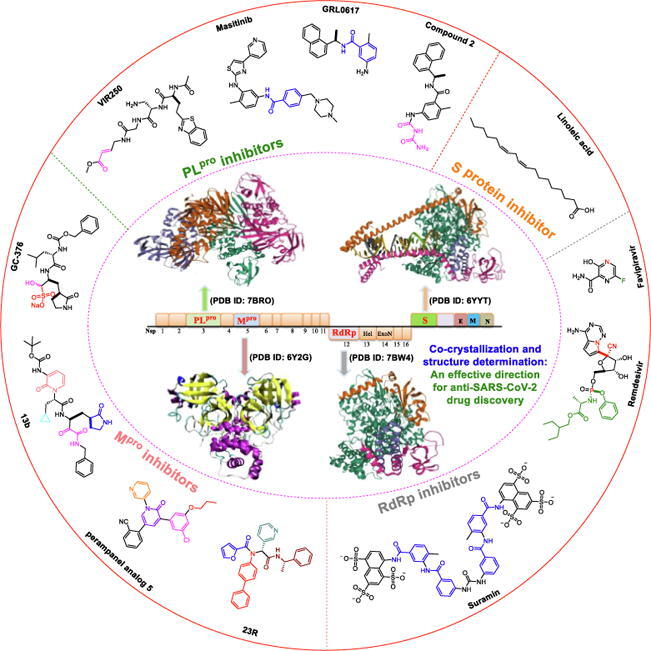
The ongoing challenge to develop new antiviral agents that are highly specific and effective against SARS-CoV-2 has spurred the medical community to explore multiple techniques and strategies, including co-crystal structure-guided precision drug discovery [12], [13], [14], [15]. Since 2020, this strategy has gained traction in validating bioactive constituents that could serve as COVID-19 agents [16], [17]. The determination of X-ray crystal structures of therapeutically relevant drug targets in pharmaceutical research represents a potentially valuable direction for anti–SARS-CoV-2 drug discovery and lead optimization. SARS-CoV-2 possesses a positive-sense RNA genome, and its genome organization is depicted in Fig. 1. Several promising targets among SARS-CoV-2 enzymes and proteins have been identified and are of significant research interest (Fig. 1), including papain-like protease (PLpro) [18], [19], main protease (Mpro) [20], [21], RNA-dependent RNA polymerase (RdRp) [22], [23], and the spike (S) glycoprotein, which binds to the angiotensin-converting enzyme 2 (ACE2)-binding domain [24], [25].
Fig. 1.

Organization of the SARS-CoV-2 genome and potential therapeutic targets. (A) Genome organization of SARS‐CoV‐2. (B) The structures of PLpro (the Protein Data Bank (PDB) ID: 7BRO). (C) The structures of Mpro (PDB ID: 6Y2G). (D) The structures of RdRp (PDB ID: 7BW4). (E) The structures of S glycoprotein (PDB ID: 6YYT).
In particular, PLpro plays an essential role in the cleavage and maturation of RNA virus polyproteins, dysregulation of the host inflammatory response, and disruption of host immune responses [26], [27]. Mpro, also called 3C-like protease (3CLpro), plays a crucial role in the polyprotein maturation process and may serve as a prime therapeutic target [28], [29], [30]. RdRp is another essential enzyme that regulates viral replication by catalyzing the RNA template–dependent development of phosphodiester bonds [31]. ACE2 is a host cell surface receptor necessary for SARS-CoV-2 infection [32]. The S protein plays a critical role in mediating membrane fusion and cell entry via interactions with the ACE2 cell surface receptor [33]. The immense therapeutic potential of targeting PLpro, Mpro, RdRp, and the S protein in the development of effective drugs has been widely explored.
Innovative drug development constitutes an important research direction, and results to date have been outstanding, with several interesting articles already published from the perspectives of potential drugs and drug targets [34], [35], [36], [37], [38], mechanisms of action and inhibition [26], [39], [40], structure-based drug design [41], [42], [43]. Other articles were identified by virtual screening of the ZINC, TCMSP, ChEMBL, Pubchem, NPASS, and Drug bank databases [44], [45], [46], [47], [48]. However, to our knowledge, no systematic reviews of inhibitor-target co-crystal complexes of SARS-CoV-2 have been published. In the current review, we focused on medicinal chemistry efforts based on analyses of X-ray co-crystal structures of the abovementioned targets in complex with newly identified small-molecule inhibitors as a means of supporting the precision design and screening of anti–COVID-19 agents through the characterization of ligand-bound targets. COVID-19 inhibitors of significance from the perspective of medicinal chemistry are prefaced by a brief introduction summarizing the relevant pharmacology, discovery process (drug design), preparation methods, and mechanisms of action. The main objective of this report is to provide a ‘co-crystal structure-guided drug discovery protocol’ to facilitate the development of safe and effective drugs to combat COVID-19.
2. Natural products and analogues as SARS-CoV-2 inhibitors
Natural products provide a rich and novel source for drug development and play an integral role as starting points in the development of therapeutic strategies for treating various complex diseases due to their tremendous structural diversity and unique chemical compositions [49]. In the current race to identify safe and efficacious drugs for COVID-19, natural products with a range of valuable bioactivities have attracted significant attention in terms of evaluating and validating their therapeutic effects [45], [46], [47]. As a result of continuing research, several natural products have been highlighted thus far as promising drug leads to combat COVID-19 [50], [51], [52]. As a result of continuing research, several natural products have been highlighted thus far as promising drug leads to combat COVID-19 [53], [54], [55], [56], [57], [58].
Scutellariae radix (Scutellaria baicalensis) is a well-known herbal medicine used to treat lung injury [59] and inflammatory diseases [60]. It is a widely used nutritional supplement with an impressive safety profile. Liu and co-workers recently demonstrated that crude scutellariae radix extract inhibits SARS-CoV-2 replication (half-maximal effective concentration [EC50] of 0.74 μg/mL) with almost no toxicity (selectivity index [SI] > 675.7) against Vero E6 cells [61]. Baicalein is the major component of scutellariae radix and exhibits broad-spectrum antiviral effects (Fig. 2A) [62], [63]. As expected, baicalein was shown to be effective in inhibiting Mpro activity (half-maximal inhibitory concentration [IC50] of 0.39 μM) [61]. Du and co-workers reported that baicalein can be used to treat acute lung injury, as it improves respiratory function, inhibits inflammatory cell infiltration, and downregulates the release of cytokines [64]. A docking model showed that the 6-OH group of baicalein plays a pivotal role in terminating RNA replication via hydrogen bonding interactions with L141 [61]. To elucidate the underlying molecular mechanisms of baicalein, active sites were explored. Su and co-workers generated a crystal structure of baicalein-Mpro at a resolution of 2.2 Å (PDB ID: 6M2N) [65]. Further analysis revealed that the non-peptidomimetic inhibitor baicalein was ensconced in the core of the substrate-binding pocket via multiple interactions. For example, hydroxyl groups of baicalein form hydrogen bond interactions with Leu141/Gly143 and Ser144/His163 of Mpro to stabilize the tetrahedral transition state of the proteolytic reaction. The enone carbonyl group of baicalein forms hydrogen bond interactions with Glu166, and the free aromatic rings of baicalein form π-π interactions with Cys145 and His41 as well as hydrophobic interactions with Gln189, Arg188, Met49, Cys44, and His41 to prevent substrate access to the active site, rather than covalently blocking catalytic Cys145. These interactions could underlie the observed potent activity (IC50 of 0.94 μM, EC50 of 2.94 μM, and SI > 212) of baicalein against SARS-CoV-2 Mpro [65]. Shuanghuanglian oral liquids extracted from three types of traditional Chinese medicines (including Scutellaria baicalensis Georgi) effectively blocked the replication of SARS-CoV-2 in Vero E6 cells (IC50 of 0.064 μM), highlighting the significant contributory role of this active component in the biological activity of Chinese herbal treatments [65]. Importantly, the crystal structure of the Mpro-baicalein complex provided direct data that could facilitate elucidation of the molecular mechanisms underlying the beneficial effects of traditional medicines.
Fig. 2.

Natural products as SARS-CoV-2 inhibitors. (A) Chemical structures of baicalein and X-ray crystal structure of baicalein with SARS-CoV-2 Mpro (PDB ID: 6M2N) [68]. (B) Chemical structures of myricetin and X-ray crystal structure of myricetin with SARS-CoV-2 Mpro (PDB ID: 7B3E) [68]. (C) Chemical structures of shikonin and X-ray crystal structure of shikonin with SARS-CoV-2 Mpro (PDB ID: 7CA8) [74]. (D) Chemical structures of sinefungin and X-ray crystal structure of sinefungin with SARS-CoV-2 Nsp10-Nsp16 (PDB ID: 6YZ1) [82]. (E) Chemical structures of linoleic acid and cryo-electron microscopy structure of linoleic acid with SARS-CoV-2 S protein (EMD ID: 11145) [84].
Myricetin, a flavonol monomer, is a natural product isolated from the “medicine food homology” species Chinese bayberry (Myrica rubra Sieb. et Zucc.) and exhibits a range of pharmacological effects against acute lung injury [66], inflammatory diseases, and infections with pathogenic microbes [67]. Kuzikov and co-workers reported that myricetin inhibits the synthesis of SARS-CoV-2 Mpro in vitro (IC50 of 0.22 μM) [68]. To optimize precision drug design, Su and co-workers generated a crystal structure of the Mpro-myricetin complex at a resolution of 2.1 Å (PDB ID: 7DPP) [68] (Fig. 2B), which unambiguously revealed that myricetin is covalently bound to catalytic Cys145. Su et al. also showed unprecedented inhibitor-target binding patterns, specifically, a covalent bond between the Cys145 sulfur and the 2′ position of myricetin, which was distinct from baicalein and other reported flavonoid structures [68]. In addition, Kuzikov and co-workers reported another crystal structure of the Mpro-myricetin complex with the same covalent binding mode, at 1.77-Å resolution (PDB ID: 7B3E) [69]. Further analysis of the X-ray structure of the myricetin-Mpro complex revealed that the binding pocket is only partially occupied by myricetin, presenting an important signal and ideal lead for structure-based drug design. Based on molecular dynamics (MD) simulations and molecular mechanics Poisson-Boltzmann surface area (MMPBSA) calculations, Sharma et al. [70] revealed that baicalein forms a stable combination with non-structural protein 15 (Nsp15) with low binding energy (−65.663 kJ/mol).
The natural naphthoquinone shikonin, derived from Lithospermum erythrorhizon Sieb. et Zucc (Fig. 2C), has numerous pharmacological properties that support its further development as a therapeutic agent [71]. This agent has been shown to exert anti-inflammatory, anti-fungal, and anti-HIV effects [72]. Yang et al. revealed that shikonin inhibits the replication of SARS-CoV-2 Mpro in vitro (EC50 of 15.75 μM) [73]. Li and co-workers [74] initially solved the crystal structure of the shikonin-Mpro complex at 2.45 Å (Fig. 2C). Crystallography data showed that the His41-Cys145 catalytic dyad undergoes large conformational changes upon complex formation, leading to significant differences with other reported structures [75]. Li and co-workers recently revealed that two novel hydrogen-bonding interactions (one with Gln189 and Thr190, another with Met165, His164, and Cys145) and a π-π stacking interaction with His41 play critical roles in the binding of shikonin with Mpro [74]. These findings clearly indicate a distinct binding pattern suggestive of binding site diversity. However, Ma and co-workers recently reported that shikonin may not be a target-specific Mpro inhibitor, as its activity declines markedly in the presence of reducing reagents [76]. Further elucidation of the different binding modes should provide a solid foundation for effective COVID-19 drug design.
The genome of SARS-CoV-2 contains ~ 29 800 bases encoding 16 nonstructural proteins (Nsp1-Nsp16) essential for viral replication [77]. For example, Nsp16 plays a critical role in immune evasion during virus replication [78]. The natural nucleoside antibiotic sinefungin is a structural analogue of S-adenosylmethionine (SAM), which serves as a key methyl group donor to numerous biomolecules [79]. SAM-dependent 2′-O-RNA methyltransferase (MTase) is a potential relevant drug target in COVID-19 chemotherapy [80]. The function of MTase is associated with Nsp16, which requires Nsp10 as a cofactor for activity [80], [81]. Mahalapbutr et al. [80] also reported that sinefungin exhibits very high susceptibility to Nsp16 (especially through electrostatic interactions with the 2′-OH and N3 of the RNA’s adenosine moiety) based on atomistic MD simulations and free-energy calculations. To establish the mechanisms by which sinefungin inhibits the Nsp16 MTase at a molecular level, Krafcikova and co-workers [82] determined the crystal structure of SARS-CoV-2 Nsp10-Nsp16 complexed with sinefungin at a resolution of 2.4 Å (PDB ID: 6YZ1) (Fig. 2D). The co-crystallization structure showed that sinefungin binds the SAM-binding pocket localized in a canyon within Nsp16 and forms several interactions with specific residues (nucleoside-binding pocket: Asp99, Asn101, and Asp114; methionine-binding pocket: Asn43, Asp130, and Lys170). These data provide an important starting point for structure-based inhibitor discovery.
Linoleic acid, an essential free fatty acid, is an important modulator of the inflammatory response [83]. Toelzer and co-workers [84] recently demonstrated that a combination of linoleic acid (50 μM) and the RdRp inhibitor remdesivir (20 to 200 nM) exert a synergistic inhibitory effect in human Caco-2 ACE2 + cells in vitro. To clarify the underlying inhibitory mechanism of action of linoleic acid, the cryo-electron microscopic (cryo-EM) structure of S protein complexed with linoleic acid was determined at 2.85-Å resolution (Electron Microscopy Data [EMD] ID: 11145) (Fig. 2E) [84]. Further analysis of the linoleic acid binding pocket within the S protein revealed that the hydrocarbon tail of linoleic acid binds to hydrophobic amino acids, whereas the acidic head group interacts with a positively charged anchor (Arg408 and Gln409) to irreversibly lock the S protein. The S protein hydrophobic pocket with a tube-like shape fits well with linoleic acid, resulting in reduced ACE2 interactions, thus setting the stage for an intervention strategy based on linoleic acid binding to the S protein. Further studies are warranted to establish whether linoleic acid exerts anti–COVID-19 effects in vivo. Note that dynamic-nonequilibrium MD studies showed that the linoleic acid site forms a stable combination with the functional regions of the S protein, but variation in the S protein could be exploited to tune a targeted allosteric response [85].
Calpain inhibitor II, developed by the Cerro group [78], has shown potential utility against SARS-CoV-2 infection [87]. Initial research on the synthesis of calpain inhibitor II commenced with structural modification of leupeptin (a naturally occurring and relatively non-specific inhibitor of thiol proteases) by replacing the guanidine group with a methyl group to create calpain inhibitor I (Fig. 3), which exhibited more-potent and selective blockade of calpain activity [86]. An even more important contributor to antiviral drug design is the 4′-S substituted calpain inhibitor II, which exhibited more-potent blockade of calpain activity [86]. Calpain inhibitor II, a broad-spectrum antiviral agent, can be used to treat infections with multiple viruses, such as endemic human coronavirus 229E (HCoV-229E) (EC50 of 0.08 μM) and human coronavirus OC43 (EC50 of 1.82 μM) [87]. As for SARS-CoV-2, Wang et al. [88] confirmed that calpain inhibitor II is highly effective against Mpro (IC50 of 0.97 μM).
Fig. 3.

Leupeptin and its analogues as inhibitors of SARS-CoV-2 and the reaction mechanism [89].
Wang and co-workers [89] also reported the crystal structure of calpain inhibitor II–Mpro at 1.65-Å resolution (PDB ID: 6XA4). Further analysis of the complex structure revealed that stabilized thiohemiketal with the (S) configuration occupies the oxyanion hole, which is formed by Gly143, Cys145, and, in part, Ser144 (Fig. 3). Additionally, the structure exhibited several other interactions with active site residues, including multiple hydrogen bonds of the amide group with His164, Met165, and Glu166 (Fig. 3). Interestingly, a weak hydrogen bond between the sulfur group and His163 was detected, which could explain the stronger activity of calpain inhibitor II (IC50 = 0.97 μM) relative to calpain inhibitor I (IC50 = 8.60 μM) [89].
The introduction of X-ray crystallography technology has provided a highly successful strategy for exploration of natural products and their analogues, improving our understanding of drug interactions at the molecular level based on structures of complexes, to facilitate the design of effective COVID-19 therapies. While natural products have demonstrated potential effectiveness, clinical trial evidence of their utility as anti–COVID-19 agents is currently lacking. Future research will therefore focus on obtaining stronger evidence to support the clinical utility of natural products. In this scenario, strategies for the large-scale manufacture of natural products and analogues, such as shikonin, sinefungin, and calpain inhibitor II, are urgently required.
3. FDA-approved drugs as SARS-CoV-2 inhibitors
Remdesivir, an RdRp inhibitor, was the first and only drug against COVID-19 approved by the U.S. Food and Drug Administration (FDA) in 2020 [90], [91], [92], [93]. The highly potent 1′-CN-substituted GS-441524, inspired by the natural product tubercidin, is important in the design of anti–SARS-CoV-2 agents [94]. Triphosphate GS-443902 is established as the active form of GS-441524. However, monophosphate conversion of GS-441524 to GS-441524 monophosphate is extremely difficult [95], highlighting the necessity of developing a prodrug that can overcome this delivery bottleneck. To achieve better cellular uptake in vivo, monophosphorylated remdesivir was developed by Gilead Sciences (Fig. 4A) [96]. The GS-5734 1′–CN group plays a pivotal role in terminating RNA replication by disrupting the exoribonuclease proofreading activity and inhibiting the RdRp polymerization activity [97].
Fig. 4.

Design, mechanism of action, and synthesis of the covalent RdRp inhibitor remdesivir [101].
To increase the efficiency of remdesivir, researchers at Gilead Science initially developed strategies for preparation of the P-stereogenic prodrug using a scalable process through chiral preparative high-performance liquid chromatography (HPLC) or chiral resolution [96], which inevitably led to waste of resources. For further improvement of efficiency, an elegant asymmetric strategy (Fig. 4B) for producing remdesivir via chiral bicyclic imidazole-catalyzed dynamic kinetic asymmetric transformation (DyKAT) with excellent stereoselectivity (SP:RP = 22:1) was recently developed by Zhang and colleagues [98]. In the clinical context, development of a large-scale synthesis method for remdesivir is urgently needed. As remdesivir is a phosphoramidate prodrug, conversion into the triphosphate GS-443902 is required. Similar to many prodrugs, the postulated activation pathway of remdesivir is divided into four sequential steps (Fig. 5): cell entry (via increased hydrophobicity with the aid of a phosphorylated group), removal of the masking group (via enzymatic and chemical demasking), phosphorylation (via NMP kinases), and incorporation into the growing SARS-CoV-2 RNA strand. In cells, the triphosphate form, GS-443902, can block SARS-CoV-2 replication by evading the “proofreading” of viral RNA sequences [99].
Fig. 5.

Bioconversion of the covalent RdRp inhibitor remdesivir.
Wakchaure et al. [100] showed that remdesivir has high binding energy (−29.7 kCal/mol) with respect to RdRp, and MD simulations also revealed that remdesivir binds in the catalytic site via the formation of hydrogen bond interactions. Xu et al. [101] reported the cryo-EM structure of RdRp-remdesivir (using its triphosphate metabolite GS-443902) at 2.5-Å resolution (PDB ID: 7BV2). The cryo-EM structure unambiguously demonstrated that remdesivir monophosphate (IV), positioned at the center of the catalytic site of the primer RNA, covalently binds to the 1 + position of the template strand, thus terminating chain elongation (Fig. 4A). Three strong H-bonds with active site residues (ribose –OH groups: Asp623, Ser682, and Asn691; sugar 2′–OH: Asn691) were identified (Fig. 4A). Notably, the implicit, cryptic, and allosteric binding sites provide a versatile platform for designing high-potency inhibitors with kinetic selectivity [102], [103], [104]. Based on thermodynamic profiling, Srivastava et al. found that triphosphate GS-443902 inactivates RdRp by not only interfering with binding in the initial catalytic site but also by significantly blocking the nucleoside 5′-triphosphate entrance site [105]. Based on MMPBSA calculations, Khan et al. [106] proposed that remdesivir inhibits SARS-CoV-2 via a multi-target mechanism, as it binds to Mpro (−7.8 kCal/mol), membrane protein (−7.4 kCal/mol), and RDRP (−7.1 kCal/mol).
Remdesivir exhibits broad-spectrum activity against multiple viruses in vitro, such as SARS-CoV, Middle East respiratory syndrome coronavirus (MERS-CoV), respiratory syncytial virus, Ebola virus (EBOV), and HCoV-229E, with EC50 values of 0.069 μM, 0.090 μM, 0.021 μM, 0.012 μM, and 0.024 μM, respectively [107], [108], [109], [110], [111]. Moreover, remdesivir reportedly protects rhesus monkeys from MERS-CoV infection [112] and displays clinical safety and efficacy against EBOV infection [113]. Although remdesivir does not appear to be highly effective in patients with COVID-19 [114], it has the obvious advantage of rapid clinical translation to a rational prodrug strategy against SARS-CoV-2. In addition, multiple lines of evidence suggest that combinations of remdesivir and other agents (such as baricitinib [115] and dexamethasone [116]) exert synergistic therapeutic effects against COVID-19.
Drug repurposing, a time-saving process, is a viable strategy to quickly, safely, and successful discover clinically approved drugs for COVID-19 treatment [117], [118], [119], [120]. Favipiravir, a promising repurposed prodrug, was identified by Toyama Chemical Co., Ltd., and approved in Japan in 2014 for the treatment of influenza virus infections [121]. To date, widespread research attention has focused on the utility of favipiravir as a specific anti–COVID-19 agent [122]. Favipiravir is an orally bioavailable prodrug that exhibits potent activity against COVID-19 (EC50 of 61.88 μM) with moderate selectivity (therapeutic window > 6.46) [123]. Favipiravir was recently shown to protect Syrian hamsters from SARS-CoV-2 infection, with a strong dose effect [124]. Abdelnabi and co-workers reported that a combination of favipiravir and molnupiravir exerted a marked synergistic inhibitory effect in a SARS-CoV2 hamster infection model [125]. Several clinical trials have highlighted the efficacy of favipiravir against COVID-19 [126], [127], [128], [129]. For example, Cai and co-workers reported that favipiravir significantly improved recovery rate (91.43%) and shortened viral clearance time (4 days), with fewer adverse events (11.4%) in patients with mild to moderate COVID-19 (N = 80) [129].
To elucidate the mechanisms underlying SARS-CoV-2 polymerase activity, Naydenova and co-workers examined the structure of SARS-CoV-2 RdRp in complex with favipiravir (using its triphosphate metabolite favipiravir–ribonucleoside triphosphate [RTP]) at 2.5-Å resolution via cryo-EM (PDB ID: 7AAP; EMD-11692) (Fig. 6) [130]. The cryo-EM structure unambiguously showed that favipiravir-RTP pairs with the 1 + nucleotide of the template strand via noncovalent interactions (side-chains and two catalytic Mg2+ ions) to terminate chain extension (Fig. 6). Peng and colleagues [131] reported an alternative structural snapshot (PDB ID: 7CTT; EMD ID: 30469) distorted in one direction, suggesting another productive conformation at the catalytic site. These distinct cryo-EM states may account for the slow, weak, and inefficient incorporation of the inhibitor into the RNA primer strand, leading to the high EC50 (61.88 μM).
Fig. 6.

Bioconversion of the non-covalent RdRp inhibitor favipiravir [130].
Similar to the prodrug remdesvir, favipiravir must be converted into the active form, favipiravir-RTP. The activation mechanism of favipiravir involves four sequential steps (Fig. 6): cell entry, phosphoribosylation, phosphorylation, and non-covalent incorporation into the growing SARS-CoV-2 RNA strand [132]. Notably, favipiravir-RTP binds non-covalently to the polymerase in a manner distinct from that of remdesivir-RTP. Furthermore, Celik et al. [133] used MD simulations to highlight the binding site differences of favipiravir and its active triphosphate metabolites (favipiravir-RTP) with RdRp. Their results demonstrated that favipiravir-RTP forms a more-stable complex than favipiravir. This finding indicates an unusual binding mode, which provides an important basis for non-covalent RdRp inhibitor discovery.
Free-energy perturbation (FEP) calculations based on molecular simulations and statistical mechanics represents a promising approach to guide structural modifications in drug discovery research [134], [135], [136]. Based on this method, Jorgensen’s group [137] recently showed that the non-covalent SARS-CoV-2 Mpro inhibitor perampanel analogue 5 effectively treats SARS-CoV-2 infection by inhibiting Mpro activity (IC50 of 0.14 μM) and inducing significant suppression of viral replication in Vero E6 cells (EC50 of 1.5 μM), with moderate cytotoxicity (SI = 14.7). Perampanel, a poorly effective inhibitor of Mpro with an IC50 of 100–250 μM, was an initial hit compound selected for redesign and further optimization based on its relatively simple structure and FEP modeling studies [138]. As shown in Fig. 7A, perampanel analogue 2 was identified as a highly potent target via initial docking analyses using FEP calculations [138], [139].
Fig. 7.

Structure-based design and synthesis of perampanel analogues 2, 4, and 5 [137], [139].
As expected, the anti–SARS-CoV-2 activity of perampanel analogue 2 was markedly improved, with an IC50 of 9.99 μM, which was further enhanced by a factor of ~ 2 upon addition of a second chlorine atom to perampanel analogue 2 to generate perampanel analogue 4. Jorgensen et al. [137] determined the crystal structure of a complex of SARS-CoV-2 Mpro and perampanel analogue 4 at a resolution of 1.6 Å (PDB ID: 7L10), which unambiguously revealed that the chlorophenyl edge packs well against the imidazole ring of His41 and that the other meta-chlorine near Gln189 should be replaced with an alkyl or alkoxy group. These data provided valuable guidance for the synthesis of propoxy perampanel analogue 5, leading to a notable improvement in the IC50 value from 4.02 μM to 0.14 μM for perampanel analogue 4. Examination of the crystal structure of Mpro in complex with perampanel analogue 5 (1.8 Å resolution, PDB ID: 7L11) revealed that the terminal methyl group extended into the hydrophobic region at the juncture of Met165, Leu167, and Pro168. In addition, a combination of perampanel analogue 5 and remdesivir exhibited a significant therapeutic effect against COVID-19, with a synergy volume/antagonism volume ratio of 30.8/0 μM2 %. Jorgensen et al. [137] additionally explored several structurally novel and non-peptidic inhibitors with IC50 values in the 20 nM range identified from FEP calculations and crystal structures.
To increase the efficiency of target perampanel analogues 2, 4, and 5, Jorgensen et al. [137] further developed a general route for synthesis, as shown in Fig. 7C. Briefly, Suzuki cross-coupling of 5-bromo-2-methoxypyridine with (2-cyanophenyl) boronic acid yielded S1, followed by deprotection to produce intermediate S2, which was subjected to Chan-Lam coupling to yield intermediate S3. Bromination of S3 yielded key intermediate S4, which was subjected to Suzuki cross-coupling with aryl boronic acids to generate target perampanel analogues 2, 4, and 5. Notably, upscaling of the total synthesis strategy remains an urgent requirement.
4. Candidate drugs and analogues as SARS-CoV-2 inhibitors
A wide range of drug candidates may be effective against COVID-19 via broad-spectrum effects, both in vitro and in vivo [140], [141]. These candidate drugs provide a basis to further explore and develop SARS-CoV-2 inhibitors, which could reduce the period of treatment and minimize costs. The peptidomimetic α-ketoamide inhibitor 11r, an antiviral agent, can be used to inhibit multiple viruses, such as SARS-CoV (EC50 of 1.4 μM), enterovirus A71 (EC50 of 0.8–0.9 μM), coxsackievirus B3 (EC50 of 0.45 μM), and MERS-CoV (EC50 of 0.0004 μM) [142]. Hilgenfeld et al. showed that 11r, a specific Mpro inhibitor, is effective against SARS-CoV-2 infection (EC50 of 0.18 μM), thus presenting a lead for further development [20]. To improve its plasma half-life, lead 11r was modified by hiding the P3-P2 amide bond within a pyridone ring. As expected, the plasma half-life in mice increased from 18 min to 1 h by preventing the cellular protease–mediated cleavage of the amide bond. In addition, to enhance solubility and antiviral activity, compound 13b (EC50 of 0.67 μM) was produced by replacing the hydrophobic cinnamyl and cyclohexyl moieties with a low-hydrophobic Boc group and smaller cyclopropyl group, respectively. The researchers reported total synthesis of compound 13r on the ~ 50-mg scale [20]. The key components of the strategy were nucleophilic substitution (SN2) and nucleophilic addition of isocyanides to aldehydes (Fig. 8).
Fig. 8.

Structure-based design and synthesis of the peptidomimetic inhibitor 13b [20].
Notably, compound 13b exhibited significantly improved plasma protein binding (reduced from the 99% of 11r to 90%) and plasma half-life in mice (increased from the 18 min of 11r to 1.8 h). Compound 13b exhibited favorable pharmacokinetic properties in mice, including good tropism to the lungs after subcutaneous administration, suitability for direct administration via inhalation, long-term efficacy, and good tolerability without adverse reactions. The hydrophobic Boc group appeared to play an essential role in crossing of the cell membrane and binding to viral Mpro, without which the compound was almost inactive [20]. The above studies offer a platform for research into more-effective pyridone-containing anti-SARS-CoV-2 drugs to suppress disease progression and support the importance of structure-based approaches in the design and optimization of SARS-CoV-2 inhibitors. Rungrotmongkol et al. [143] reported that compound 13b was a suitable template for structural modifications at the P1′ and P4 sites to enhance interactions.
The Hilgenfeld group [20] determined the crystal structure of 13b-Mpro at 1.95-Å resolution (PDB ID: 6Y2F), which unambiguously revealed the presence of a stabilized thiohemiketal generated via nucleophilic attack of Cys145 over the α-carbonyl of the ketoamide warhead. Moreover, several interactions with active site residues (such as the amide oxygen of 13b forming hydrogen bond interactions with Gly143, Cys145, and partly Ser144 to form the “oxyanion hole”; the α-keto group of 13b forming a covalent bond with catalytic Gly145 to yield the thiohemiketal group) were identified. Apparently, there is a large space (distance > 3.6 Å) between Thr190, Gln189, and the pyridone ring of compound 13b, suggesting that a group larger than the pyridone ring could be designed based on the P3 moieties. In addition, a small model of the thiol moiety (active center of Mpro) with inhibitor 13b was generated (Fig. 9) [21]. In this model, the C-S bond can be formed directly (without deprotonation of the thiol group), and water molecules play an important role in the kinetics, as the energy barrier decreases by 9.0 kCal/mol for the intermediate when assisted with an explicit water molecule, which was facilitated via a 6-membered transition state instead of a constrained 4-membered state. Furthermore, Kumari et al. [144] used MD simulations to highlight the binding site differences of 13b with monomeric and dimeric Mpro. Their results showed that dimeric Mpro exhibits higher affinity for 13b via residue interactions between S1 and F140, E166, and H172.
Fig. 9.

Transition states of direct C-S bond formation between the inhibitor 13b and the thiol moiety.
Chirality is one of the most crucial attributes for a vast majority of pharmaceutical compounds in the natural world and chiral bioactive substances play a key role in disease control [145]. Chiral agents have attracted significant research attention due to enantiomers often possessing differential binding affinities for SARS-CoV-2 enzymes and proteins.
Kitamura and co-workers [146] recently demonstrated that the non-covalent SARS-CoV-2 Mpro inhibitor 23R can be used to treat the virus infection. The compound inhibited Mpro (IC50 of 0.31 μM) and exerted significant suppressive effects in Vero E6 cells (EC50 of 1.27 μM, SI > 78.7). Initial research on 23R began with structural modification of the SARS-CoV Mpro agent ML188 (R), which was separated using chiral stationary-phase supercritical fluid chromatography from ML188 enantiomers (S/R). In 2013, Jacobs and co-workers [147] demonstrated that the lead compound ML188 (S/R) and its single stereoisomer ML188 (R) effectively inhibited replication of SARS-CoV Mpro with IC50 values of 4.8 μM and 1.5 μM, respectively. However, the single stereoisomer ML188 (S) was inactive and had no inhibitory effect on SARS-CoV replication. The X-ray crystal structure of the Mpro-ML188 (R) complex (PDB ID: 3V3M) revealed that an optimal shape complementary to SARS-CoV was formed through several key interactions (Fig. 10A) [147]. Among these, the 3-pyridyl group occupied the S1-subpocket via a critical hydrogen bond with the His163 side chain. Knowledge of the structure of this non-covalent SARS-CoV Mpro inhibitor could further guide effective inhibitor design.
Fig. 10.

Structure-based design and synthesis of the non-covalent SARS-CoV-2 Mpro inhibitor 23R [146], [147].
Based on the high-affinity binding (between the hydrogen bond and His-163 side chain) strategy, 39 compounds (a mixture of enantiomers or diastereomers) were prepared using the Ugi four-component reaction (Ugi-4CR), which provides an expeditious strategy for lead optimization [146]. Among these compounds, 23 (S/R) significantly inhibited viral replication, with an IC50 of 0.66 μM and low toxicity (SI > 303). This elegant process of 23 (S/R) synthesis is depicted in Fig. 10B. After separation of the single stereoisomers, 23R and 23S, via reverse-phase HPLC, IC50 values were determined as 0.31 μM for the diastereomer 23R and 5.61 μM for the less-active 23S diastereomer [146]. To further elucidate the binding forces, Kitamura and co-workers [146] generated a crystal structure of 23R complexed with Mpro at 2.6 Å resolution (PDB ID: 7KX5). The P1 pyridinyl ring occupied the S1 pocket, forming a close (2.9 Å) hydrogen bond with the His163 side chain. Another point of particular interest is the location of the terminal α-methylbenzyl group between the S2 and S4 sites, which represents a previously unreported mechanism of binding pocket formation. The S4 pocket remained mainly unoccupied by 23R, indicating the presence of sufficient space that could be explored for further drug development. The COVID-19 pandemic has highlighted the urgent need for rapid structure-based drug discovery and development, and this strategy achieved expedited design of a non-covalent Mpro inhibitor through coupling of co-crystal structures, structure-based design, and Ugi-4CR methodologies.
5. Other active compounds as SARS-CoV-2 inhibitors
Numerous small-molecule inhibitors have shown promising results as efficient therapeutic agents for SARS-CoV-2. Earlier, Hoffman and co-workers [148] at Pfizer Worldwide Research and Development highlighted the efficacy of the novel Mpro inhibitor, PF-00835231, in treatment of infections. Potent activity of this compound against hCoV 229E (EC50 of 0.090 μM), SARS-CoV (EC50 of 4.8 μM) and SARS-CoV-2 (EC50 of 0.13 μM) with no observable cytotoxicity was reported. Examination of the X-ray crystal structure of the Mpro-PF-00835231 complex (PDB ID: 6XHM) revealed that the stabilized tetrahedral carbinol complex is generated via nucleophilic attack of Cys145 over the carbonyl carbon of the hydroxymethylketone warhead [148]. Furthermore, compound PF-00835231 exhibited synergistic activity against COVID-19 in combination with remdesivir in HeLa-ACE2 cells. Preclinical experiments confirmed that the absorption, distribution, metabolism, and excretion profile as well as safety and activity profiles of PF-00835231 were suitable to warrant further development as a potent Mpro inhibitor. Clinical trials of PF-00835231 have been registered (NCT04627532 and NCT04535167) for in-depth studies of the compound’s anti–SARS-CoV-2 activity. Additionally, the Dittmann group [149] reported that statistically, PF-00835231 is a more-potent inhibitor than remdesivir. In addition to the abovementioned active compounds, several other small-molecule inhibitors with known crystal structures have been shown to exhibit significant anti–SARS-CoV-2 activity (Table 1). Further research is required to establish the clinical safety and efficacy of these inhibitor candidates, however.
Table 1.
Other small-molecule SARS-CoV-2 inhibitors with known crystal structures.
| No. | Name | Structure | EC50 or IC50 (μM) | Target | PDB ID | Refs |
|---|---|---|---|---|---|---|
| 1 | Boceprevir | 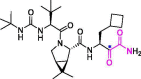 |
4.13 | Mpro | 6XQU | [150], [151], [152] |
| 2 | Calpain inhibitor XII | 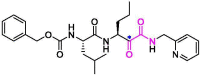 |
0.45 | Mpro | 6XFN | [89], [153], [154] |
| 3 | Calpeptin | 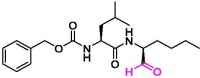 |
1.56 | Mpro | 7AKU | [155] |
| 4 | Carmofur | 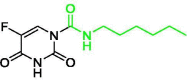 |
1.82 | Mpro | 7BUY | [156], [157] |
| 5 | Ebselen | 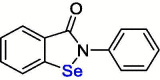 |
4.67 | Mpro | 7BAK | [158], [159] |
| 6 | GC-373 | 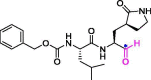 |
0.40 | Mpro | 6WTK | [160] |
| 7 | GC-376 | 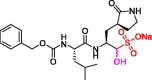 |
0.030 | Mpro | 6WTJ | [89], [160], [161] |
| 8 | GRL0617 | 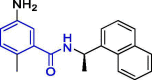 |
2.20 | PLpro | 7CMD | [19], [162], [163], [164] |
| 9 | GS-441524 | 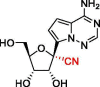 |
0.70 | RdRp | 7BF6 | [165], [90] |
| 10 | Isofloxythepin | 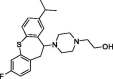 |
4.80 | Mpro | 7AY7 | [155] |
| 11 | Masitinib | 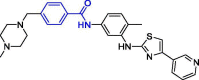 |
2.50 | PLpro | 7JU7 | [166], [167] |
| 12 | MI-23 | 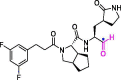 |
0.0076 | Mpro | 7D3I | [168] |
| 13 | ML188 | 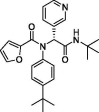 |
2.5 | Mpro | 7L0D | [146], [169] |
| 14 | MPI1 | 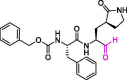 |
0.100 | Mpro | 7JPZ | [170] |
| 15 | MPI3 | 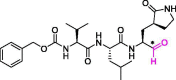 |
0.0085 | Mpro | 7JQ0 | [170] |
| 16 | MPI4 | 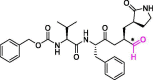 |
0.015 | Mpro | 7JQ1 | [170] |
| 17 | MPI5 | 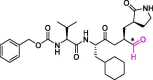 |
0.033 | Mpro | 7JQ2 | [170] |
| 18 | MPI6 | 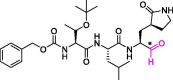 |
0.060 | Mpro | 7JQ3 | [170] |
| 19 | MPI7 | 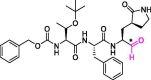 |
0.047 | Mpro | 7JQ4 | [170] |
| 20 | MPI8 | 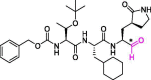 |
0.105 | Mpro | 7JQ5 | [170] |
| 21 | Narlaprevir | 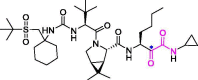 |
5.10 | Mpro | 6XQT | [150], [152] |
| 22 | N3 | 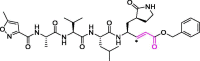 |
16.77 | Mpro | 6LU7 | [73], [171] |
| 23 | Pelitinib | 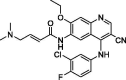 |
1.25 | Mpro | 7AXM | [155] |
| 24 | PF-00835231 | 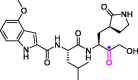 |
0.13 | Mpro | 6XHM | [172] |
| 25 | Suramin | 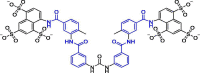 |
0.26 | RdRp | 7D4F | [173], [174] |
| 26 | Telaprevir | 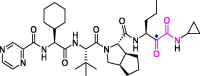 |
18.0 | Mpro | 6XQS | [150] |
| 27 | UAWJ246 | 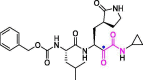 |
0.045 | Mpro | 6XBG | [89] |
| 28 | UAWJ247 | 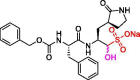 |
0.042 | Mpro | 6XA4 | [89] |
| 29 | UAWJ248 | 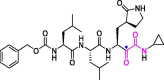 |
0.012 | Mpro | 6XBI | [89] |
| 30 | VIR250 | 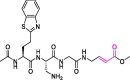 |
not reported | PLpro | 6WUU | [27] |
| 31 | VIR251 | 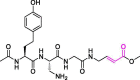 |
not reported | PLpro | 6WX4 | [27] |
| 32 | 5 h | 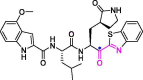 |
4.2 | Mpro | 7JKV | [175] |
| 33 | 11a | 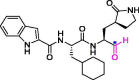 |
0.053 | Mpro | 6LZE | [75], [176] |
| 34 | 11b | 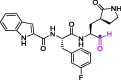 |
0.040 | Mpro | 6M0K | [75] |
| 35 | 15 l | 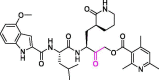 |
0.019 | Mpro | 7MBI | [177] |
| 36 | 2 | 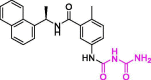 |
5.10 | PLpro | 7JIT | [19] |
| 37 | 3 | 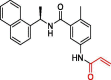 |
6.40 | PLpro | 7JIV | [19] |
| 38 | 14 | 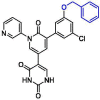 |
0.128 | Mpro | 7L12 | [137] |
| 39 | 21 | 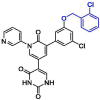 |
0.018 | Mpro | 7L13 | [137] |
| 40 | 26 | 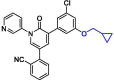 |
0.17 | Mpro | 7L14 | [137] |
6. Conclusion and future perspectives
COVID-19 is an ongoing global health crisis, with a current estimated 4,180,161 fatalities worldwide. Efficacious anti–SARS-CoV-2 drugs are not yet available, despite remdesivir having been granted full authorization in October 2020 by the U.S. FDA [114]. Over the past year, the scientific community has made remarkable inroads into developing promising anti–COVID-19 agents by employing multiple techniques and strategies. Among these approaches, the generation of X-ray crystal structures of relevant drug targets has provided effective guidance for anti–SARS-CoV-2 drug discovery and lead optimization.
Theoretically, components associated with each stage of the SARS-CoV-2 replication cycle could be comprehensively explored as promising therapeutic targets. Current anti–SARS-CoV-2 drug discovery and development strategies primarily focus on preventing viral replication via the targeting of Mpro, PLpro, or RdRp and blocking binding of the S protein to ACE2 receptors. To improve the precision design and screening of anti–SARS-CoV-2 drugs, we focused on all available X-ray co-crystal structures of the abovementioned targets in complex with newly identified small-molecule inhibitors, including natural products, FDA-approved drugs, and candidate drugs. The determined co-crystal structures offer a direct molecular-level perspective that could help elucidate the underlying mechanisms of action and provide guidance for future structure-based drug design. In the case of compound 13b, for example, a larger group at the P3 moiety could be designed to further improve the anti–SARS-CoV-2 activity in view of the space between Thr190, Gln189, and the pyridone ring of 13b. On the other hand, examination of the structures revealed no strong or durable effects for some non-covalent inhibitors (such as favipiravir, 23R, and perampanel analogue 5), supporting the need for further research focusing on covalent inhibitors to reduce “off-target” risks. In addition, combinations of several small molecules (such as linoleic acid and PF-00835231) with remdesivir have shown significant additive/synergistic anti–SARS-CoV-2 activities in vitro, presenting candidate drug combinations that may be effective in treating COVID-19 patients. Furthermore, the high mutation rate of SARS-CoV-2, resulting in the generation of novel variants, has led to increased pathogenicity [178], [179]. This may be one of the reasons for the failure of inhibitors against SARS-CoV-2. In this case, we suggest that collaborating groups should pay close attention to conserved binding sites available near the binding pocket during structure-based drug development, as this highly conserved region has promising druggable value but often goes unnoticed [180], [181], [182]. Despite substantiation of in vitro effectiveness and extensive clarification of associated mechanisms and interactions, direct clinical evidence of therapeutic efficacy for many of these agents is lacking at present. Nevertheless, we suggest that small-molecule inhibitors (including drug combinations) represent useful agents to prevent and control the COVID-19 pandemic.
CRediT authorship contribution statement
Zhonglei Wang: Conceptualization, Writing – original draft, Writing - review & editing, Visualization. Liyan Yang: Conceptualization, Writing - review & editing. Xian-En Zhao: Conceptualization, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the project of the PhD research start-up funds of Qufu Normal University, China (Grant No. 614901, and 615201). The authors, therefore, gratefully acknowledge the Qufu Normal University for the financial supports.
Contributor Information
Zhonglei Wang, Email: wangzl16@tsinghua.org.cn.
Liyan Yang, Email: yangly@iccas.ac.cn.
Xian-En Zhao, Email: xianenzhao@163.com.
References
- 1.https://covid19.who.int/, assessed on 29 July 2021.
- 2.Eurosurveillance Editorial Team Note from the editors: World Health Organization declares novel coronavirus (2019-nCoV) sixth public health emergency of international concern. Euro Surveil. 2020;25:200131e. doi: 10.2807/1560-7917.ES.2020.25.5.200131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldman J.D., Lye D.C.B., Hui D.S., Marks K.M., Bruno R., Montejano R. Remdesivir for 5 or 10 days in patients with severe Covid-19. N Engl J Med. 2020;383:1827–1837. doi: 10.1056/NEJMoa2015301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang G., Yang M.L., Duan Z.L., Liu F.L., Jin L., Long C.B. Dalbavancin binds ACE2 to block its interaction with SARS-CoV-2 spike protein and is effective in inhibiting SARS-CoV-2 infection in animal models. Cell Res. 2021;31:17–24. doi: 10.1038/s41422-020-00450-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan S., Yin X., Meng X., Chan J.F.W., Ye Z.W., Riva L. Clofazimine broadly inhibits coronaviruses including SARS-CoV-2. Nature. 2021;593:418–423. doi: 10.1038/s41586-021-03431-4. [DOI] [PubMed] [Google Scholar]
- 6.Han Y., Duan X., Yang L., Nilsson-Payant B.E., Wang P., Duan F. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature. 2021;589:270–275. doi: 10.1038/s41586-020-2901-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White K.M., Rosales R., Yildiz S., Kehrer T., Miorin L., Moreno E. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science. 2021;371:926–931. doi: 10.1126/science.abf4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li R., Hou Y., Huang J., Pan W., Ma Q., Shi Y. Lianhuaqingwen exerts anti-viral and anti-inflammatory activity against novel coronavirus (SARS-CoV-2) Pharmacol Res. 2020;156 doi: 10.1016/j.phrs.2020.104761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J., Wang Y.K., Gao Y., Hu L.S., Yang J.W., Wang J.R. Protection against COVID-19 injury by Qingfei Paidu decoction via anti-viral, anti-inflammatory activity and metabolic programming. Biomed Pharmacother. 2020;129 doi: 10.1016/j.biopha.2020.110281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.WHO Solidarity Trial Consortium Repurposed antiviral drugs for Covid-19—Interim WHO solidarity trial results. N Engl J Med. 2021;384:497–511. doi: 10.1056/NEJMoa2023184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamontagne F., Agoritsas T., Siemieniuk R., Rochwerg B., Bartoszko J., Askie L. A living WHO guideline on drugs to prevent covid-19. BMJ. 2021;372 doi: 10.1136/bmj.n526. [DOI] [PubMed] [Google Scholar]
- 12.Erlanson D.A. Many small steps towards a COVID-19 drug. Nat Commun. 2020;11:5048. doi: 10.1038/s41467-020-18710-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang S., Yang M., Hong Z., Zhang L., Huang Z., Chen X. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharma Sin B. 2020;10:1228–1238. doi: 10.1016/j.apsb.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mengist H.M., Fan X., Jin T. Designing of improved drugs for COVID-19: Crystal structure of SARS-CoV-2 main protease Mpro. Signal Transduc Tar. 2020;5:67. doi: 10.1038/s41392-020-0178-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ionescu M.I. An overview of the crystallized structures of the SARS-CoV-2. Protein J. 2020;39:600–618. doi: 10.1007/s10930-020-09933-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rut W., Groborz K., Zhang L., Sun X., Zmudzinski M., Pawlik B. SARS-CoV-2 Mpro inhibitors and activity-based probes for patient-sample imaging. Nat Chem Biol. 2021;17:222–228. doi: 10.1038/s41589-020-00689-z. [DOI] [PubMed] [Google Scholar]
- 17.Huynh T., Wang H., Luan B. In silico exploration of the molecular mechanism of clinically oriented drugs for possibly inhibiting SARS-CoV-2's main protease. J Phys Chem Lett. 2020;11:4413–4420. doi: 10.1021/acs.jpclett.0c00994. [DOI] [PubMed] [Google Scholar]
- 18.Fu L., Ye F., Feng Y., Yu F., Wang Q., Wu Y. Both boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat Commun. 2020;11:4417. doi: 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osipiuk J., Azizi S.A., Dvorkin S., Endres M., Jedrzejczak R., Jones K.A. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat Commun. 2021;12:743. doi: 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poater A. Michael acceptors tuned by the pivotal aromaticity of histidine to block COVID-19 activity. J Phys Chem Lett. 2020;11:6262–6265. doi: 10.1021/acs.jpclett.0c01828. [DOI] [PubMed] [Google Scholar]
- 22.Peng Q., Peng R., Yuan B., Zhao J., Wang M., Wang X. Structural and biochemical characterization of the nsp12-nsp7-nsp8 core polymerase complex from SARS-CoV-2. Cell Rep. 2020;31 doi: 10.1016/j.celrep.2020.107774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Byléhn F., Menéndez C.A., Perez-Lemus G.R., Alvarado W., de Pablo J.J. Modeling the binding mechanism of remdesivir, favilavir, and ribavirin to SARS-CoV-2 RNA-dependent RNA polymerase. ACS Central Sci. 2021;7:164–174. doi: 10.1021/acscentsci.0c01242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hillen H.S., Kokic G., Farnung L., Dienemann C., Tegunov D., Cramer P. Structure of replicating SARS-CoV-2 polymerase. Nature. 2020;584:154–156. doi: 10.1038/s41586-020-2368-8. [DOI] [PubMed] [Google Scholar]
- 25.Tegally H., Wilkinson E., Giovanetti M., Iranzadeh A., Fonseca V., Giandhari J. Emergence of a SARS-CoV-2 variant of concern with mutations in spike glycoprotein. Nature. 2021;592:438–443. doi: 10.1038/s41586-021-03402-9. [DOI] [PubMed] [Google Scholar]
- 26.Klemm T., Ebert G., Calleja D.J., Allison C.C., Richardson L.W., Bernardini J.P. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020;39 doi: 10.15252/embj.2020106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rut W, Lv Z, Zmudzinski M, Patchett S, Nayak D, Snipas SJ, et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti-COVID-19 drug design. Sci Adv 2020;6:eabd4596. [DOI] [PMC free article] [PubMed]
- 28.Rathnayake AD, Zheng J, Kim Y, Perera KD, Mackin S, Meyerholz DK, et al. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV-infected mice. Sci Transl Med 2020;12:eabc5332. [DOI] [PMC free article] [PubMed]
- 29.Bharadwaj S., Dubey A., Yadava U., Mishra S.K., Kang S.G., Dwivedi V.D. Exploration of natural compounds with anti-SARS-CoV-2 activity via inhibition of SARS-CoV-2 Mpro. Brief Bioinform. 2021;22:1361–1377. doi: 10.1093/bib/bbaa382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kneller D.W., Phillips G., O'Neill H.M., Jedrzejczak R., Stols L., Langan P. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat Commun. 2020;11:3202. doi: 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368:779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 33.Wang Q., Zhang Y., Wu L., Niu S., Song C., Zhang Z. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020;181:894–904.e9. doi: 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gil C., Ginex T., Maestro I., Nozal V., Barrado-Gil L., Cuesta-Geijo M.Á. COVID-19: Drug targets and potential treatments. J Med Chem. 2020;63:12359–12386. doi: 10.1021/acs.jmedchem.0c00606. [DOI] [PubMed] [Google Scholar]
- 35.Xu J., Xue Y., Zhou R., Shi P.Y., Li H., Zhou J. Drug repurposing approach to combating coronavirus: Potential drugs and drug targets. Med Res Rev. 2021;41:1375–1426. doi: 10.1002/med.21763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu Y., Li Z., Zhao Y.S., Huang Y.Y., Jiang M.Y., Luo H.B. Therapeutic targets and potential agents for the treatment of COVID-19. Med Res Rev. 2021;41:1775–1797. doi: 10.1002/med.21776. [DOI] [PubMed] [Google Scholar]
- 37.Cannalire R., Cerchia C., Beccari A.R., Leva F.S.D., Summa V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: State of the art and future opportunities. J Med Chem. 2021 doi: 10.1021/acs.jmedchem.0c01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Banerjee R., Perera L., Tillekeratne L.M.V. Potential SARS-CoV-2 main protease inhibitors. Drug Discov Today. 2021;26:804–816. doi: 10.1016/j.drudis.2020.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tian L., Qiang T., Liang C., Ren X., Jia M., Zhang J. RNA-dependent RNA polymerase (RdRp) inhibitors: The current landscape and repurposing for the COVID-19 pandemic. Eur J Med Chem. 2021;213 doi: 10.1016/j.ejmech.2021.113201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y., Liang C., Xin L., Ren X., Tian L., Ju X. The development of Coronavirus 3C-Like protease (3CLpro) inhibitors from 2010 to 2020. Eur J Med Chem. 2020;206 doi: 10.1016/j.ejmech.2020.112711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tripathi N., Tripathi N., Goshisht M.K. COVID-19: Inflammatory responses, structure-based drug design and potential therapeutics. Mol Divers. 2021 doi: 10.1007/s11030-020-10176-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiong M., Su H., Zhao W., Xie H., Shao Q., Xu Y. What coronavirus 3C-like protease tells us: From structure, substrate selectivity, to inhibitor design. Med Res Rev. 2021;41:1965–1998. doi: 10.1002/med.21783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Picarazzi F., Vicenti I., Saladini F., Zazzi M., Mori M. Targeting the RdRp of emerging RNA viruses: The structure-based drug design challenge. Molecules. 2020;25:5695. doi: 10.3390/molecules25235695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pinto G.P., Vavra O., Marques S.M., Filipovic J., Bednar D., Damborsky J. Screening of world approved drugs against highly dynamical spike glycoprotein of SARS-CoV-2 using caverdock and machine learning. Comput Struct Biotec. 2021;19:3187–3197. doi: 10.1016/j.csbj.2021.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pan B., Fang S., Zhang J., Pan Y., Liu H., Wang Y. Chinese herbal compounds against SARS-CoV-2: Puerarin and quercetin impair the binding of viral S-protein to ACE2 receptor. Comput Struct Biotec. 2020;18:3518–3527. doi: 10.1016/j.csbj.2020.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh N., Villoutreix B.O. Resources and computational strategies to advance small molecule SARS-CoV-2 discovery: Lessons from the pandemic and preparing for future health crises. Comput Struct Biotec. 2021;19:2537–2548. doi: 10.1016/j.csbj.2021.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jade D., Ayyamperumal S., Tallapaneni V., Nanjan C.M.J., Barge S., Mohan S. Virtual high throughput screening: Potential inhibitors for SARS-CoV-2 PLPRO and 3CLPRO proteases. Eur J Pharmacol. 2021;901 doi: 10.1016/j.ejphar.2021.174082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nallusamy S., Mannu J., Ravikumar C., Angamuthu K., Nathan B., Nachimuthu K. Exploring phytochemicals of traditional medicinal plants exhibiting inhibitory activity against main protease, Spike glycoprotein, RNA-dependent RNA polymerase and non-structural proteins of SARS-CoV-2 through virtual screening. Front Pharmacol. 2021;12 doi: 10.3389/fphar.2021.667704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li G., Lou H.X. Strategies to diversify natural products for drug discovery. Med Res Rev. 2018;38:1255–1294. doi: 10.1002/med.21474. [DOI] [PubMed] [Google Scholar]
- 50.Zhang L., Song J., Kong L., Yuan T., Li W., Zhang W. The strategies and techniques of drug discovery from natural products. Pharmacol Therapeut. 2020;216 doi: 10.1016/j.pharmthera.2020.107686. [DOI] [PubMed] [Google Scholar]
- 51.Atanasov A.G., Zotchev S.B., Dirsch V.M., Supuran C.T. Natural products in drug discovery: Advances and opportunities. Nat Rev Drug Discov. 2021;20:200–216. doi: 10.1038/s41573-020-00114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harvey A.L., Edrada-Ebel R., Quinn R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov. 2015;14:111–129. doi: 10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]
- 53.Wang Z., Yang L. Turning the tide: Natural products and natural-product-inspired chemicals as potential counters to SARS-CoV-2 infection. Front Pharmacol. 2020;11:1013. doi: 10.3389/fphar.2020.01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Christy M.P., Uekusa Y., Gerwick L., Gerwick W.H. Natural products with potential to treat RNA virus pathogens including SARS-CoV-2. J Nat Prod. 2021;84:161–182. doi: 10.1021/acs.jnatprod.0c00968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang L., Wang Z. Natural products, alone or in combination with FDA-approved drugs, to treat COVID-19 and lung cancer. Biomedicines. 2021;9:689. doi: 10.3390/biomedicines9060689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang J., Tao G., Liu J., Cai J., Huang Z., Chen J.X. Current prevention of COVID-19: Natural products and herbal medicine. Front Pharmacol. 2020;11 doi: 10.3389/fphar.2020.588508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Z., Yang L. Chinese herbal medicine: Fighting SARS-CoV-2 infection on all fronts. J Ethnopharmacol. 2021;270 doi: 10.1016/j.jep.2021.113869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khalifa S.A.M., Yosri N., El-Mallah M.F., Guo R.G.Z., Musharraf S.G., Du M. Screening for natural and derived bioactive compounds in preclinical and clinical studies: One of the frontlines of fighting the coronaviruses pandemic. Phytomedicine. 2021;85 doi: 10.1016/j.phymed.2020.153311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hu L., Wang Y., Sun H., Xiong Y., Zhong L., Wu Z. An untargeted metabolomics approach to investigate the wine-processed mechanism of Scutellariae radix in acute lung injury. J Ethnopharmacol. 2020;253 doi: 10.1016/j.jep.2020.112665. [DOI] [PubMed] [Google Scholar]
- 60.Chen Q.Y., Wang C.Q., Yang Z.W., Tang Q., Tan H.R., Wang X. Differences in anti-inflammatory effects between two specifications of Scutellariae Radix in LPS-induced macrophages in vitro. Chin J Nat Medicines. 2017;15:515–524. doi: 10.1016/S1875-5364(17)30077-8. [DOI] [PubMed] [Google Scholar]
- 61.Liu H., Ye F., Sun Q., Liang H., Li C., Li S. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. J Enzym Inhib Med Ch. 2021;36:497–503. doi: 10.1080/14756366.2021.1873977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu B.Y., Li L., Liu G.L., Ding W., Chang W.G., Xu T. Baicalein attenuates cardiac hypertrophy in mice via suppressing oxidative stress and activating autophagy in cardiomyocytes. Acta Pharmacol Sin. 2021;42:701–714. doi: 10.1038/s41401-020-0496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang J., Deng Y., Cheng B., Huang Y., Meng Y., Zhong K. Protective effects and molecular mechanisms of baicalein on thioacetamide-induced toxicity in zebrafish larvae. Chemosphere. 2020;256 doi: 10.1016/j.chemosphere.2020.127038. [DOI] [PubMed] [Google Scholar]
- 64.Song J., Zhang L., Xu Y., Yang D., Zhang L., Yang S. The comprehensive study on the therapeutic effects of baicalein for the treatment of COVID-19 in vivo and in vitro. Biochem Pharmacol. 2021;183 doi: 10.1016/j.bcp.2020.114302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Su H.X., Yao S., Zhao W.F., Li M.J., Liu J., Shang W.J. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol Sin. 2020;41:1167–1177. doi: 10.1038/s41401-020-0483-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu H., Qi Q., Yan X. Myricetin ameliorates sepsis-associated acute lung injury in a murine sepsis model. N-S Arch Pharmacol. 2021;394:165–175. doi: 10.1007/s00210-020-01880-8. [DOI] [PubMed] [Google Scholar]
- 67.Song X., Tan L., Wang M., Ren C., Guo C., Yang B. Myricetin: A review of the most recent research. Biomed Pharmacother. 2021;134 doi: 10.1016/j.biopha.2020.111017. [DOI] [PubMed] [Google Scholar]
- 68.Su H., Yao S., Zhao W., Zhang Y., Liu J., Shao Q. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat Commun. 2021;12:3623. doi: 10.1038/s41467-021-23751-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kuzikov M., Costanzi E., Reinshagen J., Esposito F., Vangeel L. Identification of inhibitors of SARS-CoV-2 3CL-Pro enzymatic activity using a small molecule in vitro repurposing screen. ACS Pharmacol Transl Sci. 2021;4(3):1096–1110. doi: 10.1021/acsptsci.0c00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sharma J., Bhardwaj V.K., Singh R., Rajendran V., Purohit R., Kumar S. An in-silico evaluation of different bioactive molecules of tea for their inhibition potency against non structural protein-15 of SARS-CoV-2. Food Chem. 2021;346 doi: 10.1016/j.foodchem.2020.128933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kumar A., Shashni S., Kumar P., Pant D., Singh A., Verma R.K. Phytochemical constituents, distributions and traditional usages of Arnebia euchroma: A review. J Ethnopharmacol. 2021;271 doi: 10.1016/j.jep.2021.113896. [DOI] [PubMed] [Google Scholar]
- 72.Chen X., Yang L., Zhang N., Turpin J.A., Buckheit R.W., Osterling C. Shikonin, a component of chinese herbal medicine, inhibits chemokine receptor function and suppresses human immunodeficiency virus type 1. Antimicrob Agents Ch. 2003;47:2810–2816. doi: 10.1128/AAC.47.9.2810-2816.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 74.Li J., Zhou X., Zhang Y., Zhong F., Lin C., McCormick P.J. Crystal structure of SARS-CoV-2 main protease in complex with the natural product inhibitor shikonin illuminates a unique binding mode. Sci Bull. 2021;66:661–663. doi: 10.1016/j.scib.2020.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dai W., Zhang B., Jiang X.M., Su H., Li J., Zhao Y. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ma C., Hu Y., Townsend J.A., Lagarias P.I., Marty M.T., Kolocouris A. Ebselen, disulfiram, carmofur, PX-12, tideglusib, and shikonin are nonspecific promiscuous SARS-CoV-2 main protease inhibitors. ACS Pharmacol Transl Sci. 2020;3:1265–1277. doi: 10.1021/acsptsci.0c00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ahmed F., Sharma M., Al-Ghamdi A.A., Al-Yami S.M., Al-Salami A.M., Refai M.Y. A comprehensive analysis of cis-acting RNA elements in the SARS-CoV-2 genome by a bioinformatics approach. Front Genet. 2020;11 doi: 10.3389/fgene.2020.572702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Viswanathan T., Arya S., Chan S.H., Qi S., Dai N., Misra A. Structural basis of RNA cap modification by SARS-CoV-2. Nat Commun. 2020;11:3718. doi: 10.1038/s41467-020-17496-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bhattacharya A., Sharma M., Pakkinathan C., Rosen B.P., Leprohon P., Ouellette M. Genomewide analysis of mode of action of the s-adenosylmethionine analogue sinefungin in leishmania infantum. mSystems. 2019;4:e00416–e419. doi: 10.1128/mSystems.00416-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mahalapbutr P., Kongtaworn N., Rungrotmongkol T. Structural insight into the recognition of s-adenosyl-l-homocysteine and sinefungin in SARS-CoV-2 Nsp16/Nsp10 RNA cap 2’-O-methyltransferase. Comput Struct Biotec. 2020;18:2757–2765. doi: 10.1016/j.csbj.2020.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.El Hassab M.A., Ibrahim T.M., Al-Rashood S.T., Alharbi A., Eskandrani R.O., Eldehna W.M. In silico identification of novel SARS-COV-2 2′-O-methyltransferase (nsp16) inhibitors: Structure-based virtual screening, molecular dynamics simulation and MM-PBSA approaches. J Enzym Inhib Med Ch. 2021;36:727–736. doi: 10.1080/14756366.2021.1885396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Krafcikova P., Silhan J., Nencka R., Boura E. Structural analysis of the SARS-CoV-2 methyltransferase complex involved in RNA cap creation bound to sinefungin. Nat Commun. 2020;11:3717. doi: 10.1038/s41467-020-17495-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Simopoulos A.P., Serhan C.N., Bazinet R.P. The need for precision nutrition, genetic variation and resolution in Covid-19 patients. Mol Aspects Med. 2021;77 doi: 10.1016/j.mam.2021.100943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Toelzer C., Gupta K., Yadav S.K.N., Borucu U., Davidson A.D., Williamson M.K. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science. 2020;370:725–730. doi: 10.1126/science.abd3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Oliveira A.S.F., Shoemark D.K., Ibarra A.A., Davidson A.D., Berger I. The fatty acid site is coupled to functional motifs in the SARS-CoV-2 spike protein and modulates spike allosteric behaviour. bioRxiv. 2021 doi: 10.1101/2021.06.07.447341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.del Cerro S., Larson J., Oliver M.W., Lynch G. Development of hippocampal long-term potentiation is reduced by recently introduced calpain inhibitors. Brain Res. 1990;530:91–95. doi: 10.1016/0006-8993(90)90660-4. [DOI] [PubMed] [Google Scholar]
- 87.Hu Y., Ma C., Szeto T., Hurst B., Tarbet B., Wang J. Boceprevir, calpain inhibitors II and XII, and GC-376 have broad-spectrum antiviral activity against coronaviruses. ACS Infect Dis. 2021;7:586–597. doi: 10.1021/acsinfecdis.0c00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sacco MD, Ma C, Lagarias P, Gao A, Townsend JA, Meng X, et al. SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci Adv 2020;6:eabe0751. [DOI] [PMC free article] [PubMed]
- 90.Li Y., Cao L., Li G., Cong F., Li Y., Sun J. Remdesivir metabolite GS-441524 effectively inhibits SARS-CoV-2 infection in mouse models. J Med Chem. 2021 doi: 10.1021/acs.jmedchem.0c01929. [DOI] [PubMed] [Google Scholar]
- 91.Kokic G., Hillen H.S., Tegunov D., Dienemann C., Seitz F., Schmitzova J. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat Commun. 2021;12:279. doi: 10.1038/s41467-020-20542-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang Z., Yang L. GS-5734: a potentially approved drug by FDA against SARS-CoV-2. New J Chem. 2020;44:12417–12429. [Google Scholar]
- 93.Malin J.J., Suárez I., Priesner V., Fätkenheuer G., Rybniker J. Remdesivir against COVID-19 and other viral diseases. Clin Microbiol Rev. 2021;34:e00162–e220. doi: 10.1128/CMR.00162-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Metobo S.E., Xu J., Saunders O.L., Butler T., Aktoudianakis E., Cho A. Practical synthesis of 1′-substituted tubercidin C-nucleoside analogs. Tetrahedron Lett. 2012;53:484–486. [Google Scholar]
- 95.Warren T.K., Jordan R., Lo M.K., Ray A.S., Mackman R.L., Soloveva V. Therapeutic Efficacy of The Small Molecule GS-5734 Against Ebola virus in rhesus monkeys. Nature. 2016;531:381–385. doi: 10.1038/nature17180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Siegel D., Hui H.C., Doerffler E., Clarke M.O., Chun K., Zhang L. Discovery and synthesis of a phosphoramidate prodrug of a pyrrolo[2,1-f][triazin-4-amino] adenine C-nucleoside (GS-5734) for the treatment of Ebola and emerging viruses. J Med Chem. 2017;60:1648–1661. doi: 10.1021/acs.jmedchem.6b01594. [DOI] [PubMed] [Google Scholar]
- 97.Zhang L., Zhang D., Wang X., Yuan C., Li Y., Jia X. 1′-Ribose cyano substitution allows Remdesivir to effectively inhibit nucleotide addition and proofreading during SARS-CoV-2 viral RNA replication. Phys Chem Chem Phys. 2021;23:5852–5863. doi: 10.1039/d0cp05948j. [DOI] [PubMed] [Google Scholar]
- 98.Wang M., Zhang L., Huo X., Zhang Z., Yuan Q., Li P. Catalytic asymmetric synthesis of the anti-COVID-19 drug remdesivir. Angew Chem Int Edit. 2020;59:20814–20819. doi: 10.1002/anie.202011527. [DOI] [PubMed] [Google Scholar]
- 99.Yan V.C., Muller F.L. Advantages of the parent nucleoside GS-441524 over remdesivir for Covid-19 treatment. ACS Med Chem Lett. 2020;11:1361–1366. doi: 10.1021/acsmedchemlett.0c00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wakchaure P.D., Ghosh S., Ganguly B. Revealing the inhibition mechanism of RNA-dependent RNA polymerase (RdRp) of SARS-CoV-2 by remdesivir and nucleotide analogues: A molecular dynamics simulation study. J Phys Chem B. 2020;124(47):10641–10652. doi: 10.1021/acs.jpcb.0c06747. [DOI] [PubMed] [Google Scholar]
- 101.Yin W., Mao C., Luan X., Shen D.D., Shen Q., Su H. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science. 2020;368:1499–1504. doi: 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sztain T., Amaro R., McCammon J.A. Elucidation of cryptic and allosteric pockets within the SARS-CoV-2 main protease. J Chem Inf Model. 2021 doi: 10.1021/acs.jcim.1c00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zimmerman M.I., Porter J.R., Ward M.D., Singh S., Vithani N., Meller A. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nature Chem. 2021;13:651–659. doi: 10.1038/s41557-021-00707-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zuzic L, Samsudin F, Shivgan AT, Raghuvamsi PV, Marzinek JK, Boags A, Uncovering cryptic pockets in the SARS-CoV-2 spike glycoprotein. 2021.doi: https://doi.org/10.1101/2021.05.05.442536. [DOI] [PMC free article] [PubMed]
- 105.Srivastava M., Mittal L., Kumari A., Asthana S. Molecular dynamics simulations reveal the interaction fingerprint of remdesivir triphosphate pivotal in allosteric regulation of SARS-CoV-2 RdRp. Front Mol Biosci. 2021;8 doi: 10.3389/fmolb.2021.639614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Khan F.I., Kang T., Ali H., Lai D. Remdesivir strongly binds to RNA-dependent RNA polymerase, membrane protein, and main protease of SARS-CoV-2: Indication from molecular modelling and simulations. Front Pharmacol. 2021;12 doi: 10.3389/fphar.2021.710778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sheahan TP, Sims AC, Graham RL, Menachery VD, Gralinski LE, Case JB, et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med 2017;9:eaal3653. [DOI] [PMC free article] [PubMed]
- 108.Sheahan T.P., Sims A.C., Leist S.R., Schäfer A., Won J., Brown A.J. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat Commun. 2020;11:222. doi: 10.1038/s41467-019-13940-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brown A.J., Wona J.J., Grahama R.L., Dinnon K.H., III, Simsa A.C., Feng J.Y. Broad spectrum antiviral remdesivir inhibits human endemic and zoonotic deltacoronaviruses with a highly divergent RNA dependent RNA polymerase. Antiviral Res. 2019;169 doi: 10.1016/j.antiviral.2019.104541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tchesnokov E.P., Raeisimakiani P., Ngure M., Marchant D., Götte M. Recombinant RNA-dependent RNA polymerase complex of Ebola virus. Sci Rep. 2018;8:3970. doi: 10.1038/s41598-018-22328-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McMullan L.K., Flint M., Chakrabarti A., Guerrero L., Lo M.K., Porter D. Characterisation of infectious Ebola virus from the ongoing outbreak to guide response activities in the Democratic Republic of the Congo: A phylogenetic and in vitro analysis. Lancet Infect Dis. 2019;19:1023–1032. doi: 10.1016/S1473-3099(19)30291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.de Wit E., Feldmann F., Cronin J., Jordan R., Okumura A., Thomas T. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc Natl Acad Sci USA. 2020;117:6771–6776. doi: 10.1073/pnas.1922083117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jacobs M., Rodger A., Bell D.J., Bhagani S., Cropley I., Filipe A. Late Ebola virus relapse causing meningoencephalitis: A case report. Lancet. 2016;388:498–503. doi: 10.1016/S0140-6736(16)30386-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schwartz I.S., Heil E.L., McCreary E.K. Remdesivir: A pendulum in a pandemic. BMJ. 2020;371 doi: 10.1136/bmj.m4560. [DOI] [PubMed] [Google Scholar]
- 115.Kalil A.C., Patterson T.F., Mehta A.K., Tomashek K.M., Wolfe C.R., Ghazaryan V. Baricitinib plus remdesivir for hospitalized adults with Covid-19. N Engl J Med. 2021;384:795–807. doi: 10.1056/NEJMoa2031994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Garibaldi BT, Wang K, Robinson ML, Zeger SL, Roche KB, Wang MC, et al. Effectiveness of remdesivir with and without dexamethasone in hospitalized patients with COVID-19. medRxiv 2020. DOI: 10.1101/2020.11.19.20234153.
- 117.Li G., Ruan S., Zhao X., Liu Q., Dou Y., Mao F. Transcriptomic signatures and repurposing drugs for COVID-19 patients: Findings of bioinformatics analyses. Comput Struct Biotec. 2021;19:1–15. doi: 10.1016/j.csbj.2020.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bairi K.E., Trapani D., Petrillo A., Page C.L., Zbakh H., Daniele B. Repurposing anticancer drugs for the management of COVID-19. Eur J Cancer. 2020;141:40–61. doi: 10.1016/j.ejca.2020.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shende P., Khanolkar B., Gaud R.S. Drug repurposing: New strategies for addressing COVID-19 outbreak. Expert Rev Anti-Infe. 2021;19:689–706. doi: 10.1080/14787210.2021.1851195. [DOI] [PubMed] [Google Scholar]
- 120.Yuan S., Chan J.F.W., Chik K.K.H., Chan C.C.Y., Tsang J.O.L., Liang R. Discovery of the FDA-approved drugs bexarotene, cetilistat, diiodohydroxyquinoline, and abiraterone as potential COVID-19 treatments with a robust two-tier screening system. Pharmacol Res. 2020;159 doi: 10.1016/j.phrs.2020.104960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shiraki K., Daikoku T. Favipiravir, an anti-influenza drug against life-threatening RNA virus infections. Pharmacol Therapeut. 2020;209 doi: 10.1016/j.pharmthera.2020.107512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.McCullough P.A. Favipiravir and the need for early ambulatory treatment of SARS-CoV-2 infection (COVID-19) Antimicrob Agents Ch. 2020;64:e02017–e2020. doi: 10.1128/AAC.02017-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Driouich J.S., Cochin M., Lingas G., Moureau G., Touret F., Petit P.R. Favipiravir antiviral efficacy against SARS-CoV-2 in a hamster model. Nat Commun. 2021;12:1735. doi: 10.1038/s41467-021-21992-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Abdelnabi R., Foo C.S., Kaptein S.J.F., Zhang X., Langendries L., Vangeel L. The combined treatment of molnupiravir and favipiravir results in a marked potentiation of antiviral efficacy in a SARS-CoV-2 hamster infection model. bioRxiv. 2021 doi: 10.1101/2020.12.10.419242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Udwadia Z.F., Singh P., Barkate H., Patil S., Rangwala S., Pendse A. Efficacy and safety of favipiravir, an oral RNA-dependent RNA polymerase inhibitor, in mild-to-moderate COVID-19: A randomized, comparative, open-label, multicenter, phase 3 clinical trial. Int J Infect Dis. 2021;103:62–71. doi: 10.1016/j.ijid.2020.11.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Doi Y, Hibino M, Hase R, Yamamoto M, Kasamatsu Y, Hirose M, et al. A prospective, randomized, open-label trial of early versus late favipiravir in hospitalized patients with COVID-19. Antimicrob Agents Ch 2020;64:e01897-20. [DOI] [PMC free article] [PubMed]
- 128.Chen C., Huang J., Cheng Z., Wu J., Chen S., Zhang Y. Favipiravir versus arbidol for COVID-19: A randomized clinical trial. medRxiv. 2020 doi: 10.1101/2020.03.17.20037432. [DOI] [Google Scholar]
- 129.Cai Q., Yang M., Liu D., Chen J., Shu D., Xia J. Experimental treatment with favipiravir for COVID-19: An open-label control study. Engineering. 2020;6:1192–1198. doi: 10.1016/j.eng.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Naydenova K., Muir K.W., Wu L.F., Zhang Z., Coscia F., Peet M.J. Structure of the SARS-CoV-2 RNA-dependent RNA polymerase in the presence of favipiravir-RTP. Proc Natl Acad Sci USA. 2021;118 doi: 10.1073/pnas.2021946118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Peng Q., Peng R., Yuan B., Wang M., Zhao J., Fu L. Structural basis of SARS-CoV-2 polymerase inhibition by favipiravir. Innovation. 2021;2 doi: 10.1016/j.xinn.2021.100080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Joshi S., Parkar J., Ansari A., Vora A., Talwar D., Tiwaskar M. Role of favipiravir in the treatment of COVID-19. Int J Infect Dis. 2021;102:501–508. doi: 10.1016/j.ijid.2020.10.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Celik I., Erol M., Duzgun Z. In silico evaluation of potential inhibitory activity of remdesivir, favipiravir, ribavirin and galidesivir active forms on SARS-CoV-2 RNA polymerase. Mol Divers. 2021 doi: 10.1007/s11030-021-10215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lee T.S., Allen B.K., Giese T.J., Guo Z., Li P., Lin C. Alchemical binding free energy calculations in AMBER20: Advances and best practices for drug discovery. J Chem Inf Model. 2020;60:5595–5623. doi: 10.1021/acs.jcim.0c00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cournia Z., Allen B., Sherman W. Relative binding free energy calculations in drug discovery: Recent advances and practical considerations. J Chem Inf Model. 2017;57:2911–2937. doi: 10.1021/acs.jcim.7b00564. [DOI] [PubMed] [Google Scholar]
- 136.Wang L., Wu Y., Deng Y., Kim B., Pierce L., Krilov G. Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J Am Chem Soc. 2015;137:2695–2703. doi: 10.1021/ja512751q. [DOI] [PubMed] [Google Scholar]
- 137.Zhang C.H., Stone E.A., Deshmukh M., Ippolito J.A., Ghahremanpour M.M., Tirado-Rives J. Potent noncovalent inhibitors of the main protease of SARS-CoV-2 from molecular sculpting of the drug perampanel guided by free energy perturbation calculations. ACS Cent Sci. 2021;7:467–475. doi: 10.1021/acscentsci.1c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ghahremanpour M.M., Tirado-Rives J., Deshmukh M., Ippolito J.A., Zhang C.H., de Vaca I.C. Identification of 14 known drugs as inhibitors of the main protease of SARS-CoV-2. ACS Med Chem Lett. 2020;11:2526–2533. doi: 10.1021/acsmedchemlett.0c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pelly S., Liotta D. Potent SARS-CoV-2 direct-acting antivirals provide an important complement to COVID-19 vaccines. ACS Cent Sci. 2021;7(3):396–399. doi: 10.1021/acscentsci.1c00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.McKee D.L., Sternberg A., Stange U., Laufer S., Naujokat C. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol Res. 2020;157 doi: 10.1016/j.phrs.2020.104859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Li Z., Yang L. Underlying mechanisms and candidate drugs for COVID-19 based on the connectivity map database. Front Genet. 2020;11 doi: 10.3389/fgene.2020.558557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang L., Lin D., Kusov Y., Nian Y., Ma Q., Wang J. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: Structure-based design, synthesis, and activity assessment. J Med Chem. 2020;63:4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- 143.Somboon T., Mahalapbutr P., Sanachai K., Maitarad P., Lee V.S., Hannongbua S. Computational study on peptidomimetic inhibitors against SARS-CoV-2 main protease. J Mol Liq. 2021;322 doi: 10.1016/j.molliq.2020.114999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kumari A., Mittal L., Srivastava M., Asthana S. Binding mode characterization of 13b in the monomeric and dimeric states of SARS-CoV-2 main protease using molecular dynamics simulations. J Biomol Struct Dyn. 2021 doi: 10.1080/07391102.2021.1927844. [DOI] [PubMed] [Google Scholar]
- 145.Saha D., Kharbanda A., Yan W., Lakkaniga N.R., Frett B., Li H.Y. The exploration of chirality for improved druggability within the human kinome. J Med Chem. 2020;63:441–469. doi: 10.1021/acs.jmedchem.9b00640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kitamura N., Sacco M.D., Ma C., Hu Y., Townsend J.A., Meng X. Expedited approach toward the rational design of noncovalent SARS-CoV-2 main protease inhibitors. J Med Chem. 2021 doi: 10.1021/acs.jmedchem.1c00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jacobs J., Grum-Tokars V., Zhou Y., Turlington M., Saldanha S.A., Chase P. Discovery, synthesis, and structure-based optimization of a series of N-(tert-Butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J Med Chem. 2013;56:534–546. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hoffman R.L., Kania R.S., Brothers M.A., Davies J.F., Ferre R.A., Gajiwala K.S. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J Med Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.de Vries M., Mohamed A., Prescott R.A., Valero-Jimenez A., Desvignes L., O’Connor R. A comparative analysis of SARS-CoV-2 antivirals in human airway models characterizes 3CLpro inhibitor PF-00835231 as a potential new treatment for COVID-19. J Virol. 2021;95:e01819–e1820. doi: 10.1128/JVI.01819-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kneller D.W., Galanie S., Phillips G., O'Neill H.M., Coates L., Kovalevsky A. Malleability of the SARS-CoV-2 3CL Mpro active-site cavity facilitates binding of clinical antivirals. Structure. 2020;28:1313–1320. doi: 10.1016/j.str.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Oerlemans R., Ruiz-Moreno A.J., Cong Y., Kumar N.D., Velasco-Velazquez M.A., Neochoritis C.G. Repurposing the HCV NS3-4A protease drug boceprevir as COVID-19 therapeutics. RSC Med Chem. 2021;12:370–379. doi: 10.1039/d0md00367k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Manandhar A., Blass B.E., Colussi D.J., Almi I., Abou-Gharbia M., Klein M.L. Targeting SARS-CoV-2 M3CLpro by HCV NS3/4a inhibitors: In silico modeling and in vitro screening. J Chem Inf Model. 2021;61:1020–1032. doi: 10.1021/acs.jcim.0c01457. [DOI] [PubMed] [Google Scholar]
- 153.Rawson J.M.O., Duchon A., Nikolaitchik O.A., Pathak V.K., Hu W.S. Development of a cell-based luciferase complementation assay for identification of SARS-CoV-2 3CLpro inhibitors. Viruses. 2021;13:173. doi: 10.3390/v13020173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu C., Boland S., Scholle M.D., Bardiot D., Marchand A., Chaltin P. Dual inhibition of SARS-CoV-2 and human rhinovirus with protease inhibitors in clinical development. Antivir Res. 2021;187 doi: 10.1016/j.antiviral.2021.105020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Guenther S., Reinke P.Y.A., Fernandez-Garcia Y., Lieske J., Lane T.J., Ginn H.M. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science. 2021;372:642–646. doi: 10.1126/science.abf7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jin Z., Zhao Y., Sun Y., Zhang B., Wang H., Wu Y. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat Struct Mol Biol. 2020;27:529–532. doi: 10.1038/s41594-020-0440-6. [DOI] [PubMed] [Google Scholar]
- 157.Gao J., Zhang L., Liu X., Li F., Ma R., Zhu Z. Repurposing low-molecular-weight drugs against the main protease of severe acute respiratory syndrome coronavirus 2. J Phys Chem Lett. 2020;11:7267–7272. doi: 10.1021/acs.jpclett.0c01894. [DOI] [PubMed] [Google Scholar]
- 158.Amporndanai K., Meng X., Shang W., Jin Z., Rogers M., Zhao Y. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat Commun. 2021;12:3061. doi: 10.1038/s41467-021-23313-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Weglarz-Tomczak E., Tomczak J.M., Talma M., Burda-Grabowska M., Giurg M., Brul S. Identification of ebselen and its analogues as potent covalent inhibitors of papain-like protease from SARS-CoV-2. Sci Rep. 2021;11:3640. doi: 10.1038/s41598-021-83229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vuong W., Khan M.B., Fischer C., Arutyunova E., Lamer T., Shields J. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat Commun. 2020;11:4282. doi: 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Caceres C.J., Cardenas-Garcia S., Carnaccini S., Seibert B., Rajao D.S., Wang J. Efficacy of GC-376 against SARS-CoV-2 virus infection in the K18 hACE2 transgenic mouse model. Sci Rep. 2021;11:9609. doi: 10.1038/s41598-021-89013-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gao X., Qin B., Chen P., Zhu K., Hou P., Wojdyla J.A. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm Sin B. 2021;11:237–245. doi: 10.1016/j.apsb.2020.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fu Z., Huang B., Tang J., Liu S., Liu M., Ye Y. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat Commun. 2021;12:488. doi: 10.1038/s41467-020-20718-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jamalan M., Barzegari E., Gholami-Borujeni F. Structure-based screening to discover new inhibitors for papain-like proteinase of SARS-CoV-2: An in silico study. J Proteome Res. 2021;20:1015–1026. doi: 10.1021/acs.jproteome.0c00836. [DOI] [PubMed] [Google Scholar]
- 165.Ni X., Schröder M., Olieric V., Sharpe M.E., Hernandez-Olmos V., Proschak E. Structural insights into plasticity and discovery of remdesivir metabolite GS-441524 binding in SARS-CoV-2 macrodomain. ACS Med Chem Lett. 2021;12:603–609. doi: 10.1021/acsmedchemlett.0c00684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Drayman N, DeMarco JK, Jones KA, Azizi SA, Froggatt HM, Tan K, et al. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science DOI: 10.1126/science.abg5827. [DOI] [PMC free article] [PubMed]
- 167.Martínez-Ortega U., Figueroa-Figueroa D.I., Hernández-Luis F., Aguayo-Ortiz R. In silico characterization of masitinib interaction with SARS-CoV-2 main protease. ChemMedChem. 2021 doi: 10.1002/cmdc.202100375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Qiao J., Li Y.S., Zeng R., Liu F.L., Luo R.H., Huang C. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lockbaum G.J., Reyes A.C., Lee J.M., Tilvawala R., Nalivaika E.A., Ali A. Crystal structure of SARS-CoV-2 main protease in complex with the non-covalent inhibitor ML188. Viruses. 2021;13:174. doi: 10.3390/v13020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yang K.S., Ma X.R., Ma Y., Alugubelli Y.R., Scott D.A., Vatansever E.C. A quick route to multiple highly potent SARS-CoV-2 main protease inhibitors. ChemMedChem. 2021;16:942–948. doi: 10.1002/cmdc.202000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Arafet K., Serrano-Aparicio N., Lodola A., Mulholland A.J., González F.V., Świderek K. Mechanism of inhibition of SARS-CoV-2 Mpro by N3 peptidyl michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity. Chem Sci. 2021;12:1433–1444. doi: 10.1039/d0sc06195f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hoffman R.L., Kania R.S., Brothers M.A., Davies J.F., Ferre R.A., Gajiwala K.S. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J Med Chem. 2020;63(21):12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yin W., Luan X., Li Z., Zhou Z., Wang Q., Gao M. Structural basis for inhibition of the SARS-CoV-2 RNA polymerase by suramin. Nat Struct Mol Biol. 2021;28:319–325. doi: 10.1038/s41594-021-00570-0. [DOI] [PubMed] [Google Scholar]
- 174.Salgado-Benvindo C., Thaler M., Tas A., Ogando N.S., Bredenbeek P.J., Ninaber D.K. Suramin inhibits SARS-CoV-2 infection in cell culture by interfering with early steps of the replication cycle. Antimicrob Agents Ch. 2020;64:e00900–e920. doi: 10.1128/AAC.00900-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hattori S.I., Higashi-Kuwata N., Hayashi H., Allu S.R., Raghavaiah J., Bulut H. A small molecule compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat Commun. 2021;12:668. doi: 10.1038/s41467-021-20900-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ramos-Guzmán C.A., Ruiz-Pernía J.J., Tuñón I. Multiscale simulations of SARS-CoV-2 3CL protease inhibition with aldehyde derivatives. Role of protein and inhibitor conformational dynamics in the reaction mechanism. ACS Catal. 2021;11:4157–4168. doi: 10.1021/acscatal.0c05522. [DOI] [PubMed] [Google Scholar]
- 177.Bai B., Belovodskiy A., Hena M., Kandadai A.S., Joyce M.A., Saffran H.A. Peptidomimetic α-acyloxymethylketone warheads with six-membered lactam P1 glutamine mimic: SARS-CoV-2 3CL protease inhibition, coronavirus antiviral activity, and in vitro biological stability. J Med Chem. 2021 doi: 10.1021/acs.jmedchem.1c00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhang L., Jackson C.B., Mou H., Ojha A., Peng H., Quinlan B.D. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat Commun. 2020;11:6013. doi: 10.1038/s41467-020-19808-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ozono S., Zhang Y., Ode H., Sano K., Tan T.S., Imai K. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat Commun. 2021;12:848. doi: 10.1038/s41467-021-21118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Trigueiro-Louro J., Correia V., Figueiredo-Nunes I., Gíria M., Rebelo-de-Andrade H. Unlocking COVID therapeutic targets: A structure-based rationale against SARS-CoV-2, SARS-CoV and MERS-CoV Spike. Comput Struct Biotec. 2020;18:2117–2131. doi: 10.1016/j.csbj.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Shah M., Ahmad B., Choi S., Woo H.G. Mutations in the SARS-CoV-2 Spike RBD are responsible for stronger ACE2 binding and poor anti-SARS-CoV mAbs cross-neutralization. Comput Struct Biotec. 2020;18:3402–3414. doi: 10.1016/j.csbj.2020.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Long C., Romero M.E., Rocco D.L., Yu J. Dissecting nucleotide selectivity in viral RNA polymerases. Comput Struct Biotec. 2021;19:3339–3348. doi: 10.1016/j.csbj.2021.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]