

Abstract
Introduction:
The past decades have witnessed a remarkable improvement in the health of patients with Gaucher disease, the inherited deficiency of the lysosomal enzyme glucocerebrosidase, resulting from the availability of enzyme replacement and substrate reduction therapies. Especially in pediatric populations, early diagnosis and initiation of treatment is essential to achieving optimal outcomes.
Areas Covered:
The authors review the literature pertaining to the effectiveness of currently available therapies and describe new pharmacotherapies under development, especially for young patients.
Expert opinion:
For pediatric patients with non-neuronopathic Gaucher disease, there may be new therapeutic options on the horizon in the form of gene therapy or small molecule glucocerebrosidase chaperones. These have the potential to result in a cure for systemic disease manifestations and/or to reduce the cost and convenience of treatment. For children with neuronopathic Gaucher disease, the challenge of targeting therapy to the central nervous system is being explored through new modalities including brain targeted gene therapy, in-utero therapy, brain-penetrant small molecule chaperones, and other methods that convey enzyme across the blood-brain barrier. Indeed, these are exciting times for both pediatric patients with Gaucher disease and those with other lysosomal storage disorders.
Keywords: Glucocerebrosidase, Gaucher disease, Neuronopathic, Enzyme replacement therapy, Substrate reduction therapy, Small molecule chaperones, Gene therapy
1.0. Introduction
Gaucher disease (GD) is a lysosomal storage disorder (LSD), resulting from an insufficiency of the enzyme glucocerebrosidase (GCase; E.C. 3.2.1.45) due to biallelic pathogenic variants in the gene GBA1. The buildup of glucosylceramide and glucosylsphingosine in the lysosomes of macrophages due to deficient or defective GCase leads to inappropriate lipid accumulation within macrophages in the spleen, liver, bone marrow and other tissues. GD is characterized by wide phenotypic heterogeneity and is traditionally separated into three subtypes based on severity: non-neuronopathic or type 1 GD (GD1; OMIM #230800), acute neuronopathic or type 2 GD (GD2; OMIM #230900), and chronic neuronopathic or type 3 GD (GD3; OMIM #231000).
The onset of disease manifestations for this disorder may occur at any time throughout the lifespan, with an earlier presentation generally correlating with increased disease severity. In GD1, clinical findings, which may be mild to severe, can begin in childhood, adolescence, or adulthood, and may include anemia, thrombocytopenia, bone fractures or pain, hepatosplenomegaly, bleeding, or leukopenia. The symptoms can be non-specific and overlap with more common disorders, such as leukemia, viral infections, or other malignancies, often resulting in a lengthy diagnostic odyssey [1]. In GD2, symptom onset occurs perinatally or in infancy, and manifestations may include hydrops fetalis, congenital ichthyosis, anemia, thrombocytopenia, hepatosplenomegaly, and feeding difficulties related to an abnormal swallow [2–4]. GD2 remains a devastating and progressive neurodegenerative disease, culminating in death in infancy or early childhood. In GD3, manifestations may include visceral involvement, myoclonic epilepsy, ataxia, background slowing on electroencephalogram, learning disabilities, and impaired horizontal saccadic ocular movements [5,6]. The wide phenotypic heterogeneity in GD1 is mirrored in GD3, with some patients having impaired eye movements as their sole neurological feature, while others have cognitive impairment or severe systemic manifestations resulting in a shortened lifespan. Due to the metabolic burden that GD places on pediatric patients, they may also exhibit short-stature, growth delay or delayed puberty, in addition to other disease manifestations [7,8]. Importantly, with the introduction of newborn and genetic carrier screening programs, the age of presentation does not necessarily correlate with disease severity. For example, children with no or very mild clinical manifestations diagnosed via screening should be closely monitored rather than immediately initiating GD-specific interventions.
GD is among the most common LSDs, with an incidence in 1:40,000 to 60:000 [9]. The carrier rate is higher in the Ashkenazi Jewish population, 1:16, leading to a GD incidence of 1:850 in this population [9,10]. In populations where the GBA1 pathological variant p.Asn409Ser (N370S) is frequent, mainly in Europe, North America, and among those of Ashkenazi Jewish descent, GD1 is the predominant form of the disease. In Asia and parts of Africa, the more predominant mutation is p.Leu483Pro (L444P), and GD2 and GD3 make up a higher portion of reported cases, reaching 60% of GD cases in some populations [1,11,12].
The visceral manifestations of the disease, including hepatosplenomegaly, thrombocytopenia, and anemia, are well managed with currently approved Gaucher therapies (Tables 1 and 2). The oldest approved therapy, enzyme replacement therapy (ERT), developed and validated by physician-scientists working at the National Institutes of Health in the 1970s-90s [13], acts as a replacement for the patient’s own endogenous enzyme. Substrate reduction therapy (SRT), currently only approved for adult patients, is a more recent therapy and primarily works to reduce the production of glucocerebroside (GluCer), allowing a patient’s residual GCase enzyme to keep pace and avoid further accumulation [14].
Table 1:
Compassionate Programs and Other Treatment Support Options
| Treatment (Trade name/Manufacturer) | Type* | Company/Organization | Link |
|---|---|---|---|
| Imiglucerase (Cerezyme) | Compassionate Use/Copay assistance | Sanofi-Genzyme) | https://www.cerezyme.com/access-copay-assistance |
| Velaglucerase alfa (VPRIV) | Compassionate Use/Copay assistance | Takeda | https://www.vpriv.com/patient-support/onepath-product-support |
| Taliglucerase alfa (Elelyso) | Compassionate Use/Copay assistance | Pfizer | https://www.elelyso.com/personal-support |
| Eliglustat (Cerdelga) | Compassionate Use/Copay assistance | Sanofi-Genzyme | https://www.cerdelga.com/patient-support-and-resources |
| Miglustat (Zavesca) | Compassionate Use/Copay assistance | Actelion | https://zavesca.com/patient-patient-support.html |
| Non-specific | Copay assistance | National Gaucher Foundation (NGF) | https://www.gaucherdisease.org/financial-support/care-program/ |
| Non-specific | Copay assistance | National Organization of Rare Diseases (NORD) | https://rarediseases.org/wp-content/uploads/2020/11/Gaucher-PAP-FAQ-10-2020.pdf |
| Non-specific | Copay assistance/Expanded access | International Gaucher Alliance (IGA) | https://gaucheralliance.org/gb/about_iga/humanitarian_aid |
Table 2.
Current Therapies for Gaucher Disease
| Treatment (Trade name/Manufacturer) | Type* | Age Approved** | Method of Administration | Dosing |
|---|---|---|---|---|
| Imiglucerase (Cerezyme, Sanofi-Genzyme) | ERT | 2 and older | Intravenous | 15U/kg - 60U/kg |
| Velaglucerase alfa (VPRIV, Takeda) | ERT | 4 and older | Intravenous | 15U/kg - 60U/kg |
| Taliglucerase alfa (Pfizer Elelyso) | ERT | 4 and older | Intravenous | 30U/kg - 60U/kg |
| Eliglustat (Cerdelga, Sanofi-Genzyme) | SRT | 18 and older | Oral | 84 mg once or twice daily depending on CYP2D6 Metabolizer status |
| Miglustat (Zavesca, Actelion Pharmaceuticals) | SRT | 18 and older | Oral | 100 mg 3 times per day, with dose modification possible if needed due to side effects |
ERT - Enzyme Replacement Therapy, SRT - Substrate Reduction Therapy
Age approved based on clinical trial safety data. All ERT formulations are routinely used in patients under age 2 or 4 if clinically indicated.
Increased attention to treatments for all forms of GD has resulted from the recent appreciation of a link between the enzyme GCase and the development of a seemingly unrelated disorder, Parkinson disease [15]. Variants in GBA1 are now recognized as the most common known genetic risk factor for Parkinson disease and related disorders [16,17]. Thus, therapies developed for this rare disorder may prove beneficial for this far more common complex disease.
A majority of patients with GD experience disease manifestation in childhood, based on data from a disease registry reported by the International Collaborative Gaucher Group (ICGG), with GD2 and GD3 manifesting exclusively and predominately in infancy and childhood, respectively [10,18,19]. A childhood diagnosis of symptomatic GD often indicates that pathologic amounts of substrate have already accumulated, and thus children often have a more severe presentation than adults with GD1 diagnosed later in life. Therefore, in such patients, a prompt diagnosis and administration of treatment is imperative for maintaining good quality of life and normal life expectancy [20–24]. In a 2006 study of 887 patients with GD1 described in the ICGG database, patients diagnosed in childhood presented with more severe abnormalities at their time of diagnosis. The researchers found that anemia and hepatosplenomegaly were most common among patients with GD1 diagnosed by age 6 years, and that bone involvement was more prevalent in patients diagnosed in their teenage years [19]. Additionally, many patients diagnosed in childhood experienced growth retardation, thrombocytopenia, and bone crises [25,26]. While pulmonary involvement is relatively uncommon in patients with GD1, lung disease is frequently seen in children with GD2 and GD3 [27]. One limitation of data collected through the ICGG, a pharmaceutical company-sponsored registry, is that it is more likely to include patients requiring therapy, resulting in a potential bias to more severely affected individuals.
The goal of this paper is to review how GD is diagnosed in the pediatric population, to discuss both currently approved and potential future therapies, and to highlight relevant current ethical issues impacting each of these areas.
2.0. Diagnosing Gaucher disease
This section will discuss the clinical manifestations of Gaucher disease in pediatric patients, newborn screening programs, establishing the diagnosis, and the physical and psychosocial impact of Gaucher disease in childhood.
2.1. Establishing the diagnosis of Gaucher disease in childhood
The diagnosis of GD in children with clinical manifestation of the disease should be supported by medical history, family history, physical examination, and laboratory tests that confirm low residual enzyme activity, as well as by the analysis of GBA1 variants [28]. Asymptomatic children with GD are generally diagnosed during family screening, prenatal testing and newborn screening. The recent increases in childhood diagnoses have been attributed to the more widespread use of such screening [28–30]. The expanded use of next-generation sequencing as a diagnostic tool in children has also led to the earlier recognition of GD in some cases. Post-diagnosis, children should have genotyping performed, usually by sequencing of GBA1, as well as baseline blood tests and organ and bone assessments in order to determine disease severity and identify possible complications. These evaluations can also inform appropriate treatment plans [28]. It should be emphasized that a bone marrow evaluation is not necessary to establish the diagnosis and can usually be avoided in children. In addition, for non-neuronopathic GD patients diagnosed via newborn screening programs, one can generally wait at least one year before assessment and X-rays are not required for diagnostic purposes. Also, both due to cost concerns and and to avoid the unnecessary use of sedation, abdominal ultrasound rather than magnetic resonance imaging (MRI) assessments can be performed for early follow-up of visceral manifestations [31,32].
2.1.1. Newborn screening
Newborn screening (NBS) has garnered international support as an important public health measure to aid in the early diagnosis and treatment of potentially fatal metabolic, endocrine, and hematologic disorders [33]. Screening rates for specific diseases vary from country to country, with rates as high as 99% in Chile and Singapore and less than 10% in Bangladesh and the Dominican Republic [34]. Following the development of tandem mass spectrometry (MS/MS) as an ideal method to screen for metabolic disorders, interest in adding the LSDs to standard NBS protocols has grown, especially in Japan, Korea, Taiwan, and the United States [34]. LSDs represent an ideal class of disorders for NBS as their markers can be detected in dried blood spots (DBS), an inexpensive and easily attainable biospecimen, and, for many of these disorders, approved treatments now exist [35].
Pilot NBS for LSDs other than GD were initially undertaken in Italy and Taiwan [36–39]. Since then, GD has been added to LSD NBS pilot studies in several states including California, Illinois, Missouri, New York and Washington [40–43]. Moreover, Illinois, Missouri, New Jersey, and Tennessee now routinely include GD in their NBS. However, it has not been added to the Department of Health and Human Services’ Recommended Uniform Screening list. Based on these pilot studies, the population incidence of GD ranges from 1:4,374 in New York City, which has a high Ashkenazi Jewish population, to 1:61,600 in Missouri [40,44].
2.1.2. The impact of early diagnosis
An early diagnosis of GD can avoid a tumultuous diagnostic odyssey for families; yet, given the vast clinical heterogeneity in GD, diagnosis via NBS is complicated by several ethical considerations. Given that GD2 is typically lethal in early childhood, it may not fit the criteria for NBS, where the goal is to diagnose diseases where early intervention can impact the overall prognosis. Also, NBS purposefully excludes late-onset disorders due to the implications surrounding patient consent and unknown benefits of a very early diagnosis [35]. Even though many patients with GD1 present in childhood, some patients do not develop symptoms until adulthood, and many never reach medical attention. Thus, it is essential that families be made aware of the subtleties of this diagnosis. Having the infrastructure for continual support of diagnosed newborns and their families such as genetic counselors and physicians with experience treating GD is essential to creating a sustainable NBS system [34]. A study, based on interviews with geneticists, genetic counselors, and biochemical lab directors on their views of NBS for different LSDs, revealed similar concerns regarding the screening. Of note, the healthcare providers in this study disagreed with regards to the role of NBS in family planning but showed universal dissatisfaction with how new conditions are added to NBS panels [45]. Conversely, when 91 patients with LSDs, 22 with GD, were surveyed on their opinions regarding NBS for LSDs, participants felt most concerned about issues surrounding insurability and least concerned about the jeopardization of children’s autonomy. Interestingly, nine of the respondents with GD thought their health would be better had they been diagnosed via NBS, yet only three of them felt they would be more satisfied with their lives [45].
According to recommendations published in 2013 by a group of international GD experts, ERT is recommended for every child and adolescent with clinical signs of GD1 or GD3 [28]. Utilizing ERT for patients with GD2 can help reduce visceral symptoms; however, there is little evidence that ERT can ameliorate neurological manifestations. In very young children, differentiating between the types of GD can be challenging due to limited genotype-phenotype correlation and heterogeneity in phenotypic presentations. However, distinguishing between the types and beginning treatment promptly after symptom onset is important due to the potential long-term benefits of starting care early. Children who begin ERT promptly experience reduction of spleen and liver size, improvement in anemia and thrombocytopenia, and a reduced likelihood of developing future skeletal complications [26,28,46]. Care of patients with GD2 often depends on parental preferences and can also be primarily supportive, including palliative care, feeding tubes and/or tracheostomy [28].
2.2. Psychosocial aspects of Gaucher disease in childhood
Having GD can take a psychological toll on children. Certain disease manifestations such as organomegaly, delayed puberty, and growth retardation may affect body image and self-esteem while eliciting feelings of insecurity and isolation, which can lead to the development of behavioral problems [8,23,47–49]. Additionally, chronic pain and fatigue may interfere with normal socialization and school performance [50,51]. In a recent study, American children with GD1 self-reported a significantly lower health-related quality of life (HRQoL) across all health dimensions compared to healthy controls. Younger patients with GD reported lower HRQoL scores as well as less optimal psychological functioning than young adult patients aged 18-30 years [52]. Patient and parent-reported HRQoL scores in another study of pediatric patients with GD1 conducted in Spain were higher than those in the American study. The authors postulate that the higher-than-expected HRQoL scores were related to the efficacy of ERT, their solid healthcare infrastructure, and low baseline disease severity [53].
2.3. Monitoring children with Gaucher disease
After the diagnosis of GD is made, patients should be initially referred to a center well versed in treating GD and closely followed whether treatment is started at the time of diagnosis or deferred. Expanded access, compassionate use, and financial assistance programs exist to help patients receive these very expensive therapies and patient/community organizations act as an invaluable resource (Table 1). Use of a pediatric GD severity scoring system, focused on GD1 [54] or nGD [55] may prove valuable in initial assessments and continued monitoring of disease progression or stability. While no one system has been universally adopted, consistent single center or case use may still be clinically useful.
Decisions on when to start treatment may be aided by measuring certain biomarkers in addition to assessing pathological phenotypes. Biomarkers that have previously been utilized in monitoring GD, such as ferritin, tartrate-resistant acid phosphatase, and angiotensin-converting enzyme are not specific to GD [56]. Chitotriosidase and CCL18 are secreted by activated macrophages, which include Gaucher cells, yet are still not GD-specific and are not central to disease pathology. Also, about 10% of the population carry a variant resulting in deficient chitotriosidase [57–59]. After the diagnosis of GD is made, decisions regarding when to start treatment may be aided by measuring certain biomarkers. Biomarkers previously utilized in monitoring GD, such as ferritin, tartrate-resistant acid phosphatase, and angiotensin-converting enzyme are not specific to GD [56]. Chitotriosidase and CCL18 are secreted by activated macrophages, which include Gaucher cells, yet they are also not GD-specific and are not central to the disease pathology. Also, about 10% of the population carry a variant resulting in deficient chitotriosidase [57–59].
Glucosylsphingosine (lyso-Gb1) is a direct metabolite of glucosylceramide, one of the substrates that accumulates due to GCase deficiency. In vitro and in vivo studies have documented the role of sphingolipid accumulation in GD pathophysiology. An analysis of deceased patients with GD showed accumulation of glucosylsphingosine in the spleen, liver, and in patients with GD2 and GD3, cerebrum and cerebellar cortex [60]. Levels of lyso-Gb1 have been shown to be significantly higher in patients with GD compared to healthy controls, and a few studies have found correlations between lyso-Gb1 levels and GD disease severity, including presence or absence of neurological symptoms [56,61,62]. In addition, lyso-Gb1 levels have been shown to decrease after initiating treatment. Taken together, these data suggest that lyso-Gb1 may have clinical utility in monitoring GD progression.
Recently, a systemic literature review assessing the value of lyso-Gb1 in adult and children patients with GD found that evidence reported in 74 original research articles supported the use of lyso-Gb1 as a disease-monitoring biomarker for GD [56]. Some evidence supports lyso-Gb1 as a prognostic biomarker, but additional studies are required [56].
A recent study used ultra-performance liquid chromatography coupled to time-of-flight mass spectrometry to analyze the plasma of 16 patients with GD1 blood for biomarkers and characterized four analogs of lyso-Gb1. This group suggested the potential benefits of screening for multiple biomarkers in order to monitor GD [63]. Another recent study utilized liquid chromatography–mass spectrometry (LC-MS/MS) to quantify lyso-Gb1 in DBS samples of patients who were deemed high-risk based on enzymatic activity assays. Patients with GD, confirmed via GBA1 sequencing and enzymatic activity analysis, all had significantly elevated lyso-Gb1 levels, demonstrating the utility of analyzing lyso-Gb1 levels in DBS using LC-MS/MS in monitoring GD [64].
In a study assessing the levels of lyso-Gb1 specifically in children with GD, an association between GD severity and lyso-Gb1 levels was found, consistent with adult data [61]. Significantly higher levels of lyso-Gb1 were found in children with severe GD1 compared to milder GD1 cases. Children who eventually started ERT had significantly higher levels of lyso-Gb1 compared to untreated children [61].
3.0. Therapy for Gaucher disease
Alongside advances in the diagnoses and screening of GD, there has also been great progress in therapeutic development. This section will review currently approved therapies for GD (Table 2), as well as newer therapies now being developed (Table 3, Figures 1 and 2).
Table 3.
Current Clinical Trials Enrolling patients with Gaucher Disease
| Name (Investigational product) | Type | Details | Eligibility | Identifier | Location |
|---|---|---|---|---|---|
| GuardOne (AVR-RD-02, AvroBio) | Gene Therapy | Phase 1/2 Lentiviral Vector | Type 1 GD, Ages 16-35 | NCT04145037 | Calgary, Canada; Melbourne, Australia |
| PROVIDE (PR001, Prevail Therapeutics/Eli Lilly) | Gene Therapy | Phase 1/2 AAV9 Vector | Type 2 GD, Ages up to 24 months | NCT04411654 | New York, NY, USA; Pittsburgh, PA, USA; Minneapolis, MN, USA; Oakland, CA, USA |
| PROPEL (PR001A, Prevail Therapeutics/Eli Lilly) | Gene Therapy | Phase 1/2a | Parkinson disease patients with at least one pathogenic GBA1 mutation | NCT04127578 | Chicago, IL, USA; Orlando FL, USA; New York, NY, USA |
| ELIKIDS (Eliglustat, Sanofi-Genzyme) | Drug Therapy | Phase 3 | Type 1 and 3 GD, Ages 2-18, For safety and efficacy in pediatric patients | NCT03485677 | Argentina, Canada, France, Italy, Japan, Russian Federation, Spain, Sweden, Turkey, United Kingdom |
| Ambroxol | Drug Therapy | Clinical Trial | Type 1 GD, Ages 18-75, suboptimal response to ERT | NCT03950050 | Jerusalem, Israel |
| LEAP2IT (venglustat, Sanofi-Genzyme) | Drug Therapy | Phase 3 | Type 1 and 3 GD, Ages 12 and older, in combination with Cerezyme | NCT02843035 | New Haven, CT, USA; Dallas, TX, USA; Mainz, Germany; Minato-Ku, Japan; Cambridge, London, Salford, United Kingdom |
| N-acetylcysteine | Drug Therapy | Clinical Trial | Type 1 GD, Ages 18 and older | NCT02583672 | Minneapolis, MN, USA; New York, NY, USA |
GD – Gaucher Disease, ERT – Enzyme Replacement Therapy
Figure 1. Therapeutic strategies to enhance glucocerebrosidase activity.
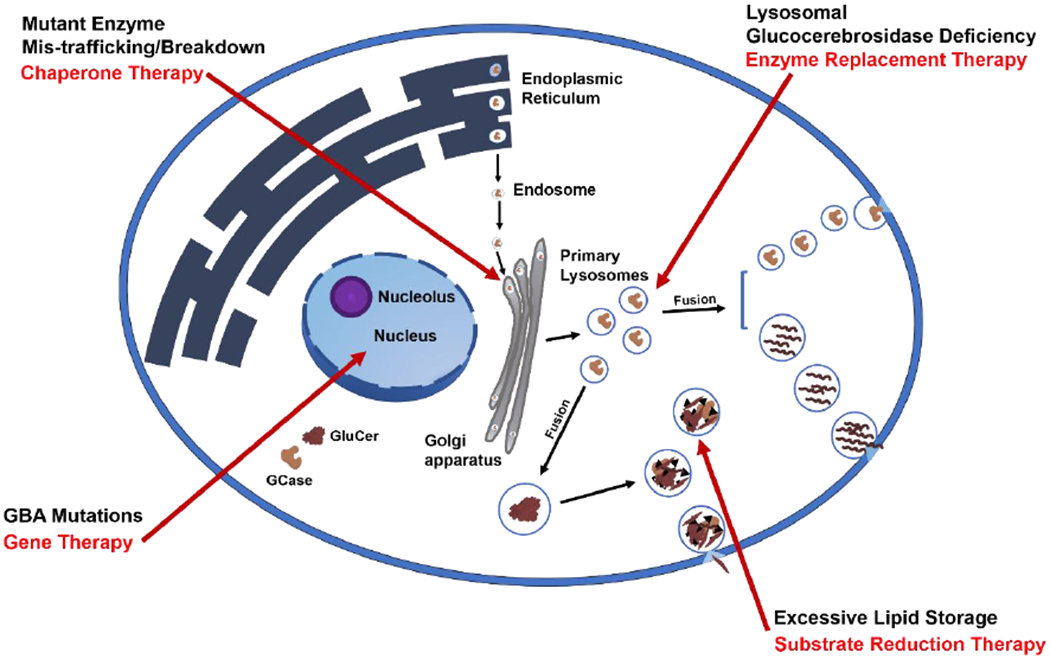
Mutations in GBA1 lead to misfolded glucocerebrosidase within the endoplasmic reticulum (ER), leading to protein mis-trafficking or degradation. Enzyme Replacement Therapy is currently used to introduce fully-functioning enzyme into the cell. Substrate reduction therapy targets the synthesis of glucosylceramide to prevent substrate accumulation. Gene therapy targets the host genome to endogenously restore glucocerebrosidase activity. Small-molecule chaperones bind to mutant glucocerebrosidase, stabilizing and facilitating the transport of the mutant enzyme to lysosomes. GCase, glucocerebrosidase. GluCer, glucosylceramide.
Figure 2. Approaches to delivering Gaucher disease therapies to the brain.
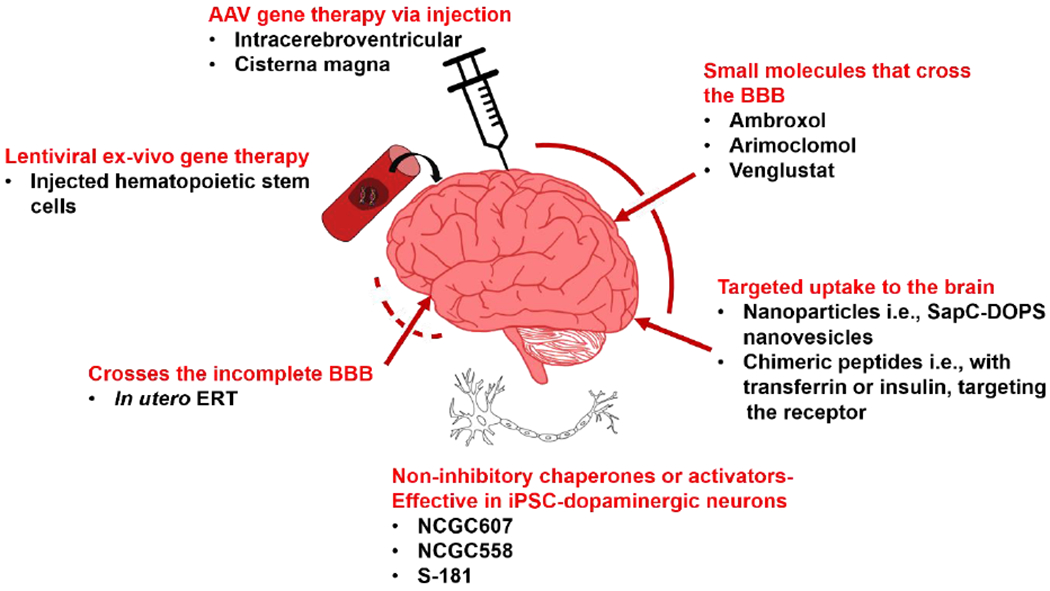
Currently approved treatment modalities do not cross the BBB, and therefore do not alleviate neurological manifestations of GD. Several newer strategies are shown. AAV, adeno-associated virus; BBB, blood-brain barrier; ERT, enzyme replacement therapy; iPSC, induced pluripotent stem cell; SapC-DOPS, saposin C-dioleoylphosphatidylserine
3.1. Current therapies
Prior to the approval of ERTs, patients with GD1 underwent procedures such as blood transfusions, splenectomies or severe cytopenia, and joint replacement surgeries [65]. However, complications occurred after many of these procedures. Some patients developed blood transfusion dependence, iron overload, and increases in toxic glycosphingolipids as a result of transfused red and white blood cells. Splenectomy did reverse symptoms relating to pressure, but subsequent studies reported accelerated bone disease and complications involving other organs [65,66]. It has been reported that bone marrow failure, liver failure, crippling skeletal disease, pulmonary hypertension, and premature death followed some splenectomies [65]. Bone marrow transplantation has been performed on a few subjects with GD [67–69]. While it does appear to correct the visceral manifestations of GD, it had little to no impact on neurological manifestation in GD2 and GD3. Because of the availability of other therapeutic options, and the known risk of this procedure, it is seldom performed. It is also important to appreciate that some patients, especially those with genotype p.Asn409Ser/p.Asn409Ser (N370S/N370S), managed quite well prior to the advent of ERT, with many seldom requiring medical attention.
3.1.1. Enzyme replacement therapy
In 1964, Christian de Duve first suggested that lysosomal storage diseases might be treated by enzyme replacement. Dr. Roscoe O. Brady, having discovered that deficient GCase led to the development of GD, first proposed a strategy to replace the missing enzyme by supplementing deficient enzyme [70,71]. The proof-of-concept of ERT was initially established in vitro in the monogenic disorder I-cell disease [72]. However, implementing ERTs for patients with GD1 required the production of purified, concentrated enzyme from human origins. Over three decades, the development of ERTs focused on assessing the efficacy of placenta-derived GCase, alglucerase, and then finally recombinant GCase, or imiglucerase. The recombinant protein, generated using Chinese hamster ovary (CHO) cells in the mid-1990s, continues to serve as the most widely used GCase in clinics globally. Several other forms of the recombinant enzyme, including taliglucerase and velaglucerase alpha, which are produced in plant cells and human fibrosarcoma lines respectively, subsequently received approval and are used to treat GD [73,74] (Table 2). The enzyme preparations are all administered intravenously. Some patients receive infusions at clinics, hospitals, or at home through either self-administration or administration by visiting nurses.
Both pediatric and adult patients with GD1 respond well to the currently available ERTs, which significantly improve hematological parameters, reduce hepatosplenomegaly, and partially prevent or ameliorate bone disease, including bone marrow infiltration, bone pains/crises and osteoporosis/osteopenia [75–77]. Since splenectomy appeared to increase the risk for bone disease, the removal of the spleen should be avoided except for rare cases of grossly infarcted, massive or ruptured spleens that are considered life-threatening [78].
Despite the high cost, most Gaucher centers begin children with symptomatic GD on higher dosages of ERT, starting at 60 U/kg every other week, compared to 30-60 U/kg every other week for adults. While in early years, some groups advocated for frequent infusions of much lower doses of ERT, this is now rarely implemented. Qiu et al reported a retroactive analysis comparing the efficacy of low dose (7.5-15 U/kg/2 weeks) and high dose (40 U/kg/2 weeks) imiglucerase therapy for Chinese patients in response to the 2009 ERT shortage. Measuring blood counts, organ volumes, and bone syndromes, this study found no dose-dependent differences in visceral enlargement or anemia. While 20 U/kg/2 weeks was considered adequate to reverse organomegaly and cytopenia, it was less effective for patients with skeletal involvement [79]. Additionally, a report on two pediatric patients with GD3 [80] found that despite long-term, high-dose ERT, the patients developed avascular necrosis, which was also reported in previous studies of rare treated adults with GD1 [81,82]. In these cases, no relationship was found between the development of the bone infarction and ERT dose and duration, highlighting a limitation in the current ERT treatment, specifically in targeting certain tissues such as bone, which may not effectively take up the exogenous enzyme [80].
Nonetheless, ERT has been found to be a safe treatment for patients of all ages across all GD subtypes. In reports from clinical trials, pharmacovigilance programs by each respective manufacturer, and from drug or disease registries, there have been no reported deaths or permanent damage due to ERT administration [31]. In very rare cases, anaphylactic shock has been reported, typically presenting early in the treatment, and this is not considered a reason for discontinuation of therapy. In most cases, modulating the infusion rate, treating with antihistamines or simply switching to another ERT resolves these events [31].
ERT during the SARS-CoV-2 pandemic has prompted more questions regarding the administration of available treatments [83,84]. During SARS-CoV-2, many patients tried to avoid hospitals and clinics to prevent exposure to the virus. The availability of home infusion nurses was also compromised due to nursing shortages or reallocation. It is generally not recommended to stop infusions [83–85]. However, reports during a Cerezyme ERT shortage in 2009 demonstrated that patients who were extremely stable under chronic therapy were able to tolerate some breaks in treatment, and an evaluation performed two years after the shortage found that patients who had experienced a 6-month treatment gap did not develop any irreversible complications [86,87]. A survey conducted during the SARS-CoV-2 pandemic in Spain found that one-fourth of patients with GD receiving ERT at hospitals reported dose interruptions [83]. Another recent report evaluated determinants of SARS-CoV-2 infection in GD patients, both adults and children, during the peak of the pandemic in the New York City metropolitan area. Here, it was suggested that GD does not confer an increased risk for infection, despite additional risk factors [85]. It is recommended breaks in treatment be limited in nature, only when there is no other recourse, and that treatment should resume at the earliest possible opportunity.
3.1.2. Substrate reduction therapy
Substrate reduction therapy targeting pathways that produce glucocerebroside was first approved by the Food and Drug Administration (FDA) to treat GD in 2003 [88,89]. Developed using glucosylceramide synthase inhibitors, SRT treatment aims to reduce accumulating glycosphingolipids. The two currently approved oral SRTs for the treatment of GD, miglustat and eliglustat tartrate, are intended exclusively for adult patients with GD1 (Table 2) [90,91]. Compared to intravenously administered ERT, the orally taken SRT rapidly diffuses into various tissues and has the potential advantage of improving bone complications through direct drug delivery to bone compartments. A recent study assessed the possible use of RANK pathway components, major effectors at multiple levels of the bone regeneration cycle, as markers for bone disease progression in GD [92]. Here, they reported a reduction in osteoclastogenic biomarkers in a cohort of patients on SRT compared to an ERT cohort. While further evidence is required, the probable reduction in osteoclast activity with SRT suggests that this therapy might be useful in treating patients with specific bone complications [92].
Eliglustat, the most commonly used SRT, has been used as a long-term treatment for adults with GD1. Patients considering this therapy must be evaluated for their CYP2D6 status using an FDA-cleared test, to determine whether they are CYP2D6 extensive metabolizers, intermediate metabolizers, or poor metabolizers [86,91,93,94]. to assess patient eligibility and recommended dosage. Thus, assessment of drug-drug interaction with other medications can be vital, especially if considered for pediatric patients. Side effects may include general gastrointestinal upset as well as headache, back/arm/leg pain, dizziness or weakness, and eliglustat may be contraindicated in those who have hepatic impairment, cardiac arrhythmias related to prolonged PR, QTc or QRS intervals, or based on CYP2DG metabolizer status and other concurrent medications. Currently, there are clinical trials of eliglustat for pediatric patients with GD1 and GD3 to evaluate the safety and pharmacokinetics of eliglustat in pediatric patients, either alone or in combination with Cerezyme (Table 3). For this study, anticipated to be completed by March 2023, treatment will be analyzed over two one-year treatment periods [95].
Miglustat (Zavesca®, Actelion), first approved in 2003 for use in GD1 when ERT is not appropriate, is contraindicated in those with neurological symptoms, pregnant or planning pregnancy, and patients with renal or hepatic involvement. Due to its side effect profile, which can include moderate to severe GI distress, tremor, or peripheral neuropathy, its current use is limited in GD [96]. Another newer version of SRT currently under investigation, Venglustat, is able to penetrate the CNS, and could specifically be useful in GD3. Trials have begun in adults with GD3 and will now include adolescents (Table 3) [97]. Preliminary results from this trial, reported prior to peer review, indicate a favorable safety profile and initial indications of positive neurological effects as measured by increase in certain brain regions of participants with GD3 [98].
3.2. New therapies
Several new therapeutic approaches currently under consideration or development are illustrated in Figures 1 and 2.
3.2.1. Small molecule chaperones
Small molecule chaperones, including iminosugars, ambroxol, and other competitive glucocerebrosidase inhibitors and non-inhibitory chaperones or allosteric activators, are currently under investigation as a treatment for GD. These prototype drugs have potential particularly for neurological manifestations, given that some of these molecules can penetrate the blood-brain barrier (BBB) and facilitate proper enzyme folding and translocation to lysosomes [99].
3.2.1.1. Ambroxol
One candidate chaperone, Ambroxol, is an available drug already approved for other purposes, although not in the United States. Ambroxol was first identified in a screen testing a library of approved drugs for their impact on GCase activity [100]. Originally used to treat airway mucus hypersecretion and hyaline membrane disease in newborns, this drug is now in clinical trials for adult patients with GD1, as well as GBA1-heterozygotes with associated Parkinson disease [101,102]. The phase II trial focuses on expanding its efficacy for GD by assessing its pharmacokinetics. Ambroxol has currently been identified as a pharmacological GCase chaperone effective for six different GCase mutations: p.Asn227Ser, p.Phe252Ile, p.Gly232Trp, p.Arg159Trp, p.Gly241Arg and p.Asn409Ser, where in vitro and in vivo studies indicate a response to Ambroxol at concentrations of 0.3–30 μmol/L [100,103,104]. Currently, patients with GD1 reporting suboptimal responses to ERT are eligible for an ongoing study to assess Ambroxol as an alternative GD treatment option (Table 3) [101]. However, a larger placebo-controlled trial is necessary to clearly establish its efficacy.
Already there is considerable anecdotal data regarding Ambroxol in the pediatric population from scattered case reports. One report described a 5-year-old female patient with GD1 (genotype p.Leu395Trpfs*8/p.Arg392Trp) with hepatosplenomegaly, anemia, thrombocytopenia, bone lesions including aseptic necrosis of the femoral heads bilaterally, and elevated plasma chitotriosidase activity. The authors reported that the administration of Ambroxol (10mg/kg/day initially for 6 months, then increased to 15 mg/kg/day for 2.5 years) was safe, and resulted in remodeling of the sphericity of the femoral head bilaterally [105]. Ambroxol has also been administered in neuronopathic forms of GD with some indications of neurological improvement, particularly in seizure control and gait [103,104,106–109].
3.2.1.2. Isofagomine
Another small molecule chaperone, isofagomine, was developed by Amicus Therapeutics to bind and stabilize misfolded p.Asn409Ser GCase. While there was evidence that this molecule could increase enzymatic activity in vitro in patient fibroblasts [110], clinical trials were halted in 2009 due to poor cell penetration and the lack of improvement [111].
3.2.1.3. Histone deacetylase inhibitors
Another set of molecules, known as histone deacetylase inhibitors (HDACIs), have been studied as a potential class of medications to treat GD, as well as other protein misfolding diseases such as Niemann-Pick type C disease, Huntington disease, and cystic fibrosis. A known HDACi, SAHA, and a unique HDACi, LB-205, reported in a 2011 study, were found to increase GCase activity in fibroblasts from patients with GD1 and GD2 by modulating two molecular chaperones, heat-shock protein (HSP) 90 and HSP 70 [112].
3.2.1.4. Non-inhibitory chaperones
A high-throughput screen for small molecule chaperones of GCase, performed using a sample of patient spleen as the source of p.Asn409Ser mutant GCase from, resulted in the identification of the first non-inhibitory chaperones [113]. A lead molecule, NCGC758, increased GCase activity and reversed lipid storage in patient-derived induced pluripotent stem cells (iPSC)-macrophages, restoring the impaired macrophage function [114]. A second non-inhibitory chaperone, NCGC607 was reported to increase GCase activity and to reverse lipid storage in patient-derived iPSC-dopaminergic neurons [115]. Another small molecule modulator, S-181, was found to increase the activity of mutant and wild-type GCase in an iPSC-dopaminergic model carrying the c.84insG mutation, as well as in a murine model [116]. This molecule was generated upon examining the structure activity relationship of quinazoline inhibitors and recent GCase crystallography experiments that suggest that GCase can exist as a dimer in vivo. In solution and as a crystal, it was found that GCase has a butterfly-shaped dimer structure, and this interface yields an allosteric binding pocket that can be targeted by small molecule activators [117].
Another study tested Arimoclomol, a small molecule that increases the levels of HSP70, a chaperone that helps to fold glucocerebrosidase. This HSP amplifier can cross the BBB, and was successfully used to enhance the folding, maturation activity and localization of mutant GCase in patient cells and in a neuronal model of GD [118].
3.2.2. In utero Enzyme Replacement Therapy
To date, attempts at administering ERT directly to the brain using intrathecal or intraventricular routes have not proved successful. However, researchers have postulated that administering ERT prenatally via umbilical vein injections could potentially impact the course of GD2. While our understanding of the BBB and its formation is still limited, it is known that the BBB develops in utero, and is fully formed at birth [119]. Therefore, the efficacy of any in utero therapy for GD2 would be dependent on administering the therapy early enough in fetal development to allow passage through an incomplete BBB. This approach could have utility for families who have previously had an affected child, but otherwise, the practicality of in utero interventions may be limited.
A 2020 study by Nguyen et al. used in utero ERT (IUERT) to treat mouse models of the LSD Mucopolysaccharidosis type VII (MPS7) which is caused by deficient levels of b-glucuronidase [120]. The authors showed that IUERT penetrates the BBB by comparing levels of b-glucuronidase activity in microglia of treated and untreated MPS7 mice. They further demonstrated that continuous IUERT treatment reduced neuronal inflammation, improved grip strength, and reversed organomegaly in these mice. An upcoming phase 1 clinical trial will enroll pregnant women carrying fetuses confirmed to have GD2, along with other LSDs, to study the utility of IUERT in humans (Table 3) [121]. The implementation of this technology requires prior genetic counseling, as families eligible for IUERT should also be informed of the option of prenatal diagnosis and pre-implantation diagnosis (PGD). Providers should be educated in severity procedures like PGD and IUERT, as well as related psychosocial considerations.
3.2.3. Gene therapy
Gene therapy is another potential strategy for the treatment of GD. Over the years, research into gene therapy for GD has focused on two out of the five main classes of viral vector systems (adenoviruses, adeno-associated virus (AAV), herpes simplex-1 viruses, retroviruses, and lentiviruses). These vector systems can be further categorized into two groups according to whether they integrate into the host genome or persist as extrachromosomal episomes [122]. The two systems currently being investigated for GD are AAVs, non-integrating vectors, and lentiviruses which do integrate [122]. Both can mediate persistent transgene expression in non-proliferating and proliferating cells [123,124].
In animal models, gene therapy for GD has shown progress. Using a mouse model of neuronopathic GD, researchers injected a recombinant AAV vector encoding GBA1 into the ventricle of 16-day gestation fetuses [125]. Their mouse model carried a loxP-flanked neomycin disruption of the murine gba, followed by Cre recombinase. AAV treatment restored neuronal GCase expression in this gba knockout mouse model. Assessment of the long-term efficacy of this AAV therapy indicated that at age 70 days, treated knockout mice appeared normal and fertile. In contrast, untreated knockout mice developed forelimb paralysis, followed by tetraparesis, necessitating sacrifice at 14 and 15 days of age. GCase activity in treated knockout mice was comparable to wild-type (WT). However, at 100 days of age, the treated knockout mice had a poorer performance on grid walk and rotarod tests and weighed significantly less than WT littermates. Furthermore, this intracerebroventricular treatment did not lead to alleviation of visceral pathology, failing to prevent Gaucher cell infiltration in spleen, liver, and lungs. Intravenous injection, however, prevented splenomegaly and Gaucher cell infiltration within spleen, liver, and lungs, suggesting that both modes of administration may be necessary [125].
Currently, one company, Prevail Therapeutics (now Eli Lilly), is running a phase I/II clinical trial using their AAV9 gene therapy, PR001, for young patients with neuronopathic GD [126]. This study aims to deliver a healthy copy of GBA1 into the cisterna magna as a one-time injection (Table 3). After dosing, PR001 therapy will be assessed for safety, tolerability, immunogenicity, biomarkers, and efficacy, over a period of 12 months. While this study specifically focuses on GD2 and GBA1-associated Parkinson disease, gene therapy may serve as a potential treatment for all forms of GD. A second company, Avrobio, has developed lentiviral-based gene therapy for GD1 [127]. Avrobio’s trial is an ex-vivo therapy, where hematopoietic stem cells are harvested from the blood or bone marrow and injected with lentivirus carrying wild-type GBA1 (Table 3). The modified hematopoietic stem cells are then infused back to disperse throughout the body and begin expressing the corrected GCase enzyme. A third company, Freeline Therapeutics, is initiating a study using liver-targeted AAV therapy for GD1. Pre-clinical data suggests that a single injection of the AAV-based GBA1 construct can produce fully-functioning GCase enzyme and prevent the accumulation of lipid substrates. The functioning GCase is found at long-term sustained steady-state levels within the bloodstream and is taken up by macrophages in key organs [128].
3.2.4. Nanovesicles
Another novel approach is to successfully deliver ERT directly to the brain. Sun et al are developing a strategy utilizing BBB-penetrating nanovesicles. Termed SapC-DOPS-GCase, these vesicles penetrate the BBB utilizing surface phosphatidylserine on blood vessels and other cells including neurons, astrocytes, and microglia [129]. Upon intravenous administration, functional GCase is transported within SapC-DOPS nanovesicles, preserving GCase function and stabilizing the enzyme upon uptake into cells via the mannose-receptor independent pathways. Assessing their therapeutic strategy in GD mouse models, they found significant improvements in neurodegeneration, CNS inflammation, survival, and neurological phenotype [129].
4.0. Special considerations for pediatric therapy
The Food and Drug Administration has highlighted several approaches [130] regarding the safe and effective use of drugs in pediatric populations. In order to permit extrapolating adult efficacy data towards pediatric populations, information must be provided that demonstrates a similar course of disease and effect of the drug within both pediatric and adult patients. Ultimately, the developmental changes within pediatric populations have to be taken into account prior to any implementation of current treatments suited for adult populations. For example, developmental changes that occur within the muscle, fat, skin, and water content and the degree of vascularization can influence the degree of absorption of drugs delivered via subcutaneous, percutaneous, or intramuscular absorption [131]. In addition, how these drugs are distributed once absorbed can be affected by changes in body composition. Once distributed, changes in the child’s metabolizing capacity will affect the bioavailability and elimination of the drug, specific to the intestinal and hepatic metabolic processes involved [132].
In 2017, the FDA published guidelines highlighting drug development for pediatric rare diseases, using GD as a model [130]. Exploring the pharmacokinetics, treatment-induced changes in different disease manifestations, and clinical response to treatment can enable the extrapolation of adult efficacy data and benefit pediatric Gaucher therapy development with regards to efficiency and reducing testing burden to patients. Nonetheless, additional clinical trials must be performed, especially those focused on GD2, where patients die in early infancy.
Other issues arise when developing therapy that must be administered prenatally. If treatment specifically for neuronopathic GD becomes available, umbilical vein injections such as those used with IUERT and in utero-gene therapy (IUGT) could be a feasible delivery method. However, the International Fetal Transplantation and Immunology Society (IFeTIS) consensus statement on IUGT declared that IUGT should be considered solely when a reliable prenatal diagnosis can be achieved and if there is reliable evidence of genotype-phenotype correlation [133]. This is not always the case with neuronopathic GD. Furthermore, the IFeTIS emphasized the importance of non-directive counseling of the parents, maternal safety, and potential that typically fatal in-utero phenotypes, when ameliorated by in utero therapy, may produce a severely disabled child.
There are also special safety and ethical considerations for any gene therapy within pediatric populations which reinforce the necessity of pediatric clinical trials and further research on questions surrounding informed consent in this population [134,135]. Additionally, because gene therapy targets somatic cells, pediatric patients who undergo gene therapy may still need to receive counseling once they start family planning later on in life.
5.0. Conclusion
GD continues to present a pharmacological challenge. Great strides have been made over the past 30 years with the development of four different ERT and two SRT treatment options, with SRT therapy likely to be available for pediatric use in the near future. However, none of these therapies address all aspects of GD, and the currently available drugs remain expensive and inconvenient life-long therapies not suitable for everyone and not always available in resource-poor regions of the world. Gene therapy, small molecule chaperones and novel therapeutic delivery systems are at the forefront of current research efforts to address this still potentially devastating disease.
6.0. Expert opinion
The increasing number of treatments available and under development for the lysosomal storage disorders in general, and GD in particular, reflect a monumental achievement of the research community. Continued activity in developing drugs that enhance glucocerebrosidase levels and activity are in part driven by the Parkinson disease community, based on the recently appreciated reciprocal relationship between levels of glucocerebrosidase and the aggregate-prone protein alpha-synuclein. This association is infusing new energy into the field, as glucocerebrosidase-enhancing treatments may also have efficacy for Parkinson disease, a common complex disorder which currently lacks effective disease-altering therapeutics.
While currently patients with non-neuronopathic GD are benefiting greatly from the available therapies, the drugs remain extremely expensive, often inconvenient, and a life-long treatment, but not a cure. The high cost of each of the enzyme preparations, as well as the available oral drugs, is a considerable drain on health care systems, and on a global scale, many of the neediest cases are unable to get treatment. Furthermore, no treatment is currently available that definitively impacts the CNS sequela of the disease, and physicians have little to offer families of infants with the most devastating form of the disease, GD2. While brain-directed treatments for those with neuronopathic GD still remains elusive, new therapeutic modalities such as small molecule chaperones, nanovesicles, protein modification, and gene therapy are productive areas of interest. Many of these approaches remain in the realm of basic science, however, several new therapies or new applications of older therapies have reached the clinical trial phase of development.
While the influx of interest and research in GD over the last decade due to the discovery of the link between GBA1 and Parkinson disease is particularly exciting and certainly a net benefit to many patients with GD, this has not necessarily translated to improvement in treatments for the youngest patients. Pediatric pharmacotherapy research, as driven by industry, experiences additional challenges due to considerations such as the cost of trials and efficacy and safety concerns in vulnerable populations. This remains one of the larger barriers to improved therapies in this area for this age group. Furthermore, GD2 is a rare form of a rare disorder with a short life expectancy, and it is often difficult to recruit an adequate number of patients for clinical trials.
In the past few years, advances in the field of gene therapy have been remarkable, and overall, the number of clinical trials using this modality is rising exponentially. Since GD1 can be reversed by bone marrow transplantation, it is likely that gene therapy will ultimately be successful in patients with GD1. However, currently this is a treatment with significant risks, and overall, the population of patients with non-neuronopathic GD is faring well on currently available treatments. One may predict that in the coming decades, once the details of gene therapy are resolved in other disorders, and the associated risks mitigated, gene therapy may provide patients with GD1 with a full cure. For neuronopathic GD, the new trials administering AAV-gene therapy directly to the cisterna magnum will need to be followed closely. It is yet to be determined whether the damage and neurodegeneration seen in GD2 is fully reversible, or whether the destructive course is already established in utero. There is also the risk that changing a devastating acute disorder into one with chronic neurological damage may not serve this community well. Here, in utero therapy may be the better option. Additionally, an important resource for families with known pathogenic GBA1 variants or families with children who have been diagnosed with GD is genetic counseling to discuss prenatal genetic testing options and, potentially, in utero therapies for future pregnancies.
The potential use of small molecule chaperones is particularly exciting. This strategy may provide an inexpensive and safe treatment that could be available to patients around the world. The fact that many of these molecules are brain-penetrant suggests that this mode may provide a needed therapy for patients with GD3. Since Ambroxol is known to be a safe and widely available drug, it is the low-hanging fruit, yet it is essential to conduct rigorous placebo-controlled clinical trials rather than relying on anecdotal case-based reports. Non-competitive chaperones are also an attractive approach and once optimized, may be easier to dose.
Another pressing issue that needs to be addressed in the field concerns the need for an integrated disease registry that records outcomes for all patients with GD, treated and non-treated, receiving the various forms of therapy. The initial registries have been funded by pharmaceutical companies, and primarily were comprised of patients receiving the manufacturer’s own therapy. While such registries are expensive and challenging to maintain, registries not tied to pharma can ultimately provide essential information regarding disease outcomes relevant to the entire patient community.
There is no doubt that this is an exciting time for the field of GD. The prognosis for children diagnosed in the 21st Century far exceeds previous expectations. The variety of approaches and the renewed energy in the field suggests that the future is bright, and that improved therapeutics are on the horizon.
Article Highlights Box:
During the past decades, efficacious new therapies have been developed for children with Gaucher disease which reverse or prevent many of the non-neurological manifestations.
However, the currently available therapies require life-long administration, are very costly, and do not cross the blood-brain-barrier.
Newborn screening programs, community carrier screening, and an increased awareness of the disorder have contributed to a more frequent identification of Gaucher disease in childhood, sometime before any disease manifestations are apparent.
Children with Gaucher disease should be monitored regularly, and if clinical manifestations develop, should be placed on Enzyme Replacement Therapy.
Glucosylsphingosine (lyso-Gb1) is a useful biomarker for evaluating the need for and response to treatment.
New therapies under development for Gaucher disease, including potentially the neuronopathic forms, are small molecule chaperones, gene therapy, in utero Enzyme Replacement Therapy and gene therapy, nanovesicle enzyme delivery, and brain-penetrant Substrate Reduction Therapy.
Acknowledgments
Funding:
The authors are supported by intramural funding from the National Institutes of Health via the National Human Genome Research Institute.
Abbreviations used:
- GD:
Gaucher disease
- LSD:
lysosomal storage disorder
- GD1:
type 1 Gaucher disease
- GD2:
type 2 Gaucher disease
- GD3:
type 3 Gaucher disease
- GCase:
glucocerebrosidase
- ERT:
enzyme replacement therapy
- SRT:
substrate reduction therapy
- GluCer:
glucosylceramide
- ICGC:
International Collaborative Gaucher Group
- HRQoL:
health-related quality of life
- NBS:
newborn screening
- MS/MS:
tandem mass spectrometry
- lyso-Gb1:
glucosylsphingosine
- LC:
liquid chromatography
- DBS:
dried blood spot
- CHO:
Chinese hamster ovary
- FDA:
Food and Drug Administration
- BBB:
blood-brain barrier
- HDACIs:
histone deacetylase inhibitors
- HSP:
heat shock protein
- IUERT:
in utero ERT
- MPS7:
Mucopolysaccharidosis type VII
- AAV:
adeno-associated virus
- WT:
wildtype
- IFeTIS:
International Fetal Transplantation and Immunology Society
- IUGT:
in utero-gene therapy
- SapC:
saposin C
- DOPS:
dioleoylphosphatidylserine
- iPSC:
induced pluripotent stem cells
Footnotes
Declaration of Interest:
The Sidransky laboratory has received support from F.Hoffman-La Roche Ltd under a Cooperative Research and Development Agreement with the National Human Genome Research Institute. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures:
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Annotated bibliography
(particularly relevant papers are highlighted with 1 or 2 stars)
- [1].Weinreb NJ, Charrow J, Andersson HC. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med. 2002;113:112–119. [DOI] [PubMed] [Google Scholar]
- [2].Seehra G, Solomon B, Ryan E. Five-parameter evaluation of dysphagia: A novel prognostic scale for assessing neurological decline in Gaucher disease type 2. Mol Genet Metab. 2019;127:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Roshan Lal T, Seehra GK, Steward AM, et al. The Natural History of Type 2 Gaucher Disease in the 21st Century: A Retrospective Study. Neurology. 2020; 10.1212/WNL.0000000000010605. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Updated Natural History of type 2 Gaucher disease
- [4].Schiffmann R, Sevigny J, Rolfs A, et al. The definition of neuronopathic Gaucher disease. J Inherit Metab Dis. 2020;43:1056–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Comprehensive and consensus definition of neuronopathic Gaucher disease
- [5].Poffenberger CN, Inati S, Tayebi N, et al. EEG abnormalities in patients with chronic neuronopathic Gaucher disease: A retrospective review. Mol Genet Metab. 2020;131:358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sidransky E Gaucher Disease: Insights from a Rare Mendelian Disorder. Discov Med. 2012;14:273–281. [PMC free article] [PubMed] [Google Scholar]
- [7].Hughes D, Mikosch P, Belmatoug N. Gaucher Disease in Bone: From Pathophysiology to Practice. J Bone Min Res. 2019;34:996–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kauli R, Zaizov R, Lazar L, et al. Delayed growth and puberty in patients with Gaucher disease type 1: natural history and effect of splenectomy and/or enzyme replacement therapy. Isr Med Assoc J. 2000;2:158–163. [PubMed] [Google Scholar]
- [9].Nalysnyk L, Rotella P, Simeone JC, et al. Gaucher disease epidemiology and natural history: a comprehensive review of the literature. Hematology. 2017;22:65–73. [DOI] [PubMed] [Google Scholar]
- [10].Charrow J, Andersson HC, Kaplan P, et al. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. 2000;160:2835–2843. [DOI] [PubMed] [Google Scholar]; * Description of large Gaucher disease patient registry
- [11].Jeong SY, Park SJ, Kim HJ. Clinical and genetic characteristics of Korean patients with Gaucher disease. Blood Cells Mol Dis. 2011;46:11–14. [DOI] [PubMed] [Google Scholar]
- [12].Tylki-Szymanska A, Vellodi A, El-Beshlawy A, et al. Neuronopathic Gaucher disease: demographic and clinical features of 131 patients enrolled in the International Collaborative Gaucher Group Neurological Outcomes Subregistry. J Inherit Metab Dis. 2010;33:339–346. [DOI] [PubMed] [Google Scholar]; * Description of neuropathic Gaucher disease registry
- [13].Barton NW, Brady RO, Dambrosia JM. Replacement therapy for inherited enzyme deficiency–macrophage-targeted glucocerebrosidase for Gaucher’s disease. N Engl J Med. 1991;324:1464–1470. [DOI] [PubMed] [Google Scholar]; * First report of ERT
- [14].Cox T, Lachmann R, Hollak C. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet (London, England; 2000. [DOI] [PubMed] [Google Scholar]; * First report of SRT
- [15].Aflaki E, Westbroek W, Sidransky E. The complicated relationship between Gaucher disease and Parkinsonism: Insights from a rare disease. Neuron. 2017;93:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Review of GBA1-associated Parkinson disease
- [16].Sidransky E, Nalls MA, Aasly JO, et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N Engl J Med. 2009;361:1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nalls MA, Duran R, Lopez G, et al. A Multicenter Study of Glucocerebrosidase Mutations in Dementia With Lewy Bodies. JAMA Neurol. 2013;70:727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Grabowski GA, Petsko GA, Kolodny EH. Gaucher disease. In: Valle D, Beaudet AL, Vogelstein B, et al. , editors. Online Metab Mol Bases Inherit Dis. New York, NY: McGraw Hill; 2011. [Google Scholar]; ** Comprehensive review of GD
- [19].Kaplan P, Andersson HC, Kacena KA, et al. The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Arch Pediatr Adolesc Med. 2006;160:603–608. [DOI] [PubMed] [Google Scholar]; * Summary of children with GD1
- [20].Charrow J, Andersson HC, Kaplan P, et al. Enzyme replacement therapy and monitoring for children with type 1 Gaucher disease: consensus recommendations. J Pediatr. 2004;144:112–120. [DOI] [PubMed] [Google Scholar]
- [21].Weinreb NJ, Deegan P, Kacena KA, et al. Life expectancy in Gaucher disease type 1. Am J Hematol. 2008;83:896–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Maas M, Hangartner T, Mariani G, et al. Recommendations for the assessment and monitoring of skeletal manifestations in children with Gaucher disease. Skelet Radiol. 2008;37:185–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis and assessment. Consens Statements Eur J Pediatr. 2004;163:58–66. [DOI] [PubMed] [Google Scholar]; * Consensus report of neuronopathic GD
- [24].Andrade-Campos M, Alfonso P, Irun P, et al. Diagnosis features of pediatric Gaucher disease patients in the era of enzymatic therapy, a national-base study from the Spanish Registry of Gaucher Disease. Orphanet J Rare Dis. 2017;12:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Katz K, Mechlis-Frish S, Cohen IJ, et al. Bone scans in the diagnosis of bone crisis in patients who have Gaucher disease. J Bone Jt Surg Am. 1991;73:513–517. [PubMed] [Google Scholar]
- [26].Elstein D, Abrahamov A, Dweck A, et al. Gaucher disease: pediatric concerns. Paediatr Drugs. 2002;4:417–426. [DOI] [PubMed] [Google Scholar]
- [27].Brady RO, Barton NW, Grabowski GA. The role of neurogenetics in Gaucher disease. Arch Neurol. 1993;50:1212–1224. [DOI] [PubMed] [Google Scholar]
- [28].Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013;172:447–458. [DOI] [PubMed] [Google Scholar]; ** Management recommendations for children
- [29].Yang AC, Bier L, Overbey JR, et al. Early manifestations of type 1 Gaucher disease in presymptomatic children diagnosed after parental carrier screening. Genet Med. 2017;19:652–658. [DOI] [PubMed] [Google Scholar]; * Children with GD diagnosed through screening programs
- [30].Anderson S Newborn Screening for Lysosomal Storage Disorders. J Pediatr Health Care. 2018;32:285–294. [DOI] [PubMed] [Google Scholar]
- [31].Revel-Vilk S, Szer J, Mehta A, et al. How we manage Gaucher disease in the era of choices. Br J Haematol. 2018;182:467–480. [DOI] [PubMed] [Google Scholar]; * Management approach for GD
- [32].Degnan AJ, Ho-Fung VM, Ahrens-Nicklas RC, et al. Imaging of non-neuronopathic Gaucher disease: recent advances in quantitative imaging and comprehensive assessment of disease involvement. Insights Imaging [Internet]. 2019. [cited 2021 Mar 4];10. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6616606/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Petros M Revisiting the Wilson-Jungner criteria: how can supplemental criteria guide public health in the era of genetic screening? Genet Med. 2012;14:129–134. [DOI] [PubMed] [Google Scholar]
- [34].Therrell BL, Padilla CD, Loeber JG, et al. Current status of newborn screening worldwide: 2015. Semin Perinatol. 2015;39:171–187. [DOI] [PubMed] [Google Scholar]; * An overview of newborn screening for LSDs
- [35].Wasserstein MP, Caggana M, Bailey SM, et al. The New York pilot newborn screening program for lysosomal storage diseases: Report of the First 65,000 Infants. Genet Med. 2019;21:631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Spada M, Pagliardini S, Yasuda M, et al. High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chien YH, Chiang SC, Zhang XK, et al. Early detection of Pompe disease by newborn screening is feasible: results from the Taiwan screening program. Pediatrics. 2008;122:39–45. [DOI] [PubMed] [Google Scholar]
- [38].Liao HC, Chiang CC, Niu DM, et al. Detecting multiple lysosomal storage diseases by tandem mass spectrometry–a national newborn screening program in Taiwan. Clin Chim Acta. 2014;431:80–86. [DOI] [PubMed] [Google Scholar]
- [39].Kemper AR, Hwu WL, Lloyd-Puryear M, et al. Newborn screening for Pompe disease: synthesis of the evidence and development of screening recommendations. Pediatrics. 2007;120:1327–1334. [DOI] [PubMed] [Google Scholar]
- [40].Sanders KA, Gavrilov DK, Oglesbee D, et al. A Comparative Effectiveness Study of Newborn Screening Methods for Four Lysosomal Storage Disorders. Int J Neonatal Screen. 2020;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hopkins PV, Campbell C, Klug T, et al. Lysosomal storage disorder screening implementation: findings from the first six months of full population pilot testing in Missouri. J Pediatr. 2015;166:172–177. [DOI] [PubMed] [Google Scholar]
- [42].Burton BK, Charrow J, Hoganson GE, et al. Newborn Screening for Lysosomal Storage Disorders in Illinois: The Initial 15-Month Experience. J Pediatr. 2017;190:130–135. [DOI] [PubMed] [Google Scholar]
- [43].Elliott S, Buroker N, Cournoyer JJ, et al. Pilot study of newborn screening for six lysosomal storage diseases using Tandem Mass Spectrometry. Mol Genet Metab. 2016;118:304–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hopkins PV, Klug T, Vermette L, et al. Incidence of 4 Lysosomal Storage Disorders From 4 Years of Newborn Screening. JAMA Pediatr. 2018;172:696–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lisi EC, McCandless SE. Newborn Screening for Lysosomal Storage Disorders: Views of Genetic Healthcare Providers. J Genet Couns. 2016;25:373–384. [DOI] [PubMed] [Google Scholar]
- [46].Elstein D, Abrahamov A, Itzchaki M, et al. Commentary: low-dose high-frequency enzyme replacement therapy prevents fractures without complete suppression of painful bone crises in patients with severe juvenile onset type I Gaucher disease. Blood Cells Mol Dis. 1998;24:303–305306–308. [DOI] [PubMed] [Google Scholar]
- [47].Bembi B, Ciana G, Mengel E, et al. Bone complications in children with Gaucher disease. Br J Radiol. 2002;75Suppl 1:37–44. [DOI] [PubMed] [Google Scholar]
- [48].Kaplan P, Mazur A, Manor O, et al. Acceleration of retarded growth in children with Gaucher disease after treatment with alglucerase. J Pediatr. 1996;129:149–153. [DOI] [PubMed] [Google Scholar]
- [49].Zevin S, Abrahamov A, Hadas-Halpern I, et al. Adult-type Gaucher disease in children: genetics, clinical features and enzyme replacement therapy. Q J Med. 1993;86:565–573. [PubMed] [Google Scholar]
- [50].Damiano AM, Pastores GM, Ware JE. The health-related quality of life of adults with Gaucher’s disease receiving enzyme replacement therapy: results from a retrospective study. Qual Life Res. 1998;7:373–386. [DOI] [PubMed] [Google Scholar]
- [51].Wilson AC, Palermo TM. Physical activity and function in adolescents with chronic pain: a controlled study using actigraphy. J Pain. 2012;13:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Alioto AG, Gomez R, Moses J, et al. Quality of life and psychological functioning of pediatric and young adult patients with Gaucher disease, type 1. Am J Med Genet A. 2020;182:1130–1142. [DOI] [PubMed] [Google Scholar]
- [53].Remor E, Baldellou A. Health-related quality of life in children and adolescents living with Gaucher disease and their parents. Health Psychol Behav Med. 2018;6:79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kallish S, Kaplan P. A disease severity scoring system for children with type 1 Gaucher disease. Eur J Pediatr. 2013;172:39–43. [DOI] [PubMed] [Google Scholar]
- [55].Davies EH, Mengel E, Tylki-Szymanska A, et al. Four-year follow-up of chronic neuronopathic Gaucher disease in Europeans using a modified severity scoring tool. J Inherit Metab Dis. 2011;34:1053–1059. [DOI] [PubMed] [Google Scholar]
- [56].Revel-Vilk S, Fuller M, Zimran A. Value of glucosylsphingosine (Lyso-Gb1) as a biomarker in Gaucher disease: A systematic literature review. Int J Mol Sci. 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Use of LysoGB1 as a disease biomarker-review
- [57].Raskovalova T, Deegan PB, Mistry PK, et al. Accuracy of chitotriosidase activity and CCL18 concentration in assessing type I Gaucher disease severity. A systematic review with meta-analysis of individual participant data. Haematologica [Internet]. 2020. [cited 2021 Jan 2]; Available from: https://haematologica.org/article/view/9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Boot RG, Renkema GH, Verhoek M, et al. The human chitotriosidase gene. Nature of inherited enzyme deficiency. J Biol Chem. 1998;273:25680–25685. [DOI] [PubMed] [Google Scholar]
- [59].Aerts JMFG, Kallemeijn WW, Wegdam W, et al. Biomarkers in the diagnosis of lysosomal storage disorders: proteins, lipids, and inhibodies. J Inherit Metab Dis. 2011;34:605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Orvisky E, Park JK, LaMarca ME, et al. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: correlation with phenotype and genotype. Mol Genet Metab. 2002;76:262–270. [DOI] [PubMed] [Google Scholar]
- [61].Hurvitz N, Dinur T, Cohen MB, et al. Glucosylsphingosine (Lyso-gb1) as a biomarker for monitoring treated and untreated children with gaucher disease. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Cozma C, Cullufi P, Kramp G, et al. Treatment Efficiency in Gaucher Patients Can Reliably Be Monitored by Quantification of Lyso-Gb1 Concentrations in Dried Blood Spots. Int J Mol Sci [Internet]. 2020. [cited 2021 Jan 2];21. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7369829/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Menkovic I, Boutin M, Alayoubi A, et al. Identification of a Reliable Biomarker Profile for the Diagnosis of Gaucher Disease Type 1 Patients Using a Mass Spectrometry-Based Metabolomic Approach. Int J Mol Sci [Internet]. 2020. [cited 2021 Jan 2];21. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7660648/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tang C, Jia X, Tang F, et al. Detection of glucosylsphingosine in dried blood spots for diagnosis of Gaucher disease by LC-MS/MS. Clin Biochem. 2021;87:79–84. [DOI] [PubMed] [Google Scholar]
- [65].Mistry PK, Lopez G, Schiffmann R, et al. Gaucher Disease: Progress and Ongoing Challenges. Mol Genet Metab. 2017;120:8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Current management challenges in GD
- [66].Krasnewich D, Dietrich K, Bauer L, et al. Splenectomy in Gaucher disease: new management dilemmas. Blood. 1998;91:3085–3087. [PubMed] [Google Scholar]
- [67].Tsai P, Lipton JM, Sahdev I, et al. Allogenic bone marrow transplantation in severe Gaucher disease. Pediatr Res. 1992;31:503–507. [DOI] [PubMed] [Google Scholar]
- [68].Rappeport JM, Ginns EI. Bone-marrow transplantation in severe Gaucher’s disease. N Engl J Med. 1984;311:84–88. [DOI] [PubMed] [Google Scholar]
- [69].Ringdén O, Groth CG, Erikson A, et al. Ten years’ experience of bone marrow transplantation for Gaucher disease. Transplantation. 1995;59:864–870. [PubMed] [Google Scholar]
- [70].Brady RO, Kanfer JN, Shapiro D. Metabolism of glucocerebrosides II. Evidence of an enzymatic deficiency in Gaucher’s disease. Biochem Biophys Res Commun. 1965;18:221–225. [DOI] [PubMed] [Google Scholar]
- [71].Ries M Enzyme replacement therapy and beyond—in memoriam Roscoe O. Brady, M.D. (1923–2016). J Inherit Metab Dis. 2017;40:343–356. [DOI] [PubMed] [Google Scholar]
- [72].Neufeld EF. Enzyme replacement therapy – a brief history. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Dis Perspect 5 Years FOS [Internet]. Oxford: Oxford PharmaGenesis; 2006. [cited 2020 Oct 23]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK11588/. [Google Scholar]
- [73].Burrow TA, Grabowski GA. Velaglucerase alfa in the treatment of Gaucher disease type 1. Clin Investig. 2011;1:285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Zimran A, Brill-Almon E, Chertkoff R, et al. Pivotal trial with plant cell-expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood. 2011;118:5767–5773. [DOI] [PubMed] [Google Scholar]
- [75].Rosenthal DI, Doppelt SH, Mankin HJ, et al. Enzyme replacement therapy for Gaucher disease: skeletal responses to macrophage-targeted glucocerebrosidase. Pediatrics. 1995;96:629–637. [PubMed] [Google Scholar]
- [76].Poll LW, Maas M, Terk MR, et al. Response of Gaucher bone disease to enzyme replacement therapy. Br J Radiol. 2002;75Suppl 1:A25–36. [DOI] [PubMed] [Google Scholar]
- [77].Gupta P, Pastores G. Pharmacological treatment of pediatric Gaucher disease. Expert Rev Clin Pharmacol. 2018;11:1183–1194. [DOI] [PubMed] [Google Scholar]
- [78].Stone DL, Ginns EI, Krasnewich D, et al. Life-threatening splenic hemorrhage in two patients with Gaucher disease. Am J Hematol. 2000;64:140–142. [DOI] [PubMed] [Google Scholar]
- [79].Qiu Z, Zhang B, Song Y, et al. Maintenance Efficacy of Low Dose Imiglucerase for Gaucher Disease [Internet]. PREPRINT; 2020. [cited 2021 Jan 3]. Available from: https://www.researchsquare.com/article/rs-25075/v1. [Google Scholar]
- [80].Potnis KC, Flueckinger LB, Ha CI, et al. Bone manifestations in neuronopathic Gaucher disease while receiving high-dose enzyme replacement therapy. Mol Genet Metab. 2019;126:157–161. [DOI] [PubMed] [Google Scholar]
- [81].Fost M de, Noesel CJM van, Aerts JMFG, et al. Persistent bone disease in adult type 1 Gaucher disease despite increasing doses of enzyme replacement therapy. Haematologica. 2008;93:1119–1120. [DOI] [PubMed] [Google Scholar]
- [82].Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore). 2011;90:52–60. [DOI] [PubMed] [Google Scholar]
- [83].Andrade-Campos M, Escuder-Azuara B, de Frutos LL, et al. Direct and indirect effects of the SARS-CoV-2 pandemic on Gaucher Disease patients in Spain: Time to reconsider home-based therapies? Blood Cells Mol Dis. 2020;85:102478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Mistry P, Balwani M, Barbouth D, et al. Gaucher disease and SARS-CoV-2 infection: Emerging management challenges. Mol Genet Metab. 2020;130:164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Fierro L, Nesheiwat N, Naik H, et al. Gaucher disease and SARS-CoV-2 infection: Experience from 181 patients in New York. Mol Genet Metab [Internet]. 2020. [cited 2021 Jan 3]; Available from: http://www.sciencedirect.com/science/article/pii/S1096719220305576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Zimran A, Goldblatt J, Szer J. Should eliglustat be first line therapy for patients with type 1 Gaucher disease? Definitions of safety and efficacy. Blood Cells Mol Dis. 2018;68:14–16. [DOI] [PubMed] [Google Scholar]
- [87].Goldblatt J, Fletcher JM, McGill J, et al. Enzyme replacement therapy “drug holiday”: results from an unexpected shortage of an orphan drug supply in Australia. Blood Cells Mol Dis. 2011;46:107–110. [DOI] [PubMed] [Google Scholar]
- [88].McCormack PL, Goa KL. Miglustat. Drugs. 2003;63:2427–2434. [DOI] [PubMed] [Google Scholar]
- [89].DailyMed - ZAVESCA- miglustat capsule [Internet]. [cited 2021 Jan 3]. Available from: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=817892d1-ee12-4632-85fc-57ccdf16d7b8.
- [90].Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother. 2013;47:1182–1193. [DOI] [PubMed] [Google Scholar]
- [91].Bennett LL, Turcotte K. Eliglustat tartrate for the treatment of adults with type 1 Gaucher disease. Drug Des Devel Ther. 2015;9:4639–4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Limgala RP, Goker-Alpan O. Effect of Substrate Reduction Therapy in Comparison to Enzyme Replacement Therapy on Immune Aspects and Bone Involvement in Gaucher Disease. Biomolecules [Internet]. 2020. [cited 2021 Jan 3];10. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7226435/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Goker-Alpan O Optimal therapy in Gaucher disease. Ther Clin Risk Manag. 2010;6:315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Balwani M, Burrow TA, Charrow J, et al. Recommendations for the use of eliglustat in the treatment of adults with Gaucher disease type 1 in the United States. Mol Genet Metab. 2016;117:95–103. [DOI] [PubMed] [Google Scholar]
- [95].Sanofi. Open Label, Two Cohort (With and Without Imiglucerase), Multicenter Study to Evaluate Pharmacokinetics, Safety, and Efficacy of Eliglustat in Pediatric Patients With Gaucher Disease Type 1 and Type 3 [Internet]. clinicaltrials.gov; 2020. [cited 2020 Dec 30]. Report No.: NCT03485677. Available from: https://clinicaltrials.gov/ct2/show/NCT03485677.; * Clinical trial of Eliglustat in Pediatric Patients
- [96].Zavesca (miglustat) 100 mg hard capsules - Summary of Product Characteristics (SmPC) - (emc) [Internet]. [cited 2021 Mar 5]. Available from: https://www.medicines.org.uk/emc/product/39/smpc.
- [97].Genzyme, a Sanofi Company. A 3-part Study to Evaluate the Efficacy and Safety of Venglustat in Combination With Cerezyme in Adult and Pediatric Patients With Gaucher Disease Type 3 (GD3) With Open-label Long-term Treatment [Internet]. clinicaltrials.gov; 2020. [cited 2020 Dec 30]. Report No.: NCT02843035. Available from: https://clinicaltrials.gov/ct2/show/NCT02843035.; * Clinical trial of Venglustat in both adult and pediatric patients
- [98].Schiffmann R, Cox TM, Dedieu J. Venglustat combined with imiglucerase positively affects neurological features and brain connectivity in adults with Gaucher disease type 3. Abstract - 17th Annual WORLDSymposium. Molecular Genetics and Metabolism 2021;132;S95. [Google Scholar]
- [99].Jung O, Patnaik S, Marugan J, et al. Progress and potential of non-inhibitory small molecule chaperones for the treatment of Gaucher disease and its implications for Parkinson disease. Expert Rev Proteomics. 2016;13:471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]; * An overview of non-inhibitory small molecule chaperones
- [100].Maegawa GHB, Tropak MB, Buttner JD, et al. Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J Biol Chem. 2009;284:23502–23516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Shaare Zedek Medical Center. Ambroxol Therapy for Patients With Type 1 Gaucher Disease and Suboptimal Response to Enzyme Replacement Therapy [Internet]. clinicaltrials.gov; 2019. [cited 2020 Dec 30]. Report No.: NCT03950050. Available from: https://clinicaltrials.gov/ct2/show/NCT03950050.
- [102].Mullin S, Smith L, Lee K, et al. Ambroxol for the Treatment of Patients With Parkinson Disease With and Without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol. 2020;77:427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Kim Y-M, Yum M-S, Heo SH, et al. Pharmacologic properties of high-dose ambroxol in four patients with Gaucher disease and myoclonic epilepsy. J Med Genet. 2020;57:124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Narita A, Shirai K, Itamura S, et al. Ambroxol chaperone therapy for neuronopathic Gaucher disease: A pilot study. Ann Clin Transl Neurol. 2016;3:200–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Jiang W, Yi M, Maegawa GHB, et al. Ambroxol improves skeletal and hematological manifestations on a child with Gaucher disease. J Hum Genet. 2020;65:345–349. [DOI] [PubMed] [Google Scholar]
- [106].Charkhand B, Scantlebury MH, Narita A, et al. Effect of Ambroxol chaperone therapy on Glucosylsphingosine (Lyso-Gb1) levels in two Canadian patients with type 3 Gaucher disease. Mol Genet Metab Rep. 2019;20:100476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Chu S-Y, Chien C-C, Hwu W-L, et al. Early initiation of high-dose oral ambroxol in combination with enzyme replacement therapy in a neuropathic Gaucher infant. Blood Cells Mol Dis. 2020;81:102402. [DOI] [PubMed] [Google Scholar]
- [108].Ciana G, Dardis A, Pavan E, et al. In vitro and in vivo effects of Ambroxol chaperone therapy in two Italian patients affected by neuronopathic Gaucher disease and epilepsy. Mol Genet Metab Rep. 2020;25:100678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Pawlinski L, Krawczyk M, Fiema M, et al. Dual-action ambroxol in treatment of chronic pain in Gaucher Disease. Eur J Pain. 2020;24:992–996. [DOI] [PubMed] [Google Scholar]
- [110].Steet RA, Chung S, Wustman B, et al. The iminosugar isofagomine increases the activity of N370S mutant acid β-glucosidase in Gaucher fibroblasts by several mechanisms. Proc Natl Acad Sci. 2006;103:13813–13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Amicus Therapeutics Announces Preliminary Results of Phase 2 Study with Plicera(TM) for Gaucher Disease | Amicus Therapeutics [Internet]. [cited 2021 Jan 3]. Available from: https://ir.amicusrx.com/news-releases/news-release-details/amicus-therapeutics-announces-preliminary-results-phase-2-study.
- [112].Yang C, Rahimpour S, Lu J, et al. Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones. Proc Natl Acad Sci. 2013;110:966–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Goldin E, Zheng W, Motabar O, et al. High throughput screening for small molecule therapy for Gaucher disease using patient tissue as the source of mutant glucocerebrosidase. PLOS ONE. 2012;7:e29861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Aflaki E, Stubblefield BK, Maniwang E, et al. Macrophage models of Gaucher disease for evaluating disease pathogenesis and candidate drugs. Sci Transl Med. 2014;6:240ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Aflaki E, Borger DK, Moaven N, et al. A New Glucocerebrosidase chaperone reduces α-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and parkinsonism. J Neurosci. 2016;36:7441–7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Burbulla LF, Jeon S, Zheng J, et al. A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson’s disease. Sci Transl Med. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Zheng J, Chen L, Skinner OS, et al. β-Glucocerebrosidase modulators promote dimerization of β-Glucocerebrosidase and reveal an allosteric binding site. J Am Chem Soc. 2018;140:5914–5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Fog-Tonnesen CK, Zago P, Malini E, et al. Arimoclomol as a potential therapy for neuronopathic Gaucher Disease. bioRxiv. 2018;281824. [Google Scholar]
- [119].Goasdoué K, Miller SM, Colditz PB, et al. Review: The blood-brain barrier; protecting the developing fetal brain. Placenta. 2017;54:111–116. [DOI] [PubMed] [Google Scholar]
- [120].Nguyen Q-H, Witt RG, Wang B, et al. Tolerance induction and microglial engraftment after fetal therapy without conditioning in mice with mucopolysaccharidosis type VII. Sci Transl Med [Internet]. 2020. [cited 2021 Jan 3];12. Available from: https://stm.sciencemag.org/content/12/532/eaay8980. [DOI] [PubMed] [Google Scholar]
- [121].UCSF. UCSF Mucopolysaccharidosis Trial: In Utero Enzyme Replacement Therapy for Lysosomal Storage Diseases [Internet]. [cited 2021 Jan 3]. Available from: https://clinicaltrials.ucsf.edu/trial/NCT04532047. [Google Scholar]; * Clinical trial of IUERT
- [122].Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4:346–358. [DOI] [PubMed] [Google Scholar]
- [123].Nakai H, Storm TA, Kay MA. Increasing the size of rAAV-mediated expression cassettes in vivo by intermolecular joining of two complementary vectors. Nat Biotechnol. 2000;18:527–532. [DOI] [PubMed] [Google Scholar]
- [124].Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18:358–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Massaro G, Mattar CNZ, Wong AMS, et al. Fetal gene therapy for neurodegenerative disease of infants. Nat Med. 2018;24:1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Prevail Therapeutics. An Open-label, Phase 1/2 Study to Evaluate the Safety and Efficacy of single-dose PR001A in infants with Type 2 Gaucher disease [Internet]. clinicaltrials.gov; 2020. [cited 2020 Dec 30]. Report No.: NCT04411654. Available from: https://clinicaltrials.gov/ct2/show/NCT04411654.; * Clinical trial testing AAV Gene therapy for GD2
- [127].AvroBio. An Adaptive, Open-Label, Multinational Phase 1/2 Study Of The Safety and Efficacy of Ex Vivo, Lentiviral Vector-Mediated Gene Therapy AVR-RD-02 for Subjects With Type 1 Gaucher Disease [Internet]. clinicaltrials.gov; 2020. [cited 2020 Jun 1]. Report No.: NCT04145037. Available from: https://clinicaltrials.gov/ct2/show/NCT04145037.; * Clinical trial testing Lentiviral Gene Therapy for GD1
- [128].Miranda CJ, Canavese M, Chisari E, et al. Liver-Directed AAV Gene Therapy for Gaucher Disease. Blood. 2019;134:3354–3354. [Google Scholar]
- [129].Sun Y, Liou B, Chu Z, et al. Systemic enzyme delivery by blood-brain barrier-penetrating SapC-DOPS nanovesicles for treatment of neuronopathic Gaucher disease. EBioMedicine. 2020;55:102735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Research C for DE and. Pediatric Rare Diseases--A Collaborative Approach for Drug Development Using Gaucher Disease as a Model; Draft Guidance for Industry [Internet]. US Food Drug Adm. FDA; 2020. [cited 2021 Jan 3]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/pediatric-rare-diseases-collaborative-approach-drug-development-using-gaucher-disease-model-draft.
- [131].SJY MD, JVAMP FRCP, editors. Neonatal and Pediatric Pharmacology: Therapeutic Principles in Practice. Philadelphia; 2010. [Google Scholar]
- [132].Leeder JS. Translating pharmacogenetics and pharmacogenomics into drug development for clinical pediatrics and beyond. Drug Discov Today. 2004;9:567–573. [DOI] [PubMed] [Google Scholar]
- [133].Almeida-Porada G, Waddington SN, Chan JKY, et al. In Utero Gene Therapy Consensus Statement from the IFeTIS. Mol Ther J Am Soc Gene Ther. 2019;27:705–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Botkin JR, Belmont JW, Berg JS, et al. Points to Consider: Ethical, Legal, and Psychosocial Implications of Genetic Testing in Children and Adolescents. Am J Hum Genet. 2015;97:6–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Buckland KF, Bobby Gaspar H. Gene and cell therapy for children — New medicines, new challenges? Adv Drug Deliv Rev. 2014;73:162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]